
Engineering enzyme microenvironments for enhanced biocatalysis
Louis
Lancaster
 a,
Walaa
Abdallah
b,
Scott
Banta
*b and
Ian
Wheeldon
*a
a,
Walaa
Abdallah
b,
Scott
Banta
*b and
Ian
Wheeldon
*a
aDepartment of Chemical and Environmental Engineering, University of California, Riverside, Riverside, CA 92521, USA. E-mail: iwheeldon@engr.ucr.edu
bDepartment of Chemical Engineering, Columbia University, New York, New York 10027, USA. E-mail: sbanta@columbia.edu
First published on 25th May 2018
Abstract
Protein engineering provides a means to alter protein structure leading to new functions. Much work has focused on the engineering of enzyme active sites to enhance catalytic activity, however there is an increasing trend towards engineering other aspects of biocatalysts as these efforts can also lead to useful improvements. This tutorial discusses recent advances in engineering an enzyme's local chemical and physical environment, with the goal of enhancing enzyme reaction kinetics, substrate selectivity, and activity in harsh conditions (e.g., low or high pH). By introducing stimuli-responsiveness to these enzyme modifications, dynamic control of activity also becomes possible. These new biomolecular and protein engineering techniques are separate and independent from traditional active site engineering and can therefore be applied synergistically to create new biocatalyst technologies with novel functions.
 Louis Lancaster | Louis Lancaster is a PhD candidate in Chemical and Environmental Engineering at the University of California, Riverside, where he also received a bachelor's degree in Chemical Engineering. He works under the guidance of Professor Ian Wheeldon on the development of catalytic cascades that incorporate biocatalysis and electrocatalysis for use in multi-step reaction pathways. |
 Walaa Abdallah | Walaa Abdallah is a fourth-year chemical engineering graduate student in Professor Scott Banta's Lab in Columbia University, USA. She received her Bachelor and Master of Science in Chemical Engineering from Manhattan College. Her research interests include protein engineering, enzyme kinetics, and multi-step catalysis. |
 Scott Banta | Scott Banta PhD is a Professor of Chemical Engineering at Columbia University. He received his BSE degree from the University of Maryland, Baltimore County, and his MS and PhD degrees from Rutgers University. He began his faculty career in the Department of Chemical Engineering at Columbia University in 2004 and his research has focused on the engineering of proteins and peptides for various applications in areas including biocatalysis, bioelectrocatalysis, biomaterials, gene and drug delivery, biosensing, and bioenergy. His group is also developing new biotechnology platforms for energy harvesting and conversion. |
 Ian Wheeldon | Ian Wheeldon is an Associate Professor of Chemical and Environmental Engineering at the University of California, Riverside. He received his PhD in Chemical Engineering from Columbia University, and completed two years of postdoctoral training at Harvard Medical School and the Wyss Institute for Biologically Inspired Engineering at Harvard University. Dr Wheeldon received a Master's of Applied Science from the Royal Military College of Canada, and a Bachelor's of Applied Science from Queen's University, Canada. His research is focused on synthetic biology and biocatalysis. |
Key learning points(1) Enzymes are an attractive option for many catalytic applications due to their high activity, selectivity, and ease of engineering.(2) Catalytic function can be enhanced by creating a local environment around an enzyme or active site that differs from the bulk environment. (3) Engineering the local electrostatic charge can alter substrate selectivity, increase reaction rate, and broaden pH-activity profiles. (4) Dynamic enzyme activity can be created by engineering local environments that change in response to external cues. |
Introduction
Proteins are a diverse and important class of polymeric biomolecules, serving a variety of vital functions in biological systems. One critical function is biocatalysis: enzymes participate in nearly every biochemical reaction necessary for life, including redox processes in energy conversion, the biosynthesis of essential metabolites, transcription and translation of genetic information, and the transport of critical molecules. Proteins also serve as natural structural materials with nano- and microscale chemical and physical features; collagen, spider silk, cytoskeletal actin, and mussel adhesives are just a few examples.Protein engineering is a rapidly maturing field. The standard techniques of rational design were published ∼30 years ago,1 but the past decade has witnessed a stunning growth in the field thanks to advances in other biological pursuits. Low cost DNA synthesis, advances in computational protein design and modelling, new high throughput screening methods, and high-resolution mass-spectrometry are pushing the field forward at a rapid pace.2
Historically, a major thrust in the field of protein engineering has been the development of new catalysts and catalytic processes. Native enzymes possess selectivity unparalleled by their non-biological counterparts, including chemo-, enantio-, and site-selectivity. In addition, enzyme turnover rates on a per site basis can far exceed those attainable with homo- and heterogeneous catalysis. These properties have become highly optimized for their specific environments through natural selection over the course of hundreds of millions of years, so it is no surprise that most enzymes do not perform as well, if at all, outside of their natural cellular milieu. Combinatorial engineering strategies like directed evolution have been very successful in producing enzymes with enhanced stabilities and other properties that make them useful in large-scale applications.3 Similarly, rational design and immobilization strategies have been widely used to enhance stability, turnover, and substrate selectivity.4
A more recent trend in enzyme engineering is an approach that focuses on controlling the nano- and microscale environment of an enzyme or multienzyme complex. A primary goal is to promote optimal conditions in close proximity to the enzyme despite unfavourable or changing conditions in the bulk environment. This would enable increased reaction rates in extreme conditions (e.g., low or high pH), tunable kinetic parameters to fit a desired application, increased molecular efficiency in one-pot and cascade reactions, and dynamic control of activity within multistep reaction systems. Importantly, the protein and biomolecular techniques used to modify enzymes and control the local environment are independent from rational design and directed evolution of the active site and can therefore be applied synergistically with traditional enzyme engineering.
The goals of microenvironment engineering are not dramatically different from those of traditional enzyme immobilization, but the focus is shifted from engineering the support and enzyme attachment to the modification of the local environment of freely diffusing enzymes and enzyme complexes. Over the past three decades, the market share of immobilized enzymes has dramatically decreased, in part due to activity losses from diffusional limitations and from the immobilization procedure themselves.5 Decreased costs of protein production have also partially mitigated the primary advantage of immobilization, that of enzyme recovery and recycling. The microenvironment engineering techniques described here avoid many of the challenges with immobilization. For example, DNA and polymer nanostructures have been used to alter the local electrostatic field and enhance substrate binding,6,7 and protein microcompartment encapsulation can extend the working pH range, control substrate selectivity, and minimize diffusional effects for desired substrates.8,9
In this tutorial review, we aim to provide a concise and informative guide to emerging protein and biomolecular engineering approaches for the development of nano- and microstructured biocatalysts. The discussion begins with examples of engineering enzyme kinetic parameters through modifications targeted outside of the active site. This is followed by discussions of current efforts to control substrate selectivity, extend the working pH-range for high enzyme activity, and dynamically control activity. As many of these techniques are new, their demonstrations are with model enzymatic systems. As such, our discussions focus on the methods used to enable the new techniques and the enhancements brought about in the studied model system.
Each section is not meant to be a comprehensive review, but instead uses select examples to highlight current research trends and approaches to engineering enzyme biocatalysts and their local chemical and physical environments. While our discussions focus on model systems, microenvironment engineering is broadly applicable to current challenges in chemical synthesis, biosensing, enzyme therapeutics, and energy conversion and storage. These approaches are not limited to single reactions but have also proved valuable in designing dynamic activity in cascade reactions and enhancing the flux down multienzyme reaction pathways. We direct readers to other, more comprehensive reviews for a full discussion on the kinetics of multienzyme complexes.10–13
Enhancing reaction kinetics (KM and kcat)
Traditional approaches to enzyme engineering by rational design generally target residues in or very near active sites, changing important amino acids to alter substrate binding affinity, molecular selectivity or catalytic reaction rate. Directed evolution has been particularly successful for the generation of enzymes with activities towards new substrates, enhancing selectivity, or stability, also through active site mutations as well as mutations distributed throughout the protein molecule. These enzyme engineering strategies are well-developed, have been discussed thoroughly elsewhere, and do not fall into the scope of this tutorial.14–17 This section focuses on recent examples in which enzymes were modified with non-mutation based methods to alter the local nano/microscale structure and environment, resulting in improved kinetic parameters (e.g., KM and kcat).One approach that our research groups have explored is the rational design of intermolecular interactions between a nanostructured enzyme and its substrates.6,12,18 This approach was inspired by enzymes like superoxide dismutase (SOD), one of the fastest known enzymes, that exploits electrostatic interactions between a charged substrate and oppositely charged residues on the surface of the enzyme. In the case of SOD, its superoxide substrate is directed to the opening of the active site tunnel by a positively charged patch of surface residues.19 Our engineering strategy mimics this effect by introducing substrate–enzyme binding interactions far from the active site that result in increased local substrate concentrations, effectively reducing the apparent Michaelis constant (KM,app) and driving higher catalytic rates at low bulk substrate concentrations (Fig. 1).
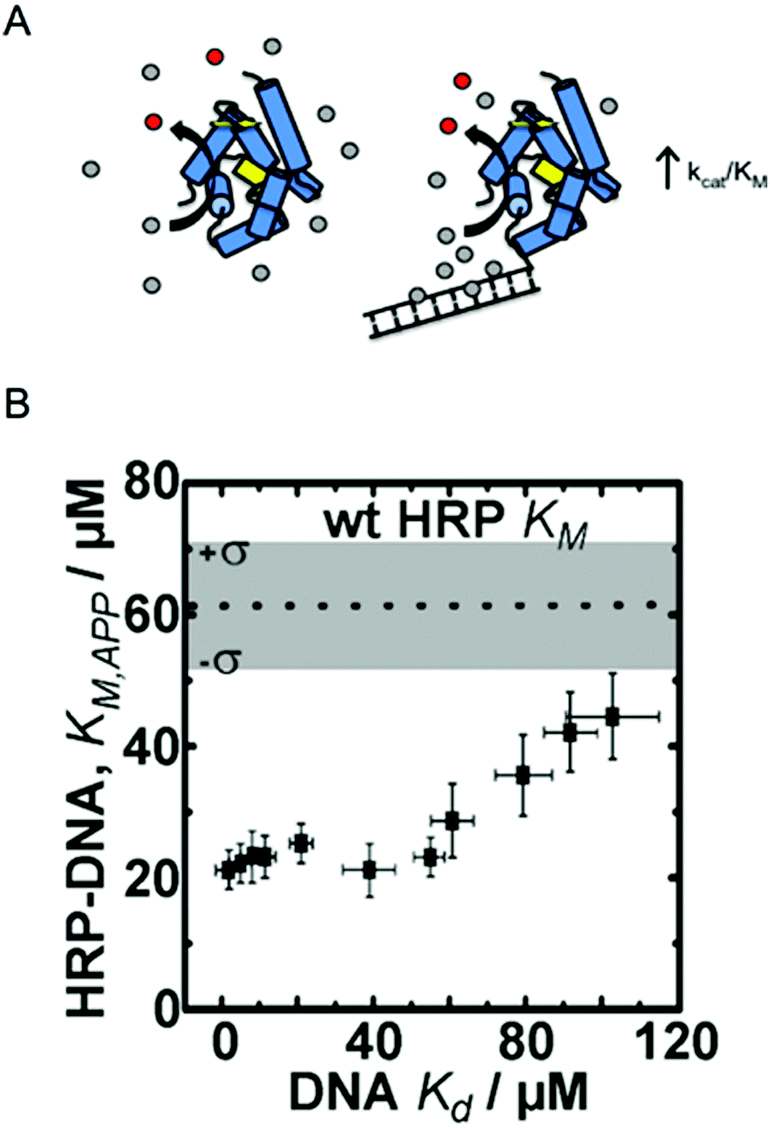 | ||
| Fig. 1 Enzyme engineering through the rational design of intermolecular interactions. (A) A schematic diagram of the approach. Conjugation of a DNA fragment with micromolar binding affinity to the enzyme's substrate can increase the local substrate concentration and increase catalytic efficiency. Reproduced with permission from ACS Catal., 2015, 5(4), 2149–2153. Copyright 2015, American Chemical Society.6 (B) The relationship between DNA binding of the HRP substrate TMB and the apparent KM of HRP–DNA. The DNA sequence-dependent binding of TMB allows for creation HRP–DNA nanostructures with TMB binding affinities ranging from ∼1 to ∼100 μM. Reproduced with permission from ChemBioChem, 2016, 17(15), 1430–1436.18 | ||
Working with the model enzyme horseradish peroxidase (HRP), we created a series of enzyme–DNA structures. HRP was used because it oxidizes a range of different substrates that we identified as micromolar range binders to double stranded DNA.12 DNA was selected as the binding interface because recent advancements in DNA nanotechnology have enabled precise control over molecular level chemical and physical features and user-defined sequences are easily obtained through commercial vendors.20,21 DNA was also selected as the structural modifier because it is known to have affinity towards many different small molecules, including anti-cancer drugs, polymer precursors, chemical nerve agents, and DNA imaging fluorophores among others.22–24
To demonstrate our approach, we first screened multiple HRP substrates for DNA binding affinity using Autodock simulation software. Two substrates were selected based on the binding energies predicted by the software, which were later confirmed with ligand binding assays.6,22 Tetramethylbenzidine (TMB) was selected for its DNA sequence-dependent binding and the common colorimetric substrate ABTS was selected as a control since it has no binding affinity to double stranded (ds) DNA.6 Conjugation of 20 bp dsDNA fragments to free primary amine groups on the surface of HRP by standard bifunctional crosslinking chemistry yielded HRP–DNA structures with ∼1 dsDNA fragment per enzyme. The conjugation of DNA fragments of varying sequence and structure (dsDNA and DNA DX tiles; see ref. 20) produced a series of HRP–DNA complexes with a range of binding affinity to TMB (∼1 to 100 μM). Full kinetic analysis of the HRP–DNA structures revealed that the binding affinity of the DNA to TMB decreased KM,app from 60 ± 16 μM to 23 ± 3 μM (a 2.6-fold decrease), but did not significantly alter kcat (∼80 s−1). As shown in Fig. 1B, the effect increased with stronger TMB binding, reaching a maximum effect when the Kd of TMB to DNA was less than ∼20 μM. This trend was not observed with the control substrate ABTS, which has no affinity to the HRP conjugated DNA. Kinetic analysis of HRP–DNAs with 4-aminophenol (4-AP) as a substrate confirmed the effect of the designed intermolecular interactions. The KM of 4-AP was reduced from 7 ± 1 mM to 2.9 ± 0.5 mM when a DNA DX tile with a measured Kd of 2 ± 1 μM to TBM was attached.18
Short dsDNA fragments were also attached to the alcohol dehydrogenase D (AdhD) from Pyrococcus furiosus to explore whether the DNA binding effect could be used to enhance cofactor utilization. We had previously engineered AdhD to accept the NAD(H) mimic nicotinamide mononucleotide (NMN(H)) in place of NAD(H).25 This provided an enzyme system to test our enzyme–DNA strategy because NMN(H) has micromolar binding affinity to DNA, while NAD(H) does not.6 Conjugation of a 20 bp dsDNA fragment to AdhD did not significantly alter kinetic parameters with NAD+ and 2,3-butanediol as co-substrates, but binding of NMN+ was enhanced (Kd,app for NMN+ decreased from 534 ± 30 μM to 294 ± 18 μM). In this case, the DNA-cofactor binding effect did not enhance catalysis as the increased local cofactor concentration led to substrate inhibition, a characteristic common to ordered bi–bi NAD(H) cofactor dependent enzymes.26
With both HRP and AdhD, the result of attaching a designed DNA nanostructure was increased substrate binding. AhdD–DNA constructs exhibited a reduced Kd,app towards the cofactor mimic NMN+. With HRP–DNA, KM,app for the substrate TMB was reduced. These experimental results were supported by Brownian dynamic simulations. Simulations of HRP–DNA suggested that the presence of DNA results in an increased local residence time of the substrate, as well as an increased substrate on-rate (kON).18 The latter effect was also confirmed experimentally with a 3.7-fold increase in kON of TMB to HRP–DNA in comparison to unmodified HRP.
Another successful approach to increasing the local substrate concentration is creating enzyme structures with covalently bound but mobile substrates. This approach was taken by Fu et al. in designing multienzyme complexes with cofactor swing arms.27 The strategy was inspired by naturally occurring chemical swing arms that channel substrates from one active site to another in multienzyme complexes.28 One natural example is the pyruvate dehydrogenase complex, which catalyses the oxidative decarboxylation of pyruvate to acetyl coenzyme A in a three-step cascade. Fu et al.'s engineered complex of glucose-6-phosphate dehydrogenase (G6pDH) and malate dehydrogenase (MDH) was created by tethering the enzymes to a DNA DX tile functionalized with a poly(T)20 swing arm modified with an NAD(H) moiety. The cofactor swing arm resulted in efficient cascade catalysis (90-fold greater than with freely diffusing NAD(H)), in large part due to an increase in local cofactor concentration. Titration experiments with free NAD(H) suggested that the swing arm bound cofactor creates an effective concentration of ∼20 μM when one NAD(H) swing arm is present for each enzyme pair.
In addition to engineering KM, enzyme nanostructures have also been successful in boosting reaction turnover. One recent example showed that the turnover of HRP can be enhanced when modified with large, flexible dsDNA.29 In this case, multiple long dsDNA strands were appended to HRP by initiator-triggered hybridization chain reaction. The result was HRP crowded by dsDNA, which increased kcat by >3-fold while maintaining a KM value equivalent to unmodified HRP. It is important to note here that the substrate (ABTS) does not have DNA binding affinity. This same effect was also observed with a series of model enzymes encapsulated in DNA nanocages, tightly packed dsDNA structures that were used to encapsulate single enzymes and two-step enzyme cascades.30 Kinetic analysis of nanocage–enzyme structures revealed increased turnover of HRP, MDH, G6pDH, lactate dehydrogenase (LDH), and glucose oxidase (GOx). The increases in kcat ranged from a high of 9.6-fold for HRP, to a low of 4.1-fold for LDH.
A similar example modified β-lactamase (βLac) with up to four strands of 48.5 kbp lamda phage DNA (λ DNA). The result was again an increase in kcat (∼2-fold enhancement) with no significant effect on KM.31 This example is discussed in more detail later in the dynamic activity subsection of this review, because the authors were able to show control of enzyme activity by altering the structure of the conjugated DNA.
In the KM-engineering examples discussed above, there is a clear mechanism that drives the effect. The introduction of new substrate binding interactions or the covalent attachment of cofactor in close proximity to the enzyme results in an increase in effective local concentration. This increase in substrate (or cofactor) concentration creates an environment where KM,app is lower than the intrinsic KM of the enzyme. The mechanism of turnover enhancement is less clear. Increased turnover was not observed when small DNA fragments were used (e.g., 20 bp dsDNA or 1 DNA DX tile, see ref. 6 and 18), but turnover was increased with larger DNA modifications (e.g., λ DNA, see ref. 29 and 31). This suggests that the high charge density of the DNA backbone likely plays a significant role in altering the local environment. For example, the negative charge of the phosphate backbone may decrease local pH.32 Controlling the pH through enzyme-nanostructures is addressed in detail in the following section.
Tuning pH-activity profile
A common challenge in enzyme catalysis is a limited pH range for high activity. This is a problem for both single enzyme reactions as well as multienzyme cascades, which require optimization of all reaction steps within the same bulk environment. This subsection reviews select examples of enzyme engineering focused on tuning enzyme pH-activity profiles.Chymotrypsin (ChT) is a commonly used protease in LC-MS peptide fingerprinting and other protein degradation applications. Substrate specificity towards charged peptides and its use at low pH (pH 5 and below) has recently been explored through engineering the local environment. For example, Murata et al. reported the ability to graft charged polymers onto the enzyme's surface in a controlled manner.7 Positively charged poly(quaternary ammonium) molecules were grown on the surface of ChT by atom transfer radical polymerization (ATRP) to produce ChT–pQA. Polymer growth occurred from initiator molecules conjugated to free lysine residues (ChT has 14 surface lysines), thus creating a dense cationic shell around the enzyme. By controlling the stoichiometry of surface initiators, enzymes were modified with varying quantities of polymer molecules and excess charge, the largest of which had a surface area 40-fold greater than unmodified ChT, and a substantially increased positive charge.
The reaction mechanism of proteolytic cleavage is dependent on the stabilization of peptide substrates in the catalytic triad, serine, aspartic acid, and histidine. The protonation state of the histidine is critical; it must be deprotonated for the reaction to proceed. With a pKa of 7.3, the protonation state is highly sensitive at near neutral pH with low activity under acidic conditions.33 The cationic shell of ChT–pQA effectively reduces the pKa of the histidine, increasing catalytic activity at lower pH. As judged by a 5-fold increase in kcat/KM, the pH optimum was shifted from pH 8 (unmodified ChT) to pH 5 (ChT–pQA). The polymer-modified enzyme was also determined to be more thermostable, exhibit higher activity towards negatively charged substrates, and to have a measurable binding affinity towards charged inhibitors.7
Enzyme/polymer modifications have also been used to engineer pH-activity profiles of two-step reaction cascades. In this example, D-amino acid oxidase (DAAO) and cytochrome C (Cyt C) were used to catalyse the oxidation of alanine and the removal of coproduced hydrogen peroxide (by DAAO and Cyt C, respectively; Fig. 2).34 The challenge of this cascade is that DAAO exhibits optimum activity between pH 8 and 9, while Cyt C is active under slightly acidic conditions (pH ∼4.5). Conjugation of negatively charged poly(methacrylic acid) to Cyt C created an anion shell around the protein that maintained a local environment of low pH. The engineered cascade achieved rates upward of 10-fold higher than the unmodified enzymes.
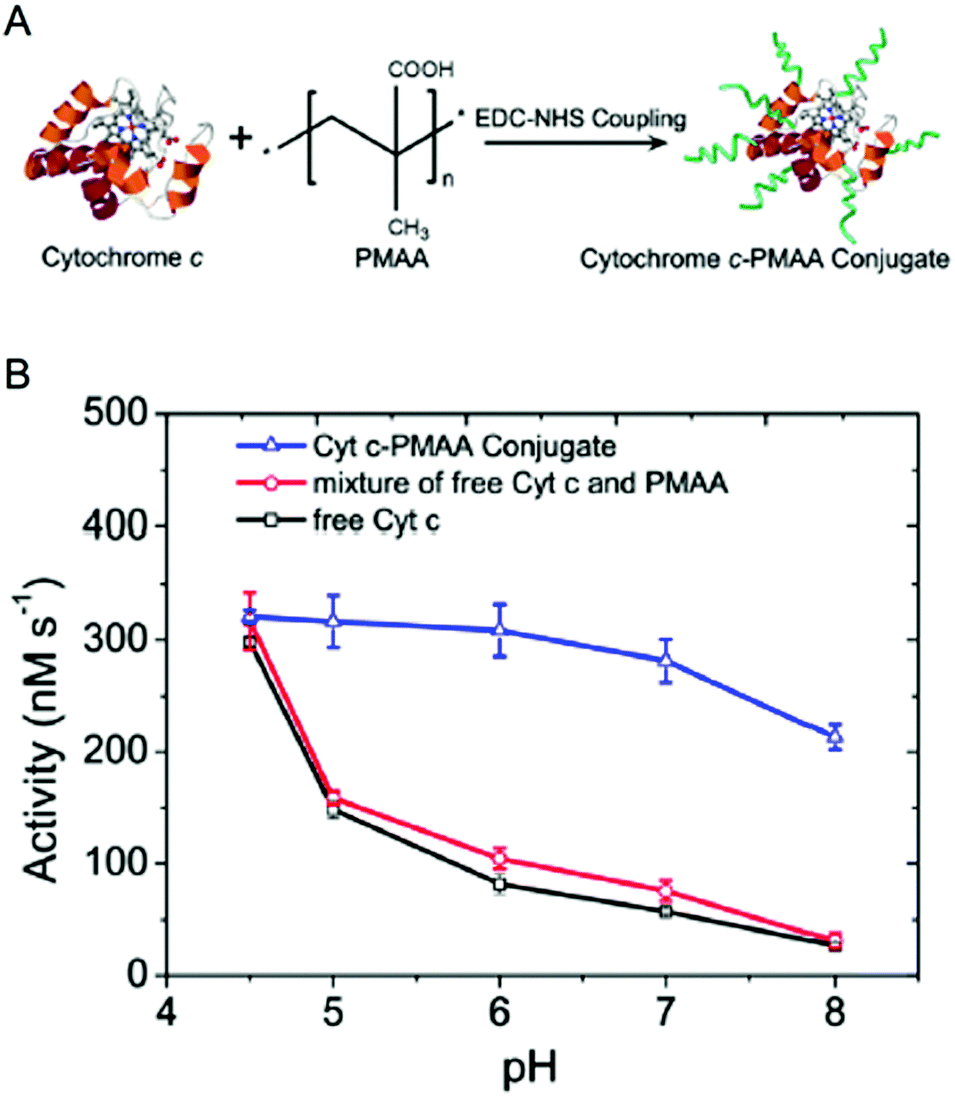 | ||
| Fig. 2 Engineering pH-activity profiles. (A) Poly(methacrylic acid) modification of cytochrome C. (B) The pH-activity profile of cytochrome C towards the oxidation of Amplex Red in the presence of hydrogen peroxide. The negatively charged polymer shell around cytochrome C maintains a low local pH resulting in higher activity in more alkaline solutions. Reproduced with permission from ACS Catal., 2017, 7(3), 2047–2051. Copyright 2017, American Chemical Society.34 | ||
Similar to the poly(methacrylic acid) modifications to Cyt C, many of the enzyme–DNA examples discussed in the previous subsection result in nanostructures where enzymes are crowded by negative charge. A recent analysis of the effect of DNA on the local pH (i.e., the protonation state of charged residues) shows that DNA nanostructures can alter the pKa of a given residue within a few nanometers of the structure.32 These calculations were used to demonstrate that many of the observed enhancements in multistep enzyme nanostructures are potentially due to pH matching (as described above in the DAAO/Cyt C example) and not due to reduced interenzyme distances or enzyme proximity. A recent work by Yan and coworkers directly tested this effect by encapsulating enzymes (both single enzymes and two-step enzyme cascades) in DNA nanocages.30 The results of this study show that the DNA nanostructure is largely responsible for increased cascade activity by enhancing turnover of the enzyme components, and that enzyme proximity has only a minor effect on cascade throughput. More accurate calculations and further experiments are still needed, but the effect of altered pH due to highly charged local environments (from charged polymers or from DNA) has substantial explanatory power to describe the mechanism(s) of turnover enhancement in enzyme structures.
Here we describe a number of current examples that use polymer and DNA modifications to control the local enzyme environment, but the general concept was shown over 50 years ago. In a 1964 publication, Goldstein et al. showed that modification of the protease trypsin with a maleic acid/ethylene copolymer shifted the pH optimum 2.5 pH units more alkaline, under conditions of low ionic strength.35 This concept is being applied again using current biomolecular and protein engineering techniques to enhance the pH-activity profiles of various enzymes and multienzyme complexes.
Engineering substrate specificity
The previous sections discussed the use of charged molecules (e.g., DNA, cationic and anionic polymers) to modify the local enzyme environment with the goals of enhancing substrate binding, catalytic turnover, and pH-activity profile. Similar strategies, as well as others that restrict substrate access by electrostatics or steric hindrance, with the goal of engineering substrate specificity are discussed here. These strategies are generally not directed at increasing turnover of a given substrate, but instead promoting the reaction of one substrate over another by controlling access to an enzyme's active site.One recent example uses a protein microcompartment, or nanocage, to create an electrostatic barrier between a protease and its charged peptide substrates.36 The cage-forming protein lumazine synthase from Aquifex aeolicus (AaLS-13) assembles into an icosahedral structure with pores interspersed through the shell. The pores are sufficiently sized such that small globular proteins and peptide substrates can cross the protein barrier, which through a series of mutations maintains an overall negative charge (Fig. 3A).
 | ||
| Fig. 3 Tuning protease substrate specificity with protein microcompartment encapsulation. (A) Encapsulation of TEVp protease fused to supercharged GFP (GFP(+36)) in a lumazine synthase (AaLS-13) microcompartment. A series of differently charged peptide substrates is shown: tev-K7E0 is positively charged (++), K4E3 is zwitterionic (+−), and tev-K1E6 is negatively charged (−−). (B) Encapsulated GFP(+36)–TEVp turnover with the series of peptides, blue is tev-K7E0, purple is rev-K4E3, and red is tev-K1E6. Adapted with permission from J. Am. Chem. Soc., 2018, 140, 860–863. Copyright 2018, American Chemical Society.36 | ||
The high negative charge of the protein shell creates a technical challenge for loading enzyme cargo. One successful solution was the fusion of supercharged GFP to the enzyme of interest,8,37 in this case the protease TEVp. Fusion of GFP(+36) to TEVp resulted in the loading of up to 72 fusion proteins per nanocage; however, optimal reaction kinetics were found with only four GFP(+36)–TEVp molecules per compartment.
Proteolytic activity of encapsulated GFP(+36)–TEVp, unmodified TEVp, and GFP(+36)–TEVp was measured with a series of peptide substrates. This series included peptides with overall negative charge (tev-K1E6), overall positive charge (tev-K7E0), a neutral peptide (tev-K1), and a zwitterionic peptide (tev-K4E3). Before being fused to GFP(+36), TEVp showed no preference for any of the substrates. Fusion of GFP(+36) to TEVp began to shift substrate selectivity towards the anionic peptide; electrostatic attraction of the peptide resulted in a 4-fold decrease in KM,app. When assembled in the AaLS-13 nanocage, cleavage of the neutral peptide by GFP(+36)–TEVp was reduced by 6-fold, activity toward the zwitterionic substrate was reduced by 7-fold, and the anionic peptide by 100-fold. Conversely, the rate of hydrolysis of the cationic substrate increased by 5-fold.
The ChT–pQA system discussed in the previous section creates a similar microenvironment to the protein nanocage, but in this case with a positively charged barrier. That is, ChT (chymotrypsin) was encapsulated in a positively charged pQA shell. In addition to modifying the pH-activity profile, the charged shell also created a microenvironment that preferences anionic protease inhibitors while disfavouring the binding of positively charged inhibitors. A negatively charged inhibitor bound 3.7-fold more strongly to ChT–pQA than to unmodified ChT. The effect with a positive inhibitor was significantly more dramatic with a 27-fold decrease in binding when ChT was modified with pQA.7
The protein microcompartment strategy has also been demonstrated with engineered viral capsids. The Tullman-Ercek group tuned the electrostatic charge around the capsid pores to control the flux of substrates and products of an alkaline phosphatase in and out of the nanoreactor.9 By altering pore charge the effective KM of PhoA decreased nearly 2-fold, demonstrating the ability to tune enzyme kinetics through protein encapsulation.
Other microenvironment engineering strategies have also been demonstrated. A series of papers by Rotello and coworkers use amino-acid-functionalized gold nanoparticles to create a charged environment local to ChT.38,39 When ChT was bound to particles decorated with glutamic acid residues, activity towards cationic substrates increased 3-fold. Activity was cut in half for neutral substrates, and anionic substrate activity was reduced by 95%. Analysis of these experiments led to the conclusion that the glutamic acid modifications influenced the diffusion of substrate molecules to the nanoparticle-bound protease. Positively charged substrates could access the immobilized enzyme, while diffusion of negatively charged peptides in the local environment was significantly restricted.
An alternative strategy to tuning substrate specificity is to control active site access via conditional steric hindrance. This concept was recently explored using a light-activated conformational switch conjugated near the active site of a lipase from Bacillus thermocathenolatus (BTL2).40 In this work, two different small molecules that undergo light-induced structural changes were attached separately to five different residues in and around the substrate binding pocket. Azobenzene transitions between cis and trans under UV and visible light, respectively, while ring opening and closing of iodoacetate–spiropyran follows the same light switching pattern. These systems were explored to alter and control the native enantioselectivity of BTL2.
The most successful construct was the conjugation of azobenzene to P295, a residue outside of the binding pocket. The trans conformer leaves the active site unobstructed and kinetic analysis showed that the native selectivity towards the R enantiomer of 2-butyryloxy-2-phenylacetic acid remained intact. Under UV light, the cis isomer partially blocks substrate binding leading to a preference for the S enantiomer. This light-induced selectivity switching resulted in a ∼60% change in enantiomeric excess from R to S, when calculated at reaction yields lower than 25%. Other constructs also showed changes in selectivity but not to the extent of the P295 conjugate. Docking simulations supported the experimental results. Enzyme–azobenzene conjugates that had larger shifts in enantioselectivity also showed corresponding changes in enantiomer binding energies. The S isomer showed increased binding affinity for the active site when the ligand was in its UV-induced conformation, and the R isomer was preferred when the ligand was in its relaxed state.
The authors of this work describe the opening to BTL2's active site as a “lid aperture”. The photochromic molecules were attached to P295, a residue that acts as a hinge for the peptide lid that creates the opening. The cis/trans conformation of azobenzene alters the position of the lid, thus controlling stereoselectivity. This effect is supported by the analysis of other lipase lid structures over the past 20 years that show a relationship between lid structure, access to the active site, and substrate selectivity.41,42
A second example of controlled selectivity through steric blocking is monobody-mediated control of β-galactosidase activity.43 BgaD-D from Bacillus circulans exhibits trans-galactosylation activity, converting lactose into galacto-oligosaccharides of varying chain length. Unmodified BgaD-D concatenates lactose units with no specificity towards tri-, tetra-, and longer chain length oligosaccharides. Partial blocking of the active site with an engineered binding protein (i.e., the monobody), suppressed nearly all activity towards tetrasaccharides and longer oligosaccharides.
Dynamic enzyme activity
Common to the enzyme engineering efforts described in the previous sections is the use of protein and biomolecular techniques to control the local chemical and physical environment. As a result, the effects are often interrelated: for example, creating a charged shell around an enzyme can both increase the effective pH range and alter substrate selectivity. Similarly, exploiting conformational change in photoactive molecules to control substrate binding also crosses the boundaries between the sections of this review. That is, the control of selectivity (and activity towards a given substrate) is dynamic.Dynamic regulation of enzymatic activity is an important observation in natural metabolic pathways as this is a means for pathways to employ feedback loops and other control strategies in response to environmental stimuli without waiting for the slow responses required by changes in gene expression. Engineers have been developing dynamically controlled protein switches44,45 to enable new sensing, regulation and cellular control strategies. Protein engineering can be used to introduce classic allosteric regulation into enzymes, or the conformational dynamics of one protein domain can be used to influence the activity of another. Another emerging route towards the dynamic control of enzyme function is through the introduction of dynamic control of the local chemical environment.
In our previous work, we have engineered cofactor selectivity in various aldo–keto reductases including the thermostable AdhD from P. furiosus, but none of our previous approaches allowed for dynamic regulation.26,46 Recently, we inserted a calcium-dependent peptide from the fifth block of the RTX domain of the adenylate cyclase toxin from Bordetella pertussis into AdhD.47 The RTX peptide is intrinsically disordered in the absence of calcium and folds into a β-roll structure upon calcium binding.48,49 Previous work with AdhD had demonstrated that mutations made in the substrate binding loops can alter cofactor and substrate selectivity.50 Using this information, the RTX peptide was inserted into substrate binding loop A, which is distal to the binding pocket, resulting in the chimeric enzyme β-AdhD (Fig. 4A). The hypothesis was that upon calcium binding the β-roll structure would alter the local environment of the active site, resulting in calcium-dependent activity.
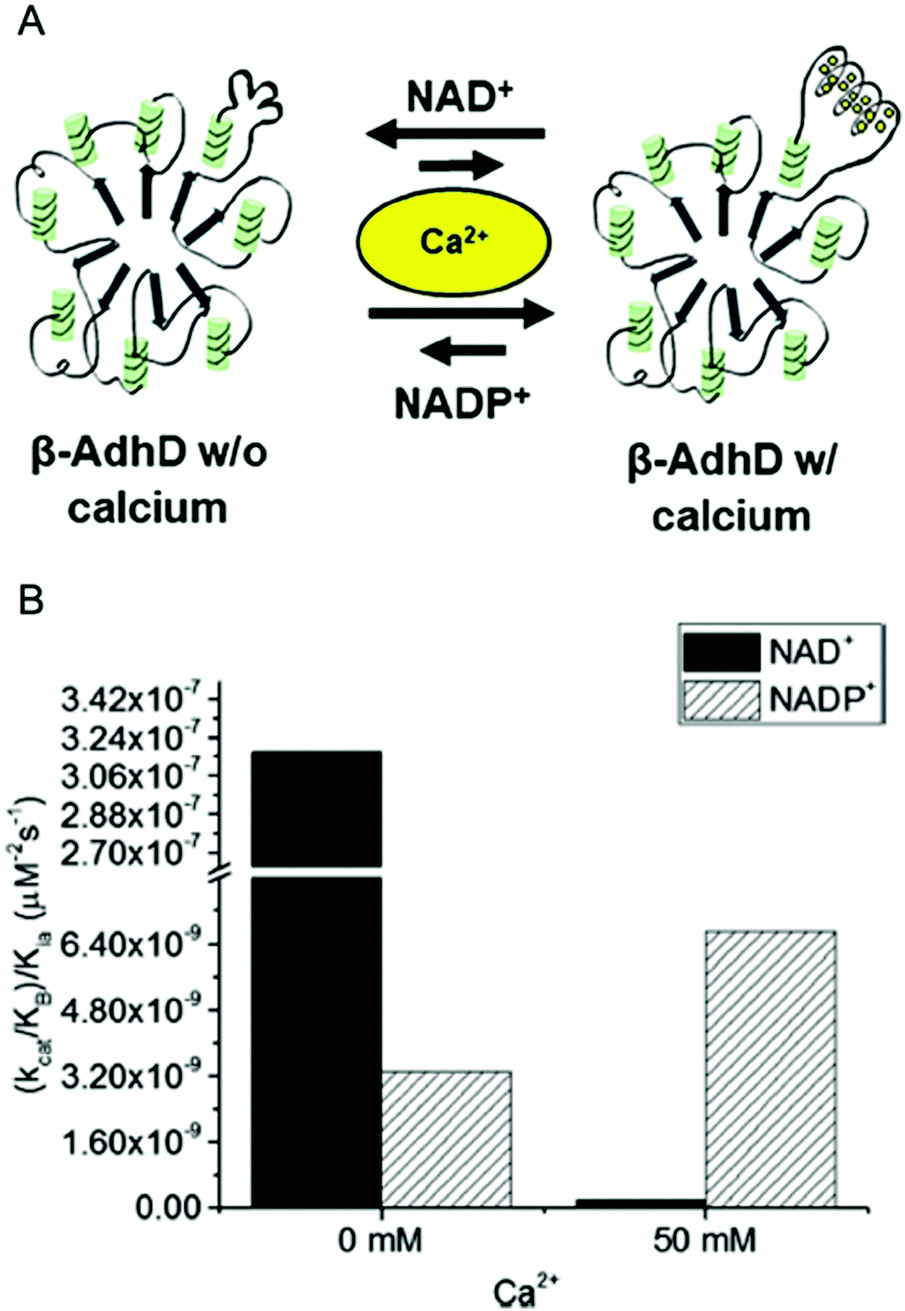 | ||
| Fig. 4 Calcium-regulated activity of β-AdhD. (A) A schematic of the calcium-dependent activity and cofactor switching of β-AdhD, an engineered alcohol dehydrogenase D (AdhD) from P. furiosus. The 5th block of the RTX domain of the adenylate cyclase toxin from Bordetella pertussis was inserted into a substrate binding loop. The addition of calcium ions induces folding of the RTX domain into a β-roll. (B) Apparent catalytic efficiency of β-AdhD with 0 and 50 mM calcium with both NAD+ and NADP+. Reproduced with permission from ACS Catal., 2018, 8, 1602–1613. Copyright 2018, American Chemical Society.47 | ||
Kinetic analysis of β-AdhD revealed that insertion of the RTX domain decreased overall activity but introduced the ability to control cofactor selectivity. Upon calcium addition, β-AdhD's NADP+-dependent activity increased, while NAD+-dependent activity was substantially reduced (Fig. 4B). Kinetic analysis with NAD+ as a cofactor suggests that the folded RTX domain perturbs the formation of the enzyme–cofactor–substrate complex. When the calcium concentration exceeded the dissociation constant for the RTX domain, a decrease in affinity for NAD+ was observed. However, the reason why this affected catalysis with NAD+ but not NADP+ was not readily apparent. Further analysis revealed that calcium is a competitive inhibitor to NAD+ in the wild type enzyme. The RTX domain in β-AdhD also allows calcium to act as a non-competitive inhibitor by altering active site geometry. Thus, in β-AdhD the effect of calcium both in the active site and in the RTX domain resulted in calcium serving as a competitive and non-competitive inhibitor of NAD+-dependent activity. Catalysis with NAD+ can proceed effectively in the absence of calcium, while cofactor utilization transitions to a preference for NADP+ in the presence of calcium.
This approach to dynamic regulation of cofactor-dependent activity is also biomimetic. It is becoming clear that many proteins exhibit dynamic structures to accommodate their diverse functions. Observed dynamic effects range from allosteric regulation of protein function by small structural changes upon ligand binding to folding–unfolding transitions in the increasingly important family of intrinsically disordered proteins.51,52 Changing structural conformation allow proteins to alter their chemical functionality and increasingly, these dynamic structural perturbations are being explored for engineering applications.
Another way to control activity is through enzyme modifications using dynamic DNA structures. One example is the attachment of λ DNA to βLac, an example described above that resulted in enhanced catalytic turnover. In addition to increased kcat, the large 48.5 kbp pieces of λ DNA attached to the enzyme also enabled dynamic control over the rate enhancement.31 Structural analysis of the modified enzyme showed that upon the addition of 50 μM spermine (SPM4+), the λ DNA fragments change conformation. As the strands became compact, enzymatic function resembled that of the unmodified βLac. DNA compaction and the effect this had on catalysis were shown to be reversible, for example the addition of NaCl allowed the λ DNA to unfold and restore upward of ∼80% of the pre-compacted activity.
The last example of dynamic activity is one that controls the rate of a two-step reaction cascade by modifying the structure of a multienzyme complex. A model GOx/HRP reaction cascade was attached to a DNA nano-tweezer; GOx at the end of one arm and HRP at the end of the other.53 In the closed conformation, the enzymes are in close proximity. The addition of “fuel” DNA strands forces a change in the shape of the nanostructure, pulling the enzymes apart (the open state). The system can be cycled from open to closed through the repeated addition of “fuel” and “antifuel” strands, opening and closing the structure, respectively.
In the closed state, the rate of the coupled reaction is high. The change in structure and the separation of the enzymes slows the conversion of GOx produced hydrogen peroxide to water with the concomitant oxidation of ABTS. Kinetic analysis of the same GOx/HRP cascade immobilized inside DNA nanocages suggests that the change in cascade rate is due to a combination of effects, including a change in local chemical environment and a change in interenzyme distance. It is possible that in the closed state, the enzymes are crowded by DNA increasing HRP reaction rate as was observed with the examples discussed at the beginning of this review (see ref. 29–31). Interenzyme distance may also play a role in the transient kinetics.54
Conclusion and outlook
Decades of rational protein engineering design efforts have focused on creating mutations in or near active sites, binding pockets, interfaces, and similar regions that influence substrate–enzyme interactions. However, it is common in combinatorial engineering work, including directed evolution, to identify positive mutations at locations distant from the active site. The reasons for the beneficial impacts of these mutations are often not clear. As more and more biomolecular engineering efforts are aimed at altering the local environment of enzymes and their active sites it is becoming clear that these types of approaches can be just as important and beneficial as traditional enzyme engineering. It also seems likely that distant cryptic mutations found in directed evolution selections play important roles in these effects.This tutorial review presents a set of experimental work that we feel represents the current trends in engineering the local environment of enzymes. The goal of these works is to enhance enzyme catalysis: reaction rates, pH-activity profiles, and substrate selectivity. Biomolecular and protein engineering strategies to control the local environment have also enabled dynamic enzyme activity that responds to external cues. Combined, these examples also show how nanoscale engineering focused on enzyme modification outside of the active site can enhance catalysis in a controlled and rational manner. These examples also demonstrate how altering the local environment can have interrelated effects on catalytic function, for example, simultaneously broadening pH-activity profiles and altering substrate selectivity.
Individually, these examples present novel strategies in their respective efforts in enzyme engineering. But as these works were viewed and discussed in the context of one another, some similarities become increasingly clear. This is particularly true for controlled local charge. Substrate selectivity and pH-activity profiles were both altered by modifying the electrostatics in close proximity to the enzyme of interest. Similarly, DNA crowding increased turnover, possibility due to an enhanced pH-activity profile. DNA nanostructures were also able to create new intermolecular binding interactions between a modified enzyme and its substrates to increased substrate binding. With respect to dynamic control, two different strategies are apparent. One alters the architecture or structure of nanoengineered enzymes to transiently modify the local environment (e.g., local electrostatics). The other regulates access to an active site by engineering conformational changes close near the active site. As the available biomolecular and protein engineering tools continue to develop, it may be possible to better integrate the enhancements that result from a given strategy, as well as independently tune the desired effects.
Looking to the future, we believe that engineering the nano- and microscale environment around enzymes and their active sites will enable new technologies for multistep reaction cascades, tuneable activity, and other dynamic systems that can adapt to changing environmental conditions.55 These technologies could benefit chemical manufacturing by enabling biocatalysis in harsh bulk environments and increasing synthesis efficiency by combining multiple reaction steps into a single pot. Biosensing and enzyme therapeutics would benefit from enhanced substrate selectivity and higher turnover at low substrate concentrations. When combined with the power of traditional rational design and combinatorial enzyme engineering approaches, microenvironment engineering promises to have a significant impact on the future of biocatalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Army Research Office MURI grant W911NF1410263 for funding.Notes and references
- F. H. Arnold, Curr. Opin. Biotechnol., 1993, 4, 450–455 CrossRef PubMed.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef PubMed.
- F. H. Arnold, Acc. Chem. Res., 1998, 31, 125–131 CrossRef.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- R. DiCosimo, J. McAuliffe, A. J. Poulose and G. Bohlmann, Chem. Soc. Rev., 2013, 42, 6437–6474 RSC.
- Y. N. Gao, C. C. Roberts, J. Zhu, J. L. Lin, C. E. A. Chang and I. Wheeldon, ACS Catal., 2015, 5, 2149–2153 CrossRef.
- H. Murata, C. S. Cummings, R. R. Koepsel and A. J. Russell, Biomacromolecules, 2014, 15, 2817–2823 CrossRef PubMed.
- R. Zschoche and D. Hilvert, J. Am. Chem. Soc., 2015, 137, 16121–16132 CrossRef PubMed.
- J. E. Glasgow, M. A. Asensio, C. M. Jakobson, M. B. Francis and D. Tullman-Ercek, ACS Synth. Biol., 2015, 4, 1011–1019 CrossRef PubMed.
- H. O. Spivey and J. Ovadi, Methods, 1999, 19, 306–321 CrossRef PubMed.
- I. Wheeldon, S. D. Minteer, S. Banta, S. C. Barton, P. Atanassov and M. Sigman, Nat. Chem., 2016, 8, 299–309 CrossRef PubMed.
- J. L. Lin and I. Wheeldon, ACS Catal., 2013, 3, 560–564 CrossRef.
- Y. F. Zhang and H. Hess, ACS Catal., 2017, 7, 6018–6027 CrossRef.
- C. Jackel, P. Kast and D. Hilvert, Annu. Rev. Biophys., 2008, 37, 153–173 CrossRef PubMed.
- A. S. Bommarius and M. F. Paye, Chem. Soc. Rev., 2013, 42, 6534–6565 RSC.
- D. L. Trudeau, M. A. Smith and F. H. Arnold, Curr. Opin. Chem. Biol., 2013, 17, 902–909 CrossRef PubMed.
- M. T. Reetz, J. Am. Chem. Soc., 2013, 135, 12480–12496 CrossRef PubMed.
- Y. Gao, C. C. Roberts, A. Toop, C. E. Chang and I. Wheeldon, ChemBioChem, 2016, 17, 1430–1436 CrossRef PubMed.
- E. D. Getzoff, J. A. Tainer, P. K. Weiner, P. A. Kollman, J. S. Richardson and D. C. Richardson, Nature, 1983, 306, 287–290 CrossRef PubMed.
- X. J. Li, X. P. Yang, J. Qi and N. C. Seeman, J. Am. Chem. Soc., 1996, 118, 6131–6140 CrossRef.
- M. R. Jones, N. C. Seeman and C. A. Mirkin, Science, 2015, 347, 1260901 CrossRef PubMed.
- Y. Gao, S. Or, A. Toop and I. Wheeldon, Langmuir, 2017, 33, 2033–2040 CrossRef PubMed.
- G. E. Kellogg, J. N. Scarsdale and F. A. Fornari, Nucleic Acids Res., 1998, 26, 4721–4732 CrossRef PubMed.
- Y. Ma, J. Zhang, G. Zhang and H. He, J. Am. Chem. Soc., 2004, 126, 7097–7101 CrossRef PubMed.
- E. Campbell, M. Meredith, S. D. Minteer and S. Banta, Chem. Commun., 2012, 48, 1898–1900 RSC.
- E. Campbell, I. R. Wheeldon and S. Banta, Biotechnol. Bioeng., 2010, 107, 763–774 CrossRef PubMed.
- J. Fu, Y. R. Yang, A. Johnson-Buck, M. Liu, Y. Liu, N. G. Walter, N. W. Woodbury and H. Yan, Nat. Nanotechnol., 2014, 9, 531–536 CrossRef PubMed.
- R. N. Perham, Annu. Rev. Biochem., 2000, 69, 961–1004 CrossRef PubMed.
- J. Collins, T. Zhang, S. W. Oh, R. Maloney and J. Fu, Chem. Commun., 2017, 53, 13059–13062 RSC.
- Z. Zhao, J. Fu, S. Dhakal, A. Johnson-Buck, M. Liu, T. Zhang, N. W. Woodbury, Y. Liu, N. G. Walter and H. Yan, Nat. Commun., 2016, 7, 10619 CrossRef PubMed.
- S. Rudiuk, A. Venancio-Marques and D. Baigl, Angew. Chem., Int. Ed., 2012, 51, 12694–12698 CrossRef PubMed.
- Y. Zhang, S. Tsitkov and H. Hess, Nat. Commun., 2016, 7, 13982 CrossRef PubMed.
- S. P. Edgcomb and K. P. Murphy, Proteins, 2002, 49, 1–6 CrossRef PubMed.
- Y. F. Zhang, Q. Wang and H. Hess, ACS Catal., 2017, 7, 2047–2051 CrossRef.
- L. Goldstein, Y. Levin and E. Katchalski, Biochemistry, 1964, 3, 1913–1919 CrossRef PubMed.
- Y. Azuma, D. L. V. Bader and D. Hilvert, J. Am. Chem. Soc., 2018, 140, 860–863 CrossRef PubMed.
- Y. Azuma, R. Zschoche, M. Tinzl and D. Hilvert, Angew. Chem., Int. Ed., 2016, 55, 1531–1534 CrossRef PubMed.
- C. C. You, S. S. Agasti, M. De, M. J. Knapp and V. M. Rotello, J. Am. Chem. Soc., 2006, 128, 14612–14618 CrossRef PubMed.
- C. C. You, M. De, G. Han and V. M. Rotello, J. Am. Chem. Soc., 2005, 127, 12873–12881 CrossRef PubMed.
- A. Bautista-Barrufet, F. Lopez-Gallego, V. Rojas-Cervellera, C. Rovira, M. A. Pericas, J. M. Guisan and P. Gorostiza, ACS Catal., 2014, 4, 1004–1009 CrossRef.
- J. Uppenberg, N. Ohrner, M. Norin, K. Hult, G. J. Kleywegt, S. Patkar, V. Waagen, T. Anthonsen and T. A. Jones, Biochemistry, 1995, 34, 16838–16851 CrossRef PubMed.
- P. Grochulski, Y. Li, J. D. Schrag and M. Cygler, Protein Sci., 1994, 3, 82–91 CrossRef PubMed.
- S. Tanaka, T. Takahashi, A. Koide, S. Ishihara, S. Koikeda and S. Koide, Nat. Chem. Biol., 2015, 11, 762–764 CrossRef PubMed.
- J. H. Ha and S. N. Loh, Chemistry, 2012, 18, 7984–7999 CrossRef PubMed.
- V. Stein and K. Alexandrov, Trends Biotechnol., 2015, 33, 101–110 CrossRef PubMed.
- K. Solanki, W. Abdallah and S. Banta, Biotechnol. J., 2016, 11, 1483–1497 CrossRef PubMed.
- W. Abdallah, K. Solanki and S. Banta, ACS Catal., 2018, 8, 1602–1613 CrossRef.
- B. Bulutoglu and S. Banta, Toxins, 2017, 9, 289 CrossRef PubMed.
- L. Bumba, J. Masin, P. Macek, T. Wald, L. Motlova, I. Bibova, N. Klimova, L. Bednarova, V. Veverka, M. Kachala, D. I. Svergun, C. Barinka and P. Sebo, Mol. Cell, 2016, 62, 47–62 CrossRef PubMed.
- E. Campbell, S. Chuang and S. Banta, Protein Eng., Des. Sel., 2013, 26, 181–186 CrossRef PubMed.
- J. Guo and H. X. Zhou, Chem. Rev., 2016, 116, 6503–6515 CrossRef PubMed.
- P. E. Wright and H. J. Dyson, Nat. Rev. Mol. Cell Biol., 2015, 16, 18–29 CrossRef PubMed.
- L. Xin, C. Zhou, Z. Yang and D. Liu, Small, 2013, 9, 3088–3091 CrossRef PubMed.
- J. Fu, M. Liu, Y. Liu, N. W. Woodbury and H. Yan, J. Am. Chem. Soc., 2012, 134, 5516–5519 CrossRef PubMed.
- L. Lancaster, D. P. Hickey, M. S. Sigman, S. D. Minteer and I. Wheeldon, Chem. Commun., 2018, 54, 491–494 RSC.
| This journal is © The Royal Society of Chemistry 2018 |