
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A computational study of CH4 storage in porous framework materials with metalated linkers: connecting the atomistic character of CH4 binding sites to usable capacity†
Ehud
Tsivion
 ac,
Jarad A.
Mason
c,
Miguel. I.
Gonzalez
c,
Jeffrey R.
Long
acd and
Martin
Head-Gordon
*bc
ac,
Jarad A.
Mason
c,
Miguel. I.
Gonzalez
c,
Jeffrey R.
Long
acd and
Martin
Head-Gordon
*bc
aMaterials Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA
bChemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA
cDepartment of Chemistry, University of California, Berkeley, California 94720, USA. E-mail: mhg@cchem.berkeley.edu
dDepartment of Chemical and Biomolecular Engineering, University of California, Berkeley, California 94720, USA
First published on 29th March 2016
Abstract
To store natural gas (NG) inexpensively at adequate densities for use as a fuel in the transportation sector, new porous materials are being developed. This work uses computational methods to explore strategies for improving the usable methane storage capacity of adsorbents, including metal–organic frameworks (MOFs), that feature open-metal sites incorporated into their structure by postsynthetic modification. The adsorption of CH4 on several open-metal sites is studied by calculating geometries and adsorption energies and analyzing the relevant interaction factors. Approximate site-specific adsorption isotherms are obtained, and the open-metal site contribution to the overall CH4 usable capacity is evaluated. It is found that sufficient ionic character is required, as exemplified by the strong CH4 affinities of 2,2′-bipyridine-CaCl2 and Mg, Ca-catecholate. In addition, it is found that the capacity of a single metal site depends not only on its affinity but also on its geometry, where trigonal or “bent” low-coordinate exposed sites can accommodate three or four methane molecules, as exemplified by Ca-decorated nitrilotriacetic acid. The effect of residual solvent molecules at the open-metal site is also explored, with some positive conclusions. Not only can residual solvent stabilize the open-metal site, surprisingly, solvent molecules do not necessarily reduce CH4 affinity, but can contribute to increased usable capacity by modifying adsorption interactions.
Introduction
Natural gas (NG) is a naturally occurring mixture of hydrocarbons, consisting mostly of methane (CH4). NG is predominantly used for domestic, commercial and industrial applications such as heating, cooking and electric power production. NG is also used as an alternative fuel source for the transportation sector,1,2 and has several advantages over gasoline. Methane has the highest ratio of hydrogen to carbon of all fossil fuels and thus has the highest gravimetric energy density. Because of this, NG burns cleaner than gasoline and emits lower levels of greenhouse gases through its life cycle,3 although capture of waste CO2 at the source is a critical separation challenge in light of climate change considerations. Additionally, recent advances in horizontal drilling and hydraulic fracturing technologies are expected to increase NG production and sustain its low price.4The main challenge in using NG as a fuel for transportation is that it has a much lower volumetric energy density at ambient conditions (0.04 MJ L−1) than gasoline (32.4 MJ L−1). Therefore, NG cannot be practically used for passenger vehicle applications in its standard state. Instead, NG is currently stored on-board the vehicle in either compressed (CNG) or liquefied (LNG) forms; however, these require expensive and bulky equipment for storage, compression and thermal isolation. A different approach to on-board storage involves having the NG stored in an adsorbed state inside a porous material with a high surface area. In the adsorbed state, the effective volume of the gas molecules is considerably smaller than in the gaseous state, which enables storage of larger quantities in lower volumes and at lower pressures. Over the last several decades, studies on adsorbed natural gas (ANG) have focused mostly on activated carbons;5 however, in recent years metal–organic frameworks (MOFs) have emerged as promising storage media.6
MOFs are a family of compounds consisting of metal ions or clusters coordinated to organic ligands (linkers), which form extended network structures.7 Due to their tunable pore sizes, MOFs have attracted attention for their potential use as gas-storage media. Furthermore, MOF composition and structure can be modified and tuned, as evidenced by their many other potential applications, including in catalysis8,9 and chemical separations.6,10,11
In 2012, the US Department of Energy (DOE) outlined the latest targets for ANG storage, which require a volumetric density of 263 v STP/v at maximum pressure of 35 bar, equivalent to the same density of CNG at 250 bar and 25 °C. For activated carbons, the most recent reports on volumetric CH4 capacities are in the range of 130–170 v STP/v, well below the DOE target.12 But even for MOFs, a recent study indicates there exists an upper bound for the usable capacity at 65 bar of approximately 200 v STP/v, also well below the DOE target.13,14 Known MOFs with the highest CH4 capacities, such as MOF-5,15,16 HKUST-1,17 UTSA-76a18 and MOF-519,19 are already at or close to that boundary. Evidently, to get closer to the DOE target, the attraction between CH4 and its host material should be increased beyond what is currently achievable by MOFs or activated carbons, for example by binding multiple CH4's to a given site.
A promising strategy for augmenting the CH4 capacity of MOFs is by introduction of open-metal sites into their structure.20–22 Most open-metal sites have a significantly stronger interaction energy with CH4 than other adsorption sites on the pore surface of a MOF, and therefore, MOFs containing high concentrations of open-metal sites are expected to have higher CH4 capacities. It is important to note that the usable (also known as “deliverable”) CH4 capacity is more important than the total CH4 capacity for ANG applications. The usable capacity is defined as the difference between the amount of CH4 adsorbed at the maximum adsorption pressure, typically 35–65 bar, and the amount that is still adsorbed at the minimum desorption pressure. For ANG storage, the minimum desorption required for gas to flow from the fuel tank to the combustion engine depends on the specific requirements of each vehicle, but 5.8 bar is often assumed for initial comparisons between materials. The optimal adsorption enthalpy (ΔHads) for maximizing usable CH4 capacity at ambient conditions has been shown to be about −17 kJ mol−1.23 Higher enthalpies result in over-adsorption of the CH4 at low pressures, while lower enthalpies result in under-adsorption at higher pressures. In addition to an optimal ΔHads, sufficiently high densities of open-metal sites are needed to achieve high usable capacities.
Insertion of open-metal sites into MOFs can be realized by the post-synthetic modification (PSM) approach,22,24,25 whereby the MOF is first synthesized and then afterwards the open-metal site functionality is introduced. The separation of the synthesis process into two (or more) steps can be necessary, since the open-metal sites are chemically active and can prevent the formation of the MOF. While many MOFs have open-metal sites as part of their frameworks, the PSM approach is more widely applicable to any MOF that has metal chelating sites present on the organic linkers. Furthermore, there is an additional flexibility in potential types and composition of open-metal sites, such that open-metal sites with rationally tailored properties are possible. An important challenge related to the practical use of open-metal sites is the likely presence of solvent molecules: at the end of MOF fabrication, the open-metal sites are usually passivated by the presence of coordinated solvent molecules or charge-balancing anions. In order for the MOF to realize its full CH4 adsorption potential, these solvent molecules must be fully removed during activation.26 The activation of strongly interacting open-metal sites is expected to be challenging (or even impossible) due to the strong metal–solvent interaction, which can be on the order of −100 kJ mol−1, several times stronger than the expected interaction with CH4.
Although this introduction is focused on MOFs, the open-metal sites insertion approach, as well as the issues related to the presence of solvent, are completely general and are relevant to any other porous framework material, such as covalent–organic frameworks27,28 (COFs) and porous-aromatic frameworks29,30 (PAFs).
In this study, the adsorption of CH4 on several types of open-metal sites, both known and hypothetical, is explored. Also, the presence of solvent molecules, and their effect on CH4 adsorption, is studied in detail. By analyzing the nature of the underlying interactions, we attempt to provide an intellectual framework for understanding metal site-CH4 chemistry and provide guidelines for the design of effective NG storage materials. The selection of the studied metal-ions is mostly driven by practical considerations: a suitable ion should have an adequate adsorption energy, be lightweight, cheap and environmentally benign. In these respects, the alkaline-earth metals Mg and Ca seem more adequate than the first-row transition metals, since due to their strong ionic character, they are expected to have strong interactions with CH4, and are also earth-abundant and non-toxic. Additionally, due to their closed shell character, they are not expected to be involved in any strong covalent interactions with, or to activate, the C–H bonds of CH4.
The manuscript is organized as follows: the Model and computational details section reviews and discusses: (1) the cluster model, (2) the thermodynamic model for adsorption isotherms, (3) usable capacity, (4) energy decomposition analysis, and (5) computational details of the DFT calculations. The Results section is divided into several parts, each examining the CH4 adsorption on a specific type of system: (1) bare metal ions, (2) catecholate linker metal-sites, (3) bipyridine linker metal-sites, (4) nitrilotriacetic acid metal-sites, and (5) solvent effects in catecholate metal-sites. The Discussion and conclusion section is divided into (1) expected usable capacities and (2) conclusion.
Model and computational details
Cluster model
In this study, the CH4 interactions with open-metal sites are studied using a cluster model, which consists of a single adsorption site, made from a single metal ion complexed to a linker molecule (i.e., a metalated linker) with one or more adsorbed CH4 molecules. MOFs are composed of well-defined ligand subunits that, to a reasonable approximation, maintain their individual precursor (pre-MOF) structures. Since the CH4 adsorption site has approximately the same geometry as in its pre-MOF molecular state, cluster models are able to provide valuable computational description of, and insights into, the interaction between CH4 and the adsorption site. Moreover, cluster models enable an accurate computational description of linker–metal–methane–solvent systems, utilizing higher level electronic structure theory methods than can be applied to periodic MOF models. For these reasons, cluster models have been successfully used in recent studies to model small molecule adsorption in MOFs.9,31–36 In spite of these successes, cluster models have yet to be extensively used and tested for prediction of adsorption isotherms and usable capacities, as described in below.A cluster model is a good approximation of the MOF environment only in the vicinity of an open-metal site and its linker, as at larger distances other effects introduced by the MOF environment become significant. We therefore truncate our model after the first coordination/solvation shell of the metal. Other adsorption sites besides the open-metal sites, are also present in the MOF and contribute to the overall CH4 capacity. Especially at higher occupancy (high pressures), a CH4 molecule adsorbed on an open-metal site can interact with CH4 on other adsorption sites, or gaseous CH4, resulting in slightly increased adsorption energy. However, this study is focused on understanding open-metal site-CH4 interactions, which are only weakly dependent on the extended MOF environment, and therefore a cluster-model approach is appropriate. An estimate of the usable CH4 capacity, useful for qualitative comparisons between different open-metal sites, can be made on the basis of these calculations, and is described below.
Thermodynamic model of adsorbed CH4
To connect between the cluster model calculations and experimental observables, such as usable capacity, a simple equilibrium model for estimating metalated linker site occupancy and the associated site adsorption isotherms will be used. The model assumes that each metalated linker (L) can adsorb up to a maximum of S methane molecules (the first solvation shell). A metalated linker with i adsorbed molecules is denoted by Li. Methane cannot adsorb on the linker to form Li unless Li−1 already exists. Thus a sequence of binding equilibria exists: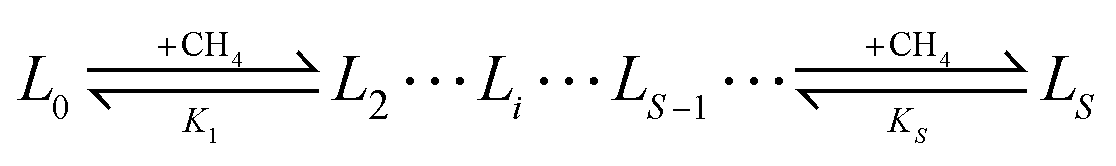 | (1) |
The associated equilibrium constants, Ki, are straightforwardly given by:
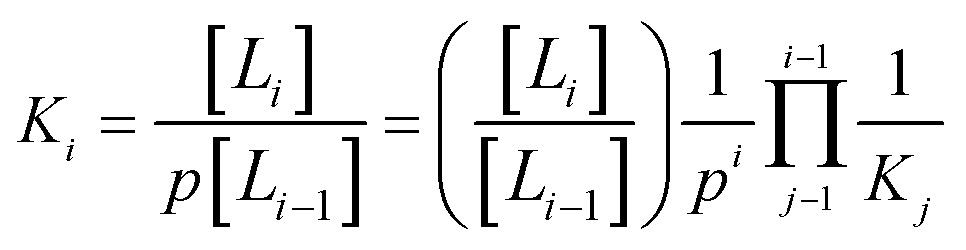 | (2) |
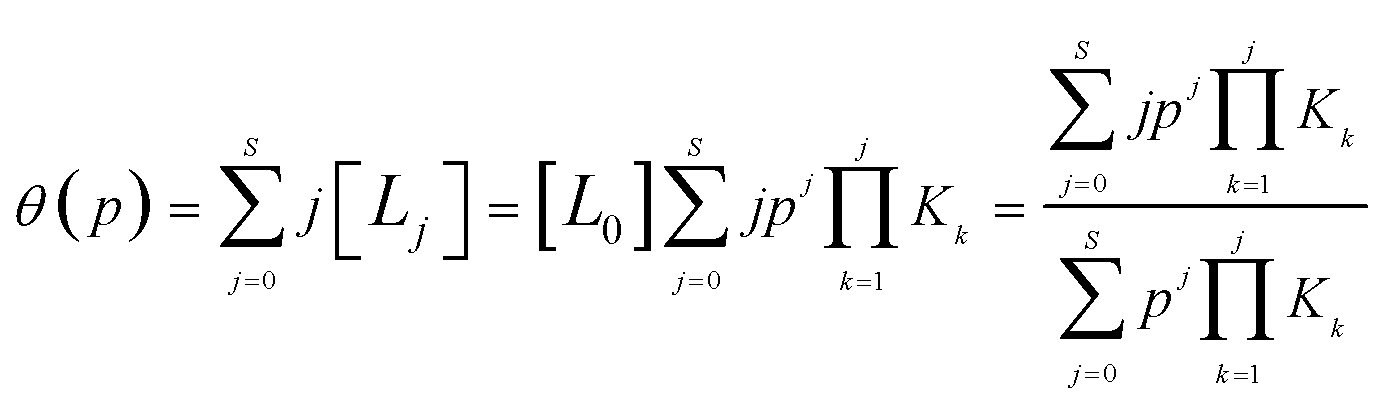 | (3) |
The equilibrium constants Ki are evaluated using the standard expression, Ki = exp(−ΔGi/RT), where ΔGi is the differential free energy of adsorption of the ith CH4 molecule. The evaluation of ΔGi requires both the adsorption entropy, ΔSi, and the adsorption enthalpy, ΔHi. The nuclear motion contributions are usually evaluated in quantum chemical (QC) calculations by assuming that the adsorbent–adsorbate complex is a collection of harmonic oscillators with only vibrational degrees of freedom. When the adsorbing molecule interacts weakly with the adsorbent, the adsorbed molecule retains approximately 2/3 of its translational and rotational freedom,37 however, the harmonic approximation, commonly used in QM calculation of calculation of the thermodynamic partition function, cannot take into account that certain lower-frequency motions, common for inter-molecular physical interaction, should be treated as hindered internal rotations or translations, rather than as vibrations.38 Since the translational degrees of freedom have much higher entropy than the vibrational, and vibrations have much higher internal energy than translations, the treatment of motions as vibrations, rather than hindered translations, results in an underestimation of the entropy and overestimation of the (positive) internal energy of the complex.
Since rigorous evaluation of ΔGi is unfeasible, we calculate ΔUivia the difference of electronic energies from QC calculations and adopt simple, though reasonable, approximations for the remaining contributions. As discussed in the ESI,† ΔSi is assumed to be independent of i, and set to the value of −9.5R which represent an intermediate value measured for materials for adsorptive storage applications.23,39 The value of ΔHi is assumed to be given by ΔUi + ΔUvib − RT, based on the pV contribution, and the change in vibrational energy. Upon adsorption of a CH4 molecule, a weak “physical” bond (50 to 150 cm−1) is formed by restriction of the CH4 movement through conversion of one translational degree of freedom into one “bonding” vibration, whose contribution ΔUvib is, to a good approximation, 2.5 kJ mol−1 at ambient conditions.
We assume that contributions to the MOF usable capacity from second layer coverage is of minor importance at the pressure range considered here of less than 65 bar, as we truncate adsorption upon closing of the first solvent shell (a characteristic number, S molecules for a given binding site). Furthermore, this model does not attempt to simulate adsorption isotherms for any specific MOF, as this requires larger models that are individually tailored for specific MOFs. Rather, the model enables qualitative comparison of the usable capacity associated with different binding site designs, in a given framework. As described below, we will choose existing pre-metalated frameworks for this purpose.
Usable capacity
An internal combustion engine in a passenger vehicle operates across a range of CH4 pressures. When the ANG fuel tank is full, the pressure is maximal (pmax).As the NG is desorbed and flows into the engine, the pressure gradually drops down to a minimal pressure, which is needed by the engine to operate (pmin). In this work, pmax is chosen as either 35 bar, which is the maximal pressure that is obtained using a single-step compressor, or pmax = 65 bar, which is also used in the literature. The value of pmin is taken to be 5.8 bar.
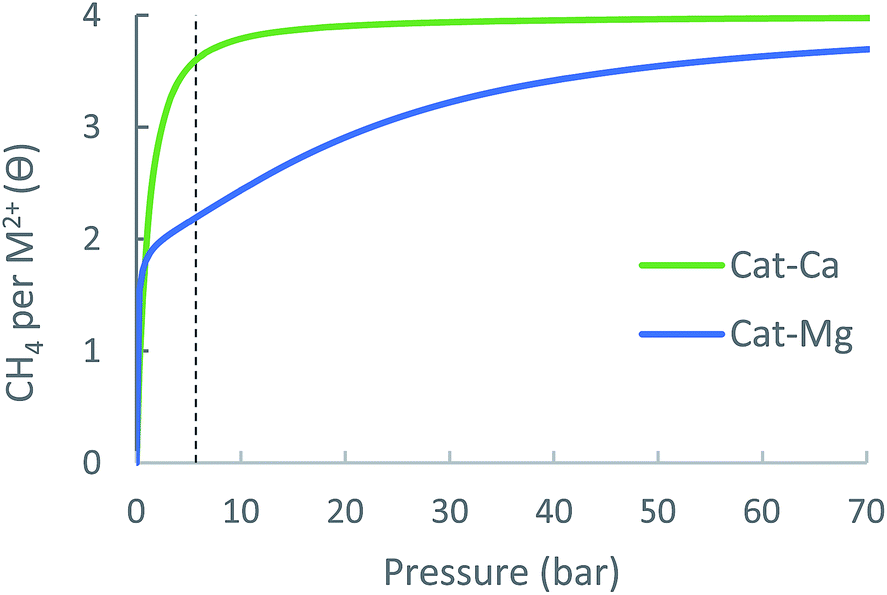
An ideal storage material (or a metal-site) should be able to swing between high surface coverage at pmax and low surface coverage at pmin. To illustrate this consideration, Fig. 1 shows adsorption curves of three different ANG materials A, B and C, each having a different NG adsorption enthalpy, ΔHads(A) > ΔHads(B) > ΔHads(C). Material A is a strong NG adsorbent: at pmax its surface is completely covered, i.e., a large amount of CH4 is adsorbed. However, at pmin the coverage of A remains high such that only a small amount of CH4 is released to the engine and the usable capacity is sub-optimal. Material C is a weak CH4 adsorbent: at pmax peak coverage is not reached, and C contains a relatively small amount of CH4 and is therefore not able to store NG optimally. Material B has an optimal ΔHads for CH4 storage at ambient conditions: although it does not reach full coverage at pmax, its ability to sufficiently release CH4 at pmin gives B a better usable capacity than A and C. Notably, for cases such as A, where usable capacity is limited by over-attraction for CH4, heating could be applied to the storage tank at lower pressures to facilitate the release of strongly adsorbed CH4 molecules, thereby increasing usable capacity.
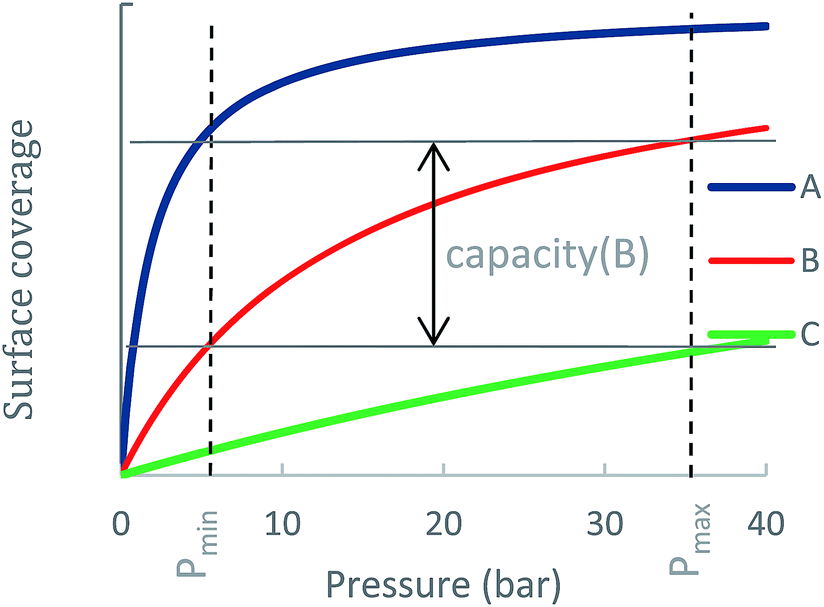 | ||
| Fig. 1 Usable capacity of three different ANG materials A, B and C, where ΔHads(A) > ΔHads (B) > ΔHads (C). The usable capacity is proportional to the difference between surface coverage at maximal and minimal pressures. The usable capacity of B is larger than A and C since A is over-adsorbing and C is under-adsorbing. | ||
To distinguish the usable capacity of the MOF from the contribution of the individual metal-site that are calculated, we define the metal-site's usable occupancy as the difference between the number of adsorbed molecules on a metal-site at pmax and at pmin:
| Δθuo = θpmax − θpmin |
To increase the readability of the paper, the expected usable site occupancy of the open-metal sites is compared to that of the clusters in the parent MOFs (see Tables 3, 5, 6 and 9). This is done by rewriting the Langmuir equation, to obtain θcluster, cluster occupancy values that are comparable to θoms, the occupancy of the open-metal sites. Further details are found in the ESI.† The ESI† also contains a section which evaluates the methodology used here for estimation of usable capacities, to experimental gas-measurement results for the well-characterised MOF-5 system. Quantitative agreement is obtained between the model and experiment.
We emphasize that the interactions reported are the computationally calculated energies, and not the predicted experimentally measurable enthalpies. Conversely, the predicted adsorption isotherms and derived site occupancies, do incorporate certain data and assumptions regarding the thermodynamic nature of the systems, and therefore can in principle be compared to experiment, subject to limitations in modeling discussed already.
Energy decomposition analysis
The physisorption of CH4 is essentially non-chemical because it does not involve the formation or breakage of chemical bonds. Therefore, standard wave-function analysis concepts, such as partial charges and bond orders, cannot provide an adequate picture of the key contributions to physisorption. Instead, we employ “Energy Decomposition Analysis” (EDA),40,41 as implemented in the Q-Chem quantum chemistry package,42 which decomposes the interaction energy of two or more molecules, into three contributions: (1) frozen (FRZ), (2) polarization (POL) and (3) charge transfer (CT).| EInteraction = EFRZ + EPOL + ECT | (4) |
In more detail, the EDA uses the “SCF-MI”43,44 approach for obtaining an “Absolutely Localized Molecular Orbital” (ALMO) wave-function ΨALMO. The ALMOs prohibit CT by fragment-blocking the MO coefficient matrix. The FRZ term is the energy required to bring infinitely separated molecules into the complex geometry, using the frozen MOs of the fragments: 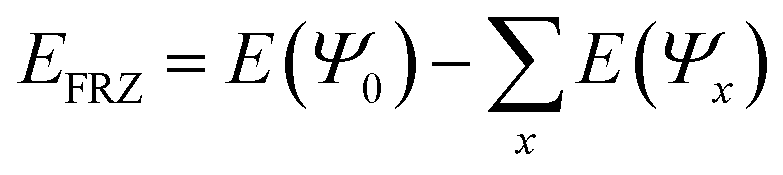 . The POL term is evaluated as the difference between the energy of the optimized ALMO wave-function and the frozen wave-function: EPOL = E(ΨALMO) − E(Ψ0). The CT term is evaluated as the energy difference between the energy of the fully converged SCF wave-function of the complex and the CT-excluded ALMO energy: ECT = E(ΨSCF) − E(ΨALMO). The positive energy related to the geometric distortion of the molecule in its complex geometry with respect to its isolated geometry is called the “geometric distortion” (GD) energy. For the sake of readability, only the parts of the EDA which are essential for the discussion are shown while the full EDA is found in the ESI.†
. The POL term is evaluated as the difference between the energy of the optimized ALMO wave-function and the frozen wave-function: EPOL = E(ΨALMO) − E(Ψ0). The CT term is evaluated as the energy difference between the energy of the fully converged SCF wave-function of the complex and the CT-excluded ALMO energy: ECT = E(ΨSCF) − E(ΨALMO). The positive energy related to the geometric distortion of the molecule in its complex geometry with respect to its isolated geometry is called the “geometric distortion” (GD) energy. For the sake of readability, only the parts of the EDA which are essential for the discussion are shown while the full EDA is found in the ESI.†
This work also employs complementary occupied-virtual orbital pairs (COVPs)45 for visualization of the intermolecular CT interactions. The COVPs are a chemical representation of inter-molecular CT in simple terms of donor–acceptor orbital pairs that provide a compact representation of the most significant donor–acceptor orbital interactions.
Computational details
All calculations are carried out using the meta-GGA B97M-V density functional,46 which utilizes the VV10 non-local correlation functional for its treatment of dispersion interactions.47 The VV10 term relies only on electronic densities to compute the dispersion interactions, without any predetermined atom-specific parameters. Therefore, this dispersion treatment is more transferrable compared to other dispersion treatment approaches, and can be applied successfully to a wider range of systems, not specifically included in benchmark datasets. A recent benchmark by Herbert and coworkers on intermolecular interactions involving ions demonstrated that B97M-V performs very well for anion-neutral dimers, cation-neutral dimers and ion pairs.48The structures are initially optimized using the 6-31g* basis set and validated to be a minimum on the energy potential surface using a standard frequency analysis, validating that the correct conformation of the structures is obtained. These geometries are then further optimized using the triple-zeta def2-tzvp basis set49 which are then used, along with the larger quadruple-zeta def2-qzvp basis set,50 for evaluation of interaction energies and EDA analysis. The reported adsorption energies and charge transfer interactions are counterpoise corrected for basis set superposition error. The insensitivity of the geometry optimizations to additional diffuse functions was tested on the CH4 dimer, cat-Mg and cat-Ca, using the diffuse def2-tzvpd basis, which resulted in similar structures and interaction.
Due to the shallow nature of the potential energy surface of the weakly interacting molecules, there is an inherent challenge in the exact identification of the global minimum structures. This is particularly true for partially solvated structures. Thus, two different geometry optimizations from different initial guesses, may converge into geometrically similar structures but having slightly different energies, of about ±1 kJ mol−1, which can be considered as an additional uncertainty of these calculations.
Results
Methane clusters formed by metallic ions
We start the study of CH4 adsorption by looking at the simplest models of CH4 molecules interacting with a bare ion, forming a solvation shell. Due to the strict electrostatic nature of the alkali or alkaline-earth metal ions, these clusters can provide useful insights into the interaction of the CH4 with the strong electrostatic fields induced by the ions. It is emphasized, that in spite of the simplicity of the models, this is not a purely theoretical case-study, as doping of porous materials with lithium ions is a well-recognized strategy for enhancing the adsorption of CH4, as well as other species.51–53The panels in Fig. 2 show the first solvation shell formed by CH4 molecules around Be2+, Mg2+, Ca2+ and Li+ ions, and the corresponding EDA is shown in Table 1. Since the CH4⋯CH4 interactions are relatively weak, on the order of −2 kJ mol−1, the dominant interactions driving the formation of the clusters are the cation–CH4 interactions.
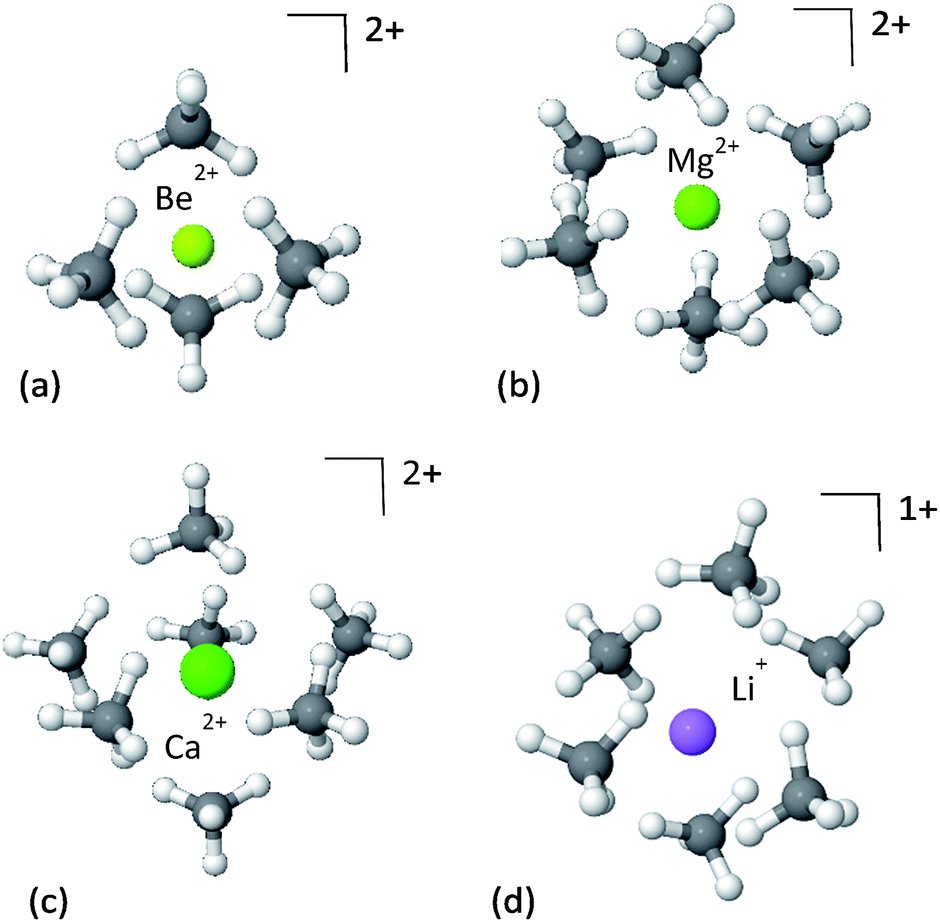 | ||
| Fig. 2 First solvation shell of methane clusters formed around bare ions: (a) Be2+–4CH4, (b) Mg2+–6CH4, (c) Ca2+–7CH4 and (d) Li+–6CH4. Although the Li+ complex is considerably less charged than the Mg2+ complex, they are able to coordinate the same number of CH4 molecules due to their similar radii. However, their adsorption energies are different. | ||
| Be2+(CH4)4 | Mg2+(CH4)6 | Ca2+(CH4)7 | Li+(CH4)6 | |
|---|---|---|---|---|
| FRZ | 11.0 | 0.0 | 4.2 | −0.2 |
| POL | −234.2 | −103.6 | −64.8 | −24.7 |
| CT | −36.2 | −11.2 | −15.1 | −5.1 |
| Total | −259.3 | −114.8 | −75.7 | −30.0 |
The strongest attractive force in these clusters is polarization of the CH4, induced by the electrostatic fields of the cations. This is unsurprising given the chemical stability of methane, which means CT will be relatively small. The strength of the polarization is correlated with the ionic radius, such that the smaller the radius, the stronger the polarization. The ionic radii of Be2+, Mg2+ and Ca2+ are 59, 86 and 114 pm, respectively,54 as compared to the corresponding polarization energies of −234.2, −103.6 and −64.8 kJ mol−1. Smaller radii enable closer proximity of CH4 to the metal ion, where the electrostatic field of the ion is much stronger, and thus the induced polarization of the methane is also stronger.
Perhaps surprisingly, in spite of its smaller charge, Li+ coordinates the same number of CH4 molecules as Mg2+. This implies that the maximal coordination number of the ion is better correlated to ionic radius than charge, and reflects methane packing effects. This is supported by the close similarity between the ionic radius of lithium (90 pm) and that of magnesium (86 pm). However, because of its smaller charge the electrostatic fields near Li+ are weaker than near Mg2+, and the resulting polarization in the CH4 molecules is considerably less.
Although less important than polarization, charge transfer has a significant contribution to the interaction energy of the clusters. Its absolute measure is close for the Mg2+ and Ca2+ clusters (−11.3 and −15.1 kJ mol−1), however, it is relatively more important for Ca2+ where it accounts for 20% of the attractive energy. COVP analysis (Fig. 3) indicates that charge is transferred mostly from the C–H bonds of the CH4 into high-valence orbitals on the metal. These CT interactions are closely related to the covalent (CT) interactions between C–H groups and transition metals, also known as “agostic interactions” in the context of the chemistry of C–H bond.55 However, for the cases discussed here, the vacant d-orbitals of the main group ions are too diffuse and high in energy for strong C–H(CH4) to d(M) orbital interactions. Therefore, the ion mostly polarizes the CH4 without activating the C–H bonds—a desirable property for storage applications. As could be expected,56 CT is considerably stronger for Be2+, which could be related to the fact that the valence orbitals of Be2+ are the 2s and 2p shells,57 and not d orbitals (see Fig. 3c). The small radius and strong polarization of Be2+ also contribute to physical proximity, which increases CT. It is also interesting to note, that CT per CH4 is similar for Mg2+ and Ca2+, which implies that CT is not necessarily proportional to the polarization induced by the ion.
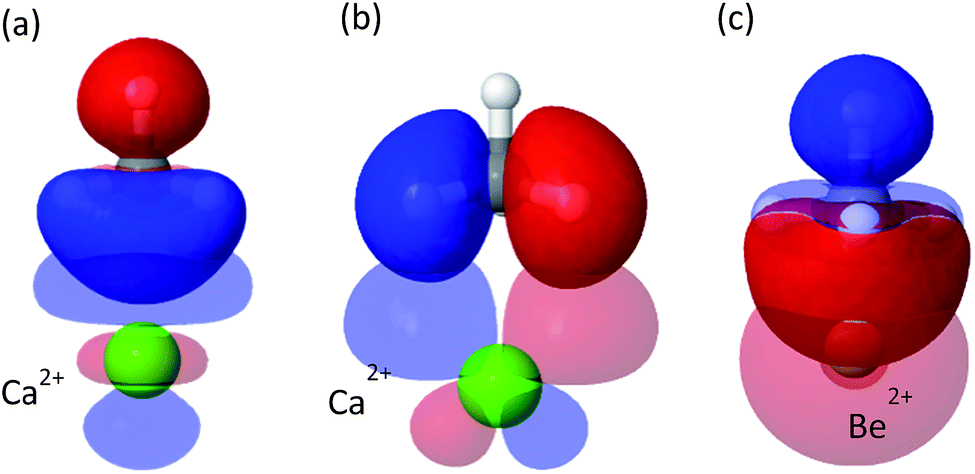 | ||
| Fig. 3 Significant donor–acceptor pairs for the charge-transfer interaction from CH4 to Ca2+ and Be2+. Panel (a) shows a σ-symmetry donation from C–H(CH4) to Ca2+ with associated energy lowering of ΔE = −4.3 kJ mol−1 and panel (b) shows π-symmetry donation from C–H(CH4) to Ca2+ with energy lowering of ΔE = −2.6 kJ mol−1. Panel (c) shows CT from CH4 to Be2+ where the C–H bonds donate into the 2s orbital. | ||
Under realistic conditions, the metal ion is never completely free and would be either solvated with its electrostatic field shielded by the solvent or be in close proximity, or coordinated, to some balancing counter-ion or ligand. In other words, under realistic conditions the metal ion would have a reduced electrostatic field and carry a reduced charge – both of which are expected to weaken the interaction with CH4. Additionally, there are steric constraints associated with direct coordination.
As an illustration of the consequences of these effects, the maximal coordination number of the Ca2+ ion is reduced to 6CH4 molecules, instead of 7, as will be shown below.
The catecholate (cat) linker
The MOF linkers discussed first are based on the catecholate (cat) ligand, which is a commonly used chelating agent throughout coordination chemistry. A key feature of the catecholate-(Mg, Ca) linker, as shown in Fig. 4, is that the arrangement of the electrostatic charges around the metal ion results in a strong dipole moment. Moreover, the catecholate is a bidentate ligand, such that the metal ion coordination sphere still has 4 vacant coordination sites. Metalated catecholate linkers have been previously studied in the context of H2 storage58–60 and shown to be capable of strong physical interactions with H2.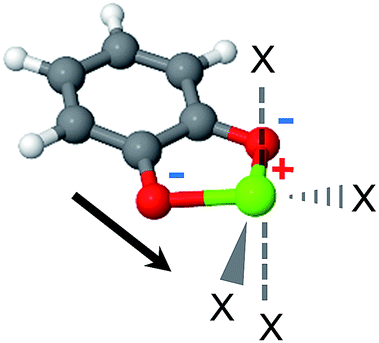 | ||
| Fig. 4 The catechol-Mg metalated linker. The electrostatic dipole moment (arrow) is formed by the two negatively charged O atoms and the positive metal. ‘X’ marks potential CH4 adsorption sites. The positive and negative formal charges are marked by red and blue colors, respectively. | ||
MOFs containing catechol ligands were recently synthesized and metalated by Fe3+, Cr3+ and Pd2+.61–63 The specific metalations discussed here have yet to be attained, however, there is experimental evidence64 for the preparation of catecholate complexes with Mg2+, and other studies, focused on the role of melamine in biological Ca2+ regulation, indicate that both Ca2+ and Mg2+ form complexes with catechol even in aqueous solutions.65–69
The optimized structures of the clusters of CH4 with catechol-(Mg, Ca) complexes are shown in Fig. 5, and the corresponding EDA is provided in Table 2. It can be seen that cat-Mg and cat-Ca share similar structural characteristics for their interaction with methane molecules. In both cases, CH4 gradually fills the coordination sphere of the ion, forming a tetrahedral structure for 2CH4 and an octahedral structure for 4CH4. However, the interaction energy change (ΔEads) for the addition of each CH4 molecule shows different trends: it is decreasing for cat-Mg while only slightly decreasing for cat-Ca.
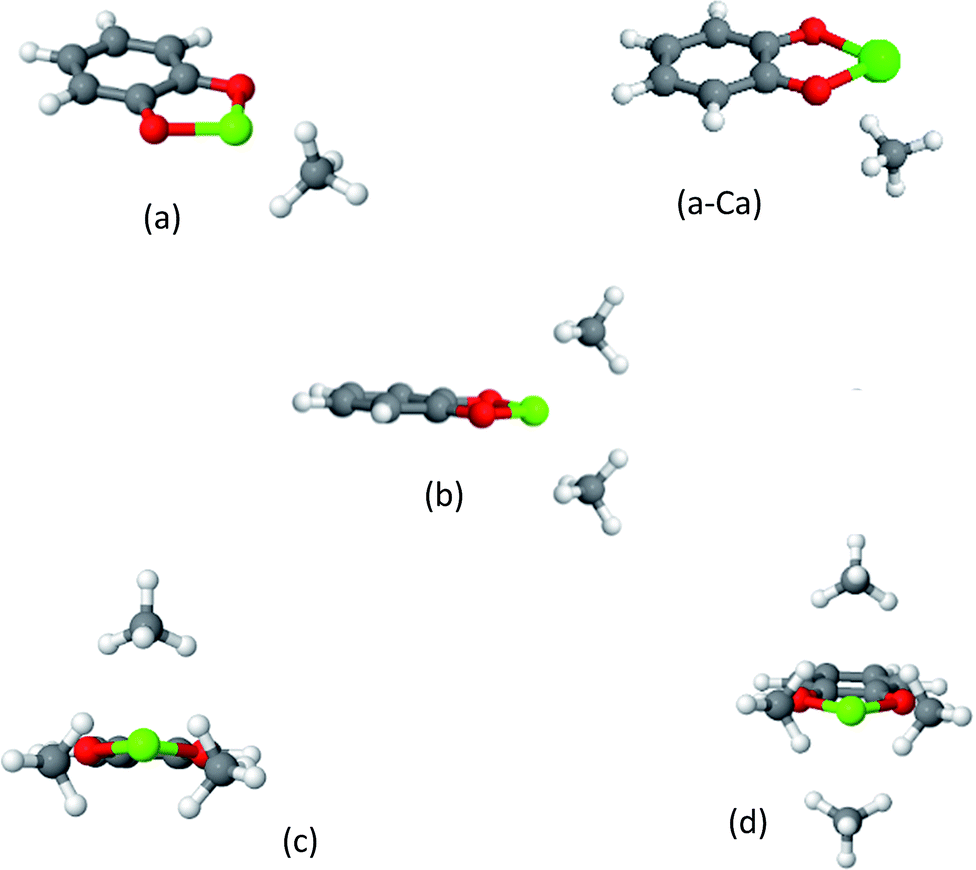 | ||
| Fig. 5 Clusters of CH4 on cat-Mg. Cat-Ca forms similar structures, except for the case of the 1CH4 molecule, which is shown in panel (a-Ca). The strong electrostatic response of the CH4 in (a) can be deduced from its alignment along the dipole moment of cat-Mg. As shown in panel (b), two CH4 molecules complete tetrahedral coordination around Mg or Ca, while three and four CH4 molecules partially complete or complete an octahedral coordination environment, as shown in panels (c) and (d). | ||
| N th CH4 | Cat-Mg | Cat-Ca | ||
|---|---|---|---|---|
| ΔPOL | ΔEads | ΔPOL | ΔEads | |
| 1 | −49.9 | −40.7 | −19.9 | −26.7 |
| 2 | −33.6 | −27.0 | −17.6 | −24.7 |
| 3 | −17.9 | −14.6 | −16.0 | −21.7 |
| 4 | −1.0 | −17.1 | −16.4 | −22.2 |
The EDA can provide useful insights into this behaviour. For both cat-Mg and cat-Ca the dominant attractive interaction is the polarization, i.e., the electrostatic response of the CH4 to the electrostatic fields of the ion. The polarization interaction for cat-Mg is expected to be larger than for cat-Ca, due to the smaller size of the Mg2+ ion—as was demonstrated above for the Mg2+ and Ca2+ clusters. Indeed, the polarization energy for the first coordinated CH4 molecule is much stronger for cat-Mg than for cat-Ca (−49.9 vs. −19.9 kJ mol−1), which also results in them having different geometries. Since Mg is small with respect to methane, it can be efficiently shielded by CH4, such that the electrostatic field of the Mg2+ ion is decreased significantly with the addition of each CH4.
Indeed, the fourth coordinated CH4 is polarized only by −1.0 kJ mol−1vs. −49.9 kJ mol−1 for the first. The radius of Ca2+ is larger than Mg2+; and therefore, CH4 does not as efficiently shield its electrostatic field.
The importance of the electrostatic interaction between the CH4 and the static charges of the cat-Ca is demonstrated by examining the structure of the cat-Ca cluster with a single CH4 molecule in Fig. 5(a-Ca): the two positive H atoms in CH4 are aligned under the two negative O atoms in cat-Ca, while the negative C atom in CH4 is aligned under the positive Ca2+ ion.
CT interactions (see Fig. 6) are approximately the same for both cat-Mg and cat-Ca, on the order of approximately −4 to −5 kJ mol−1. Charge is transferred from the C–H bonds of CH4 into high valence orbitals on the metal, and charge is transferred back from π orbitals of the oxygens to vacant anti-bonding C–H* orbitals in CH4. Both backward and forward contributions are of approximately similar importance. Compared to the clusters studied above, the CT interactions are considerably weaker. Due to the presence of the coordinating catecholate linker: the acceptor orbitals of the metal ion are now already partly occupied prior to methane coordination.
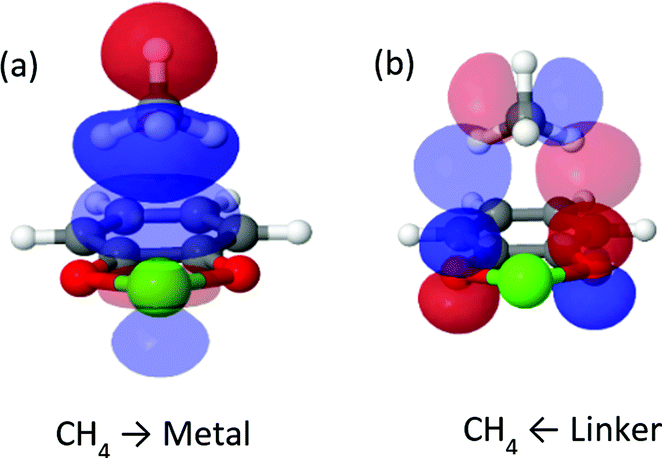 | ||
| Fig. 6 Most important complementary occupied-virtual pair orbitals in cat-Ca. Panel (a) shows forward donation from methane to metal, panel (b) shows backward donation. | ||
 | ||
| Fig. 7 Expected metal-site adsorption isotherms for the cat-(Mg, Ca) at 298.15 K. | ||
| Cat-Mg | Cat-Ca | MOF-5 | |
|---|---|---|---|
| Δθuo (35 bar) | 1.13 | 0.34 | 1.6 |
| Δθuo (65 bar) | 1.47 | 0.37 | 2.6 |
For cat-Mg, however, the third and fourth CH4 molecules are adsorbed with weaker energies of −14.6 and −17.1 kJ mol−1, which are suitable for methane storage and thus contribute significantly to the usable site occupancy. Thus, at 35 bar the usable site occupancy of cat-Mg is 1.13, more than three times that of cat-Ca (0.34). We note that if a larger value for ΔSads is used, the usable site occupancy of cat-Ca increases, due to increased attenuation of its relatively strong interactions.
The ability of the metalated cat linkers to adsorb a large number of CH4 molecules does not come without challenges: due to their strong partial charge and strong dipole moment, the exposed metal ions could be attracted to negatively charged areas in the MOF framework, thus destabilizing it towards collapse of the porous structure. Also, solvent molecules present during synthesis could be difficult to remove during activation and block the CH4 adsorption sites. Later, we will return to the question of how tightly bound residual solvent molecules at the open-metal site may impact usable occupancy.
The 2,2′-bipyridine (bpy) linker
2,2′-Bipyridine (bpy) is one of the most widely used ligands in coordination chemistry due to its strong affinity for metals. It is also commonly used as a MOF linking element, and there are several reports in the literature of MOFs containing metalated bpy units.21,70–72Fig. 8 shows a metalated bpy linker with two accompanying chloride anions for overall electrical neutrality. Assuming a square planar exposed metal site of this type, one expects two potential CH4 adsorption sites at the axial positions. Since the metal ion is coordinated by four negatively charged centers, which potentially reduce the strong-polarizing electrostatic fields, the adsorption mechanism of CH4 to the metal site is expected to be mostly frozen-electrostatic with the attraction of CH4 to the negatively charged counterions and positive metal. The most common metalation of bpy to date, as reported in three papers,21,71,72 is by PdCl2 and PtCl2, which results in a square planar complex of the type shown in Fig. 8. However, these complexes show only a weak interaction with CH4, exhibiting a binding energy of about 10 kJ mol−1, which is insufficient for CH4 storage. Here, we study the bpy-CuCl2, bpy-ZnCl2 and bpy-CaCl2 complexes and analyze their CH4 adsorption properties.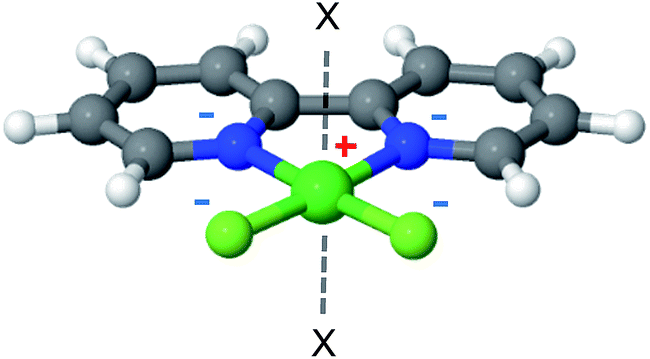 | ||
| Fig. 8 2,2′-Bipyridine metalated linker. ‘X’ marks potential CH4 adsorption sites. | ||
While MOF metalation by bpy-CuCl2 was recently demonstrated,73 there is also experimental evidence from both solution74,75 and the gas-phase76 for complexation of Ca2+ and Zn2+ with bpy. We note, that for practical purposes using larger pyridyl ligands, such as 1,10-phenanthroline would result in stronger metal-linker binding,75,76 and is another potential future synthetic target.
The optimized structures of the complexes are shown in Fig. 9; bpy-ZnCl2 is tetrahedral, bpy-CuCl2 and bpy-CaCl2 are predicted to be moderately distorted square planar complexes. Due to the larger size of the Ca2+ cation in bpy-CaCl2, it is less efficiently coupled (coordinated) to the bpy ligand, and therefore maintains some of its pre-coordinated ionic nature. The resulting geometry of the bpy-CaCl2 complex is intermediate between a fully coordinated tetrahedral structure, dominated by covalent interactions, and a planar structure dominated by the ionic interactions between the F− counterions and the positive H of the bpy. When two CH4 molecules are adsorbed on the “naked” complex (not shown in the figure), the electrostatic interactions between the positive Ca2+ ion and the negative nitrogen atoms are screened, shifting the equilibrium towards the planar structure and reducing the “twisting” of the Cl− counterions and shifting towards a planar position.
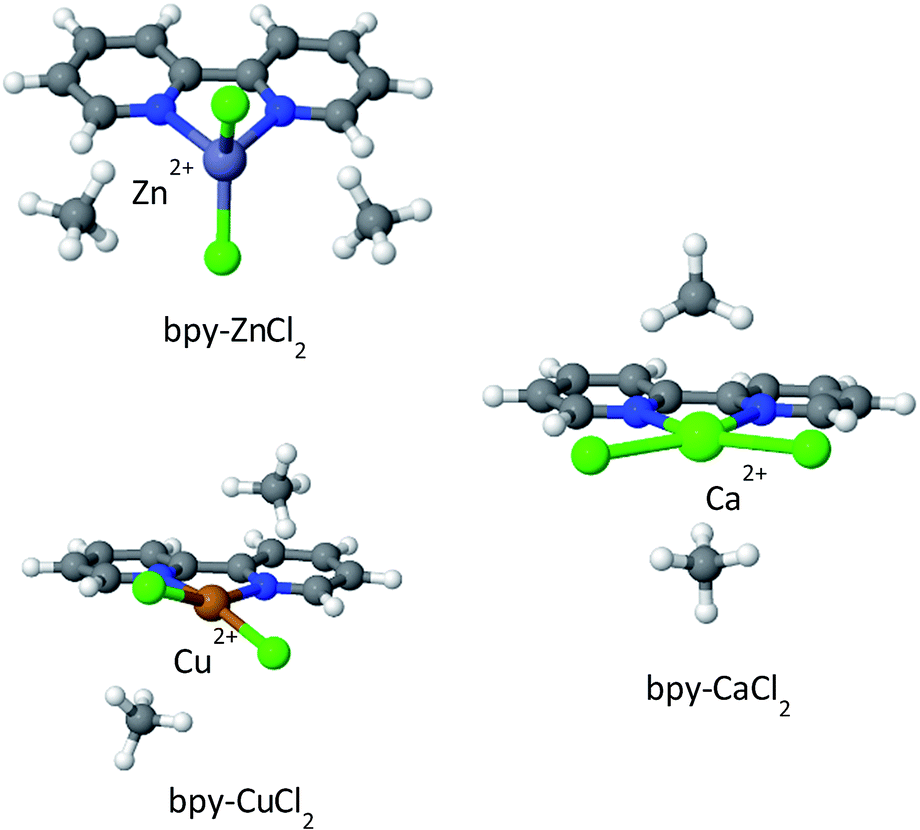 | ||
| Fig. 9 CH4 adsorbed on metalated bpy linkers. In the absence of adsorbed CH4 molecules, the structure of bpy-CaCl2 is close to that shown for bpy-CuCl2. | ||
Similarly, bpy-CuCl2 which is also non-planar, is expected to have some small ionic contribution to its character.
Overall binding energies for one and two CH4 molecules, as well as the EDA, are shown in Table 4. The EDA indicates that bpy-CuCl2 and bpy-ZnCl2 have similar characteristics: both ΔCT and ΔPOL are on the order of −2.5 to −4 kJ mol−1, however, the frozen-electrostatic interactions are stronger for bpy-CuCl2 than for bpy-ZnCl2 by −3.2 kJ mol−1 for the first adsorbed CH4 molecule. The weaker frozen-electrostatic interaction for bpy-ZnCl2 is explained by the tetrahedral geometry of the Zn ion and thus being shielded, both electrostatically and sterically, by negative charges—the positive ion is therefore not available for attracting the negative carbon atom of CH4. Moreover, the ionic character of bpy-CuCl2, increases the frozen-electrostatic attraction forces for CH4. Increased ionic character is even more pronounced in bpy-CaCl2 and therefore the adsorption of CH4 to bpy-CaCl2 is much stronger than to bpy-CuCl2 by about −6.9 kJ mol−1 and is characterized by elevated charge-transfer and polarization interactions, due to the partially exposed Ca2+ ion. The frozen interactions for bpy-CaCl2 are relatively small, due to increased (positive) steric repulsion.
| Nth CH4 | bpy-CuCl2 | bpy-ZnCl2 | bpy-CaCl2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ΔFRZ | ΔPOL | ΔCT | ΔEads | ΔFRZ | ΔPOL | ΔCT | ΔEads | ΔFRZ | ΔPOL | ΔCT | ΔEads | |
| 1 | −7.1 | −2.8 | −3.2 | −12.9 | −3.9 | −2.6 | −3.8 | −10.1 | −1.2 | −11.8 | −9.5 | −19.8 |
| 2 | −4.3 | −4.1 | −3.7 | −10.2 | −3.9 | −2.5 | −3.9 | −10.1 | −3.9 | −11.4 | −9.5 | −21.3 |
We note that the interaction with CH4 could be increased, by about −1 kJ mol−1, by using F− counter-ions that are more electronegative than Cl−.
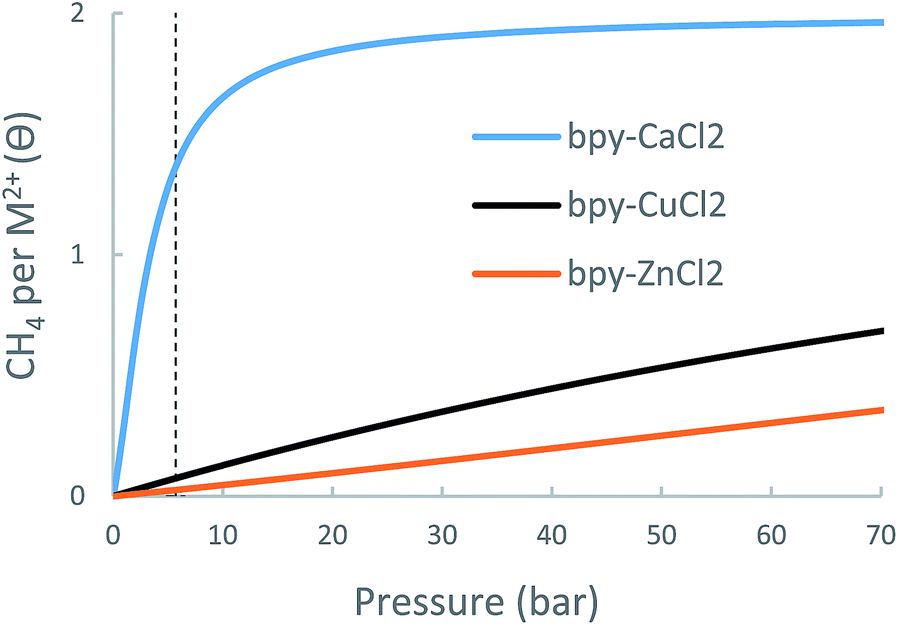 | ||
| Fig. 10 Calculated metal-site adsorption isotherms for the bpy-(CuCl2, ZnCl2, CaCl2) at 298.15 K. | ||
| bpy-CuCl2 | bpy-ZnCl2 | bpy-CaCl2 | UiO-67-bpy | |
|---|---|---|---|---|
| Δθuo (35 bar) | 0.32 | 0.15 | 0.54 | 2.4 |
| Δθuo (65 bar) | 0.57 | 0.30 | 0.58 | 3.4 |
While CH4 storage bpy-CaCl2 is non-optimal due to over-attraction and bpy-CuCl2 and bpy-ZnCl2 suffer from under-attraction, it is very possible that other forms of bpy-metalation would result in intermediate-strength interactions and better CH4 storage performance.
The NTA ligand
To date, post-synthetic metalation of both the 2,2′-bipyridine and catechol linkers has been demonstrated experimentally. To open future synthetic opportunities, it is compelling to extend the reach of the open-metal site-CH4 adsorption strategy by searching for other possible metalated sites that would have optimal CH4 adsorptive properties. bpy and cat represent limiting cases of CH4 adsorbing coordination compounds: the cat complexes have a “bent” geometry which offers a high number of coordinated CH4 molecules, but are very chemically active, while the bpy complexes have lower chemical activity but are not sufficiently attractive and can coordinate no more than two CH4 molecules. Other coordination geometries, such as a trigonal (pyramidal) geometry, can potentially be located mid-way, providing high usable capacity and a sufficient attraction and reasonable chemical reactivity. Trigonal coordination could be achieved by using a ligand such the nitrilotriacetic acid (NTA), which is a tri- or tetradentate tripod ligand.77 Due to the higher coordination number of the NTA complex with the metal, it is expected to have weaker attractive interactions with CH4. Although no MOF containing NTA has yet been prepared, we investigated it here as a model system for evaluating the CH4 storage potential of trigonal metal-coordination. We note that a partially coordinated calcium ion is known to exist in nature at the catalytic core of photo-system II.78The optimized structures of the NTA-Ca and NTA-Mg are shown in Fig. 11 and the EDA is shown in Table 6. The coordination of the ion with the terminal oxygen atoms forms an exposed sorbent surface above the trigonal planar complex in the case of Mg and an exposed sorbent surface above a trigonal pyramidal complex in the case of Ca. The trigonal pyramidal complex results from the large volume of the Ca2+ ion, which prevents the NTA ligand from fully “encapsulating” it, thereby leaving it partially exposed. The EDA indicates that for both NTA-Mg and NTA-Ca, the polarization and charge transfer interactions are the most dominant, as a result of the exposed coordination of the metal.
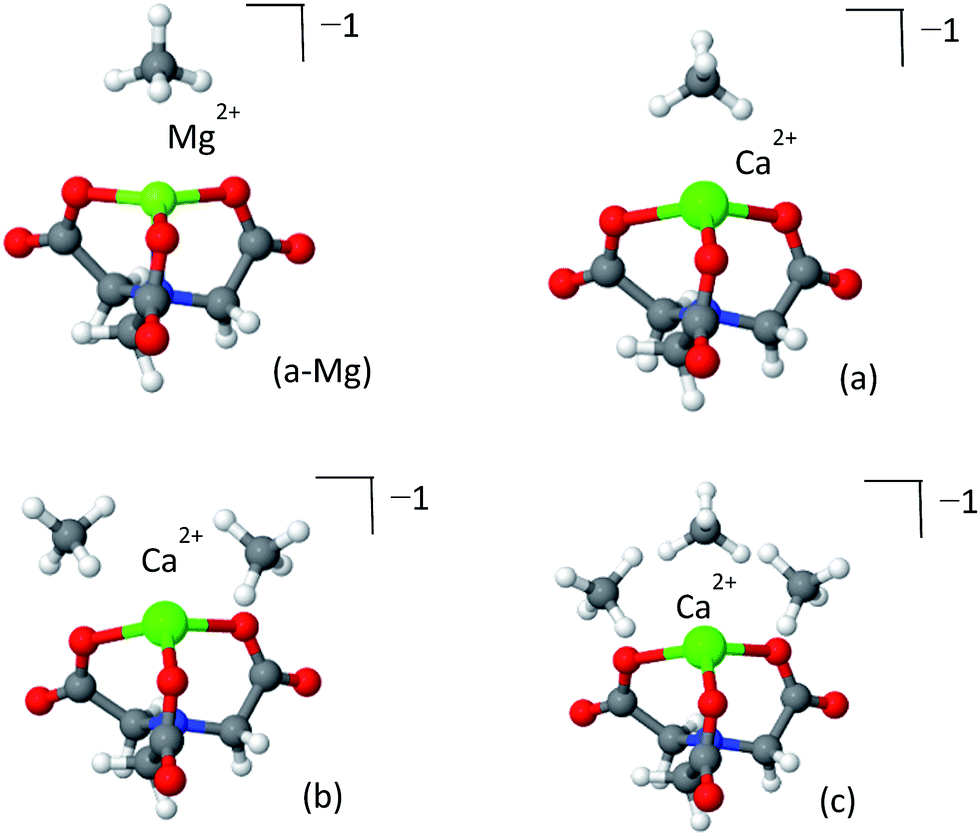 | ||
| Fig. 11 Clusters of NTA-Mg (panel a-Mg) and NTA-Ca (panels a–c) with CH4. The first adsorbed CH4 NTA-Mg blocks the adsorption site, while three CH4 can adsorb to the pyramidal NTA-Ca. | ||
| Nth CH4 | NTA-ligand | |||
|---|---|---|---|---|
| ΔFRZ | ΔPOL | ΔCT | ΔEads | |
| 1 (Mg) | −1.4 | −14.8 | −5.4 | −20.4 |
| 1 (Ca) | −2.2 | −9.7 | −6.3 | −17.4 |
| 2 (Ca) | −3.4 | −8.1 | −6.4 | −17.0 |
| 3 (Ca) | −3.8 | −9.1 | −6.5 | −18.2 |
The pyramidal-exposed geometry of the NTA-Ca complex is a unique feature that can significantly increase its CH4 capacity. The pyramidal binding site has three faces, each of which can adsorb a single CH4 molecule (Fig. 12). Because the faces of the pyramid are pointed at different directions, the adsorbed CH4 molecules are sufficiently separated from each other, by 4.0 Å, such that steric repulsion are minimal. With such small occupancy dependent effects, the bonding characteristics of all adsorbed CH4, as seen in the EDA, are very similar, which is clearly promising for sorbent purposes.
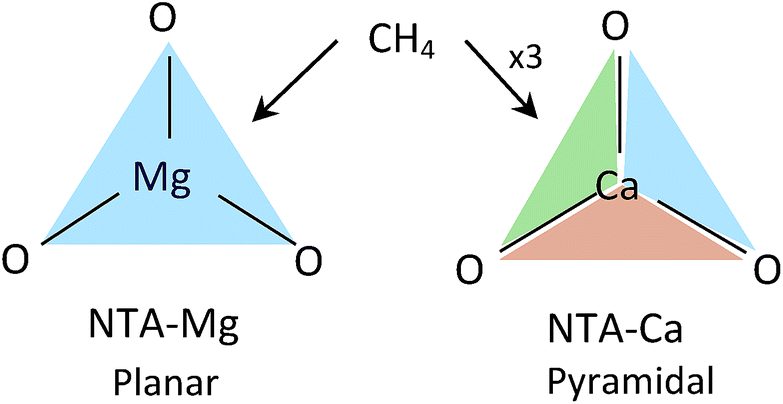 | ||
| Fig. 12 CH4 adsorption in planar vs. pyramidal exposed surfaces. Each face (colored triangle) can adsorb one CH4 molecule. | ||
Due to the small size of the Mg2+ ion, the NTA ligand fully coordinates it to yield a trigonal-planar NTA-Mg complex. The first adsorbed CH4 molecule is located on-top of the Mg2+ ion, where the induced polarization interactions are the strongest. Being smaller, Mg2+ is a better polarizer than Ca2+, such that the adsorption energy of a single CH4 for NTA-Mg is stronger by −3.0 kJ mol−1 and the polarization component is larger by −5.1 kJ mol−1 than for NTA-Ca. However, the single methane completely obstructs the binding site for subsequent molecules, such that only one can be effectively adsorbed.
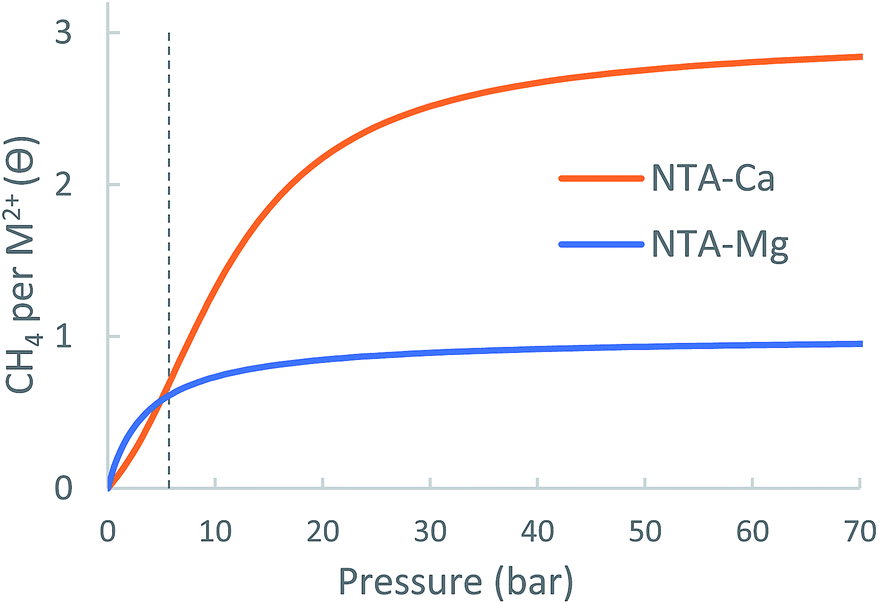 | ||
| Fig. 13 Calculated metal-site adsorption isotherms of the NTA-(Mg, Ca) at 298.15 K. | ||
| NTA-Ca | NTA-Mg | MOF-5 | |
|---|---|---|---|
| Δθuo (35 bar) | 1.90 | 0.29 | 1.6 |
| Δθuo (65 bar) | 2.12 | 0.33 | 2.4 |
Since no NTA-linker based MOF is known to exist, usable capacity estimates for this open metal site embedded in a framework cannot not be provided. However, the NTA-Ca system clearly demonstrates the tremendous potential of using partially coordinated calcium ion for the purpose of CH4 storage.
The effect of solvent on methane adsorption
Although cat metalated linkers have good CH4 adsorption abilities, their realization in experiment could face difficulties due to the high reactivity of the almost completely exposed metal ion centre. As already explained above, the exposed ions can attract guest molecules that may be deposited in the MOF during its preparation and processing, such as solvent molecules. These species are attracted to the metal ion several times more strongly than CH4, thereby effectively poisoning it toward CH4 adsorption. Furthermore, the removal of a typical solvent molecule from cat-(Mg, Ca) would require energies of about −100 kJ mol−1 (the binding energy of methanol and acetonitrile), which is much higher than the thermal energy present at room temperature, or even at 150–250 °C, and approaches the thermal stability limit of most MOFs. Hence the presence of solvent molecules is difficult to avoid, and would be typically viewed as a significant potential deterrent to pursuing the synthetic realization of such sites.However, we would like to point out a different, and potentially much more optimistic perspective. The presence of solvent molecules at the open-metal site could actually be advantageous. The exposed metal ions will be attracted to the negatively charged component of the MOF, thus decreasing the overall framework stability, but the presence of residual solvent molecules could potentially stabilize the MOF by making the metal centre more chemically viable. Considering these prospects, it is intriguing to explore the effect of the presence of solvent on the adsorption of CH4. Here, we study the CH4 adsorption of cat-(Mg, Ca) in the presence of a single methanol (MeOH) and acetonitrile (MeCN) solvent molecules, which are often used in the final washing step of the MOF before its activation. Note that, due to the large number of possible solvents and number of solvent molecules, a more thorough study of solvent effects is beyond the scope of this work.
The resulting structures for cat-Mg are shown in Fig. 14, and the EDA of the clusters and of the solvent–metal interaction, compared to the solvent-free complex, are shown in Table 8. The structures of cat-Ca are given in the ESI.†
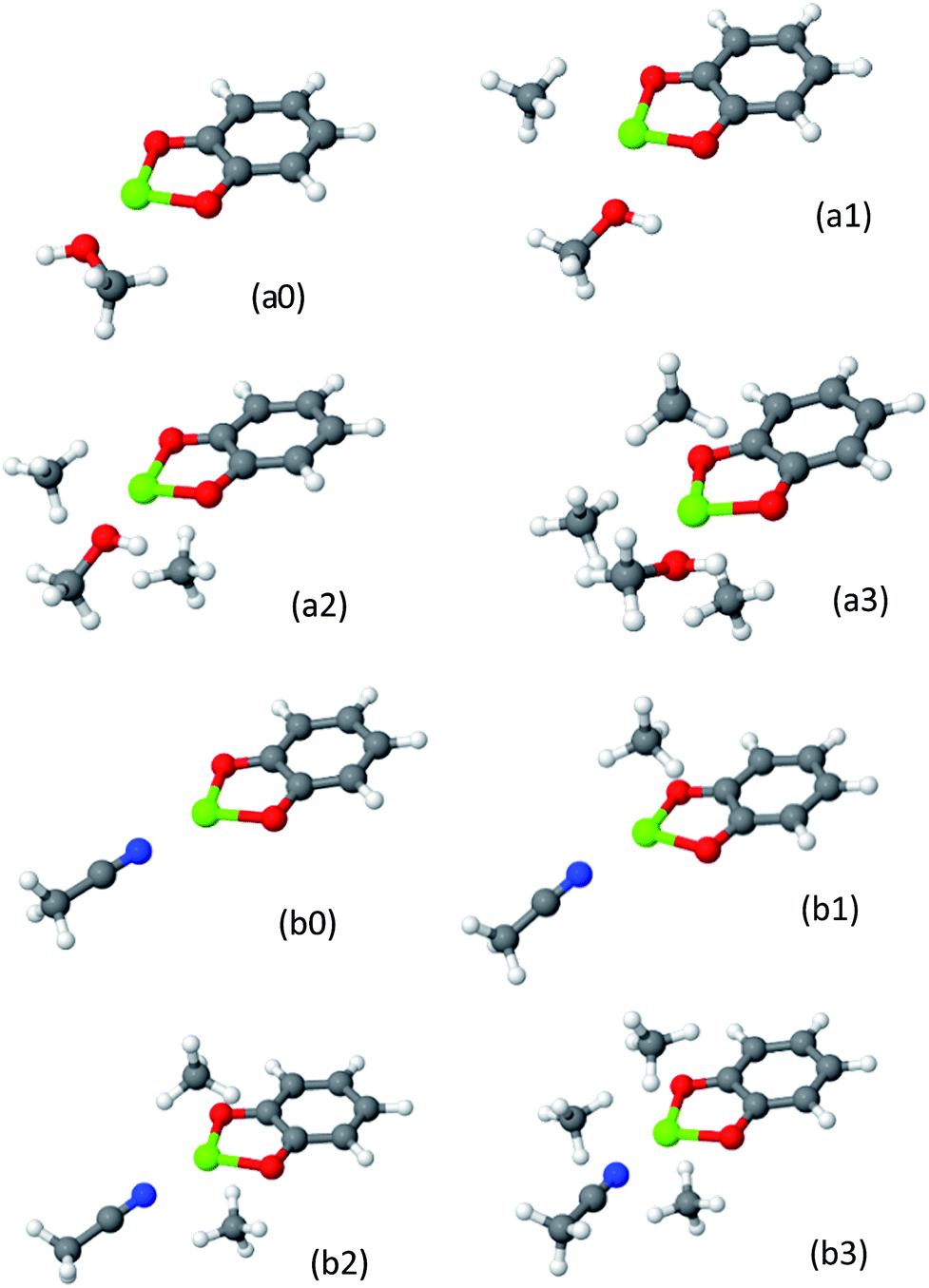 | ||
| Fig. 14 CH4 adsorbed on metalated cat-Mg linkers in the presence of solvent molecule. Panels (a0–a3) and (b0–b3) show MeOH and MeCN solvents-complexes, consecutively. As one solvent molecule is coordinated to the metal, no more than three CH4 molecules can have significant contribution to the CH4 usable site occupancy. | ||
| N(CH4) | Cat-Mg | |||||
|---|---|---|---|---|---|---|
| ΔEPOL | ΔEads | |||||
| — | MeOH | MeCN | — | MeOH | MeCN | |
| 1 | −49.9 | −36.7 | −21.1 | −40.7 | −20.7 | −17.3 |
| 2 | −33.6 | −13.2 | −13.0 | −27.0 | −19.9 | −15.5 |
| 3 | −17.9 | −4.2 | −3.6 | −14.6 | −16.5 | −12.5 |
| Total | −101.4 | −54.2 | −37.7 | −82.3 | −57.1 | −45.3 |
| N(CH4) | Cat-Ca | |||||
|---|---|---|---|---|---|---|
| ΔEPOL | ΔEads | |||||
| — | MeOH | MeCN | — | MeOH | MeCN | |
| 1 | −19.9 | −18.2 | −16.7 | −26.7 | −23.2 | −22.6 |
| 2 | −17.6 | −15.5 | −14.4 | −24.7 | −22.5 | −17.6 |
| 3 | −16.0 | −13.8 | −13.9 | −21.7 | −20.3 | −21.0 |
| Total | −53.5 | −47.6 | −45.0 | −73.2 | −65.9 | −61.2 |
The maximum number of CH4 molecules that are substantially attracted to the solvent@cat-(Mg, Ca) complex is three, compared to four when no solvent is present. This implies that, as expected, the solvent molecule occupies a single site in the metal ion coordination sphere.
The comparison of the EDA of cat-Mg and cat-Ca sheds light on the effect of the solvent. The solvent shields the ion and prevents it from inflicting strong polarizing forces on the adsorbed CH4 molecule. The shielding effect is stronger for cat-Mg than for cat-Ca, since Mg2+ is smaller than Ca2+, it tends to polarize its environment and is more easily shielded. As can be seen in Table 8, for cat-Mg the adsorption energy (ΔEads) for three adsorbed CH4 molecules in the presence of MeOH, is reduced by −25 kJ mol−1, but only by −7 kJ mol−1 for cat-Ca. The EDA also shows that this large reduction for cat-Mg is mostly due to a −47 kJ mol−1 decrease in the polarization component, as a result of solvent shielding.
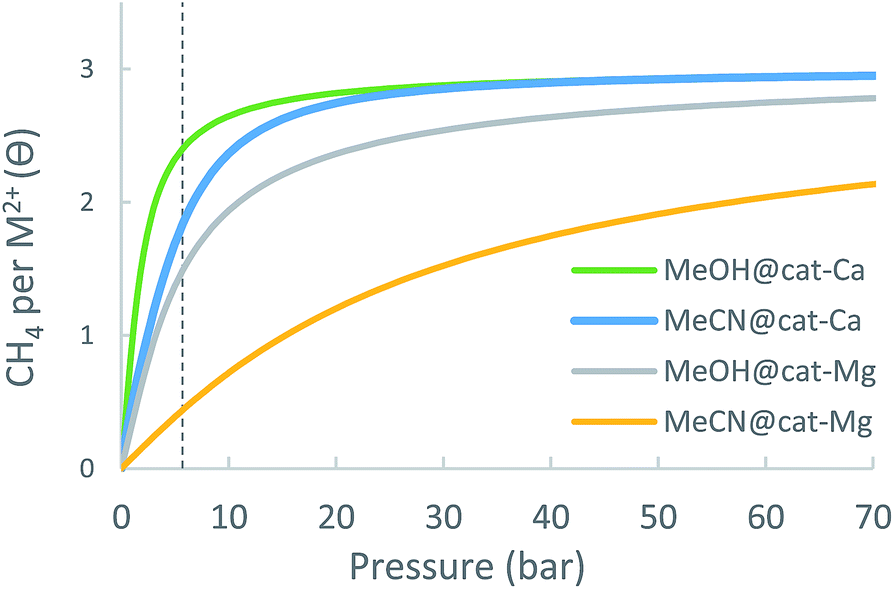 | ||
| Fig. 15 Calculated adsorption isotherms of cat-(Mg, Ca) metal-site with coordinated solvent molecule (MeOH/MeCN) at 298.15 K. | ||
| Cat-Mg | MeOH@cat-Mg | MeCN@cat-Mg | MOF-5 | |
|---|---|---|---|---|
| Δθuo (35 bar) | 1.13 | 1.10 | 1.20 | 1.6 |
| Δθuo (65 bar) | 1.47 | 1.26 | 1.64 | 2.6 |
| Cat-Ca | MeOH@cat-Ca | MeCN@cat-Ca | MOF-5 | |
|---|---|---|---|---|
| Δθuo (35 bar) | 0.34 | 0.49 | 1.03 | 1.6 |
| Δθuo (65 bar) | 0.37 | 0.53 | 1.09 | 2.6 |
Discussion and conclusion
Expected usable capacity of linker-metalated MOFs
From estimating the properties of the individual metal-site, we now proceed to estimating the potential contribution to the usable capacity of actual MOFs. The reader is provided with rough estimates of the usable capacity of MOFs modified to accommodate open-metal sites of the type studied here. We use existing experimental data for pre-metalated MOFs and estimate its post-modification usable capacity. Particularly, MOF-5 (Zn4O(bdc)3) is used to demonstrate the increase in usable capacity by catechol linker metalation and UiO-67-bpy (Zr6O4(OH)4(bpydc)6) to demonstrate bpy linker metalation.We assume that the linkers of the parent MOF had been metalated or completely replaced by new metalated-linkers. It is also assumed that the volume and surface area of the parent MOF are identical to the modified MOF and that the contribution of new open-metal sites to the overall CH4 storage, is additive to the capacity of the parent MOF. This idealized approach allows us to provide an upper bound on the usable capacity. Of course the actual numbers must be smaller, due to reduction in the MOF surface area upon metalation, possible steric overlap between CH4 molecules adsorbed on different sites and steric repulsion from the MOF itself, especially at high site-occupancy. There can also be significant materials limitations, such as the fact that we employ the crystalline density, whereas the material at present is only available as a powder at densities that are several times lower, as demonstrated for MOF-5.79 However, the important question is to identify the upper limit to potential usable capacity, and thus the motivation for future experimental efforts.
The maximal addition of usable CH4 capacity (Δnoms), in v[STP]/v units, as a result of the introduction of the open-metal sites to the structure is evaluated as follows:
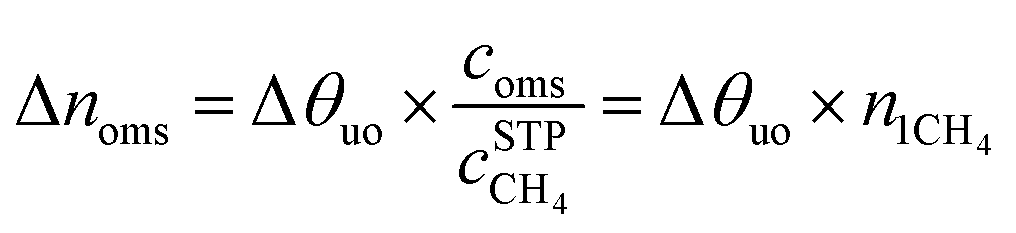 | (5) |
The main results are shown in Table 10. As seen in the last row of Table 10, several metalated MOFs can potentially cross the current limitation of 200 v/v at 65 bar. Metalation by cat-Mg is the most beneficial with a contribution of about 60 v/v at 35 bar. At higher pressure of 65 bar this contribution increases by about 18 v/v. Due to the small usable site occupancy, metalation of UiO-67-bpy by CaCl2 results in a small increase of 26 v/v at 35 bar. At higher pressure this contribution is increased only to about 28 v/v such that the total expected usable capacity of UiO-67-bpy-CaCl2 at 65 bar is about 191 v/v.
| MOF-5 | MOF-5 | MOF-5 | UiO-67-bpy | |
|---|---|---|---|---|
| Parent linker | bdc | bdc | bdc | bpydc |
| Modified linker | Cat-Mg | Cat-Ca | MeCN@cat-Ca | bpy-CaCl2 |
| Parent capacity [v/v] 35 bar | 114 | 114 | 114 | 115 |
| Metalated linker capacity [v/v] 35 bar | 60 | 18 | 54 | 26 |
| Expected capacity [v/v] 35 bar | 174 | 132 | 168 | 141 |
| Parent capacity [v/v] 65 bar | 179 | 179 | 179 | 163 |
| Metalated linker capacity [v/v] 65 bar | 78 | 19 | 58 | 28 |
| Expected capacity [v/v] 65 bar | 257 | 198 | 237 | 191 |
The usable capacity of MOF-5-cat-Ca can be improved if a residual solvent molecule (MeCN) is present, which increases its usable capacity by 36 v/v, from 132 v/v to 168 v/v at 35 bar. At maximum pressure of 65 bar the usable capacity of MOF-5-cat-Ca@MeCN can possibly reach 237 v/v.
The main implications of these numbers is that material designs of these types have considerable potential, even allowing for the inevitable limitations of real materials, including lack of monolithic single crystals, residual solvent and other trace contaminants in real natural gas. Finally, while designing an adsorbent that can operate at ambient temperature is preferred, we note that these materials with strong CH4 binding sites would benefit significantly from using waste heat on-board a vehicle to maximize the usable capacity by increasing the desorption temperature.
Conclusion
Metal–organic frameworks (MOFs) offer tremendous surface area for gas storage, but the simultaneous demands of high surface area, adequate thermal and mechanical stability, and the ideal binding energies for multiple methane molecules at a single site are nearly impossible to satisfy. Rather, it may be better to divide and conquer, and address the binding site requirements by post-synthetic modifications, such as the introduction of open-metal sites, as decorations to relatively rigid organic linkers. This work has been concerned with the possibilities and limitations of such sites for the purposes of methane storage. Our main conclusions are as follows:(1) Adequate interaction energy is a necessary condition for an effective CH4 storage. The results obtained here imply, that in order to have a sufficient CH4 adsorption ability, the metal ion should not only be (partially) exposed, but should also have sufficient ionic character, i.e., the charge separation of the metal ion and its coordination environment should be large. However, ionic character in molecules is closely related to deviation from covalency, which tends to spread the electronic densities in between the atoms, preventing accumulation of charge on specific atoms. Preparation of such species poses considerable synthetic challenges: for instance, for the bpy linker, only the bpy-CaCl2 complex has significant interaction with CH4—which is made possible by its weak coordination by the linker, which could prevent the Ca2+ ion from forming a stable complex. In cat-Mg and cat-Ca, which both have promising CH4 adsorption properties, the metal ion is almost completely exposed, being coordinated by only two oxygen atoms. Cat-Mg and cat-Ca are therefore expected to be highly reactive and can potentially affect the MOFs structural stability.
(2) While adequate interaction energy is a necessary condition for CH4 adsorption, the overall uptake of the open-metal site is also related to the site geometry, where pyramidal, as in NTA-Ca, or “bent”, as in cat-(Mg, Ca), can accommodate more CH4 molecules that are adsorbed with approximately the same energy. The formation of geometrically exposed sites, where the coordination sphere of the metal-ions is exposed along more than one axis, can result in a substantial increase in the capacity of the site.
(3) The usable capacity of the catechol based cat-Mg open-metal sites benefits mostly from the large number of coordinated methane molecules: although the first adsorbed CH4 is bound too strongly, the subsequent ones are more weakly bound and contribute to effective adsorption. Even in the presence of solvent which greatly reduces the adsorption energy, the overall usable capacity remains similar, or increases. Too strongly bound CH4 molecules in the non-solvated case become storage-effective in the presence of solvent, which compensates for the reduced effectiveness of the more weakly attracted CH4.
(4) Another important point is the ratio between total adsorbed CH4 and the storage-usable adsorbed CH4. Since effectively not much more than a single CH4 contributes to the usable capacity of the cat-(Ca, Mg) site, their usable capacity is low with respect to their total capacity, such that a significant portion of the volume of the MOF is expected to be “wasted” on non-usable capacity. For instance, cat-Mg has a ratio of only 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 usable to total adsorbed CH4. The bpy-CaCl2 and NTA-Ca open-metal sites are more effective in that sense, where relatively more methane molecules per open-metal site contribute to the usable capacity of the MOF and the ratio between usable and total adsorbed CH4 is higher. NTA-Ca is especially effective with a ratio of 9:10 usable to total adsorbed CH4.
:4 usable to total adsorbed CH4. The bpy-CaCl2 and NTA-Ca open-metal sites are more effective in that sense, where relatively more methane molecules per open-metal site contribute to the usable capacity of the MOF and the ratio between usable and total adsorbed CH4 is higher. NTA-Ca is especially effective with a ratio of 9:10 usable to total adsorbed CH4.
(5) Another interesting finding is the unique properties of the Ca2+ ion: for some systems, calcium is very resilient to its coordination environment and is able to maintain its CH4 adsorbing properties in the presence of solvent or when coordinated by the tridentate NTA ligand. This seems to be a result of its large ionic radius, such that the coordinating environment is not able to shield its electrostatic charge, which is dispersed over a large area. When the calcium ion remains partially exposed it can adsorb one or more CH4 with more-than sufficient energy, depending on the steric availability in its coordination sphere.
(6) While the exact effect of solvent molecule(s) is difficult to determine and specific recommendations cannot be given, this study demonstrates that the presence of residual solvent is not necessarily detrimental for CH4 adsorption applications. On the contrary, solvent molecules can be used to modify the interaction between the open-metal site and the adsorbed CH4 molecule, especially for attenuating excessively strong binding into the desired range. The large variety of solvent molecules implies that other effects could be expected for the complex-solvent–CH4 system. These are left for future investigations, perhaps in response to future specific experimental developments in the field.
Acknowledgements
This research was supported by the Advanced Research Projects Agency – Energy (ARPA-E), U.S. Department of Energy with additional support from the National Science Foundation, grant number CHE-1363342. We thank Narbe Mardirossian, Matthew Kapelewski and Dr Eric Bloch for helpful discussions.Notes and references
- Alternative Fuels Data Center: Natural Gas [Internet]. 2015 [cited 2015 Feb 12]. Available from: http://www.afdc.energy.gov/fuels/natural_gas.html.
- List of natural gas vehicles – Wikipedia, the free encyclopedia [Internet]. 2015 [cited 2015 Feb 12]. Available from: http://en.wikipedia.org/wiki/List_of_natural_gas_vehicles.
- Argonne GREET Model [Internet]. 2015 [cited 2015 Feb 12]. Available from: https://greet.es.anl.gov/.
- Annual Energy Outlook 2014-Energy Information Administration [Internet]. 2015 [cited 2015 Feb 12]. Available from: http://www.eia.gov/forecasts/aeo/.
- N. D. Parkyns, D. F. Quinn and J. W. Patrick, Porosity in carbons, Edw Arnold Lond., 1995, p. 291 Search PubMed.
- B. Li, H.-M. Wen, W. Zhou and B. Chen, Porous Metal–Organic Frameworks for Gas Storage and Separation: What, How, and Why?, J. Phys. Chem. Lett., 2014, 5(20), 3468–3479 CrossRef CAS PubMed.
- H.-C. Zhou, J. R. Long, O. M. Yaghi, J. R. Long and O. M. Yaghi, Introduction to Metal–Organic Frameworks, Chem. Rev., 2012, 112(2), 673–674 CrossRef CAS PubMed.
- M. Yoon, R. Srirambalaji and K. Kim, Homochiral Metal–Organic Frameworks for Asymmetric Heterogeneous Catalysis, Chem. Rev., 2012, 112(2), 1196–1231 CrossRef CAS PubMed.
- D. J. Xiao, E. D. Bloch, J. A. Mason, W. L. Queen, M. R. Hudson and N. Planas, et al., Oxidation of ethane to ethanol by N2O in a metal–organic framework with coordinatively unsaturated iron(II) sites, Nat. Chem., 2014, 6(7), 590–595 CrossRef CAS PubMed.
- J.-R. Li, J. Sculley and H.-C. Zhou, Metal–Organic Frameworks for Separations, Chem. Rev., 2012, 112(2), 869–932 CrossRef CAS PubMed.
- K. Lee, W. C. Isley, A. L. Dzubak, P. Verma, S. J. Stoneburner and L.-C. Lin, et al., Design of a Metal–Organic Framework with Enhanced Back Bonding for Separation of N2 and CH4, J. Am. Chem. Soc., 2014, 136(2), 698–704 CrossRef CAS PubMed.
- M. E. Casco, M. Martínez-Escandell, E. Gadea-Ramos, K. Kaneko, J. Silvestre-Albero and F. Rodriguez-Reinoso, High-Pressure Methane Storage in Porous Materials: Are Carbon Materials in the Pole Position?, Chem. Mater., 2015, 27(3), 959–964 CrossRef CAS.
- C. M. Simon, J. Kim, D. A. Gomez-Gualdron, J. S. Camp, Y. G. Chung and R. L. Martin, et al., The materials genome in action: identifying the performance limits for methane storage, Energy Environ. Sci., 2015, 8(4), 1190–1199 CAS.
- Y. Peng, V. Krungleviciute, I. Eryazici, J. T. Hupp, O. K. Farha and T. Yildirim, Methane Storage in Metal–Organic Frameworks: Current Records, Surprise Findings, and Challenges, J. Am. Chem. Soc., 2013, 135(32), 11887–11894 CrossRef CAS PubMed.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Design and synthesis of an exceptionally stable and highly porous metal–organic framework, Nature, 1999, 402(6759), 276–279 CrossRef CAS.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Reticular synthesis and the design of new materials, Nature, 2003, 423(6941), 705–714 CrossRef CAS PubMed.
- S. S.-Y. Chui, S. M.-F. Lo, J. P. Charmant, A. G. Orpen and I. D. Williams, A chemically functionalizable nanoporous material [Cu3(TMA)2(H2O)3]n, Science, 1999, 283(5405), 1148–1150 CrossRef CAS PubMed.
- B. Li, H.-M. Wen, H. Wang, H. Wu, M. Tyagi and T. Yildirim, et al., A Porous Metal–Organic Framework with Dynamic Pyrimidine Groups Exhibiting Record High Methane Storage Working Capacity, J. Am. Chem. Soc., 2014, 136(17), 6207–6210 CrossRef CAS PubMed.
- F. Gándara, H. Furukawa, S. Lee and O. M. Yaghi, High Methane Storage Capacity in Aluminum Metal–Organic Frameworks, J. Am. Chem. Soc., 2014, 136(14), 5271–5274 CrossRef PubMed.
- B. Chen, X. Zhao, A. Putkham, K. Hong, E. B. Lobkovsky and E. J. Hurtado, et al., Surface Interactions and Quantum Kinetic Molecular Sieving for H2 and D2 Adsorption on a Mixed Metal–Organic Framework Material, J. Am. Chem. Soc., 2008, 130(20), 6411–6423 CrossRef CAS PubMed.
- E. D. Bloch, D. Britt, C. Lee, C. J. Doonan, F. J. Uribe-Romo and H. Furukawa, et al., Metal insertion in a microporous metal–organic framework lined with 2,2′-bipyridine, J. Am. Chem. Soc., 2010, 132(41), 14382–14384 CrossRef CAS PubMed.
- W. M. Bloch, A. Burgun, C. J. Coghlan, R. Lee, M. L. Coote and C. J. Doonan, et al., Capturing snapshots of post-synthetic metallation chemistry in metal–organic frameworks, Nat. Chem., 2014, 6(10), 906–912 CrossRef CAS PubMed.
- J. A. Mason, M. Veenstra and J. R. Long, Evaluating metal–organic frameworks for natural gas storage, Chem. Sci., 2014, 5(1), 32–51 RSC.
- S. M. Cohen, Postsynthetic Methods for the Functionalization of Metal–Organic Frameworks, Chem. Rev., 2012, 112(2), 970–1000 CrossRef CAS PubMed.
- J. D. Evans, C. J. Sumby and C. J. Doonan, Post-synthetic metalation of metal–organic frameworks, Chem. Soc. Rev., 2014, 43(16), 5933–5951 RSC.
- J. E. Mondloch, O. Karagiaridi, O. K. Farha and J. T. Hupp, Activation of metal–organic framework materials, CrystEngComm, 2013, 15(45), 9258–9264 RSC.
- X. Feng, X. Ding and D. Jiang, Covalent organic frameworks, Chem. Soc. Rev., 2012, 41(18), 6010–6022 RSC.
- S.-Y. Ding and W. Wang, Covalent organic frameworks (COFs): from design to applications, Chem. Soc. Rev., 2013, 42(2), 548–568 RSC.
- T. Ben and S. Qiu, Porous aromatic frameworks: synthesis, structure and functions, CrystEngComm, 2013, 15(1), 17–26 RSC.
- C. Pei, T. Ben and S. Qiu, Great Prospects for PAF-1 and its derivatives, Mater. Horiz., 2015, 2(1), 11–21 RSC.
- T. B. Lee, D. Kim, D. H. Jung, S. B. Choi, J. H. Yoon and J. Kim, et al., Understanding the mechanism of hydrogen adsorption into metal organic frameworks, Catal. Today, 2007, 120(3–4), 330–335 CrossRef CAS.
- K. Sillar, A. Hofmann and J. Sauer, Ab initio study of hydrogen adsorption in MOF-5, J. Am. Chem. Soc., 2009, 131(11), 4143–4150 CrossRef CAS PubMed.
- N. Planas, A. L. Dzubak, R. Poloni, L.-C. Lin, A. McManus and T. M. McDonald, et al., The Mechanism of Carbon Dioxide Adsorption in an Alkylamine-Functionalized Metal–Organic Framework, J. Am. Chem. Soc., 2013, 135(20), 7402–7405 CrossRef CAS PubMed.
- P. Verma, X. Xu and D. G. Truhlar, Adsorption on Fe–MOF-74 for C1–C3 Hydrocarbon Separation, J. Phys. Chem. C, 2013, 117(24), 12648–12660 CAS.
- E. D. Bloch, M. R. Hudson, J. A. Mason, S. Chavan, V. Crocellà and J. D. Howe, et al., Reversible CO Binding Enables Tunable CO/H2 and CO/N2 Separations in Metal–Organic Frameworks with Exposed Divalent Metal Cations, J. Am. Chem. Soc., 2014, 136(30), 10752–10761 CrossRef CAS PubMed.
- K. Sillar and J. Sauer, Ab Initio Prediction of Adsorption Isotherms for Small Molecules in Metal–Organic Frameworks: The Effect of Lateral Interactions for Methane/CPO-27-Mg, J. Am. Chem. Soc., 2012, 134(44), 18354–18365 CrossRef CAS PubMed.
- C. T. Campbell and J. R. Sellers, The entropies of adsorbed molecules, J. Am. Chem. Soc., 2012, 134(43), 18109–18115 CrossRef CAS PubMed.
- D. G. Truhlar, A simple approximation for the vibrational partition function of a hindered internal rotation, J. Comput. Chem., 1991, 12(2), 266–270 CrossRef CAS.
- S. K. Bhatia and A. L. Myers, Optimum Conditions for Adsorptive Storage, Langmuir, 2006, 22(4), 1688–1700 CrossRef CAS PubMed.
- R. Z. Khaliullin, E. A. Cobar, R. C. Lochan, A. T. Bell and M. Head-Gordon, Unravelling the Origin of Intermolecular Interactions Using Absolutely Localized Molecular Orbitals, J. Phys. Chem. A, 2007, 111(36), 8753–8765 CrossRef CAS PubMed.
- P. R. Horn, E. J. Sundstrom, T. A. Baker and M. Head-Gordon, Unrestricted absolutely localized molecular orbitals for energy decomposition analysis: theory and applications to intermolecular interactions involving radicals, J. Chem. Phys., 2013, 138(13), 134119 CrossRef PubMed.
- Y. Shao, Z. Gan, E. Epifanovsky, A. T. B. Gilbert, M. Wormit and J. Kussmann, et al., Advances in molecular quantum chemistry contained in the Q-Chem 4 program package, Mol. Phys., 2015, 113(2), 184–215 CrossRef CAS.
- E. Gianinetti, M. Raimondi and E. Tornaghi, Modification of the Roothaan equations to exclude BSSE from molecular interaction calculations, Int. J. Quantum Chem., 1996, 60(1), 157–166 CrossRef CAS.
- R. Z. Khaliullin, M. Head-Gordon and A. T. Bell, An efficient self-consistent field method for large systems of weakly interacting components, J. Chem. Phys., 2006, 124(20), 204105 CrossRef PubMed.
- R. Z. Khaliullin, A. T. Bell and M. Head-Gordon, Analysis of charge transfer effects in molecular complexes based on absolutely localized molecular orbitals, J. Chem. Phys., 2008, 128(18), 184112 CrossRef PubMed.
- N. Mardirossian and M. Head-Gordon, Mapping the genome of meta-generalized gradient approximation density functionals: the search for B97M-V, J. Chem. Phys., 2015, 142(7), 074111 CrossRef PubMed.
- O. A. Vydrov and T. V. Voorhis, Nonlocal van der Waals density functional: the simpler the better, J. Chem. Phys., 2010, 133(24), 244103 CrossRef PubMed.
- K. U. Lao, R. Schäffer, G. Jansen and J. M. Herbert, Accurate Description of Intermolecular Interactions Involving Ions Using Symmetry-Adapted Perturbation Theory, J. Chem. Theory Comput., 2015, 11(6), 2473–2486 CrossRef CAS PubMed.
- D. Rappoport and F. Furche, Property-optimized Gaussian basis sets for molecular response calculations, J. Chem. Phys., 2010, 133(13), 134105 CrossRef PubMed.
- F. Weigend, F. Furche and R. Ahlrichs, Gaussian basis sets of quadruple zeta valence quality for atoms H–Kr, J. Chem. Phys., 2003, 119(24), 12753–12762 CrossRef CAS.
- J. Lan, D. Cao and W. Wang, High Uptakes of Methane in Li-Doped 3D Covalent Organic Frameworks, Langmuir, 2010, 26(1), 220–226 CrossRef CAS PubMed.
- Z. Xiang, D. Cao, W. Wang, W. Yang, B. Han and J. Lu, Postsynthetic Lithium Modification of Covalent-Organic Polymers for Enhancing Hydrogen and Carbon Dioxide Storage, J. Phys. Chem. C, 2012, 116(9), 5974–5980 CAS.
- Z. Yang and D. Cao, Effect of Li Doping on Diffusion and Separation of Hydrogen and Methane in Covalent Organic Frameworks, J. Phys. Chem. C, 2012, 116(23), 12591–12598 CAS.
- R. D. Shannon, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32(5), 751–767 CrossRef.
- M. Brookhart, M. L. Green and G. Parkin, Agostic interactions in transition metal compounds, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(17), 6908 CrossRef CAS PubMed.
- K. Eskandari, Characteristics of beryllium bonds; a QTAIM study, J. Mol. Model., 2012, 18(8), 3481–3487 CrossRef CAS PubMed.
- K. D. Jordan and J. Simons, Comment on the electronic structure of small beryllium and magnesium clusters and their anions, J. Chem. Phys., 1980, 72(4), 2889–2890 CrossRef CAS.
- R. B. Getman, J. H. Miller, K. Wang and R. Q. Snurr, Metal Alkoxide Functionalization in Metal–Organic Frameworks for Enhanced Ambient-Temperature Hydrogen Storage, J. Phys. Chem. C, 2011, 115(5), 2066–2075 CAS.
- L. Wang, Y. Sun and H. Sun, Incorporating magnesium and calcium cations in porous organic frameworks for high-capacity hydrogen storage, Faraday Discuss., 2011, 151, 143–156 RSC.
- S. K. Brand, Y. J. Colón, R. B. Getman and R. Q. Snurr, Design strategies for metal alkoxide functionalized metal–organic frameworks for ambient temperature hydrogen storage, Microporous Mesoporous Mater., 2013, 171, 103–109 CrossRef CAS.
- K. K. Tanabe, C. A. Allen and S. M. Cohen, Photochemical Activation of a Metal–Organic Framework to Reveal Functionality, Angew. Chem., Int. Ed., 2010, 49(50), 9730–9733 CrossRef CAS PubMed.
- H. Fei, J. Shin, Y. S. Meng, M. Adelhardt, J. Sutter and K. Meyer, et al., Reusable Oxidation Catalysis Using Metal-Monocatecholato Species in a Robust Metal–Organic Framework, J. Am. Chem. Soc., 2014, 136(13), 4965–4973 CrossRef CAS PubMed.
- H. Fei and S. M. Cohen, Metalation of a Thiocatechol-Functionalized Zr(IV)-Based Metal–Organic Framework for Selective C–H Functionalization, J. Am. Chem. Soc., 2015, 137(6), 2191–2194 CrossRef CAS PubMed.
- V. T. Athavale, L. H. Prabhu and D. G. Vartak, Solution stability constants of some metal complexes of derivatives of catechol, J. Inorg. Nucl. Chem., 1966, 28(5), 1237–1249 CrossRef CAS.
- J. A. Stone and D. Vukomanovic, Electrospray and collisionally activated dissociation study of the association of pyocyanin with alkali metal cations and doubly charged alkaline earth and zinc cations1, Int. J. Mass Spectrom., 2001, 210–211, 341–359 CrossRef.
- L. Hong, Y. Liu and J. D. Simon, Binding of Metal Ions to Melanin and Their Effects on the Aerobic Reactivity, Photochem. Photobiol., 2004, 80(3), 477–481 CrossRef CAS PubMed.
- L. Hong and J. D. Simon, Insight into the Binding of Divalent Cations to Sepia Eumelanin from IR Absorption Spectroscopy, Photochem. Photobiol., 2006, 82(5), 1265–1269 CrossRef CAS PubMed.
- W. D. Bush and J. D. Simon, Quantification of Ca2+ binding to melanin supports the hypothesis that melanosomes serve a functional role in regulating calcium homeostasis, Pigm. Cell Res., 2007, 20(2), 134–139 CrossRef CAS PubMed.
- P. A. Riley and M. R. L. Stratford, Oxidative calcium release from catechol, Bioorg. Med. Chem. Lett., 2015, 25(7), 1453–1454 CrossRef CAS PubMed.
- F. Carson, S. Agrawal, M. Gustafsson, A. Bartoszewicz, F. Moraga and X. Zou, et al., Ruthenium Complexation in an Aluminium Metal–Organic Framework and Its Application in Alcohol Oxidation Catalysis, Chem.–Eur. J., 2012, 18(48), 15337–15344 CrossRef CAS PubMed.
- H. Fei and S. M. Cohen, A robust, catalytic metal–organic framework with open 2,2′-bipyridine sites, Chem. Commun., 2014, 50(37), 4810–4812 RSC.
- S. Øien, G. Agostini, S. Svelle, E. Borfecchia, K. A. Lomachenko and L. Mino, et al., Probing Reactive Platinum Sites in UiO-67 Zirconium Metal–Organic Frameworks, Chem. Mater., 2015, 27(3), 1042–1056 CrossRef.
- M. I. Gonzalez, E. D. Bloch, J. A. Mason, S. J. Teat and J. R. Long, Single-Crystal-to-Single-Crystal Metalation of a Metal–Organic Framework: A Route toward Structurally Well-Defined Catalysts, Inorg. Chem., 2015, 54(6), 2995–3005 CrossRef CAS PubMed.
- G. Anderegg, Pyridin derivate als Komplexbildner V. Die Metallkomplexe von 1,10-Phenanthrolin und α,α′-Dipyridyl, Helv. Chim. Acta, 1963, 46(6), 2397–2410 CrossRef CAS.
- S. Capone, A. De Robertis, C. De Stefano and R. Scarcella, Thermodynamics of formation of magnesium, calcium, strontium and barium complexes with 2,2′-bipyridyl and 1,10-phenanthroline, at different ionic strengths in aqueous solution, Talanta, 1985, 32(8), 675–677 CrossRef CAS PubMed.
- M. Satterfield and J. S. Brodbelt, Relative Binding Energies of Gas-Phase Pyridyl Ligand/Metal Complexes by Energy-Variable Collisionally Activated Dissociation in a Quadrupole Ion Trap, Inorg. Chem., 2001, 40(21), 5393–5400 CrossRef CAS PubMed.
- B. L. Barnett and V. A. Uchtman, Structural investigations of calcium-binding molecules. 4. Calcium binding to aminocarboxylates. Crystal structures of Ca(CaEDTA). 7H2O and Na(CaNTA), Inorg. Chem., 1979, 18(10), 2674–2678 CrossRef CAS.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å, Nature, 2011, 473(7345), 55–60 CrossRef CAS PubMed.
- Y. Ming, J. Purewal, D. Liu, A. Sudik, C. Xu and J. Yang, et al., Thermophysical properties of MOF-5 powders, Microporous Mesoporous Mater., 2014, 185, 235–244 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Evaluation of usable capacity, additional EDA information, additional structural information, synthesis Zr6O4(OH)4(bpydc)6 and measurements of CH4 adsorption, comparison to experimental results for MOF-5. See DOI: 10.1039/c6sc00529b |
| This journal is © The Royal Society of Chemistry 2016 |