
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A triblock terpolymer vs. blends of diblock copolymers for nanocapsules addressed by three independent stimuli†
Shahed
Behzadi
a,
Markus
Gallei
*b,
Johannes
Elbert‡
b,
Michael
Appold
b,
Gunnar
Glasser
a,
Katharina
Landfester
a and
Daniel
Crespy
*ac
aMax Planck Institute for Polymer Research, D-55128 Mainz, Germany. E-mail: crespy@mpip-mainz.mpg.de
bMacromolecular Chemistry Department, Technische Universität Darmstadt, D-64287 Darmstadt, Germany. E-mail: M.Gallei@mc.tu-darmstadt.de
cVidyasirimedhi Institute of Science and Technology, 555 Moo 1 Payupnai, Wangchan, Rayong 21210, Thailand
First published on 14th April 2016
Abstract
Stimuli-responsive micro- to nano-scale containers have gained increasing attention due to their unique potential to selectively release payloads under specific environmental conditions. We report here novel triple stimuli-responsive nanocapsules that selectively respond to changes in temperature, pH value, and redox potential. The nanocapsules were prepared from either a triple responsive triblock terpolymer or a blend of responsive diblock copolymers, both synthesized by sequential anionic polymerization. We then compared the release performance of nanocapsules under oxidative conditions and changes of temperature or pH value. Our results reveal the close correlation between the release properties of stimuli-responsive nanocontainers and the microstructure of the polymer shell. In fact, the microphase separation between the responsive diblock copolymers across the shell significantly hinders the triggered release of the payload from the nanocapsules. These results demonstrate that the fine morphology of triblock terpolymers can be exploited to achieve the triggered release of payloads from polymer nanocontainers upon application of three different external triggers.
Introduction
The control of the transport of molecules and ions through a membrane is crucial in release systems applied for biomedicine and anticorrosion. Polymer micro- and nano-scale containers are release systems that allow for the encapsulation of large amount of payloads, their protection from the surrounding environment, and control of their release profiles. Nevertheless, artificial membranes are far less advanced compared to natural membranes and therefore research on controlled release of molecules through artificial membranes is necessary. The triggered release of payloads from containers in response to external stimuli has sparked considerable interest among researchers since they have the potential for many different applications.1–8 Tremendous efforts have been devoted to obtain a triggered release by modifying the physiochemical properties of the shell. These efforts include the use of stimuli-responsive functional groups that display sharp transitions of properties to certain stimuli from an external environment. Miniature containers consisting of stimulus-responsive shells are promising for applications such as biomedicine,9–13 anticorrosion,14,15 sensors,16 and water purification.17 Most of these studies are focused on one trigger to release payloads. However, practical applications occur usually in complex environments. In some cases, containers responsive to multiple stimuli are necessary for selective release of several payloads. An example is combination therapy, where multiple medications are needed to fight one disease through the synergetic effect of drugs.18,19 In other cases, pulsatile release of payloads from miniature containers is required, namely for treating chronic diseases to relieve patients’ pain caused by frequent injections.Block copolymers, consisting of two or more polymer segments that are covalently connected, feature the intrinsic capability to undergo microphase separation that yields fascinating structures in bulk or selective solvents.20,21 For example, multi-compartmentalized vesicles, polygonal bilayer sheets and wormlike micelles were synthesized from ABC triblock terpolymers and their self-assembled structures were studied in water.22–24 Furthermore, a variety of interesting stimuli-responsive containers have been generated by exploiting block copolymers as shell-forming materials.25–39 For instance, pH/temperature- and pH/redox-responsive nanocapsules (NCs) were synthesized from diblock copolymers and the selective release of two different payloads from NCs has been studied.34 In another study, triple-responsive diblock polymer vesicles were synthesized with multiple biomolecular-layer structures through self-assembly.38 However, there are still major challenges associated with these studies, including non-controlled leakage of the encapsulated substance (∼30%) and relatively low efficiency for triggered release in response to voltage (∼45%) and temperature changes (∼35%). Triple-responsive micelles synthesized from diblock copolymers are other successful examples for multi stimuli-responsive materials.27,33 A fascinating example for multi-stimuli-responsive micelles is the micelle formation by triple-responsive disulfide linked diblock copolymers.27 Another one is the triple-stimuli micelle synthesized by self-assembly of amphiphilic diblock copolymers in water.36
However in these examples, the micelles do not encapsulate a liquid core as it is the case for polymer nanocapsules. Bearing in mind that at least one polymer needs to remain hydrophobic to hold the shape of the polymer nanocontainer. Recently, vesicles of ferrocene-containing triblock terpolymers were synthesized and their redox-responsive release was studied.40 Although the hydrophobic segment of the triblock terpolymer guarantees the structural integrity of these vesicles after oxidation, there was only one addressable segment present, i.e. the ferrocene containing segment.40 Triple-responsive nanocontainers can be in principle fabricated using three responsive homopolymers, two dual-responsive diblock copolymers, or a triple-responsive triblock terpolymer. We can anticipate that the preparation of triple-responsive containers with a blend of stimulus-responsive homopolymers will most probably result in phase separation inside the shell. The buried homopolymers will therefore not be addressable by one of the external stimuli applied to the nanocontainers. Another disadvantage of using a blend of stimulus-responsive homopolymers for the preparation of containers is the decrease of container stability due to the absence of covalent linkages between different compartments. To overcome this issue, we propose to use a triple responsive triblock terpolymer to build the shell of nanocapsules. To the best of our knowledge, no triple-responsive nanocontainers or nanoparticles consisting of a triple responsive triblock terpolymer have been reported to date. Alternatively, we compare the performance of such nanocapsules with nanocapsules prepared with a blend of responsive diblock copolymers. In both types of samples, we synthesized the polymer so that the shells have exactly the same chemical composition in terms of the molar amount of the block segments. To fulfil this goal, the triblock poly(vinylferrocene)-b-poly(methyl methacrylate)-b-poly(N,N-dimethylaminoethyl methacrylate) (PVFc-b-PMMA-b-PDMAEMA) and blends of the corresponding diblock copolymers of PVFc-b-PMMA and PDMAEMA-b-PMMA were synthesized. Herein, PVFc-b-PMMA-b-PDMAEMA is presented as the first triple-responsive shell material that combines responsivity to oxidation and changes in pH value and temperature. The copolymers were used to fabricate nanocapsules and the triggered release of the encapsulated payload upon pH change, redox reagent, and temperature was investigated. A detailed insight into the correlation between the release properties and shell microstructure of the NCs was obtained using a combination of characterization methods.
Results and discussion
Preparation and characterization of triblock terpolymer nanocapsules
The triblock terpolymers (see Fig. 1 and the Experimental section (Table 3)) were used for the preparation of NCs by the solvent evaporation process from miniemulsion droplets.41 This technique has been successfully applied for synthesis of NCs from different polymers including functional block copolymers42 and for the encapsulation of various payloads.43 The final morphology of a colloidal system consisting of different non-miscible phases strongly depends on the interfacial tension between these phases.44 A triblock terpolymer and hexadecane (a non-solvent for the copolymer) were dissolved in dichloromethane containing a small amount of Nile-Red (NR) and then mixed with a basic aqueous solution of cetyltrimethylammmonium chloride (CTMA-Cl) by stirring and ultrasonication to form a miniemulsion (Fig. 1a). After the evaporation of dichloromethane, the terpolymer precipitated at the droplet surface and formed a shell around the liquid core composed of hexadecane. These dispersions were later used for stimuli-responsive release experiments. The TEM images showed evidence of a core–shell structure for the NCs (Fig. 1b–d). In the TEM images some NCs appeared collapsed inside the vacuum chamber of the electron microscope. The obtained NCs displayed hydrodynamic diameters between 190 and 225 nm while their polydispersity indexes were measured between 0.22 and 0.29 (Table 1) as determined by DLS measurements. The relatively high polydispersity index of the NCs is inherent to the preparation method used and not the result of droplet coalescence or aggregation.45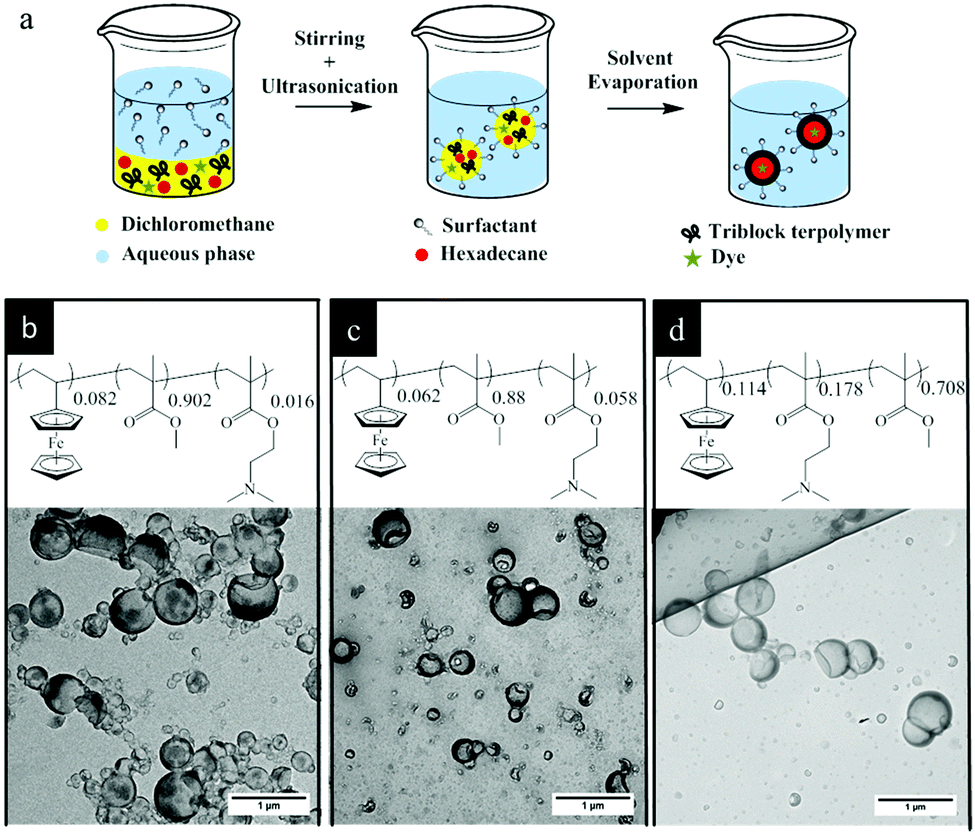 | ||
| Fig. 1 (a) Schematics for the preparation of NCs by solvent evaporation miniemulsion. TEM images of NCs prepared with triblock terpolymers PVFc8.2-b-PMMA90.2-b-PDMAEMA1.6 (b), PVFc6.2-b-PMMA88-b-PDMAEMA5.8 (c), and PVFc11.4-b-PDMAEMA17.8-b-PMMA70.8 (d). | ||
| NCs shell | Hydrodynamic radius (nm) | Polydispersity index |
|---|---|---|
| PVFc8.2-b-PMMA90.2-b-PDMAEMA1.6 | 225 | 0.29 |
| PVFc6.2-b-PMMA88-b-PDMAEMA5.8 | 193 | 0.22 |
| PVFc11.4-b-PDMAEMA17.8-b-PMMA70.8 | 190 | 0.29 |
Stimuli-responsive release of the payload from triblock terpolymer nanocapsules
The presence of a responsive unit in a shell is not a guarantee that the shell will allow for a release of payload upon activation of the shell by an external stimulus. Therefore, we used different compositions of triblock terpolymers to achieve a significant triggered release of an encapsulated dye due to the presence of different morphologies. It is expected that an increase in the length of a responsive block results in the increase of the shell responsivity to the stimuli. Therefore, triblock terpolymers with different block lengths were synthesized to determine the appropriate composition of the triblock terpolymer to yield an efficient stimulus-responsive release from the NCs. The stimuli-induced release of the payload was determined by monitoring the decrease of NR fluorescence with time in the aqueous release medium after application of stimuli. We took the advantage of the fact that the fluorescence intensity of NR decreases largely in polar media.46 The released amount of NR was calculated on the basis of the fact that the NR fluorescence intensity in an aqueous medium decreases upon application of stimuli due to the opening of NCs.47 The NC dispersions were dialyzed before applying the pH change, redox agent, and temperature to remove any residual non-encapsulated dye. It is widely accepted that PVFc can be oxidized by a variety of oxidants.48 The oxidation of the ferrocene complex to cationic iron species is associated with an increase of the polarity.49,50 This feature can be applied for the acceleration of payload release. The response of NC dispersions was determined by addition of H2O2 at various concentrations to the dispersions and monitoring the decrease of NR fluorescence in the aqueous release medium at t = 24 h. In general, the release of NR was more pronounced when H2O2 concentration increased from 4.4 to 35 wt% (Fig. 2a and b). Upon increasing of H2O2 concentration from 4.4 to 35 wt%, there was an observable increment in the NR release up to 54%, 85%, and 91% from the NCs with 6.2, 8.2, and 11.4 mol% of PVFc, respectively (Fig. 2a and b). At concentration of H2O2 equal to 4.4 wt%, there was a significant difference (∼30%) between the NR release from the NCs with 11.4 mol% PVFc and the NCs with 6.2 and 8.2 mol% of PVFc. This difference gradually decreased at intermediate concentrations of H2O2. Upon a further increase in H2O2 concentration to 35 wt%, there was no significant difference between the release from the NCs with 8.2 and 11.4 mol% PVFc (Fig. 2a and b). Therefore, the redox-responsive release from NCs largely depended on the ratio of the oxidizing agent/amount of responsive units in the copolymer. NCs with 11.4 mol% of PVFc displayed more response at the lowest concentration of H2O2. The significant increase in H2O2 concentration from 4.4 to 35 wt% led to an excess of H2O2 and, therefore, the difference in mol% of PVFc between 8.2 and 11.4% was no longer a controlling parameter. This explains that the same release from NCs with 8.2 and 11.4 mol% PVFc at 35 wt% H2O2 concentration was observed. However, the increase in H2O2 concentration was effective until the amount of ferrocene moieties of PVFc remains sufficient for the oxidation reaction to ferrocenium. At H2O2 concentration equal to 35 wt%, the released amount of NR from the NCs with 6.2 mol% of PVFc was ∼55% whereas it was ∼85% for the NCs with 8.2 mol% of PVFc. This significant difference implies that the small amount of ferrocene units of the NCs with 6.2 mol% of PVFc did not induce sufficient change to trigger the release of the dye. Therefore, a further increasing H2O2 concentration will not lead to a significant rise in the dye release. These findings can be explained by closed spherical-type morphology for the PVFc-domain, hampering the formation of a percolative pathway for the oxidized PVFc and therefore inducing a lack of payload release.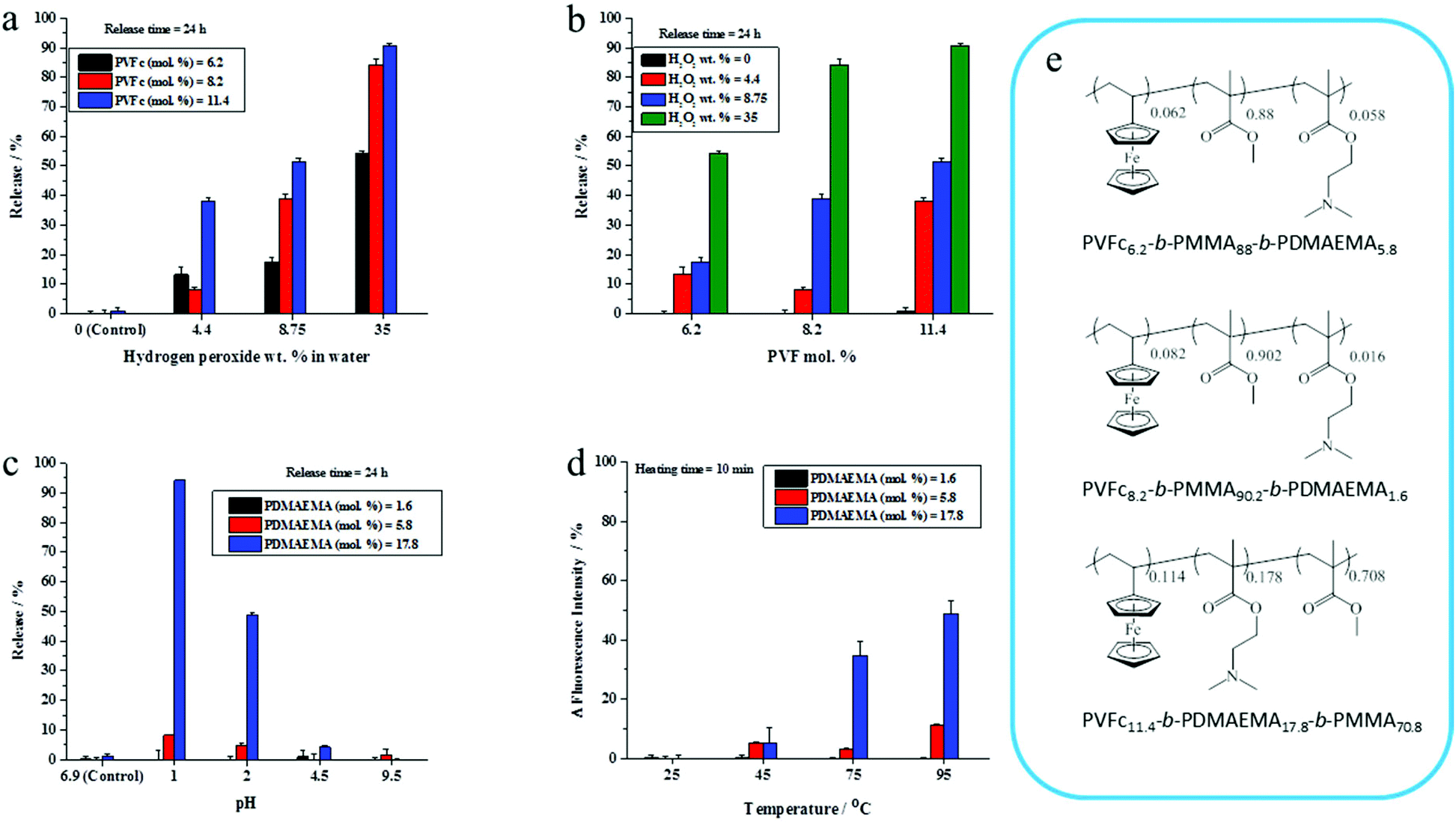 | ||
| Fig. 2 The released amount of NR out of the NCs prepared from the triblock terpolymer with different compositions at different concentrations of H2O2 (a and b), different pH values (c). Temperature response for the NCs at t = 10 min (d). (e) Chemical structure of the triblock terpolymers used for the preparation of the NCs. | ||
PDMAEMA with tertiary amine groups in their side-chain can respond to pH and temperature.51 Under acidic conditions, the tertiary amine groups are protonated and PDMAEMA (pKa ∼ 7.2) is water-soluble.52 Furthermore, PDMAEMA displays a hydrophilic-to-hydrophobic transition above its lower critical solution temperature (LCST) (∼42 °C).33 Therefore, the opening of the capsules can be triggered through a change in the shell polarity upon application of pH values or temperature changes. Dispersions of the NCs were acidified to different pH values and the shell opening was correlated with the change of NR fluorescence intensity in the aqueous release medium. NC shells containing 1.6 and 5.8 mol% of PDMAEMA did not respond significantly to acidification. Conversely, the NR fluorescence intensity for the NCs with 17.8 mol% of PDMAEMA decreased at pH values ≤ 2 compared to their control samples for release experiments, where the pH value was measured at ∼6.9, respectively (Fig. 2c). The temperature responsiveness of NCs was reported in terms of changes in NR fluorescence intensity. No effect of the temperature on the NR fluorescence intensity was observed for NC shells containing 1.6 and 5.8 mol% PDMAEMA. For NC shells containing 17.8 mol% PDMAEMA, NR fluorescence intensity increased upon heating the dispersion above 75 °C. This is attributed to the PDMAEMA chains that undergo a molecular transition from a coiled to a dense globular structure. In other words, the environment of the dye was changed to hydrophobic, which resulted in an increase of fluorescence intensity at higher temperatures (Fig. 2d). Therefore, we selected for the studies in the ensuing section NCs which were prepared from the triblock terpolymer containing PVFc and PDMEMA at molar percentages 11.4 and 17.8, respectively. Indeed, these polymers significantly responded to the pH change, redox reagent, and temperature. In the rest of the manuscript, the NCs prepared from the triblock copolymer PVFc11.4-b-PMMA70.8-b-PDMAEMA17.8 is denominated as NC-TC.
Stimuli-responsive release of the payload from nanocapsules consisting of blends of diblock copolymers
As mentioned in the introduction, triple stimuli-responsive release can also be in principle realized with a blend of two responsive diblock copolymers as the shell material. Therefore, PVFc-b-PMMA and PDMAEMA-b-PMMA were synthesized to prepare NCs in order to gain additional insights into release profiles accompanied by changes in morphology. The molar percentages of PVFc and PDMAEMA were calculated so that there were possibilities to prepare polymer shells with the same composition of shells. Furthermore, the molar amount of the different blocks was the same in the triblock terpolymer (NC-TC) and in the blend of diblock copolymers (NC-DC) (see Fig. 3 and the Experimental section (Table 3)). TEM images of sample NC-DC showed evidence of a core–shell structure (Fig. 3b and c). The presence of small darker areas was observed on the surface of sample NC-DC in the TEM micrographs (Fig. 3c) whereas they did not exist in the TEM image of sample NC-TC (Fig. 3d). The collapse of NCs due to the evaporation of the liquid core in the vacuum of SEM provides the opportunity to have a detailed insight to identify the observed small darker areas (Fig. 3e and f). These small darker areas appeared as small domains and located on the interior surface of sample NC-DC (Fig. 3e). Therefore, microphase separation in sample NC-DC is evidenced by the presence of darker areas in TEM and the solid domains in SEM micrographs. The NC-TC displayed a patchy structure morphology consisting of very small grains on the exterior surface of the NCs (Fig. 3f), which revealed the presence of heterogeneities on a nanoscale on the NC-TC surface. Additional to the observed morphology of the NC-TC, we investigated the bulk morphology of the corresponding triblock terpolymer PVFc11.4-b-PDMAEMA17.8-b-PMMA70.8 by using TEM (Fig. S1†). TEM images were obtained without further staining revealing spheres in matrix morphology. The darker appearing spherical domains can be assigned to poly(vinylferrocene).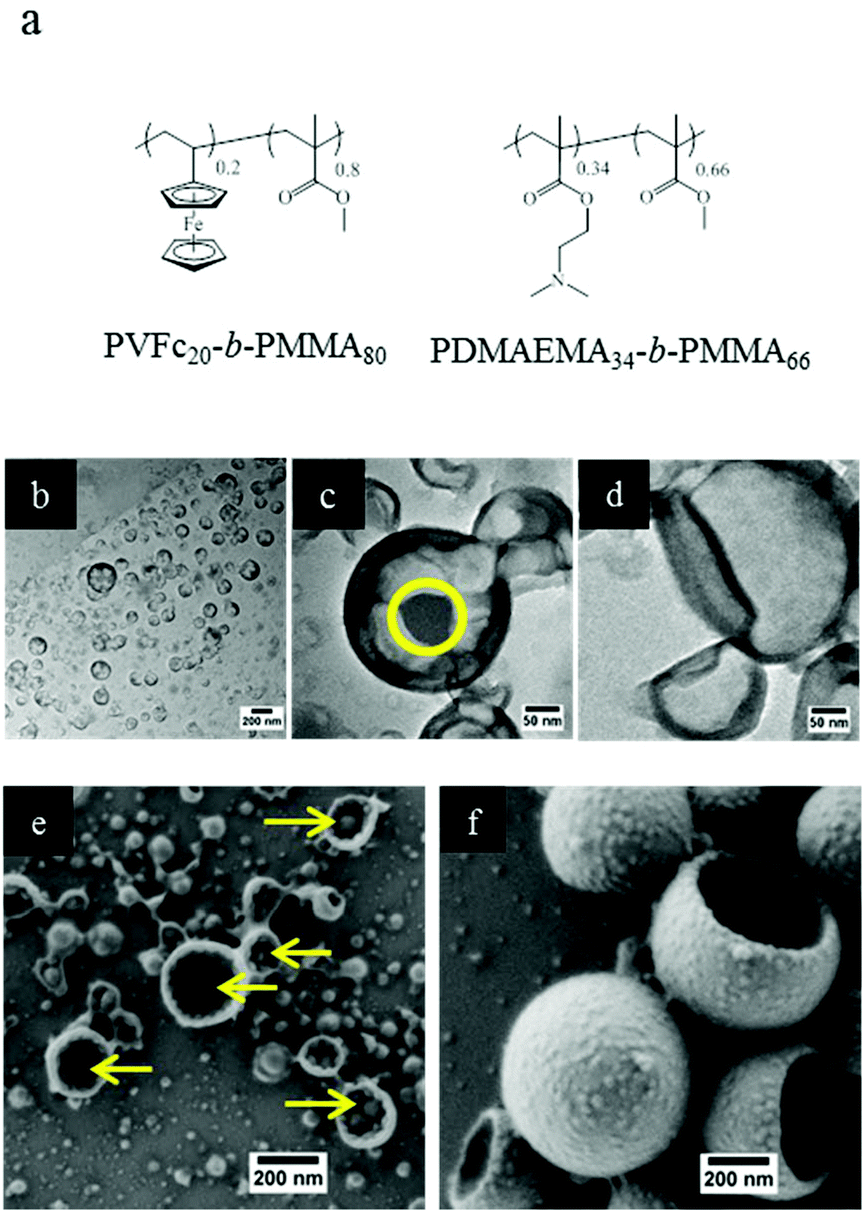 | ||
| Fig. 3 (a) Chemical structure of the diblock copolymers used for the preparation of sample NC-DC. TEM images (b and c) and SEM image (e) of the NCs prepared with the mixture of diblock copolymers, i.e. PVFc20-b-PMMA80 and PDMAEMA34-b-PMMA66. (d) TEM image and (f) SEM image of the NCs prepared from PVFc11.4-b-PDMAEMA17.8-b-PMMA70.8. Yellow circle and arrows indicate small dark areas and solid small domains in TEM and SEM images of the NCs prepared from PVFc11.4-b-PDMAEMA17.8-b-PMMA70.8. | ||
It is widely accepted that PDMAEMA accepts protons under acidic conditions and thus the polymer chains tends to expand due to coulombic repulsion.51 Thereby, one can expect an increase in the hydrodynamic radius of NCs due to gradual protonation and consequent swelling of the PDMEAMA block under acidification if this block is present on the surface of the NCs. The dispersions of both types of NCs were subjected to different pH values and the change of the hydrodynamic radius was followed (Table 2). In the case of sample NC-TC, there was no observable change in the hydrodynamic radius at pH 9.5 and 6.5 while an increase of ∼14 nm was observed at pH 4.5. The hydrodynamic radius of NC-DC exhibited a sharp increase (∼42 nm) when the pH value was decreased from 9.5 to 6.5. Upon further decrease to a pH value of 4.5, the hydrodynamic radius of NC-DC did not display a significant increase compared to their size at pH 6.5 and reached ∼169 nm. The more pronounced increase in size for NC-DC (∼46 nm) than for NC-TC (∼14 nm) upon acidification suggests that a larger number of pH-responsive units are accessible on the surface of NC-DC in comparison with the surface of NC-TC while both samples have the same shell composition. The non-responsive blocks ensured the structural integrity of the NCs by preventing dissolution of the block copolymers in the aqueous phase upon acidification. The zeta potentials of the NC dispersions were also measured at different pH values to monitor changes in the surface charge of the NCs caused by the protonation of PDMAEMA block segments on the NC surface (Table 2). At pH 6.5, the zeta potential of sample NC-TC was ∼43 mV and it slightly increased to ∼48 mV upon further decreasing the pH to 4.5. Moreover, there was no observable change in the zeta potential of sample NC-TC when the pH value increased from 6.5 to 9.5. The zeta potential of sample NC-DC was ∼36 mV at pH 9.5 while upon further decreasing the pH to 6.5 and 4.5, it increased to ∼45 and ∼50 mV, respectively. The zeta potential results imply that PDMEAMA block segments were present on the surface of NC-TC and NC-DC since the NC surface charge increased due to the PDMEAMA protonation upon acidification of the dispersions. It can also be concluded from the zeta potential measurements that the quantity of PDMEAMA block segments present on the surface of NC-TC was lower than that of NC-DC since the latter displayed more significant changes in its surface charge upon pH change while the shell has the same composition in both types of samples. Although the increment in zeta potential for sample NC-TC upon acidification is not large, this slight increase can be correlated with the investigations reported below with electron microscopy.
| Entry | Hydrodynamic radius (nm) | Zeta potential (mV) | ||||
|---|---|---|---|---|---|---|
| pH = 4.5 | pH = 6.5 | pH = 9.5 | pH = 4.5 | pH = 6.5 | pH = 9.5 | |
| NC-TC | 204 ± 122 | 190 ± 102 | 191 ± 117 | 48 ± 11 | 43 ± 5 | 43 ± 6 |
| NC-DC | 169 ± 61 | 165 ± 69 | 123 ± 48 | 50 ± 8 | 45 ± 7 | 36 ± 7 |
In order to further investigate the microstructure, we carried out selective staining on the dried nanocapsules. Indeed, the reaction between selective agents and a polymer system can enhance the electron density contrast for TEM studies. In this study, iodomethane and chlorotrimethylsilane were utilized as staining agents to demonstrate whether the NC surface contains PVFc and PDMAEMA segment blocks. Before that, PDMAEMA-b-PMMA NCs and PVFc-b-PMMA NCs were synthesized and used to determine how iodomethane and chlorotrimethylsilane change the TEM contrast of these NCs in the reaction with PVFc and PDMAEMA block segments (Fig. S2 and S3†). Upon utilization of iodomethane as a staining agent, the surface of NC-TC became dark and exhibited some black spots (Fig. 4a) whereas no significant change was observed for NC-DC (Fig. 4b). Moreover, the surface of NCs prepared from the PVFc-b-PMMA block copolymer became dark after staining with iodomethane (Fig. 4c) while the TEM micrograph of NCs prepared from the PDMAEMA-b-PMMA block copolymer did not display any contrast change (Fig. S3a†). These results imply that PVFc is present on the surface of NC-TC and not on the surface of NC-DC since iodomethane reacts with PVFc units by an oxidation reaction. Upon application of chlorotrimethylsilane, changes in the TEM contrast and morphology of NC-TC, NC-DC, and NCs prepared from the PDMAEMA-b-PMMA block copolymer were observed (Fig. 4d–f). However, there was no observable difference in the TEM micrograph of NCs prepared from the PVFc-b-PMMA block copolymer after staining with chlorotrimethylsilane (Fig. S3b†). Staining experiments with chlorotrimethylsilane suggest that PDMAEMA existed on the surface of NC-DC since chlorotrimethylsilane reacts with PDMAEMA units. One can conclude from the staining results that the shell of sample NC-DC contained two domains: the PDMEAMA-b-PMMA diblock copolymer as the exterior layer and the PVFc-b-PMMA block copolymer as the interior layer (Fig. 5).
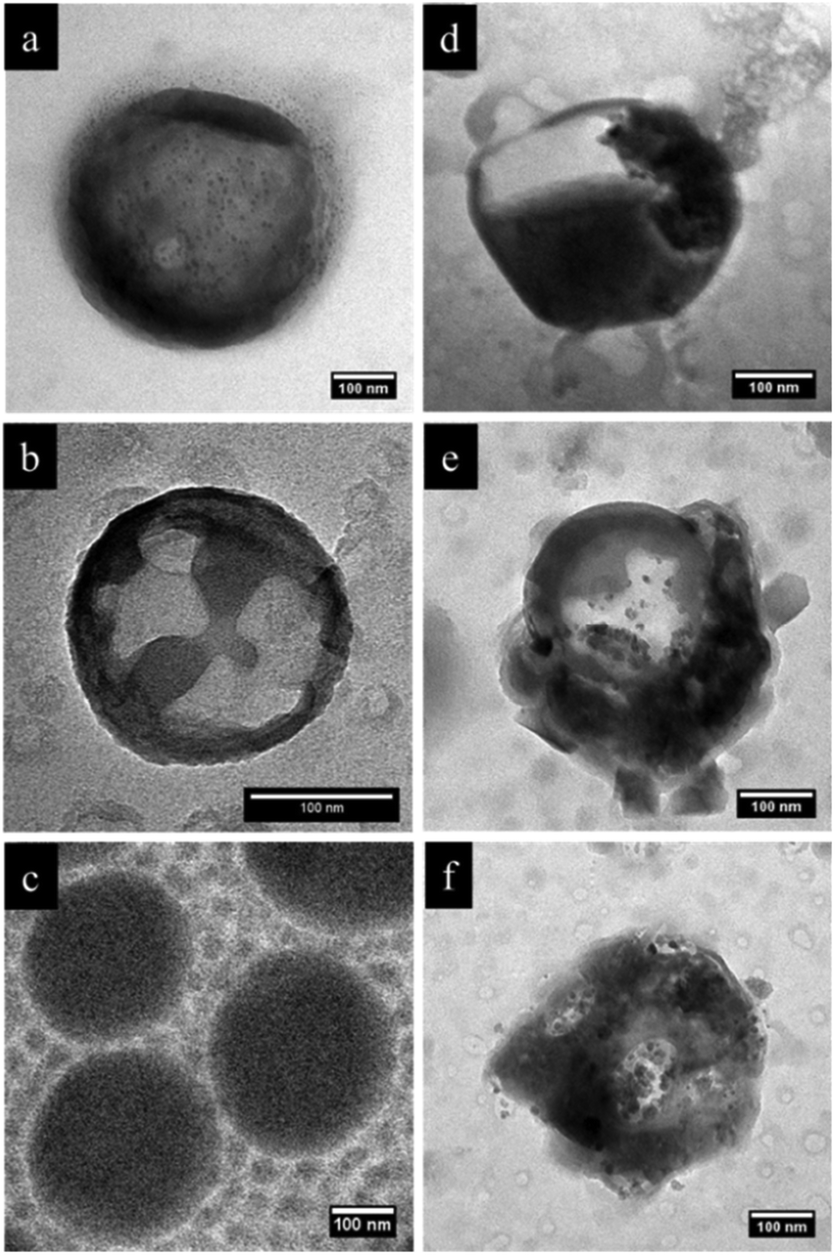 | ||
| Fig. 4 TEM images of NCs prepared from (a) the PVFc11.4-b-PDMAEMA17.8-b-PMMA70.8 triblock copolymer (sample NC-TC), (b) the mixture of PVFc20-b-PMMA80and PDMAEMA34-b-PMMA66block copolymers (sample NC-DC), and (c) the PVFc20-b-PMMA80block copolymer after selective staining experiments with iodomethane. TEM images of (d) sample NC-TC, (e) sample NC-DC, and (f) NCs prepared from the PDMAEMA34-b-PMMA66block copolymer after selective staining experiments with chlorotrimethylsilane. | ||
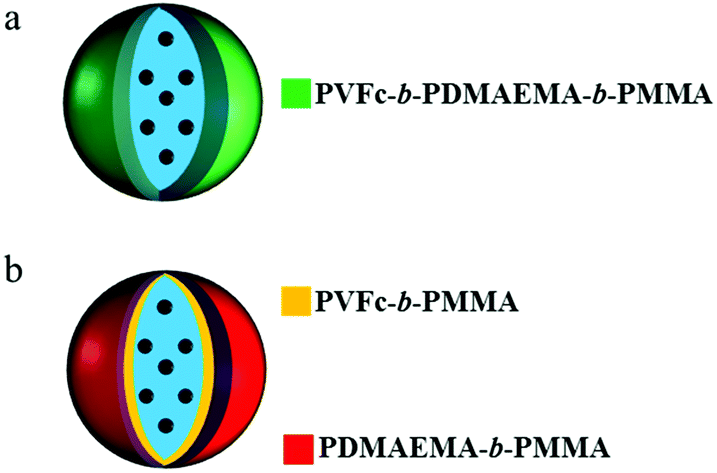 | ||
| Fig. 5 Schematic illustration of the configuration of shell-forming materials for (a) NCs prepared from the PVFc11.4-b-PDMAEMA17.8-b-PMMA70.8 triblock terpolymer and (b) NCs prepared from the mixture of PVFc20-b-PMMA80 and PDMAEMA34-b-PMMA66 block copolymers. Microphase separation inside the shell is the consequence of the latter one whereas a fine distribution of the segment blocks is achieved for the first one. | ||
The response of NC-DC dispersions to oxidation, changes of pH and temperature, was investigated after removing the non- encapsulated dye by dialysis. The change of the NR fluorescence intensity was followed to monitor the NCs opening (Fig. 6). The overall release of the payload is the sum of two contributions – burst and (slow) diffusive release.53 We studied pH and redox-induced release of NR from NC-DC and NC-TC at t = 10 and 1440 min. There was a remarkable increase in NR release from 38 to 77% for sample NC-TC and from 20 to 90% for sample NC-DC upon increasing the concentration of H2O2 from 4.4 to 35 wt% (Fig. 6a and b). At H2O2 concentration of 8.75 wt%, the NR release out of NC-DC was ∼30% whereas this value for NC-TC reached ∼52%. Furthermore, the ratio between burst and diffusive release (Rb/d) for NC-TC and NC-DC depended on H2O2 concentration. Rb/d increased from ∼0.5 to ∼3 for the NC-TC while it was almost constant (∼2.2) for sample NC-DC. The dispersions of NC-TC and NC-DC did not exhibit observable NR release at basic pH whereas the release slightly increased at intermediate pH values (4.5 and 6.5) and reached ∼4% for NC-TC and t ∼ 8% for NC-DC (Fig. 6c and d). Upon further decrease of pH value to 2 and below, the release reached ∼24% in the case of sample NC-DC, while about 95% of the encapsulated NR was released at pH 1.0 for sample NC-TC at t = 1440 min. Moreover, these results showed that diffusive release played a key role in the efficient release at pH 1.0. No effect of temperature on the fluorescence intensity of NR was observed for sample NC-DC (Fig. 6e).
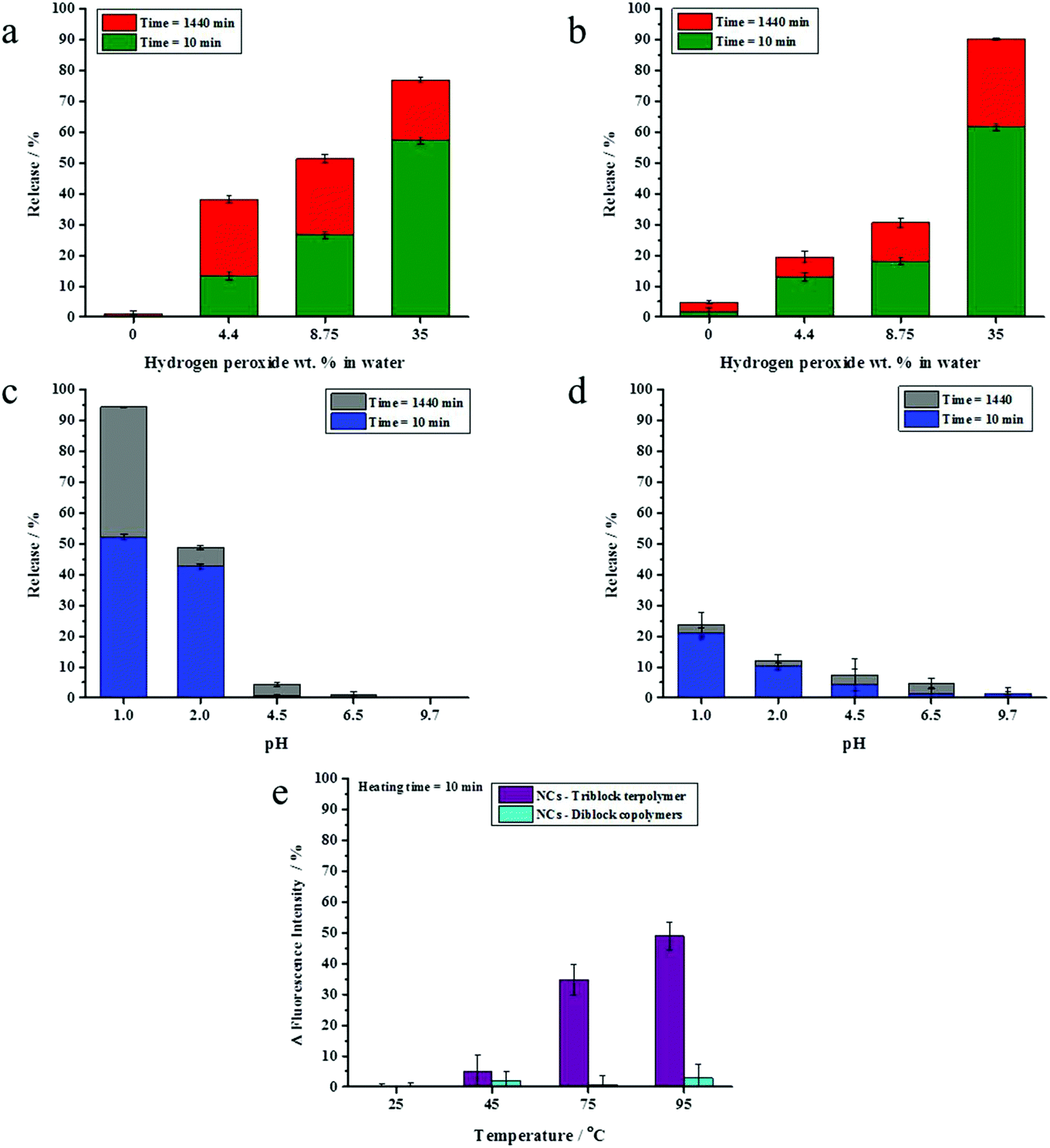 | ||
| Fig. 6 Released amount of NR at different H2O2 concentrations out of the NCs consisting of PVFc11.4-b-PDMAEMA17.8-b-PMMA70.8(a) and the mixture of PVFc20-b-PMMA80and PDMAEMA34-b-PMMA66 (b) at t = 10 and 1440 min. The released amount of NR at different pH values for NCs prepared from the aforementioned triblock terpolymer (c) and the mixture of diblock copolymers (d) at t = 10 and 1440 min. (e) The temperature responsiveness of both types of NCs at t = 10 min. | ||
These results show that the NCs of a blend of diblock copolymers did not significantly respond to the pH change and temperature. In contrast, the NCs prepared from the triblock terpolymer exhibited a responsive behaviour to pH change and temperature. Because the composition of the NCs is the same, the difference in the release profile must be associated with differences in the microstructure of the polymer shell (Fig. 3 and 5). Microphase separation (Fig. 3 and 5) between PVFc-b-PMMA and PDMAEMA-b-PMMA diblock copolymers in NC-DC shells can explain the observed differences in stimuli-induced release of the payload between NC-DC and NC-TC. In the case of NC-TC, the PVFc and PDMAEMA blocks were distributed on the NC surface and across the shell. The carefully designed triblock terpolymer precisely targeted the desired triple stimuli-responsive release of the payload. In the case of NC-DC, the phase separation occurred between PVFc-b-PMMA and PDMAEMA-b-PMMA diblock copolymers, with PDMAEMA-b-PMMA and PVFc-b-PMMA block copolymers forming the exterior and the interior layer of the shell, respectively (Fig. 5b). The interior layer played the role of a barrier that hinders the function of PDMAEMA as a pH-responsive unit (Fig. 5b). In other words, the core was not accessible for the trigger. Furthermore, the presence of higher relative amount of PDMAEMA block segments on the shell surface of NC-DC resulted in the protonation and the subsequent swelling of PDMAEMA-b-PMMA diblock copolymer even at intermediate pH-values (∼6.9) (Table 2). Therefore, the PDMAEMA-b-PMMA diblock copolymer, as the exterior layer, did not suppress the penetration and subsequent access of H2O2 to the PVFc segment block of the interior layer and consequently the triggered release of NR at pH ∼6.9. Although the selective staining and release experiments provide an excellent explanation for the observed differences between the release properties of NC-TC and NC-DC, these two experiments cannot be exactly compared. Indeed, the selective staining experiments were carried on a dried sample for TEM measurements whereas the release experiments were performed in dispersion. However, the results revealed that the observed differences between the release properties of NC-TC and NC-DC are due to a difference in shell morphology.
Conclusions
Novel triple stimuli-responsive NCs consisting of PVFc-b-PMMA-b-PDMAEMA triblock terpolymers were prepared by a solvent evaporation process from miniemulsion droplets. The NCs could encapsulate the dye in the core and perfectly protect it from non-controlled leakage. The NC dispersions exhibited high colloidal stability in aqueous release medium. The triggered release of the payload was achieved because the NC shell was addressable by three different stimuli: pH change, oxidizing agent, and temperature. Alternatively, NCs prepared with a blend of responsive diblock copolymers, where their shells had exactly the same chemical composition in terms of molar amount of blocks with NCs prepared from triblock copolymers. However, microphase separation occurred between the diblock copolymers in the NC shell, which resulted in formation of shells with non-homogeneous distribution of the diblock copolymers across the shell. Our results imply that PVFc-b-PMMA and PDMAEMA-b-PMMA diblock copolymers formed the interior and exterior layers across the shell, respectively. The exterior layer was not addressable more since its access to the core was hindered by the interior layer. This configuration of diblock copolymers across the shell, therefore, made these NCs stimulus-responsive instead of triple stimuli-responsive. This study revealed how the microstructure of container shells dictates the release properties of payloads from polymer NCs and pave the way to precisely design NCs with addressable and high functional shells for applications in which selective delivery of one or several payloads upon application of different stimuli is necessary. The effect of the shell microstructure on collaborative release of payloads from NCs triggered by multiple external stimuli will be a part of a future study.Experimental
Materials
Dichloromethane (Sigma-Aldrich, 99.99%), hexadecane (HD, Sigma-Aldrich, 99%), Nile-red (Sigma-Aldrich), hexadecyltrimethylammonium chloride (CTMA-Cl, Alfa Aesar, 96%), sodium dodecyl sulfate (SDS, Alfa Aesar, 99%), hydrogen peroxide (H2O2, Sigma-Aldrich, 35%), sodium hydroxide pellets (Sigma-Aldrich, 97%), hydrochloric acid (HCl) solution (Sigma-Aldrich, 37%), iodomethane (Merck), and chlorotrimethylsilane (Alfa Aesar, 98%) were used as received. Demineralized water was used for preparation of all dispersions. All other solvents and reagents were purchased from Fisher Scientific and used as received, unless otherwise stated. Tetrahydrofuran (THF) was distilled from sodium/benzophenone under reduced pressure (cryo-transfer) prior to the addition of 1,1-diphenylethylene (DPE) and n-BuLi as well as a second cryo-transfer. Methyl methacrylate and N,N-dimethylaminoethyl methacrylate (DMAEMA) were purified by 2–fold distillation over calcium hydride (CaH2). In both cases, trioctyl aluminium (25 wt% in hexane) was subsequently added dropwise until a pale yellow color appeared. Prior to use in anionic polymerization protocols, the monomers were freshly distilled from these solutions. Vinylferrocene was synthesized and purified as reported elsewhere.54Exemplary synthesis of PVFc6.2-b-PMMA88-b-PDMAEMA5.8
In an ampule equipped with a stirring bar 200 mg (1.89 mmol, 33 eq.) VFc and 60 mg (1.41 mmol, 50 eq.) LiCl are dissolved in dry 5 ml THF. The solution is cooled to −12 °C, and 34 μL (0.03 mmol, 1 eq.) n-BuLi in hexane (1.6 M) is added quickly. After 16 h, an aliquot of the solution is taken from the ampule for characterization of the PVFc-block and terminated by adding methanol. Then, 20 mg (0.12 mmol, 4 eq.) DPE and 6 mg (0.06 mmol, 2 eq.) DMSB are added to the PVFc macro-anions. Meanwhile 1.1 g (11.2 mmol, 400 eq.) MMA is dissolved in 45 mL dry THF and cooled to −78 °C. The polymerization of the MMA is started by fast addition of the PVFc-macroinitiator. After 1 h of reaction time, a sample of the solution is taken from the ampule for characterization of the diblock copolymer and terminated by adding methanol. Then, 130 mg (0.8 mmol, 27 eq.) DMAEMA is added and after stirring at −78 °C for 1 h, 0.1 mL methanol is added to terminate the polymerization. The polymer solution is added to 250 ml water and stirred at 70 °C to precipitate the polymer. The polymer is collected by filtration and dried in vacuo. Yield: 1.3 g (90%).The other polymers were synthesized by the same procedure in comparable yields. The amounts of the three monomers were varied according to the desired copolymer compositions. In the case of the triblock copolymer with a different block order (PVFc11.4-b-DMAEMA17.8-b-PMMA70.8), DMAEMA was initiated with the PVFc-macroinitiator followed by addition of MMA. In the case of PDMAEMA34-b-PMMA66, diphenylhexyl lithium was used instead of the PVFc-macroinitiator. Molecular weights of the triblock terpolymers and diblock copolymers are presented in Table 3.
| Entry | Apparent average molecular weight (g mol−1) | |||
|---|---|---|---|---|
| PVFc (Mn)a | PVFc (Mw)a | PMMA (Mw)b | PDMAEMA (Mw)b | |
| a Determined by SEC-MALLS in THF. b Calculated from 1H-NMR spectra. | ||||
| PVFc8.2-b-PMMA90.2-b-PDMAEMA1.6 | 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 700 |
17800 |
88200b |
2500 |
| PVFc6.2-b-PMMA88-b-PDMAEMA5.8 | 8500 | 25800 |
173000b |
17900 |
| PVFc11.4-b-PDMAEMA17.8-b-PMMA70.8 | 8500 | 25800 |
75200b |
29600 |
| PVFc20-b-PMMA80 | 9200 | 37400 |
70600b |
— |
| PDMAEMA34-b-PMMA66 | — | — | 89700a |
30500 |
Preparation of the nanocapsules
50 mg of polymer and 50 mg of hexadecane were dissolved in 2.5 g of dichloromethane containing 1 mg of Nile-red (NR). The organic phase was added to 20 g of an aqueous solution with concentrations of 0.05 mg mL−1 and 0.1 mmol L−1 of hexadecyltrimethylammonium chloride (CTMA-Cl) and NaOH, respectively. The mixture was stirred at 1100 rpm for 1 h in a closed glass vial to obtain a macroemulsion. The emulsion was then subjected to ultrasonication under ice-cooling for 2 min in a 30 s pulse per 10 s pause regimen (Branson W450-D sonifier with a 1/2 in. tip). Afterward, dichloromethane was evaporated at room temperature while stirring at 500 rpm for 16 h. The dispersions were then dialyzed against their continuous phases for 1 day to remove the non-encapsulated dye. For the synthesis of NCs prepared from the PVFc-b-PMMA block copolymer, an aqueous solution with a concentration of 0.05 mg mL−1 sodium dodecyl sulfate (SDS) was used.Release experiments
To determine the responsiveness of the NCs, dispersions were mixed with aqueous solutions containing CTMA-Cl (0.05 mg mL−1) and HCl at different concentrations (10−5 to 0.1 mol L−1) with volume ratios 1:5 and stirred for 24 h. To measure the response of NCs to oxidation, the dispersions were mixed with hydrogen peroxide solutions at different concentrations (35 to 4.4 wt%) containing CTMA-Cl (0.05 mg mL−1) with a volume ratio 1:5 and stirred for 24 h. The response of the NCs to temperature was measured by heating the dispersions at different temperatures (45 to 95 °C) for 10 min. Control samples of the dispersions were also mixed with their continuous phase without addition of an acid or an oxidant at room temperature.
Characterization methods
The prepared NCs were characterized with DLS, SEC, SEM, TEM, and zeta potential, while fluorescence analysis was utilized to measure the release of NR (see the ESI†).Acknowledgements
M. G. and M. A. acknowledge the German Research Foundation (DFG GA 2169/1-1) and the Fonds der Chemischen Industrie (FCI) for partial financial support of this work. The authors thank Daniel Scheid for support with TEM measurements of the triblock terpolymer bulk samples.References
- L. Y. Chu, T. Yamaguchi and S.-I. Nakao, Adv. Mater., 2002, 14, 386 CrossRef CAS.
- Y. Zhu, J. Shi, W. Shen, X. Dong, J. Feng, M. Ruan and Y. Li, Angew. Chem., Int. Ed., 2005, 117, 5213 CrossRef.
- E. R. Gillies and J. M. J. Fréchet, Bioconjugate Chem., 2005, 16, 361 CrossRef CAS PubMed.
- A. S. Angelatos, B. Radt and F. Caruso, J. Phys. Chem. B, 2005, 109, 3071 CrossRef CAS PubMed.
- S. Giri, B. G. Trewyn, M. P. Stellmaker and V. S. Y. Lin, Angew. Chem., Int. Ed., 2005, 44, 5038 CrossRef CAS PubMed.
- G. K. Such, E. Tjipto, A. Postma, A. P. Johnston and F. Caruso, Nano Lett., 2007, 7, 1706 CrossRef CAS PubMed.
- A. P. R. Johnston, G. K. Such and F. Caruso, Angew. Chem., Int. Ed., 2010, 49, 2664 CrossRef CAS PubMed.
- A. P. Esser-Kahn, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Macromolecules, 2011, 44, 5539 CrossRef CAS.
- Y. Bae, S. Fukushima, A. Harada and K. Kataoka, Angew. Chem., Int. Ed., 2003, 42, 4640 CrossRef CAS PubMed.
- C. D. L. H. Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276 RSC.
- G. R. Hendrickson, M. H. Smith, A. B. South and L. A. Lyon, Adv. Funct. Mater., 2010, 20, 1697 CrossRef CAS.
- J. Kim, J. Yoon and R. C. Hayward, Nat. Mater., 2010, 9, 159 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991 CrossRef CAS PubMed.
- L.-P. Lv, Y. Zhao, N. Vilbrandt, M. Gallei, A. Vimalanandan, M. Rohwerder, K. Landfester and D. Crespy, J. Am. Chem. Soc., 2013, 135, 14198 CrossRef CAS PubMed.
- D. Shchukin and H. Möhwald, Science, 2013, 341, 1458 CrossRef CAS PubMed.
- Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605 CrossRef CAS PubMed.
- A. K. Kota, G. Kwon, W. Choi, J. M. Mabry and A. Tuteja, Nat. Commun., 2012, 3, 1025 CrossRef PubMed.
- R. K. Jain, Nat. Med., 2001, 7, 987 CrossRef CAS PubMed.
- J. B. Fitzgerald, B. Schoeberl, U. B. Nielsen and P. K. Sorger, Nat. Chem. Biol., 2006, 2, 458 CrossRef CAS PubMed.
- F. S. Bates and G. H. Fredrickson, Annu. Rev. Phys. Chem., 1990, 41, 525 CrossRef CAS PubMed.
- M. W. Matsen and F. S. Bates, Macromolecules, 1996, 29, 1091 CrossRef CAS.
- Z. Li, M. A. Hillmyer and T. P. Lodge, Nano Lett., 2006, 6, 1245 CrossRef CAS PubMed.
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Macromolecules, 2012, 45, 5081 CrossRef CAS.
- C. J. Mable, N. J. Warren, K. Thompson, O. Mykhaylyk and S. P. Armes, Chem. Sci., 2015, 6, 6179 RSC.
- L.-Y. Chu, S.-H. Park, T. Yamaguchi and S.-I. Nakao, Langmuir, 2002, 18, 1856 CrossRef CAS.
- S. Berger, H. Zhang and A. Pich, Adv. Funct. Mater., 2009, 19, 554 CrossRef CAS.
- A. Klaikherd, C. Nagamani and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 4830 CrossRef CAS PubMed.
- M. Motornov, Y. Roiter, I. Tokarev and S. Minko, Prog. Polym. Sci., 2010, 35, 174 CrossRef CAS.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101 CrossRef PubMed.
- M. J. Robb, L. A. Connal, B. F. Lee, N. A. Lynd and C. J. Hawker, Polym. Chem., 2012, 3, 1618 RSC.
- A. H. Gröschel, F. H. Schacher, H. Schmalz, O. V. Borisov, E. B. Zhulina, A. Walther and A. H. E. Müller, Nat. Commun., 2012, 3, 710 CrossRef PubMed.
- X. Huang and B. Voit, Polym. Chem., 2013, 4, 435 RSC.
- J. Dong, Y. Wang, J. Zhang, X. Zhan, S. Zhu, H. Yang and G. Wang, Soft Matter, 2013, 9, 370 RSC.
- R. H. Staff, M. Gallei, K. Landfester and D. Crespy, Macromolecules, 2014, 47, 4876 CrossRef CAS.
- C. Fleischmann, J. Gopez, P. Lundberg, H. Ritter, K. L. Killops, C. J. Hawker and D. Klinger, Polym. Chem., 2015, 6, 2029 RSC.
- G. Wu, S.-C. Chen, C.-L. Liu and Y.-Z. Wang, ACS Nano, 2015, 9, 4649 CrossRef CAS PubMed.
- Y. Zhao, L.-P. Lv, S. Jiang, K. Landfester and D. Crespy, Polym. Chem., 2015, 6, 4197 RSC.
- Y. Wu, S. Liu, Y. Tao, C. Ma, Y. Zhang, J. Xu and Y. Wei, ACS Appl. Mater. Interfaces, 2014, 6, 1470 CAS.
- D. J. Callahan, W. Liu, X. Li, M. R. Dreher, W. Hassouneh, M. Kim, P. Marszalek and A. Chilkoti, Nano Lett., 2012, 12, 2165 CrossRef CAS PubMed.
- P. Shi, Y. Qu, C. Liu, H. Khan, P. Sun and W. Zhang, ACS Macro Lett., 2015, 5, 88 CrossRef.
- K. Landfester, Adv. Mater., 2001, 13, 765 CrossRef CAS.
- R. H. Staff, M. Gallei, M. Mazurowski, M. Rehahn, R. Berger, K. Landfester and D. Crespy, ACS Nano, 2012, 6, 9042 CrossRef CAS PubMed.
- R. H. Staff, K. Landfester and D. Crespy, Recent advances in the emulsion solvent evaporation technique for the preparation of nanoparticles and nanocapsules, in Hierarchical Macromolecular Structures: 60 Years after the Staudinger Nobel Prize II, ed. V. Percec, Springer, 2013, p. 329 Search PubMed.
- S. Torza and S. G. Mason, J. Colloid Interface Sci., 1970, 33, 67 CrossRef CAS.
- R. H. Staff, D. Schaeffel, A. Turshatov, D. Donadio, H. J. Butt, K. Landfester, K. Koynov and D. Crespy, Small, 2013, 9, 3514 CrossRef CAS PubMed.
- A. K. Dutta, K. Kamada and K. Ohta, J. Photochem. Photobiol., A, 1996, 93, 57 CrossRef CAS.
- J. Jiang, X. Tong and Y. Zhao, J. Am. Chem. Soc., 2005, 127, 8290 CrossRef CAS PubMed.
- C. D'Silva, S. Afeworki, O. L. Parri, P. K. Baker and A. E. Underhill, J. Mater. Chem., 1992, 2, 225 RSC.
- J. Elbert, M. Gallei, C. Rüttiger, A. Brunsen, H. Didzoleit, B. Stühn and M. Rehahn, Organometallics, 2013, 32, 5873 CrossRef CAS.
- J. Elbert, F. Krohm, C. Rüttiger, S. Kienle, H. Didzoleit, B. N. Balzer, T. Hugel, B. Stühn, M. Gallei and A. Brunsen, Adv. Funct. Mater., 2014, 24, 1591 CrossRef CAS.
- F. Liu and M. W. Urban, Macromolecules, 2008, 41, 6531 CrossRef CAS.
- A. S. Lee, V. Bütün, M. Vamvakaki, S. P. Armes, J. A. Pople and A. P. Gast, Macromolecules, 2002, 35, 8540 CrossRef CAS.
- K. M. Gallagher and O. I. Corrigan, J. Controlled Release, 2000, 69, 261 CrossRef CAS PubMed.
- M. Gallei, R. Klein and M. Rehahn, Macromolecules, 2010, 43, 1844 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py00344c |
| ‡ Present address: Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA, 02139, USA. |
| This journal is © The Royal Society of Chemistry 2016 |