
A salen–Al/carbazole dyad-based guest–host assembly: enhancement of luminescence efficiency via intramolecular energy transfer†
Sang Woo
Kwak
a,
Hyomin
Jin
b,
Heuiseok
Shin
a,
Ji Hye
Lee
b,
Hyonseok
Hwang
b,
Junseong
Lee
 c,
Min
Kim
a,
Yongseog
Chung
a,
Youngjo
Kim
*a,
Kang Mun
Lee
*b and
Myung Hwan
Park
*d
c,
Min
Kim
a,
Yongseog
Chung
a,
Youngjo
Kim
*a,
Kang Mun
Lee
*b and
Myung Hwan
Park
*d
aDepartment of Chemistry, Chungbuk National University, Cheongju, Chungbuk 28644, Republic of Korea. E-mail: ykim@chungbuk.ac.kr
bDepartment of Chemistry, Institute for Molecular Science and Fusion Technology, Kangwon National University, Chuncheon, Gangwon 24341, Republic of Korea. E-mail: kangmunlee@kangwon.ac.kr
cDepartment of Chemistry, Chonnam National University, Gwangju 61186, Republic of Korea
dDepartment of Chemistry Education, Chungbuk National University, Cheongju, Chungbuk 28644, Republic of Korea. E-mail: mhpark98@chungbuk.ac.kr
First published on 12th April 2018
Abstract
A novel class of salen–Al/carbazole dyads (D1 and D2) was synthesized and fully identified. The emission spectra of the dyads presented intriguing dual-emission patterns via an intramolecular energy transfer (IET) state in solution. Furthermore, the IET feature of the dyads was clearly observed in the rigid state. Interestingly, the emission efficiency of the dyads was enhanced by the significant IET process from the carbazole group to the salen–Al moiety. Particularly, D1 exhibited a nearly three-fold enhanced luminescence efficiency compared to the corresponding mononuclear aluminum complexes (A1). Such an emission process of these guest–host systems was further supported by theoretical calculation.
Carbazole-based compounds have been widely utilized in prominent optoelectronic applications such as organic light-emitting diodes (OLEDs) and photovoltaic cells, due to their excellent thermal and electronic properties.1–5 These intriguing properties are mainly attributed to the rigidity and electron-abundant nature of the carbazole unit6,7 and can be further improved by systematic combination with various functional groups, thus allowing carbazole derivatives to broaden their applicability in optoelectronics.8–12
Recently, a novel class of Pt-,13–16 Au-,17,18 Cr-,19 and Ru-based20 organometallic complexes containing carbazole ligands have received much attention because they can provide positive effects by altering optical properties such as emission-colour tuning and enhancement of quantum efficiency. Yam and co-workers developed novel platinum complexes via systematic design between the platinum centre and the electron-donating carbazolyl ligands, exhibiting highly efficient multicolour emission derived from the intramolecular charge transfer (ICT) process arising from the donor–acceptor conjugate systems.13 To date, to the best of our knowledge, there have been no reported studies on aluminum–carbazole complexes for optical applications, though aluminum-based complexes have been well known as prominent optoelectronic materials. Therefore, we selected salen and carbazole ligands as proper anchoring ligands into the aluminum centre for the induction of the desired luminescence properties and the enhancement of emission efficiency via the IET process. Herein, we describe the detailed synthesis and optical properties of novel guest–host dyads (D1 and D2) based on salen–Al complexes with a carbazole unit in conjunction with computational calculations.
The synthetic routes for salen–Al/carbazole dyads (D1 and D2) are shown in Scheme 1. These dyads were prepared by the reaction of the salen–Al precursors (M1: (3-tBu-salen)Al-Me and M2: (3,5-tBu-salen)Al(CH3)) with 4-(2,7-di-tBu-9H-carbazol-9-yl)phenol (C2) in moderate yields (68% for D1 and 64% for D2). D1 and D2 were fully characterized by 1H and 13C NMR spectroscopy (Fig. S1–S4 in the ESI†) and elemental analysis. The solid-state molecular structure of D1 was analysed by the single-crystal X-ray diffraction method (Fig. 1). Single crystals of D1 suitable for X-ray diffraction studies were obtained from the slow diffusion of MeOH into a THF solution. The structural parameters including selected bond lengths (Å) and angles (°) for D1 are presented in Tables S1 and S2 of the ESI.† The phenoxy group connects the Al atom with a carbazole group as the key group, indicating that D1 is an assembly dyad with a five-coordinate aluminum centre. The trigonality parameter21,22 (τ) value for D1 is 0.47, indicating that the geometry around the Al centre of D1 is an intermediate between trigonal bipyramidal (tbp) and square pyramidal (sp) structures. Furthermore, the carbazole-centred plane is significantly distorted with respect to the phenoxy group linked to the Al centre, owing to the dihedral angle of 54.6°. This structural feature confirms that the delocalization of electron density between the salen–Al moiety and the carbazole group in the assembly dyad cannot occur.
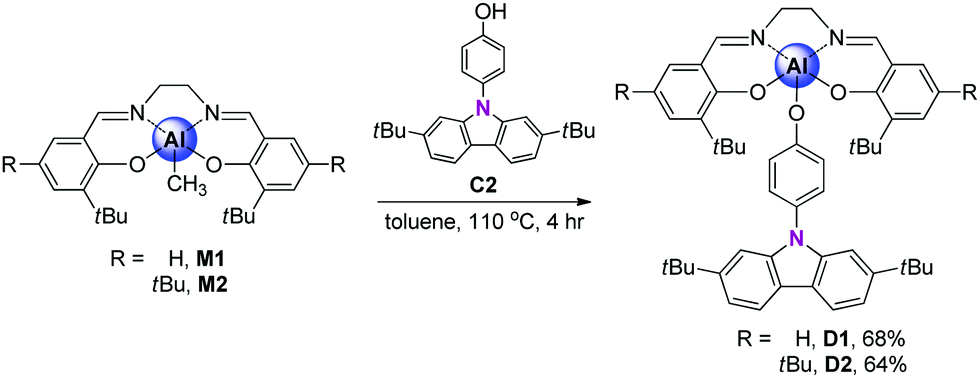 | ||
| Scheme 1 Synthetic routes for salen–Al/carbazole dyads. | ||
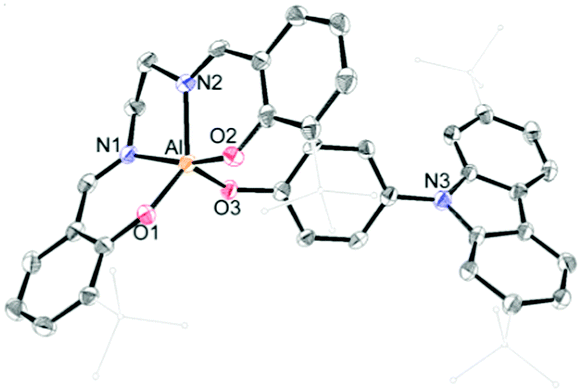 | ||
| Fig. 1 X-ray crystal structures of D1 (40% thermal ellipsoids). The H atoms are omitted and t-butyl groups are simply drawn for clarity. | ||
To investigate the photophysical properties of the assembly dyads (D1 and D2), UV-vis absorption (Fig. 2 and Table 1) and photoluminescence (PL) measurements (Fig. 3 and Table 1) were performed in THF at room temperature. D1 and D2 exhibited a dominant low-energy absorption band at ca. 346 nm and 358 nm, respectively, which is associated with the major salen–Al-centred π–π* transition that is also observed for closely related aluminum compounds (λabs = 348 nm for A1 and 359 nm for A2, Table 1) without the carbazole unit. The high-energy absorptions in the region of ca. 288 nm are assignable to the carbazole-centred π–π* transition that is also present in the absorption spectra of C2 and other carbazole-based compounds.13,14,16,18–20
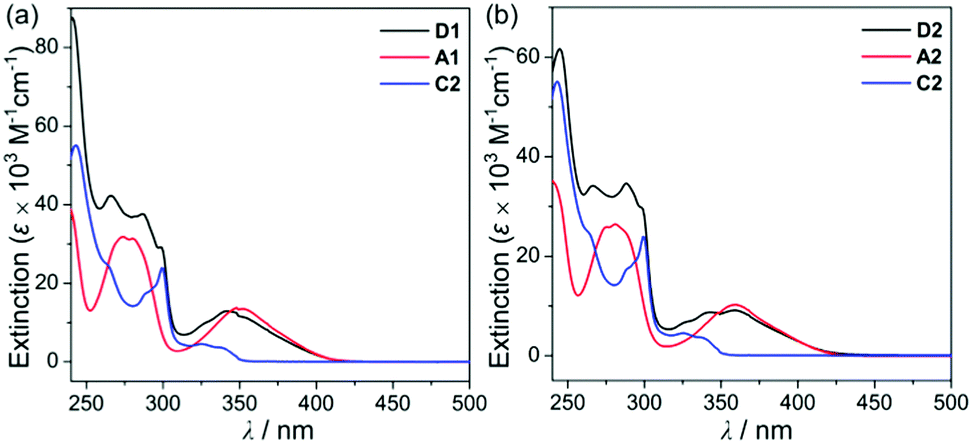 | ||
| Fig. 2 (a) UV-vis absorption spectra in THF (5.0 × 10−5 M) for (a) D1, A1, and C2 and (b) D2, A2, and C2. | ||
| Comp. | λ abs /nm (ε × 10−3 M−1 cm−1) | λ em /nm | Φ PL | τ /ns | |
|---|---|---|---|---|---|
| 380 nm | 460 nm | ||||
| a Measured using a 5.0 × 10−5 M THF solution. b Absolute PL quantum yield measured in film state (5 wt% doped on PMMA). c λ ex = 310 nm. d λ ex = 316 nm. | |||||
| A1 | 348 (13.8) | 459c | 0.026c | — | 3.923 |
| 280 (31.4) | |||||
| A2 | 359 (10.3) | 478d | 0.117d | — | 6.923 |
| 281 (26.4) | |||||
| C2 | 326 (4.60) | 360c,d | — | 0.38 | — |
| 299 (23.9) | |||||
| D1 | 346 (12.8) | 364c | 0.072c | 5.0 | 7.7 |
| 288 (37.4) | 458c | ||||
| D2 | 358 (9.10) | 360c | 0.244c | 6.4 | 8.2 |
| 288 (34.7) | 475c | ||||
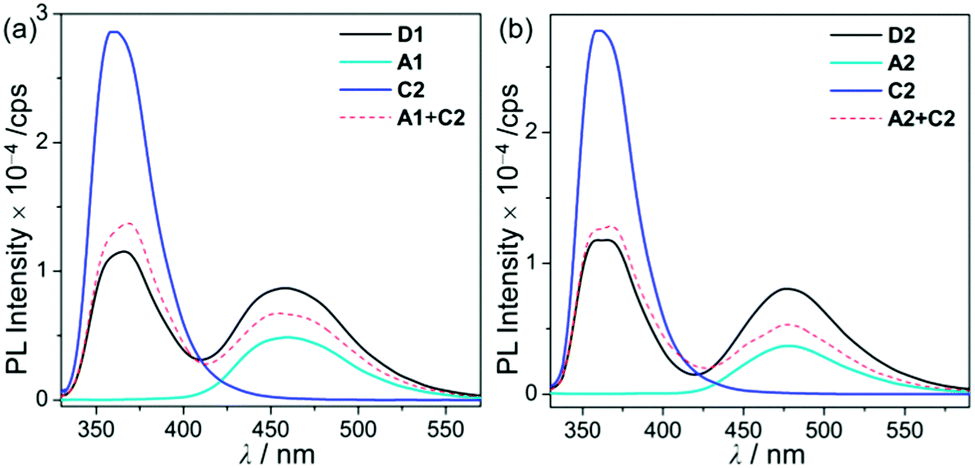 | ||
| Fig. 3 PL spectra in THF (5.0 × 10−5 M) for (a) D1, A1, and C2 (λex = 310 nm) and (b) D2, A2, and C2 (λex = 316 nm) at room temperature. | ||
Hence, the absorption spectra of D1 and D2 are quite similar to the arithmetic sum of the absorption bands for the corresponding mononuclear compounds, strongly suggesting that the electronic transitions of these assembly dyads independently occur on the salen–Al moiety and the carbazole group. The PL spectra of D1 and D2 present specific dual-emission patterns when excited at 316 nm and 310 nm, respectively (Fig. 3 and Table 1). These excited values are the minimum points in each excitation graph for dyads and can only partially excite the carbazole moieties (Fig. S5 in the ESI†). A comparison of the emission intensities of D1 and D2 with their relevant mononuclear aluminum (A1 or A2) and carbazole compound (C2) under the same conditions reveals that the intensity of the carbazole moiety (λem = 360 nm for C2) was drastically reduced and simultaneously, the intensity of the salen–Al part (λem = 459 nm for A1 and 478 nm for A2) increased nearly two-fold compared to that of the mononuclear Al compound (A1 or A2). Particularly, the fact that the PL intensity of each part changed with no significant alteration in the emission wavelengths shows the relevance of the partial IET from the carbazole group to the salen–Al moiety. These IET features are also evident from the emission spectra of their corresponding mononuclear compounds (A1 + C2 (λex = 310 nm) and A2 + C2 (λex = 316 nm)) in a solution with an equimolar mixture (Fig. 3). However, the IET characteristics of the mixture are not as efficient as those of the guest–host dyads (D1 and D2). Additional consideration of significant overlaps between the emission band of the carbazole compound (C2) and the absorption bands of the salen–Al compounds (A1 or A2) also confirms this phenomenon (Fig. S8 in the ESI†). Thus, the mechanism of the IET between the carbazole moiety (as the donor) and the salen–Al moieties (as the acceptor) in the dyads (D1 and D2) is the Förster mechanism via through-space interaction. The emission decay lifetime measurements (Fig. S9–S12 in the ESI†) of these dyads indicate that the emissions at 380 nm assignable to carbazole-centred fluorescence (5.0 ns for D1 and 6.4 ns for D2) are slightly shorter than those at 460 nm (ca. 8.0 ns). To check the rate constant (kIET) for the IET process in the dyads (D1 and D2) and the equimolar systems (A1 + C2 or A2 + C2), we additionally carried out the decay lifetime measurements (at 380 nm and 460 nm) for the equimolar mixtures (Fig. S12 and S13). From the results (6.5 ns for A1 + C2, 8.3 ns for A2 + C2), the kIET values for A1 + C2 and A2 + C2 at 380 nm, which is the emission region of the energy donor (C2), were calculated as 1.5 × 108 s−1 and 1.2 × 108 s−1, respectively, resulting that the values are lower than those of dyad complexes (2.0 × 108 s−1 for D1 and 1.6 × 108 s−1 for D2). These results clearly indicate that the IET process in the dyads is faster and more effective than that in the physically mixed systems,24 further implying the IET from the carbazole group to the salen–Al moiety. Interestingly, such an IET phenomenon of D1 and D2 is observed in the rigid state. The only intense emission assigned to the salen–Al moiety appeared in the film state (5 wt% doped in poly(methyl methacrylate) (PMMA)), indicating that the IET feature completely occurs (Fig. 4).
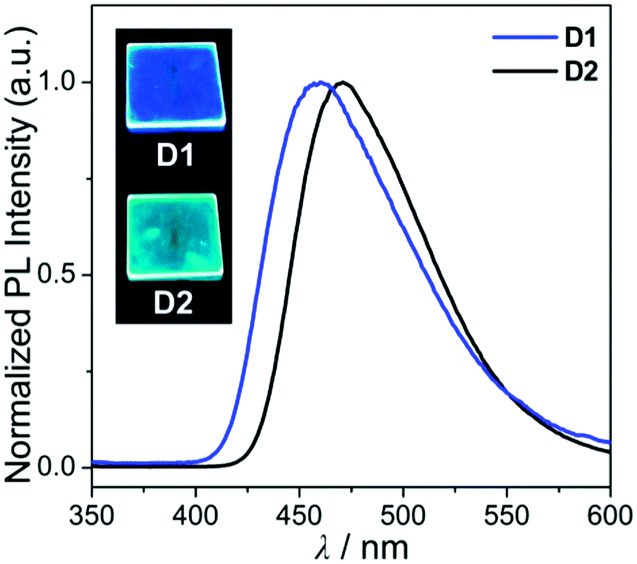 | ||
| Fig. 4 PL spectra of the film (5 wt% doped on PMMA) for D1 and D2. The insets are photographs of the film under a handheld UV lamp (λex = 365 nm). | ||
In the PMMA film, importantly, the obtained quantum efficiency (ΦPL) of these guest–host dyads (D1 and D2) clearly indicates that the emission efficiency can be tuned by the nature of the IET feature (Table 1). D1 (ΦPL = 0.072) and D2 (ΦPL = 0.244) containing a key carbazole group had higher quantum efficiencies than A1 (ΦPL = 0.026) and A2 (ΦPL = 0.117), respectively. Notably, ΦPL of D1 was enhanced approximately three-fold compared to that of A1. Accordingly, both guest–host dyads showed significantly higher luminescence efficiency than their corresponding aluminium complexes without any key carbazole groups. These findings highlight the significance of the IET effect of the guest–host dyads on ΦPL.
To better elucidate the origin of the electronic transitions of the dyad complexes, time-dependent density functional theory (TD-DFT) calculations on the ground (S0) and first excited state (S1) optimized structures for the two guest–host dyads were performed using the B3LYP functional and 6-31G(d) basis sets (Fig. 5, Table 2, and Fig. S14–S17 in the ESI†). The computed electronic transitions for the dyads in their S0 and S1 states are in good agreement with the experimentally observed absorption and emission bands. The calculation results for the optimized structures in the S0 state demonstrate that the major contributions (fcalc > 0.03) to the low-energy absorptions below 370 nm are mainly associated with HOMO−1 → LUMO transitions (λabs = 376 nm for D1 and 385 nm for D2; Fig. 5 and Table 2). HOMO−1 is dominantly localized on the salen–Al moiety (94.6% for D1 and 97.7% for D2) and LUMO also predominantly occupies the salen–Al parts (99.4% for D1 and D2, Tables S4 and S8 in the ESI†), indicating that the absorptions originate from the salen–Al-centred π–π* transition. On the other hand, the largest electronic transitions (fcalc ≈ 0.8) for the absorption regions above 370 nm are mainly attributed to the HOMO−2 → LUMO+1 transition. The orbital contribution of HOMO−2 and LUMO+1 is completely localized on the carbazole moiety (>99%, Tables S4 and S8 in the ESI†), due to which the absorption process corresponds to a carbazole-centred π–π* transition. Hence, these findings for the calculated absorption transitions clearly indicate that the main absorption processes in D1 and D2 occur independent of the salen–Al-centred and carbazole-centred π–π* transitions, respectively. The same features are also present in the calculation results for the S1-optimized structures of the dyads (Fig. 5 and Table 2), which were used to clarify the intrinsic character in emission bands. The main contribution to low-energy emissions below 450 nm is the HOMO−1 → LUMO transition, which could be assigned to the salen–Al-centred emission, whereas the high-energy emissions above 350 nm are closely related to the HOMO−2 → LUMO+1 transition attributed to the carbazole-centred emission. While the molecular orbital distributions of HOMO−1 and LUMO are concentrated in the salen–Al parts (>99%, Tables S6 and S10 in the ESI†), those of HOMO−2 and LUMO+1 are localized on the carbazole parts (>99%, Tables S6 and S10 in the ESI†). Consequently, the insights obtained from the computed results for the S1-optimized structures of D1 and D2 strongly suggest that the experimentally observed dual-emissions in the THF solution resulted from the independently bounded transition in the salen–Al centre and carbazole moieties, respectively. Along with the experimental results, DFT calculations further confirm that the dyads only exhibit partial IET from the carbazole group to the salen–Al moiety.
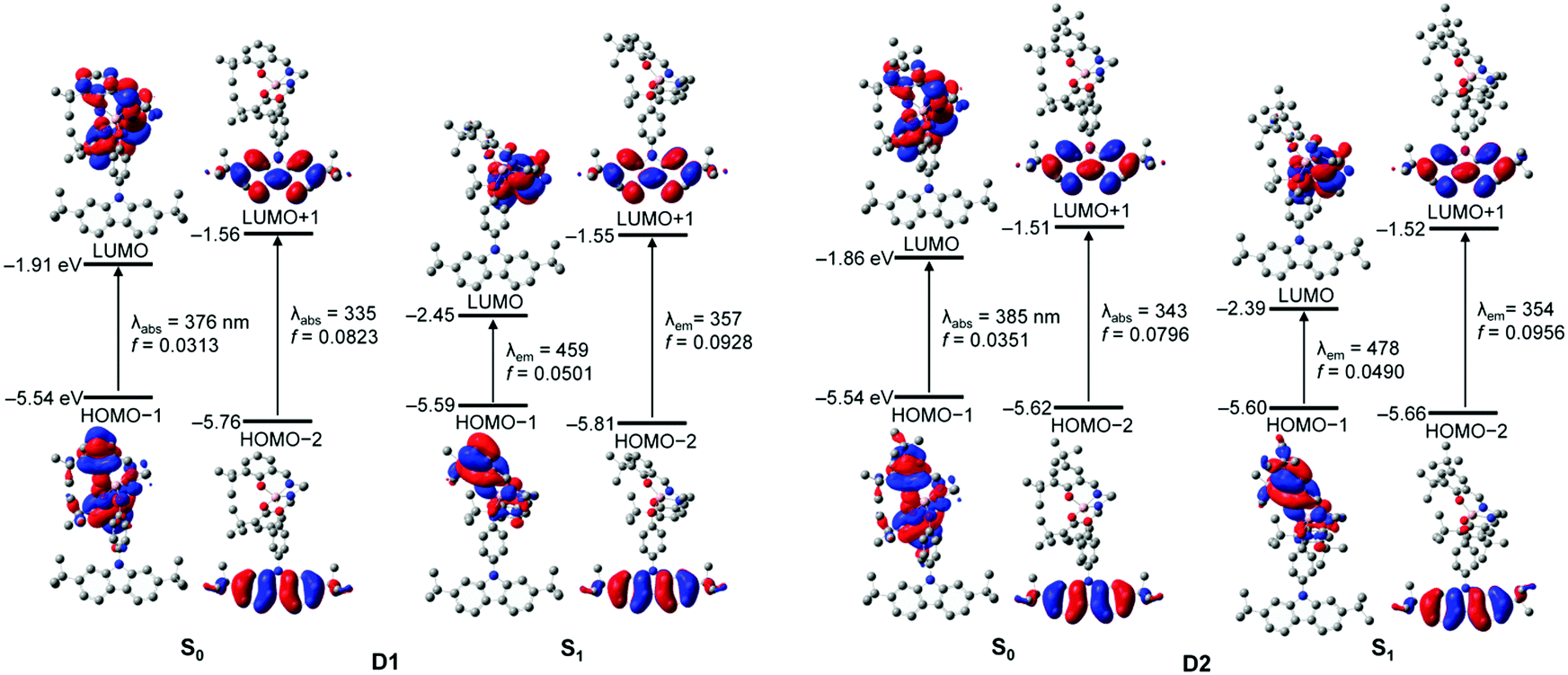 | ||
| Fig. 5 Frontier molecular orbitals for D1 and D2 at their ground (S0) and first singlet (S1) state optimized geometries in THF and the major electronic transitions from TD-DFT calculations (isovalue = 0.04). | ||
| λ calc/nm | f calc | Assignment | ||
|---|---|---|---|---|
| a Singlet energies for the vertical transition calculated at the optimized S0 and S1 geometries. | ||||
| D1 | S0 | 375.71 | 0.0313 | HOMO−1 → LUMO (70.6%) |
| 335.32 | 0.0823 | HOMO−2 → LUMO+1 (92.0%) | ||
| S1 | 458.81 | 0.0501 | HOMO−1 → LUMO (99.9%) | |
| 356.89 | 0.0928 | HOMO−2 → LUMO+1 (93.0%) | ||
| D2 | S0 | 384.98 | 0.0351 | HOMO−1 → LUMO (97.7%) |
| 342.97 | 0.0796 | HOMO−2 → LUMO+1 (93.2%) | ||
| S1 | 477.66 | 0.0490 | HOMO−1 → LUMO (98.9%) | |
| 354.08 | 0.0956 | HOMO−2 → LUMO+1 (94.8%) | ||
In summary, novel guest–host systems (D1 and D2) based on salen–Al/carbazole dyads were prepared and fully characterized. These guest–host systems exhibited nearly two-fold enhanced luminescence efficiencies compared to their corresponding aluminum complexes through a significant IET feature from the carbazole group to the salen–Al moiety in PMMA films. The computed calculation results also showed the independently bounded transition on the salen–Al centre and carbazole moieties, respectively, further verifying the experimental results. Thus, it is anticipated that these guest–host systems based on salen–Al/carbazole dyads can constitute a novel class of optoelectronic materials in OLEDs. In particular, the dyads can be used as a promising candidate of guest–host systems in blue-emitting fluorescent OLEDs (FLOLEDs). Furthermore, the present study may provide important insights into the future development of prominent optoelectronic materials using small-molecule organometallic complexes based on their different desirable electronic features. Relevant studies on expanding novel Al-based guest–host systems are in progress.
This work was supported by the Basic Science Research Program (2016R1C1B1008452 for M. H. Park and 2015R1D1A1A01061043 for Y. Kim) and Basic Research Laboratory (2017R1A4A1015405 for K. M. Lee) through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915 RSC.
- X. Yang, G. Zhou and W.-Y. Wong, Chem. Soc. Rev., 2015, 44, 8484 RSC.
- A. W. Schmidt, K. R. Reddy and H.-J. Knölker, Chem. Rev., 2012, 112, 3193 CrossRef CAS PubMed.
- Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245 RSC.
- N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. Neagu-Plesu, M. Belletête, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732 CrossRef CAS PubMed.
- D. F. O’Brien, P. E. Burrows, S. R. Forrest, B. E. Koene, D. E. Loy and M. E. Thompson, Adv. Mater., 1998, 10, 1108 CrossRef.
- B. E. Koene, D. E. Loy and M. E. Thompson, Chem. Mater., 1998, 10, 2235 CrossRef CAS.
- Y. H. Lee, S. H. Park, J. Oh, J. W. Shin, J. Jung, S. Yoo and M. H. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 24035 Search PubMed.
- A. Venkateswararao, K. R. J. Thomas, C.-P. Lee, C.-T. Li and K.-C. Ho, ACS Appl. Mater. Interfaces, 2014, 6, 2528 Search PubMed.
- N. Prachumrak, S. Pojanasopa, S. Namuangruk, T. Kaewin, S. Jungsuttiwong, T. Sudyoadsuk and V. Promarak, ACS Appl. Mater. Interfaces, 2013, 5, 8694 CAS.
- A. Michaleviciute, E. Gurskyte, D. Y. Volyniuk, V. V. Cherpak, G. Sini, P. Y. Stakhira and J. V. Grazulevicius, J. Phys. Chem. C, 2012, 116, 20769 CAS.
- S.-i. Kato, H. Noguchi, A. Kobayashi, T. Yoshihara, S. Tobita and Y. Nakamura, J. Org. Chem., 2012, 77, 9120 CrossRef CAS PubMed.
- A. K.-W. Chan, M. Ng, Y.-C. Wong, M.-Y. Chan, W.-T. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2017, 139, 10750 CrossRef CAS PubMed.
- F. K.-W. Kong, M.-C. Tang, Y.-C. Wong, M. Ng, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2017, 139, 6351 CrossRef CAS PubMed.
- G. Li, A. Wolfe, J. Brooks, Z.-Q. Zhu and J. Li, Inorg. Chem., 2017, 56, 8244 CrossRef CAS PubMed.
- F. K.-W. Kong, M.-C. Tang, Y.-C. Wong, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2016, 138, 6281 CrossRef CAS PubMed.
- C.-H. Lee, M.-C. Tang, Y.-C. Wong, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2017, 139, 10539 CrossRef CAS PubMed.
- M.-C. Tang, D. P.-K. Tsang, M. M.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, Angew. Chem., Int. Ed., 2013, 52, 446 CrossRef CAS PubMed.
- K. Shen, X. Tian, J. Zhong, J. Lin, Y. Shen and P. Wu, Organometallics, 2005, 24, 127 CrossRef CAS.
- N. D. McClenaghan, R. Passalacqua, F. Loiseau, S. Campagna, B. Verheyde, A. Hameurlaine and W. Dehaen, J. Am. Chem. Soc., 2003, 125, 5356 CrossRef PubMed.
- D. A. Atwood and M. J. Harvey, Chem. Rev., 2001, 101, 37 CrossRef CAS PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- S. W. Kwak, B. H. Choi, J. H. Lee, H. Hwang, J. Lee, H. Kwon, Y. Chung, K. M. Lee and M. H. Park, Inorg. Chem., 2017, 56, 6039 CrossRef CAS PubMed.
- R. Ziessel and A. Harriman, Chem. Commun., 2011, 47, 611 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic data in CIF format, experimental, computational, and spectroscopic data and 1H as well as 13C NMR spectra (PDF). CCDC 1819002. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc01528g |
| This journal is © The Royal Society of Chemistry 2018 |