
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel colorimetric assay for α-methylacyl-CoA racemase 1A (AMACR; P504S) utilizing the elimination of 2,4-dinitrophenolate†
Maksims
Yevglevskis
a,
Guat L.
Lee
a,
Amit
Nathubhai
a,
Yoana D.
Petrova
a,
Tony D.
James
 b,
Michael D.
Threadgill
a,
Timothy J.
Woodman
a and
Matthew D.
Lloyd
*a
b,
Michael D.
Threadgill
a,
Timothy J.
Woodman
a and
Matthew D.
Lloyd
*a
aDrug & Target Development, Department of Pharmacy & Pharmacology, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: M.D.Lloyd@bath.ac.uk; Fax: +-44-1225-386114; Tel: +-44-1225-386786
bDepartment of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK
First published on 17th March 2017
Abstract
α-Methylacyl-CoA racemase (AMACR; P504S) regulates branched-chain fatty acid degradation, activates Ibuprofen and is a recognised cancer drug target. A novel, facile colorimetric assay was developed based on elimination of 2,4-dinitrophenolate. The assay was used to test 5 known inhibitors, determining IC50 and Ki values, reversibility and characterizing irreversible inhibition.
α-Methylacyl-CoA racemase (AMACR;‡ P504S; E.C. 5.1.99.4) performs a key role in branched-chain fatty acid β-oxidation and the pharmacological activation of Ibuprofen and related drugs.1,2 AMACR catalyses its reaction by a deprotonation/reprotonation mechanism,3–5 in which either epimer of a 2-methylacyl-CoA or 2-arylpropanoyl-CoA substrate is converted to a near 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of epimers.3,5 Conversion of R-2-methylacyl-CoAs to their S-epimers enables degradation by β-oxidation.1,6R-Ibuprofen and most related R-2-APA drugs1 are pharmacologically activated by conversion to their corresponding acyl-CoA esters, before epimerization by AMACR. Hydrolysis of the epimeric products gives a mixture of R- and S-2-APA drugs, with the R-product recycled by the same pathway. Thus, inactive R-2-APA drugs are converted into their S-enantiomers, which are potent inhibitors of cyclooxygenase.7
:1 mixture of epimers.3,5 Conversion of R-2-methylacyl-CoAs to their S-epimers enables degradation by β-oxidation.1,6R-Ibuprofen and most related R-2-APA drugs1 are pharmacologically activated by conversion to their corresponding acyl-CoA esters, before epimerization by AMACR. Hydrolysis of the epimeric products gives a mixture of R- and S-2-APA drugs, with the R-product recycled by the same pathway. Thus, inactive R-2-APA drugs are converted into their S-enantiomers, which are potent inhibitors of cyclooxygenase.7
Concentrations of AMACR are increased in all prostate cancers,8,9 and in several other cancers.10–13 Increased AMACR catalytic activity has also been reported in prostate cancer.14,15 Reducing the cellular AMACR 1A15–17 protein using siRNA or shRNA reduces proliferation of prostate cancer cell lines via a pathway which is synergistic with androgen withdrawal.15 Some advanced prostate cancer cell lines revert from androgen-independent to androgen-dependent status17 upon AMACR knock-down. Consequently, AMACR has attracted considerable interest as a novel prostate cancer drug target16,18–20 and biomarker.1,21
Despite this interest, few inhibitors of AMACR have been reported. The majority of inhibitors are rationally designed acyl-CoA derivatives,18–20,22 which do not comply with Lipinski's guidelines23 and need to be delivered as carboxylate prodrugs. Drug delivery can be limited by in vivo conversion of the prodrug to the acyl-CoA drug, although preliminary in vivo studies24 appear promising. N-Dodecyl-N-methylcarbamoyl-CoA 1, a transition-state analogue, is the most potent inhibitor reported to date.19 Several non-specific protein-modifying agents also inhibit AMACR.16 No structure–activity relationships have been reported, probably due to the difficulties in assaying enzyme activity.
AMACR catalyses the conversion of either R- or S-2-methylacyl-CoA esters into a near 1:1 mixture of epimers3,5 (Scheme 1A). Assaying AMACR activity is challenging due to the reversibility of the reaction and the difficulties in differentiating the epimeric products since the stereochemical centre undergoing a change in configuration is remote from the stereochemical centres in the CoA moiety. Several endpoint assays have been reported and used for inhibitor testing, such as those using chiral acyl-CoA substrates18,19 or their derivatives.25 Notably, Wilson et al.16 reported screening of a library of 5000 compounds using a modified radiolabelled assay, and identified several non-specific protein modifying agents that were potent inhibitors of AMACR. Their assay was used for kinetic analysis of AMACR, but it is an endpoint assay measuring substrate conversion at a single time-point and extensive manipulation of samples is required. In addition the assay is likely to be complicated by the presence of a kinetic isotope effect. A continuous circular dichroism (CD) assay for the closely related Mycobacterium tuberculosis enzyme (MCR), which follows the conversion of either R- or S-ibuprofenoyl-CoA 2 to a near racemic mixture,26 has been used for limited testing of inhibitors.22 This assay is not subject to a kinetic isotope effect, but is low-throughput as only one sample can be analysed at the same time.
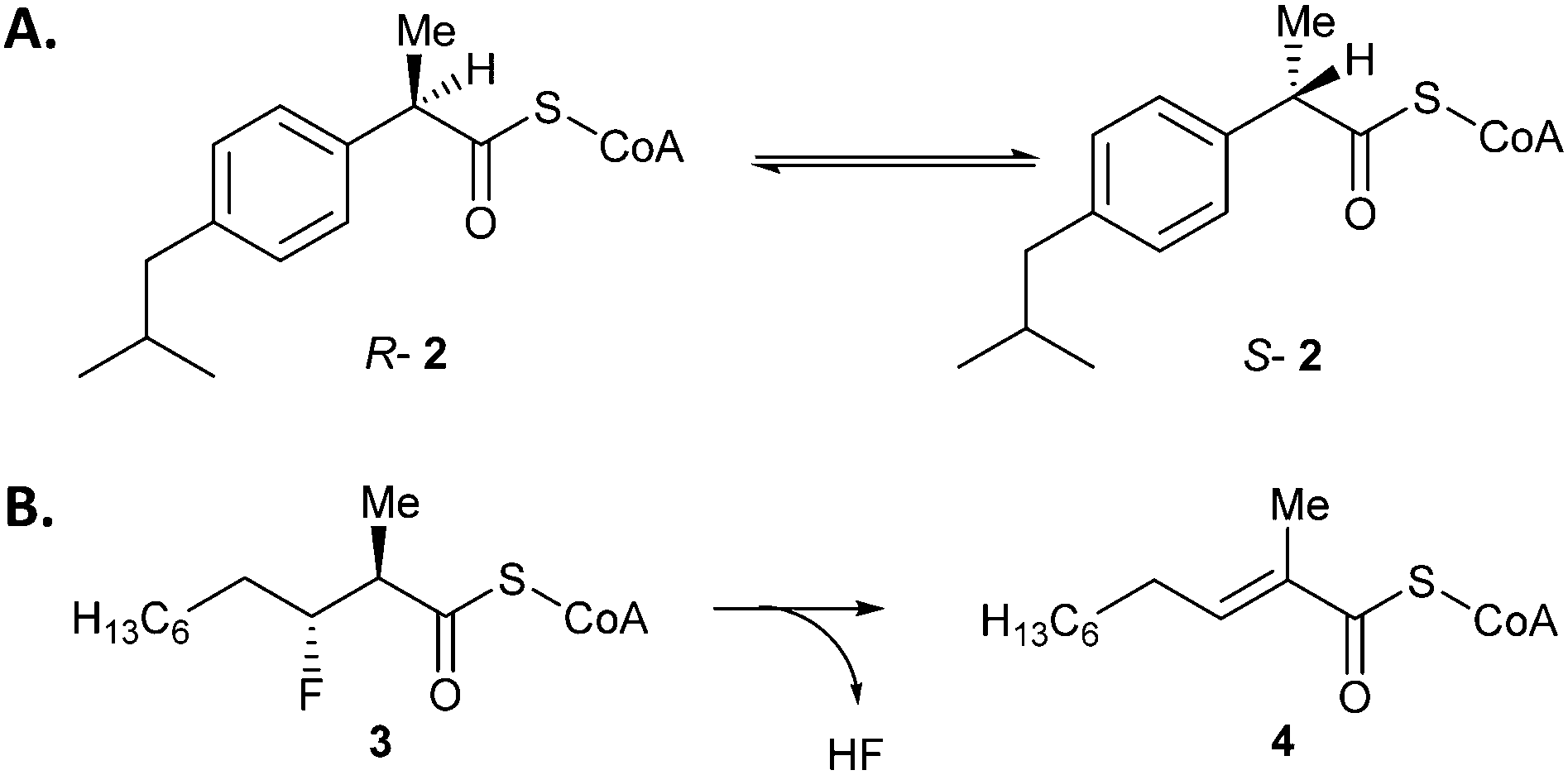 | ||
| Scheme 1 Reactions catalysed by AMACR. (A) Conversion of R- or S-2-methylacyl-CoA into an epimeric mixture, as illustrated by ibuprofenoyl-CoA 2. Assays commonly use an acyl-CoA substrate with α-3H which is ‘washed out’,16 or unlabelled substrate in the presence of 2H2O followed by 1H NMR analysis of the ‘washed in’ product.3,5 (B) The elimination of HF from (2R,3R)-3-fluoro-2-methyldecanoyl-CoA 3 to give unsaturated product 4. | ||
It has also been discovered that AMACR catalyses the elimination of fluoride from substrates such as 3-fluoro-2-methyldecanoyl-CoA (Scheme 1B), probably via an E1cb mechanism.27 This reaction has the advantage of being irreversible, and the methyl peaks of substrate and product do not overlap in the 1H spectrum allowing a simple route to determine the extent of conversion. This reaction has been used for the preliminary characterization of known inhibitors.28
The elimination of fluoride from 3-fluoro-2-methyldecanoyl-CoA led us to consider whether a colorimetric leaving group might eliminate from a suitable substrate. The pKa of HF (3.2)29 and 2,4-dinitrophenol (3.93)30 are similar, suggesting the conjugate bases will have similar leaving-group ability. Significantly, 2,4-dinitrophenolate 5 is a well-characterized chromophore (ε354 = 15300 M−1 cm−1).31 AMACR is known to accept substrates with a wide variety of side-chain structures,1 and hence the use of a substrate containing a 2,4-dinitrophenoxy- moiety as a chromogenic leaving group was investigated.
Racemic 3-(2,4-dinitrophenoxy)-2-methylpropanoyl-CoA 6 was synthesized (Scheme 2) by reaction of Sanger's reagent 7 with 2-methylpropan-1,3-diol 8 under basic conditions. Treatment of alcohol 9 with CrO3 and H2SO4 resulted in the racemic acid 10. This was coupled with CoA–SH5,27,28 to give the desired substrate 6.
 | ||
| Scheme 2 Synthesis of substrate 6. Reagents & conditions: (i) Na metal, 83%; (ii) CrO3, conc. H2SO4, acetone, 67%; (iii) carbonyldiimidazole, CH2Cl2; (iv) CoA–SH (Li+)3, 0.1 M NaHCO3aq./THF (1:1). | ||
Incubation of 6 with active AMACR resulted in an elimination reaction (Scheme 3A). The sample containing active enzyme possessed an intense yellow colour, which was absent from negative controls (Scheme 3B). 1H NMR analysis confirmed that 2,4-dinitrophenolate 5 and the predicted unsaturated product 11 were formed (Scheme 3C). Incubation of substrate 6 with active enzyme in a microtitre plate led to a rapid increase in absorbance at 354 nm, which was not observed in controls containing heat-inactivated enzyme (Scheme 3D). This reaction occurred in an enzyme concentration-dependent manner (ESI,† Fig. S1 and S2).
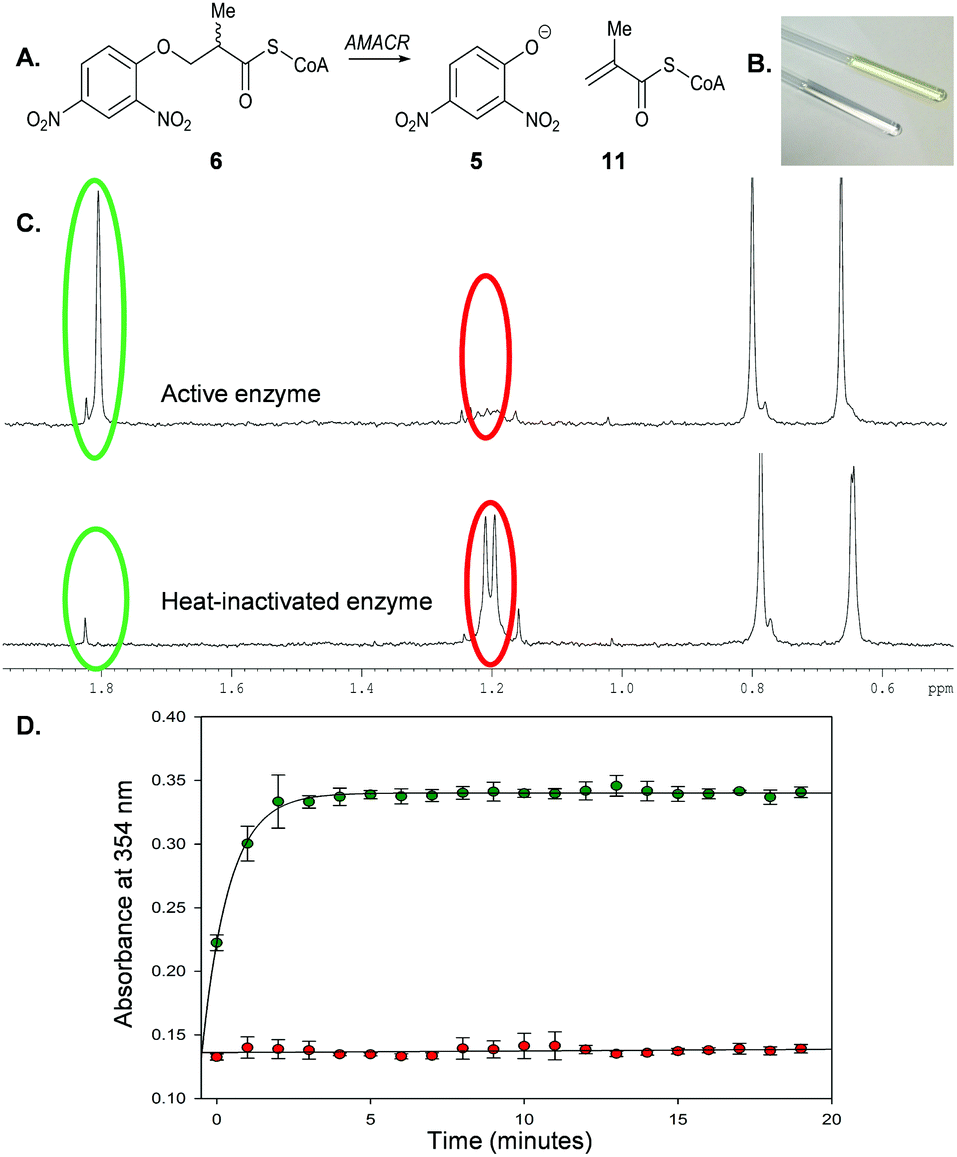 | ||
| Scheme 3 Elimination of 2,4-dinitrophenolate 5 from 3-(2,4-dinitrophenoxy)-2-methylpropanoyl-CoA 6 by AMACR. (A) Reaction catalysed by enzyme; (B) NMR tubes showing samples containing active enzyme (yellow) and heat-inactivated enzyme (clear); (C) 1H NMR spectra (500 MHz) of samples in B showing position of methyl groups of substrate 6 (red circles) and product 11 (green circles); (D) time course of reaction followed by monitoring absorbance of 2,4-nitrophenoxide 5 at 354 nm, showing negative control (heat-inactivated enzyme; red) and active enzyme (green). Assays comprised 40 μM 6 and recombinant AMACR (9.25 μg) in 100 μL NaH2PO4–NaOH, pH 7.4. Data are means ± SDM (n = 3). | ||
Assay conditions were optimized (ESI,† Fig. S3–S5) by investigating the effect of various additives on enzymatic activity. Addition of 2-mercaptoethanol, dithiothreitol or BSA had no significant effect on activity. Addition of 0.1% (v/v) Triton X-100 to the assay mixture resulted in a modest reduction of activity. This is probably due to the formation of micelles, as the amounts used are above the reported critical micellar concentrations for Triton X-100 (0.19 and 1.25 mM).32 In contrast, addition of 1.5% (w/v) N-lauroyl-sarcosine reduced activity to levels observed in negative controls. The enzyme proved to be tolerant of DMSO (ESI,† Fig. S5), up to at least 8% (v/v) (∼1.12 M). Michaelis–Menten kinetic behaviour was observed under the optimized assay conditions (ESI,† Fig. S6) and the following kinetic parameters were determined: Km = 58 μM; Vmax = 112 nmol min−1 mg−1; kcat = 0.088 s−1; kcat/Km = 1517 M−1 s−1. Optimised conditions for the assay were ∼8 μg enzyme in 200 μL NaH2PO4–NaOH aq., pH 7.4 containing 40 μM 6 and up to 8% (v/v) DMSO at 30 °C, monitoring at 354 nm. Typical rates for active enzyme and negative controls (means ± SD, n = 3) are 33 ± 1.9 and 0.72 ± 0.24 nmol min−1 mg−1. The limit of detection is ∼1.4 nmol min−1 mg−1. The dynamic range is ∼1.4–255 nmol min−1 mg−1. The sensitivity threshold is ∼0.5–1.0 nmol min−1 mg−1. The Z′* value33 for the assay was 0.906 (see ESI,† for further details on how these values were calculated). The presence of other enzymes such as branched-chain acyl-CoA oxidases (ACOXs) could potentially generate a false positive signal, but this can be avoided by using R-6 as ACOXs are known to be specific for S-2-methylacyl-CoA substrates.6
This new colorimetric assay for AMACR was validated using a series of known inhibitors for which bench-mark values are available (Fig. 1). N-Dodecyl-N-methylcarbamoyl-CoA 1 and ibuprofenoyl-CoA 2 were chosen as representative acyl-CoA inhibitors. Ebselen 12, Ebselen Oxide 13 and Rose Bengal 14 were chosen as examples of the non-specific protein modifying agents reported by Wilson et al.16 Inhibitor potency was initially assessed using dose–response curves to determine IC50 values (see Fig. 1B and C for example).
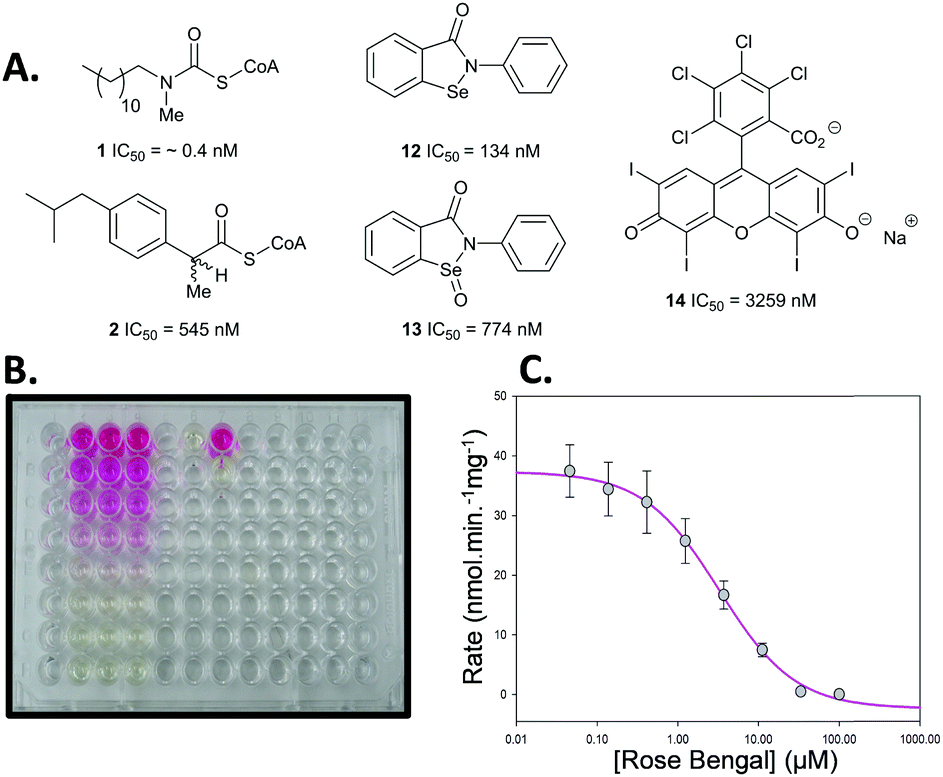 | ||
| Fig. 1 Inhibitors used in assay. (A) Chemical structures and determined IC50 values from dose–response curves; (B) example assay in 96-well plate. The pink colour results from the presence of the inhibitor Rose Bengal 14 and the yellow colour from the 2,4-dinitrophenolate 5 produced by the enzyme from substrate 6; (C) dose–response curve for Rose Bengal 14. | ||
N-Dodecyl-N-methylcarbamoyl-CoA 1 had a very low IC50 value of 0.4 nM determined by this assay, confirming its very high potency. This compares with the previous value of 98 nM, determined with an endpoint assay.19 Ibuprofenoyl-CoA 2 had a modest potency with an IC50 value of 554 nM. For both inhibitors activity was restored upon rapid dilution of the inhibitor, showing reversible inhibition. Michaelis–Menten kinetic analysis of compounds 1 and 2 showed that they were both competitive inhibitors of AMACR, with Ki values of 0.65 and 60 nM, respectively (Fig. 2 and ESI,† Fig. S7 and S8). It is noteworthy that both compounds appear to be much more potent when assayed using this method compared with other methods. Ki values of 98 nM and 56 μM were previously reported in the literature for N-dodecyl-N-methylcarbamoyl-CoA 119 and racemic ibuprofenoyl-CoA 234 (with native rat enzyme), respectively. The reasons for the difference in apparent potency are not clear, but it probably results from differences in assay conditions including micelle formation by acyl-CoA substrates and inhibitors.
 | ||
| Fig. 2 Analysis of inhibitor 1. (A) Reversibility experiment, showing enzymatic activity is restored upon dilution of concentrated enzyme with 13.5 nM 1 to 0.27 nM 1; (B) kinetic analysis to determine Ki value for 1. | ||
Analysis of the non-specific protein modification agents 12, 13 and 14, identified by Wilson et al.16 as AMACR inhibitors, confirmed their potency. Ebselen 12 gave an IC50 value of 133 nM, compared to the previously reported value16 of 2.789 μM. Rapid dilution of Ebselen 12 resulted in part restoration of activity in our assay (ESI,† Fig. S7), consistent with irreversible inhibition. This is consistent with the complex behaviour observed by Wilson et al.16
Similarly, Ebselen Oxide 13 was a potent inhibitor with an IC50 value of 774 nM compared to the previously reported value16 of 795 nM. Rapid dilution of 13 did not result in full restoration of activity, suggesting (covalent) irreversible inhibition. Further investigation showed that non-saturating time-dependent inhibition of AMACR occurred, with a second-order rate constant of 116 ± 8 M−1 s−1 (Fig. 3 and ESI,† Fig. S10), consistent with 13 being a non-specific inhibitor operating by a one-step mechanism. Compound 14 was a reversible inhibitor (IC50 = 3259 nM), compared to 10000 nM.16 Prolonged incubation caused photolytic protein degradation (ESI,† Fig. S9).
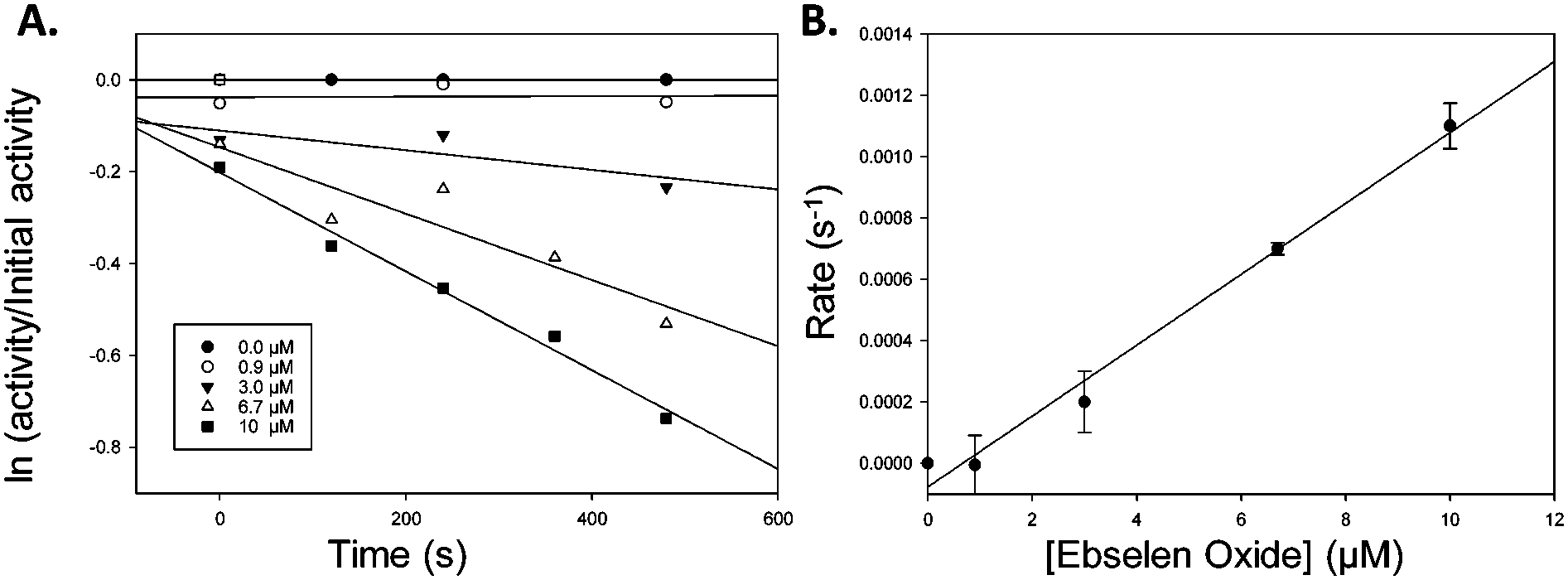 | ||
| Fig. 3 Inactivation of AMACR by Ebselen Oxide 13. (A) Time-dependent inactivation of recombinant AMACR by Ebselen Oxide 13. Concentrations of Ebselen Oxide 13 (inset) are given for the pre-incubation phase before dilution of the enzyme with substrate 6 in the assay; (B) dependence of inhibition on Ebselen Oxide 13 concentration. First order rate constants (s−1) derived from A was plotted vs. Ebselen Oxide 13 concentration in the pre-incubation phase. Data are mean ± standard error. | ||
AMACR has attracted much attention as both a novel drug target and cancer biomarker since the first reports of its involvement in prostate cancer in 2003.15 Exploitation of this discovery has been very limited due to the absence of a suitable assay. Our novel colorimetric assay provides a versatile platform for detailed characterization of inhibitors and determination of structure–activity relationships. The assay also allows for interrogation of the complex biology of AMACR and its role in lipid metabolism and cancer. Similar colorimetric assays could potentially be adapted for use with other enzymes catalysing reactions via enolate intermediates, including other racemases,35 acyl-CoA oxidases36,37 and other enzymes,38,39 several of which are of academic, medicinal, or biotechnological interest.
This work was funded by Prostate Cancer UK (S10-03 and PG14-009), a University of Bath Overseas Research Studentship, and a Biochemical Society Summer Vacation studentship (2015). The authors are members of the Cancer Research @ Bath (CR@B) network.
Notes and references
- M. D. Lloyd, M. Yevglevskis, G. L. Lee, P. J. Wood, M. D. Threadgill and T. J. Woodman, Prog. Lipid Res., 2013, 52, 220–230 CrossRef CAS PubMed.
- C. Reichel, R. Brugger, H. Bang, G. Geisslinger and K. Brune, Mol. Pharmacol., 1997, 51, 576–582 CAS.
- D. J. Darley, D. S. Butler, S. J. Prideaux, T. W. Thornton, A. D. Wilson, T. J. Woodman, M. D. Threadgill and M. D. Lloyd, Org. Biomol. Chem., 2009, 7, 543–552 CAS.
- P. Bhaumik, W. Schmitz, A. Hassinen, J. K. Hiltunen, E. Conzelmann and R. K. Wierenga, J. Mol. Biol., 2007, 367, 1145–1161 CrossRef CAS PubMed.
- T. J. Woodman, P. J. Wood, A. S. Thompson, T. J. Hutchings, G. R. Steel, P. Jiao, M. D. Threadgill and M. D. Lloyd, Chem. Commun., 2011, 47, 7332–7334 RSC.
- P. P. VanVeldhoven, K. Croes, S. Asselberghs, P. Herdewijn and G. P. Mannaerts, FEBS Lett., 1996, 388, 80–84 CrossRef CAS.
- J. A. Mitchell, P. Akarasereenont, C. Thiemermann, R. J. Flower and J. R. Vane, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 11693–11697 CrossRef CAS.
- J. Luo, S. Zha, W. R. Gage, T. A. Dunn, J. L. Hicks, C. J. Bennett, C. N. Ewing, E. A. Platz, S. Ferdinandusse, R. J. Wanders, J. M. Trent, W. B. Isaacs and A. M. De Marzo, Cancer Res., 2002, 62, 2220–2226 CAS.
- Z. Jiang, B. A. Woda, K. L. Rock, Y. D. Xu, L. Savas, A. Khan, G. Pihan, F. Cai, J. S. Babcook, P. Rathanaswami, S. G. Reed, J. C. Xu and G. R. Fanger, Am. J. Surg. Path., 2001, 25, 1397–1404 CrossRef CAS PubMed.
- Z. Jiang, G. R. Fanger, B. F. Banner, B. A. Woda, P. Algate, K. Dresser, J. C. Xu, S. G. Reed, K. L. Rock and P. G. Chu, Cancer Detect. Prev., 2003, 27, 422–426 CrossRef CAS PubMed.
- A. K. Witkiewicz, S. Varambally, R. L. Shen, R. Mehra, M. S. Sabel, D. Ghosh, A. M. Chinnaiyan, M. A. Rubin and C. G. Kleer, Cancer Epidemiol., Biomarkers Prev., 2005, 14, 1418–1423 CAS.
- C.-F. Li, F.-M. Fang, J. Lan, J.-W. Wang, H.-J. Kung, L.-T. Chen, T.-J. Chen, S.-H. Li, Y.-H. Wang, H.-C. Tai, S.-C. Yu and H.-Y. Huang, Clin. Cancer Res., 2014, 20, 6141–6152 CrossRef CAS PubMed.
- Z. Jiang, G. R. Fanger, B. A. Woda, B. F. Banner, P. Algate, K. Dresser, J. C. Xu and P. G. G. Chu, Hum. Pathol., 2003, 34, 792–796 CrossRef CAS PubMed.
- C. Kumar-Sinha, R. B. Shah, B. Laxman, S. A. Tomlins, J. Harwood, W. Schmitz, E. Conzelmann, M. G. Sanda, J. T. Wei, M. A. Rubin and A. M. Chinnaiyan, Am. J. Pathol., 2004, 164, 787–793 CrossRef CAS PubMed.
- S. Zha, S. Ferdinandusse, S. Denis, R. J. Wanders, C. M. Ewing, J. Luo, A. M. De Marzo and W. B. Isaacs, Cancer Res., 2003, 63, 7365–7376 CAS.
- B. A. P. Wilson, H. Wang, B. A. Nacev, R. C. Mease, J. O. Liu, M. G. Pomper and W. B. Isaacs, Mol. Cancer Ther., 2011, 10, 825–838 CrossRef CAS PubMed.
- K. Takahara, H. Azuma, T. Sakamoto, S. Kiyama, T. Inamoto, N. Ibuki, T. Nishida, H. Nomi, T. Ubai, N. Segawa and Y. Katsuoka, Anticancer Res., 2009, 29, 2497–2505 CAS.
- A. J. Carnell, I. Hale, S. Denis, R. J. A. Wanders, W. B. Isaacs, B. A. Wilson and S. Ferdinandusse, J. Med. Chem., 2007, 50, 2700–2707 CrossRef CAS PubMed.
- A. J. Carnell, R. Kirk, M. Smith, S. McKenna, L.-Y. Lian and R. Gibson, ChemMedChem, 2013, 8, 1643–1647 CAS.
- A. Morgenroth, E. A. Urusova, C. Dinger, E. Al-Momani, T. Kull, G. Glatting, H. Frauendorf, O. Jahn, F. M. Mottaghy, S. N. Reske and B. D. Zlatopolskiy, Chem. – Eur. J., 2011, 17, 10144–10150 CrossRef CAS PubMed.
- P.-Y. Lin, K.-L. Cheng, J. D. McGuffin-Cawley, F.-S. Shieu, A. C. Samia, S. Gupta, M. Cooney, C. L. Thompson and C. C. Liu, Biosensors, 2012, 2, 377–387 CrossRef CAS PubMed.
- M. Pal, M. Khanal, R. Marko, S. Thirumalairajan and S. L. Bearne, Chem. Commun., 2016, 52, 2740–2743 RSC.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS PubMed.
- C. Festuccia, G. L. Gravina, A. Mancini, P. Muzi, E. Di Cesare, R. Kirk, M. Smith, S. Hughes, R. Gibson, L.-Y. Lian, E. Ricevuto and A. J. Carnell, Anti-Cancer Agents Med. Chem., 2014, 14, 1031–1041 CrossRef PubMed.
- W. R. Shieh and C. S. Chen, J. Biol. Chem., 1993, 268, 3487–3493 CAS.
- D. Ouazia and S. L. Bearne, Anal. Biochem., 2010, 398, 45–51 CrossRef CAS PubMed.
- M. Yevglevskis, G. L. Lee, M. D. Threadgill, T. J. Woodman and M. D. Lloyd, Chem. Commun., 2014, 50, 14164–14166 RSC.
- M. Yevglevskis, G. L. Lee, J. Sun, S. Zhou, X. Sun, G. Kociok-Köhn, T. D. James, T. J. Woodman and M. D. Lloyd, Org. Biomol. Chem., 2016, 14, 612–622 CAS.
- D. C. Harris, Quantitative Chemical Analysis, W. H. Freeman, New York, 8th edn, 2010 Search PubMed.
- M. Y. Moridani, A. Siraki and P. J. O'Brien, Chem.-Biol. Interact., 2003, 145, 213–223 CrossRef CAS PubMed.
- S. Chafaa, J. Meullemeestre, M. J. Schwing, F. Vierling, V. Bohmer and W. Vogt, Helv. Chim. Acta, 1993, 76, 1425–1434 CrossRef CAS.
- D. Y. Yu, F. Huang and H. Xu, Anal. Methods, 2012, 4, 47–49 RSC.
- J. H. Zhang, T. D. Y. Chung and K. R. Oldenburg, J. Biomol. Screening, 1999, 4, 67–73 CrossRef PubMed.
- W. Schmitz, R. Fingerhut and E. Conzelmann, Eur. J. Biochem., 1994, 222, 313–323 CrossRef CAS PubMed.
- M. E. Tanner, Acc. Chem. Res., 2002, 35, 237–246 CrossRef CAS PubMed.
- A. R. Dewanti and B. Mitra, Biochemistry, 2003, 42, 12893–12901 CrossRef CAS PubMed.
- X. J. Liu, G. S. Deng, X. S. Chu, N. Li, L. Wu and D. Li, Bioorg. Med. Chem. Lett., 2007, 17, 3187–3190 CrossRef CAS PubMed.
- R. B. Hamed, E. T. Batchelar, I. J. Clifton and C. J. Schofield, Cell. Mol. Life Sci., 2008, 65, 2507–2527 CrossRef CAS PubMed.
- A. K. Casey, M. A. Hicks, J. L. Johnson, P. C. Babbitt and P. A. Frantom, Biochemistry, 2014, 53, 2915–2925 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details including synthesis of compounds, kinetic plots, inhibitor testing. See DOI: 10.1039/c7cc00476a |
| ‡ Abbreviations used: AMACR, α-methylacyl-CoA racemase splice variant 1A; 2-APA, 2-arylpropanoic acid (profen); BSA, bovine serum albumin; CoA, coenzyme A; MCR, 2-methylacyl-CoA racemase from M. tuberculosis. |
| This journal is © The Royal Society of Chemistry 2017 |