
Quantification of labile and stable non-polar arsenolipids in commercial fish meals and edible seaweed samples†
Ásta H.
Pétursdóttir
 *ab,
Jessica
Rodrigues de Jesus
b,
Helga
Gunnlaugsdóttir
a and
Jörg
Feldmann
*b
*ab,
Jessica
Rodrigues de Jesus
b,
Helga
Gunnlaugsdóttir
a and
Jörg
Feldmann
*b
aMatís, Research and Innovation Department, Vinlandsleid 12, 113 Reykjavik, Iceland. E-mail: astap@matis.is
bTESLA-Trace Element Speciation Laboratory, Department of Chemistry, University of Aberdeen, Aberdeen, AB24 3UE, Scotland, UK. E-mail: j.feldmann@abdn.ac.uk
First published on 17th November 2017
Abstract
This study aims at fractionation of arsenic according to its polarity into water-soluble arsenic fractions, polar and non-polar arsenolipids in herring, capelin and blue whiting fish meal and edible seaweed dulse. Changing the sequential extraction order showed a significant labile fraction of the non-polar arsenolipids (AsLps) where species transformation is considered a more likely explanation than a partitioning problem in the compounds. The majority of non-polar AsLps were not stable through water extraction for three types of fish meal (71–93% for herring, capelin and blue whiting). The non-polar AsLp fraction was minor for dulse. In 27 samples of herring and blue whiting fish meal, arsenic was mainly present in the water phase: 71% (2.8 ± 0.8 mg kg−1) and 93% (17.2 ± 1.9 mg kg−1) for herring and blue whiting on average, respectively. The polar arsenolipids in the MeOH/DCM fraction accounted for 15% and 5% (0.5–1.2 mg kg−1 As) for both herring and blue whiting, respectively. Speciation analysis of arsenolipids was undertaken for herring meal, capelin meal and dulse (red seaweed) using simultaneous HPLC-ICPMS/ESIMS for quantification and identification. Among the known arsenohydrocarbons (AsHCs), arseno fatty acids (AsFAs) and arsenosugarphospholipids (AsPLs), a novel AsFA374 was identified in dulse by arsenic detection via simultaneous protonated mass, accurate mass as well as MSMS fragmentation. Additionally, recently reported AsLp groups, arsenic containing phosphatidylcholines (AsPCs) and arseno fatty alcohols (TMAsFOHs), have been reconfirmed to occur in marine samples.
Introduction
The presence of arsenolipids has been known for decades where pioneer research on arsenic in marine food suggested that arsenolipids in cod liver oil and herring oil showed structural similarities to known phospholipids.1 Following this, the interest was sparse, partly due to analytical challenges in identifying arsenolipids. New and more sophisticated analytical techniques have refuelled the interest in arsenolipids over the recent years.2 Three main groups of arsenolipids (AsLps) have been identified, all of which consist of a dimethylarsinoyl group attached to different lipids: arsenohydrocarbons (AsHCs), arseno fatty acids (AsFAs) and arsenosugarphospholipids (AsPLs). New groups of AsLps have additionally been identified; these include arseno fatty alcohols (TMAsFOHs),3 arsenic containing phosphatidylcholines (AsPCs) and arsenic containing phosphatidylethanolamines (AsPEs).4 The AsPCs and AsPEs consist of a dimethylarsinoyl group similar to the three main groups but TMAsFOHs differ since arsenic is found as a trimethylarsenio group, Fig. 1. Methyl esters of known arsenic-fatty acids have been identified as well, but they appear to be artefacts from silica/methanol chromatography.5 The AsLps are often grouped into categories of polar and non-polar AsLps,5,6 and so far, more focus has been on polar AsLps. Different extraction methods have been used for the determination of AsLps, including partitioning between heptane and MeOH/H2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1);7,8 sequential extraction of hexane followed by MeOH/DCM,9–11 acetone and MeOH/H2O;12 and sequential extraction of MeOH/chloroform, subsequently partitioned between H2O and MeOH/chloroform and then the AsLps further partitioned between hexane and MeOH.13 Further sample clean up and purification includes partitioning between several solvents of increasing polarity using silica columns.6,11,14 With a wide choice of extractions, the order of the extraction can have an effect on the stability of AsLps in the biological sample, since sample preparation can interfere with the identification of AsLps, presumably due to their stability towards hydrolysis reactions.5,11
:1);7,8 sequential extraction of hexane followed by MeOH/DCM,9–11 acetone and MeOH/H2O;12 and sequential extraction of MeOH/chloroform, subsequently partitioned between H2O and MeOH/chloroform and then the AsLps further partitioned between hexane and MeOH.13 Further sample clean up and purification includes partitioning between several solvents of increasing polarity using silica columns.6,11,14 With a wide choice of extractions, the order of the extraction can have an effect on the stability of AsLps in the biological sample, since sample preparation can interfere with the identification of AsLps, presumably due to their stability towards hydrolysis reactions.5,11
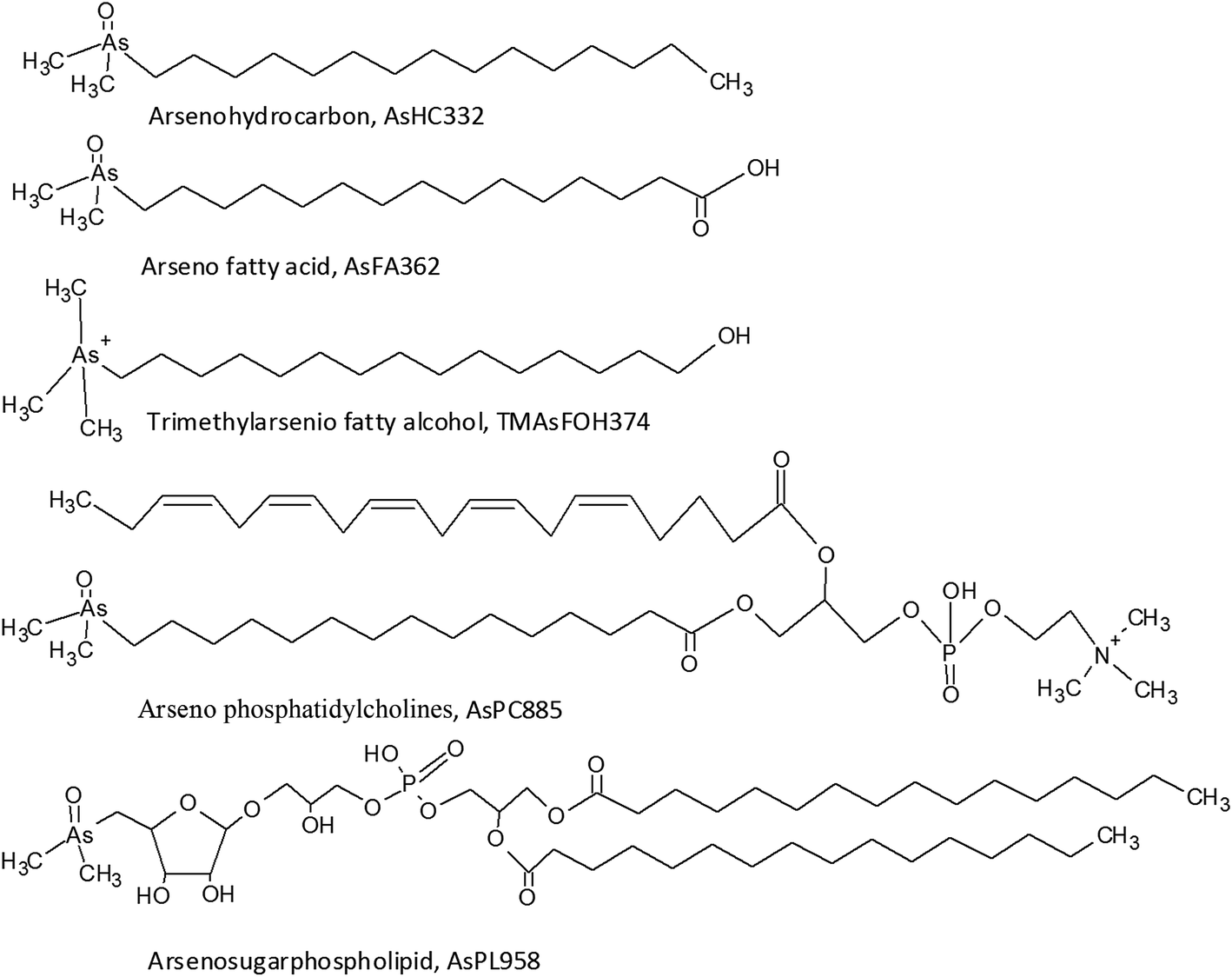 | ||
| Fig. 1 Examples of structures and proposed structures of the main groups of arsenolipids. | ||
AsLps have been identified in a range of seafood samples, including different fish oils,15–18 capelin fish meal,6 herring fillet and roe4,12 and different types of brown algae.9,10,19 Meyer et al. showed that AsHCs can exert similar toxicity as iAs in vitro in cell lines20 and showed in vivo toxicity in fruit flies and crossed their blood–brain barriers,21,22 whereas AsFAs showed much lower toxicity than AsHCs in vitro.23 Among AsHCs, the toxicity depended on the structure of the AsHC.20 Since AsHCs are often found to be the dominant AsLps, especially in fish and fish oil, there is pressure for obtaining more information on AsLps. The objective of this paper is to investigate the AsLp profile in different samples of marine origin that are often used as feed: i.e., fish meal and seaweed to investigate whether the sequence of extraction procedure would alter the AsLp profile and give indication of the robustness of a commonly used sample preparation method towards AsLp species integrity. Two sequential extraction schemes were investigated to identify labile AsLp species in contrast to partitioning problems.
Experimental procedures
Chemical and standards
All chemicals used were of analytical grade or better unless otherwise stated and double distilled water (>18 Mohm cm) was used for all sample preparation. Formic acid was supplied by Sigma-Aldrich (U.K.). Sodium dimethylarsinic acid (98% DMA) used for quantification was obtained from ChemService (U.S.A). For calibration of total As, a 1002 mg As per L certified As stock solution (as H3AsO4 in 0.5 M HNO3) was supplied by Merck (U.K.). Hexane, hydrogen peroxide (H2O2, 30%, laboratory reagent grade LR), potassium hydroxide (KOH, LR), dichloromethane (DCM), nitric acid (HNO3 69%), hydrochloric acid (HCl) and methanol (MeOH) were supplied by Fisher Scientific. Germanium used as internal standard for speciation was obtained from Aldrich Chemical Company and rhodium used as an internal standard for total As from Specpure, Alfa Aesar, (Germany).Samples
The samples included 14 herring (Clupea harengus) fish meal samples, 13 blue whiting (Micromesistius poutassou) fish meal samples, and 1 capelin (Mallotus villosus) fish meal sample from industrial partners in Iceland. Three dulse (Palmaria palmata) red seaweed samples were collected at two locations in Iceland, SW1–3.Sample preparation
:2), and the first extract was left to stand overnight. Both supernatants were then combined and evaporated to dryness (polar lipid fraction). The residue was digested for total arsenic (using 1 mL HNO3 and 2 mL H2O2), Fig. 2.
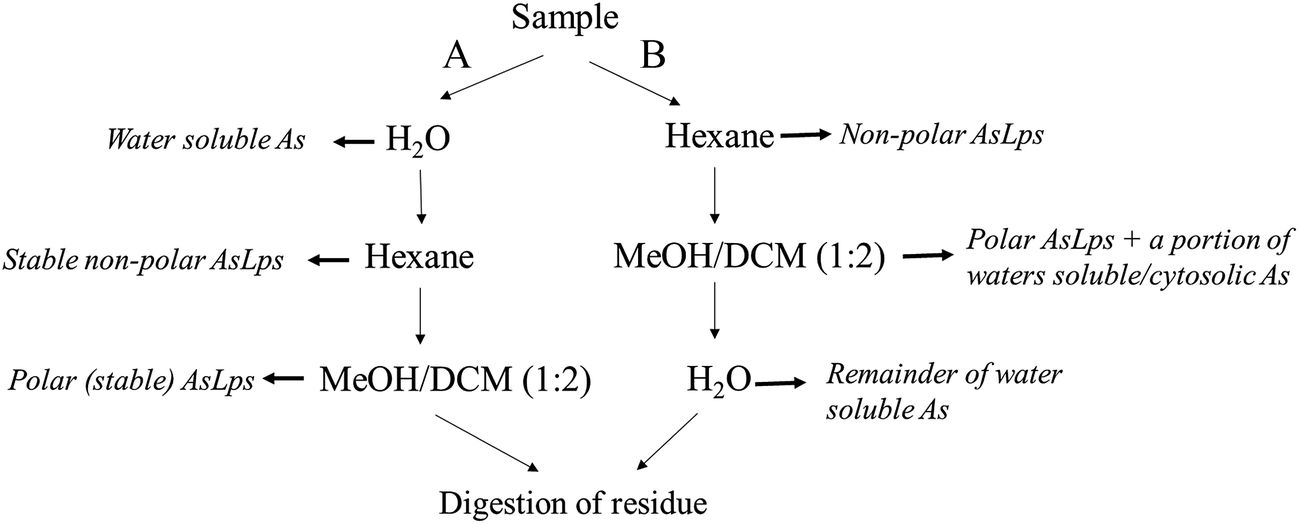 | ||
| Fig. 2 Flow diagram showing the order of extraction solvent for method A and B and how that relates to different fractions. | ||
Extraction method B was the same as A, apart from the order of extraction: hexane followed by MeOH/DCM (1:2) then water extraction and then finally the residue digested for totAs, Fig. 2.
Instrumental set-up
An Agilent 7500c was used for total arsenic determination; the ICP-MS was optimized for optimal sensitivity and stability on As on a day-to-day basis. In addition to m/z 75 for As and m/z 103 for Rh, the possible chloride interference (40Ar35Cl+) on m/z 75 was checked on m/z 77 (40Ar37Cl+ or 77Se) and on m/z 82 (82Se). An external As(V) calibration was used for the quantification of total arsenic.HPLC-ICPMS/ESI-MS was used for the speciation of AsLps. A reverse-phase column (Agilent Eclipse XDB-C18; 4.6 mm × 150 mm) with a gradient of water and methanol both in 0.1% formic acid was used for the speciation of AsLps. Gradient program: water ramping to 100% MeOH for 20 (fish meal) or 25 (seaweed) min, held at 100% MeOH for 22 min, followed by 8 min column equilibration time. The eluent was split post-column: 25% to high-resolution (HR) ICPMS (Element 2, Thermo Scientific) and 75% to ESI-MS (LTQ Orbitrap Discovery, Thermo Scientific). HR-ICPMS was used in organic mode (1570 W), in medium resolution, with platinum cones and 20% oxygen. The signal was optimized at m/z 75 to give a maximum response for As and monitored masses were m/z 31 (P), m/z 32 (S), m/z 74 (Ge) and m/z 75 (As). A response factor (100 μg As per L as DMA in 10 μg Ge per L) was used to compensate for changes in the response because of varying carbon content of the gradient, as previously described,6 making it possible to both quantify and identify species without species-specific standards. DMA was used as external standard for the quantification of arsenic species and Ge m/z 74 (10 μg L−1) was used as internal standard. The ESI-MS was operated in positive mode with a scan range of m/z 100–1800. Other parameters for the Orbitrap were 4.5 kV spray voltage, 35% normalized collision energy, 320 °C capillary temperature and 42 V capillary voltage.
Results and discussion
Quality control (QC)
Two CRMs were digested and measured for totAs concentration: Lobster Hepatopancreas Reference Material for Trace Metals TORT-2 (n = 8) and brown seaweed IAEA140 (n = 3). The measured concentrations for TORT-2 and IAEA140 were 22.1 ± 0.7 mg kg−1 compared to 21.6 ± 1.8 mg kg−1 and 42.0 ± 0.2 mg kg−1 compared to 44.3 ± 2.1 mg kg−1, respectively. The TORT-2 underwent sequential extraction method A, same as the samples, Table 1, where the mass balance added up to 83% of the certified value (18.08 ± 0.02 mg kg−1 compared to 21.6 ± 1.8 mg kg−1). The certified reference material SPS-WW2 (waste water, Spectrapure standards, Norway) was measured with each batch of fish meal sample for totAs in order to monitor the performance of the ICP-MS on a day-to-day basis, with totAs of 0.54 ± 0.03 mg kg−1 (n = 33) compared to certified 0.500 ± 0.003 mg kg−1. Additionally, a herring sample (Herr1) was included with every batch of samples to act as an in-house reference and to monitor the uncertainty, which ranged from 3 to 25% RSD, while that for hexane method A and residue method B were higher, 35% and 51%, respectively. These two fractions had the lowest concentrations of total arsenic, Table 1. A capelin fish meal sample previously analysed in the lab and reported6 was included for AsLp speciation for comparison, since CRMs certified for AsLps were not available. LOD and LOQ were calculated as 3× and 10× of SD of blank samples, respectively, where an average dilution factor was used to convert to concentration in sample.| n | Hexane | MeOH/DCM | H2O | Residue | Sum | |
|---|---|---|---|---|---|---|
| a ND – not detected. LOD – 0.003 mg kg−1, LOQ – 0.009 mg kg−1. | ||||||
| Herr1-A | 15 | 0.17 ± 0.06 | 0.75 ± 0.13 | 2.6 ± 0.2 | 0.31 ± 0.04 | 3.8 ± 0.2 |
| Herr1-B | 9 | 0.59 ± 0.02 | 2.33 ± 0.09 | 0.31 ± 0.07 | 0.18 ± 0.09 | 3.4 ± 0.2 |
| Cap1-A | 1 | 0.06 | 0.49 | 3.1 | 0.32 | 3.9 |
| Cap1-B | 1 | 0.40 | 2.4 | 0.28 | 0.37 | 3.6 |
| BW2-A | 2 | 0.017 ± 0.0001 | 0.86 ± 0.01 | 17.48 ± 0.02 | 0.37 ± 0.03 | 18.72 ± 0.01 |
| BW2-B | 2 | 0.23 ± 0.03 | 13.3 ± 0.4 | 1.65 ± 0.06 | <LOQ | 15.3 ± 1.4 |
| SW1-A | 3 | <LOQ | 0.12 ± 0.02 | 7.7 ± 0.2 | — | 7.8 ± 0.2 |
| SW1-B | 3 | <LOQ | 1.3 ± 0.2 | 6.62 ± 0.05 | — | 7.9 ± 0.2 |
| TORT-2-A | 3 | ND | 0.48 ± 0.07 | 16.3 ± 0.1 | 1.35 ± 0.07 | 18.08 ± 0.02 |
Sequential extraction
Two different sequential extractions were performed on 4 different matrices, herring fish meal, capelin fish meal, blue whiting fish meal and a dulse seaweed (Iceland), each step for measuring total arsenic concentration. Certified reference TORT-2 was also included. The sequential extractions differed in the order of solvent used, Fig. 2. Total arsenic in fish meal samples and seaweed can be found in ESI Table 1.†From Table 1, it can be seen that the order of extraction solvents has a major influence on the concentration of each fraction. A notable difference is that a large portion of the arsenic in the water-soluble fraction is extracted into the MeOH/DCM fraction. For fish meal, this includes arsenobetaine (AB); this was tested by a spiking experiment where spiked AB was found in the MeOH/DCM fraction (not shown). The seaweed samples do not contain any AB, but it appears that some water-soluble arsenic species (∼1 mg kg−1), presumably arsenosugars, are partitioned into the MeOH/DCM fraction.
An interesting trend is noticeable for the hexane fraction. When water is used for the extraction before hexane, a lower concentration of As is found in the hexane fraction for all fish meal types. This was a repeatable pattern, ESI Table 2.† Due to the very different polarities of water and hexane, this cannot be attributed to a partitioning problem of the compounds, but rather to a species transformation. Therefore, the difference between the hexane phases in the two extraction methods is referred to here as labile non-polar lipids. There is a major difference for the fish meal samples where the labile part of the hexane fraction ranges from 71 to 93% for herring and blue whiting, respectively, Fig. 3. Hence, almost all AsLps in the non-polar fraction are not stable when water extraction is applied as the first step. This reduction of arsenic in the hexane fraction following water extraction can result from the hydrolysis of non-polar AsLps. Hydrolysis of the labile AsLp short- and medium-chain fatty acids have been identified previously in fish oils and have suggested that these are hydrolysis products of their triglycerides.11
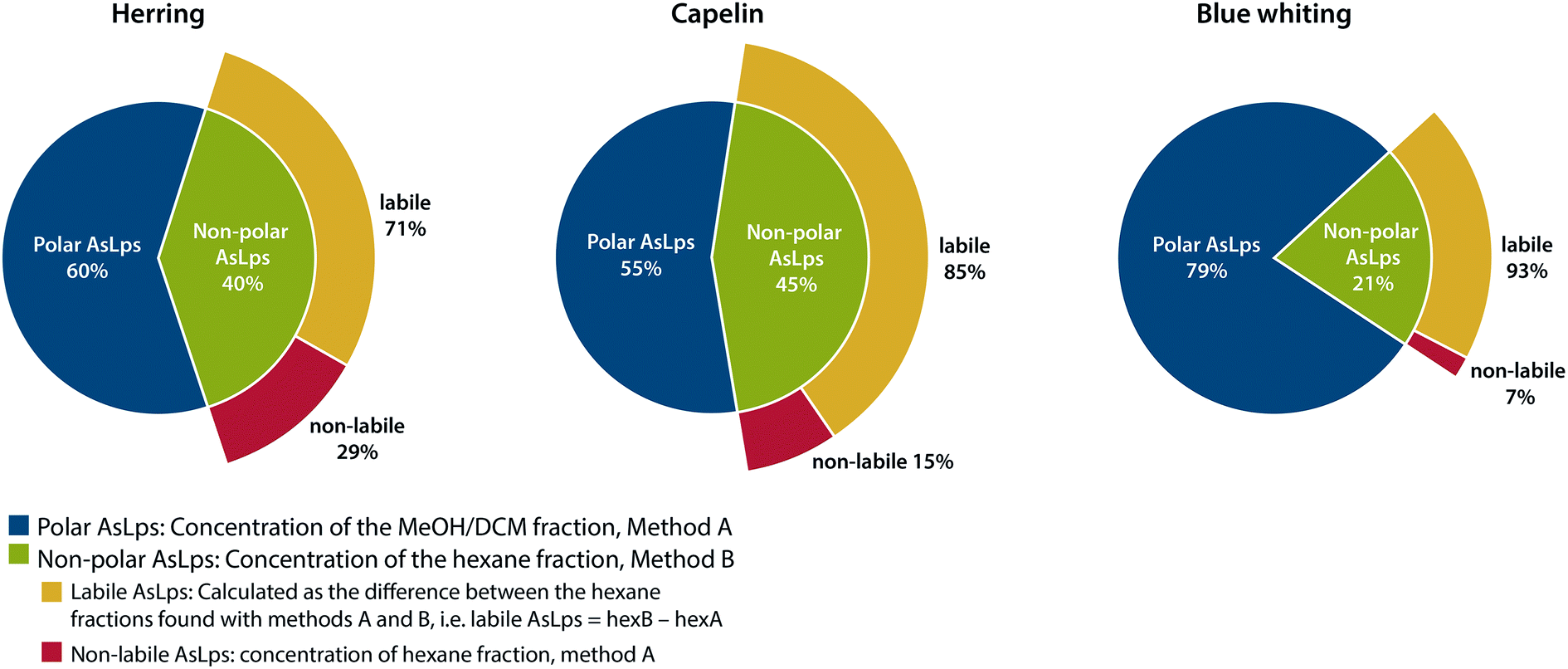 | ||
| Fig. 3 Representation of distribution of total arsenic between fractions containing polar AsLps and non-polar AsLps sub categorised into labile and non-labile AsLps. | ||
If the purpose of sequential extraction is to obtain a profile of arsenolipids using totAs, both sequences have their advantages and drawbacks. Method A, with water as the first extractant, results in an underestimation of AsLps in hexane fraction (non-polar labile lipid fraction). However, with regard to food or feed, the unstable AsLps would be in contact with water during digestion and therefore become hydrolysed when consumed. Method B, with hexane as the first extractant followed by MeOH/DCM, gives a better idea of the amount of non-polar AsLps (hexane fraction) but gives no information on the amount of AsLps in the MeOH/DCM fraction (polar lipid fraction) since the amount of water-soluble arsenic also partitioned into this relatively polar fraction by far exceeds the AsLp fraction. Both methods are needed to quantify the labile portion of the non-polar AsLps, Fig. 3.
Here, method A was used to obtain an overview of 14 samples of herring (fatty fish) and 13 samples of blue whiting (lean fish), Fig. 4. Total arsenic concentrations of the different extraction steps of all 27 fish meal samples are presented in ESI Table 1.†
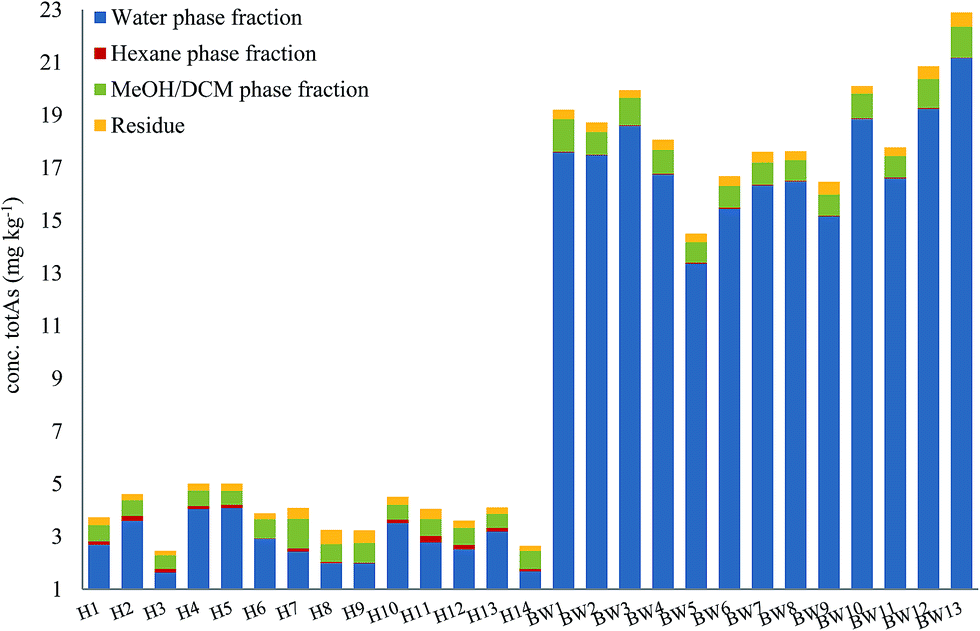 | ||
| Fig. 4 Distribution of arsenic in different fractions following sequential extraction method A (water–hexane–MeOH/DCM–residue). Graph shows an average of 2 replicates for each fraction. | ||
For both herring and blue whiting, the majority of arsenic was found in the water fraction, accounting for on average 71% and 93% of the mass balance for herring and blue whiting, respectively, Fig. 4. The hexane extraction represented a minor fraction (stable non-polar AsLps) and was barely noticeable for blue whiting (3% for herring, 0.2% for blue whiting on average). The MeOH/DCM fraction accounted for 18% and 5% on average for herring and blue whiting, respectively. For all samples, there was unaccountable arsenic found in the residue with higher percentage for herring, but similar concentration to that for blue whiting (8% for herring, 2% for blue whiting), Fig. 4.
Identification of AsLps in fish meal
To obtain structural information, capelin and herring fish meal samples were prepared with sequential extraction method B for the identification of AsLps, Table 2. Extraction method B was chosen to reduce the chances of hydrolysis of the samples.| Peak | Short name | Mol formula MH+ | Hexane | MeOH/DCM | Calc. MH+ | ||
|---|---|---|---|---|---|---|---|
| Capelin | Herring | Capelin | Herring | ||||
| Δm/z (ppm) | Δm/z (ppm) | Δm/z (ppm) | Δm/z (ppm) | ||||
| B | AsFA334 | C15H32O3As | −2.81 | −0.91 | 335.1567 | ||
| C | AsFA348 | C16H34O3As | 2.27 | 349.1724 | |||
| D | AsFA362 | C17H36O3As | −1.03 | −0.13 | 4.98 | 363.1880 | |
| F1 | AsFA436 | C23H38O3As | −0.12 | 0.16 | −0.72 | 437.2037 | |
| F3 | AsFA448 | C24H38O3As | 0.11 | 0.16 | −1.25 | 449.2037 | |
| F2 | AsFA390 | C19H40O3As | −0.53 | −1.01 | 391.2193 | ||
| H2 | AsHC404 | C23H38OAs | −2.57 | 0.44 | −2.47 | 0.78 | 405.2139 |
| I1 & I2 | AsHC332 | C17H38OAs | −2.17 | 0.26 | −2.25 | 0.68 | 333.2139 |
| H1 | TMAsFOH418 | C24H40OAs | 0.73 | 419.2295 | |||
| J2 | AsPC885 | C45H82O9NPAs | 0.62 | 886.4943 | |||
| J1 | AsHC360 | C19H42OAs | −0.97 | 0.79 | −0.43 | 0.86 | 361.2452 |
| J3 | TMAsFOH374 | C20H44OAs | −0.36 | 375.2608 | |||
| K1 | AsPC939 | C49H88O9NPAs | 1.68 | 940.5413 | |||
| K2 | AsPC985 | C53H86O9NPAs | 0.27 | 986.5256 | |||
| K3 | AsPC997 | C54H86O9NPAs | 0.50 | 998.5256 | |||
| L | AsPE1035 | C57H88O9NPAs | 0.64 | 1036.5413 | |||
Amayo et al.6 reported the identification of AsLps (3 AsHCs and 3 AsFAs) of a hexane extract (in MeOH) of capelin fish meal. The same AsHCs (AsHC404, AsHC332, AsHC360) were identified in our study in herring and capelin meal in the hexane fraction (in MeOH), but the AsFAs (AsFA363, AsFA448, AsFA436) were only found for herring, Table 2 and ESI Fig. 2.† The sample preparation carried out by Amayo et al. was on a larger scale than in this work (50 g fish meal compared to 1 g); hence, they could potentially identify species at lower concentrations for capelin. There is a much more diverse profile of AsLps found in the MeOH/DCM fraction, Table 2, although the six AsLps identified in the herring hexane fraction represent the major species for both capelin and herring, Fig. 5a and ESI Fig. 3.† This was reported previously by Taleshi et al.5 where the hexane layer (in MeOH) contained the same arsenicals as the methanol/ethanol layer but in lower concentrations.
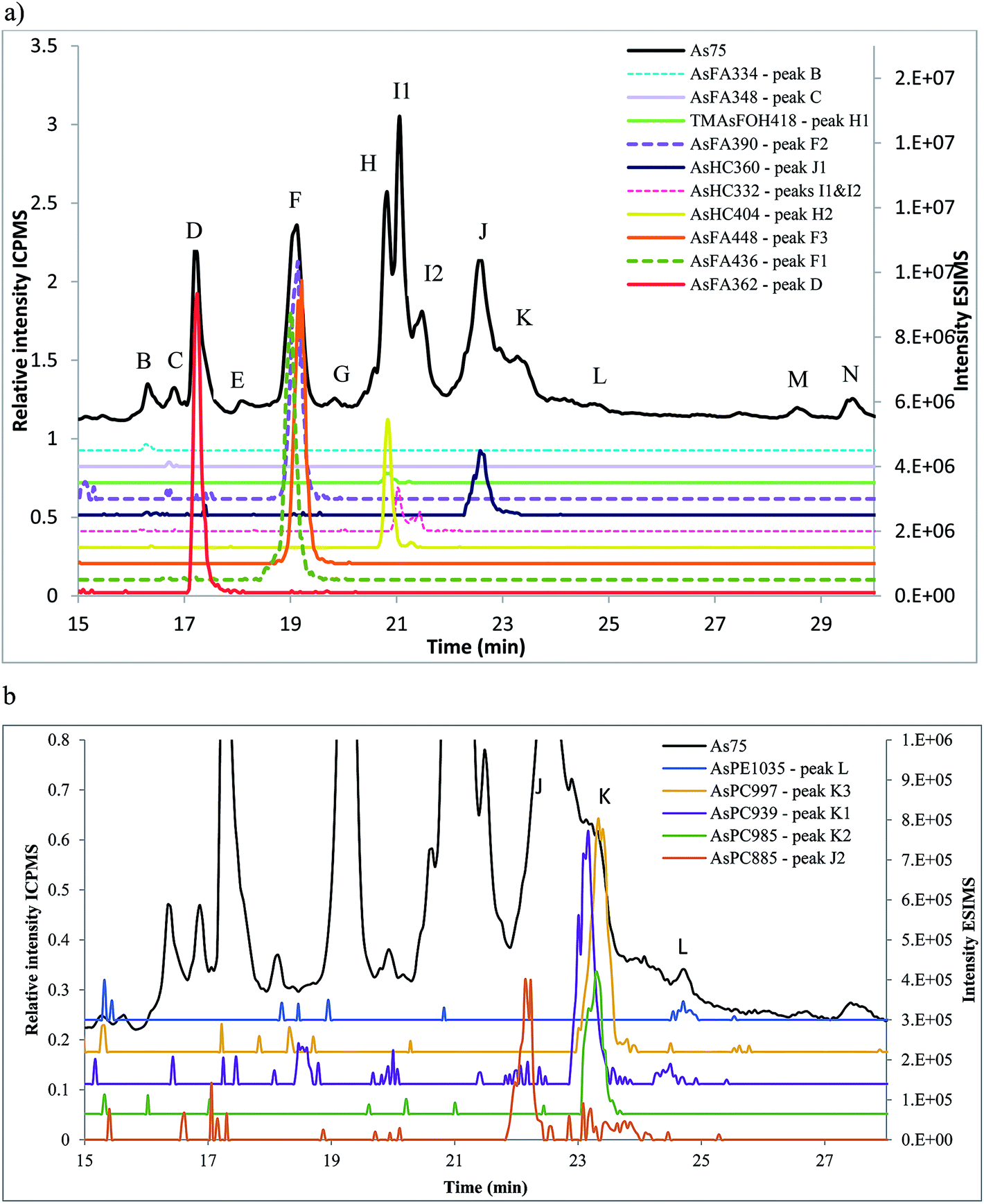 | ||
| Fig. 5 Herring fish meal (Herr1) sample analysed on Agilent Eclipse XDB-C18 column, ramping from 0 to 100% MeOH in 0.1% formic acid. Peak letters refer to the As75 signal (ICPMS, black) and numbers added for co-eluting compounds (ESIMS). (a) Signal from the ICPMS and identified AsFAs, AsHCs and TMAsFOHs by ESIMS. (b) magnified view of (a) of the signal from the ICPMS overlaid with additional ESIMS data of identified AsPCs and AsPEs. | ||
AsFA362 is a saturated AsFA, but shows a branched behaviour for capelin, indicating that different isomers exist (Fig. 5 and ESI Fig. 3†). In addition to the AsFAs and AsHCs, other classes of AsLps are identified, i.e. TMAsFOHs and AsPCs, Fig. 5. MS/MS data of m/z 375 for capelin shows a similar pattern as reported by Amayo et al., with fragments 313 and 339 indicating a Me3As+ moiety instead of the typical AsHC pattern (ESI-Fig. 4a†). Additionally, by looking at retention times, TMAsFOH374 elutes with the same retention time as AsHC360, ESI Fig. 3a,† as reported by Amayo et al.3 for capelin. Since both compounds are saturated, it is likely that because of the difference in chain length, these compounds would have slightly different elution times if they were both AsHCs. TMAsFOH374 is found based on this identification. The compound at m/z 419 in herring shows the same elution pattern as TMAsFOH418 reported by Amayo et al.,3i.e. on elution with AsHC404, Fig. 5a. MS/MS data for m/z 419 neither show typical AsHC patterns nor the same fragments as TMAsFOH374. The compound is tentatively identified as TMAsFOH418.
AsPCs is a new group of AsLps recently identified in samples of herring caviar due to their rich source of cell membrane compounds.4 Four of the five AsPCs previously identified were found as minor traces (accurate mass only) in the herring fish meal samples, along with AsPE1035, Fig. 5. In this work, the AsPCs were not concentrated enough or the collision energy was too high to have MS/MS spectra for fragmentation patterns. AsPCs and AsHC eluted close together and two heavier AsPCs eluted as a shoulder on the AsHC360 with the AsPE just shortly afterwards, Fig. 5b. These compounds were not present in the capelin sample. The four AsFAs (AsFA362, AsFA390, AsFA436, AsFA448) contained in the AsPCs were also identified unbound in the herring meal as well as in capelin.
Quantification of AsLps in fish meal
Chromatographic recovery was quantitative for the hexane fraction: 76% and 113% for herring and capelin, respectively. The column recovery was just over 200% for the MeOH/DCM fraction for both capelin and herring. It is possible that carbon based compounds co-eluting with the void volume peak enhanced the m/z 75 signal, i.e. carbon-enhancement. In addition, the peak of water-soluble arsenicals in the void was around 7 times over the calibration curve; hence, the quantification was only an estimate for this peak. The calibration curve was tailored to fit the species of interest, AsLps, where it could be noted that when the sum of AsLp species, Table 3, are compared to the total arsenic of the MeOH/DCM fraction measured with method A, Table 1, they are in good agreement: 120% and 96% for capelin and herring, respectively. The use of a response factor appears to be a suitable option for the quantification of AsLps.| Peaks | Hexane | MeOH/DCM | ||
|---|---|---|---|---|
| Capelin | Herring | Capelin | Herring | |
| a Branched behaviour. b LOD – 0.002 mg kg−1, LOQ – 0.008 mg kg−1. | ||||
| A | 0.029 | 0.024 ± 0.006 | 4.7b ± 0.6 | 4.1 ± 0.1b |
| B | — | — | 0.008 | 0.022 ± 0.002 |
| C | — | — | <LOQ | 0.012 ± 0.001 |
| D | — | <LOQ | 0.014 (D1)a | 0.079 ± 0.001 |
| 0.016 (D2)a | ||||
| E | <LOQ | — | — | <LOQ |
| F | — | 0.009 ± 0.005 | 0.045 | 0.136 ± 0.004 |
| G | — | <LOQ | — | 0.018 ± 0.002 |
| H | 0.30 | 0.22 ± 0.03 | 0.12 | 0.093 ± 0.007 |
| I | — | 0.05 ± 0.01 | 0.26 | 0.15 ± 0.02 (I1)a |
| 0.06 ± 0.02 (I2)a | ||||
| J | 0.11 | 0.13 ± 0.01 | 0.095 | 0.178 ± 0.002 |
| K | — | — | — | 0.051 ± 0.003 |
| L | — | — | — | <LOQ |
| M | <LOQ | — | <LOQ | 0.011 ± 0.001 |
| N | <LOD | <LOQ | 0.010 | 0.019 ± 0.002 |
| O | <LOQ | <LOQ | <LOQ | — |
| Sum | 0.45 | 0.45 ± 0.04 | 0.59 | 0.85 ± 0.02 |
Most of the non-polar arsenic appears to be accounted for as only 5–6% of the non-polar arsenic fraction is found in the void, Table 3. Since the same AsLps are found in both the hexane and MeOH/DCM fractions, it might seem that this is due to partitioning of the species. However, taking into consideration that the majority of the non-polar AsLps of the fish meal appear to be labile arsenicals (Fig. 3), this is unlikely. Possible candidates could be long-chain alkylated arsenoesters. Arsenoesters hydrolyse within minutes in the presence of water, although the half-life increases with chain length.24 AsFAs might be broken down products of triglycerides.11 Their occurrence in the hexane phase supports this assumption. On the other hand, it is difficult to imagine AsHCs as degradation products due to the lack of a carboxylic acid group in this species.
Identification and quantification of lipids in MeOH/DCM fraction in seaweed
Three dulse samples from Iceland (SW1–3) were measured (dulse is commonly eaten in European nations, such as Iceland and Ireland). There was an insignificant amount of AsLps in the hexane fraction of SW1 and only a low concentration in the MeOH/DCM fraction, Table 1. There appears to be a minor locational difference for the dulse samples, since the two samples collected from location 1 were similar to each other and different to that collected from location 2, Fig. 6a.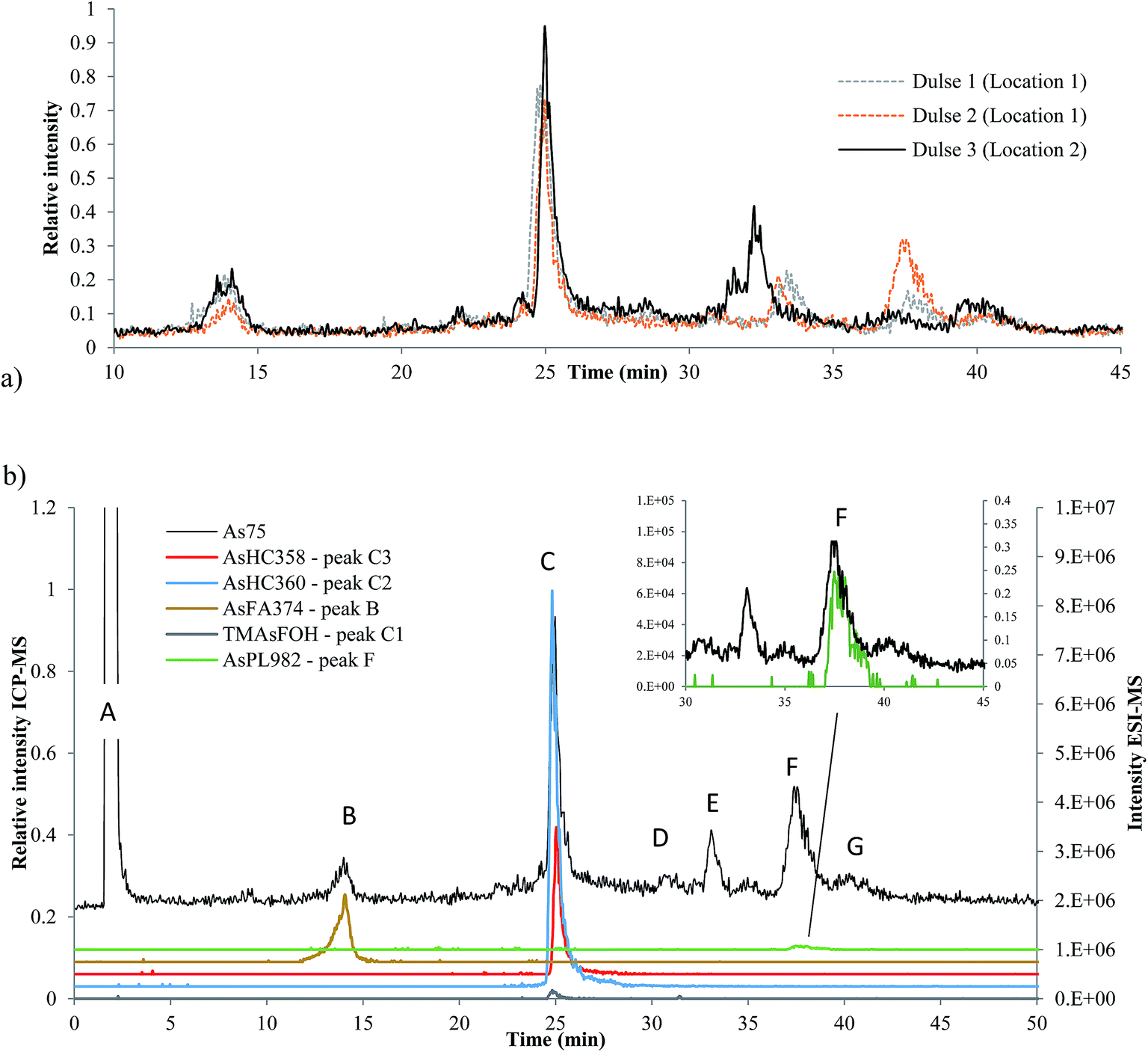 | ||
| Fig. 6 Seaweed analysed on Agilent Eclipse XDB-C18 column, ramping from 0 to 100% MeOH in 0.1% formic acid. (a) Three different dulse samples from Iceland; ICP-MS signal (b) identified peaks in dulse (SW2). Black signal ICP-MS, coloured ESI-MS. | ||
Although the column recovery for the seaweed samples was only around 60%, the sum of AsLps for SW1, Table 4, was in good agreement with totAs concentration of the MeOH/DCM fraction (method A), Table 1. The main compounds in all three Icelandic dulse samples were AsHCs (AsHC360 and AsHC358); minor peaks are attributed to compounds including AsFA and also AsPLs, Table 4. There appears to be TMAsFOH species present in the samples (peak C1) as it is more likely to be TMAsFOH374 than AsHC374 because of retention times, but the species was too low in concentration for MS/MS spectra. Typical fragmentation patterns were noted with MS/MS for the two AsHCs, ESI Table 5.†
| Short name (peak letter) | Formula (M + H) | Peak number | SW1 | SW2 | SW3 | |||
|---|---|---|---|---|---|---|---|---|
| Conc. (mg kg−1) | Δm/z (ppm) | Conc. (mg kg−1) | Δm/z (ppm) | Conc. (mg kg−1) | Δm/z (ppm) | |||
| a LOD – 0.003 mg kg−1, LOQ – 0.010 mg kg−1. | ||||||||
| A | 0.49 | 0.55 | 1.3 | |||||
| AsFA374 | C18H36O3As | B | 0.015 | −0.01 | <LOQ | −0.17 | 0.017 | 0.07 |
| TMAsFOH374 | C20H44OAs | C1 | 0.052 | 0.79 | 0.048 | 2.23 | 0.049 | 0.04 |
| AsHC360 | C19H42OAs | C2 | 1.73 | 2.04 | 1.15 | |||
| AsHC358 | C19H40OAs | C3 | 4.28 | 1.99 | 1.41 | |||
| D | <LOQ | 0.011 | ||||||
| E | 0.015 | 0.013 | 0.032 | |||||
| AsPL982 | C47H89O14AsP | F | 0.011 | 1.12 | 0.032 | 0.66 | 0.010 | |
| AsPL958 | C45H89O14AsP | G | <LOQ | −0.43 | <LOQ | 0.015 | ||
| Sum AsLps: | 0.10 | 0.12 | 0.15 | |||||
So far, AsFA have not been frequently identified in seaweed samples. Raab et al.10 reported AsFAs in brown alga (Saccharina latissima), AsFA422 and AsFA424. As reported by Garcia-Salgado et al.,19 Hijiki also had early eluting peaks of about 5% of the totAsLps content,19 potentially AsFAs. AsFA374 has not been reported before in the literature, but the signal from the ESI-MS corresponds with the early eluting peak B at 14 min, Fig. 6b. MS/MS data for AsFA374 were C4H8As, C5H10As, C6H12As and AsHC330 (ESI Table 5 and ESI Fig. 4†).
Conclusions
When using sequential extraction, there is a significant difference in the concentration of As in each fraction depending on the order of the extraction solvents. Extracting AsLps using MeOH/DCM before water extraction results in the extraction of AB and possibly other cytosolic as species. Using water first may, however, significantly transform labile non-polar AsLps, where the labile part of the hexane fraction ranges from 71 to 93% for the three fish meal types and nearly all AsLps (93%) in the blue whiting non-polar fraction are not stable through a water extraction. Sequential extractions give an idea of the distribution of As between water-soluble arsenicals and polar and non-polar AsLps without using speciation. Sequential extraction shows that the majority of As is present as water-soluble arsenic species and a significant portion of AsLps is found in the polar MeOH/DCM fraction for both herring and blue whiting or between 0.5 and 1.2 mg kg−1 As. For the herring samples, this polar AsLp fraction is in a higher proportion, since they have much lower totAs than the blue whiting samples, reflecting their fattier content compared to that of blue whiting. Sequential extraction, and therefore fractionation, can however not replace full identification using HPLC-ICPMS/ESIMS, since fractionation is somewhat arbitrary. Here sequential extraction is also used to observe the arsenolipid profile in selected samples by speciation using HPLC-ICPMS/ESIMS. The most prominent AsLp species identified in the polar fraction of the herring and capelin fish meal is AsHCs with small amounts of AsFAs identified, and the newly discovered AsPCs and AsPEs appear to be present in the herring fish meal. This work is the second to identify TMAsFOHs, AsPCs and AsPE, finding similar results as those reported in the original works.3,4 AsHCs are found in a significant portion of AsLps in both fish meal and seaweed.This is of particular interest since recent research has shown that the cytotoxicity of AsHCs is of similar magnitude as iAs.20 The need for more data on AsLps is evident, and simple methods for the determination of AsLps are of great importance to accomplish this task.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Á. Pétursdóttir thanks the Icelandic Research Fund, the Icelandic Research Fund for Graduate Students, the SORSAS Award and The College of Physical Sciences at Aberdeen University for financial support. J. Rodrigues thanks the ERASMUS Programme. The Sildarvinnslan hf (SVN) and Vinnslustodin hf (VSV) are acknowledged for the fish meal samples and Finnbogi Gudmundsson for the dulse samples. Dr Andrea Raab and Dr Dagmar Urgast are both kindly thanked for their help.References
- G. Lunde, J. Am. Oil Chem. Soc., 1968, 45, 331–332 CrossRef CAS PubMed.
- V. Sele, J. J. Sloth, A. K. Lundebye, E. H. Larsen, M. H. G. Berntssen and H. Amlund, Food Chem., 2012, 133, 618–630 CrossRef CAS.
- K. O. Amayo, A. Raab, E. M. Krupp, H. Gunnlaugsdottir and J. Feldmann, Anal. Chem., 2013, 85, 9321–9327 CrossRef CAS PubMed.
- S. A. Viczek, K. B. Jensen and K. A. Francesconi, Angew. Chem., Int. Ed., 2016, 55, 5259–5262 CrossRef CAS PubMed.
- M. S. Taleshi, G. Raber, J. S. Edmonds, K. B. Jensen and K. A. Francesconi, Sci. Rep., 2014, 4, 7492 CrossRef PubMed.
- K. O. Amayo, A. Petursdottir, C. Newcombe, H. Gunnlaugsdottir, A. Raab, E. M. Krupp and J. Feldmann, Anal. Chem., 2011, 83, 3589–3595 CrossRef CAS PubMed.
- G. Raber, S. Khoomrung, M. S. Taleshi, J. S. Edmonds and K. A. Francesconi, Talanta, 2009, 78, 1215–1218 CrossRef CAS PubMed.
- V. Sele, J. J. Sloth, B. Holmelid, S. Valdersnes, K. Skov and H. Amlund, Talanta, 2014, 121, 89–96 CrossRef CAS PubMed.
- Á. H. Pétursdóttir, K. Fletcher, H. Gunnlaugsdóttir, E. Krupp, F. C. Küpper and J. Feldmann, Environ. Chem., 2016, 13, 21–33 CrossRef.
- A. Raab, C. Newcombe, D. Pitton, R. Ebel and J. Feldmann, Anal. Chem., 2013, 85, 2817–2824 CrossRef CAS PubMed.
- E. R. Pereira, J. F. Kopp, A. Raab, E. M. Krupp, J. D. Menoyo, E. Carasek, B. Welz and J. Feldmann, J. Anal. At. Spectrom., 2016, 31, 1836–1845 RSC.
- S. Lischka, U. Arroyo-Abad, J. Mattusch, A. Kuhn and C. Piechotta, Talanta, 2013, 110, 144–152 CrossRef CAS PubMed.
- M. S. Taleshi, J. S. Edmonds, W. Goessler, M. J. Ruiz-Chancho, G. Raber, K. B. Jensen and K. A. Francesconi, Environ. Sci. Technol., 2010, 44, 1478–1483 CrossRef CAS PubMed.
- U. Arroyo-Abad, J. Mattusch, T. Reemtsma and C. Piechotta, Eur. J. Lipid Sci. Technol., 2014, 116, 1381–1387 CrossRef CAS.
- K. O. Amayo, A. Raab, E. M. Krupp and J. Feldmann, Talanta, 2014, 118, 217–223 CrossRef CAS PubMed.
- M. S. Taleshi, K. B. Jensen, G. Raber, J. S. Edmonds, H. Gunnlaugsdottir and K. A. Francesconi, Chem. Commun., 2008, 4706–4707, 10.1039/b808049f.
- M. J. Ruiz-Chancho, M. S. Taleshi, W. Goessler and K. A. Francesconi, J. Anal. At. Spectrom., 2012, 27, 501–504 RSC.
- V. Sele, H. Amlund, M. H. G. Berntssen, J. A. Berntsen, K. Skov and J. J. Sloth, Anal. Bioanal. Chem., 2013, 405, 5179–5190 CrossRef CAS PubMed.
- S. Garcia-Salgado, G. Raber, R. Raml, C. Magnes and K. A. Francesconi, Environ. Chem., 2012, 9, 63–66 CrossRef CAS.
- S. Meyer, M. Matissek, S. M. Muller, M. S. Taleshi, F. Ebert, K. A. Francesconi and T. Schwerdtle, Metallomics, 2014, 6, 1023–1033 RSC.
- S. Meyer, J. Schulz, A. Jeibmann, M. S. Taleshi, F. Ebert, K. A. Francesconi and T. Schwerdtle, Metallomics, 2014, 6, 2010–2014 RSC.
- A. C. Niehoff, J. Schulz, J. Soltwisch, S. Meyer, H. Kettling, M. Sperling, A. Jeibmann, K. Dreisewerd, K. A. Francesconi, T. Schwerdtle and U. Karst, Anal. Chem., 2016, 88, 5258–5263 CrossRef CAS PubMed.
- S. Meyer, G. Raber, F. Ebert, L. Leffers, S. M. Muller, M. S. Taleshi, K. A. Francesconi and T. Schwerdtle, Toxicol. Res., 2015, 4, 1289–1296 RSC.
- F. H. Westheimer, Science, 1987, 235, 1173–1178 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ja00333a |
| This journal is © The Royal Society of Chemistry 2018 |