
Green and practical transition metal-free one-pot conversion of substituted benzoic acids to anilines using tosyl azide†
Andivelu
Ilangovan
*,
Palaniappan
Sakthivel
and
Pandaram
Sakthivel
School of Chemistry, Bharathidasan University, Tiruchirappalli – 620 024, Tamil Nadu, India. E-mail: ilangovanbdu@yahoo.com
First published on 19th September 2016
Abstract
A simple one-pot procedure for conversion of 2-iodo/2-nitro benzoic acids and dihydropyranone fused benzoic acids into corresponding anilines, using tosyl azide, under transition metal-free conditions is explained. TsN3, is one of the more stable azides, much cheaper and easy to handle. This method is environmentally benign, adoptable for large scale preparation. Different factors such as steric and electronic effect of 2-iodo/2-nitro substituents, and the effect of conformational constraints of the fused dihydropyranone ring on co-planarity of the aromatic ring should contribute to the success of this reaction. Tosyl nitrene was found to be involved in the decarboxylation reaction. As this method is simple, scalable and requires only simple reagents we expect this method to find widespread application in organic synthesis.
Introduction
Substituted anilines are found in a wide variety of natural products and are valuable building blocks for the preparation of heterocycles, pharmaceuticals, agrochemicals, dyes and polymers.1 The functional groups such as iodo and nitro found in 2-iodoanilines2 and 2-nitroanilines3 make it amenable for multiple functionalization and construction of several bioactive natural products and complex heterocycles (Scheme 1). Consequently, developing novel methods to access polysubstituted aromatic amines has become an active research topic.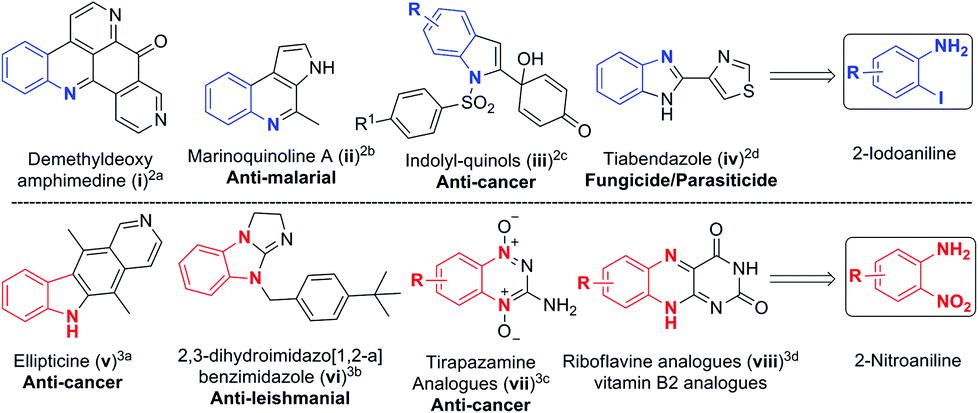 | ||
| Scheme 1 Natural products and bioactive molecules made from (2-iodo/2-nitro)anilines. | ||
Classical methods for the synthesis of anilines involve sequential electrophilic nitration and reduction under harsh conditions,4 and aromatic nucleophilic amination of compounds substituted with specific functional groups.5 Substituted anilines could also be prepared from acyclic precursors.6 Another important strategy is to form the C–N bond through coupling reactions such as Buchwald–Hartwig, Chan–Lam, and Ullmann-type in the presence of a transition metal,7 or under metal-free conditions.8 However, these reactions are mainly suitable for making secondary and tertiary anilines but not for primary anilines.
The next important and particularly useful approach for the preparation of primary anilines from carboxylic acid or its derivatives is through molecular rearrangements such as Curtius,9 Schimidt,10 Hoffman11 and Lossen12 involving an isocyanate intermediate. Although, acyl azides are useful starting materials for obtaining aniline, the drawbacks associated with it make it less attractive. Acyl azides are unstable and difficult to handle, the acid chloride or mixed anhydride13a,b used in its preparation are moisture sensitive, and phase transfer catalysts needs to be used to improve solubility of NaN3 in organic medium.13c Another and most widely used reagent in the Curtius rearrangement is diphenylphosphorazidate (DPPA).14a–c However, this reagent is toxic and moisture sensitive, and separation of desired products from the phosphorus residues is difficult and usually requires high temperature.14d Dess–Martin periodinane–NaN3 could be used to convert aldehydes to acyl azides,15 but the reaction has to be conducted below 0 °C to suppress thermal rearrangement of isocyanate. Acyl azides can also be prepared by diazotization of acylhydrazines,16 however it is not a convenient procedure. Recently, Lebel et al.17 reported a Curtius rearrangement/palladium-catalyzed indolization (Scheme 2, eqn (1)) approach for the direct synthesis of substituted indoles starting from readily available 2-iodobenzoic acid in one pot.
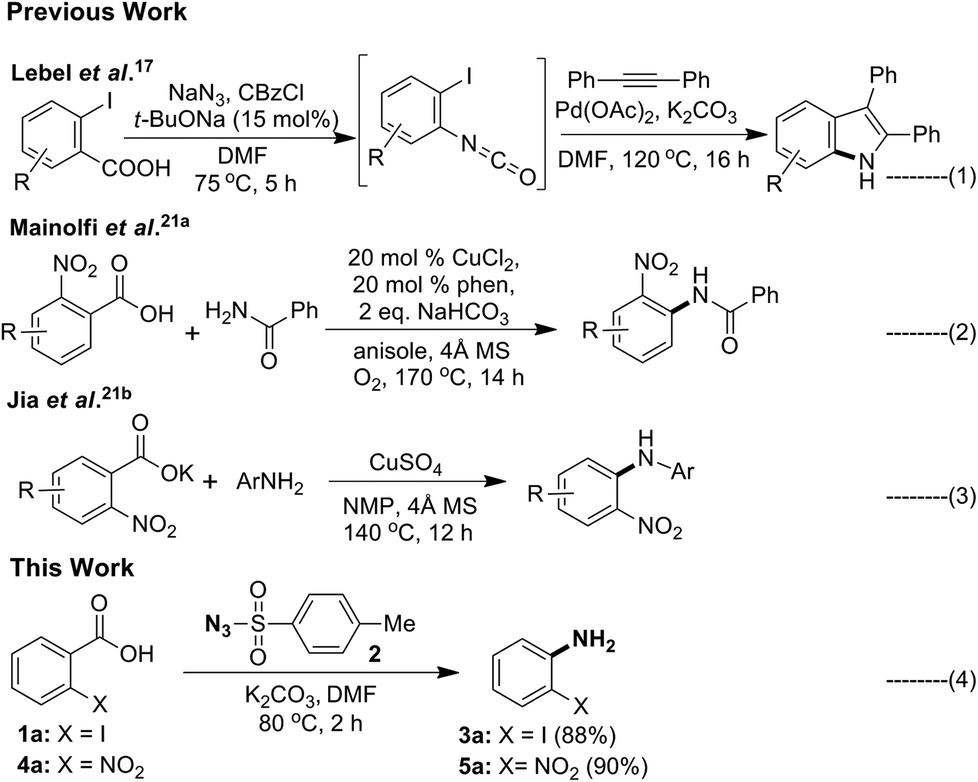 | ||
| Scheme 2 Synthetic methods for aromatic amines. | ||
Decarboxylative cross-coupling reactions are highly site selective and have been used as a new tool in C–C as well as C–heteroatom (C–S/O/N/P/halogen) bond forming reactions.18 In case of benzoic acids the ortho-nitro group19 was found to facilitate the decarboxylation. Among these transformations, C–N bond forming reactions remain largely unexplored.20 Particularly, in the case of aromatic carboxylic acids, there are only two reports available (Scheme 2, eqn (2) and (3)).21 These reports reveals that decarboxylative C–N cross coupling of ortho-nitro benzoic acid or its potassium salt with different N-nucleophiles could be carried out in the presence of copper(II)–phenanthroline complex (eqn (2))21a or CuSO4 (eqn (3))21b respectively. Interestingly, literature background reveals that decarboxylative C–N bond formation of ortho-iodo benzoic acid is unknown. Recently, several other C–H functionalisation approaches to access anilines were also reported.22
During a recent study on the synthesis of biologically active heterocycles23 we examined Pd-catalyzed ortho-C–H amidation24 of pyranochromanone acid 6a using p-toluenesulphonyl azide (tosyl azide, TsN3) and K2CO3 in DMF (Scheme 3). We were delighted to find the formation of pyranochromanone aniline t-7a instead of ortho-C–H amidation product 8. Furthermore, the same reaction took place even in the absence of palladium catalyst. Since aryl carboxylic acids are known to undergo decarboxylative functionalisation only in the presence of transition metals,18 we were surprised to observe the formation of aniline even in the absence of a transition metal. This serendipitous result impelled us to look into the scope and limitation of tosyl azide as reagent for the conversion of substituted aromatic acids in to anilines. Herein we present our results.
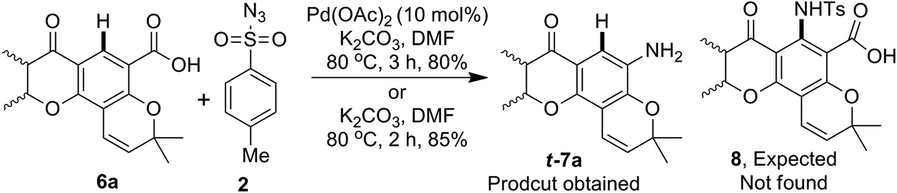 | ||
| Scheme 3 Attempted ortho C–H amidation of pyranochromanone acid 14a. | ||
Results and discussion
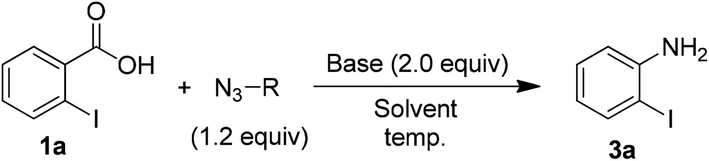
In order to examine the substrate scope and to optimize the reaction conditions, we contemplated examining this newly observed C–N bond-forming reaction using commercially available 2-iodobenzoic acid (1a) containing the both sterically hindered and electron withdrawing 2-iodo substituent.Delightfully, a mixture of o-iodobenzoic acid (1a) and tosyl azide (2), in DMF, stirred for 3 h at 80 °C, in the presence of K2CO3 (1 eq.) gave o-iodo aniline (3a), albeit in moderate yield (60%, Table 1, entry 1). Then we examined different reaction parameters and the results are summarised in Table 1. Fascinatingly, the yield of the product 3a improved to 90% when the quantity of K2CO3 was increased to 2 equiv. (entry 2). Further, increasing the quantity of K2CO3 or increasing the quantity of tosyl azide did not improve the yield (entries 3 and 4). At r.t. no reaction was observed (entry 5). Change of solvent to DMSO (entry 6) led to lower yield of aniline 3a and no reaction was observed with ACN, MeOH, THF, toluene and DCE as solvent (entry 7). Changing the base to NaHCO3 (entry 8) decreased the yield and with DBU, DABCO and TEA (entry 9) no reaction was observed. Similarly, other azide reagents such as DPPA, nosyl azide (p-nitrobenzene sulphonyl azide) and MSN3 (entries 10–12) led to decreased yield. With TMSN3 (entry 13) no reaction was observed. Based on these results, treatment of acid 1a (1.0 mmol) with tosyl azide (2, 1.2 mmol) and K2CO3 (2.0 mmol) in DMF (4 mL) at 80 °C (entry 2) was identified as the optimum conditions for further study.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Azide | Base | Solvent | Temp (°C) | Time (h) | Yieldb (%) |
| a Reaction conditions: 2-iodobenzoic acid (1a, 1 mmol), azide (1.2 mmol), base (2 mmol), solvent (4 mL). b Isolate yield. c 1.0 equiv. K2CO3 used. d 3.0 equiv. K2CO3 used. e 2.0 equiv. tosyl azide used. f NR = no reaction. | ||||||
| 1. | TsN3 | K2CO3 | DMF | 80 °C | 3 | 60c |
| 2. | TsN 3 | K 2 CO 3 | DMF | 80 °C | 2 | 90 |
| 3. | TsN3 | K2CO3 | DMF | 80 °C | 3 | 90d |
| 4. | TsN3 | K2CO3 | DMF | 80 °C | 3 | 90e |
| 5. | TsN3 | K2CO3 | DMF | r.t. | 12 | NRf |
| 6. | TsN3 | K2CO3 | DMSO | 80 °C | 12 | 20 |
| 7. | TsN3 | K2CO3 | ACN, MeOH, THF, toluene, DCE | 80 °C | 24 | NRf |
| 8. | TsN3 | NaHCO3 | DMF | 80 °C | 12 | Trace |
| 9. | TsN3 | DBU, DABCO, TEA | DMF | 80 °C | 24 | NRf |
| 10. | DPPA | K2CO3 | DMF | 80 °C | 3 | 70 |
| 11. | Nosyl azide | K2CO3 | DMF | 80 °C | 4 | 65 |
| 12. | MSN3 | K2CO3 | DMF | 80 °C | 12 | 45 |
| 13. | TMSN3 | K2CO3 | DMF | 80 °C | 12 | NRf |
Having optimized the reaction conditions, we examined the one-pot conversion of different ortho-iodobenzoic acids into the corresponding anilines and the results are portrayed in Scheme 4. Comparison of reactivity of 2-iodobenzoic acids 1b–1e, revealed that there is not much difference in rate of the reaction due to the presence of different electron-donating substituents, such as 5-OMe, 4,5-OMe, 3,5-OMe and 3,4,5-OMe, and the products 3b–3e were obtained in good yields. Interestingly, compounds 1f and 1g containing electron-withdrawing groups 5-Br and 5-NO2 furnished anilines 3f and 3g, respectively, in very good yields, but the rate of the reaction is slow compared to when electron-donating substituents are present. However, in the case of meta- and para-iodo substituted benzoic acid 1h and 1i no reaction was observed. When the reaction was tried with a halogenated benzoic acid, such as 2-bromobenzoic acid (1j), 2-chlorobenzoic acid (1k) and pentafluorobenzoic acid (1l), no conversion took place. This shows that halo substituents other than 2-iodo are ineffective.
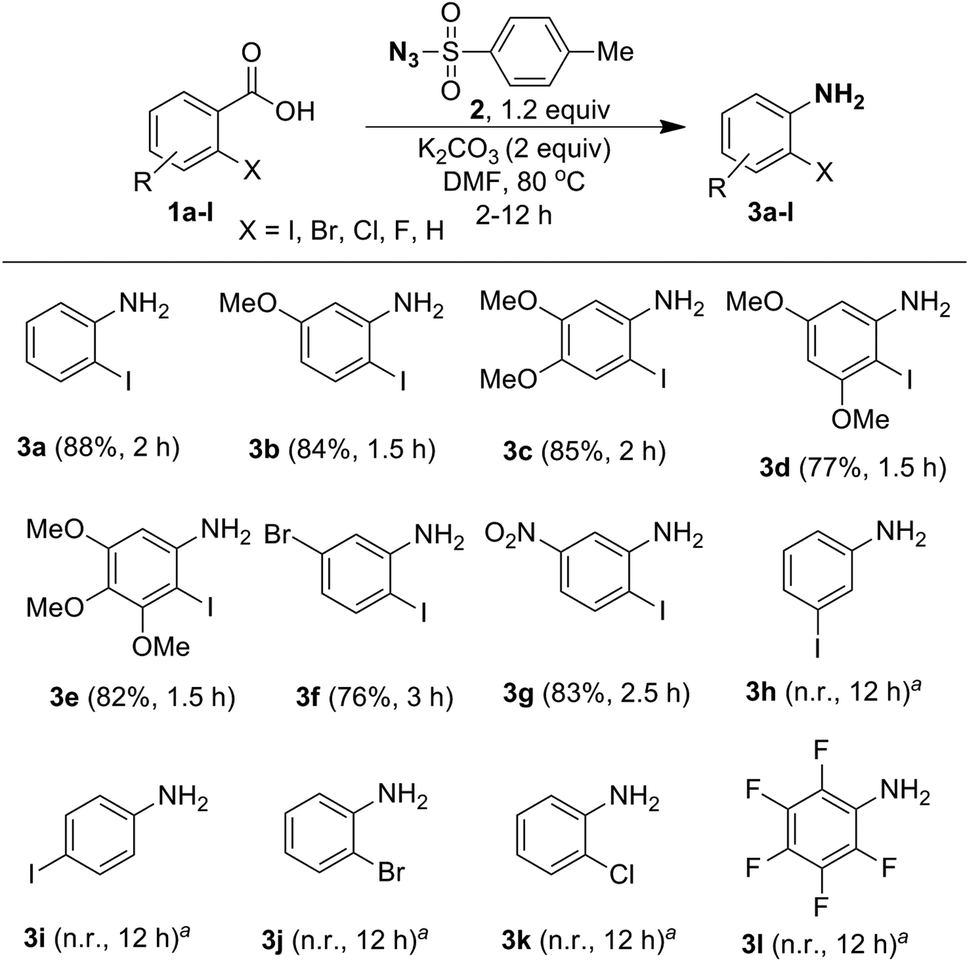 | ||
Scheme 4 One-pot synthesis of substituted ortho-iodoanilines from ortho-iodobenzoic acids using tosyl azide. Reaction conditions: halogenated benzoic acid 1 (1.0 mmol), tosyl azide (2, 1.2 mmol), K2CO3 (2.0 mmol), DMF (4 mL) at 80 °C; isolated yield; a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) no reaction. no reaction. | ||
Since ortho-nitrobenzoic acids are well known to undergo decarboxylative coupling reactions,20,21 we next explored the scope of this reaction on different ortho-nitrobenzoic acids under the optimized conditions. The results are summarized in Scheme 5. As expected, 2-nitrobenzoic acid (4a) produced aniline 5a in excellent yield. 2-Nitrobenzoic acids 4b–4d, possessing electron-donating substituents such as 5-OMe, 4,5-OMe, 3-OMe on the phenyl ring underwent facile one-pot conversion to corresponding anilines 5b–5d in high yield. In the case of methyl substitution, compared to 5-Me substituted acid 4e, the sterically more hindered 2-Me substituted acid 4f gave the corresponding aniline in excellent yield. This clearly shows that steric effect has significant influence on the rate of the reaction. For the acids 4g and 4h, containing the inductively electron-withdrawing chloro group, with substitution at the strongly influencing para position, 4h gave a complex mixture, where as 4g with substitution at the weakly influencing meta position gave product 5g albeit in low yield (25%). Similarly, the acid 4i with the strong electron-withdrawing 4-NO2 group produced only a complex mixture of products. This reveals that electron-withdrawing substituents do not favor the reaction.
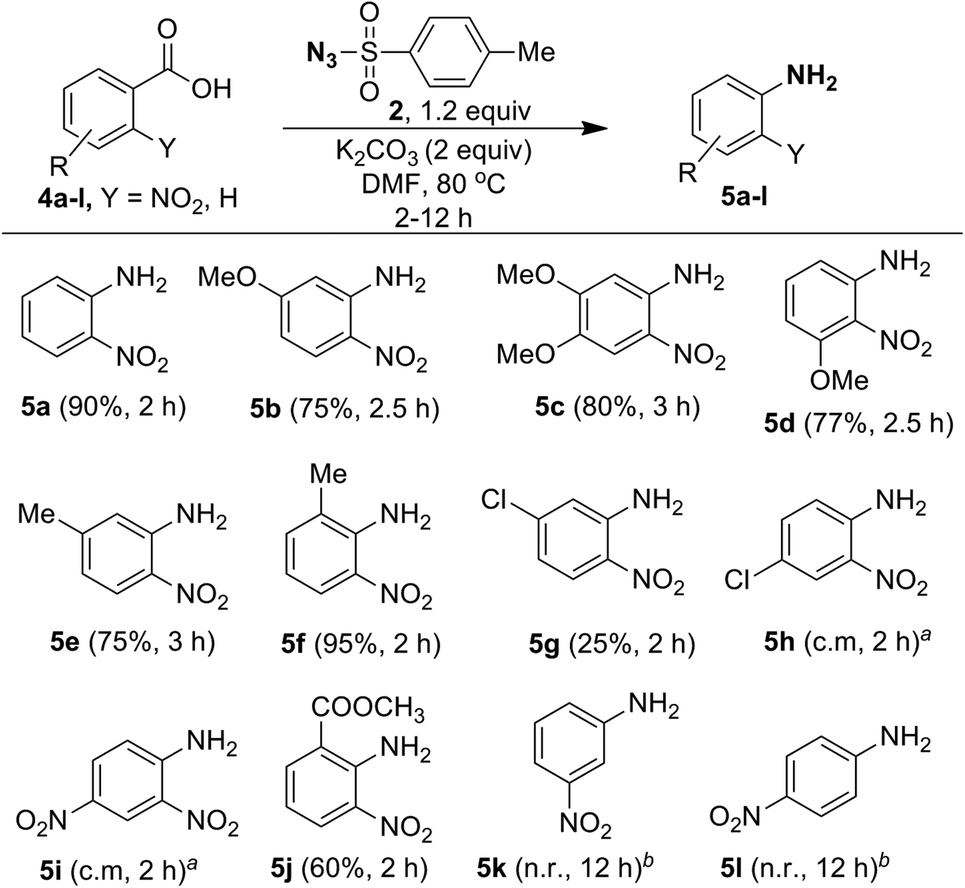 | ||
| Scheme 5 One-pot synthesis of substituted ortho-nitro anilines from ortho-nitrobenzoic acids using tosyl azide. Reaction conditions: nitrobenzoic acid 4 (1.0 mmol), tosyl azide (2, 1.2 mmol), K2CO3 (2.0 mmol), DMF (4 mL) at 80 °C; isolated yield; anot determined. Complex mixture was obtained. bNo reaction. | ||
Interestingly, although compound 4j contains the moderately electron-withdrawing 2-COOMe group, the corresponding aniline 5j was obtained in moderate yield (60%). This result, as well as results with acids 4e and 4f, shows that the steric influence is much more important than the electronic effect for this reaction. The reaction failed in the case of acids 4k (3-NO2) and 4l (4-NO2) and this reveals that ortho substitution of nitro groups is essential for the reaction to take place. It is useful to note that the methods reported for decarboxylative C–N bond formation of ortho-nitrobenzoic acids using CuCl221a or CuSO421b provides amides or secondary anilines. However, the primary anilines obtained under the present study are open for the preparation of many derivatives in addition to amide or secondary anilines.
As a part of the study we next examined conversion of pyranochromanone acids and related compounds into the corresponding aniline derivatives under the optimized conditions (Scheme 6). Similar to pyranochromanone acid 6a, substrates 6b and 6c with fused dihydropyanone ring provided the corresponding anilines tc-7b and 7c in high yield. Likewise, the O-prenyl group in acid 6d tolerated the reaction conditions well to provide aniline t-7d in good yield. The starting materials 6a–d, were taken as racemic mixture of cis and trans disatereomers and the corresponding products tc-7a (trans:cis = 55:45) and tc-7b (trans:cis = 87:13) were obtained as racemic mixture of cis and trans disatereomers. The product t-7d was obtained as a racemic mixture of trans diastereomer only. However, acid 6e, without any fused ring system, failed. It is important to note that acids 7a–d are structurally different from 2-iodo/2-nitrobenzoic acids, wherein the ortho substitution is not bulky but the reaction took place. As described by others25 we believe that the fusion of the dihydropyranone ring system would affect co-planarity of the benzene ring particularly during the decarboxylation process. Further, these results infer that for the desired conversion to take place at least one ring should have been fused to the benzoic acid ring.
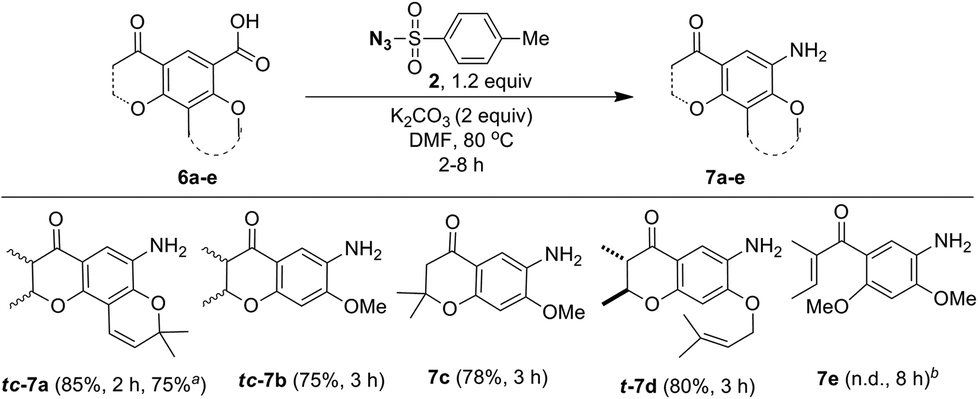 | ||
| Scheme 6 One-pot synthesis of pyranone fused anilines from pyranone fused benzoic acids using tosyl azide. Reaction conditions: substituted benzoic acid 6 (1.0 mmol), tosyl azide (2, 1.2 mmol), K2CO3 (2.0 mmol), DMF (4 mL) at 80 °C; isolate yield; areaction carried out using DPPA, Et3N, t-BuOH, 85 °C, 3 h and then TFA, DCM, 0 °C–r.t., 1 h; bnot determined. Complex mixture was obtained. tc- denotes diastereomeric mixtures of trans and cis isomers and t- denotes only trans diastereomer. | ||
Further, in order to establish a practical utility of the present method, a gram-scale experiment was carried, under the optimal reaction conditions (Scheme 7). Starting from acids 1a (5.0 g), 4a (5.0 g) and 6a (5.0 g), the desired anilines 3a (3.86 g), 5a (3.72 g) and 7a, (3.84 g) were obtained in 88%, 90% and 85% isolated yields, respectively. This is notable for both the scalability and environmental compatibility of the present method.
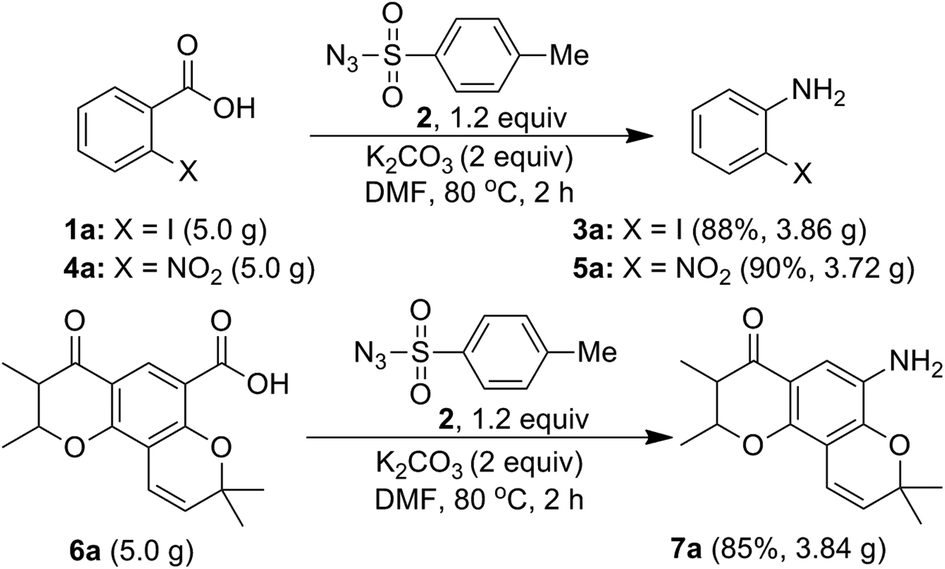 | ||
| Scheme 7 Gram-scale synthesis of substituted anilines 3a, 5a and 7a. | ||
Further, we investigated the scope of this reaction with different substituted benzoic acids 9–15, and aliphatic carboxylic acids 16–18 (Fig. 1). However, all these substrates failed to undergo reaction under the optimized conditions.
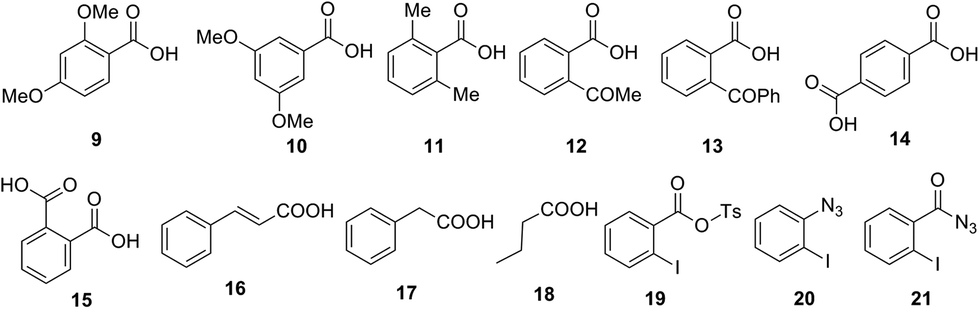 | ||
| Fig. 1 Carboxylic acid failed to undergo one-pot conversion to amine using tosyl azide and other possible intermediates. | ||
Next, we examined mechanistic aspects. To check the possibility of formation of isocynate intermediate, same as Lebel et al. (Scheme 2, eqn (1))17 we tried to trap it by adding diphenyl acetylene (22) during conversion of acid 1a to aniline 3a (Scheme 8). However, no indole 23 was formed, which confirmed the absence of a Curtius rearrangement pathway. Further none of the possible intermediates such as mixed anhydride 19, aryl azide 20 and acyl azide 21 expected through different ways of reaction between carboxylate and TsN3, could be detected in the reaction mixture (Fig. 1).
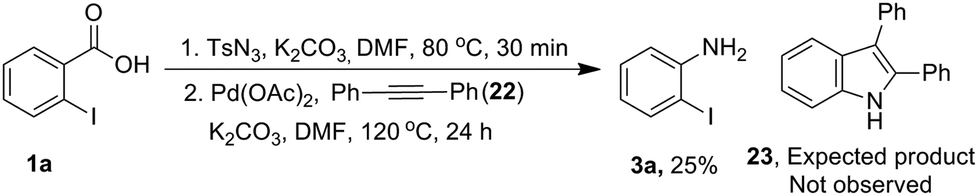 | ||
| Scheme 8 Attempted trapping of isocyanate intermediate. | ||
The foregoing discussion reveals that for the decarboxylative C–N bond formation to take place 2-iodo or 2-nitro substitution or fusion of dihydropyranone ring to benzoic acid is essential and steric effect has a significant influence on the decarboxylation. The electron-withdrawing nature and mainly the steric bulkiness of the 2-iodo or 2-nitro group may help destabilization co-planarity of the benzene as well as stabilization of the negative charge generated during the decarboxylation. Formation of the product 5f in high yield, compared product 5e highlights the importance of steric hindrance for the effective completion of the reaction. Unlike the reports from others21 using CuSO4 and CuCl2, conversion of 2-nitrobenzoic acid was observed by us without using a transition metal catalyst. This suggests that the reaction might take place through a novel pathway.
Thus we considered the involvement of tosyl nitrene.26a,b All the azides studied TsN3, DPPA, nosyl azide, MsN3 (Table 1, entries 2 and 10–12) are known to undergo thermal decomposition to form nitrene26 and provided anilines. However, TMSN3 which can't form nitrene at ≤80 °C, failed to provide aniline (Table 1, entry 13). As with carbenes the fate of nitrenes is also dependent on the solvent26a and in the present study except DMF and DMSO all other solvents failed the reaction. Also during the gram scale experiment we observed the bubble formation due to the liberation of nitrogen and CO2 gas. This confirms the involvement of tosyl nitrene in the reaction. Thus, we have observed for the first time nitrene undergoing ipso attack on the benzene ring. Based on these observations a suitable reaction mechanism is proposed as shown in Scheme 9.
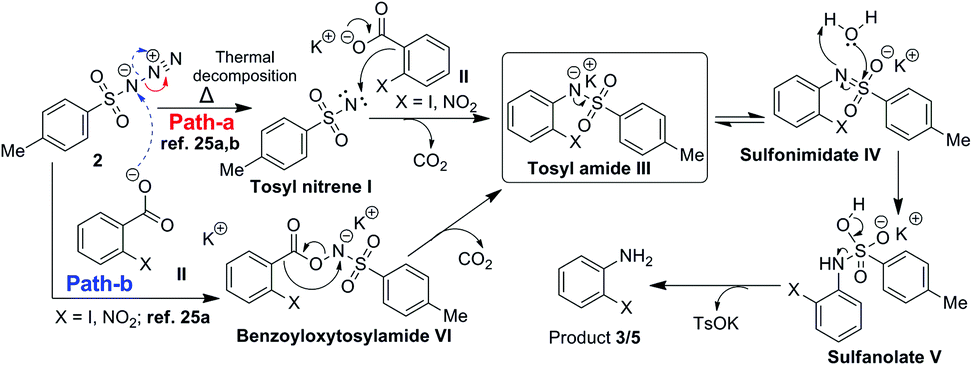 | ||
| Scheme 9 Plausible mechanism. | ||
Nitrene I formed by thermal decomposition of TsN3 (path-a), is expected undergo decarboxylative addition to potassium carboxylate II to form tosylamide III which on tautomerisation and hydrolysis should give rise to aniline 3 or 5. It is also likely that carboxylate II may also undergo reaction directly with TsN3 to form benzoyloxytosylamide VI which in turn is expected to form tosylamide III.
Conclusion
In conclusion, we have developed a useful method for one-pot conversion of 2-iodo/2-nitrobenzoic acids and pyranochromanone acids into the corresponding anilines using tosyl azide under transition metal-free conditions. 2-Iodobenzoic acids were found to undergo decarboxylative C–N coupling for the first time. This method needs only simple reagents, is environmentally benign, adoptable for large scale preparation of anilines due to the fact that TsN3 is one of the more stable azides, is much cheaper and easy to handle. Both steric and electronic effects due to 2-iodo/2-nitro substituents contribute to the success of this reaction, and the electronic effect alone by iodo or nitro substituent was found not effective. In the case of pyranochromanone acids the effect of conformational constraints of the fused dihydropyranone ring on co-planarity of the benzene ring may help the decarboxylation to take place easily. While electron-donating additional substituents improved the yield of anilines electron-withdrawing substituents decreased the yield. Mechanistic insights into this reaction shows that aromatic acids undergo ipso substitution with tosyl nitrene. Based on these results we are currently examining the reactivity of TsN3 with a different class of compounds and the results will be communicated separately.Acknowledgements
P. S. thanks UGC-RFSMS, New Delhi for the award of fellowship. We thank DST-FIST, New Delhi, India for NMR facilities at School of Chemistry, Bharathidasan University, Tiruchirappalli, India.References
- (a) B. Schlummer and U. Scholz, Adv. Synth. Catal., 2004, 346, 1599 CrossRef CAS; (b) D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338 CrossRef CAS PubMed; (c) S. Tasler and B. H. Lipshutz, J. Org. Chem., 2003, 68, 1190 CrossRef CAS PubMed; (d) A. Ricci, Amino group chemistry: from synthesis to the life sciences, John Wiley & Sons, New York, 2008 Search PubMed; (e) J. Seifert, H. Mostecká and G. Kolar, Toxicology, 1993, 83, 49 CrossRef CAS PubMed; (f) P. Demare and I. Regla, J. Chem. Educ., 2012, 89, 147 CrossRef CAS.
- (a) B. Melzer, A. Plodek and F. Bracher, J. Org. Chem., 2014, 79, 7239 CrossRef CAS PubMed; (b) J. P. Mahajan, Y. R. Suryawanshi and S. B. Mhaske, Org. Lett., 2012, 14, 5804 CrossRef CAS PubMed; (c) A. J. McCarroll, T. D. Bradshaw, A. D. Westwell, C. S. Matthews and M. F. Stevens, J. Med. Chem., 2007, 50, 1707 CrossRef CAS PubMed; (d) Y. Kim, M. R. Kumar, N. Park, Y. Heo and S. Lee, J. Org. Chem., 2011, 76, 9577 CrossRef CAS PubMed; (e) L. Åkerbladh and L. R. Odell, J. Org. Chem., 2016, 81, 2966 CrossRef PubMed; (f) B. Miao, S. Li, G. Li and S. Ma, Org. Lett., 2016, 18, 2556 CrossRef CAS PubMed.
- (a) R. B. Miller and J. G. Stowell, J. Org. Chem., 1983, 48, 886 CrossRef CAS; (b) S. Oh, S. Kim, S. Kong, G. Yang, N. Lee, D. Han, J. Goo, J. L. Siqueira-Neto, L. H. Freitas-Junior and R. Song, Eur. J. Med. Chem., 2014, 84, 395 CrossRef CAS PubMed; (c) M. P. Hay, S. A. Gamage, M. S. Kovacs, F. B. Pruijn, R. F. Anderson, A. V. Patterson, W. R. Wilson, J. M. Brown and W. A. Denny, J. Med. Chem., 2003, 46, 169 CrossRef CAS PubMed; (d) M. Baumann, I. R. Baxendale, C. H. Hornung, S. V. Ley, M. V. Rojo and K. A. Roper, Molecules, 2014, 19, 9736 CrossRef PubMed; (e) D. B. Nale and B. M. Bhanage, Green Chem., 2015, 17, 2480 RSC; (f) F. Xie, M. Zhang, H. Jiang, M. Chen, W. Lv, A. Zheng and X. Jian, Green Chem., 2015, 17, 279 RSC.
- (a) C. Wang, T. Feng and B. E. Christensen, J. Am. Chem. Soc., 1950, 72, 4887 CrossRef CAS; (b) H. U. Blaser, H. Steiner and M. Studer, ChemCatChem, 2009, 1, 210 CrossRef CAS.
- (a) F. Terrier, Modern Nucleophilic Aromatic Substitution, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2013, p. 337 Search PubMed; (b) V. Khutorianskyi, M. Sonawane, M. Pošta, B. Klepetářová and P. Beier, Chem. Commun., 2016, 52, 7237 RSC.
- (a) H. Neumann, A. Jacobi von Wangelin, S. Klaus, D. Strübing, D. Gördes and M. Beller, Angew. Chem., Int. Ed., 2003, 42, 4503 CrossRef CAS PubMed; (b) W. P. Hong, A. V. Iosub and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 13664 CrossRef CAS PubMed.
- (a) G. D. Vo and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 11049 CrossRef CAS PubMed; (b) D. S. Raghuvanshi, A. K. Gupta and K. N. Singh, Org. Lett., 2012, 14, 4326 CrossRef CAS PubMed; (c) C. Wolf, S. Liu, X. Mei, A. T. August and M. D. Casimir, J. Org. Chem., 2006, 71, 3270 CrossRef CAS PubMed; (d) J. Jiao, X.-R. Zhang, N.-H. Chang, J. Wang, J.-F. Wei, X.-Y. Shi and Z.-G. Chen, J. Org. Chem., 2011, 76, 1180 CrossRef CAS PubMed.
- (a) C. Zhu, G. Li, D. H. Ess, J. R. Falck and L. s. Kürti, J. Am. Chem. Soc., 2012, 134, 18253 CrossRef CAS PubMed; (b) N. Chatterjee and A. Goswami, Org. Biomol. Chem., 2015, 13, 7940 RSC; (c) S. Voth, J. W. Hollett and J. A. McCubbin, J. Org. Chem., 2015, 80, 2545 CrossRef CAS PubMed.
- (a) E. C. Franklin, Chem. Rev., 1934, 14, 219 CrossRef CAS; (b) H. Lebel and O. Leogane, Org. Lett., 2006, 8, 5717 CrossRef CAS PubMed.
- (a) A. Wrobleski, T. C. Coombs, C. W. Huh, S. W. Li and J. Aubé, Org. React., 2012, 78, 1 CAS; (b) X.-J. Li, J.-B. Qiao, J. Sun, X.-Q. Li and P. Gu, Org. Lett., 2014, 16, 2865 CrossRef CAS PubMed.
- (a) A. Yoshimura, M. W. Luedtke and V. V. Zhdankin, J. Org. Chem., 2012, 77, 2087 CrossRef CAS PubMed; (b) J. W. Keillor and X. Huang, Org. Synth., 2002, 234 CAS.
- (a) P. Dubé, N. F. F. Nathel, M. Vetelino, M. Couturier, C. L. Aboussafy, S. Pichette, M. L. Jorgensen and M. Hardink, Org. Lett., 2009, 11, 5622 CrossRef PubMed; (b) Y. Hoshino, M. Okuno, E. Kawamura, K. Honda and S. Inoue, Chem. Commun., 2009, 2281 RSC.
- (a) C. Dengiz, S. Özcan, E. Şahin and M. Balci, Synthesis, 2010, 1365 CAS; (b) R. Belaroussi, A. El Bouakher, M. Marchivie, S. Massip, C. Jarry, A. El Hakmaoui, G. Guillaumet, S. Routier and M. Akssira, Synthesis, 2013, 2557 CAS; (c) V. V. Sureshbabu, H. Lalithamba, N. Narendra and H. Hemantha, Org. Biomol. Chem., 2010, 8, 835 RSC.
- (a) H. Liang, Synlett, 2008, 2554 CrossRef CAS; (b) A. Flores, M. J. Camarasa, M. J. Pérez-Pérez, A. San-Félix, J. Balzarini and E. Quesada, Org. Biomol. Chem., 2014, 12, 5278 RSC; (c) S. Sunami and M. Ohkubo, Tetrahedron, 2009, 65, 638 CrossRef CAS; (d) Y. Lu and R. T. Taylor, Tetrahedron Lett., 2003, 44, 9267 CrossRef CAS.
- (a) Y. Shinomoto, A. Yoshimura, H. Shimizu, M. Yamazaki, V. V. Zhdankin and A. Saito, Org. Lett., 2015, 17, 5212 CrossRef CAS PubMed; (b) L. Marinescu, J. Thinggaard, I. B. Thomsen and M. Bols, J. Org. Chem., 2003, 68, 9453 CrossRef CAS PubMed.
- E. Ivanova, D. Borovika, I. Vozny, P. Trapencieris and R. Žalubovskis, Chem. Heterocycl. Compd., 2012, 48, 1114 CrossRef CAS.
- O. Leogane and H. Lebel, Angew. Chem., Int. Ed., 2008, 47, 350 CrossRef CAS PubMed.
- (a) L. J. Gooßen, N. Rodriguez and K. Gooßen, Angew. Chem., Int. Ed., 2008, 47, 3100 CrossRef PubMed; (b) N. Rodríguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030 RSC; (c) C. Shen, P. Zhang, Q. Sun, S. Bai, T. S. A. Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291 RSC; (d) Z. Fu, Z. Li, Y. Song, R. Yang, Y. Liu and H. Cai, J. Org. Chem., 2016, 81, 2794 CrossRef CAS PubMed; (e) G. Hu, Y. Gao and Y. Zhao, Org. Lett., 2014, 16, 4464 CrossRef CAS PubMed.
- (a) L. J. Gooßen, G. Deng and L. M. Levy, Science, 2006, 313, 662 CrossRef PubMed; (b) L. J. Goossen, N. Rodríguez, B. Melzer, C. Linder, G. Deng and L. M. Levy, J. Am. Chem. Soc., 2007, 129, 4824 CrossRef CAS PubMed; (c) L. J. Gooßen, B. Zimmermann and T. Knauber, Angew. Chem., Int. Ed., 2008, 47, 7103 CrossRef PubMed; (d) Y. Luo, X. Pan and J. Wu, Tetrahedron Lett., 2010, 51, 6646 CrossRef CAS; (e) L. J. Gooßen, B. Zimmermann and T. Knauber, Beilstein J. Org. Chem., 2010, 6, 43 Search PubMed.
- (a) W. Jia and N. Jiao, Org. Lett., 2010, 12, 2000 CrossRef CAS PubMed; (b) Y. Zhu, X. Li, X. Wang, X. Huang, T. Shen, Y. Zhang, X. Sun, M. Zou, S. Song and N. Jiao, Org. Lett., 2015, 17, 4702 CrossRef CAS PubMed; (c) C. Liu, X. Wang, Z. Li, L. Cui and C. Li, J. Am. Chem. Soc., 2015, 137, 9820 CrossRef CAS PubMed; (d) K. Kiyokawa, T. Kojima, Y. Hishikawa and S. Minakata, Chem. – Eur. J., 2015, 21, 15548 CrossRef CAS PubMed.
- (a) Y. Zhang, S. Patel and N. Mainolfi, Chem. Sci., 2012, 3, 3196 RSC; (b) W.-J. Sheng, Q. Ye, W.-B. Yu, R.-R. Liu, M. Xu, J.-R. Gao and Y.-X. Jia, Tetrahedron Lett., 2015, 56, 599 CrossRef CAS.
- (a) Y.-W. Zheng, B. Chen, P. Ye, K. Feng, W. Wang, Q.-y. Meng, L.-Z. Wu and C.-H. Tung, J. Am. Chem. Soc., 2016, 138, 10080 CrossRef CAS PubMed; (b) J. Peng, M. Chen, Z. Xie, S. Luo and Q. Zhu, Org. Chem. Front., 2014, 1, 777 RSC; (c) J. Peng, Z. Xie, M. Chen, J. Wang and Q. Zhu, Org. Lett., 2014, 16, 4702 CrossRef CAS PubMed; (d) P. Patel and S. Chang, ACS Catal., 2015, 5, 853 CrossRef CAS; (e) H. Kim, G. Park, J. Park and S. Chang, ACS Catal., 2016, 6, 5922 CrossRef CAS.
- P. Sakthivel, A. Ilangovan and M. P. Kaushik, Eur. J. Med. Chem., 2016, 122, 302 CrossRef CAS PubMed.
- (a) D. Lee and S. Chang, Chem. – Eur. J., 2015, 21, 5364 CrossRef CAS PubMed and references cited therein.
- (a) R. Grainger, J. Cornella, D. C. Blakemore, I. Larrosa and J. M. Campanera, Chem. – Eur. J., 2014, 20, 16680 CrossRef CAS PubMed; (b) M. Zabalov and R. Tiger, Russ. Chem. Bull., 2007, 56, 7 CrossRef CAS.
- (a) R. Abramovitch and R. Sutherland, Reactives Intermediates, Springer, Berlin, Heidelberg, 1970 Search PubMed; (b) B. A. Shainyan and A. V. Kuzmin, J. Phys. Org. Chem., 2014, 27, 156 CrossRef CAS; (c) G. Y. Gao, J. E. Jones, R. Vyas, J. D. Harden and X. P. Zhang, J. Org. Chem., 2006, 71, 6655 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, spectral data and copies of spectra. See DOI: 10.1039/c6qo00343e |
| This journal is © the Partner Organisations 2016 |