
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorescent chemosensors: the past, present and future
Di
Wu
a,
Adam C.
Sedgwick
 b,
Thorfinnur
Gunnlaugsson
*c,
Engin U.
Akkaya
*d,
Juyoung
Yoon
*a and
Tony D.
James
*b
b,
Thorfinnur
Gunnlaugsson
*c,
Engin U.
Akkaya
*d,
Juyoung
Yoon
*a and
Tony D.
James
*b
aDepartment of Chemistry and Nano Science, Ewha Womans University, Seoul 120-750, Korea. E-mail: jyoon@ewha.ac.kr
bDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: t.d.james@bath.ac.uk
cSchool of Chemistry and Trinity Biomedical Sciences Institute (TBSI), Trinity College Dublin, The University of Dublin, Dublin 2, Ireland. E-mail: gunnlaut@tcd.ie
dUNAM-Institute of Material Science and Nanotechnology, Bilkent University, Ankara 06800, Turkey. E-mail: eua@fen.bilkent.edu.tr
First published on 11th October 2017
Abstract
Fluorescent chemosensors for ions and neutral analytes have been widely applied in many diverse fields such as biology, physiology, pharmacology, and environmental sciences. The field of fluorescent chemosensors has been in existence for about 150 years. In this time, a large range of fluorescent chemosensors have been established for the detection of biologically and/or environmentally important species. Despite the progress made in this field, several problems and challenges still exist. This tutorial review introduces the history and provides a general overview of the development in the research of fluorescent sensors, often referred to as chemosensors. This will be achieved by highlighting some pioneering and representative works from about 40 groups in the world that have made substantial contributions to this field. The basic principles involved in the design of chemosensors for specific analytes, problems and challenges in the field as well as possible future research directions are covered. The application of chemosensors in various established and emerging biotechnologies, is very bright.
 Di Wu | Di Wu was born in Hubei province, P. R. China, in 1987. He received his Bachelor degree (2010) and PhD degree (2015) from Central China Normal University (CCNU) under the supervision of Professor Sheng Hua Liu and Jun Yin. Subsequently, he joined Prof. Juyoung Yoon's research group at Ewha Womans University (South Korea) as a postdoctoral fellow. His research interests focus on fluorescent chemosensors and new organic functional materials. |
 Adam C. Sedgwick | Adam C. Sedgwick graduated with a 1st class MChem in Chemistry for Drug Discovery from the University of Bath in 2014. During his undergraduate degree he undertook an industrial placement at BioFocus (Now Charles River) working as a medicinal chemist synthesizing compound libraries for various drug discovery applications. He is currently working towards his PhD at the University of Bath developing novel sensors for the detection of reactive oxygen species. |
 Thorfinnur Gunnlaugsson | Thorfinnur (Thorri) Gunnlaugsson MRIA, is a Professor of Chemistry in the School of Chemistry, Trinity College Dublin (TCD). His research interests lie broadly within the fields of medicinal, organic, inorganic supramolecular and materials chemistries. He is a Fellow of TCD, and was elected as a Member of The Royal Irish Academy in 2011. In 2014, he as awarded The Institute of Chemistry of Ireland (ICI) Annual Award for Chemistry (Eva Philbin Lecturer). He is the author of over 220 papers and has an H-index of 70. |
 Engin U. Akkaya | Engin U. Akkaya is a Professor at Bilkent University, Department of Chemistry and UNAM-National Nanotechnology Research Center. He is a Fellow of the Royal Society of Chemistry, and a member of the Science Academy (BA) of Turkey. His research interests include photodynamics, molecular logic gates and molecular devices, and information processing therapeutic agents. He authored 105 papers and has an H-index of 46. |
 Juyoung Yoon | Juyoung Yoon is currently Professor of Department of Chemistry and Nano Science in Ewha Womans University. His research interests include investigations of fluorescent chemosensors, molecular recognition, and new organic functional materials. He has published over 280 SCI research papers with h-index of 82. |
 Tony D. James | Tony D. James is a Professor at the University of Bath and Fellow of the Royal Society of Chemistry and holds a prestigious Royal Society Wolfson Research Merit Award In 2013 he received the Daiwa-Adrian Prize and in 2015 the Inaugural CASE Prize. His research interests include many aspects of Supramolecular chemistry, including: molecular recognition, molecular self-assembly and sensor design. He is the author of over 232 papers and has an h-index of 58. |
Key learning points(1) Strategies for the design of fluorescent chemosensors(2) Sensing mechanisms involved in the design of fluorescent chemosensors (3) Applications of fluorescent chemosensors (4) Usefulness of fluorescent chemosensors for in vitro and in vivo studies (5) Key problems and challenges in the field of fluorescent chemosensors |
1. Introduction
Compounds incorporating a binding site, a fluorophore, and a mechanism for communication between the two sites are called fluorescent chemosensors.1 If the binding sites are irreversible chemical reactions, the indicators are described as fluorescent chemodosimeters. These two definitions as well as the term “fluorescent probe” have been used interchangeably and ambiguously over the past few decades therefore we have unified the area to describe them all as fluorescent chemosensors in this review. The first fluorescent chemosensor was reported by F. Goppelsröder in 1867, and was a method for the determination of aluminum ion (Al3+) by forming a strongly fluorescent morin chelate.2 This led to the development of a number of fluorescent chemosensors for the determination of many other metal ions, over the subsequent several decades, marking in part the birth of analytical chemistry as we know it. In fact, the early fluorescent chemosensors, concentrated mainly on the detection of metal ions rather than the detection of anions or neutral species. This is due to the selective binding of metal ions in water being significantly easier than that of anions or neutral species. However, more recently and since around 1980, we have witnessed an explosive growth and development of the area catalyzed by the inspirational and pioneering work by the two fathers of modern chemosensors: de Silva and Czarnik.3 Since those pioneering days, fluorescent chemosensors have been extensively developed and the scope of applicability extended to include numerous biologically important analytes. In particular, fluorescent chemosensors for biologically and/or environmentally important cations, anions, small neutral molecules as well as biomacromolecules (such as proteins and DNA) have been developed along with a rapid advancement in microscopic imaging technologies. Analyte detection by a fluorescent chemosensor is usually achieved through one or more common photophysical mechanisms, including chelation induced enhanced fluorescence (CHEF),2 intramolecular charge transfer (ICT),3 photoinduced electron transfer (PET),4 aggregation induced emission (AIE)5 and the number of approaches is still expanding. Due to the high levels of sensitivity and in particular their ability to be used for temporal and spatial sampling for in vivo imaging applications, fluorescent chemosensors have been widely applied in a variety of fields such as biology, physiology, pharmacology, and environmental sciences. With the advent of two or multi-photon excitation and high and super-resolution fluorescence microscopy we will see an ever increasing need for highly sensitive and selective chemosensors for in vivo biological applications.There are a number of reviews that have been compiled describing fluorescent chemosensors, however, to the best of our knowledge, most of these reviews focus on either diverse fluorochromes or analytes. Only a few reviews have taken a step back and carried out an overview of progress of this field.6,7 This review summarizes progress in the development of fluorescent chemosensors over the last 50 years and introduces representative chemosensors for the detection of cations, anions, small neutral molecules as well as biomacromolecules. It contains the design principle, working mechanism and biological application of the corresponding chemosensors. Furthermore, the design principles for the construction of selective chemosensors for specific analytes and the problems and challenges encountered along the way will be discussed. We will end the review with a discussion of the future research directions and opportunities available for fluorescent chemosensors given that they are now well-established research tools.
2. Fluorescent chemosensors for cations
There are a number of metal ions that play a vital role in our daily physiological life. These include sodium (Na+) potassium (K+), calcium (Ca2+), copper (Cu+ and Cu2+) and zinc (Zn2+), among others. However, some metal ions such as lead (Pb2+), cadmium (Cd2+) and mercury (Hg2+) are toxic and cause serious health and environmental problems.2.1 Fluorescent chemosensors for alkali and alkaline earth metal ions
As mentioned above, the first recorded fluorescent chemosensor for cations dates to 1867, when Goppelsröder reported that morin forms a strongly fluorescent chelate with Al3+. In the beginning, most fluorescent chemosensors for cations were based on the coordination interactions between the hosts and the guests. For example, Sousa et al. reported two naphthalene based chemosensors 1 and 2 for the detection of alkali metal ions (Fig. 1). This resulted in the use of supramolecular chemistry in fluorescent chemosensor design.8 Interestingly, these closely related chemosensors exhibit dichotomous behavior. 1 displayed a decrease in fluorescence quantum yield, also an increase in phosphorescence quantum yield, and a slight decrease in phosphorescence lifetime when it formed a complex with alkali metal chloride salts in 95% ethanol glass at 77 K. On the contrary, complexation of 2 with potassium (K+), rubidium (Rb+), or caesium (Cs+) chloride salts caused a noticeable increase in fluorescence quantum yield, also a decrease in phosphorescence quantum yield, and a substantial decrease in phosphorescence lifetime. These observed changes can be attributed to the heavy atom effect (for Rb+ and Cs+) and/or a complexation induced change in triplet energy relative to the ground and excited singlet state energies as well as rigidification and conformational effects.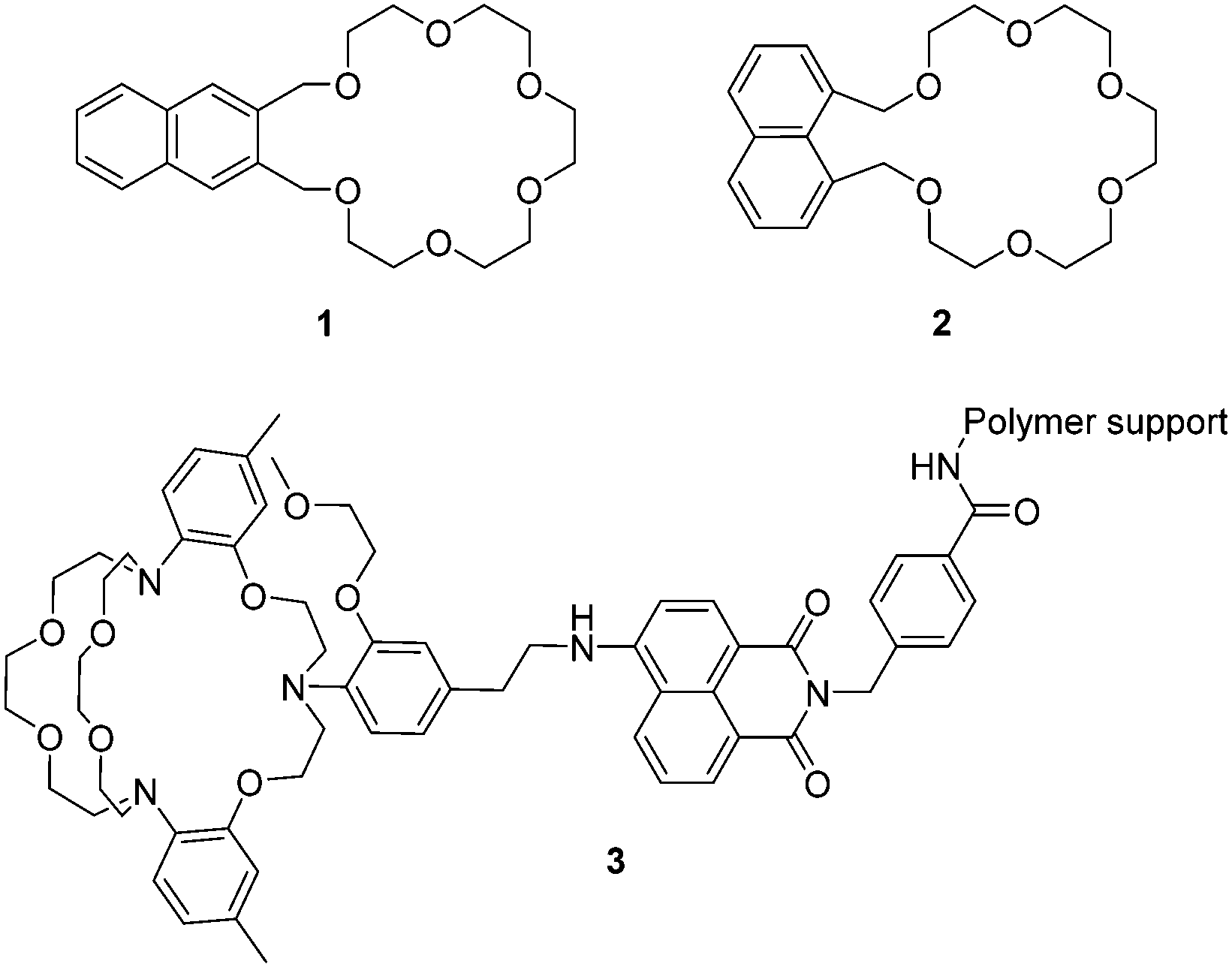 | ||
| Fig. 1 Structures of the fluorescent chemosensors 1–3. | ||
Potassium ions (K+) are one of the most important cations in living organisms because they play essential roles in many biological processes. In mammals, the concentration of K+ inside cells is about 150 mM, which is nearly 30 times higher than that in the extracellular environment. Imbalances of potassium are closely related to many diseases such as Alzheimer's disease (AD), anorexia, heart disease and diabetes. A number of fluorescent chemosensors for K+ have been developed. Currently, the 2-triazacryptand [2,2,3]-1-(2-methoxyethoxy)benzene (TAC) group, which displays a very high selectivity for detecting K+ over other physiologically relevant metal ions is the best K+-selective chelator. The TAC group was first reported by He et al. in 2003.9 They incorporated the TAC group into a 4-aminonaphthalimide based polymer via an ethylene group, in order to develop chemosensor 3 for the measurement of extracellular (serum or whole blood) potassium based on a PET mechanism (Fig. 1). The chemosensor rapidly and reversibly detects changes in potassium concentrations in whole blood samples. Furthermore, there were no interferences from clinical concentrations of Ca2+ or pH and from the interference of Na+ even at concentrations of 160 mM. Additionally, this chemosensor has been used in the Roche OPTI CCA, a commercially available whole blood analyzer, this system was developed in collaboration with de Silva (http://impact.ref.ac.uk/CaseStudies/CaseStudy.aspx?Id=38360). Although TAC based chemosensors display high selectivities and sensitivities for K+, their syntheses often require lengthy synthetic routes and harsh reaction conditions. Thus a significant demand exists for the development of more readily available ligands with the same properties displayed by TAC.
The magnesium ion (Mg2+), which has a number of critical roles such as an enzyme cofactor, a DNA conformation stabilizer and a facilitator of transmembrane ion transport, is the most abundant divalent cation in cells. Abnormal concentrations of Mg2+ ions are associated with many diseases such as migraines, diabetes, hypertension and Parkinson's disease. In order to detect cellular magnesium ions, Farruggia et al. developed two 8-hydroxyquinoline based fluorescent chemosensors 4 and 5 for the detection of Mg2+ (Fig. 2).10 Initially, these two chemosensors show very weak fluorescence due to the an intermolecular photoinduced proton transfer (PPT) and PET process. However, after binding with Mg2+, the PPT and PET process are blocked, which results in a remarkable increase in the fluorescence intensity. Chemosensors 4 and 5, with Kd of 44 and 73 μM, respectively, show high selectivity and sensitivity towards Mg2+ over other cations including Ca2+. These two chemosensors have been used to image Mg2+ in live cells. However, the limitations of these two chemosensors are that they can only be excited in the UV region, which hinders their development for practical applications.
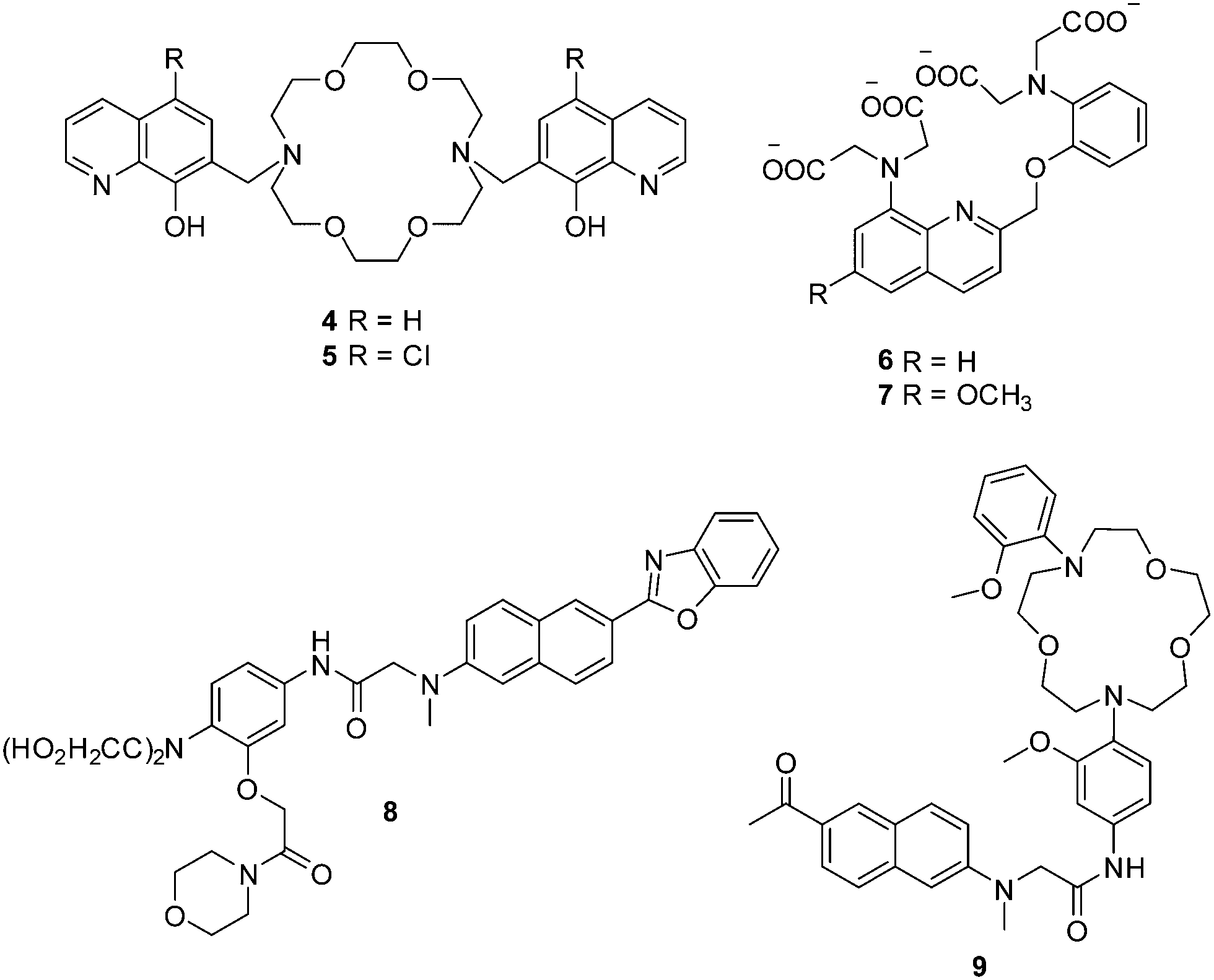 | ||
| Fig. 2 Structures of the fluorescent chemosensors 4–9. | ||
Calcium ions (Ca2+), are another important alkaline earth metal ion. Ca2+ is the most abundant element in the human body and it plays important roles in many biological processes. The monitoring of the intracellular free Ca2+ is important since imbalances of Ca2+ are related to a number of diseases such as neurodegeneration, heart disease and skeletal muscle defects. The initially reported fluorescent chemosensors for Ca2+ suffered several problems such as (1) low selectivity towards competing cations and in particularly H+ and Mg2+, (2) complex stoichiometry with Ca2+, (3) inflexibility of molecular design, or the difficulty of rationally and iteratively adjusting chelator properties with different fluorophores.11 These problems were only solved when Nobel Laureate Roger Y. Tsien developed two 1,2-bis(2-aminophenoxy) ethane-N,N,N′,N′-tetraacetic acid (BAPTA) based fluorescent chemosensors 6 and 7 for Ca2+ in 1980 (Fig. 2).11 Both of these chemosensors show good selectivity and sensitivity towards Ca2+ over Mg2+, and their application in vivo opened up a new area and understanding of cellular function and indeed revolutionized our understanding of biochemical processes within cells.
Along with the development of the field of fluorescence dyes and the advancement in microscopic imaging technologies, a series of BAPTA and derived fluorescent chemosensors for Ca2+ have been reported. In 2010, Kim, Cho and coworkers described a two photon (TP) fluorescent chemosensor 8, in which 2-(2′-morpholino-2′-oxoethoxy)-N,N-bis(hydroxycarbonylmethyl) aniline (MOBHA) was used as the Ca2+ receptor and 6-(benzo[d]oxazol-2′-yl)-2-(N,N-dimethylamino)naphthalene was used as the fluorophore (Fig. 2).12 Chemosensor 8 shows high selectivity for Ca2+ and was pH-insensitive at biologically relevant pH. To understand the Na+/Ca2+ exchange process, which is an important process vital to Ca2+ homeostasis, the Ca2+ chemosensor 8 and Na+ chemosensor 9 were applied to the simultaneously detection of Ca2+ and Na+ near the cell membrane of HeLa cells (Fig. 3). The HeLa cells labeled with 8 and 9 emitted bright two-photon excited fluorescence (TPEF) in the green channel emission (390–450 nm), corresponding to Ca2+ ions detected by 8 (Fig. 3a) and red channel emission (500–560 nm), attributed to Na+ ions detected by 9 (Fig. 3b), the merged images generated by two-photon excitation are given in Fig. 3c. These two chemosensors have been applied to monitoring Na+/Ca2+ exchange in live tissues at depths of over 100 μm (Fig. 3d–f).
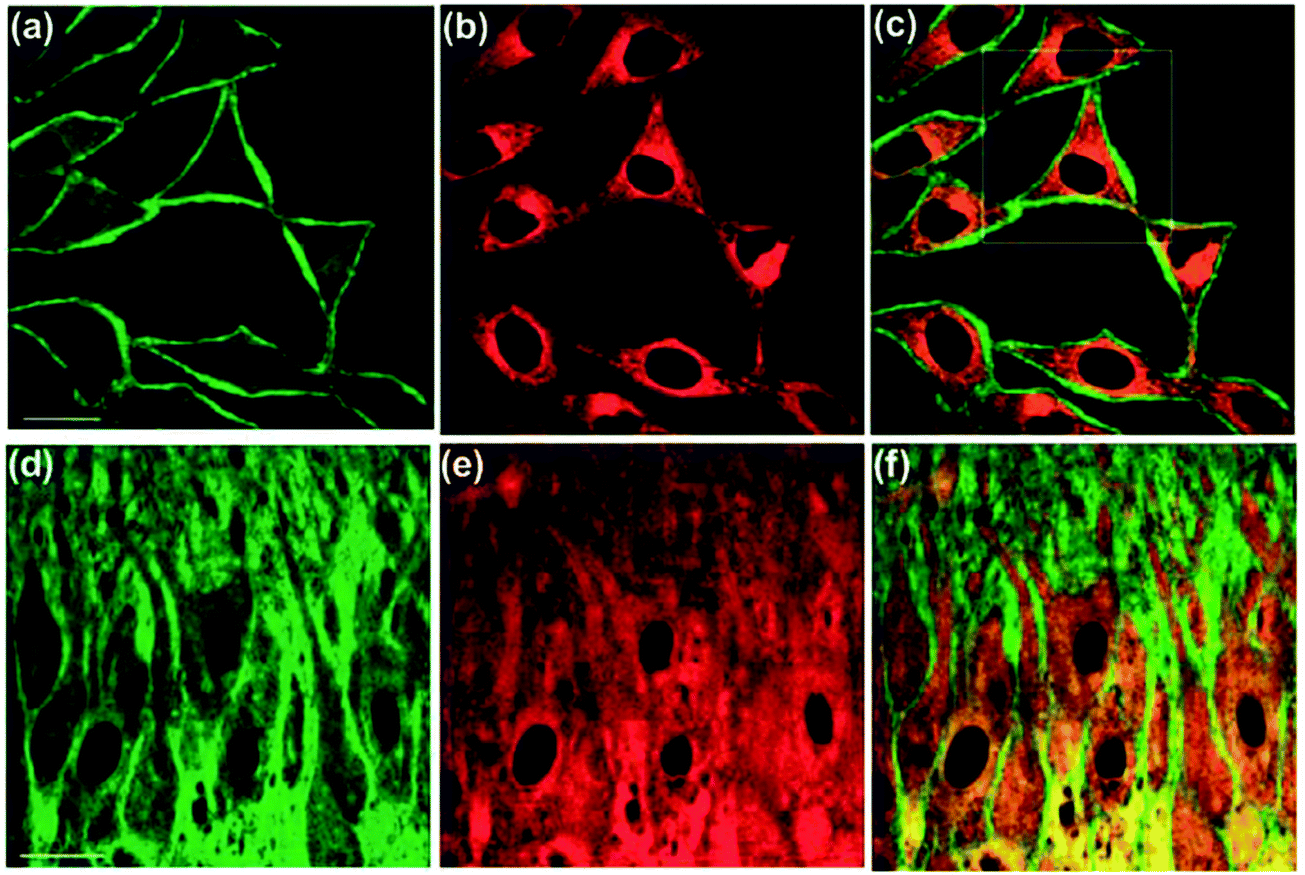 | ||
| Fig. 3 Dual-channel TPEF images of HeLa cells co-incubated with 8 and 9 collected at (a) green channel: 390–450 nm (8), (b) red channel: 500–560 nm (9), and (c) merged image of (a) and (b). TPEF images of a mice hippocampal slice co-stained with 8 and 9 collected at (d) green channel: 390–450 nm (8), (e) red channel: 500–560 nm (9) at a depth of 100–200 μm at tenfold magnification and (f) a merged image of (d) and (e). Excitation wavelength: 780 nm. Scale bars: 30 mm (a and d). Reproduced from ref. 12 with the permission of John Wiley & Sons, Inc. | ||
The proton (H+) (or hydronium ion (H3O+)) is one of the most important charged species and has a crucial role in many physiological and pathological processes including receptor-mediated signal transduction, ion transport, endocytosis, homeostasis, proliferation and apoptosis, multidrug resistance and muscle contraction. Mitochondria, an important organelle, has a critical role in cellular metabolism such as energy production, signaling, cellular differentiation, cell growth and death. The unique function of the mitochondria depends on the pH. Therefore, monitoring mitochondrial pH and in particular, changes related to mitophagy, may provide insights into mitochondrial function under physiological and pathological conditions. Sessler, Kang, Kim and coworkers developed a mitochondria-immobilized fluorescent chemosensor 10 to measure pH (Fig. 4), consisting of a piperazine-linked naphthalimide as a fluorophore with a cationic triphenylphosphonium as the mitochondrial targeting group, and a reactive benzyl chloride subunit for mitochondrial fixation.13 The chemosensor is non-fluorescent in neutral form due to the PET process. However, the PET process is inhibited at acidic pH and results in a fluorescence enhancement. 10 can be used for quantitative measurement of pH in mitochondria and real-time monitoring of mitophagy in cells. These results indicate that 10 has significant potential to be applied in biological systems, and how simple structural modifications of an established PET pH-sensor can open up new routes towards emerging biotechnologies.
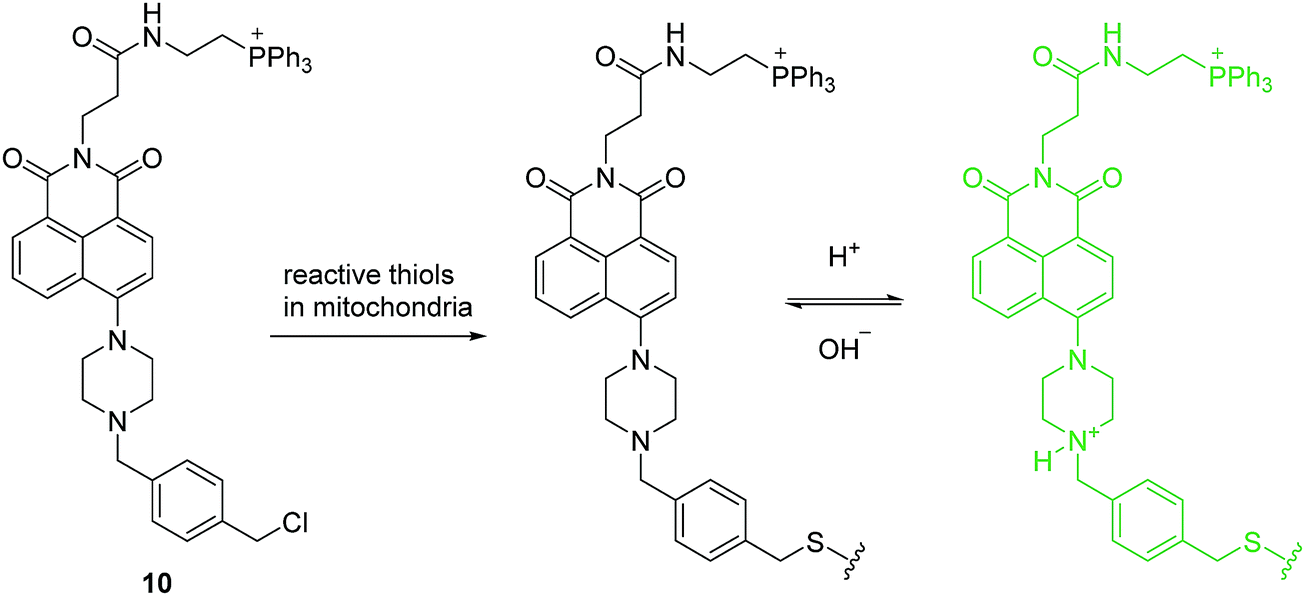 | ||
| Fig. 4 Structures of the fluorescent chemosensors 10 and the proposed mechanism for detection of pH in mitochondria. | ||
2.2 Fluorescent chemosensors for d-block metal ions
In contrast to the above-described fluorescent chemosensors for alkali and alkaline earth metal ions, which are all based on coordination interaction, some chemical reaction based fluorescent chemosensors for transition metal ions have been developed since these metal ions can trigger specific reactions. The strategies of using such reactions for sensing analytes has significantly broadened the field of chemosensors.Copper (Cu) is the third most abundant transition metal in the human body, it is an essential transition metal in living organisms because it is involved in various physiological and pathological processes. Loss of copper homeostasis is linked with diseases such as Menkes (copper deficiency), Wilson's (copper overload), Alzheimer's disease, prion disorders, neurodegeneration and cancer.
In 1997, Czarnik and co-workers reported pioneering work on a rhodamine-B derivative and its ring-opening reaction for sensing copper ion (Cu2+).14 As shown in Fig. 5, the fluorescent chemosensor 11 can undergo a selective hydrolysis reaction with Cu2+ and yield fluorescent rhodamine B as a product. This work generated a great deal of attention for the ring-opening processes of rhodamine derivatives for use as a fluorescent chemosensors.
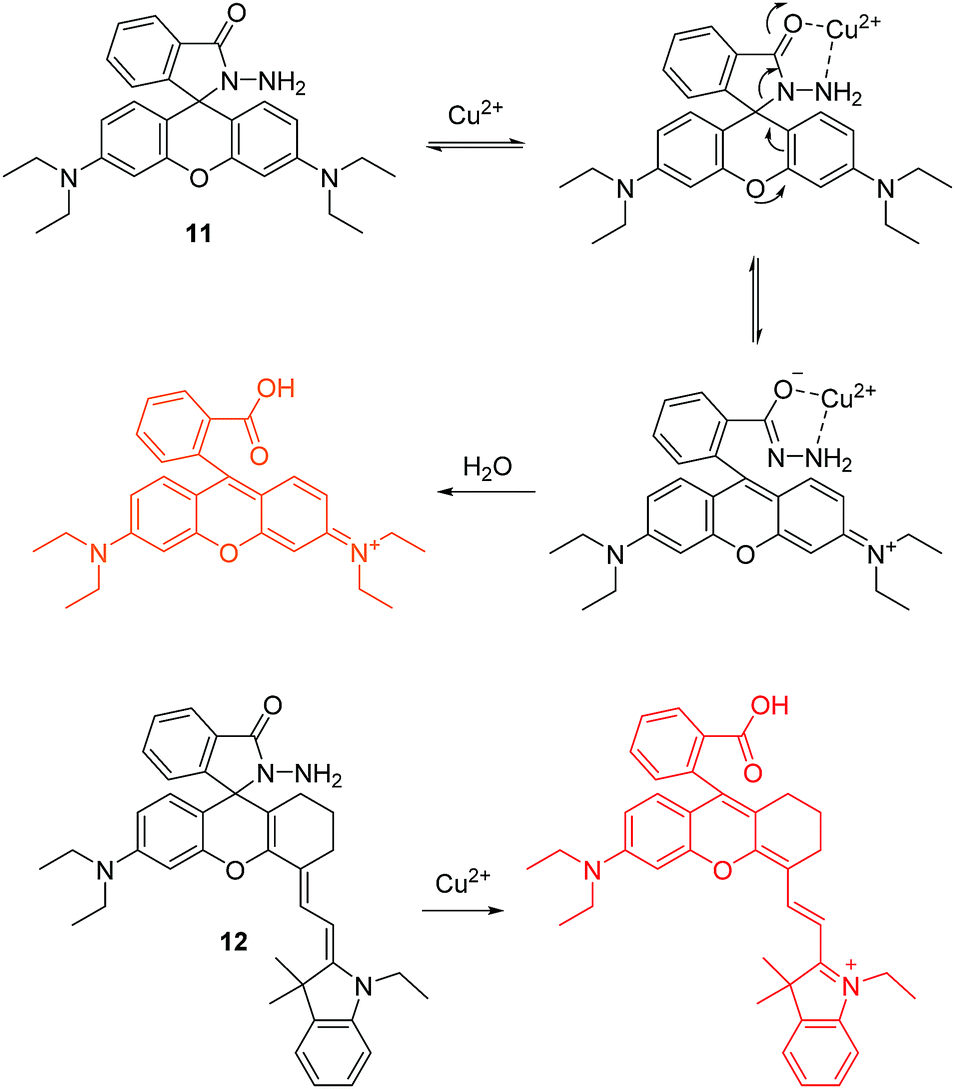 | ||
| Fig. 5 Structures and proposed mechanism of 11 and 12 for detection of Cu2+. | ||
Based on the same reaction, the Li group developed a NIR fluorescent chemosensor 12 for Cu2+ (Fig. 5).15 Undoubtedly, 12 shows high sensitivity and selectivity to Cu2+ over other related metal ions. It is particularly noteworthy that this chemosensor exhibits unique single-photon frequency upconversion luminescence (FUCL). Thus the product formed by the reaction of 12 with Cu2+ can be excited with both 670 nm and 808 nm light. Due to the low background signal associated with NIR excitation (808 nm) and NIR emission (730 nm), it has an extremely low detection limit of 3.2 ppb in aqueous solution. This is much lower than that of the Stokes’ fluorescence methods for excitation at 670 nm where the calculated detection limit is around 6.5 ppb. Significantly, this chemosensor has been applied for the diagnosis of Wilson disease in live mice and therefore offers some promise for diagnostic sensing.
Under physiological conditions, copper exists in its stable oxidized Cu2+ and reduced Cu+ states. Detection of Cu+ is just as important as the detection of Cu2+, however, many fewer fluorescent chemosensors have been developed for Cu+ than for Cu2+. The fluorescent chemosensor 13 developed by the Chang group consists of a tris[(2-pyridyl)-methyl]amine (TPA) as the binding and reaction site, and a bioluminescent D-luciferin as the reporter (Fig. 6).16 Compared with common fluorophore based chemosensors, these bioluminescent reporter based sensing platforms have low background and high signal-to-noise. Chemosensor 13 shows good sensitivity and selectivity towards Cu+ over other related species except for free Co2+ (100 μM) that gives a modest response with the chemosensor. However, the concentration of Co2+ (100 μM) is not considered physiologically relevant since most Co2+ is found tightly bound to proteins. Significantly, 13 has been used to image labile copper pools mouse model of non-alcoholic fatty liver disease. The results indicate that hepatic copper deficiency and altered expression levels of copper homeostatic proteins accompany glucose intolerance and weight gain.
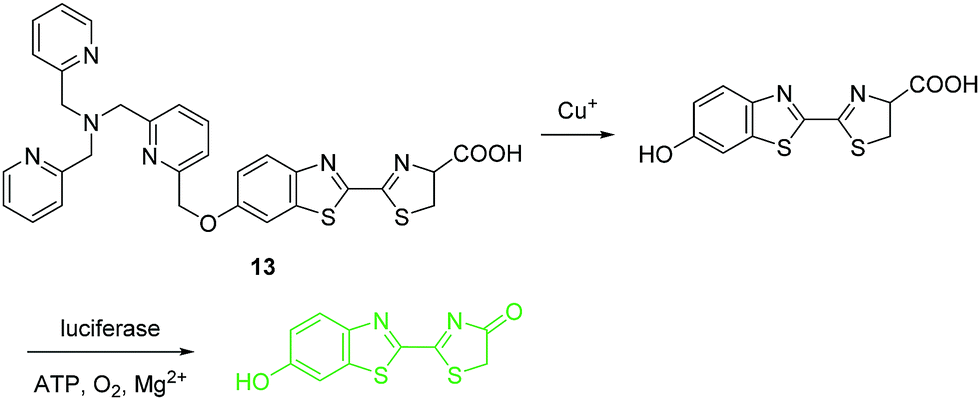 | ||
| Fig. 6 Structure and proposed mechanism of 13 for detection of Cu+. | ||
Zinc (Zn) is the second most abundant d-block metal in the human body, which is often found as pools of mobile ions in specific tissues of the body. Failure in the homeostasis of free zinc ions is closely associated with neurological diseases and free zinc ions (Zn2+) are also involved in apoptosis (programmed cell death). The majority of small-molecule fluorescent chemosensors for mobile zinc ions comprise of a fluorophore and a chelating unit containing tertiary amines. However, acidic pH can interfere with the detection of Zn2+ using these systems. To overcome these shortcomings, the Lippard group have developed a spirobenzopyran based two-photon fluorescent chemosensor 14 for Zn2+ (Fig. 7).17 Chemosensor 14 can selectively detect Zn2+ in the presence of other related metal ions over a wide range of pH from 3 to 7. The chemosensor has been applied to imaging exogenous Zn2+ in the lysosomes of HeLa cells, endogenous Zn2+ in insulin granules of MIN6 cells, and zinc-rich mossy fiber boutons in hippocampal tissue of mice. Furthermore, the relatively large two-photon absorption cross section (δ = 74 GM) and far-red emission makes it ideal for imaging zinc ions in tissue at depths of >100 μm with greater contrast than existing visible-light fluorescent chemosensors.
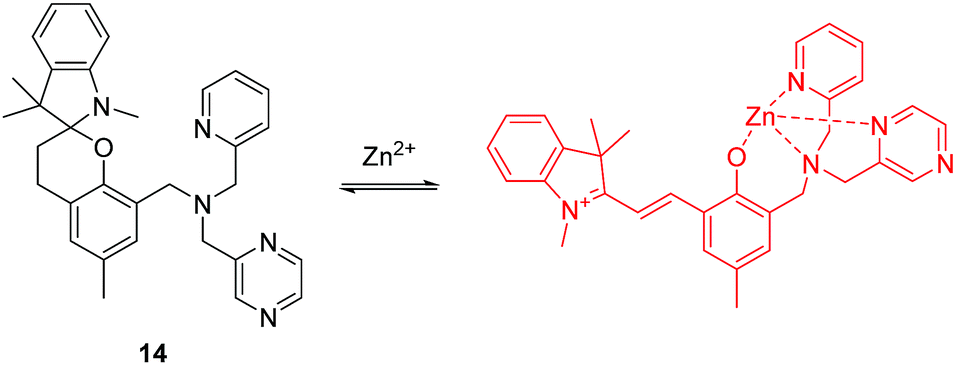 | ||
| Fig. 7 Structure and proposed mechanism of 14 for detection of Zn2+. | ||
Mercury (Hg) is one of the most prevalent deadly toxins on earth, which arises from many sources such as gold production, coal plants, thermometers, barometers and mercury lamps. In the past several decades, a huge number of fluorescent chemosensors have been developed for the detection of Hg2+. Pioneering work by Czarnik and coworkers on a desulfurisation reaction used the thiophilic character of Hg2+.18 Initially, the fluorescent chemosensor 15 is non-fluorescent due to the PET process. The addition of Hg2+ induces an enhancement in fluorescence, whereas other metal ions except for Ag+ caused no interference (Fig. 8).
 | ||
| Fig. 8 Structures and proposed mechanism of 15 and 16 for detection of Hg2+. | ||
Tae, Shin and coworkers developed a rhodamine-based fluorescent chemosensor 16 for Hg2+, which takes advantage of the known Hg2+-promoted formation of 1,3,4-oxadiazoles from thiosemicarbazoles (Fig. 8).19 Chemosensor 16 shows high sensitivity and selectivity over other metal ions including Ag+ and Pb2+, which can also promote the desulfurization reactions. The chemosensor has been used to detect exogenous Hg2+ uptake in C2C12 cells and in zebrafish in real time as well as to image accumulated Hg2+ in zebra fish organs.
Due to the close relationship between the two fields of “chemosensors” and “molecular logic”, a number of fluorescent chemosensors with two or more binding (reaction) sites have been utilized to construct molecular logic gate. For example, Akkaya and coworkers reported three styryl-Bodipy based molecular logic gates 17, 18 and 19 using Hg2+, Zn2+ and (or) Ca2+ as inputs (Fig. 9).20 With these chemosensors, the dithiaazacrown ligand is used as a Hg2+ binding site, di-2-picolylamine (DPA) is used as a Zn2+ binding site while the aza-crown ligand is used as a Ca2+ binding site in 19. Using Hg2+ and Zn2+ as inputs, the emission signaling of 17 at 570 nm responds in accordance with molecular logic gate AND function. For 18, the structure works as an AND logic gate when the absorbance is recorded at 623 nm. However, when the absorbance data is collected near the longer wavelength peak, it responds in accordance with XOR logic. Chemosensor 19 is a three-input AND logic gate using Hg2+, Zn2+ and Ca2+ as inputs when the emission signaling is recorded at 656 nm.
 | ||
| Fig. 9 Structures of the fluorescent chemosensors 17–19. | ||
3. Fluorescent chemosensors for anions
The development of anion selective chemosensors lagged behind that of cation chemosensors, due to the strong hydration of anions. However, the field of anion sensing is now a relatively mature science in line with the field of cation sensing. This was driven by the important roles anions play in biological and industrial processes as well as the need to produce new methods of sensing anionic pollutants in the environment. Over the past several decades, there have been a number of fluorescent chemosensors developed for the detection of anions, which have used host–guest interactions or chemical reactions.3.1 Fluorescent chemosensors for anions based on host–guest interaction
In 1994, Czarnik and co-workers reported an anthracene derived fluorescent chemosensor 20 for pyrophosphate (PPi) containing polyazaalkane groups (Fig. 10).2120 shows good selectivity towards PPi over other anions including phosphate (Pi), which possesses a similar structure to that of PPi. The high selectivity of 20 towards PPi results from the two polyammonium arms that are geometrically disposed for binding the six external oxygen atoms of the pyrophosphate anion. It was not until the start of this millennium that the development of fluorescent PET anion sensors that function on the bases of using charge neutral receptors was developed; this was achieved by several of the authors of this review in a concurrent manner.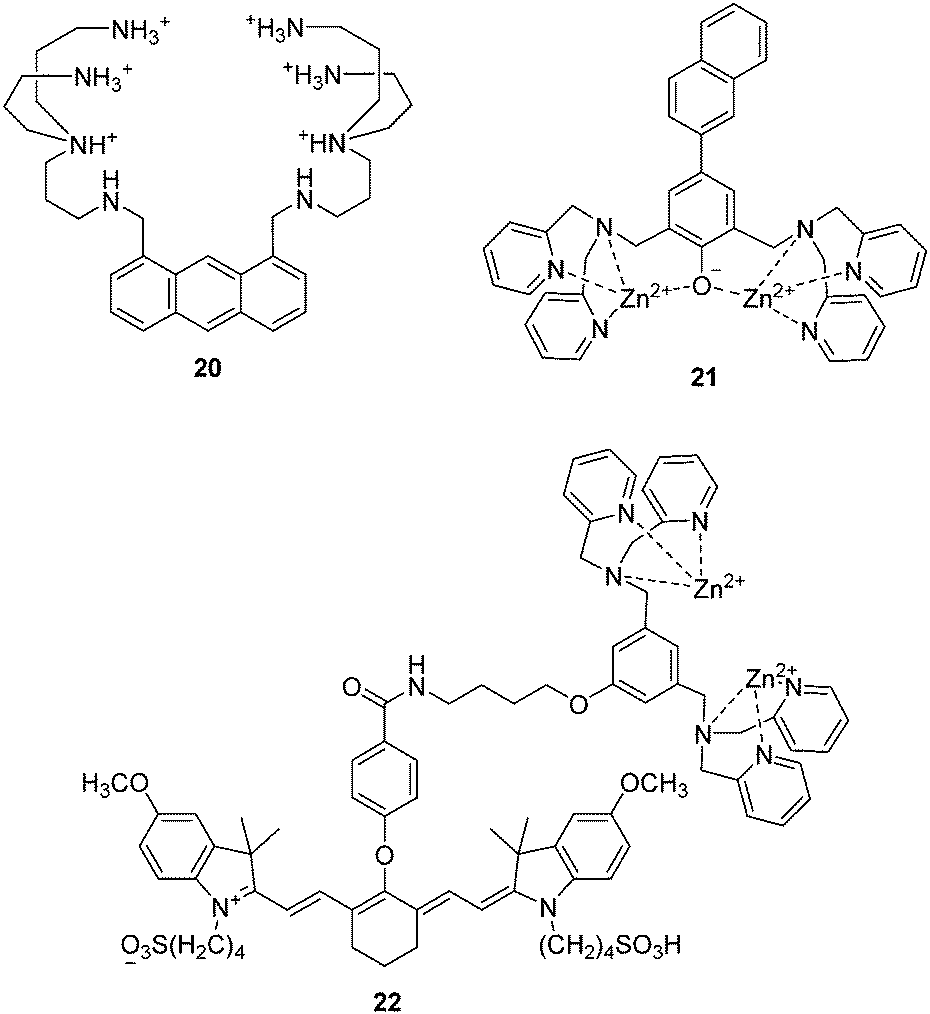 | ||
| Fig. 10 Structures of the fluorescent chemosensors 20–22. | ||
The use of a Zn2+ complex as a binding site for PPi has been found to be a particularly successful strategy due to the strong binding affinity between Zn2+ and PPi. In particular, the Hong group have extensively explored this area of chemosensor development. A representative piece of their work, contains a naphthalene derivative based Zn2+ complex 21 as shown in Fig. 10.22 The Ka for PPi of 21 was calculated to be to be 2.9 × 108 M−1, which means that 21 can detect PPi in water at nanomolar concentrations. Furthermore, 21 is able to detect less than 1 equivalent of PPi in the presence of a 50- to 250-fold excess of ATP. This is the first example of a metal complex that can discriminate PPi from ATP in aqueous solution.
Taking advantage of minimal photo-damage, deep tissue penetration and high signal-to-noise contrast of NIR fluorescent chemosensors, Smith and coworkers developed a cyanine based Zn2+ complex 22 for in vivo optical imaging of tumors and cell death events (Fig. 10).23 Zinc complex 22 can selectively accumulate in prostate and mammary tumors in two different xenograft animal models and it is now commercially available. This is an excellent example of a fluorescent chemosensor being applied for real-life practical applications.
Recently, Sessler, Anslyn, and coworkers reported two anion induced supramolecular assemblies of expanded porphyrins 23 and 24 (Fig. 11).24 Porphyrins 23 and 24 can form supramolecular polymers with several diacids, which can be used as chemosensors for both anions and organic solvents. The solubility, colour, and fluorescence of the assemblies changes dramatically when they were treated with Lewis basic anions or polar solvents, which could be caused by a decrease in the extent of aggregation. The authors have demonstrated that this system can be used as a chemosensor for identifying certain salts and various solvents by solubility, fluorescence or visible colour change.
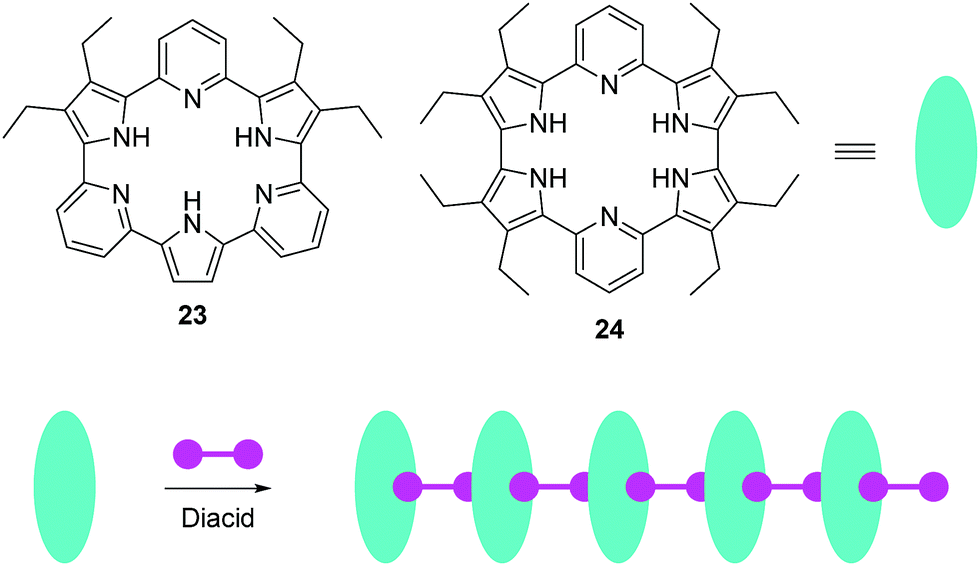 | ||
| Fig. 11 Structures of fluorescent chemosensors 23, 24 and the schematic illustrates the construction of supramolecular assemblies using 23, 24, and diacids as the building blocks. | ||
A urea and thiourea moiety can be used in the design of various fluorescent chemosensors for H-bond donors. An excellent example from the Fabbrizzi and Amendola group is the fluorescent chemosensor 25, which consists of a pyrene group as a fluorophore and a urea binding group. Chemosensor 25 displays an interesting “on1–off–on2” fluorescence response towards F− (Fig. 12).25 Initially, 25 shows typical pyrene emission and maximum at 394 nm (on1) in MeCN. However, upon addition of F−, the fluorescence decreases (off) due to an electron transfer process occurring in the locally excited complex and the conversion of locally excited complex to poorly emissive excited tautomer. Interestingly, upon further addition of F−, a yellow fluorescence turns on, while a new emission band centered at 500 nm (on2) appears, which can be ascribed to a charge-transfer emission by the deprotonated receptor; the F− deprotonation phenomena having been observed by several researchers in analogous systems. The authors demonstrated these optical features can be observed in other neutral receptors containing N–H fragments, this may provide new strategies for the design of fluorescent sensors for anions.
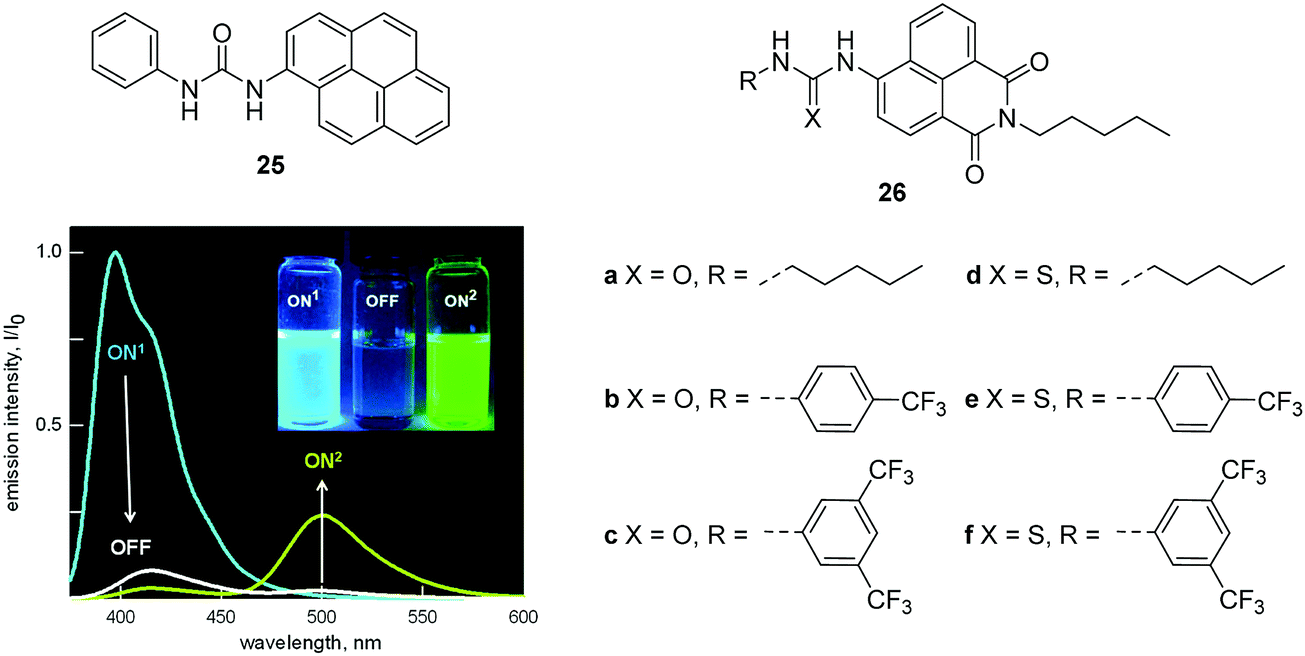 | ||
| Fig. 12 Structures of 25, 26a–f and the emission spectra taken upon addition of F− to 25 (0.01 mM) in MeCN. Reproduced from ref. 25 with the permission of the American Chemical Society. | ||
Recently, Gale and coworkers designed and synthesized a series of fluorescent anion transporters 26a–f consisting of a naphthalimide fluorophore with urea or thiourea receptors attached (Fig. 12).26 Interestingly, these transporters show two distinct localization modes within cells. The aromatic substituted transporters localize within the cytoplasm and the less lipophilic alkyl substituted transporters are over time localized in specific vesicles. Furthermore, the aromatic substituted compounds 26c–f all induce cytotoxicity in cancer cell lines, with 26f inducing apoptosis of A549 cells while alkyl substituted 26a and 26b are non-toxic towards cancer cells. These results suggest that the toxic effects can be ascribed to changes in ionic or pH gradients across intracellular membranes rather than the plasma membrane. This research is particularly important since it offers exciting new applications for fluorescent chemosensors of anions.
3.2 Fluorescent chemosensors for anions based on chemical reactions
As well as the host–guest interaction based fluorescent chemosensors for the detection of anions discussed above, a number of chemical reaction based fluorescent chemosensors for anions have been developed. These include the detection of reactive anions (reactive oxygen (ROS) and nitrogen (RNS) species). The superoxide radical (O2−˙) is generated by the one-electron reduction of molecular oxygen, which is the precursor of other ROS and RNS. Thus, elucidating the relation between O2−˙ fluxes and diseases is of great importance. The group of Yang have established a number of novel fluorescent chemosensors for ROS/RNS. In 2015, they reported a series of fluorescein based chemosensors 27a–c for O2−˙ (Fig. 13).27 It is worth noting that the trifluoromethyl group plays an important role in these chemosensors. It is a strong electron-withdrawing group and activates the sulfonate ester toward nucleophilic attack by O2−˙, yielding the free fluorophores. The trifluoromethyl group can also prevent interference from cellular reductants such as cysteine (Cys) and glutathione (GSH). All these three fluorescent chemosensors can specifically detect O2−˙ over other ROS/RNS and thiols. Furthermore, 27c contains a triphenylphosphonium group, which allows it to be used to monitor O2−˙ changes in mitochondria.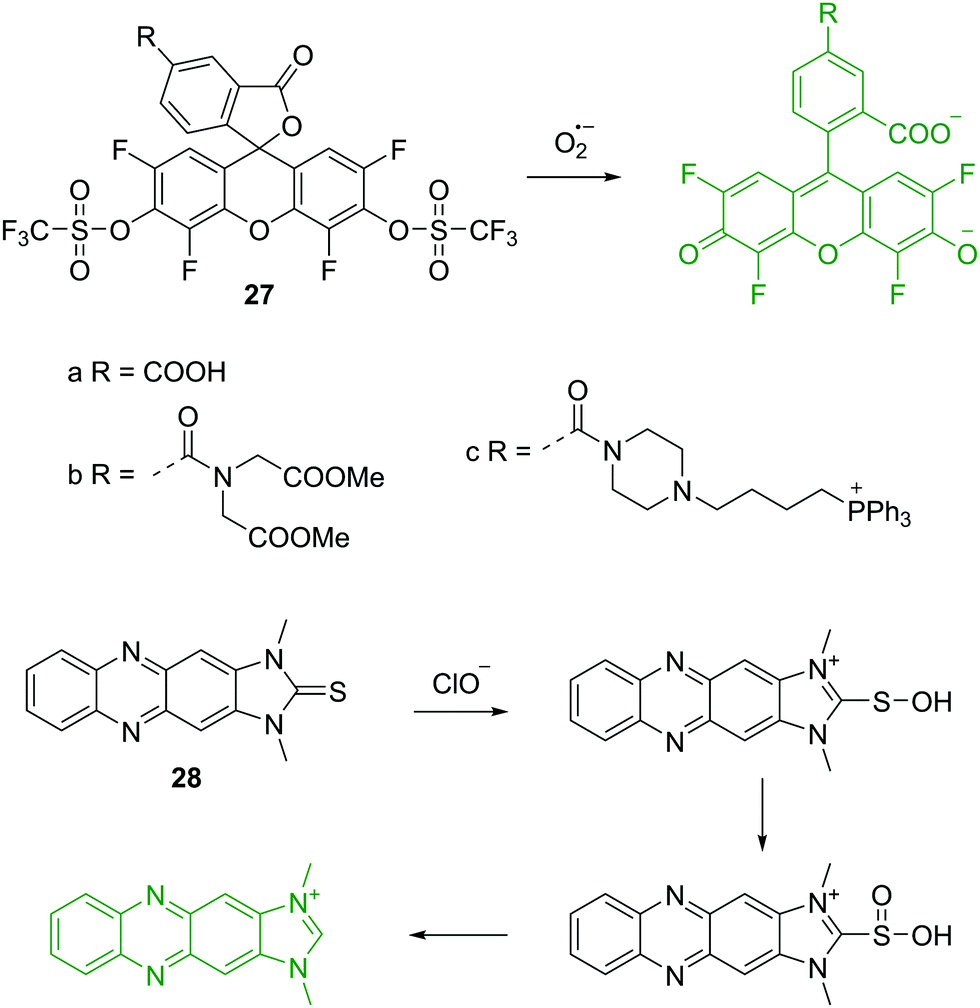 | ||
| Fig. 13 Structures and proposed mechanism of 27 and 28 for detection of O2−˙ and ClO− respectively. | ||
Hypochlorite (ClO−) is a prominent ROS, which plays an important role in regulating invading microbes. However, uncontrolled production of ClO− within phagocytes is acknowledged to be related to the start of a number of human diseases. Yoon, Kim and coworkers described a two photon fluorescent chemosensor 28 based on imidazoline-2-thiones (Fig. 13).28 Initially, 28 is non-fluorescent in PBS buffer. However, upon addition of ClO−, the fluorescence increases dramatically at 505 nm due to the formation of the corresponding imidazolium. Other ROS/RNS, do not induce observable fluorescence changes, demonstrating outstanding selectivity of the chemosensor towards ClO−. Chemosensor 28 also shows good sensitivity with a detection limit of 0.071 μM towards ClO− and has been applied to imaging ClO− in live cells and tissues.
Peroxynitrite (ONOO−) is a strong oxidant observed in physiological and pathological processes. It plays a key role in signal transduction and antimicrobial activity, however, excessive ONOO− can damage critical cell components resulting in many diseases. Recently, a boronate-based fluorescent chemosensor 29 has been developed for ONOO− (Fig. 14).29 The chemosensor displays relatively weak fluorescence due to the PET process. A large fluorescence enhancement occurs upon the addition of D-fructose. The interaction of the chemosensor with D-fructose strengthens the fluorescence signal and in addition protects the boronic acid from oxidation by other ROS/RNS. The system has good selectivity towards ONOO− over other ROS/RNS except ClO− due to its strong oxidizing ability. Additionally, the chemosensor has been used to image endogenous or exogenous ONOO− in living cells.
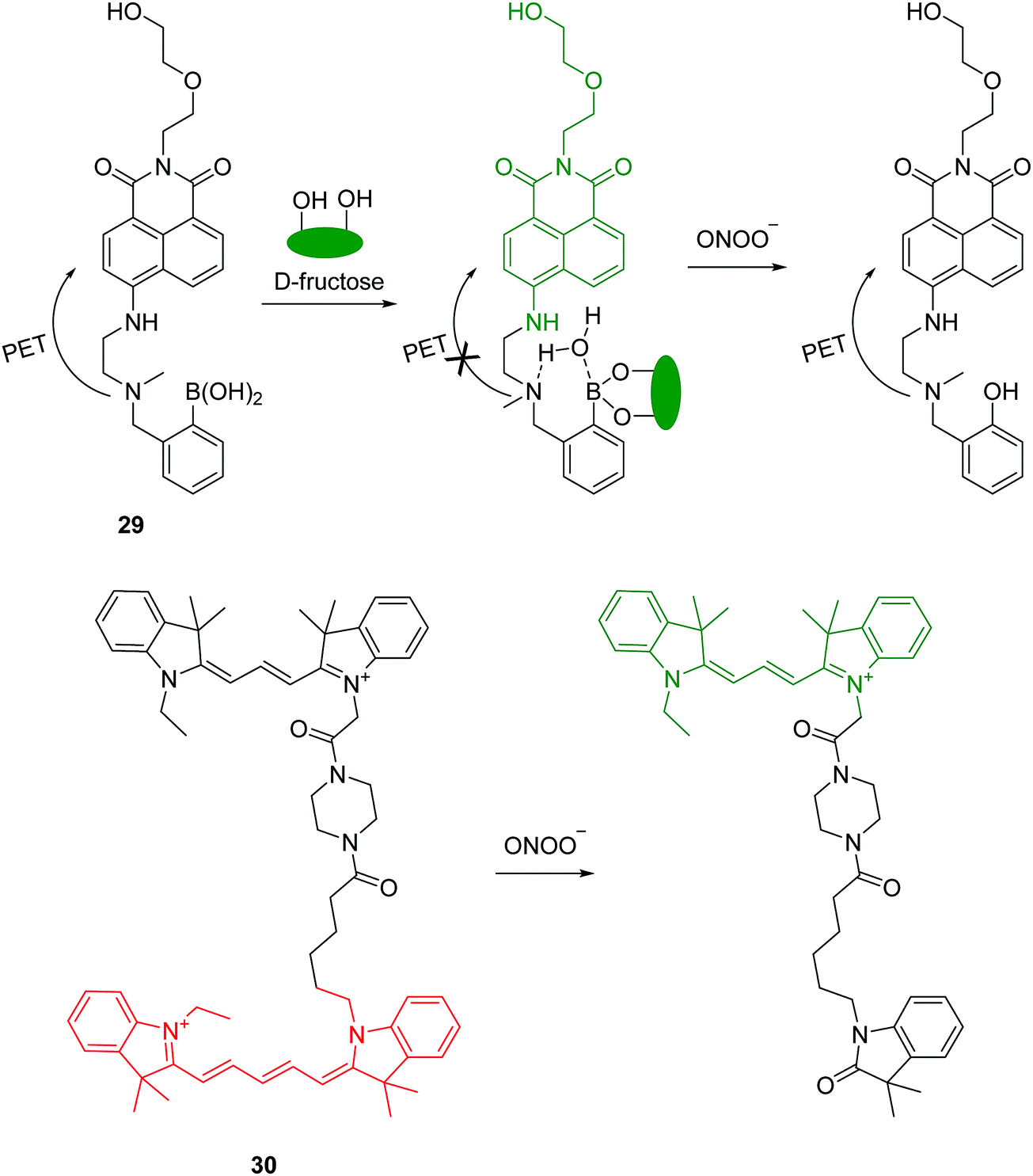 | ||
| Fig. 14 Structures and proposed mechanism of 29 and 30 for detection of ONOO−. | ||
Recently, the Qian group reported a FRET-based mitochondria-specific fluorescent chemosensor 30 for the ratiometric detection of ONOO− (Fig. 14).30 The chemosensor consists of two cyanine dyes (Cy3 and Cy5) and harnesses the differential reactivity of Cy3 and Cy5 toward ONOO−. The chemosensor displays fluorescence for Cy5 (660 nm) by FRET from Cy3 when excited at 530 nm. However, upon addition of ONOO−, a fluorescence increase at 560 nm and a decrease at 660 nm is observed which can be ascribed to the selectively oxidation of the Cy5 moiety in 30 by ONOO−. It is worth noting that both Cy3 and Cy5 moieties can be oxidized by ClO−. The chemosensor has been applied to imaging both ONOO− in live cells and the authors demonstrated that this fluorescent chemosensor can be used in semi-quantification of cellular ONOO−.
Nicotinamide adenine dinucleotide (NADH) consists of one adenine, one nicotinamide, two ribose rings, and a pair of bridging phosphate groups. Together with its oxidized form, NAD+, they are the most indispensable coenzymes and they play important roles in multiple biological processes. Inspired by the enzyme-catalyzed NADH sensing process, the group of Chang have developed two resazurin based fluorescent chemosensors 31 and 32 for NADH (Fig. 15).31 A two-step sensing mechanism was proposed as shown in Fig. 15, first, the boronic acid in 31 undergoes an esterification reaction with the diols of NADH. Second, reduction of the weakly fluorescent resazurin to the strongly fluorescent resorufin occurs. It is worth noting that the first step can facilitate the hydride transfer from NADH to 31 and accelerate the reaction in the second step. However, 31 can only work in basic conditions, which limits its applications in biological systems. To address this problem, the boronic acid in 31 was replaced by 2-(hydroxymethyl)phenylboronic acid, yielding the chemosensor 32 which can work in pH 7.4 buffer solution (Fig. 15).31 It can evaluate NADH both in vitro and in live cells. Although this chemosensor still has some drawbacks such as photoinstability and ease of wash-out, this work provides inspiration by mimicking biological processes for the design of fluorescent chemosensors.
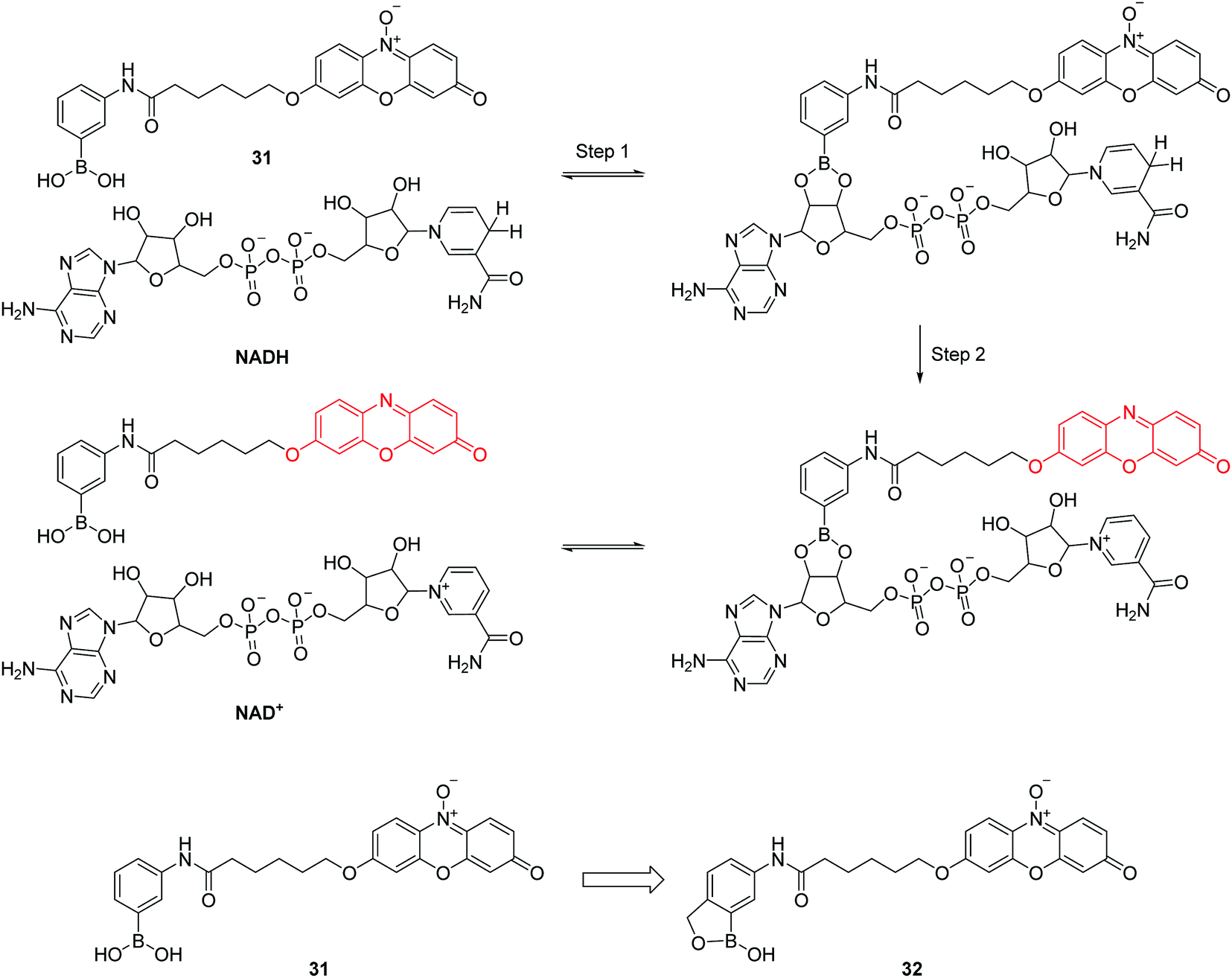 | ||
| Fig. 15 Structures of 31, 32 and proposed mechanism for detection of NADH by 31. | ||
4. Fluorescent chemosensors for small neutral molecules
Small neutral molecules such as reactive sulfur species (RSS) as well as some neutral ROS/RNS are essential for our survival since they play a vital role in a range of physiological and pathological processes. Conversely, some small neutral molecules like nitroaromatics (explosives), and nerve-gas are a threat to public health and safety. These, two important reasons have stimulated the development of a substantial number of fluorescent chemosensors for small neutral molecules over recent years.4.1 Fluorescent chemosensors for reactive sulfur species (RSS)
Intracellular thiols such as cysteine (Cys), homocysteine (Hcy) and glutathione (GSH) play key roles in biological systems. Abnormal levels of these molecules have been linked to a number of diseases, such as liver damage, leucocyte loss, psoriasis, cancer and AIDS. Accordingly, the detection of these thiol-containing biomolecules in biological samples is very important. While de Silva demonstrated the first use of PET sensors for thiols in 1998,32a it was in 2004, Martínez-Máñez and coworkers developed two squaraine based fluorescent chemosensors 33a and 33b for the detection of thiols (Fig. 16).32b Their solutions showed colour changes from blue to colorless along with fluorescence quenching in the presence of thiol-containing compounds, which is attributed to the selective addition of thiols to the cyclobutene ring in the chemosensors. These are two representative examples of thiol chemosensors that cannot distinguish Cys/Hcy and GSH.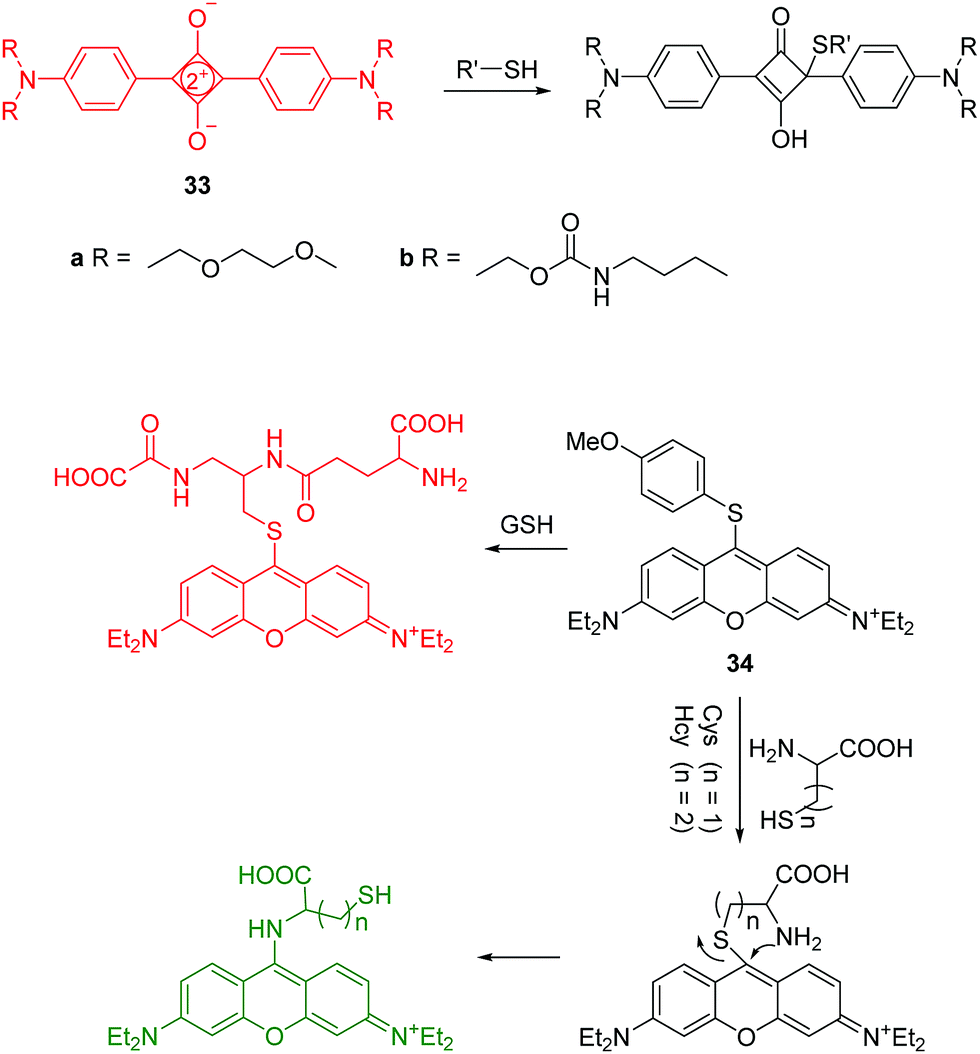 | ||
| Fig. 16 Structures and proposed mechanism of 33 and 34 for detection of thiols. | ||
The Guo group reported a pyronin B based fluorescent chemosensor 34 for the discrimination of Cys/Hcy and GSH using different emission channels (Fig. 16).33 Initially, free 34 is non-fluorescent due to the PET process from the methoxythiophenol group to the pyronin moiety. Upon treatment of 34 with GSH, a fluorescence enhancement at 622 nm occurs due to the replacement of 4-methoxythiophenol moiety by the thiol group of GSH. In the case of Cys/Hcy, an intramolecular rearrangement occurs followed by a substitution reaction, which leads to fluorescence enhancement at 546 nm. The chemosensor has been applied to imaging Cys/Hcy and GSH in live cells. Importantly, the use of the intramolecular rearrangement of Cys/Hcy with chemosensors is a typical strategy for the design of fluorescent chemosensors to discriminate Cys/Hcy and GSH.
More recently, Urano and co-workers reported two reversible fluorescent chemosensors 35a and 35b for GSH (Fig. 17).34 These two chemosensors show ratiometric fluorescence response to GSH due to the FRET process. The Si-rhodamines were selected as reaction sites because they achieve an intermolecular equilibrium with GSH. As a donor fluorophore, the O-rhodamine was selected owing to its excellent spectral overlap with the Si-rhodamines. Upon addition of GSH, the emission of the Si-rhodamines decreases while the emission of the O-rhodamine increases. The fluorescence ratio of the O-rhodamine and Si-rhodamine units resulted in Kd values of 0.6 and 3.0 mM, respectively for 35a and 35b. Chemosensor 35b has been used to image and quantify GSH in live cells. The authors have shown that these fluorescent chemosensors are revolutionary tools for investigating how GSH dynamics are regulated in a physiological context.
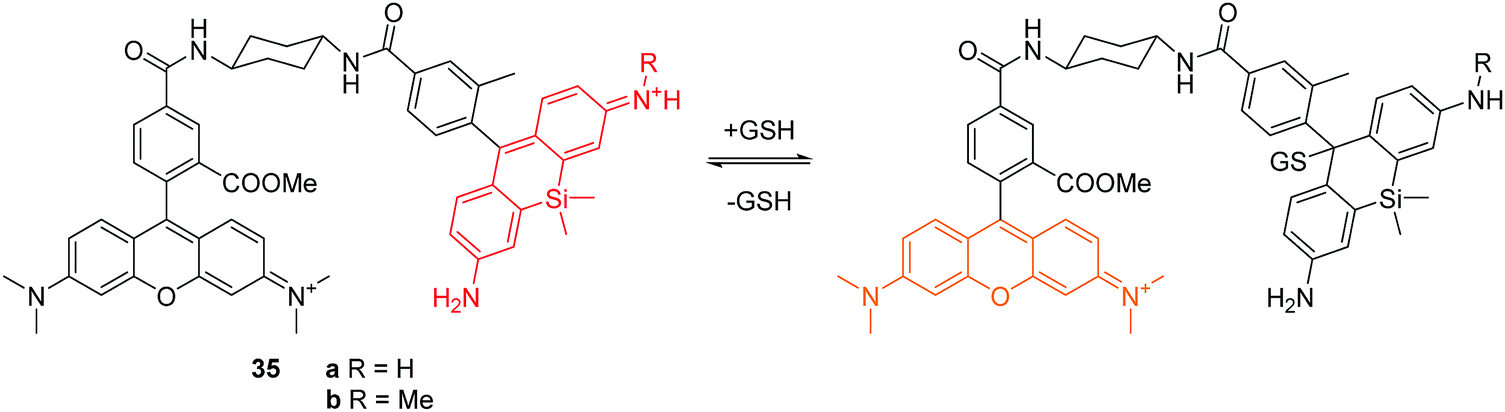 | ||
| Fig. 17 Structures and proposed mechanism of 35a and 35b for detection of GSH. | ||
Hydrogen sulfide (H2S) is the smallest member of the reactive sulfur species (RSS). It has been characterized as a crucial gaseous transmitter. However, a variety of emerging data suggest that hydrogen polysulfide (H2Sn) might be the signaling molecules instead of H2S. Given the importance of H2S and H2Sn in redox biology, the Xian group has developed several novel fluorescent chemosensors for these two species. Recently, they prepared a fluorescent chemosensor 36, which enables dual-channel discrimination between H2S and H2Sn (Fig. 18).35 The design principle for this chemosensor is that H2S selectively reacts with the azidocoumarin moiety, while H2Sn only reacts with phenyl 2-(benzoylthio)benzoate, which results in the corresponding fluorescence “turn-on”. However, the real situation is more complicated since the azide group of 36 can be partially reduced by H2Sn and the reaction of H2S with azides results in the formation of H2Sn (Fig. 18). However, due to the FRET process, the reaction with H2Sn should just produce green fluorescence from rhodol. Furthermore, less than 0.5 equivalents of H2Sn are produced from the reaction of H2S (1 equivalent) and azide, therefore the reaction with H2S can produce emission signals from both coumarin (major) and rhodol (minor). Overall, 36 can detect H2S and H2Sn from distinct emission channels. This chemosensor has been used to image H2S and H2Sn in live cells. Moreover, this work provides a strategy for developing fluorescent chemosensors that can discriminate two or more closely related species using different fluorescence channels.
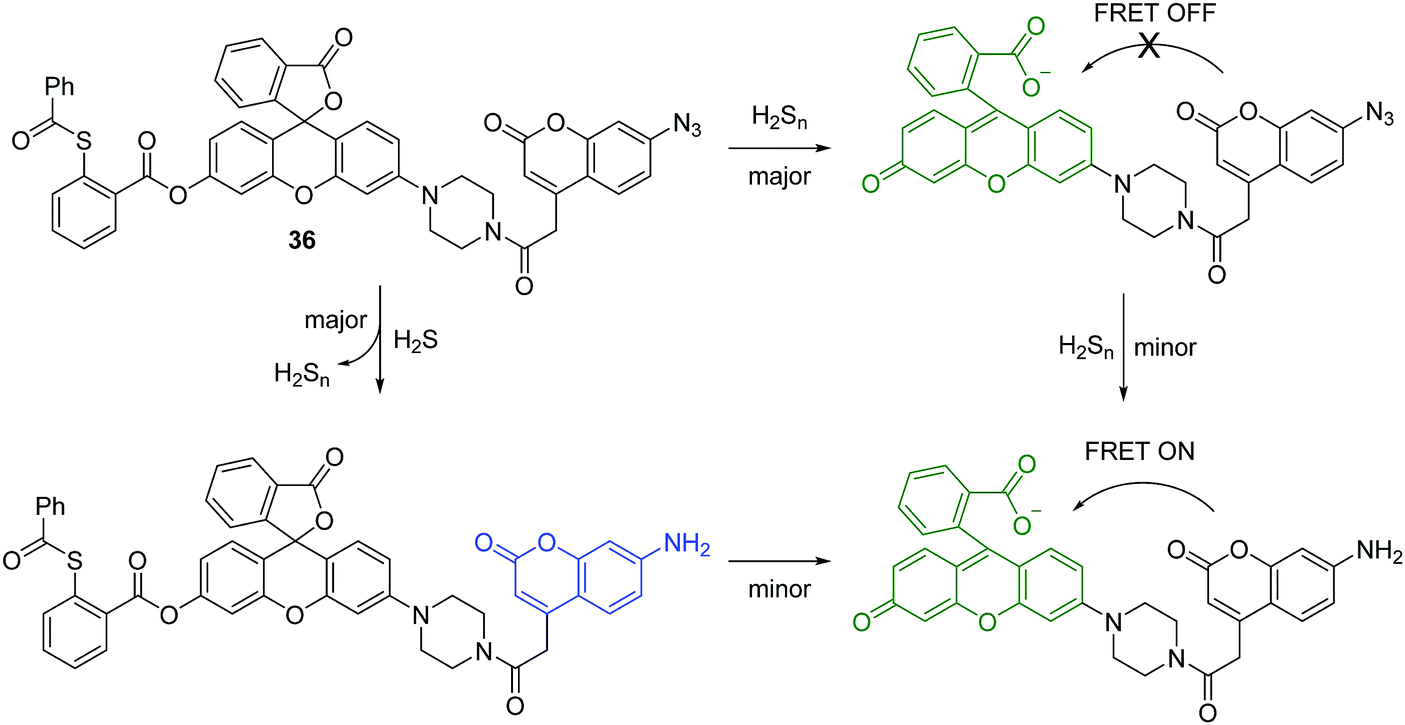 | ||
| Fig. 18 Structure and proposed mechanism of 36 for detection of H2S and H2Sn. | ||
Hydrogen selenide (H2Se) can be thought of as analogous to H2S and it has been shown to be involved in many physiological and pathological processes. There are only a few fluorescent chemosensors for H2Se that have been reported to date. Recently, the Tang group reported a hemicyanine based NIR fluorescent chemosensor 37 for H2Se, using the selective cleavage of Se–N in benzoselenadizole by H2Se through nucleophilic displacement (Fig. 19).36 Initially, the free chemosensor is non-fluorescent due to the heavy atom effect of Se. However, after the addition of H2Se, the chemosensor undergoes a ca. 10-fold “turn-on” fluorescence response. Additionally, it was successfully used for imaging endogenous H2Se in live cells and in mice.
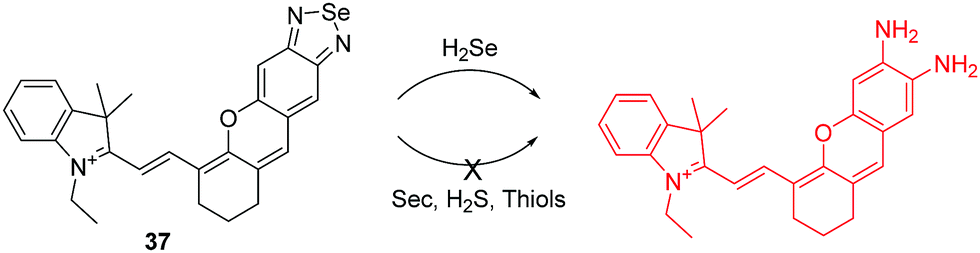 | ||
| Fig. 19 Structure and proposed mechanism of 37 for detection of H2Se. | ||
4.2 Fluorescent chemosensors for other small neutral molecules
Besides the anionic ROS/RNS, there are some neutral ROS/RNS such as hydrogen peroxide (H2O2) and nitric oxide (NO), which also play important roles in many biological processes. Lin and coworkers reported a fluorescent chemosensor 38, which can respond to H2O2, NO, and H2O2/NO with three different sets of fluorescence signals (Fig. 20).37 Upon addition of H2O2, blue emission at 460 nm with excitation at 400 nm is observed. However, when only NO is added, a rhodamine associated enhancement in emission at 580 nm is observed when excited at 550 nm. The chemosensor displays enhanced emission at just 580 nm with excitation at both 400 nm and 550 nm in the presence of both H2O2 and NO due to a FRET process. Furthermore, it was shown to be capable of simultaneously monitoring endogenous H2O2 and NO in live cells.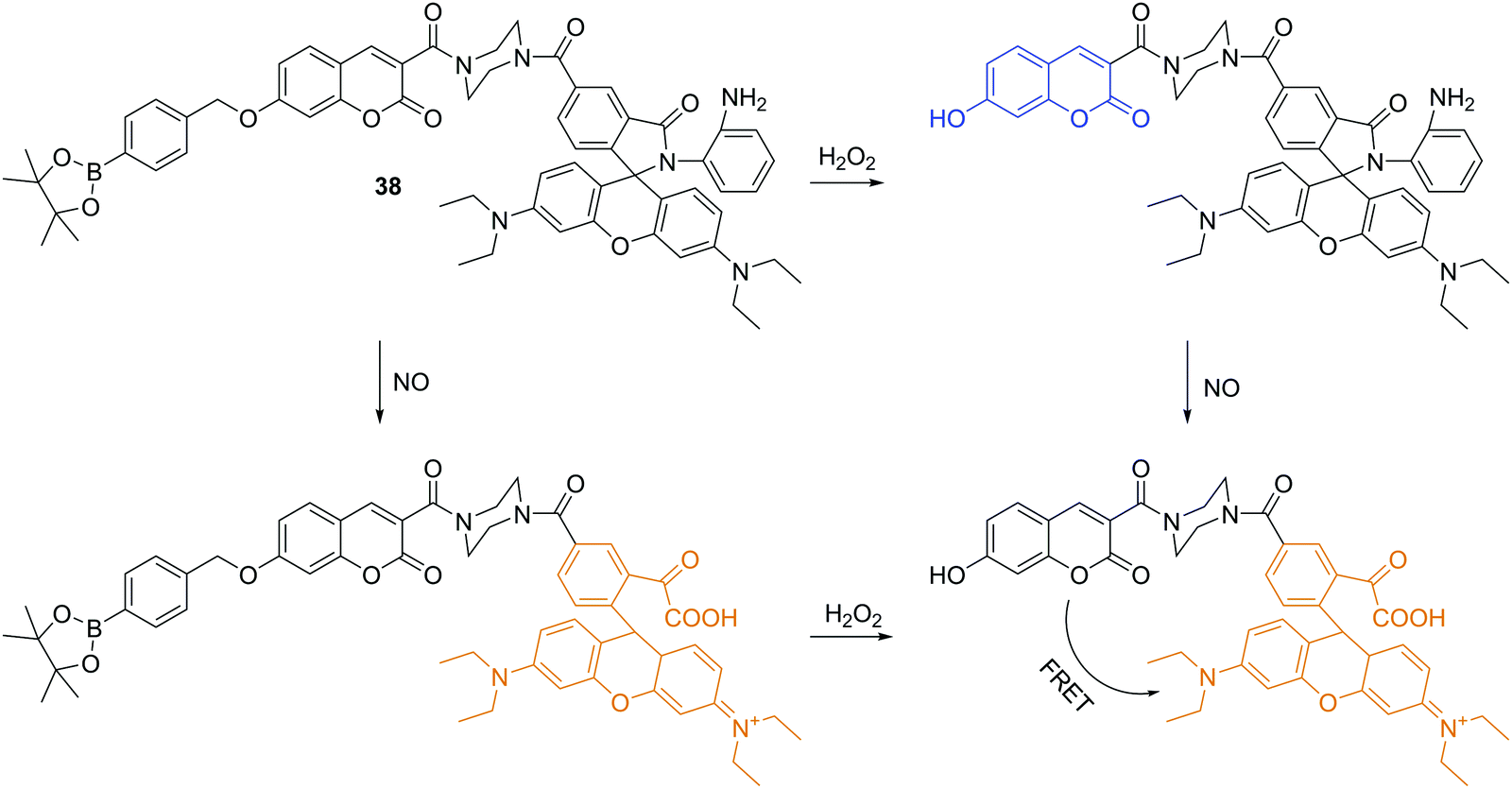 | ||
| Fig. 20 Structure and proposed mechanism of 38 for detection of H2O2 and NO. | ||
Chemical explosives such as 1,3,5-trinitroperhydro-1,3,5-triazine (RDX), picric acid (PA) and 2,4,6-trinitrotoluene (TNT) are a threat to public health and safety. Therefore, fluorescent chemosensors and sensor materials for the rapid and selective detection of chemical explosives at trace levels are of great importance. Anzenbacher Jr. and coworkers developed three pyrene based fluorescent chemosensors 39a, 39b and 39c for RDX (Fig. 21).38 These chemosensors show “turn on” fluorescence response to RDX based on different mechanisms. The fluorescence enhancement of 39a can be ascribed to protonation of the tertiary amines upon deprotonation from RDX. While, the formation of iminium cation and imine can be the reason for fluorescence enhancement of 39b and 39c, respectively. Furthermore, these three chemosensors were used to construct a fluorescent assay to discriminate different analytes.
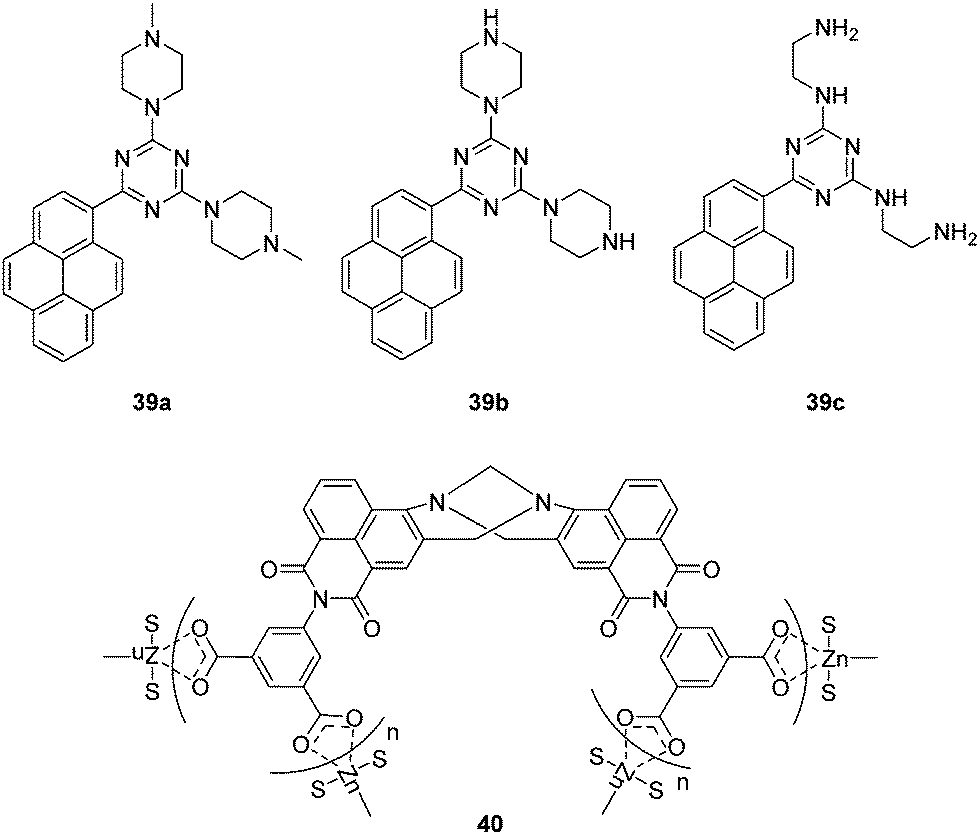 | ||
| Fig. 21 Structures of the fluorescent chemosensors 39a–c and 40. | ||
Recently, the Gunnlaugsson group have reported a supramolecular Tröger's base derived zinc coordination polymer 40 for fluorescent sensing of phenolic–nitroaromatic explosives (Fig. 21).39 The aqueous suspension of 40 displayed strong green fluorescence at 520 nm, due to the ICT transition. A selective fluorescence quenching was observed towards phenolic–nitroaromatics (4-nitrophenol (4-NP), 2,4-dinitrophenol (2,4-DNP) and PA) in the presence of other competing nitroaromatic species. Furthermore, 40 displays reversible PA sensing and the detection limit towards PA was determined to be 26.3 ppb. All these results indicate that 40 is a proficient sensor material for rapid detection of phenolic–nitroaromatics. The authors have previously demonstrated the use of Tröger's base based naphthalimide structures as cellular imaging agents and as potential anticancer agents.
Sulfur mustard (SM), a chemical warfare agent, is known to be extremely toxic, quite stable, and easy to synthesize. Discovery of fluorescent chemosensors for the selective and sensitive detection of SM is of great importance. In 2013, the Anslyn group developed fluorescent turn-on chemosensor for a sulfur mustard simulant 2-chloroethyl ethyl sulfide (CEES) based on a metal-ion indicator displacement assay (IDA) (Fig. 22).40 In this system, they developed a supramolecular system containing two units including a receptor dithiol 41 and Cd2+–indicator complex 42. Dithiol 41 can rapidly react with CEES to yield a podand 43, which exhibits high affinity to Cd2+ and hence displaces an indicator (4-methylesculetin) from 42, leading to fluorescence enhancement at 460 nm. The detection limit of CEES was found to be 0.2 μM, and can detect nerve agent levels that pose a health risk.
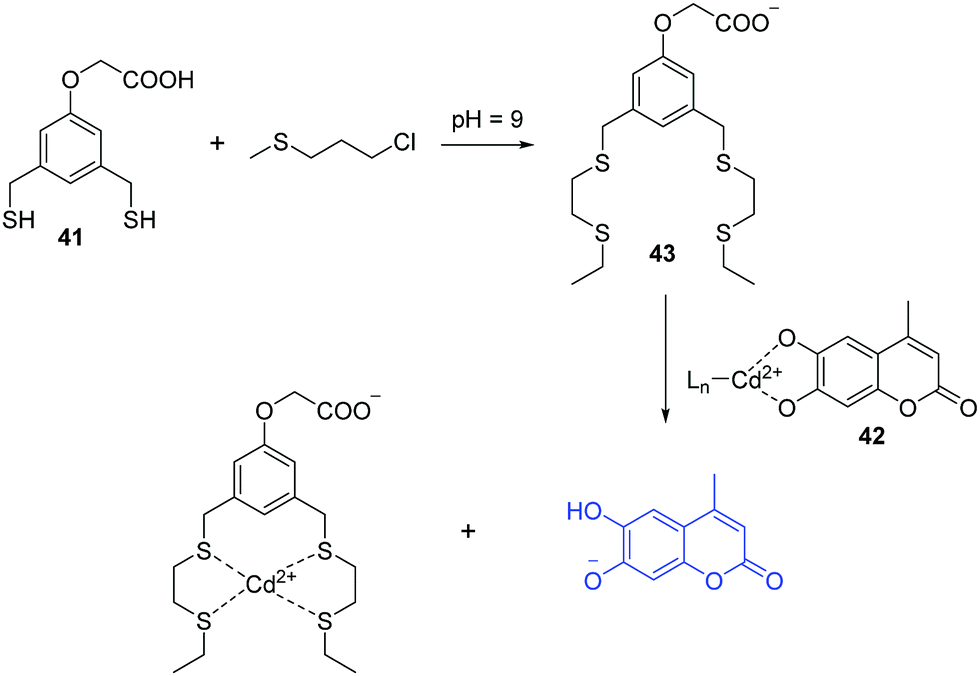 | ||
| Fig. 22 Proposed sensing mechanism for the detection of CEES. | ||
Enantioselective fluorescent chemosensors is another hot topic in the field of chemosensors. It has gained tremendous interest in recent years. The Pu group have developed a series of enantioselective fluorescent chemosensors for the recognition of some important chiral compounds. In 2015, Pu, Yu and coworkers reported a 1,1′-bi-2-naphthol (BINOL)-based bis(naphthylimine) fluorescent chemosensor 44 for the detection of chiral functional amines (Fig. 23).41 In the presence of Zn(OAc)2, 44 can react with chiral amines to release 2-naphthylamine with a blue emission (λ1 = 427 nm), which allows the substrate concentration to be determined. The combination of the remaining chiral binaphthyl unit with the chiral substrates results in significant enantioselective fluorescence enhancements at λ2 > 500 nm, which facilitates the determination of enantiomeric composition. Thus, both concentration and enantiomeric composition can be determined by one measurement of the fluorescent response of 44. This design principle represents a potentially general strategy for the development of dual responsive fluorescent sensors.
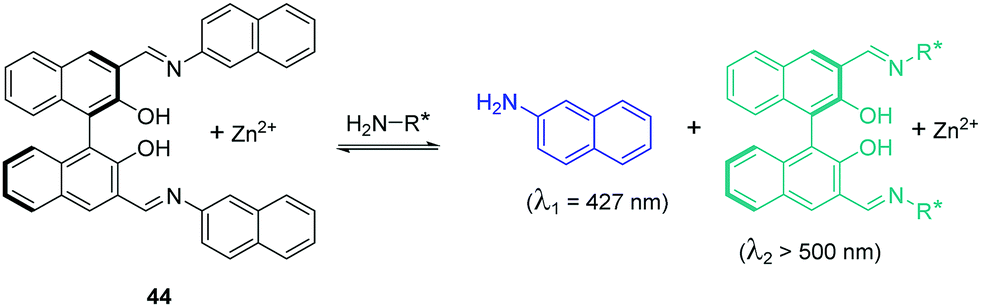 | ||
| Fig. 23 Structure and proposed mechanism of 44 for detection of chiral functional amines. | ||
5. Fluorescent chemosensors for biomacromolecules
Biomacromolecules are vital for the function of living biological systems. However, the abnormal expression of these biomacromolecules often has huge consequences. The use of fluorescence imaging techniques provides a powerful tool for studying these biomacromolecules and to fully understand their purpose in these complex biological systems due to excellent spatial and temporal resolution and high molecular specificity. The detection of biomacromolecules is a unique task as they often have large molecular weights, complex structures and a range of biological function. Over the past several decades, a number of fluorescent chemosensors have been developed, which have proven indispensable for bioimaging and the investigation of disease.Czarnik carried out pioneering work on anthrylpolyamines 45a–d to sense polyanions such as heparin, poly-L-glutamate, ds DNA (double-stranded DNA) and ss DNA (single-stranded DNA) in water (Fig. 24).42 These chemosensors display a red-shift and decrease in their emission spectra when bound to either ds DNA or to ss DNA. The chemosensor 45b is effective in binding to polyglutamate while 45c an excellent binder of heparin. They have been used to monitor the activity of pronase and heparinase, respectively.
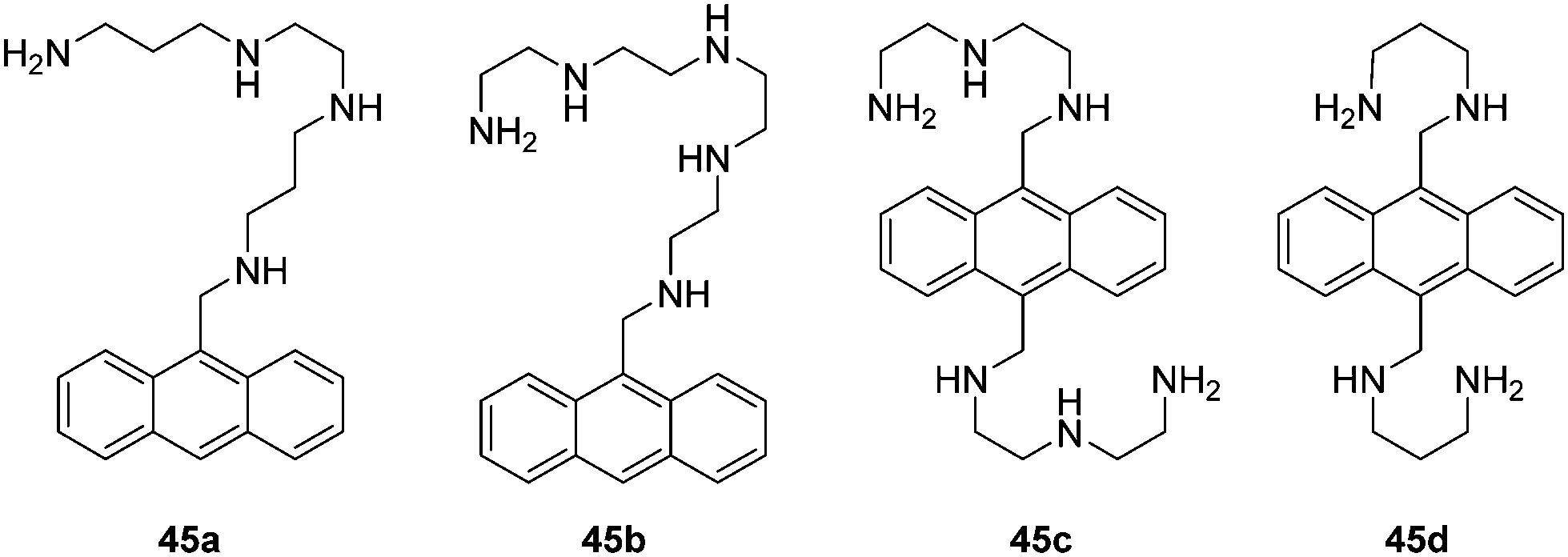 | ||
| Fig. 24 Structures of the fluorescent chemosensors 45a–d. | ||
Schmuck and co-workers reported a pyrene-based peptide beacon (fluorescent chemosensor 46) that was shown to intercalate with DNA (Fig. 25).43 In solution, the folded conformation of 46 exhibits a typical pyrene excimer emission. However, when bound to DNA the chemosensor undergoes a conformational change to the unfolded form. The change in conformation leads to a ratiometric change in fluorescence from excimer (490 nm) to monomer emission (406 nm).
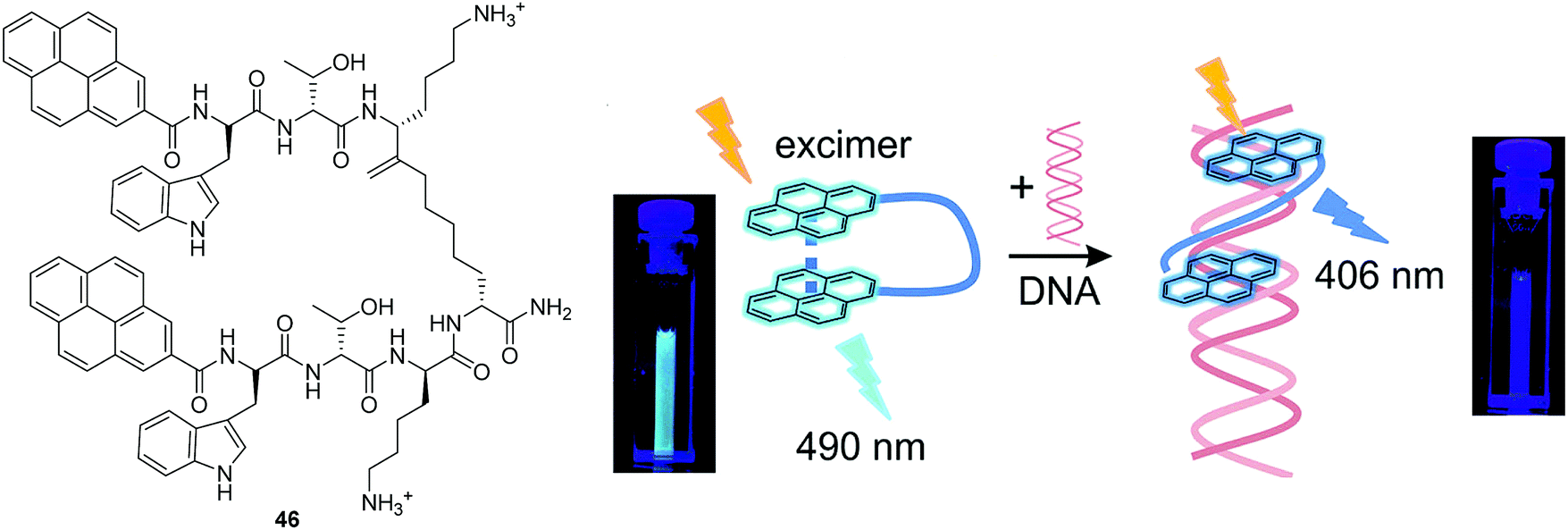 | ||
| Fig. 25 Structures of the fluorescent chemosensors 46 and the schematic illustration of 46 and Its Interaction with nucleic acid (the photographs show the corresponding cuvettes under UV light). Reproduced from ref. 43 with the permission of the American Chemical Society. | ||
For the purpose of protein labeling, Hamachi developed a fluorescent semi-synthetic chemosensor 47 based on the ligand-directed tosyl (LDT) chemistry (Fig. 26).44 In this quencher-tethered LDT (Q-LDT), the fluorophore (coumarin) is covalently attached to a protein ligand and an azoic fluorescence quencher via a labile sulfonate linkage. When the ligand binds to a target protein surface, the sulfonate undergoes nucleophilic cleavage, separating the coumarin fluorophore from the quencher. However, it still shows very weak fluorescence since the fluorophore and quencher remain close together within the ligand-binding site of the protein. However, addition of exogenous ligand leads to a fluorescence recovery as a result of the displacement of the quencher-tethered ligand (Fig. 26). This chemosensor has been applied for ligand binding assays of human carbonic anhydrase II (hCAII) and the SH2 domain in purified protein solutions as well as in crude cell lysates.
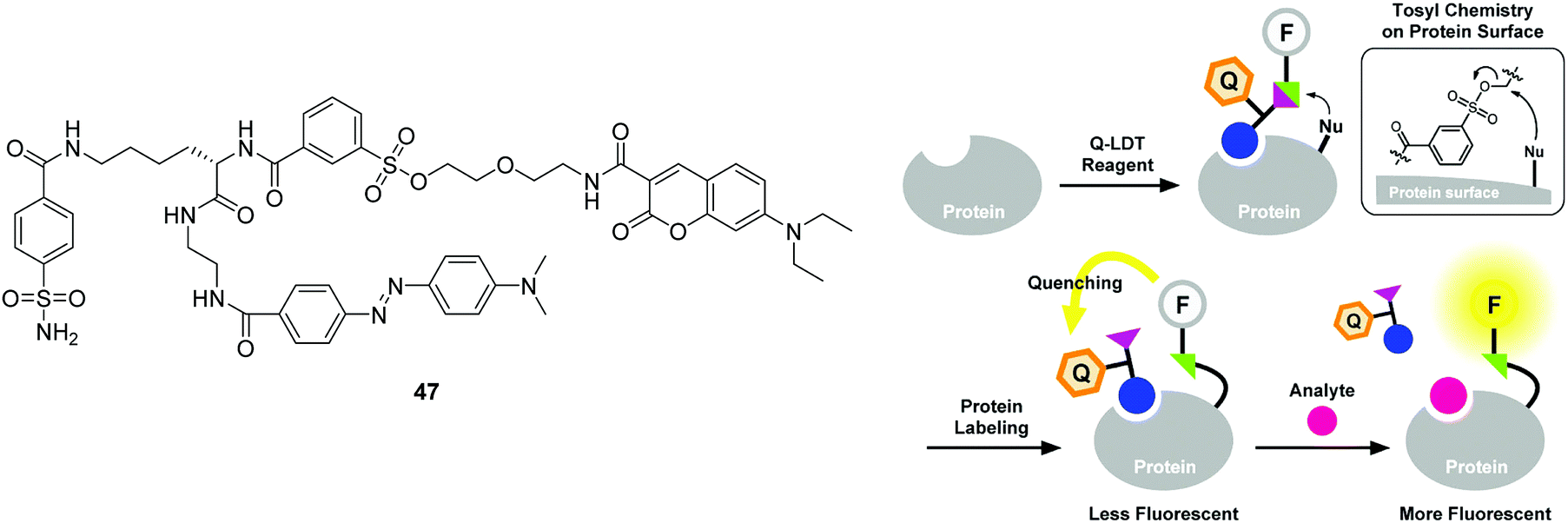 | ||
| Fig. 26 Structures of the fluorescent chemosensors 47 and schematic illustration of the strategy for the Q-LDT-mediated construction of turn-on fluorescent biosensors. Reproduced from ref. 44 with the permission of the American Chemical Society. | ||
Recently, Kikuchi and coworkers developed a fluorescent chemosensor 48 for heterochromatin protein 1 using the photoactive yellow protein (PYP) as a tag (Fig. 27).45 The chemosensor consists of a hydroxy cinnamic as the PYP ligand, fluorescein the fluorophore, and nitrobenzene the quencher moiety. The acetylated fluorescein was used in 48 because esters are membrane permeable while digested fluorescein molecules are non-permeable. However, 48 can be rapidly digested by cellular esterases yielding the anionic-48 as shown in Fig. 27. Thus this chemosensor enables no-wash selective labeling of intracellular proteins fused to the PYP tag in a desirable time frame, without adhesion or accumulation of the tag or the probe with non-targeted organelles.
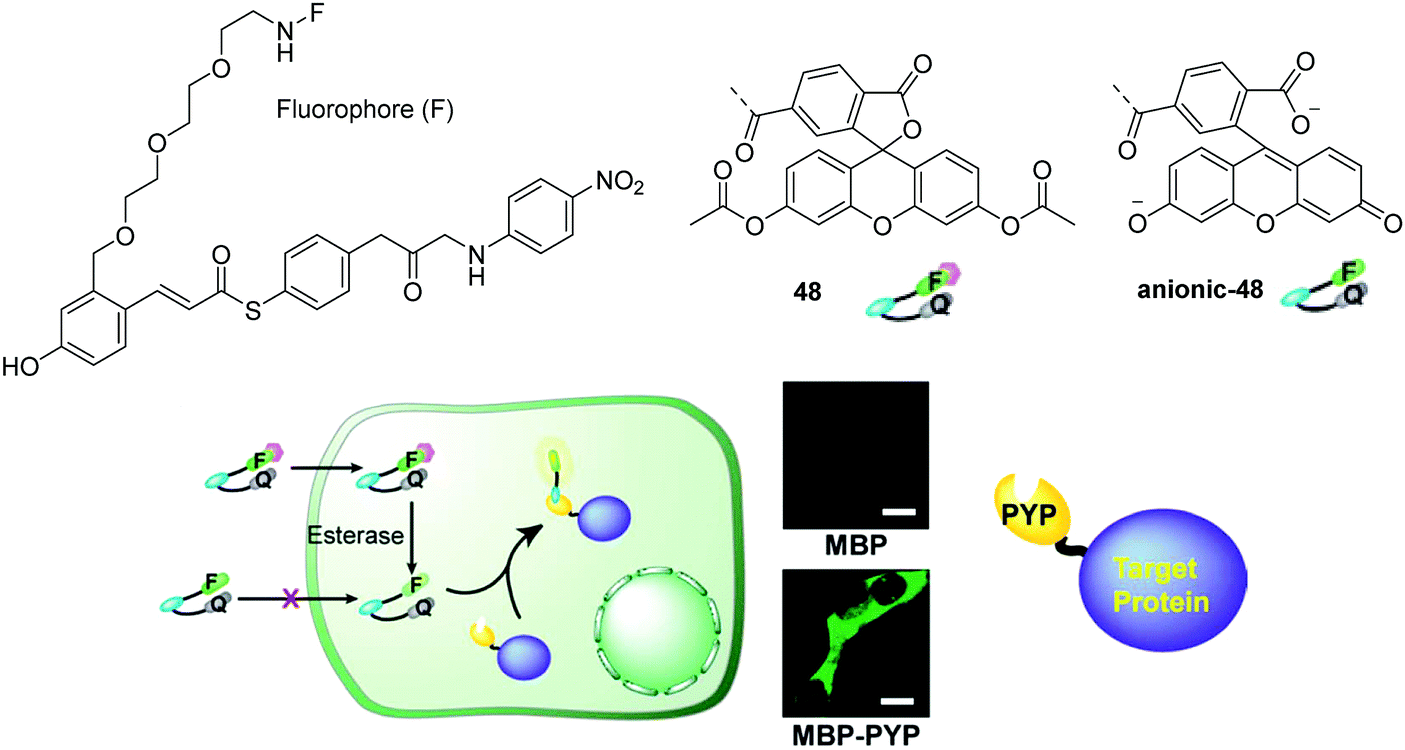 | ||
| Fig. 27 Structures of the fluorescent chemosensors 48, anionic-48 and no-wash live cell imaging of protein labeling with 48 and maltose-binding protein (MBP) (top) and MBP-PYP (bottom) expressed in HEK 293T cells. Reproduced from ref. 45 with the permission of the Royal Society of Chemistry. | ||
Alzheimer's disease (AD) is a neurodegenerative disease that has a massive effect on an individual's memory, cognitive abilities and personality.46 Amyloid-β (Aβ) plaques are considered be a key pathological biomarker for AD. Therefore, the development of a chemosensor for the detection of Aβ plaques in vivo would be highly desirable for early diagnosis and monitoring of AD. Ahn and co-workers have developed a number of two-photon dyes 49a–e as candidates for fluorescent chemosensors for Aβ plaques (Fig. 28).46 The donor–acceptor dyes developed were shown to be environmentally sensitive due to the formation of intramolecular charge transfer (ICT) excited states. In hydrophobic environments such as organic solvents, the dyes exhibited strong fluorescence. However, in highly polar solvents the probes were only weakly fluorescent. Therefore, it was believed that these chemosensors could be used for the in vivo imaging of amyloid-β (Aβ) plaques due to the cross-β sheets of the amyloid plaques providing a hydrophobic environment inside and a hydrophilic environment outside. Among those NIR dyes, 49a was shown to be a novel fluorescent chemosensor for the detection of Aβ plaques. The chemosensor which possesses a considerable two-photon absorption cross-section value at 1000 nm was shown to have the ability to penetrate the blood brain barrier (BBB) and allow in vivo imaging of Aβ in a live mouse model.
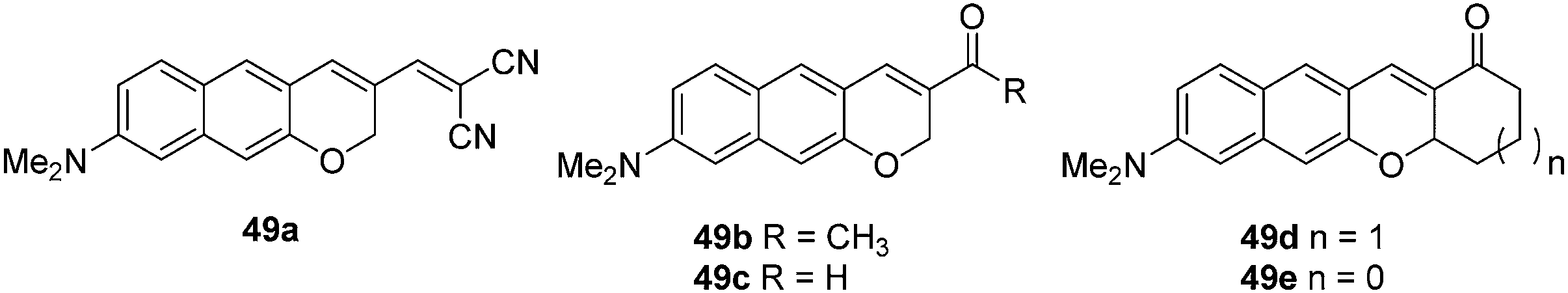 | ||
| Fig. 28 Structures of the fluorescent chemosensors 49a–e. | ||
Over 7 million people die annually as the result of cancer, with the number set to rise over the next 20 years. This highlights the need and importance of developing cancer biomarkers. A particularly useful candidate for cancer imaging is cyclooxygenase-2 (COX-2), given that different levels are expressed in tumors and in inflammatory lesions.47 Peng and coworkers reported a fluorescent chemosensor 50, which can distinguish healthy cells from cancerous cells and more importantly can distinguish cancerous cells from inflammatory cells (Fig. 29).47 In aqueous buffer, 50 is in a quenched folded-form, due to the PET process. An “off–on” fluorescence response was observed for inflammations and cancers where COX-2 is overexpressed. However the fluorescent emission is significantly different at the two sites due to different levels of COX-2 being expressed. For sites with inflammation, the fluorescence emission (615 nm) increases gradually over a COX-2 range of 0–0.12 μg mL−1. While for sites with cancer the fluorescence emission (615 nm) decreases and a new emission appears at 555 nm over a COX-2 range from 0.12–3.32 μg mL−1. Consequently, this chemosensor has been used to develop a fluorescence protocol for the selective discrimination of cancer over inflammation as shown in Fig. 29.
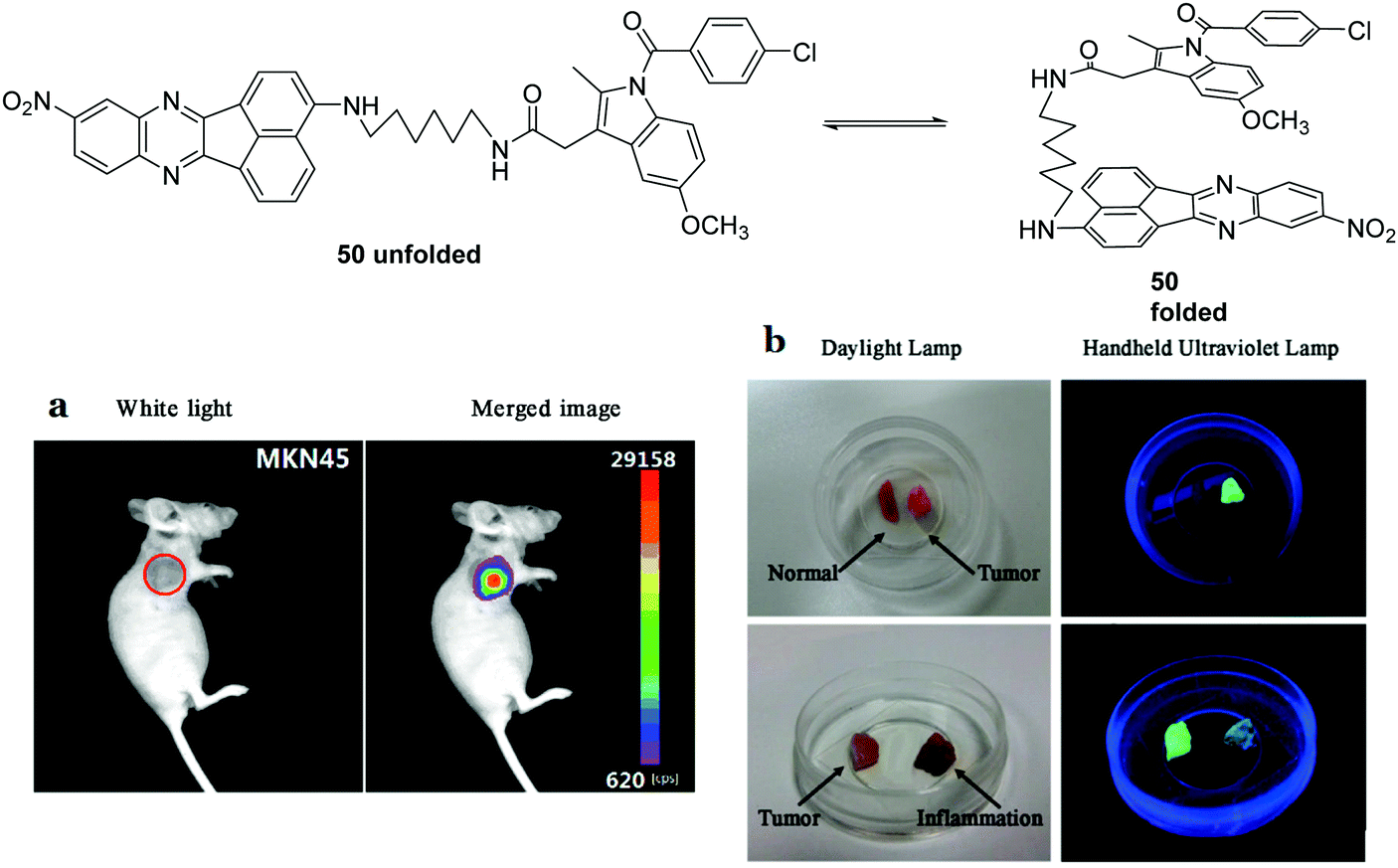 | ||
| Fig. 29 Structures of the fluorescent chemosensors 50 and imaging tumors in vivo. (a) 50 (30 μM) was injected intravenously (30 μL). The incubation time was 30 min. (b) Visualization of tumor resection by the naked eye under ultraviolet illumination. Reproduced from ref. 47 with the permission of the American Chemical Society. | ||
In contrast to fluorescent chemosensor 50, which is based on conformational changes, there have been several reaction-based fluorescent chemosensors for cancer using other biomarkers. β-Galactosidase (β-gal) is an exoglycosidase that catalyses the hydrolysis of β-galactosides to generate monosaccharides through the cleavage of the glycosidic bond. β-Gal is widely recognized as a biomarker enzyme for cell senescence and primary ovarian cancer. Zhu and coworkers recently reported a ratiometric near-infrared (NIR) fluorescent chemosensor 51 for β-gal detection (Fig. 30).48 Upon hydrolysis, there was a visual colour change from a faint yellow to a rose red, which allows the systems to be used for colorimetric detection. A 14-fold fluorescence increase was observed in the ratio (I685nm/I500nm) or a 34-fold fluorescence increase was observed at 685 nm. This NIR emission provided the opportunity for the chemosensor to be used for in situ and in vivo visualization of β-gal activity in colorectal tumor mice models. The chemosensor was successfully applied for in vivo real-time capture of β-gal activity at a tumor site as visualized using high-resolution three-dimensional imaging.
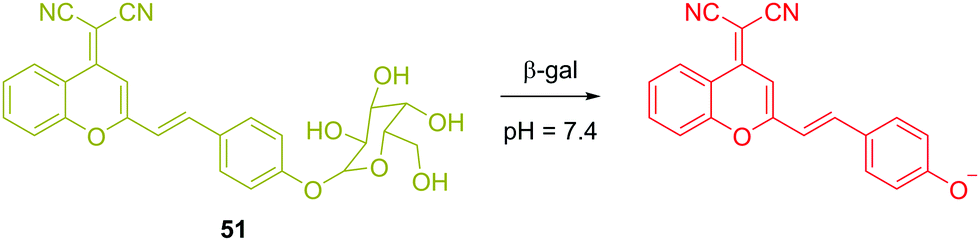 | ||
| Fig. 30 Structure and proposed mechanism of 51 for detection of β-gal. | ||
Recently, the Ma group have reported a cresyl violet based fluorescent chemosensor 52 for the detection of leucine aminopeptidase (LAP). LAP is known to be widely distributed in organisms from bacteria to humans, including various cancer cells (Fig. 31).49 The chemosensor shows a colorimetric “off–on” fluorescence response to LAP and the detection limit was determined to be 0.42 ng mL−1. Thus it can be used to monitor the concentration changes of trace amounts of LAP in different biosamples. The results indicate that cancer cells with a higher level of LAP show much stronger resistance toward cisplatin. The authors demonstrated that LAP contributes to intrinsic resistance and serves as a simple monitor to reflect the relative resistance of cancer cells.
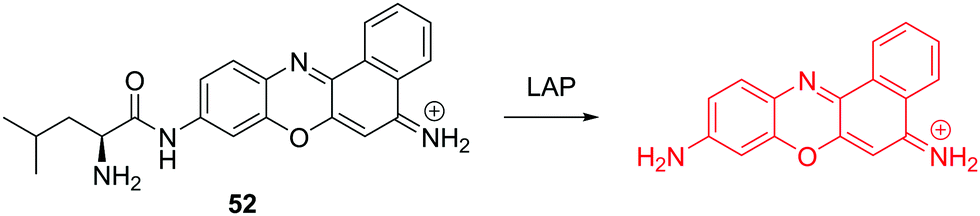 | ||
| Fig. 31 Structure and proposed mechanism of 52 for detection of LAP. | ||
Alkaline phosphatase (ALP) belongs to a subfamily of phosphatases, which are found in mammalian tissues. ALP is known as a hydrolase enzyme, which is capable of catalyzing the hydrolysis of a phosphate ester from proteins, nucleic acids and other biological molecules. It has been suggested that elevated levels of ALP are linked to a number of diseases including cancer, cardiovascular, bone and hepatic diseases.50 Nagano et al. developed an “off–on” fluorescent chemosensor 53 for the analysis of western blots (Fig. 32).50 They achieved this through the attachment of a phosphate group to the phenolic group of 2-Me-4-OMe Tokyo green (TG), which became almost non-fluorescent. The hydrolysis of the phosphate ester by ALP resulted in a strong fluorescence enhancement and the chemosensor was shown to have a high affinity for western blots. Furthermore, this design strategy may deliver a general approach for the simple and rapid detection of proteins using western blots.
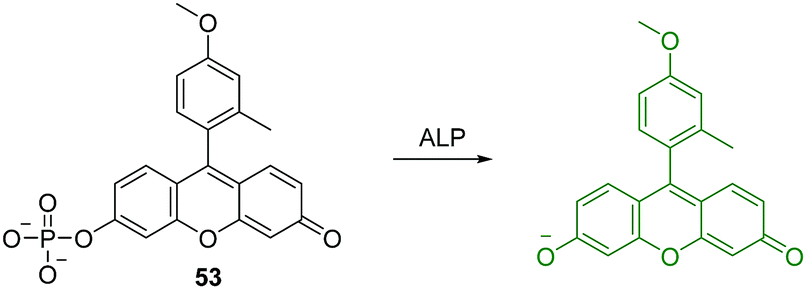 | ||
| Fig. 32 Structure and proposed mechanism of 53 for detection of ALP. | ||
6. Conclusions and outlook
The field of fluorescent chemosensors has developed significantly over the 150 years since Goppelsröder reported the first fluorescent chemosensor for Al3+. In particular, we have witnessed the explosive development of the field of fluorescent chemosensors over the past 50 years. The authors of this review believe that this phenomenal growth can in part be attributed to the pioneering research of Professor Anthony W. Czarnik's (DOB: 21-11-1957), and Professor A. Prasanna de Silva (DOB: 29-4-1952) who have inspired countless researchers through their seminal contributions to the field of chemosensors and molecular logic. We prepared this tutorial review in order to pay homage to their pivotal contributions to this field and to wish them both very happy birthdays in 2017. The review is also important since it demonstrates how a field can develop and flourish over a short 50-year period to become a recognized and established branch of chemistry. This review highlights representative examples of fluorescent chemosensors for various analytes including cations, anions, small neutral molecules and biomacromolecules from around 40 groups. Readers, requiring additional information are directed to the following recent reviews.51–54On reading this tutorial review, it may seem to young researchers that all the great problems in chemosensors research have already been solved. Nothing could be further from the truth, since we will always need “new” chemosensors for yet unknown analytes. This could be in the form of new biomarkers or trace pollutants in our air and water supplies. Also, biological and environmental analysis has increasingly stringent requirements imposed by regulatory bodies, so while a current chemosensor may work it may fall short of the required selectivity or sensitivity required for use in a specific practical application. So whether the problem requires bespoke new receptors or an improvement of existing systems, we will continue to need an increasing number of chemosensors to meet these challenges. In summary, we expect that chemosensor research will continue to expand and develop. As well as new and improved chemosensors, we anticipate that new applications or approaches to use existing fluorophores as chemosensors will emerge. For example, Gunnlaugsson and Scanlan have repurposed a naphthalimide fluorophore in the form of a “pre-probe”.55 These pre-probe chemosensors consist of a targeting group (carbohydrate) which is selectively cleaved (enzymatically) within target cancer cells to release the fluorophore allowing selective visualization. We envision that many of the other “old” chemosensors can similarly be repurposed for use in as yet unknown applications. In conclusion, the past set us going in the right direction, the present provided us with the challenges that need to be solved and the future gives us great hope that our lives will be improved by chemosensors.
Albert Einstein: “Learn from yesterday, live for today, hope for tomorrow.”
Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. D. J. and A. C. S. wish to thank the University of Bath for support. A. C. S. would like to thank the EPSRC for a Studentship. T. D. J. wishes to thank the Royal Society for a Wolfson Research Merit Award. T. G. would like to acknowledge Science Foundation Ireland (SFI) for financial support (SFI PI Award13/IA/1865). J. Y. acknowledges a grant from the National Creative Research Initiative programs of the National Research Foundation of Korea (NRF) funded by the Korean government (MSIP) (No. 2012R1A3A2048814).Notes and references
- A. W. Czarnik, Acc. Chem. Res., 1994, 27, 302–308 CrossRef CAS.
- A. W. Czarnik, Fluorescent Chemosensors for Ion and Molecule Recognition, American Chemical Society, Washington, DC, 1993 Search PubMed.
- A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed.
- B. Daly, J. Ling and A. P. de Silva, Chem. Soc. Rev., 2015, 44, 4203–4211 RSC.
- R. T. K. Kwok, C. W. T. Leung, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2015, 44, 4228–4238 RSC.
- Y. Yang, Q. Zhao, W. Feng and F. Li, Chem. Rev., 2013, 113, 192–270 CrossRef CAS PubMed.
- X. Li, X. Gao, W. Shi and H. Ma, Chem. Rev., 2014, 114, 590–659 CrossRef CAS PubMed.
- L. R. Sousa and J. M. Larson, J. Am. Chem. Soc., 1977, 99, 307–310 CrossRef CAS.
- H. He, M. A. Mortellaro, M. J. P. Leiner, R. J. Fraatz and J. K. Tusa, J. Am. Chem. Soc., 2003, 125, 1468–1469 CrossRef CAS PubMed.
- G. Farruggia, S. Iotti, L. Prodi, M. Montalti, N. Zaccheroni, P. B. Savage, V. Trapani, P. Sale and F. I. Wolf, J. Am. Chem. Soc., 2006, 128, 344–350 CrossRef CAS PubMed.
- R. Y. Tsien, Biochemistry, 1980, 19, 2396–2404 CrossRef CAS PubMed.
- H. J. Kim, J. H. Han, M. K. Kim, C. S. Lim, H. M. Kim and B. R. Cho, Angew. Chem., Int. Ed., 2010, 49, 6786–6789 CrossRef CAS PubMed.
- M. H. Lee, N. Park, C. Yi, J. H. Han, J. H. Hong, K. P. Kim, D. H. Kang, J. L. Sessler, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2014, 136, 14136–14142 CrossRef CAS PubMed.
- V. Dujols, F. Ford and A. W. Czarnik, J. Am. Chem. Soc., 1997, 119, 7386–7387 CrossRef CAS.
- Y. Liu, Q. Su, M. Chen, Y. Dong, Y. Shi, W. Feng, Z.-Y. Wu and F. Li, Adv. Mater., 2016, 28, 6625–6630 CrossRef CAS PubMed.
- M. C. Heffern, H. M. Park, H. Y. Au-Yeung, G. C. Van de Bittner, C. M. Ackerman, A. Stahl and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14219–14224 CrossRef CAS PubMed.
- P. Rivera-Fuentes, A. T. Wrobel, M. L. Zastrow, M. Khan, J. Georgiou, T. T. Luyben, J. C. Roder, K. Okamoto and S. J. Lippard, Chem. Sci., 2015, 6, 1944–1948 RSC.
- M.-Y. Chae and A. W. Czarnik, J. Am. Chem. Soc., 1992, 114, 9704–9705 CrossRef CAS.
- S.-K. Ko, Y.-K. Yang, J. Tae and I. Shin, J. Am. Chem. Soc., 2006, 128, 14150–14155 CrossRef CAS PubMed.
- O. A. Bozdemir, R. Guliyev, O. Buyukcakir, S. Selcuk, S. Kolemen, G. Gulseren, T. Nalbantoglu, H. Boyaci and E. U. Akkaya, J. Am. Chem. Soc., 2010, 132, 8029–8036 CrossRef CAS PubMed.
- D. H. Vance and A. W. Czarnik, J. Am. Chem. Soc., 1994, 116, 9397–9398 CrossRef CAS.
- D. H. Lee, S. Y. Kim and J.-I. Hong, Angew. Chem., Int. Ed., 2004, 43, 4777–4780 CrossRef CAS PubMed.
- B. A. Smith, W. J. Akers, W. M. Leevy, A. J. Lampkins, S. Xiao, W. Wolter, M. A. Suckow, S. Achilefu and B. D. Smith, J. Am. Chem. Soc., 2010, 132, 67–69 CrossRef CAS PubMed.
- Z. Zhang, D. S. Kim, C.-Y. Lin, H. Zhang, A. D. Lammer, V. M. Lynch, I. Popov, O. Š. Miljanić, E. V. Anslyn and J. L. Sessler, J. Am. Chem. Soc., 2015, 137, 7769–7774 CrossRef CAS PubMed.
- V. Amendola, G. Bergamaschi, M. Boiocchi, L. Fabbrizzi and L. Mosca, J. Am. Chem. Soc., 2013, 135, 6345–6355 CrossRef CAS PubMed.
- S. N. Berry, V. Soto-Cerrato, E. N. W. Howe, H. J. Clarke, I. Mistry, A. Tavassoli, Y.-T. Chang, R. Pérez-Tomás and P. A. Gale, Chem. Sci., 2016, 7, 5069–5077 RSC.
- J. J. Hu, N.-K. Wong, S. Ye, X. Chen, M.-Y. Lu, A. Q. Zhao, Y. Guo, A. C.-H. Ma, A. Y.-H. Leung, J. Shen and D. Yang, J. Am. Chem. Soc., 2015, 137, 6837–6843 CrossRef CAS PubMed.
- Q. Xu, C. H. Heo, G. Kim, H. W. Lee, H. M. Kim and J. Yoon, Angew. Chem., Int. Ed., 2015, 54, 4890–4894 CrossRef CAS PubMed.
- X. Sun, Q. Xu, G. Kim, S. E. Flower, J. P. Lowe, J. Yoon, J. S. Fossey, X. Qian, S. D. Bull and T. D. James, Chem. Sci., 2014, 5, 3368–3373 RSC.
- X. Jia, Q. Chen, Y. Yang, Y. Tang, R. Wang, Y. Xu, W. Zhu and X. Qian, J. Am. Chem. Soc., 2016, 138, 10778–10781 CrossRef CAS PubMed.
- L. Wang, J. Zhang, B. Kim, J. Peng, S. N. Berry, Y. Ni, D. Su, J. Lee, L. Yuan and Y.-T. Chang, J. Am. Chem. Soc., 2016, 138, 10394–10397 CrossRef CAS PubMed.
- (a) A. P. de Silva, H. Q. N. Gunaratne and T. Gunnlaugsson, Tetrahedron Lett., 1998, 39, 5077–5080 CrossRef CAS; (b) J. V. Ros-Lis, B. García, D. Jiménez, R. Martínez-Máñez, F. Sancenón, J. Soto, F. Gonzalvo and M. C. Valldecabres, J. Am. Chem. Soc., 2004, 126, 4064–4065 CrossRef CAS PubMed.
- J. Liu, Y.-Q. Sun, H. Zhang, Y. Huo, Y. Shi and W. Guo, Chem. Sci., 2014, 5, 3183–3188 RSC.
- K. Umezawa, M. Yoshida, M. Kamiya, T. Yamasoba and Y. Urano, Nat. Chem., 2017, 9, 279–286 CrossRef CAS PubMed.
- W. Chen, A. Pacheco, Y. Takano, J. J. Day, K. Hanaoka and M. Xian, Angew. Chem., Int. Ed., 2016, 55, 9993–9996 CrossRef CAS PubMed.
- F. Kong, L. Ge, X. Pan, K. Xu, X. Liu and B. Tang, Chem. Sci., 2016, 7, 1051–1056 RSC.
- L. Yuan, W. Lin, Y. Xie, B. Chen and S. Zhu, J. Am. Chem. Soc., 2012, 134, 1305–1315 CrossRef CAS PubMed.
- L. Mosca, S. K. Behzad and P. Anzenbacher Jr., J. Am. Chem. Soc., 2015, 137, 7967–7969 CrossRef CAS PubMed.
- S. Shanmugaraju, C. Dabadie, K. Byrne, A. J. Savyasachi, D. Umadevi, W. Schmitt, J. A. Kitchen and T. Gunnlaugsson, Chem. Sci., 2017, 8, 1535–1546 RSC.
- V. Kumar and E. V. Anslyn, J. Am. Chem. Soc., 2013, 135, 6338–6344 CrossRef CAS PubMed.
- K. Wen, S. Yu, Z. Huang, L. Chen, M. Xiao, X. Yu and L. Pu, J. Am. Chem. Soc., 2015, 137, 4517–4524 CrossRef CAS PubMed.
- S. A. Van Arman and A. W. Czarnik, J. Am. Chem. Soc., 1990, 112, 5376–5377 CrossRef CAS.
- J. Wu, Y. Zou, C. Li, W. Sicking, I. Piantanida, T. Yi and C. Schmuck, J. Am. Chem. Soc., 2012, 134, 1958–1961 CrossRef CAS PubMed.
- S. Tsukiji, H. Wang, M. Miyagawa, T. Tamura, Y. Takaoka and I. Hamachi, J. Am. Chem. Soc., 2009, 131, 9046–9054 CrossRef CAS PubMed.
- Y. Kamikawa, Y. Hori, K. Yamashita, L. Jin, S. Hirayama, D. M. Standley and K. Kikuchi, Chem. Sci., 2016, 7, 308–314 RSC.
- D. Kim, H. Moon, S. H. Baik, S. Singha, Y. W. Jun, T. Wang, K. H. Kim, B. S. Park, J. Jung, I. Mook-Jung and K. H. Ahn, J. Am. Chem. Soc., 2015, 137, 6781–6789 CrossRef CAS PubMed.
- H. Zhang, J. Fan, J. Wang, B. Dou, F. Zhou, J. Cao, J. Qu, Z. Cao, W. Zhao and X. Peng, J. Am. Chem. Soc., 2013, 135, 17469–17475 CrossRef CAS PubMed.
- K. Gu, Y. Xu, H. Li, Z. Guo, S. Zhu, S. Zhu, P. Shi, T. D. James, H. Tian and W.-H. Zhu, J. Am. Chem. Soc., 2016, 138, 5334–5340 CrossRef CAS PubMed.
- Q. Gong, W. Shi, L. Li and H. Ma, Chem. Sci., 2016, 7, 788–792 RSC.
- M. Kamiya, Y. Urano, N. Ebata, M. Yamamoto, J. Kosuge and T. Nagano, Angew. Chem., Int. Ed., 2005, 44, 5439–5441 CrossRef CAS PubMed.
- K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 CrossRef CAS PubMed.
- C. J. Chang, T. Gunnlaugsson and T. D. James, Chem. Soc. Rev., 2015, 44, 4176–4178 RSC.
- M. H. Lee, J. S. Kim and J. L. Sessler, Chem. Soc. Rev., 2015, 44, 4185–4191 RSC.
- J. Wu, B. Kwon, W. Liu, E. V. Anslyn, P. Wang and J. S. Kim, Chem. Rev., 2015, 115, 7893–7943 CrossRef CAS PubMed.
- E. Calatrava-Pérez, S. A. Bright, S. Achermann, C. Moylan, M. O. Senge, E. B. Veale, D. C. Williams, T. Gunnlaugsson and E. M. Scanlan, Chem. Commun., 2016, 52, 13086–13089 RSC.
| This journal is © The Royal Society of Chemistry 2017 |