
Frustrated Lewis pairs incorporating the bifunctional Lewis acid 1,1′-fc{B(C6F5)2}2: reactivity towards small molecules†
Rémi
Tirfoin
 ,
Jessica
Gilbert
,
Michael J.
Kelly
and
Simon
Aldridge
*
,
Jessica
Gilbert
,
Michael J.
Kelly
and
Simon
Aldridge
*
Inorganic Chemistry Laboratory, Department of Chemistry, University of Oxford, South Parks Road, Oxford, UK OX1 3QR. E-mail: simon.aldridge@chem.ox.ac.uk; Fax: +44(0)1865 272690; Tel: +44 (0)1865 285201
First published on 29th December 2017
Abstract
Applications of the bifunctional ferrocenediyl Lewis acid 1,1′-fc{B(C6F5)2}2 in frustrated Lewis pair (FLP) chemistry are described. The coordination (or otherwise) of a range of sterically encumbered C-, N- and P-centred Lewis bases has been investigated, with lutidine, tetramethylpiperidine, PPh3, PtBu3 and the expanded ring carbene 6Dipp being found to be sterically incapable of coordinate bond formation. The chemistry of a range of these FLPs in the presence of H2O, NH3, CO2 and cyclohexylisocyanate (CyNCO) has been investigated, with the patterns of reactivity identified including simple coordination chemistry, E–H bond cleavage and C–B insertion.
Introduction
The exploitation of frustrated Lewis pairs (FLPs) constitutes a significant development in the fields of small molecule activation and catalysis over the past decade.1–3 The cooperative use of sterically unquenched Lewis acid and Lewis basic sites facilitates transformations more typically associated with transition metal complexes.3 Since the first example of their use in the heterolytic cleavage of H2 was described in 2006,2 numerous applications have been reported in the activation of other E–H bonds (e.g. E = B, C, N, O, Si), and in the binding of small molecules, including atmospheric oxides (such as CO, CO2, NO, N2O, and SO2).3 While the Lewis acid component of a large number of these FLP systems is the commercially available perfluoroaryl borane B(C6F5)3, examples featuring other neutral boranes,4 heavier Group 13 analogues,5 borenium cations,6 and Lewis acids from the d-block have also emerged.7FLPs that contain a ferrocenyl reporter unit offer the possibility for inducing a measurable electrochemical and/or colorimetric response on small molecule capture,8,9 and therefore have potential uses in detection. Two approaches in this area are exemplified by the Fe(η5-C5Ph5)(η5-C5H4PtBu2)/B(C6F5)3 system in which the Lewis basic phosphine unit contains a ferrocenyl substituent,10 and by recent work from our laboratory utilizing the ferrocenyl borane Fe(η5-C5H5){η5-C5H4B(C6F5)2} [= FcB(C6F5)2, 1],11 a compound which was previously reported by Piers and co-workers.12a In terms of small molecule capture, the PtBu3/1 FLP binds N2O intact to generate the ambiphilic adduct tBu3P·N2O·1 (Scheme 1); while similar chemistry also occurs with PtBu3/B(C6F5)3, the formation of tBu3P·N2O·1 is signalled by a maroon-to-amber colour change and a shift of 300 mV in the FeII/III redox potential.11,13
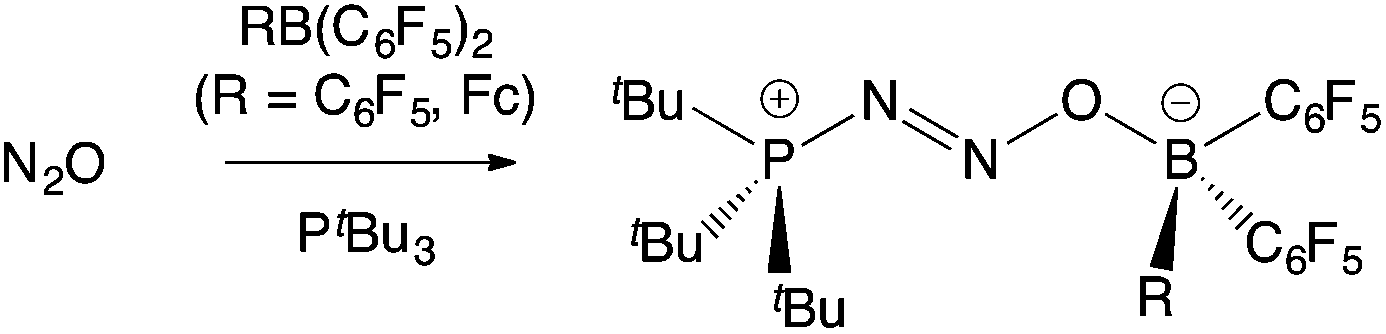 | ||
| Scheme 1 Binding of N2O by FLPs featuring PtBu3 and either B(C6F5)3 or 1.11,13 | ||
Although this N2O detection protocol illustrates the potential of ferrocenyl boranes in FLP chemistry, we wanted to probe the broader scope of this approach, by exploiting other boranes incorporating metallocene scaffolds. In particular, we hypothesized that 1,1′-fc{B(C6F5)2}2 (2) might be of interest, due to (i) its potential to act as a bifunctional (potentially chelating) Lewis acid to trap substrates bearing geometrically divergent Lewis basic sites; and (ii) its stronger Lewis acidity, in comparison to monofunctional 1 (Fig. 1).12 This observation reflects the π electron-withdrawing nature of the additional –B(C6F5)2 group, and the fact that through-space donation from the electron-rich iron centre to the individual pendant borane function(s) is less pronounced when there are two such groups.12
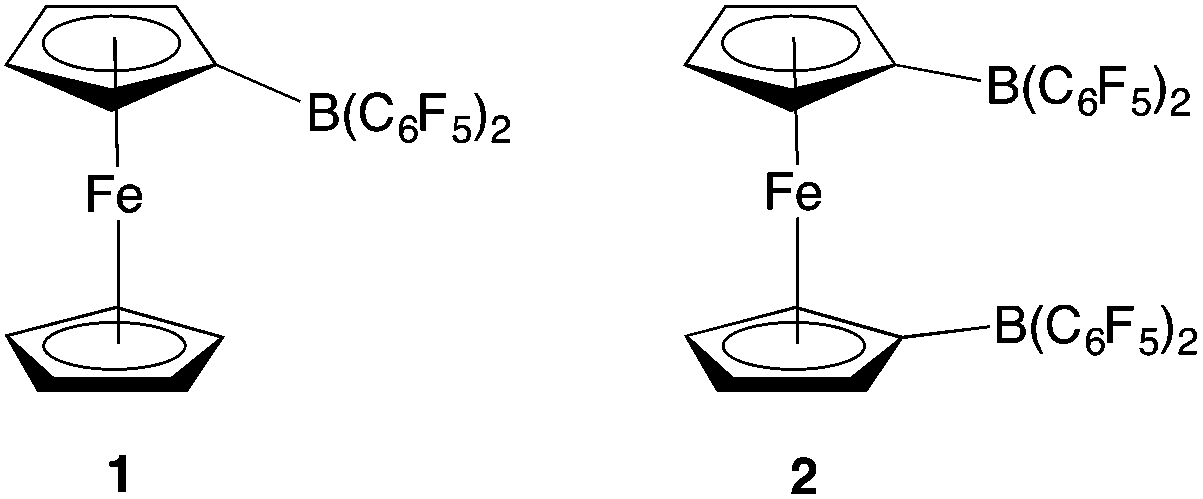 | ||
| Fig. 1 Ferrocenyl and ferrocene-diyl based Lewis acids FcB(C6F5)2 (1) and 1,1′-fc{B(C6F5)2}2 (2) [Fc = Fe(η5-C5H5)(η5-C5H4); 1,1′-fc = Fe(η5-C5H4)2]. | ||
With this in mind, we set out to explore the potential for 2 to act as the Lewis acid component in FLPs featuring a range of C-, N- and P-donors, and subsequently to probe the use of such systems in the activation of small molecules such as H2O, NH3 and CO2. We report the results of these studies in this manuscript.
Results and discussion
Alternative synthesis of 1,1′-fc{B(C6F5)2}2 (2)
Piers and co-workers have reported the synthesis of 2 from 1,1′-fc(HgCl)2 and ClB(C6F5)2 (Scheme 2).12b Generation of the ClB(C6F5)2 precursor involves the synthesis of Me2Sn(C6F5)2 from LiC6F5, and its subsequent reaction with BCl3 at elevated temperature/pressure.12c With this in mind, a more direct synthesis from LiC6F5 was targeted, drawing on the route which we have previously developed to prepare 1 from FcBBr2 (Scheme 3).14 The one-pot literature synthesis of bifunctional 1,1′-fc(BBr2)2 from ferrocene and BBr3,15 is followed by reaction with slightly greater than four equivalents of LiC6F5, generated in situ from nBuLi and C6F5Br. Although LiC6F5 is notoriously explosive above −40 °C,16 the synthetic protocol that we employ means that [as in the synthesis of Me2Sn(C6F5)2] it never warms above −78 °C, and so can be safely and reproducibly used in the synthesis of 2. Following this procedure, 2 can be isolated after extraction into hot hexane with a yield (ca. 50%) broadly comparable to that reported by Piers (up to 40%).12b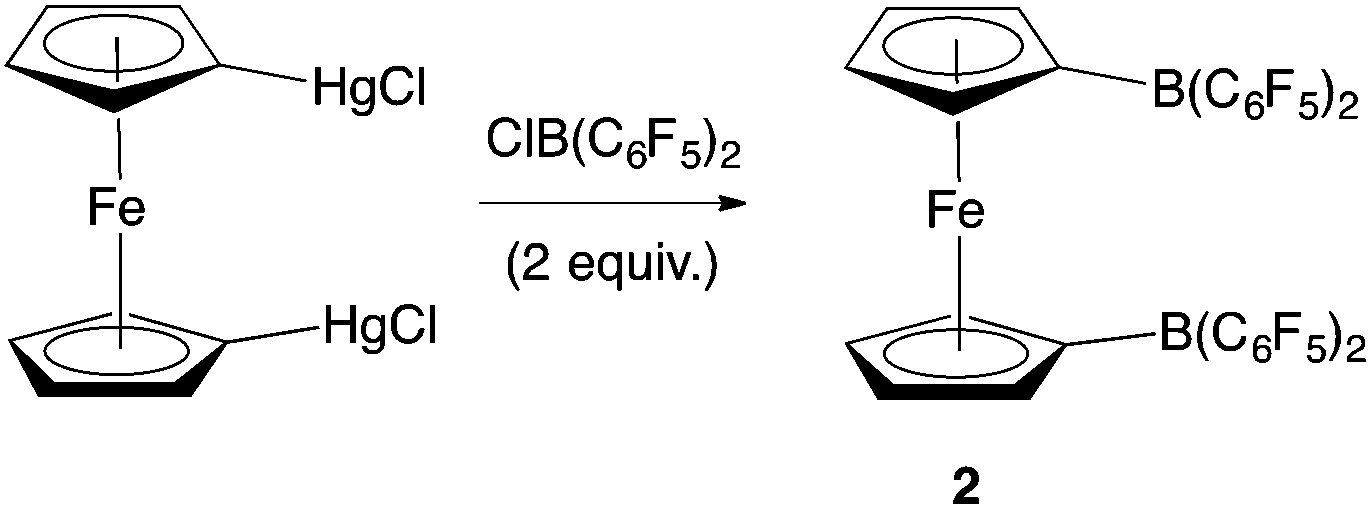 | ||
| Scheme 2 Literature synthesis of 1,1′-fc{B(C6F5)2}2 (2).12b | ||
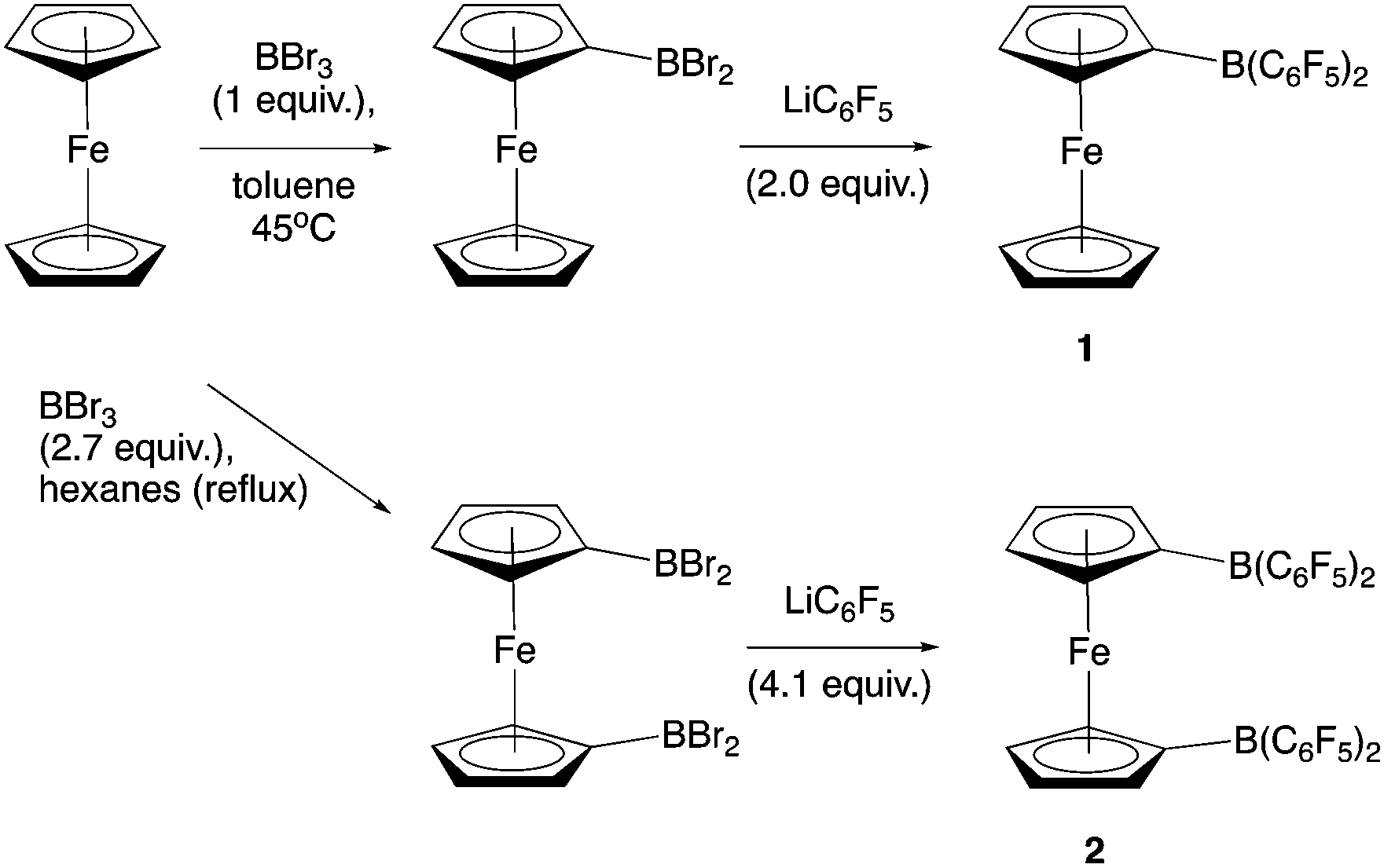 | ||
| Scheme 3 (upper) Synthesis of 1 from FcBBr2, and (lower) modification for the synthesis of 2 from 1,1′-fc(BBr2)2. Caution: The reaction temperature must be kept below −40 °C at all times when handling LiC6F5. | ||
Interaction of Lewis bases with 2
The sterically unencumbered tertiary phosphine Me3P is known to form a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adduct with 2, through the formation of two P→B coordinate bonds.12b The donor/acceptor chemistry of 1 has been explored in greater depth,11a and the combination with the much bulkier PtBu3, for example, is known to constitute an FLP. With this in mind, the interaction of 2 with a range of bulky C-, N- and P-donor Lewis bases was investigated, with the aim of identifying FLP systems for further reactivity studies.
:1 adduct with 2, through the formation of two P→B coordinate bonds.12b The donor/acceptor chemistry of 1 has been explored in greater depth,11a and the combination with the much bulkier PtBu3, for example, is known to constitute an FLP. With this in mind, the interaction of 2 with a range of bulky C-, N- and P-donor Lewis bases was investigated, with the aim of identifying FLP systems for further reactivity studies.
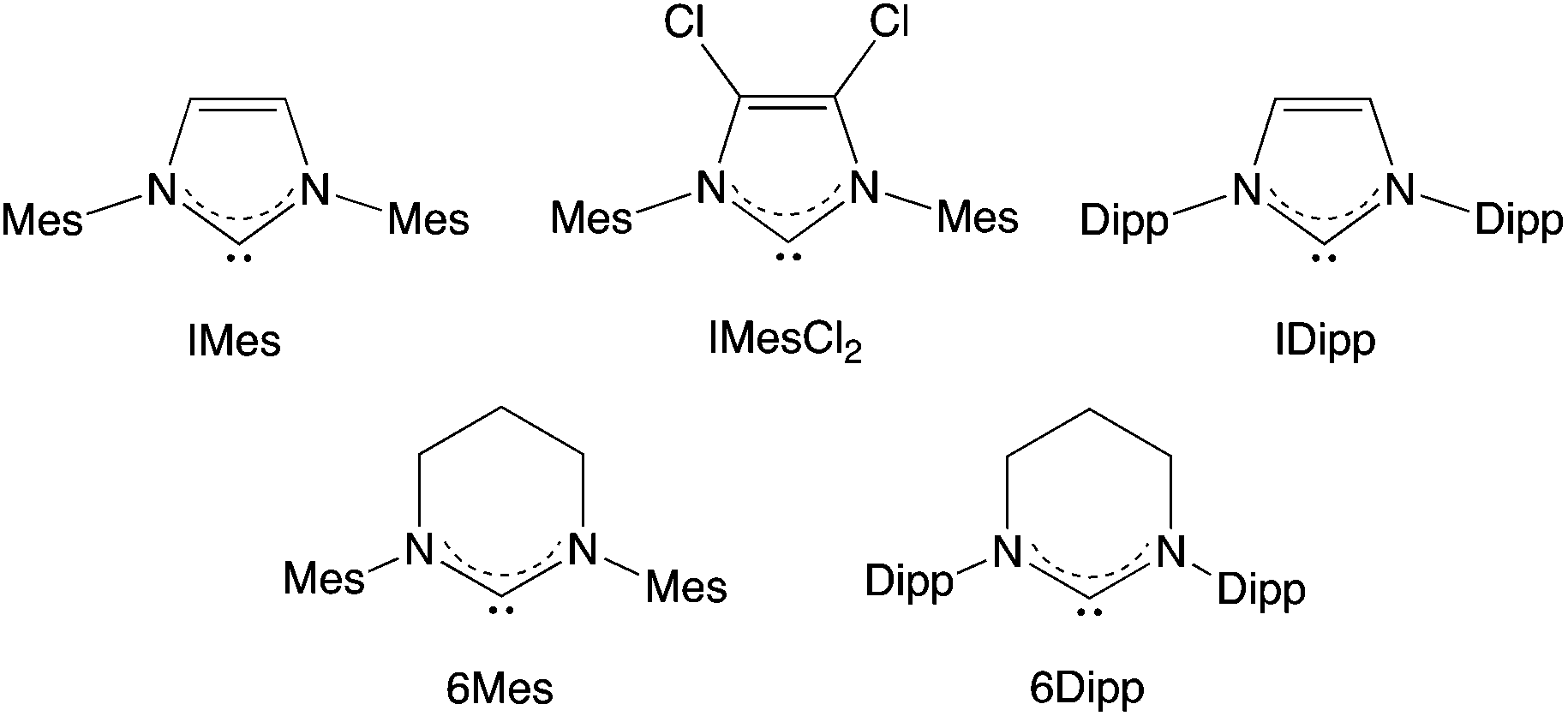 | ||
| Fig. 2 N-Heterocyclic carbenes (NHCs) utilized in the current study. | ||
The addition of IMes to 2 in a 2:1 molar ratio results in an immediate colour change from violet to orange, and the appearance of a 11B NMR resonance at δB = −14 ppm, consistent with the formation of a tetra-coordinate borate through coordination of the IMes donor.11a1H NMR spectroscopy suggests that two molecules of NHC have been assimilated and that the ‘normal’ C2-mode of carbene binding pertains, with signals at δH = 7.46 and 7.59 ppm corresponding to the backbone N(CH)2N protons (albeit with loss of the plane of symmetry). The formation of a low symmetry adduct is also consistent with the observation of ten distinct signals in the 19F NMR spectrum (indicating that the equivalence between the ortho, meta and para fluorines of the two perfluorophenyl rings had been lost); a similar pattern of 19F signals is observed in the 19F spectrum of the corresponding monofunctional adduct 1·IMes, and has been ascribed to restricted rotation about the sterically congested B–C bonds on the timescale of the NMR experiment.11a
In the case of 2, the molecular structure derived from X-ray crystallography confirms the formulation of the adduct as 1,1′-fc{B(C6F5)2·IMes}2 (2·(IMes)2, Fig. 3). The solid-state structure possesses approximate (non-crystallographic) C2 symmetry, and the fact that this adduct displays the same number of 19F NMR resonances as 1·IMes presumably reflects a similar symmetry relationship between the two –B(C6F5)2·IMes functions in solution. Bond metrics associated with the borate functions (for example B–C distances) are essentially identical to those measured for 1·IMes,11a reflecting the fact that there is relatively little geometric perturbation around the boron centre induced by the presence of the second borate function.
 | ||
| Fig. 3 Molecular structures of (left) 1,1′-fc{B(C6F5)2·IMes}2, 2·(IMes)2 and (right) 1,1′-fc{B(C6F5)2·NH3}2, 2·(NH3)2, with thermal ellipsoids set at the 35% probability level. Selected hydrogen atoms and solvate molecules omitted and mesityl groups shown in wireframe format for clarity. Selected bond lengths (Å) and angles (°): [for 2·(IMes)2] B(4)–C(60) 1.675(5), B(4)–C(3) 1.638(5), B(4)–C(14) 1.666(5), B(4)–C(47) 1.676(5), C(3)–B(4)–C(14) 113.9(3), C(3)–B(4)–C(47) 106.0(3), C(14)–B(4)–C(47) 111.2(3); [for 2·(NH3)2] B(7)–N(8) 1.624(2), B(7)–C(5) 1.601(2), B(7)–C(9) 1.648(2), B(7)–C(20) 1.639(2), B(36)–N(37) 1.627(2), B(36)–C(32) 1.600(2), B(36)–C(38) 1.649(2), B(36)–C(49) 1.638(2), C(5)–B(7)–C(9) 115.9(1), C(5)–B(7)–C(20) 113.3(1), C(9)–B(7)–C(20) 106.5(1), C(32)–B(36)–C(38) 114.4(1), C(32)–B(36)–C(49) 114.4(1), C(38)–B(36)–C(49) 108.9(1). | ||
The reaction between IMes and 2 was also examined in a 1:1 molar ratio. However, monitoring by 19F NMR suggests that this results in the formation of a mixture of the 1:1 adduct, the 2:1 adduct [i.e.2·(IMes)2], and unreacted borane starting material. Ten resonances due to 2·(IMes)2 are observed, alongside signals corresponding to the free Lewis acid and further signals, presumed to be derived from the 1:1 adduct. This inference is supported by the 11B NMR spectrum, which features a sharp signal at −13.7 ppm, consistent with the borate functions in the 2:1 adduct, alongside a broad signal consistent with uncomplexed three-coordinate borane centres. The observation of a mixture of products suggests that IMes binding at the two borane functions is essentially independent, consistent with the conformationally flexible nature of the ferrocenediyl core.
Given the diagnostic nature of the 11B and 19F signals associated with adduct formation, these measurements were then used to determine the course of the reactions of 2 with other NHCs. Thus, signals in the range δB = −10 to −13 ppm were observed on addition of two equivalents of IMesCl2, IDipp or 6Mes (with the solution in each case turning an orange colour), with the respective 19F NMR spectra displaying a similar ten-resonance pattern to that measured for 2·(IMes)2. By contrast, on addition of 6Dipp to 2, the deep maroon colour of the solution is retained, and 1H, 11B and 19F NMR spectra could be used to establish that no substantive reaction has taken place. In particular, the 11B and 19F signals are unshifted from those of the starting material 2, and the 1H NMR spectrum consists of the superposition of the signals due to 6Dipp and 2. In this case alone it appears that the NHC and 2 constitute an unquenched FLP, an observation consistent with the larger steric demands of the 6Dipp ligand (%Vbur = 50.8 vs. 36.5, 44.5 and 42.2 for IMes, IDipp and 6Mes).18,19
The combination of PCy3 and 1 has previously been shown by in situ NMR measurements to result in the formation of an FLP in benzene-d6 solution.11a However, evidence can also be obtained for the formation of a minor co-product over a period of ca. 2 weeks at room temperature (Scheme 4a). Although the overwhelmingly dominant species present in solution at this point are still 1 and PCy3, the minor product can be crystallized and shown by X-ray crystallography to be the zwitterion Fc{B(F)(C6F5)(C6F4PCy3-4)} (3), derived from SNAr attack by the phosphine at the para position of one of the perfluorophenyl rings of the Lewis acid, with accompanying migration of fluoride to the boron centre (Fig. 4). Spectroscopically, this chemistry is characterised by diagnostic signals in the 11B and 19F NMR spectra at δB = 2.0 ppm and δF = −193.7 ppm associated with the boron-bound fluoride, and a shift in the 31P NMR spectrum from the free phosphine at δP = 9.7 ppm to 39.9 ppm. Very similar NMR signals are observed for the related system derived from the analogous activation of B(C6F5)3, i.e. B(F)(C6F5)2(C6F4PCy3-4) (δB = −0.7 ppm, δP = 41.6 ppm).22
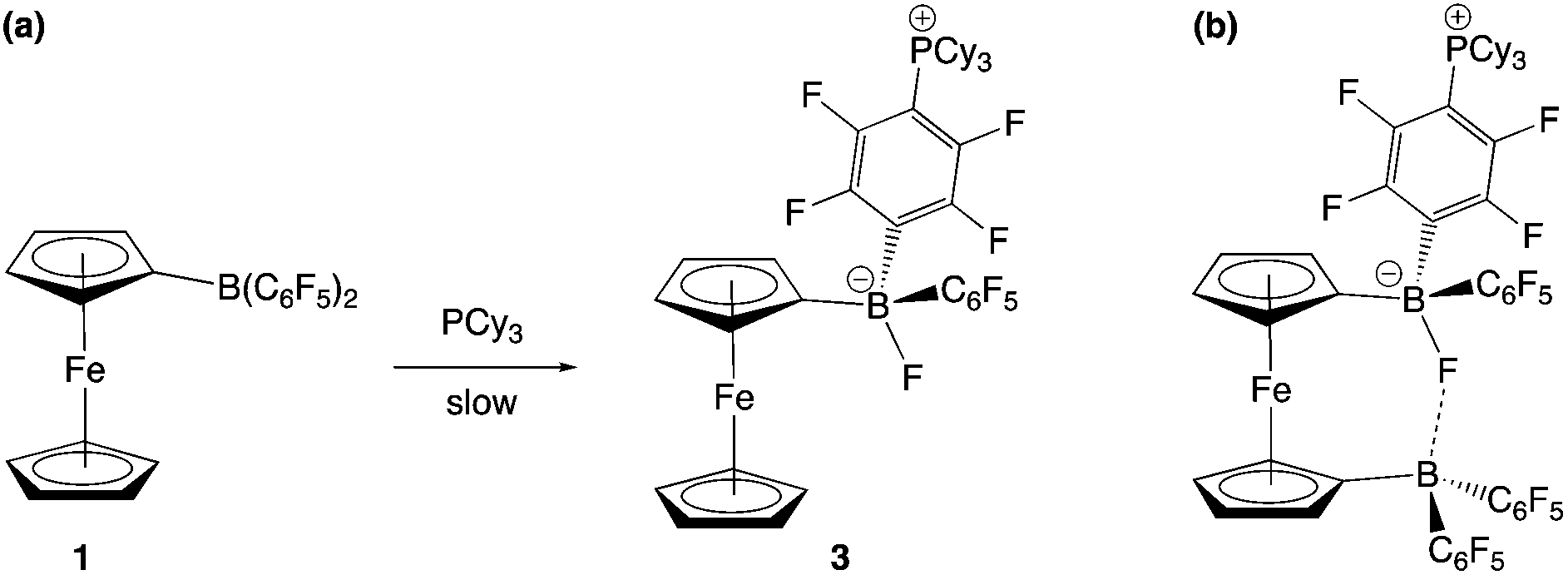 | ||
| Scheme 4 (a) Minor reaction pathway of PCy3 with 1, proceeding via nucleophilic attack at the para C–F bond of a C6F5 group to give 3. (b) Proposed structure of the product of the reaction of 2 with PCy3. | ||
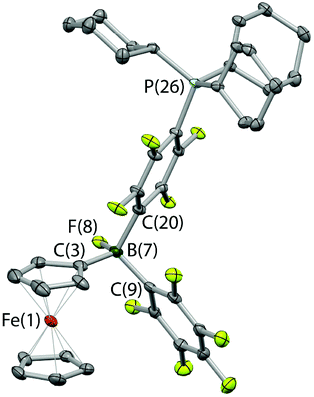 | ||
| Fig. 4 Molecular structure of Fc{B(F)(C6F5)(C6F4PCy3-4)}, 3, with thermal ellipsoids set at the 35% probability level; hydrogen atoms for clarity. Selected bond lengths (Å) and angles (°): B(7)–F(8) 1.426(2), B(7)–C(3) 1.613(3), B(7)–C(9) 1.660(3), B(7)–C(20) 1.682(3), C(3)–B(7)–C(9) 110.1(2), C(3)–B(7)–C(20) 114.0(2), C(9)–B(9)–C(20) 108.2(2). | ||
The combination of PCy3 with 2 was therefore investigated in a 2:1 molar ratio. While there was no immediate colour change on mixing, 1H, 11B, 19F and 31P NMR spectra measured after 1 h show that a reaction has taken place. A broad upfield-shifted resonance is observed in the 11B NMR spectrum at δB = 2.6 ppm, and a signal at δP = 40.1 ppm in the 31P NMR spectrum; both are suggestive of an SNAr type reaction similar to that observed for PCy3/1. The 19F NMR spectrum is more complex. In the case of Fc{B(F)(C6F5)(C6F4PCy3-4)} (3), six resonances are observed, corresponding to the boron-bound fluoride, the ortho, meta and para fluorines of the unreacted –C6F5 moiety, and two signals for the para-C6F4PCy3 moiety. If analogous reactivity were to take place at both borane functions in 2, then either six or twelve signals might be expected, depending on whether the two borate units are symmetry-related on the NMR timescale. On the other hand, if reaction were to take place at only one of the two borane units, then (superficially) nine signals would be expected, with the additional three arising from the perfluoroaryl rings associated with the unreacted borane centre. In this case twelve 19F signals are observed, but paradoxically, the 1H and 31P NMR data are consistent with the take-up of only one phosphine unit. Half of the added PCy3 remains unreacted.
Of the twelve 19F resonances observed, six can readily be assigned to fluorine environments analogous to those in 3, on the basis of their very similar pattern of chemical shifts (including a resonance at −194.9 ppm, indicative of a boron-bound fluoride). The additional six signals, come in three pairs, suggesting that they arise from the ortho, meta and para fluorines of an unreacted – B(C6F5)2 moiety, in which the equivalence of the two perfluoroaryl rings has been lost. Such inequivalence is presumably caused by restricted rotation about the B–CCp bond, which might arise either as a result of the increased steric bulk of the peripheral borate centre, or due to an interaction between the borane and the boron-bound fluoride (Scheme 4b). Some evidence for the latter comes from analysis of the magnitude of the Δδm,p splittings derived from the 19F NMR data (using in the case of the –B(C6F5)2 unit, the mean chemical shift difference between meta and para resonances). Δδm,p has a value of 10.7 ppm for starting material 2, this being reduced to 4.4 ppm for the C6F5 group of the borate function (cf. 3.6 ppm for the analogous C6F5 unit in 3). That determined for the –B(C6F5)2 group is 7.2 ppm (i.e. intermediate between borate and ‘free’ borane), and is therefore potentially suggestive of a weak secondary donor/acceptor interaction (Scheme 4b).23
Interestingly, and in contrast to PCy3/1, NMR data for PCy3/2 show no evidence of unreacted 2 (i.e. FLP formation does not occur) – an observation consistent with the enhanced electrophilicity of 2 compared to 1. It is also noteworthy that (despite the 2:1 molar ratio) PCy3 does not appear to react at the second B(C6F5)2 unit in 2. Potentially, this can be attributed to the reduced electrophilicity of the remaining –B(C6F5)2 unit on conversion of the first to an electron-donating fluoroborate function.9h
Reactions of small molecules with FLPs derived from 2
With the potential for 2 to function as an FLP in conjunction with sterically encumbered bases established, we focused on exploring the chemistry of such systems in small molecule activation. In common with 1, we find that combinations of 2 with any of PPh3, PtBu3, or 6Dipp (or for that matter the N-donors lutidine or tetramethylpiperidine) do not activate H2 either under ambient conditions or on heating to 350 K. Such observations are consistent with the electron-donating nature of the ferrocenediyl substituent and the consequently reduced Lewis acidity at boron compared to B(C6F5)3.17,24 As such, we hypothesized that the bifunctional nature of Lewis acid 2 might be better suited to the cleavage (or capture) of small molecules which yield Lewis bases that can be bound in a chelating fashion (Fig. 5). We therefore examined the reactivity of 2 towards H2O, NH3 and CO2 (plus CO2 surrogates of the type RNCO).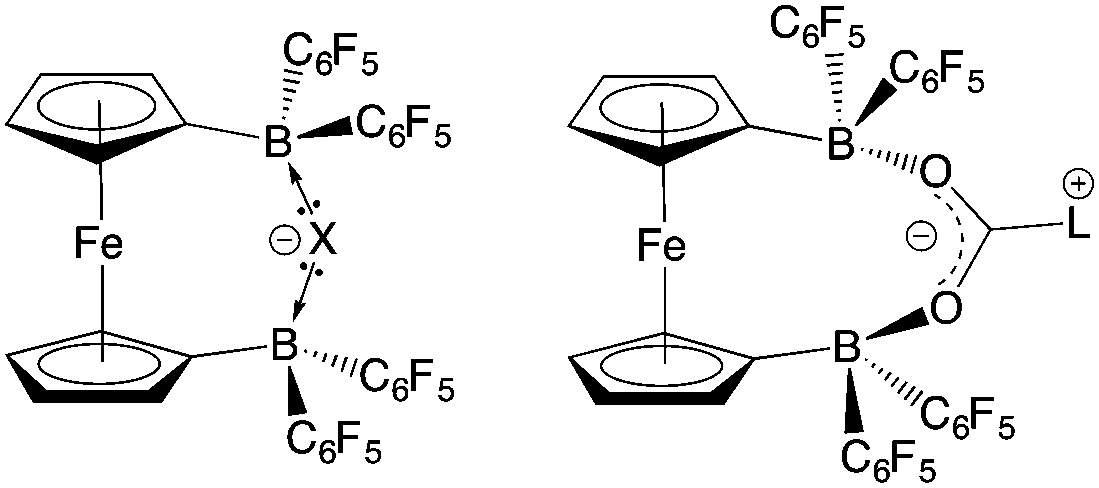 | ||
| Fig. 5 Postulated binding motifs in small molecule capture/activation using FLPs containing 2. | ||
The combination of 2 and PtBu3 reacts very readily with water via O–H bond cleavage. The dark maroon colour of 2 in benzene-d6 solution turns to a yellow-orange, and over 24 hours the formation of a yellow crystalline product is observed. In situ1H, 11B, 19F and 31P NMR studies give an indication as to the nature of the reaction taking place. Thus, the 31P spectrum shows a single peak at δP = 58.3 ppm corresponding to the phosphonium cation, [HPtBu3]+, and the 19F NMR spectrum shows three resonances, shifted from those in the starting material, and with a significant decrease in Δδm,p compared to 2 (4.4 cf. 10.7 ppm). A large upfield shift in the 11B NMR signal is also observed (to δB = 2.0 ppm), characteristic of the transformation of a three-coordinate borane to a four-coordinate borate. Moreover, single crystals suitable for X-ray crystallography could be obtained by recrystallization from chloroform. The solid-state structure so obtained confirms the nature of the product as [HPtBu3][1,1′-fc{B(C6F5)2}2(μ-OH)] (i.e. [HPtBu3][2·(μ-OH)]), featuring a hydroxide ion bridging symmetrically between the two boron centres in the anionic component (Scheme 5 and Fig. 6).25,26 From a geometric perspective, the hydroxide binding motif is similar to that observed in the zwitterionic cobaltocenium system Co(C5H4BF2)2(μ-OH) reported by Herberich and co-workers, with the B–O bond distances being comparable for the two adducts [1.569(2)/1.563(2) vs. 1.521(3)/1.556(3) Å, respectively].27
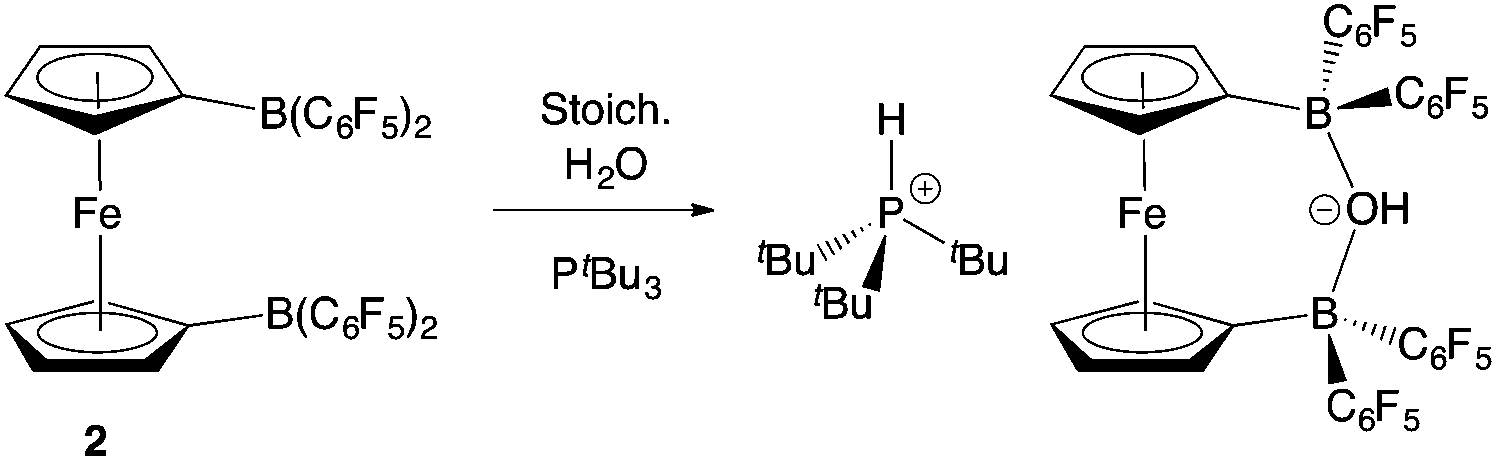 | ||
| Scheme 5 Reaction of PtBu3/2 with water.24 | ||
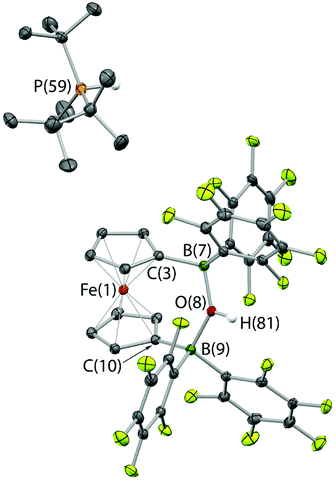 | ||
| Fig. 6 Molecular structure of [HPtBu3][1,1′-fc{B(C6F5)2}2(μ-OH)], [HPtBu3][2·(μ-OH)], with thermal ellipsoids set at the 35% probability level; hydrogen atoms (except those originating from H2O) and solvate molecules omitted for clarity. Selected bond lengths (Å) and angles (°): B(7)–O(8) 1.569(2), B(9)–O(8) 1.563(2), B(7)–C(3) 1.596(2), B(7)–C(37/48) 1.660(2)/1.664(2), B(9)–C(10)1.593(2), B(9)–C(15/26) 1.652(2)/1.662(2), B(7)–O(8)–B(9) 139.9(1), C(3)–B(7)–C(37) 109.1(1), C(3)–B(7)–C(48) 116.2(1), C(37)–B(7)–C(48) 105.7(1), C(10)–B(9)–C(15) 109.4(1), C(10)–B(9)–C(26) 114.7(1), C(15)–B(9)–C(26) 107.4(1). | ||
Given the vast disparity in the pKas of H2O and [HPtBu3]+ (ca. 31.2 and 11.4, respectively in DMSO)28,29 the cleavage of water by PtBu3/2, presumably relies very heavily from a thermodynamic viewpoint on the role of 2 as a chelating Lewis acid to sequester OH−. With this in mind, we were also interested in examining the behaviour of this FLP in the presence of ammonia (pKaca. 41 in DMSO),28 hypothesizing that a similar mode of binding might be possible with NH2−. Thus, a ‘free-pump-thaw’ degassed solution of PtBu3/2 was exposed to NH3 (at 1 atm pressure) and stirred for 4 hours. The resulting yellow crystalline product was crystallized from a concentrated diethyl ether solution. However, the isolated material does not give rise to any 31P NMR signals, and in situ measurements of the reaction mixture show a single peak at δP = 61.9 corresponding to free PtBu3. The 11B NMR spectrum of the isolated product shows a single sharp resonance at δB = −8.7 ppm, and the 19F NMR spectrum shows three signals at δF = −133.3, −156.2 and −162.7 ppm (Δδm,p = 6.5 ppm), diagnostic of a binding event occurring at both borane centres. The 1H NMR signal corresponding to the NH unit integrates for six protons, suggesting formation of the bis-ammonia adduct, 1,1′-fc{B(C6F5)2·NH3}2 [i.e.2·(NH3)2]. In order to confirm the formation of this adduct structurally, a higher yielding synthesis was developed via condensation of liquid ammonia onto solid 2. An immediate colour change was observed from deep purple to pale yellow, and (after venting the excess ammonia) crystals suitable for X-ray crystallography could be grown from diethyl ether solution. The structure so obtained (Fig. 3) confirms the formation of 2·(NH3)2. Geometrically the structure has much in common with that of 2·(IMes)2 (also Fig. 3), with the slightly less pyramidalized nature of the BC3 fragments [∑(∠C–B–C) = 336.7° (mean) cf. 330.0° (mean) for 2·(IMes)2] presumably reflecting the differing donor capabilities of ammonia and IMes.
Clearly this phosphine-based FLP system does not have sufficient unquenched reactivity to deprotonate ammonia. On the other hand, NHCs have been shown to be basic enough to deprotonate the adduct H3N·B(C6F5)3.30 Imidazolylidene donors form (unreactive) classical donor/acceptor adducts with 2, but the combination with the bulkier expanded ring NHC 6Dipp constitutes an FLP. Attempts were therefore made to activate ammonia using the 6Dipp/2 system: however, these also resulted in the isolation of 2·(NH3)2. Presumably the 2:1 adduct is formed very rapidly, and essentially irreversibly, on exposure of the FLP to ammonia; in the absence of the possibility for chelation (due to ‘blocking’ of the second Lewis acid function by NH3), 6Dipp is not sufficiently basic to deprotonate the bound NH3. Given that ItBu will deprotonate H3N·B(C6F5)3,30 and that expanded ring NHCs are typically more basic than their imidazolylidene counterparts,31 this observation again reflects the weaker intrinsic Lewis acidity of the individual boron centres in 2 compared to B(C6F5)3.12a
FLP systems featuring 1 as the Lewis acid component are not able to bind/activate CO2 (in contrast to PtBu3/B(C6F5)3, for example).31 With this in mind, analogous FLPs containing 2 were targeted, it being not only a stronger Lewis acid, but also bifunctional, and hence potentially complementary to the Lewis basic component formed in the reaction of a phosphine/NHC with CO2 (Fig. 5). However, benzene solutions containing either the PtBu3/2 or tetramethylpiperidine/2 FLP, show no propensity to bind CO2, either at room temperature or 350 K. Both tertiary phosphines and secondary amines have previously been shown to be capable of binding CO2 in conjunction with B(C6F5)3, and the failure of systems containing 2 to act in a similar manner presumably also reflects its weaker Lewis acidity.32
The 6Dipp/2 FLP features the strongest Lewis base under investigation, and it was postulated that this system might therefore be capable of binding CO2. In our hands, however, we find that this reaction does not proceed cleanly, and when using benzene as the solvent, significant precipitate is formed. In previous examples of CO2 capture by NHC-containing FLPs, an initial activation step is observed involving nucleophilic attack by the carbene at the carbon atom of CO2, before trapping of the resulting betaine derivative by the Lewis acid component.32c The reaction of 6Dipp with CO2 was therefore investigated in the absence of2 in an attempt to shed light on the course of the reaction. Exposure of a benzene solution of 6Dipp to CO2 (at ca. 1 atm pressure), results in immediate formation of an off-white precipitate. Moreover, investigation of this material by 1H NMR (in bromobenzene-d5), reveals signals characteristic of the formamidinium cation [(6Dipp)H]+, together with three multiplet resonances at 6.15, 5.14 and 5.10 ppm (integrating to 1H each), corresponding to the alkenic protons of an RCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 unit. These observations are consistent with ring opening of the 6Dipp heterocycle via deprotonation at the C5 position (Scheme 6), in a manner analogous to that observed previously for 6Dipp in the presence of other electrophiles (e.g. H+, [LAu]+).33 Although the product in this case could not be crystallographically characterized, peak envelopes at m/z = 405 and 447 in the positive and negative ESI-MS spectra, respectively, are consistent with the formation of the proposed cationic and anionic components.
CH2 unit. These observations are consistent with ring opening of the 6Dipp heterocycle via deprotonation at the C5 position (Scheme 6), in a manner analogous to that observed previously for 6Dipp in the presence of other electrophiles (e.g. H+, [LAu]+).33 Although the product in this case could not be crystallographically characterized, peak envelopes at m/z = 405 and 447 in the positive and negative ESI-MS spectra, respectively, are consistent with the formation of the proposed cationic and anionic components.
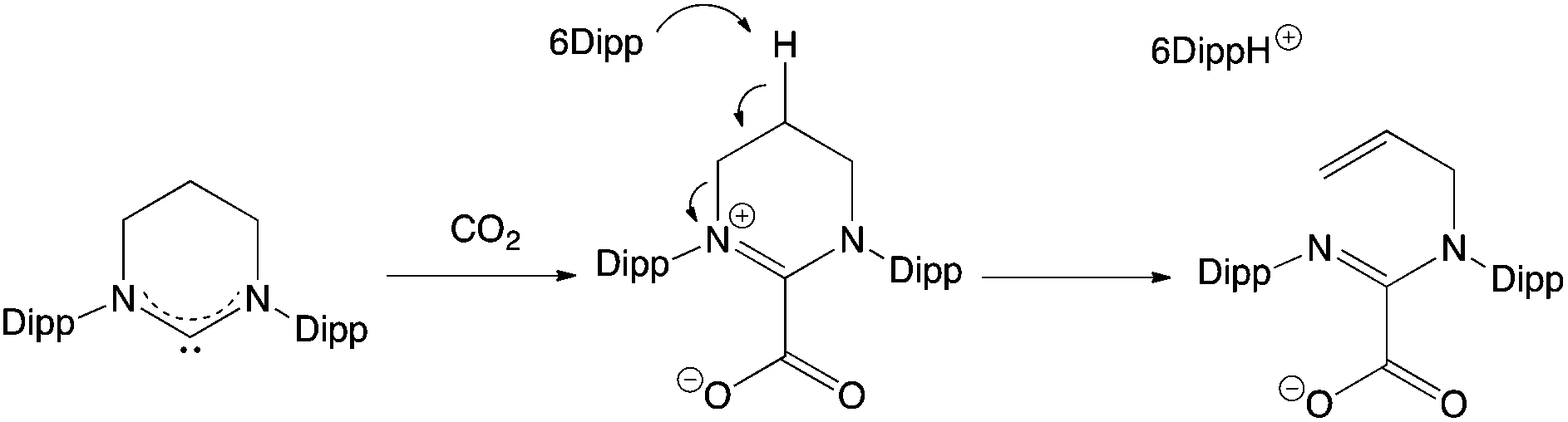 | ||
| Scheme 6 Ring opening of 6Dipp in the presence of CO2. | ||
In the absence of clean reactivity leading to the trapping of carbon dioxide by FLPs containing 2, attention was turned to more reactive CO2 surrogates, such as isocyanates. The reaction of PtBu3/2 with CyNCO gives a complex mixture of products, and so to simplify matters, the analogous chemistry employing monofunctional 1 was probed. However, as in the case of the PtBu3/2 system, the 31P NMR spectrum measured in situ implies no net involvement of the phosphine, with an unshifted resonance being observed at δP = 61.9 ppm. A control reaction (without PtBu3) generates the same product (albeit more slowly), which gives rise to signals in the 11B and 19F NMR spectra consistent with the formation of a four-coordinate boron centre. In addition, a peak envelope centred at m/z = 780 in the ESI-MS spectrum is consistent with the assimilation of two molecules of CyNCO (Scheme 7). Crystals of the product, 4, were obtained by slow cooling of a solution in benzene-d6, and X-ray crystallography was used to unambiguously determine the connectivity associated with insertion of two molecules of CyNCO into the CCp–B bond of 1 (Fig. 7).
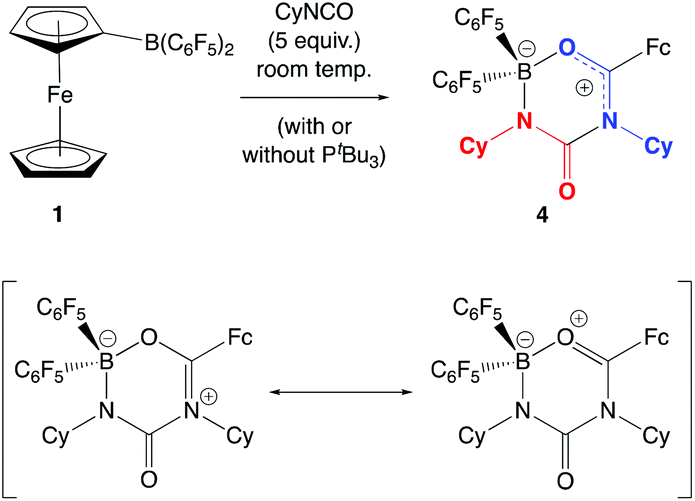 | ||
| Scheme 7 Reaction of CyNCO with the PtBu3/1 FLP (or with 1 alone), leading to the formation of 4. | ||
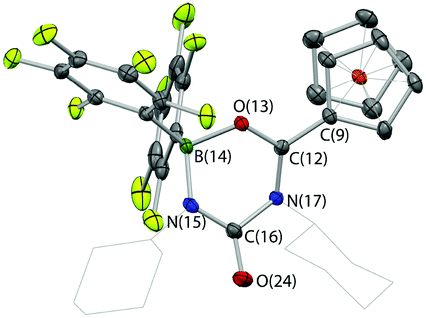 | ||
| Fig. 7 Molecular structure of 4, with thermal ellipsoids set at the 35% probability level; hydrogen atoms and solvate molecules omitted, and cyclohexyl groups shown in wireframe format for clarity. Selected bond lengths (Å): C(9)–C(12) 1.452(2), C(12)–O(13) 1.288(2), O(13)–B(14) 1.534(2), B(14)–N(15) 1.533(3), N(15)–C(16) 1.332(3), C(16)–N(17) 1.475(2), C(12)–N(17) 1.340(3), C(16)–O(24) 1.215(3). | ||
The molecular structure is based around a non-planar six-membered CNCNBO heterocycle featuring a pendant carbonyl function. Analysis of the bond lengths within the heterocycle suggest that there is some degree of delocalized π character within the O(13)–C(12)–N(17) unit [d{C(12)–O(13)} = 1.288(2) Å] and also within the amido function defined by N(15)–C(16)–O(24) [d{N(15)–C(16)} = 1.332(3) Å]. The relatively long C(16)–N(17) distance [1.475(2) Å], on the other hand, suggests less significant delocalization between these two fragments.
The insertion of heterocumulenes of this sort into reactive B–C bonds is by no means unprecedented. Cowley and co-workers have reported the insertion of dicyclohexyl carbodiimide into the B–C bond of PhBCl2 to give the benzamidinate system {PhC(CyN)2}BCl2 (Scheme 8a),34,35 and a similar initial step in the reaction of 1 with CyNCO would generate a closely-related BNCO four-membered ring via migration of the more nucleophilic ferrocenyl fragment (cf. C6F5; Scheme 8b). The more electrophilic nature of the boron centre in this intermediate then presumably facilitates nucleophilic attack by a second equivalent of the isocyanate, with subsequent ring-closure generating the observed 6-membered heterocyclic product.
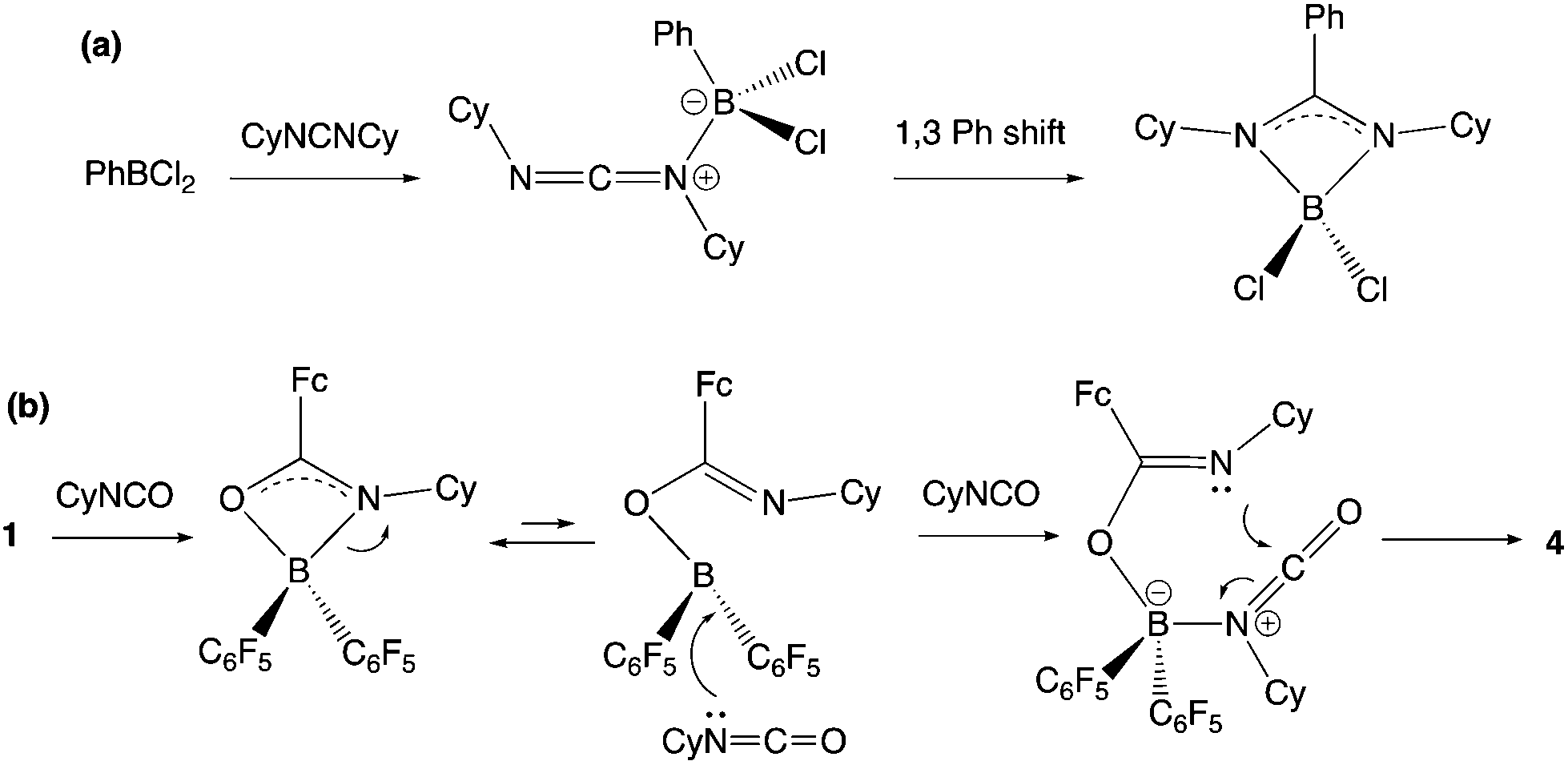 | ||
| Scheme 8 (a) Mechanism for the insertion of a carbodiimide into the B–C bond of PhBCl2 as reported by Cowley and co-workers;34 (b) potential mechanism for the double insertion of cyclohexyl isocyanate into the B–CCp bond of 1 to give 4. | ||
Experimental
Caution: Solutions containing LiC6F5 are known to explode at temperatures in excess of −40 °C.General considerations
All manipulations were carried out using standard Schlenk line or dry-box techniques under an atmosphere of argon or dinitrogen, respectively. Solvents were degassed by sparging with argon and dried by passing through a column of the appropriate drying agent using a commercially available Braun SPS. NMR spectra were recorded in chloroform-d, benzene-d6, bromobenzene-d5 or THF-d8, which were dried over molecular sieves, potassium, calcium hydride or calcium hydride respectively, and stored under argon in Teflon valve ampoules. NMR samples were prepared under argon in 5 mm Wilmad 507-PP tubes fitted with J. Young Teflon valves. 1H and 13C NMR spectra were recorded on Varian Mercury-VX-300 or Bruker AVII-500 spectrometers and referenced internally to residual protio-solvent (1H) or solvent (13C) resonances and are reported relative to tetramethylsilane (δ = 0 ppm). 11B, 19F and 31P NMR spectra were referenced with respect to Et2O·BF3, CFCl3 and 85% aqueous H3PO4, respectively. Chemical shifts are quoted in δ (ppm) and coupling constants in Hz. UV-vis spectra were collected on a Scintio UV S-2100 UV/Vis spectrometer. Elemental analyses were carried out at London Metropolitan University. BrC6F5, nBuLi, PtBu3, PPh3, lutidine, tetramethylpiperidine, ammonia and carbon dioxide were sourced commercially. 1,1′-fc(BBr2)2, FcB(C6F5)2 (1), and NHCs (IMes, IDipp, IMesCl2, 6Mes and 6Dipp) were prepared by literature routes.14,15,36Syntheses of novel compounds
General procedure for investigating the formation of Lewis base adducts of 2
The procedure is exemplified for the case of IMes in a 2:1 molar ratio. This and other Lewis bases were probed in both 2:1 and 1:1 molar ratios. In the case of 1,1′-fc{B(C6F5)2·IMes}2, 2·(IMes)2, the product was isolated and characterized by standard spectroscopic, analytical and crystallographic methods. For other Lewis bases, in situ11B and 19F NMR data (based on those measured for 2·(IMes)2) were used to determine whether or not a classical Lewis acid/base adduct was formed. IMes (0.035 g, 0.116 mmol) and 2 (0.100 g, 0.058 mmol) were dissolved in the minimum benzene required to dissolve all solid material (ca. 5 mL), and the reaction mixture stirred for 24 h at room temperature. Volatiles were then removed in vacuo, and the product recrystallized from THF/hexane as single crystals suitable for X-ray crystallography. Yield: 0.056 g, 65%.
Characterizing data for 2·(IMes)2
1H NMR (500 MHz, THF-d8, 20 °C): δ 7.59, 7.46 (d, 2JH–H = 2.1 Hz, each 2H, backbone CH of IMes), 6.99, 6.90, 6.70, 6.45 (s, each 2H, IMes aromatic CH), 3.80, 3.55, 3.22, 2.77 (br s, each 2H, CH of Cp), 2.40, 2.24, 2.20, 2.18, 1.98, 1.81 (s, each 6H, Me of IMes). 11B NMR (128 MHz, THF-d8, 20 °C): δ −14 (sharp). 19F NMR (377 MHz, THF-d8, 20 °C): δ −117.8 (d, 3JFF = 24.5 Hz, ortho-CF of C6F5), −119.0 (d, 3JFF = 24.5 Hz, ortho-CF of C6F5), −123.9 (d, 3JFF = 24.5 Hz, ortho-CF of C6F5), −128.1 (d, 3JFF = 24.5 Hz, ortho-CF of C6F5), −163.3 (t, 3JFF = 19.5 Hz, para-CF of C6F5), −164.1 (t, 3JFF = 20.0 Hz, para-CF of C6F5), −167.1 (t, 3JFF = 22.1 Hz, meta-CF of C6F5), −167.4 (t, 3JFF = 22.1 Hz, meta-CF of C6F5), −167.9 (t, 3JFF = 22.1 Hz, meta-CF of C6F5), −168.6 (t, 3JFF = 22.1 Hz, meta-CF of C6F5). Elemental microanalysis: calc. (for C76H56B2F20FeN4) C 61.53, H 3.81, N 3.78; meas. C 61.88, H 4.01, N 3.66. Crystallographic data: 2·(IMes)2·3/2(THF)·3/2(pentane), C88H83B2F20FeN4O1.5, Mr = 1678.05, orthorhombic, P 2ac 2ab, a = 11.4405(1), b = 22.3759(2), c = 32.0362(2) Å, V = 8201.0(1) Å3, Z = 4, ρc = 1.359 Mg m−3, T = 150 K, λ = 1.54180 Å. 17080 reflections collected, 16778 independent [R(int) = 0.029] used in all calculations. R1 = 0.0421, wR2 = 0.1252 for observed unique reflections [I > 2σ(I)] and R1 = 0.0428, wR2 = 0.1263 for all unique reflections. Max. and min. residual electron densities 0.84 and −0.49 e Å3. CCDC reference: 1582718.†
In situ NMR data for 2·(IMesCl2)2
11B NMR (128 MHz, benzene, 20 °C): δ −13 (sharp). 19F NMR (377 MHz, benzene, 20 °C): δ −117.0 (d, 3JFF = 16.0 Hz, ortho-CF of C6F5), −118.9 (d, 3JFF = 16.0 Hz, ortho-CF of C6F5), −122.3 (d, 3JFF = 16.0 Hz, ortho-CF of C6F5), −128.5 (d, 3JFF = 16.0 Hz, ortho-CF of C6F5), −159.3 (d, 3JFF = 22.5 Hz, para-CF of C6F5), −162.0 (d, 3JFF = 22.5 Hz, para-CF of C6F5), −164.3 (d, 3JFF = 20.9 Hz, meta-CF of C6F5), −165.4 (d, 3JFF = 20.9 Hz, meta-CF of C6F5), −166.3 (d, 3JFF = 20.9 Hz, meta-CF of C6F5), −166.5 (d, 3JFF = 20.9 Hz, meta-CF of C6F5).In situ NMR data for 2·(6Mes)2
11B NMR (128 MHz, benzene, 20 °C): δ −11 (sharp). 19F NMR (377 MHz, benzene, 20 °C): δ −112.6 (d, 3JFF = 29.7 Hz, ortho-CF of C6F5), −118.8 (d, 3JFF = 27.4 Hz, ortho-CF of C6F5), −119.9 (d, 3JFF = 24.5 Hz, ortho-CF of C6F5), −123.0 (d, 3JFF = 25.4 Hz, ortho-CF of C6F5), −153.3 (t, 3JFF = 19.4 Hz, para-CF of C6F5), −158.3 (t, 3JFF = 19.4 Hz, para-CF of C6F5), −161.2 (t, 3JFF = 21.3 Hz, meta-CF of C6F5), −163.6 (t, 3JFF = 23.6 Hz, meta-CF of C6F5), −164.2 (t, 3JFF = 23.9 Hz, meta-CF of C6F5), −165.6 (t, 3JFF = 21.4 Hz, meta-CF of C6F5).In situ NMR data for 2·(PnBu3)2
11B NMR (128 MHz, benzene, 20 °C): δ −11 (broad). 19F NMR (377 MHz, benzene, 20 °C): δ −126.2 (broad s, ortho-CF of C6F5), −157.9 (t, 3JFF = 20.3 Hz, para-CF of C6F5), −163.7 (t, 3JFF = 20.3 Hz, meta-CF of C6F5). 31P NMR (162 MHz, benzene, 20 °C): δ −2.9.In situ NMR data from the reaction of 2 with PCy3
(500 MHz, chloroform-d, 20 °C): δ 4.42, 4.38, 4.22, 4.09, 3.65, 3.29, 3.11, 3.08 (broad s, each 1H, C5H4B), 2.65 (m, 3H, Cy), 2.10–1.22 (overlapping m, 30 H, Cy). 11B NMR (128 MHz, benzene-d6, 20 °C): δ 32 (very broad), 3 (broad). 19F NMR (377 MHz, benzene-d6, 20 °C): δ −123.4 (s, 2F, C6F4), −129.3 (s, orthoCF of B(C6F5)2), −123.0 (s, orthoCF of B(C6F5)2), −131.4 (s, 2F, C6F4), −131.9 (s, orthoCF of BF(C6F4PCy3)(C6F5)), −156.3 (broad s, paraCF of B(C6F5)2), −156.7 (broad s, paraCF of B(C6F5)2), −160.1 (s, paraCF of BF(C6F4PCy3)(C6F5)), −163.5 (s, metaCF of B(C6F5)2), −163.9 (s, metaCF of B(C6F5)2), −164.5 (s, metaCF of BF(C6F4PCy3)(C6F5)), −194.9 (s, BF). 31P NMR (162 MHz, benzene-d6, 20 °C): δ 40.1.![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 11.7155(3), b = 12.6410(4), c = 15.6649(3) Å, α = 83.830(2), β = 79.824(2), γ = 66.833(3)°, V = 2097.4(1) Å3, Z = 2, ρc = 1.472 Mg m−3, T = 150 K, λ = 1.54180 Å. 8635 reflections collected, 8635 independent [R(int) = 0.019] used in all calculations. R1 = 0.0357, wR2 = 0.0824 for observed unique reflections [I > 2σ(I)] and R1 = 0.0374, wR2 = 0.0838 for all unique reflections. Max. and min. residual electron densities 0.54 and −0.51 e Å3. CCDC reference: 954137.†
, a = 11.8245(4), b = 13.6477(4), c = 15.8558(4) Å, α = 84.214(2), β = 88.541(2), γ = 87.111(3)°, V = 2542.0(1) Å3, Z = 2, ρc = 1.583 Mg m−3, T = 150 K, λ = 1.54180 Å. 27110 reflections collected, 10487 independent [R(int) = 0.026] used in all calculations. R1 = 0.0326, wR2 = 0.804 for observed unique reflections [I > 2σ(I)] and R1 = 0.0351, wR2 = 0.0822 for all unique reflections. Max. and min. residual electron densities 0.31 and −0.39 e Å3. CCDC reference: 1576950.†
, a = 10.4515(3), b = 13.5721(3), c = 14.2616(4) Å, α = 94.087(2), β = 93.027(3), γ = 100.948(2)°, V = 1976.6(1) Å3, Z = 2, ρc = 1.722 Mg m−3, T = 150 K, λ = 1.54180 Å. 41166 reflections collected, 8188 independent [R(int) = 0.021] used in all calculations. R1 = 0.0259, wR2 = 0.0656 for observed unique reflections [I > 2σ(I)] and R1 = 0.0270, wR2 = 0.0664 for all unique reflections. Max. and min. residual electron densities 0.32 and −0.34 e Å3. CCDC reference: 1576951.†
, a = 10.9221(1), b = 11.1191(1), c = 17.0196(2) Å, α = 94.976(1), β = 95.697(1), γ = 112.976(1)°, V = 1875.7(1) Å3, Z = 2, ρc = 1.520 Mg m−3, T = 150 K, λ = 1.54180 Å. 55367 reflections collected, 8562 independent [R(int) = 0.030] used in all calculations. R1 = 0.0389, wR2 = 0.0842 for observed unique reflections [I > 2σ(I)] and R1 = 0.0648, wR2 = 0.0926 for all unique reflections. Max. and min. residual electron densities 0.67 and −0.57 e Å3. CCDC reference: 1576592.†
, a = 11.7155(3), b = 12.6410(4), c = 15.6649(3) Å, α = 83.830(2), β = 79.824(2), γ = 66.833(3)°, V = 2097.4(1) Å3, Z = 2, ρc = 1.472 Mg m−3, T = 150 K, λ = 1.54180 Å. 8635 reflections collected, 8635 independent [R(int) = 0.019] used in all calculations. R1 = 0.0357, wR2 = 0.0824 for observed unique reflections [I > 2σ(I)] and R1 = 0.0374, wR2 = 0.0838 for all unique reflections. Max. and min. residual electron densities 0.54 and −0.51 e Å3. CCDC reference: 954137.†
, a = 11.8245(4), b = 13.6477(4), c = 15.8558(4) Å, α = 84.214(2), β = 88.541(2), γ = 87.111(3)°, V = 2542.0(1) Å3, Z = 2, ρc = 1.583 Mg m−3, T = 150 K, λ = 1.54180 Å. 27110 reflections collected, 10487 independent [R(int) = 0.026] used in all calculations. R1 = 0.0326, wR2 = 0.804 for observed unique reflections [I > 2σ(I)] and R1 = 0.0351, wR2 = 0.0822 for all unique reflections. Max. and min. residual electron densities 0.31 and −0.39 e Å3. CCDC reference: 1576950.†
, a = 10.4515(3), b = 13.5721(3), c = 14.2616(4) Å, α = 94.087(2), β = 93.027(3), γ = 100.948(2)°, V = 1976.6(1) Å3, Z = 2, ρc = 1.722 Mg m−3, T = 150 K, λ = 1.54180 Å. 41166 reflections collected, 8188 independent [R(int) = 0.021] used in all calculations. R1 = 0.0259, wR2 = 0.0656 for observed unique reflections [I > 2σ(I)] and R1 = 0.0270, wR2 = 0.0664 for all unique reflections. Max. and min. residual electron densities 0.32 and −0.34 e Å3. CCDC reference: 1576951.†
, a = 10.9221(1), b = 11.1191(1), c = 17.0196(2) Å, α = 94.976(1), β = 95.697(1), γ = 112.976(1)°, V = 1875.7(1) Å3, Z = 2, ρc = 1.520 Mg m−3, T = 150 K, λ = 1.54180 Å. 55367 reflections collected, 8562 independent [R(int) = 0.030] used in all calculations. R1 = 0.0389, wR2 = 0.0842 for observed unique reflections [I > 2σ(I)] and R1 = 0.0648, wR2 = 0.0926 for all unique reflections. Max. and min. residual electron densities 0.67 and −0.57 e Å3. CCDC reference: 1576592.†
Crystallography
Data for 2·(IMes)2, [HPtBu3][2·(μ-OH)], 2·(NH3)2, 3 and 4 were collected on a Oxford Diffraction Supernova diffractometer at 150 K. Data collection and reduction were carried out using CrysAlisPro or Denzo/Scalepack, structure solution using Superflip, and refinement using CRYSTALS or SHELXT-2014/7.37 Complete details of all structures are contained within the respective CIFs which have been deposited with the CCDC (reference numbers: 954137, 1576950–1576952 and 1582718).†Conclusions
The bifunctional Lewis acid 1,1′-fc{B(C6F5)2}2 (2) is shown to form frustrated Lewis pairs with the sterically encumbered N-donor bases lutidine and tetramethylpiperidine, together with PPh3, PtBu3 and the expanded ring carbene 6Dipp. In the case of PCy3, attack at the para-CF bond of one of the C6F5 groups leads to nucleophilic aromatic substitution with accompanying migration of fluoride to boron. The more facile nature of this chemistry in comparison to that observed for the mono-functional analogue FcB(C6F5)2 (1) can be attributed to the more electron deficient nature of the -B(C6F5)2 functions in 2.The PtBu3/2 FLP can be shown to cleave one of the O–H bonds in water to give a tertiary phosphonium salt in which the anion features a symmetrically bound ‘inverse-chelated’ B–O(H)–B unit. Attempts to bring about analogous N–H bond activation with ammonia are frustrated by the weaker Brønsted acidity of the N–H bond and/or the inability to generate a similar B–N(H)2–B motif due to the (less reversible) formation of strong H3N→B donor/acceptor bonds. Neither PtBu3/1 or PtBu3/2 shows any propensity to activate carbon dioxide, in contrast to the combination of PtBu3 and the stronger Lewis acid B(C6F5)3. Attempts to use the stronger Lewis base 6Dipp lead to ring opening of the carbene brought about by coordination to the electrophilic C-atom of CO2, while the use of the more reactive heterocumulene CyNCO can be shown to lead to insertion into the C–B bond linking the borane function to the ferrocene backbone.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding from the EPSRC (studentship to MJK: EP/P505216/1), and Dr Jamie Hicks (Oxford) for help with crystallography.Notes and references
- Initial identification of steric frustration in Lewis acid/base complex formation: H. C. Brown, H. I. Schlesinger and S. Z. Cardon, J. Am. Chem. Soc., 1942, 64, 325 CrossRef CAS.
- Initial demonstration of the use of FLPs in small molecule activation: G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124 CrossRef CAS PubMed.
- For a recent review, see: D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400 CrossRef CAS PubMed.
- See, for example: T. J. Herrington, A. J. W. Thom, A. J. P. White and A. E. Ashley, Dalton Trans., 2012, 41, 9019 RSC.
- See, for example: C. Appelt, J. C. Slootweg, K. Lammertsma and W. Uhl, Angew. Chem., Int. Ed., 2013, 52, 4256 CrossRef CAS PubMed.
- See, for example: J. M. Farrell, J. A. Hatnean and D. W. Stephan, J. Am. Chem. Soc., 2012, 134, 15728 CrossRef CAS PubMed.
- See, for example: A. M. Chapman, M. F. Haddow and D. F. Wass, J. Am. Chem. Soc., 2011, 133, 18463 CrossRef CAS PubMed.
- C. R. Wade, A. E. J. Broomsgrove, S. Aldridge and F. P. Gabbaï, Chem. Rev., 2010, 110, 3958 CrossRef CAS PubMed.
- (a) C. Dusemund, K. R. A. S. Sandanayake and S. Shinkai, J. Chem. Soc., Chem. Commun., 1995, 333 RSC; (b) H. Yamamoto, A. Ori, K. Ueda, C. Dusemund and S. Shinkai, Chem. Commun., 1996, 407 RSC; (c) C. Bresner, S. Aldridge, I. A. Fallis, C. Jones and L.-L. Ooi, Angew. Chem., Int. Ed., 2005, 44, 3606 CrossRef CAS PubMed; (d) K. Venkatasubbaiah, L. N. Zakharov, W. S. Kassel, A. L. Rheingold and F. Jäkle, Angew. Chem., Int. Ed., 2005, 44, 5428 CrossRef CAS PubMed; (e) C. Bresner, J. K. Day, N. D. Coombs, I. A. Fallis, S. Aldridge, S. J. Coles and M. B. Hursthouse, Dalton Trans., 2006, 3660 RSC; (f) J. K. Day, C. Bresner, I. A. Fallis, L.-L. Ooi, D. J. Watkin, S. J. Coles, L. Male, M. B. Hursthouse and S. Aldridge, Dalton Trans., 2007, 3486 RSC; (g) K. Venkatasubbaiah, I. Nowik, R. H. Herber and F. Jäkle, Chem. Commun., 2007, 2154 RSC; (h) J. K. Day, C. Bresner, N. D. Coombs, I. A. Fallis, L.-L. Ooi and S. Aldridge, Inorg. Chem., 2008, 47, 793 CrossRef CAS PubMed; (i) A. E. J. Broomsgrove, D. Addy, C. Bresner, I. A. Fallis, A. L. Thompson and S. Aldridge, Chem. – Eur. J., 2008, 14, 7525 CrossRef CAS PubMed; (j) T. Pakkirisamy, K. Venkatasubbaiah, W. S. Kassel, A. L. Rheingold and F. Jäkle, Organometallics, 2008, 27, 3056 CrossRef CAS; (k) K. Venkatasubbaiah, T. Pakkirisamy, R. A. Lalancette and F. Jäkle, Dalton Trans., 2008, 4507 RSC; (l) I. R. Morgan, A. Di Paolo, D. Vidovic, I. A. Fallis and S. Aldridge, Chem. Commun., 2009, 7288 RSC; (m) A. E. J. Broomsgrove, D. Addy, A. Di Paolo, I. R. Morgan, C. Bresner, V. Chislett, I. A. Fallis, A. L. Thompson, D. Vidovic and S. Aldridge, Inorg. Chem., 2010, 49, 157 CrossRef CAS PubMed; (n) C. Bresner, C. J. E. Haynes, D. A. Addy, A. E. J. Broomsgrove, P. Fitzpatrick, D. Vidovic, A. L. Thompson, I. A. Fallis and S. Aldridge, New J. Chem., 2010, 34, 1652 RSC; (o) I. R. Morgan, A. E. J. Broomsgrove, P. Fitzpatrick, D. Vidovic, A. L. Thompson, I. A. Fallis and S. Aldridge, Organometallics, 2010, 29, 4762 CrossRef CAS; (p) I. Siewert, P. Fitzpatrick, A. E. J. Broomsgrove, M. Kelly, D. Vidovic and S. Aldridge, Dalton Trans., 2011, 40, 10345 RSC; (q) I. Siewert, D. Vidovic and S. Aldridge, J. Organomet. Chem., 2011, 696, 2528 CrossRef CAS; (r) M. J. Kelly, A. E. J. Broomsgrove, I. R. Morgan, I. Siewert, P. Fitzpatrick, J. Smart, D. Vidovic and S. Aldridge, Organometallics, 2013, 32, 2674 CrossRef CAS; (s) R. Tirfoin and S. Aldridge, Dalton Trans., 2013, 42, 12836 RSC; (t) P. Thilagar, D. Murillo, J. Chen and F. Jäkle, Dalton Trans., 2013, 42, 665 RSC; (u) R. Tirfoin, J. A. B. Abdalla and S. Aldridge, Dalton Trans., 2015, 44, 13049 RSC; (v) R. Tirfoin, J. A. B. Abdalla and S. Aldridge, Chem. – Eur. J., 2015, 21, 11813 CrossRef CAS PubMed.
- A. Ramos, A. J. Lough and D. W. Stephan, Chem. Commun., 2009, 1118 RSC.
- (a) M. J. Kelly, J. Gilbert, R. Tirfoin and S. Aldridge, Angew. Chem., Int. Ed., 2013, 52, 14094 CrossRef CAS PubMed. See also: (b) Z. Mo, E. L. Kolychev, A. Rit, J. Campos, H. Niu and S. Aldridge, J. Am. Chem. Soc., 2015, 137, 12227 CrossRef CAS PubMed.
- (a) B. E. Carpenter, W. E. Piers, M. Parvez, G. P. A. Yap and S. J. Rettig, Can. J. Chem., 2001, 79, 857 CrossRef CAS; (b) B. E. Carpenter, W. E. Piers and R. McDonald, Can. J. Chem., 2001, 79, 291 CrossRef CAS; (c) D. J. Parks, W. E. Piers and G. P. A. Yap, Organometallics, 1998, 17, 5492 CrossRef CAS.
- E. Otten, R. C. Neu and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 9918 CrossRef CAS PubMed.
- M. J. Kelly, R. Tirfoin, J. Gilbert and S. Aldridge, J. Organomet. Chem., 2014, 769, 11 CrossRef CAS.
- B. Wrackmeyer, U. Dörfler, W. Milius and M. Herberhold, Z. Naturforsch., B: J. Chem. Sci., 1996, 51, 851 CAS.
- P. L. Coe, R. Stephens and J. C. Tatlow, J. Chem. Soc., 1962, 3227 RSC.
- See also: S. Kronig, E. Theuergarten, D. Holschumacher, T. Bannenberg, C. G. Daniluc, P. G. Jones and M. Tamm, Inorg. Chem., 2011, 50, 7344 CrossRef CAS PubMed.
- (a) P. deFreḿont, N. M. Scott, E. D. Stevens and S. P. Nolan, Organometallics, 2005, 24, 2411 CrossRef; (b) J. J. Dunsford, K. J. Cavell and B. Kariuki, Organometallics, 2012, 31, 4118 CrossRef CAS.
- %Vbur: (a) A. C. Hillier, W. J. Sommer, B. S. Yong, J. L. Petersen, L. Cavallo and S. P. Nolan, Organometallics, 2003, 22, 4322 CrossRef CAS; (b) L. Cavallo, A. Correa, C. Costabile and H. Jacobsen, J. Organomet. Chem., 2005, 690, 5407 CrossRef CAS; (c) A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 1759 CrossRef CAS; (d) F. Ragone, A. Poater and L. Cavallo, J. Am. Chem. Soc., 2010, 132, 4249 CrossRef CAS PubMed; (e) H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841 RSC.
- G. C. Welch, R. Prieto, M. A. Dureen, A. J. Lough, O. A. Labeodan, T. Höltrichter-Rössmann and D. W. Stephan, Dalton Trans., 2009, 1559 RSC.
- See, for example: T. Beringhelli, D. Maggioni and G. D'Alfonso, Organometallics, 2001, 20, 4927 CrossRef CAS.
- G. C. Welch, L. Cabrera, P. A. Chase, E. Hollink, J. D. Masuda, P. Wei and D. W. Stephan, Dalton Trans., 2007, 3407 RSC.
- See also M. J. Sgro, J. Dömer and D. W. Stephan, Chem. Commun., 2012, 48, 7253 RSC.
- (a) G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880 CrossRef CAS PubMed; (b) V. Sumerin, F. Schulz, M. Nieger, M. Leskela, T. Repo and B. Rieger, Angew. Chem., Int. Ed., 2008, 47, 6001 CrossRef CAS PubMed; (c) S. J. Geier and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 3476 CrossRef CAS PubMed.
- 2 captures/cleaves H2O over a relatively short timeframe (ca. 1 h). Prolonged exposure to excess water, however, leads to the formation of boric acid, pentafluorobenzene and ferrocene, each of which could be characterized by multinuclear NMR. Such chemistry mirrors the behaviour of 1 towards water, which also proceeds via B–C bond cleavage.12a.
- For an earlier example of O–H bond cleavage in water by an FLP, see: R. Roesler, W. E. Piers and M. Parvez, J. Organomet. Chem., 2003, 680, 218 CrossRef CAS.
- G. E. Herberich, U. Englert, A. Fischer and D. Wiebelhaus, Organometallics, 1998, 17, 4769 CrossRef CAS.
- http: //evans.rc.fas.harvard.edu/pdf/evans_pKa_table.pdf . Retrieved 24 September 2017.
- M. R. Netherton and G. C. Fu, Org. Lett., 2001, 3, 4295 CrossRef CAS PubMed.
- P. A. Chase and D. W. Stephan, Angew. Chem., Int. Ed., 2008, 47, 7433 CrossRef CAS PubMed.
- A. M. Magill, K. J. Cavell and B. F. Yates, J. Am. Chem. Soc., 2004, 126, 8717 CrossRef CAS PubMed.
- (a) C. M. Mömming, E. Otten, G. Kehr, R. Fröhlich, S. Grimme, D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 48, 6643 CrossRef PubMed; (b) T. Voss, T. Mahdi, E. Otten, R. Fröhlich, G. Kehr, D. W. Stephan and G. Erker, Organometallics, 2012, 31, 2367 CrossRef CAS; (c) E. Theuergarten, T. Bannenberg, M. D. Walter, D. Holschumacher, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2014, 43, 1651 RSC.
- N. Phillips, R. Tirfoin and S. Aldridge, Chem. – Eur. J., 2014, 20, 3825 CrossRef CAS PubMed.
- N. J. Hill, J. A. Moore, M. Findlater and A. H. Cowley, Chem. Commun., 2005, 5462 RSC.
- See also: (a) G. A. Pierce, D. Vidovic, D. L. Kays, N. D. Coombs, A. L. Thompson, E. D. Jemmis, S. De and S. Aldridge, Organometallics, 2009, 28, 2947 CrossRef CAS; (b) S. De, G. A. Pierce, D. Vidovic, D. L. Kays, N. D. Coombs, E. D. Jemmis and S. Aldridge, Organometallics, 2009, 28, 2961 CrossRef CAS.
- (a) IMes: A. J. Arduengo III, H. V. R. Dias, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1992, 114, 5530 CrossRef; (b) IMesCl2: A. J. Arduengo III, F. Davidson, H. V. R. Dias, J. R. Goerlich, D. Khanis, W. J. Marshall and T. K. Prakasha, J. Am. Chem. Soc., 1997, 119, 12742 CrossRef; (c) IDipp: A. J. Arduengo IiI, R. Krafczyk, R. Schmutzler, H. A. Craig, J. R. Goerlich, W. J. Marshall and M. Unverzagt, Tetrahedron, 1999, 55, 14523 CrossRef; (d) 6Mes, 6Dipp: M. Iglesias, D. J. Beetstra, J. C. Knight, L. Ooi, A. Stasch, S. Coles, L. Male, M. B. Hursthouse, K. J. Cavell, A. Dervisi and I. A. Fallis, Organometallics, 2008, 27, 3279 CrossRef CAS.
- (a) Z. Otwinowski and W. Minor, in Processing of X-Ray Diffraction Data Collected in Oscillation Mode, Methods Enzymol, ed. C. W. Carter and R. M. Sweet, Academic Press, New York, 1997, pp. 307–326 Search PubMed; (b) A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343 CrossRef; (c) L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef CAS; (d) P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS; (e) R. I. Cooper, A. L. Thompson and D. J. Watkin, J. Appl. Crystallogr., 2010, 43, 1100 CrossRef CAS; (f) A. L. Thompson and D. J. Watkin, J. Appl. Crystallogr., 2011, 44, 1017 CrossRef CAS; (g) G. M. Sheldrick, SHELX-2014 package, 2014 Search PubMed.
Footnote |
| † CCDC 954137, 1576950–1576952 and 1582718. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt04136e |
| This journal is © The Royal Society of Chemistry 2018 |