
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fragmentation reactions using electrospray ionization mass spectrometry: an important tool for the structural elucidation and characterization of synthetic and natural products
Daniel P.
Demarque†
 a,
Antonio E. M.
Crotti†
b,
Ricardo
Vessecchi
b,
João L. C.
Lopes
a and
Norberto P.
Lopes
*a
a,
Antonio E. M.
Crotti†
b,
Ricardo
Vessecchi
b,
João L. C.
Lopes
a and
Norberto P.
Lopes
*a
aDepartamento de Física e Quimica, Faculdade de Ciências Farmacêuticas de Ribeirão Preto, Universidade de São Paulo, Av. Café, s/n, Ribeirão Preto, SP, Brazil. E-mail: npelopes@gmail.com; Fax: +55 16 3315 4178; Tel: +55 16 3315 4158
bDepartamento de Química, Faculdade de Filosofia, Ciências e Letras de Ribeirão Preto, Av. Bandeirantes, 3900, CEP 14040-901, Ribeirão Preto, SP, Brazil
First published on 16th December 2015
Abstract
Covering: up to 2015
Over the last decade, the number of studies reporting the use of electrospray ionization mass spectrometry (ESI-MS) in combination with collision cells (or other activation methods) to promote fragmentation of synthetic and natural products for structural elucidation purposes has considerably increased. However, the lack of a systematic compilation of the gas-phase fragmentation reactions subjected to ESI-MS/MS conditions still represents a challenge and has led to many misunderstood results in the literature. This review article exploits the most common fragmentation reactions for ions generated by ESI in positive and negative modes using collision cells in an effort to stimulate the use of this technique by non-specialists, undergraduate students and researchers in related areas.
 Daniel P. Demarque | Daniel Pecoraro Demarque obtained a B.Sc. degree and a M.Sc. degree in Pharmacy at the Federal University of Mato Grosso do Sul dealing with chemometric design strategies to optimization and technological development of vegetable extracts. Currently he is pursuing his Ph.D. degree in Pharmaceutical Sciences at the University of São Paulo in Ribeirão Preto in the NPPNS (Núcleo de Pesquisa em Produtos Naturais e Sintéticos). His research is focused in mass spectrometry applications to identification of natural products, MALDI imaging of small molecules and analytical quantification of biological samples. |
 Antonio E. M. Crotti | Prof. Antonio Eduardo Miller Crotti obtained a B.Sc. in Chemistry at the University of Franca and M.Sc. degree and a Ph.D. in Chemistry at the University of São Paulo. Currently, he is professor of Organic Chemistry at the Department of Chemistry of School of Phylosophy, Sciences and Letters of Ribeirão Preto at the University of São Paulo. His research focus is on the biological activities and mass spectrometric studies of natural and synthetic organic compounds. |
 Ricardo Vessecchi | Prof. Ricardo Vessecchi received his B.S. in chemistry from University of São Paulo in 2002, his M.Sc. in pharmaceutical sciences and he received his Ph.D. in chemistry having carried out work in the area of theoretical chemistry under the supervision of Professor S. E. Galembeck at Department of Chemistry, FFCLRP-USP, in 2009. In 2009, he joint to the NPPNS as postdoctoral researcher combining theoretical chemistry and mass spectrometry. Currently, he is Professor at FFCLRP-USP in the Physical Chemistry area. His research interests are dedicated to the applications of computational chemistry at gas-phase ion chemistry, mass spectrometry and fragmentation mechanisms. |
 João Luis Callegari Lopes | Prof. João Luis Callegari Lopes obtained a B.Sc. degree in Pharmacy and a Ph.D. in Chemistry at the University of São Paulo. He is retired as a Full Professor of Organic Chemistry at the Physics and Chemistry Department of the School of Pharmaceutical Sciences of Ribeirão Preto – University of São Paulo since 2014, but still developing his activities as a Senior Professor. His research deals with organic chemistry, with emphasis on natural products chemistry and mass spectrometry. |
 Norberto P. Lopes | Prof. Norberto Peporine Lopes obtained a B.Sc. and M.Sc. degree in Pharmacy and a Ph.D. in Chemistry at the University of São Paulo. At present, he is Full Professor of Organic Chemistry at the University of São Paulo in Ribeirão Preto and head of the Physics and Chemistry Department of the School of Pharmaceutical Sciences of Ribeirão Preto – University of São Paulo. Currently, he is Fellow of the Royal Society of Chemistry and board member of the Brazilian Chemical Society and the Brazilian Society of Mass Spectrometry. His research deals with organic chemistry, with emphasis on natural products chemistry and mass spectrometry. |
1 Introduction
The development of electrospray ionization (ESI) represents an important advance in mass spectrometry. This technique allows thermolabile and high-molecular-weight compounds, such as biopolymers and proteins, to be ionized and transferred to the gas phase, thus developing new applications in molecular biology, medicine and plant metabolomics.1–3 The influence of ESI in chemistry was recognized in 2002 when John B. Fenn (one of the scientists behind ESI development) won the Nobel Prize in Chemistry. Since then, the coupling of liquid chromatography to mass spectrometry (LC-MS), which was possible because of the nature of the ESI process (an atmospheric pressure ionization method), has expanded the diversity of molecules that can be analysed by mass spectrometry, including a variety of small organic molecules.4The scheme shown in Fig. 1 represents an overview of MS/MS spectra results. Initially, the analyte is infused into the ESI source for the charge generation (Fig. 1A); here, protonated/deprotonated molecules can be produced by Brönsted–Lowry acid–base reactions (protonation/deprotonation) and/or via the Lewis acid–base mechanism (adduct formation by interactions between cations or anions with neutral molecules). These “even electron ions” (EE+ or EE−) (Fig. 1B) typically display low internal energy content; consequently, little or no fragmentation is often observed in a MS spectrum of these ions.2 The radical ions (“odd electron ions” or OE˙+) produced by electron ionization (EI) have much more internal energy, and their fragment ions can be seen in a MS spectrum.2 Exceptions have been extensively described in the ESI source by the loss of one electron for some characteristic compounds, affording odd electron ions, which are called molecular ions (Fig. 1B).5 The molecular ion formation is more prone to occur in positive mode than radical anion formation in negative mode.6–9 Considering all the ionization possibilities presented in Fig. 1B and the balance of the species formation, the structure elucidation step identifies the nature of the ionization process in the ESI source occurring with the analyte under study. The balance of molecular ions (M˙+), protonated molecules ([M + H]+) and cationized species (i.e., [M + Na]+) may provide relevant information about the redox potential, proton affinity and stereochemistry of the analyte.3,5
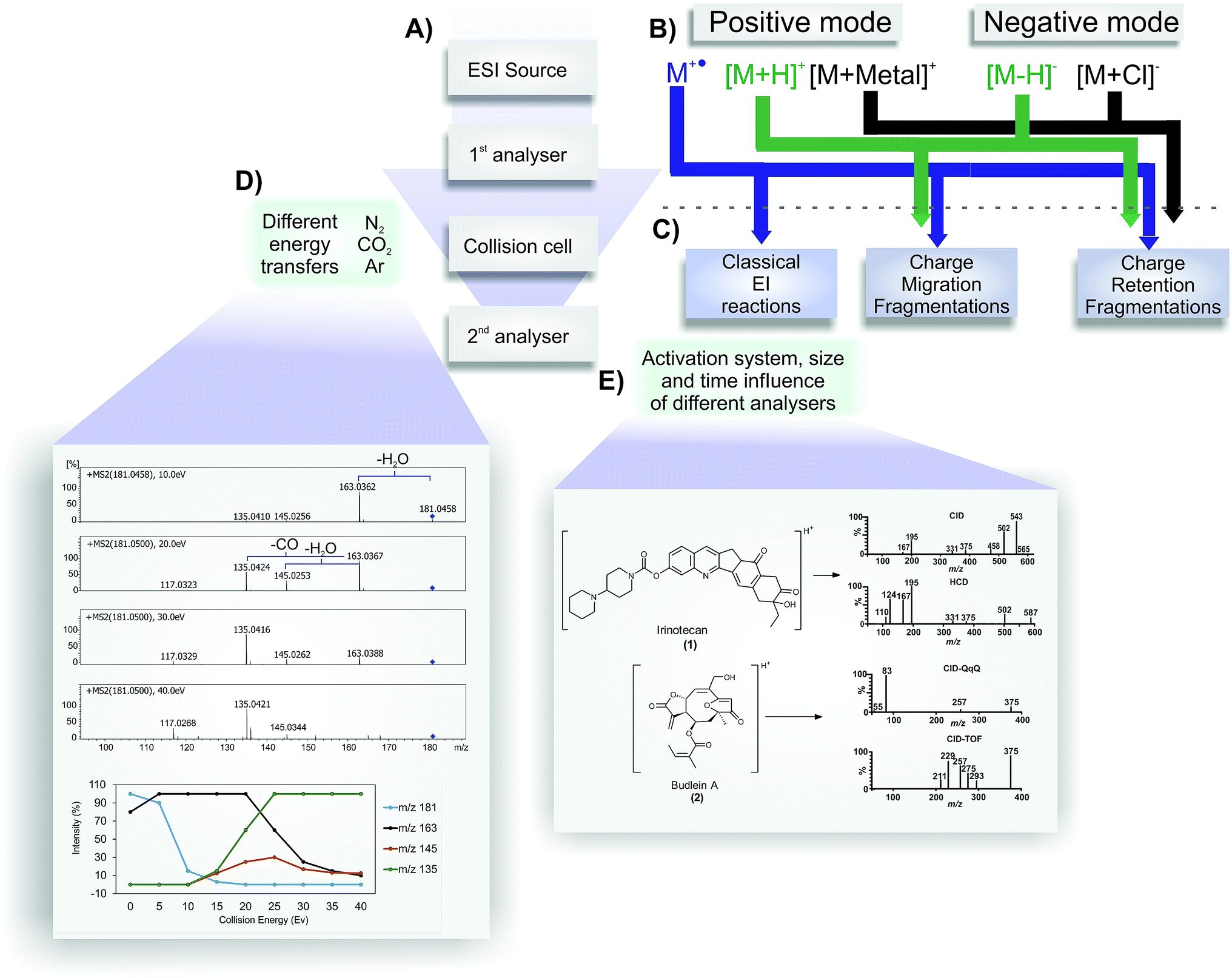 | ||
| Fig. 1 Representative overview of MS/MS spectra results. (A) Representation of mass spectrometer structures. The ions are generated in the ESI source in positive or negative mode (B). Depending of the ion type ((M˙+), [M + H]+, [M + metal]+, [M − H]−, [M − Cl]−), after passing to the collision cell, they may follow classical EI reactions, charge migration fragmentations or charge retention fragmentations (C) (negative radical anions may occur in specific cases and are not represented). Different collision cells (E), different collision gases and different collision energies (D) generate different spectra, as shown in the comparison of irinotecan (1) in CID and HCD and budlein A (2) in CID-QqQ and CID-TOF (E). An example of the influence of different collision energies can be seen in the fragmentation of 2,3-dihydroxycinnamic acid in positive mode. | ||
As a soft ionization method – which does not typically promote higher source fragmentation (exception for molecular ions) – ESI is commonly used in mass spectrometers equipped with collision cells (or other activation methods) to induce fragmentation (Fig. 1A). A collision cell (or other related MS component) generates a MS2 (or MS/MS) spectrum, and the fragmented ions generated are detected in the second mass analyser. These fragments can be used for structure elucidation purposes. To achieve a better understanding of the reactions that may occur under collision-induced dissociation conditions, a brief explanation and comparison between their operational parameters are provided because it is possible to have the same fragmentation reactions using a variety of activation methods.
In tandem mass spectrometry (MS/MS), ions generated by ESI can be selected in the first stage of MS to collide with molecules of an inert collision gas (Fig. 1D).10 This step is very fast (between 10−14 to 10−16 s) and leads to an increase in the ion's internal energy, promoting it to an excited state.11 Immediately after the ion excitation, the unimolecular decomposition is initiated and, as result, the activated selected ions (the precursor ions) are converted into fragment ions (product ions) in a collision energy-dependent manner following three major classes of reactions (Fig. 1C).4,10,12 The fragmentation of odd electron ions (molecular ions) are well described in several important textbooks about electron ionization (EI-MS), such as the McLafferty compendium; therefore, it is not discussed in the present review.13 The fragmentation of protonated or deprotonated molecules (positive and negative modes, respectively) can be classified into two major groups of reactions: charge retention fragmentations and charge-migration fragmentations (Fig. 1C). Cationized molecules (and other related species) normally fragment following charge retention fragmentation reactions. There are few examples of charge-migration fragmentations, most of them restricted to sulfate and phosphate compounds.14,15 However, the restriction of a major group of fragmentation reactions for cationized species does not indicate that more structure information can be achieved from protonated species (by the possible occurrence of both groups of reactions). For some classes of compounds, especially ionophores (polyketide antibiotics), the fragmentation reactions of sodiated species are much more informative than those of its protonated molecules.16 Knowledge about the precursor ion structure and its fragmentation reactions plays a key role in the structure elucidation of natural and synthetic compounds based only on ESI-MS/MS. Finally, Fig. 1D shows three examples of gases that are typically used in collisional activation, providing additional relevant information. As expected when setting up the instrument conditions, the efficiency to transfer the energy must be different for each gas, according to the law of conservation of momentum.11 Thus, the applied collision gas must be considered for a comparative analysis of MS/MS spectra because different gases can cause differences in the ion intensities in the MS/MS spectrum.
Another important piece of information to be used for a reliable analysis by mass spectrometry is the type of mass analyser used in tandem mass spectrometry. MS/MS spectra can gain information about aspects of the fragmentation processes that occur by using different mass spectrometers (Fig. 1E). Thus, the conducted experiments using analysers such as Triple Quadrupole (QqQ), Quadrupole-Time of Flight (QTOF), Ion Trap (IT), Orbitraps, Fourier Transform Mass Spectrometry (FTMS), and Higher Energy Collisional Dissociation (HCD) fragmentation from trap analysers can induce less fragmentation ions.17–21
The energy involved during the dissociation process can be used to support the formation suggestion for the most specific ions. Experiments using different activation methods, i.e., conventional low-energy CID, electron capture dissociation (ECD) and electron transfer dissociation (ETD), or different analysers, i.e., sequential or tandem mass spectrometers, can result in the observation of different ions in the MS/MS or MSn spectra.
The delay between the collision and the fragment detection determines the intensity of the ions and provides a characteristic spectrum. The formation of metastable ions (ions formed in the source that are decomposed before detection),22 rearrangements, proton migration and other mechanisms can occur during the time-scale of the experiments. It is important to observe that long-time analysers may show a difference in the fragmentation behaviours compared with fast procedures. This may be due to the competition of kinetic and thermodynamic factors; thus, the most stable ion is occasionally not the most intense ion (in fast procedures), probably because several initial fragmentation reactions were required to afford it. The combination between energy-resolved plotting, survival-yield methods and computational calculations of fragmentation barriers is an important tool to characterize some fragmentation processes and non-homogeneous fragmentation behaviours in ESI-MS/MS. However, these studies require time, computational cost and accurate structural characterization. The use of the concepts of reactivity and ion stability can also be important to propose fragmentation mechanisms based on the main fragmentation reactions, as will be discussed in this article.
Other activation methods can eventually be used to provide structure information in addition to that obtained from ESI-CID. For example, the recently published results using natural products were evaluated in studies with the isomers of budlein A (2) and centratherin by Sartori and co-workers,21 showing that HCD elicits complementary fragmentation pathways in addition to the CID available in Q-TOF and QqQ; indeed, it produces diagnostic ions in the MS/MS spectra that aid the elucidation of metabolite structure. Illustrative schematic spectra are presented in Fig. 1E. HCD can improve the fragmentation in the low mass region, as observed in CID using QqQ. Thus, HCD would be a complementary source for elucidation and characterization. Fig. 1E displays two studies using CID (QqQ and QTOF) and HCD (Orbitrap). The same effect can be observed for irinotecan (1) fragmentation in CID and HCD (Fig. 1E). Finally, trap machines sometimes can have a blind zone that is approximately 15% of the total mass. The classical method used to identify microcystin in ESI (positive mode) is the observation of the side-chain cleavages with the elimination of Adda moieties.23,24 This key fragment typically occurs in positive mode at 135 mass units; however, for microcystin with a mass of over than 1000 mass units, sometimes it is not possible to observe this key ion in trap machines, which can lead to an incorrect interpretation for inexperienced users. Thus, it is important to realise that different analysers can show different results for the same molecule, and use the correct strategy to have the maximum results.
Although different fragments can be seen according to the collision cell type and mass analyser, the reactions compiled in this work can be observed in all collisional cells coupled to ESI sources with different analysers. It is also important to note that the fragmentations described in this review were selected to display a protocol for possible fragmentation that occurs in mass spectrometry analysis of natural and synthetic compounds.
2 Fragmentation reactions rationalization for structural elucidation
Understanding the fragmentation reactions of the precursor ion is essential for the structural characterization of organic compounds using ESI-CID-MS/MS data. Initial studies have devoted special attention to standardize the reactions under CID conditions, primarily those from atmospheric pressure ionization techniques.25,26 These studies showed major reactions, and the mechanisms were derived from the functional groups, which are the fundamental keys to the mechanisms. For studies with natural and synthetic products, however, the overall system must be considered, so that the mechanism involved cannot be suggested based solely on the functional groups.In the structure elucidation process based on ESI-MS/MS data, the knowledge of the fragmentation reactions involved in the formation of the peaks of the mass spectrum may provide important information about the molecular structure and the connectivity between the atoms. Isomer differentiation can be performed using this technique, demonstrating its potential and utility as a structure elucidation tool.27,28
Although there is an increasing number of studies reporting proposed fragmentations, the lack of a systematic compilation has led to errors in mechanism determinations. Efforts have been focused on defining the most important terms used in mass spectrometry; however, compilations that include the major fragmentation reaction classes remain scarce.22 The use of the pioneers' reference materials is extremely valuable for the interpretation of the data collected here.
When performing the rationalization of a fragmentation pathway, it is important to remember that the proposed fragmentation processes must be based on organic chemistry principles. In the scientific literature, inadequate arrow representations, such as those shown in Fig. 2, can be extensively observed. Two different curved arrow systems are available to represent the electron movement in the mechanism of retro-heteroene reactions (Fig. 4.3 and Section 5.4). However, considering that the highest electron density is at the X atom, the representation shown in Fig. 2a is the most appropriate; the representation in Fig. 2b is not recommended because it depicts the electrons moving towards the heteroatom, which is highly unlikely.
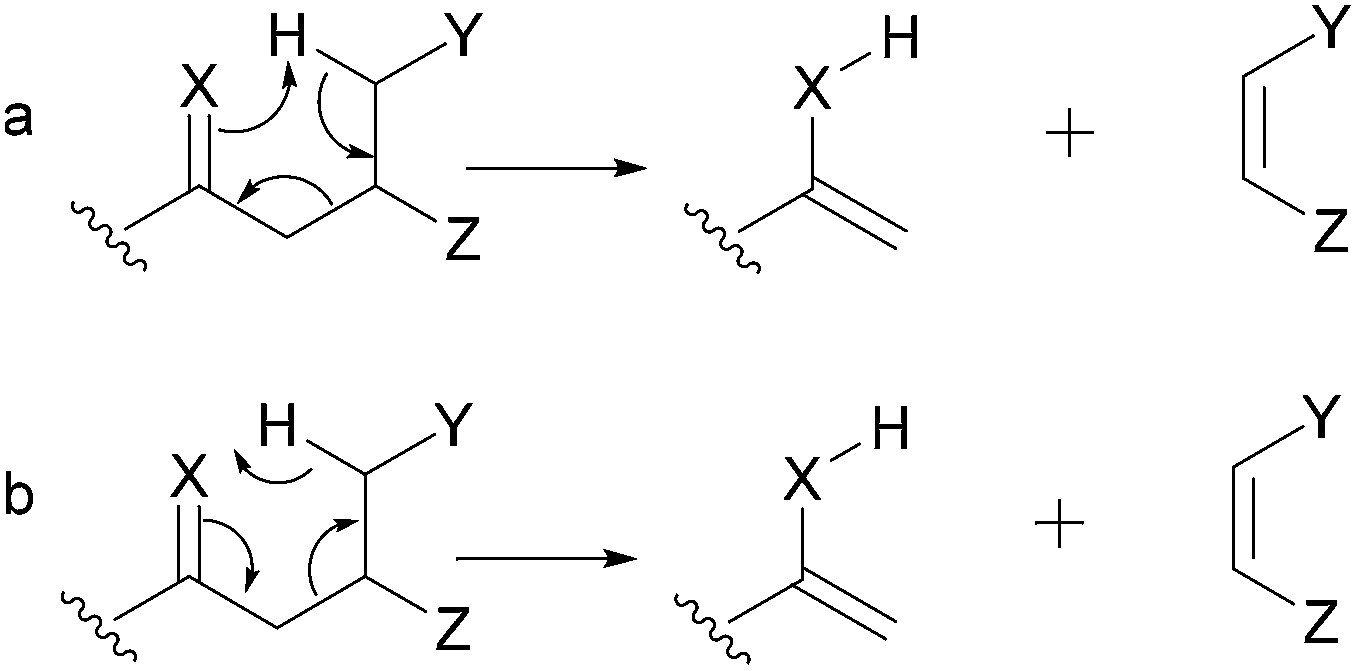 | ||
| Fig. 2 Representations used to represent retro-heteroene (X = heteroatom) reactions. | ||
Other mechanisms can lead to misunderstandings when isobaric fragment ions are generated (e.g., ions displaying identical m/z values). Fig. 3 shows two possible mechanisms leading to the same fragments: McLafferty-type rearrangements and remote hydrogen rearrangements. The energetically most favoured process under CID conditions will depend on the chemical structure. For example, the m/z 173 fragment ion of deprotonated dicaffeoylquinic acid (3) can result from a remote hydrogen rearrangement from m/z 191 (route a, Fig. 3, fragment (3.2)).29 Alternatively, an isomeric fragment ion can be formed directly from the precursor ion by a McLafferty-type rearrangement (route b, Fig. 3, fragment (3.1)). Although the six-membered transition state involved in the McLafferty-type rearrangement (route a) is lower in energy than that in the four-centred transition state of remote hydrogen rearrangements (route b), the formation of the m/z 173 (3.3) fragment ion through route a is favoured because the process is driven by the formation of a double bond conjugated with the carbonyl group. Therefore, the basic organic chemistry concepts must be considered not only to establish the fragmentation pathways but also to choose the most likely one.
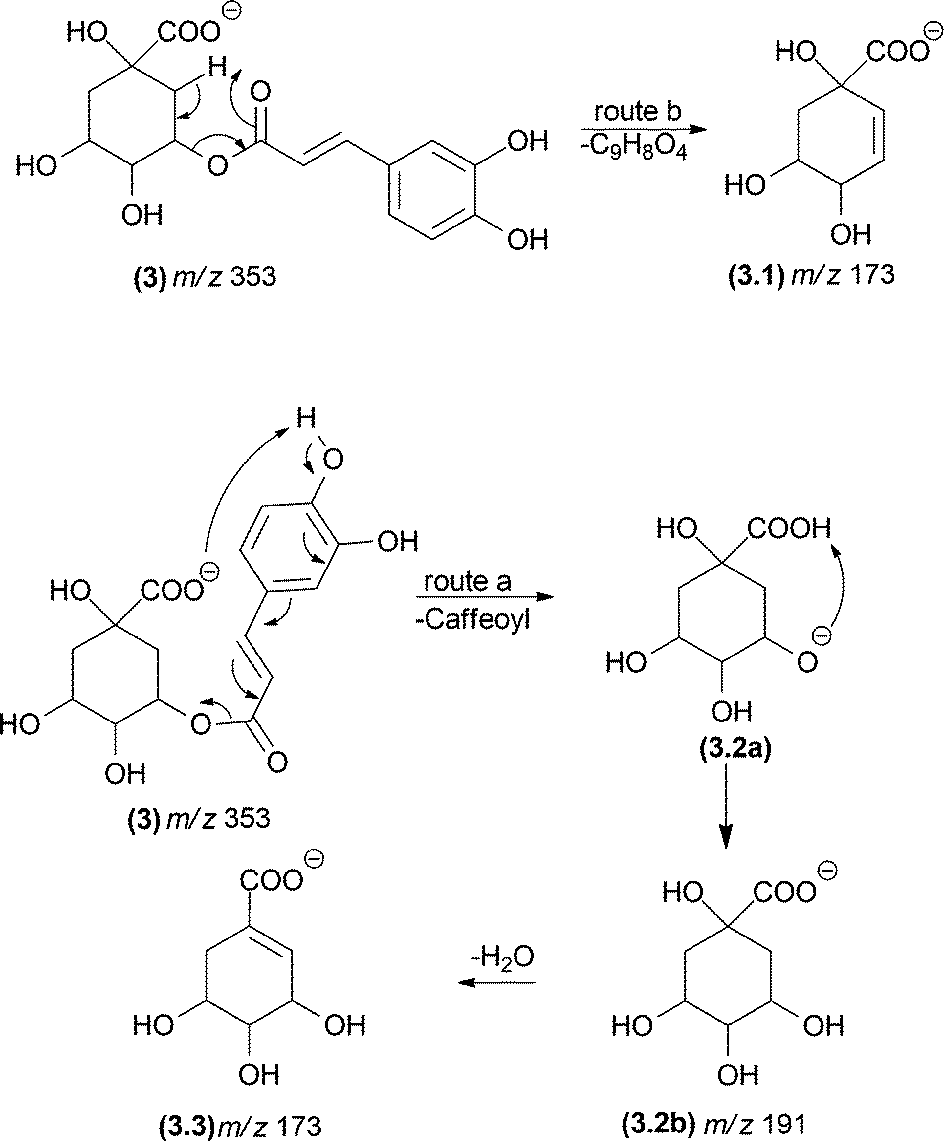 | ||
| Fig. 3 Competition between a remote hydrogen rearrangement and a McLafferty-type rearrangement in dicaffeoylquinic acids. | ||
To contribute to a rationalization of the fragmentation process, this review article aims to systematize the gas-phase fragmentation reactions of organic micromolecules (with a molecular mass of less than 5.000 u). For this purpose, the gas-phase fragmentation reactions were systematically divided, and examples from the literature are provided.
Here, the fragmentation reactions were divided into two main groups: charge retention fragmentations (CRF) and charge-migration fragmentations (CMF). The terminology used illustrates the hypothetical even electron ions ABCDE+ or ABCDE−, where the letters represent an atom, a functional group, or even several groups, such as an alkyl chain. This terminology is similar to that adopted by Fred W. McLafferty, which classifies the gas-phase fragmentation reactions of ions generated by electron ionization (EI) and chemical ionization (CI).13
3 Charge retention fragmentations (CRF)
Charge retention fragmentations (CRF) are composed of a class of reactions that results in fragment ions with the charge located at the site identical to its precursor ion. Because CRF typically occurs at a location that is physically remote from the location of the charge and without its direct participation in the mechanism, the term “remote site fragmentation” has also been used to refer to this class of gas-phase fragmentation reactions.30 The charge must be stable in its location and must not be able to migrate to the reaction site; in addition, it has minimal to no involvement in the reaction.11 The distance from the charge site and the reaction site may influence the competition between the CRF and CMF reactions.31 Normally, CRF reactions proceed through concerted mechanisms and may occur even at low collision energies, depending on the chemical structure, ionization method, mass collision target and other equipment parameters.32CRF can be classified into nine main reaction mechanisms: remote hydrogen rearrangements (Fig. 4.1), retro-Diels–Alder (RDA) reactions (Fig. 4.2), retro-ene reactions (Fig. 4.3), retro-heteroene reactions (Fig. 4.4), charge remote fragmentations (Fig. 4.5), aromatic eliminations (Fig. 4.6), other pericyclic processes (Fig. 4.7), carbon monoxide eliminations from cyclic carbonyl compounds (Fig. 4.8) and radical eliminations (Fig. 4.9).
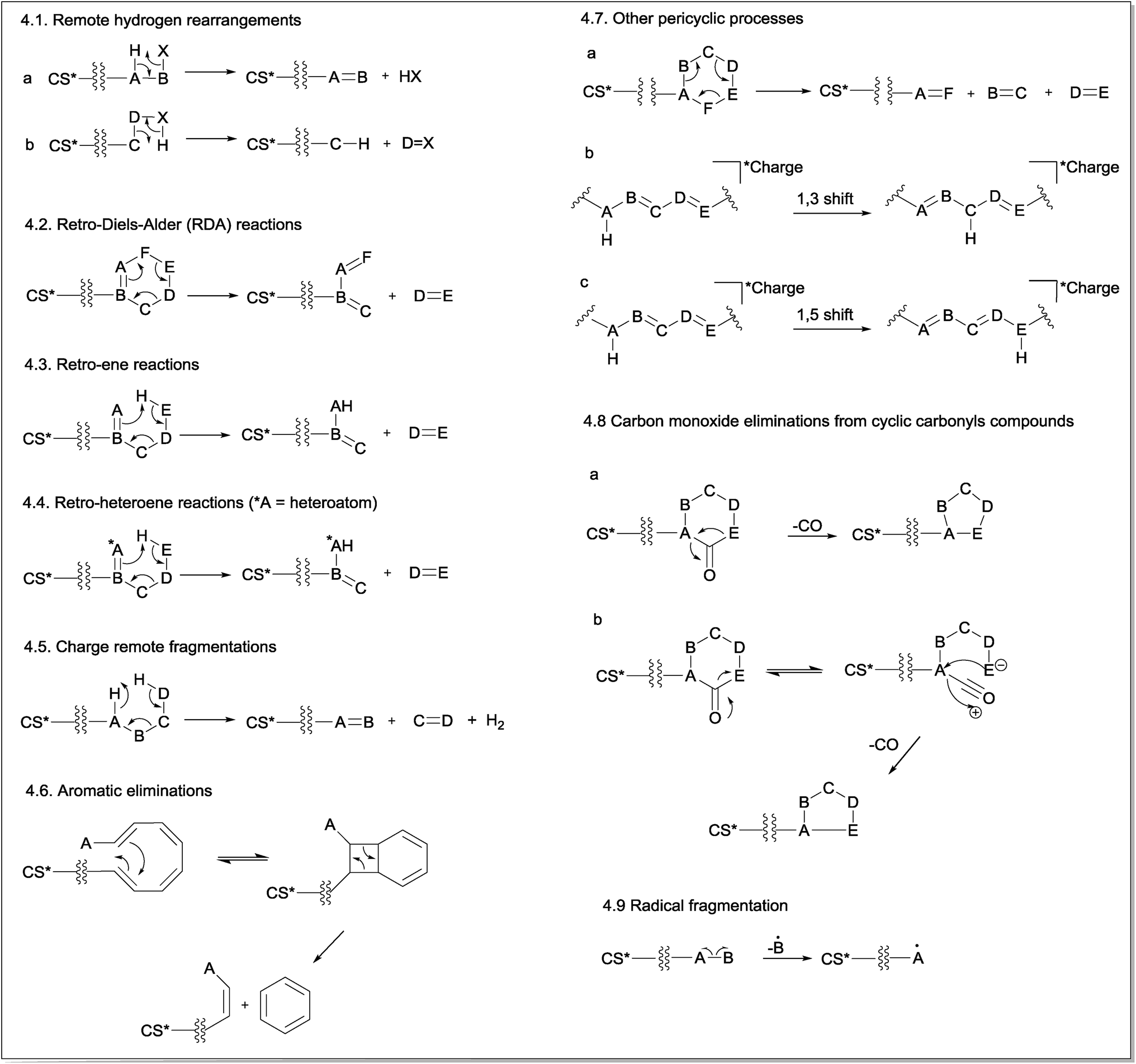 | ||
| Fig. 4 General mechanism of charge retention fragmentations (CRF) (CS* = charge site, which can be a positive or a negative charge). | ||
The first mechanism of CRF is the remote hydrogen rearrangement (Fig. 4.1). This mechanism involves an intramolecular 1,2-elimination (or intramolecular β-elimination) with the consequent formation of a new π bond in the detected fragment ion (Fig. 4.1a) or in the eliminated neutral molecule (Fig. 4.1b).
The second general reaction presented in Fig. 4.2 involves a simultaneous reorganization of bonds via a cyclic transition state. This type of reaction is called a “pericyclic reaction” because the electrons flow around the outside of the ring. Cycles containing a double bond can fragment via retro-Diels–Alder (RDA) fragmentation reactions in which a six-membered ring containing the double bond fragments undergoes a pericyclic mechanism to yield a diene and a dienophile in a process that is the reverse of the Diels–Alder reaction.
The stereospecificity and the concerted nature of the gas-phase RDA fragmentation mechanism were investigated by Denekamp and co-workers.33 The authors reported that cis-fused bicyclic systems afforded only cis-double bonds in the charged or neutral cyclic product, originating from the non-cyclohexene portion of the precursor (the original dienophile) (Fig. 5a). However, the expected formation of the trans double bond in the charged or neutral cyclic compound from trans-fused bicyclic systems, originating from the non-cyclohexane portion of the precursor (the original dienophile), does not occur because of the high energy requirements of a trans double bond in small-sized rings (Fig. 5b). These results corroborated the concerted nature of RDA fragmentations and the involvement of a favourable six-membered transition state. However, the authors also found that for select compounds, the RDA reactions occurred with partial stereospecificity or with no stereospecificity. They concluded that the nature of the RDA fragmentation reactions is strongly dependent on the structure of the precursor ion.
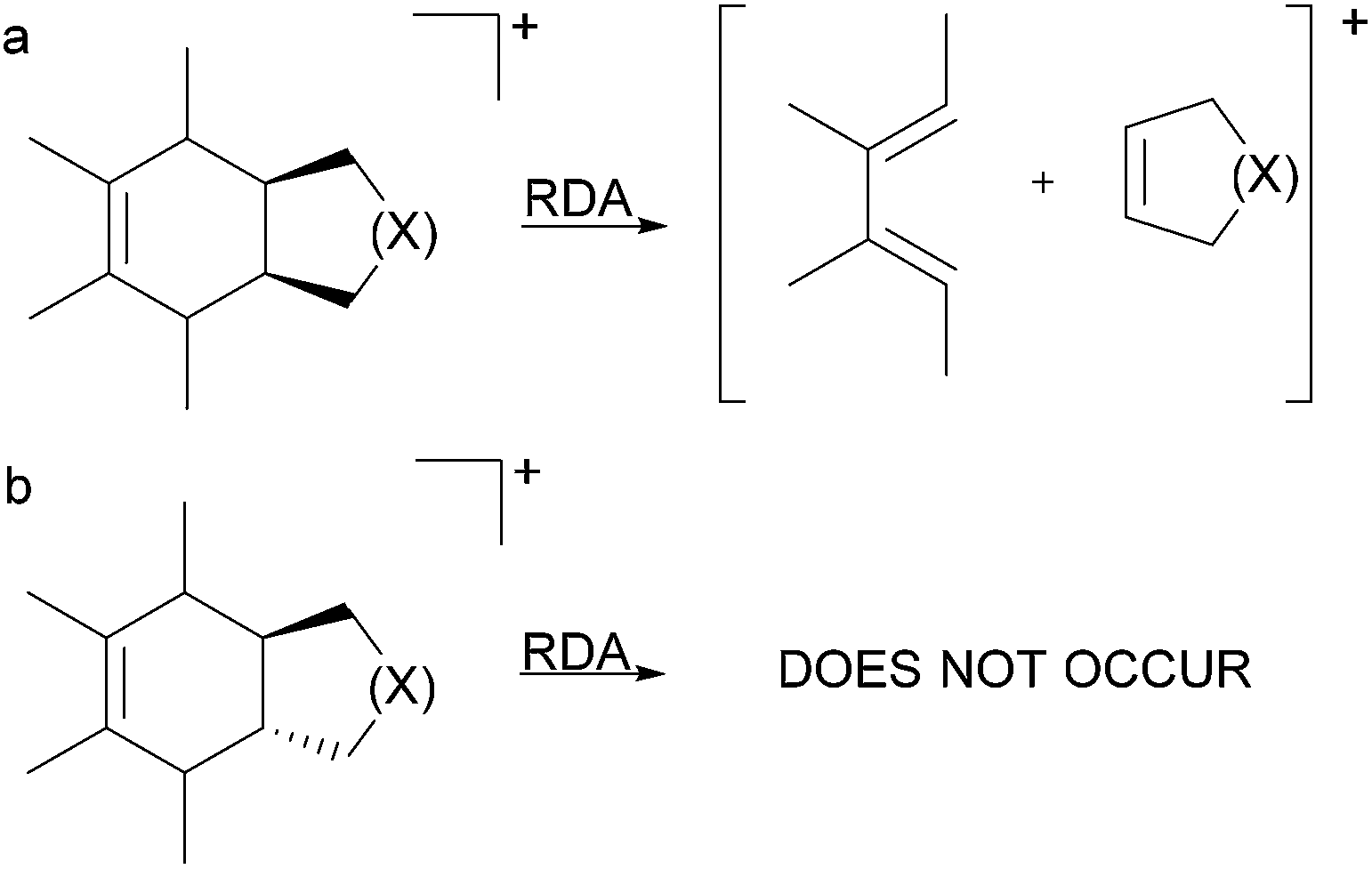 | ||
| Fig. 5 RDA fragmentation in small cis- and trans-fused bicyclic rings. | ||
Fig. 4.3 presents the retro-ene fragmentation reactions, which involve a transfer of a γ-hydrogen to an unsaturated carbon via a six-membered transition state to yield both ene and enophil as fragments. In most cases, these reactions proceed via a concerted mechanism.34 Retro-heteroene fragmentation reactions (Fig. 4.4) are similar to retro-ene reactions but differ in that an X–H bond (where X is a heteroatom, usually oxygen) is formed instead of a C–H bond.
Fig. 4.5 shows a general fragmentation mechanism called “charge remote fragmentation”. Although this term has been used to refer to every reaction occurring away from the charge site, in this review article, this term was used to describe a class of charge-retention fragmentation reactions that involves a six-membered cyclic transition state and leads to the elimination of H2 and an olefin (as first described by Adams and Gross (1986)).35–37
Another CRF explored in this review is a fragmentation reaction that requires at least four conjugated double bonds and a relative conformational mobility for its occurrence (Fig. 4.6). This reaction leads to aromatic ring eliminations from polyene systems; thus, it is named aromatic eliminations. Considering that the energy barrier for the isomerization of the double bonds is relatively low, polyenes ionized by ESI can assume the conformation required for this reaction under CID conditions so that a disrotatory ring-closure allowed by orbital symmetry occurs.38,39 Subsequently, a pericyclic reaction will realize a six-membered ring. Coughlan and co-workers showed that this mechanism occurred using theoretical calculations.40
In addition to retro-Diels–Alder (RDA), retro-ene and retro-heteroene reactions, a non-classic pericyclic cycloreversion process involving cyclohexane rings has also been reported under CID conditions. In this concerted process, three σ-bonds are broken, and three new π-bonds are formed, resulting in the opening of the six-membered ring (Fig. 4.7a). However, cycloreversion in compounds possessing a cyclohexane ring in their structures is not a classical cycloreversion process because no synthetic procedures based on the simultaneous use of three alkenes to produce a cyclohexane were found. However, the simultaneous elimination of H2O and two alkenes (for alcohols with more than 4Cs) through a six-membered transition state similar to that involved in the cycloreversion reactions in cyclohexane are well described in electron ionization processes.41
The sigmatropic rearrangements are pericyclic processes in which a bond is broken while another bond is formed across a π-system (Fig. 4.7b and c). In summary, an atom (or a group of atoms) σ-bonded to a carbon may rearrange its position through one or more π-electron systems.42 Although this reaction has been reported to occur in ESI-CID experiments, it does not produce any fragment ion because the total number of σ-bonds and π-bonds in the identical structure is maintained. However, this process can produce more stable ions or lead to a reorganization of the system to favour subsequent fragmentation processes.
A common fragmentation involving carbonyl compounds is the CO elimination from cyclic carbonyl compounds. In principle, two mechanisms are possible for this type of CO elimination. The first one (Fig. 4.8a) is a single-step mechanism that involves simultaneous (concerted) bond breaking and bond making events. This mechanism is the most often cited in the literature, notably for cyclic ketones. A second possible mechanism involves an equilibrium between the cyclic and ring-opened structures, followed by an internal attack (Fig. 4.8b). However, there is no theoretical evidence to support this mechanism to date.
As previously discussed, select molecules can lose one electron under ESI conditions, producing molecular ions (odd-electron ions).43 In addition, for select compounds, their even-electron ions generated in the ESI source can also produce radical cations (odd-electron ions) by homolytic cleavage under CID conditions. The fragmentation of radical species depends on the charge and radical site, which is important for the initiation of the proposed mechanism. Ions that exhibit separation between charge site and radical site are labelled as distonic ions (ions containing radical and ionic charges in different atoms in the same molecule). Fig. 4.9 shows the mechanism for radical fragmentation.
4 Charge migration fragmentations (CMF)
The CMF are fragmentation reactions in which the charge is displaced from the precursor ion. In general, CRF gas-phase reactions of negative ions are similar to those of positive ions because they occur in positions remote from the charge site. However, CMF in negative ions significantly differ from those of positive ions. In positive ions, CMF reactions usually eliminate a leaving group for which the charge site was initially the location of a neutral molecule. However, the neutral loss from negative ions typically occurs at the part of the ion structure that was initially charged (Fig. 6). In this review, the charge-migration fragmentation of positive and negative ions is discussed separately. | ||
| Fig. 6 Charge-migration fragmentation (CMF) reactions in positive (a) and negative (b) ions. | ||
4.1 CMF for positive ions
The first fragmentation reaction presented in the CMF section for positive ions is a simple inductive cleavage involving the heterolysis of a B–C bond in which AB bears the positive charge (Fig. 7.1.1). The mechanism of this gas-phase fragmentation reaction is similar to the ionization step of E1 and SN1 mechanisms; however, in this case, the solvent stabilizing effect on the resulting fragment ion does not exist. For this reason, the occurrence of this class of CMF reactions is strongly dependent on the stability of the product ion. Eliminations of CO, H2O or ROH by simple inductive cleavages can be favoured when they result in resonance-stabilized allylic and benzylic ions.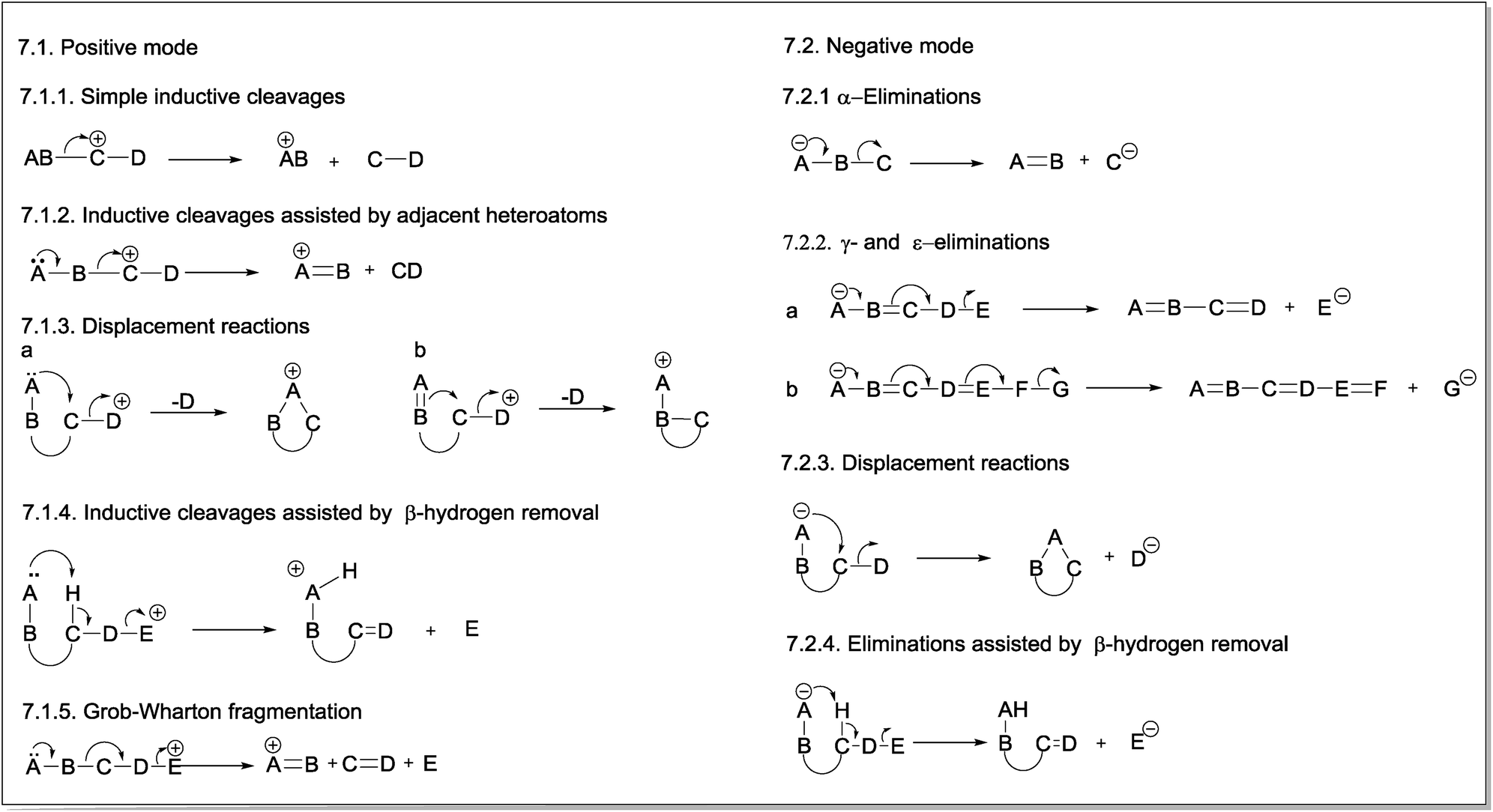 | ||
| Fig. 7 General mechanisms of charge migration fragmentations (CMF) in positive (7.1) and negative (7.2) modes. | ||
Inductive cleavages can also occur when a heteroatom bearing a lone pair is adjacent to the charge site in the resulting fragment ion. This heteroatom can overlap the p orbitals of the vacant orbital in the carbon, thereby stabilizing the structure of the fragment ion (Fig. 7.1.2).13
The third CMF presented in positive ions are displacement reactions, which are intramolecular processes in which the lone pair of a heteroatom (Fig. 7.1.3a) or, eventually, the π-electrons of an unsaturated group (Fig. 7.1.3b) of a neighbouring group (e.g., a spatially close group) assists the elimination of the group containing the positive charge, becoming a neutral molecule.13 The mechanism of these gas-phase fragmentation reactions is similar to the anchimeric assistance that forms intermediate ions (e.g., phenonium ions) in the solution phase. In this case, however, no further nucleophilic attack is noted on the structure of the resulting cyclic ion.
A heteroatom with a lone pair can also assist the inductive cleavage that leads to the elimination of the group bearing the positive charge as a neutral group by removal of the β-hydrogen (Fig. 7.1.4). The concerted nature of the reaction mechanism of this class of fragmentation suggests a similarity to the E2 mechanism occurring in solution. In this case, however, the process is unimolecular: the base and the leaving group (i.e., the charged group in the precursor ion, which will be eliminated as a neutral group) are in the same molecule.44 As a result of the elimination assisted by β-hydrogen removal, a new π-bond is formed in the resulting fragment ion.
Fig. 7.1.5 presents the last fragmentation proposed in this review for positive ions: the Grob–Wharton fragmentation. This reaction breaks an aliphatic or alicyclic chain into three fragments.45,46 The group containing the positive charge (E) is eliminated as a neutral molecule, whereas a new π-bond is formed between the C and D atoms, which are also eliminated. The positive charge in the product ion is stabilized by the lone electron pair of the heteroatom, and the occurrence of this reaction depends on the stability of the product ion. Considering that two bonds are formed and two other bonds are broken in a Grob–Wharton fragmentation, the energy requirements for this reaction suggest a concerted mechanism because the stabilizing effect of the solvent on cationic or anionic species (as reported for Grob–Wharton fragmentations in solution) does not operate in the gas phase.
4.2 CMF for negative ions
The simplest fragmentation reaction involving negative ions is an α-elimination in which the pair of electrons from the atom bearing the negative charge (A) is used to form a new π-bond with the adjacent atom (B) (Fig. 7.2.1). The bond between this atom and the next atom (C) is then broken; thus, this latter atom becomes negatively charged. Therefore, α-eliminations are the simplest fragmentation process occurring with negative ions in the gas phase.The second reaction for negative ions is the “γ-elimination”. This term was adopted to refer to a general class of reactions in which the neutral eliminated group has conjugated double bonds (Fig. 7.2.2). The group that will bear the negative charge in the product ion is usually in a γ-position; however, ε- or even ζ-eliminations can also occur, depending on the number of double bonds between the group with the negative charge in the precursor ion (A) and the group that will bear the negative charge in the product ion (E).
Displacement reactions can also occur with negative ions under CID conditions. The nucleophilic group involved in anchimeric assistance must be spatially close to the electrophilic site. In contrast to positive mode, the negative charge is transferred to the group being eliminated (Fig. 7.2.3).
Heteroatoms with lone electron pairs and other negatively charged groups that provide anchimeric assistance in displacement reactions can also act as Lewis bases and assist the formation of product ions by removing the β-hydrogen from the group that will bear the negative charge. This class of fragmentation reactions results in the formation of a new π-bond in the eliminated molecule (Fig. 7.2.4).
In this review article, examples for each type of CRF and CMF were selected to show the process of these reactions and to exemplify the characterization/elucidation of small molecules by MS/MS.
5 CRF examples
5.1 Remote hydrogen rearrangements
The most typical remote hydrogen rearrangements are those involved in dehydration reactions in compounds containing a hydroxyl group in their structures, such as saponins47 (4) and neoclerodane diterpenes48 (5). The elimination of sugar from glycosylated compounds, such as neoclerodane diterpenes (6),48 also occurs through remote hydrogen rearrangement (Fig. 8).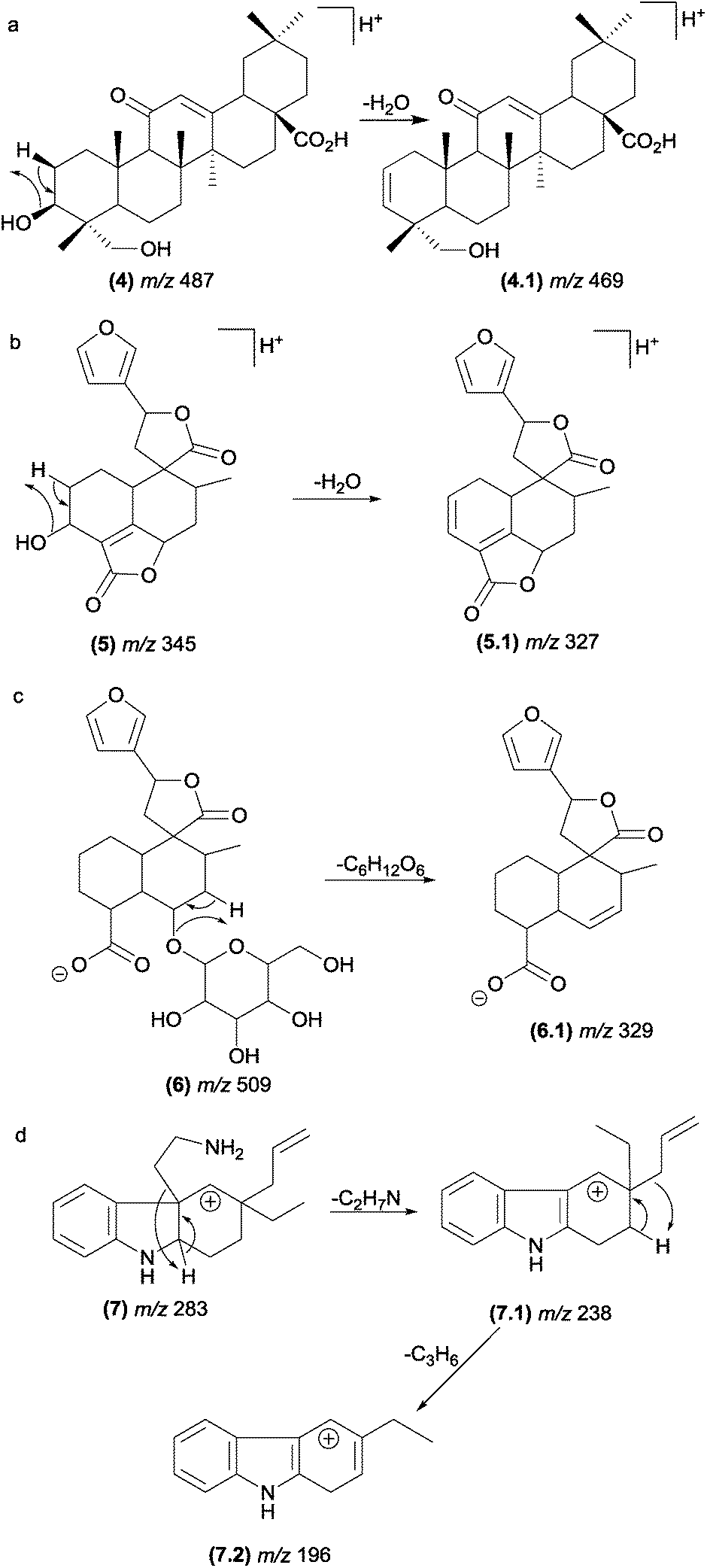 | ||
| Fig. 8 Remote hydrogen rearrangements involved in the dehydration of saponins (a), diterpenes (b), glycosylated diterpenes and protonated plumeran alkaloids with the loss of a chain side (d). | ||
Although these reactions are one of the most prevalent fragmentations in natural products, the relative intensities of the product ions in the MS/MS spectrum are not always high. The entropy factor places steric restrictions on the product formation because of the tighter four-membered transition state involved in this mechanism.13 However, these reactions can be energetically favoured when the π bond formed is adjacent to the charge site. Aguiar and co-workers reported that remote hydrogen rearrangements are the major CID fragmentation of protonated plumeran alkaloids isolated from Aspidosperma spruceanum.49 For these alkaloids (7), the rearrangements are driven by charge stabilization, and they result in resonance-stabilized ion structures (Fig. 8d). Remote hydrogen rearrangements have also been reported for phosphatidylglycerol,50 proteins51–56 and cyclic ethers.57
5.2 Retro-Diels–Alder (RDA) reactions
RDA reactions play a key role in the identification of flavonoids and their derivatives.58–68 The analysis of the resulting fragments allows determination of the groups attached to the structural core of flavonoids and the structural variation in this class of compounds. Rijke and co-workers69 considered RDA reactions as the most important fragmentation reaction for flavones, dihydroflavones, flavonols and isoflavones. The fragmentation in the C-ring may occur in all subclasses of flavonoids, and the RDA mechanism may follow pathways a, b and/or c (Fig. 9), according to the structural subclass.70,71 The double bond present in the C-ring of flavones (10), flavonols, chalcones and isoflavones leads to an additional RDA mechanism compared with dihydroflavones and dihydroflavonols.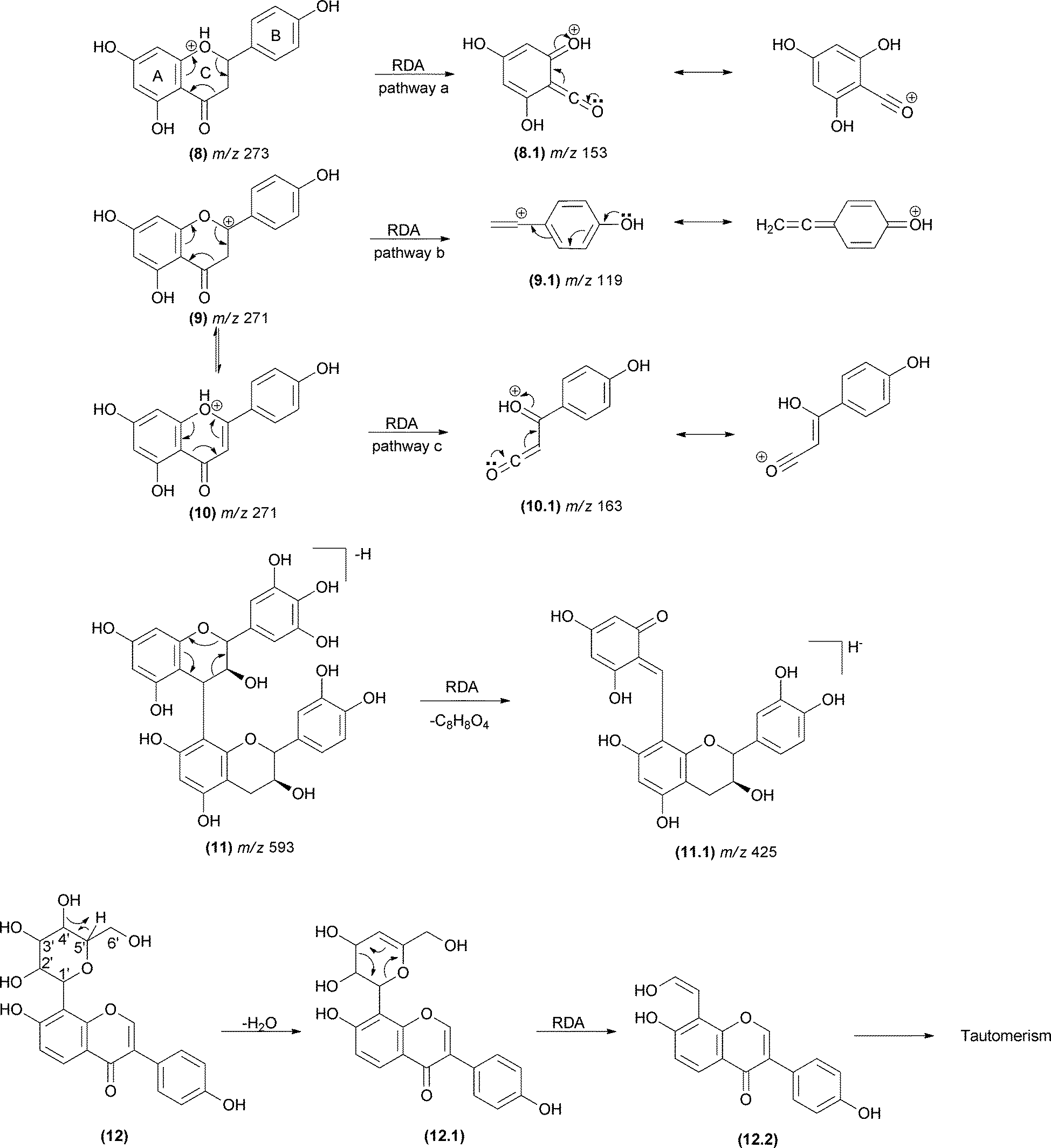 | ||
| Fig. 9 RDA mechanisms involving the C-ring of flavonoids (8–10) and proanthocyanidins (10) and sugar units in natural products (12). The double bond present in the C-ring of flavones, flavonols, chalcones and isoflavones leads to an additional pathway (pathway c). | ||
In proanthocyanidins (11) – the condensed tannins – the group attached to the A-ring is usually another catechol unit, which plays an important role in the elucidation of proanthocyanidin dimers (Fig. 9).72–74 The sugar unit of natural products may also fragment by RDA (12). Typically, a H2O loss precedes the RDA fragmentation by remote hydrogen rearrangement, forming an unsaturated moiety in the sugar unit (12.1), which enables the RDA process (Fig. 9). It is important to observe the preference of unsaturation formation between carbons 4′ and 5′ to form a more substituted alkene. Occasionally, the dehydrated fragment is not detected. The resulting enol fragment (12.2) presumably equilibrates to the keto form.75
The RDA fragmentation reactions are also reported for terpenoids that display a cyclohexene moiety (Fig. 10). In limonoids (13), the RDA occurs in the lactone ring as indicated in Fig. 10a.76 In this case, the elimination of a carbonyl fragment added to the elimination of a conjugated alkene (13.1) favoured the RDA mechanism. In saponins (14), the RDA occurs in the aglycone moiety, resulting in the major fragment ions of m/z 203 (14.2) and m/z 191 (14.1).77
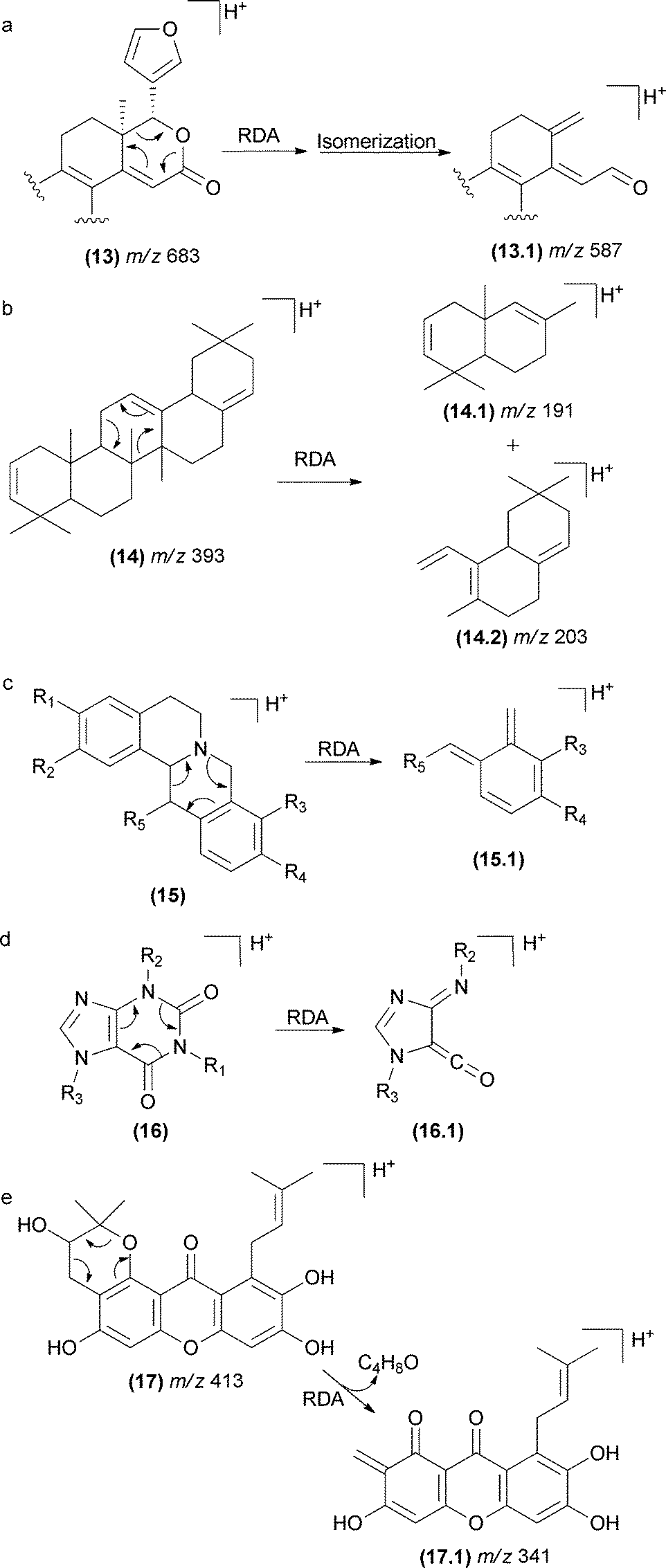 | ||
| Fig. 10 RDA fragmentation in terpenoids limonoids (a), saponins (b), isoquinolinealkoloids (c), caffeine (d), and polyprenylated xanthones (e). | ||
This class of fragmentation reactions has also been reported for isoquinoline alkaloids found in Chinese herbs78–80 (15). The double bond of the aromatic ring gives rise to the RDA fragmentation in this case. Other cases include caffeine81 (16), polyprenylated xanthones from Garcinia species82 (17), iridoids porphyrins83 and spirolide marine toxins (Fig. 10).84,85
5.3 Retro-ene reactions
The retro-ene fragmentation pathways can be found in deprotonated marine toxins (dinophysistoxins) (Fig. 11). In these structure types, these reactions occur with the participation of the exocyclic double bond at the E-ring (18). The negative charge is likely placed at the carboxylate moiety, but other fragment ions suggest that the charge can migrate to other less acidic sites due to the CID process. In addition to the retro-ene reaction, other typical charge-retention fragmentations, such as RDA at the B-ring, were also reported for these compounds.86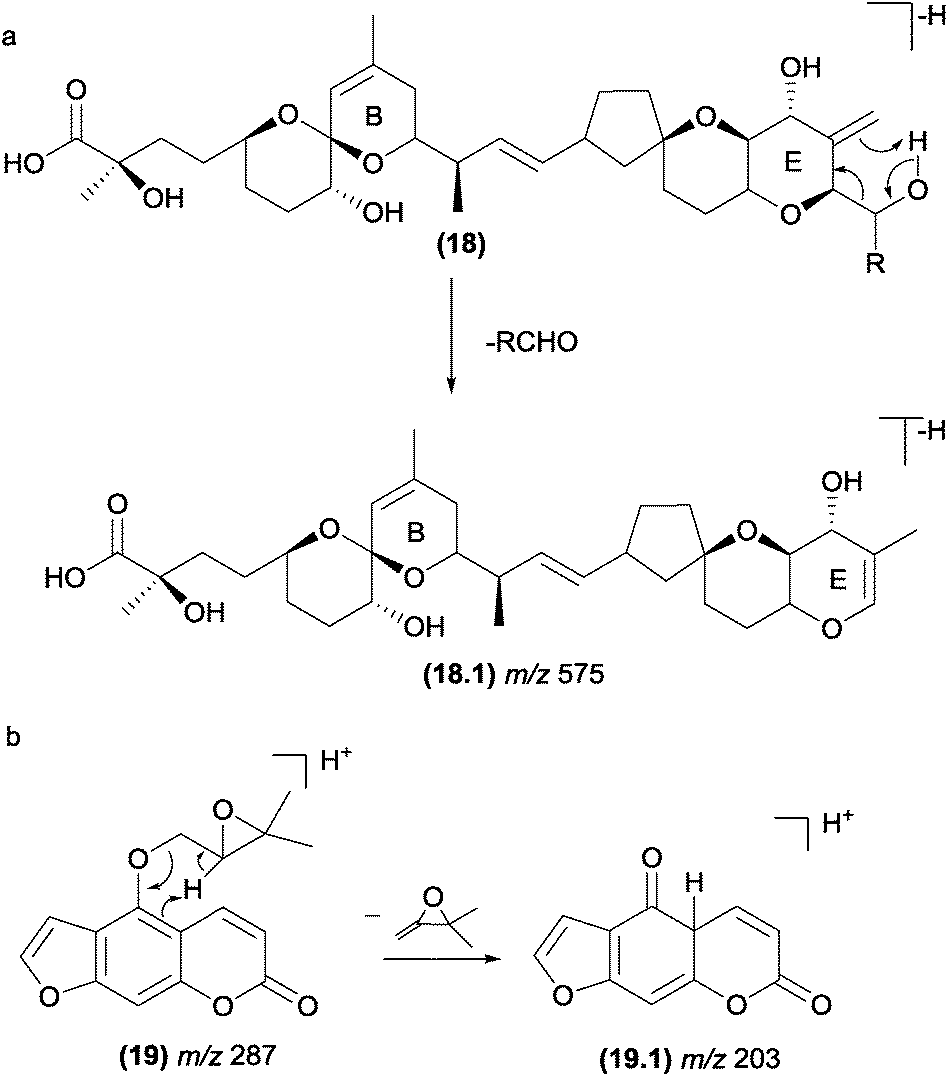 | ||
| Fig. 11 Retro-ene fragmentation reactions in marine toxins (a) and furanocoumarins (b). | ||
In natural products with a prenyl side chain, such as flavonoids, coumarins, and quinonoids, the elimination of the prenyl moiety involves a retro-ene reaction. Li and co-workers87 reported that retro-ene reactions involved in the elimination of the oxygenated prenyl chain from protonated prenylated furanocoumarins (19) occurs through the participation of the double bond of the aromatic ring and results in the fragment ion of m/z 203 (Fig. 11b). The authors demonstrated that this fragmentation pattern could help to identify and differentiate a variety of substituents on C-5 and C-8.
A retro-ene reaction was also reported for long-chain unsaturated fatty acids, which helped to determine the double bond position and has been discussed in several mass spectrometry studies.88–92
5.4 Retro-heteroene reactions
In retro-heteroene reactions when A* (Fig. 4.4) is an oxygen atom, retro-heteroene fragmentations are composed of two major groups of reactions: McLafferty-type rearrangements93 and retro-Aldol reactions.94 These reactions are common in natural products possessing carbonyl groups in their structures, such as ketones, esters, carboxylic acids and amides. Part of the driving force for these reactions is provided by the formation of the extremely strong O–H bond.13McLafferty-type rearrangements have been reported for a diversity variety of natural products possessing carbonyl groups. This reaction is also involved in the formation of the fragment ions of polyether ionophore antibiotics (20) from Streptomyces sp. AMC 23 (Fig. 12a).93,95 Li and co-workers reported the occurrence of McLafferty-type hydrogen rearrangements for the elimination of the prenyl chain from prenylated furanocoumarins.87 In Fig. 12b, a gas-phase Claisen rearrangement is followed by a McLafferty-type rearrangement, leading to the elimination of the prenyl chain. This process is energetically favoured by re-aromatization of the B-ring. Other terpenoids with side chains can also exhibit McLafferty-type eliminations.21,96
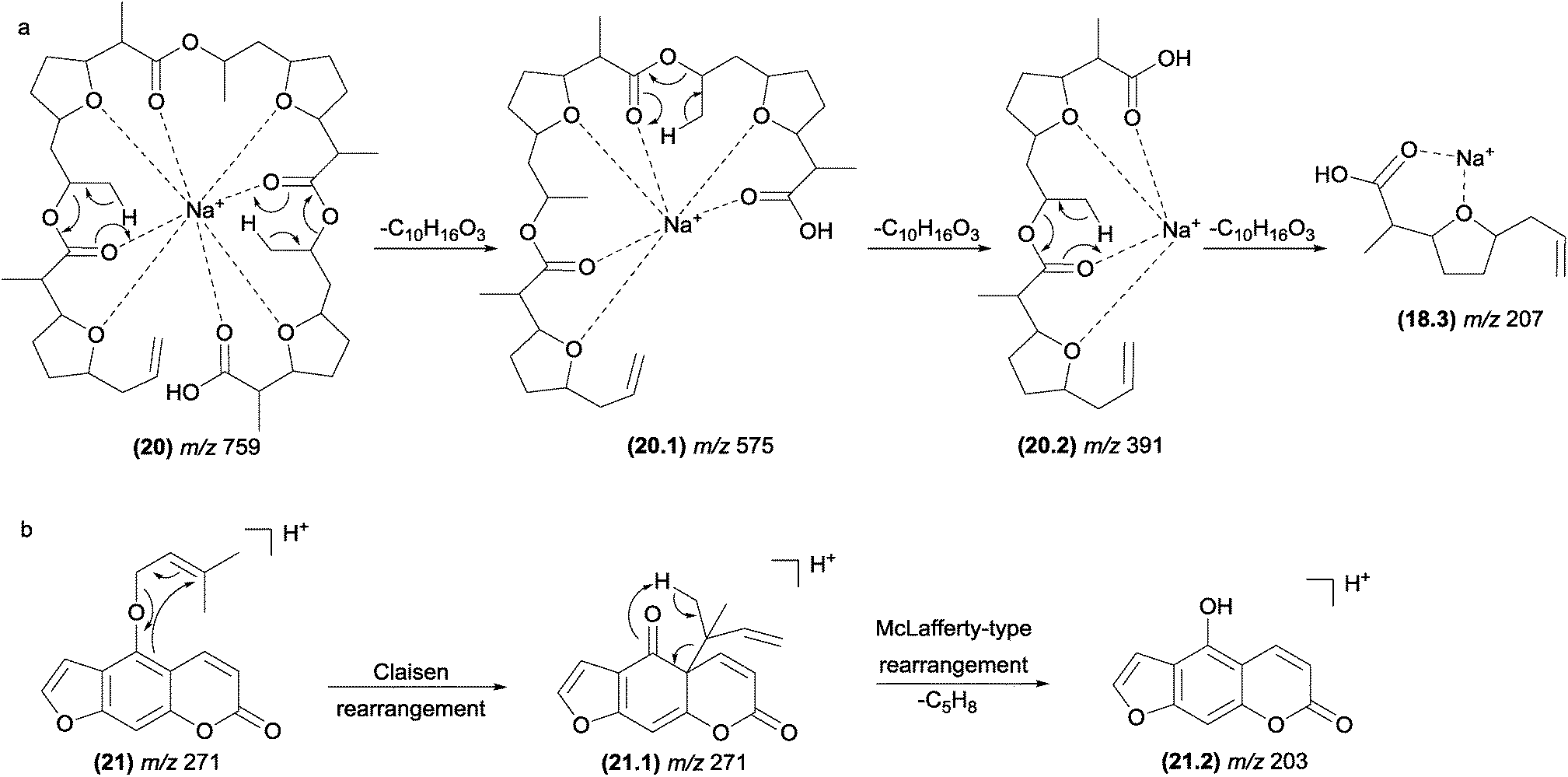 | ||
| Fig. 12 McLafferty-type rearrangements in polyether ionophore antibiotics (a) and furanocoumarins (b). | ||
In furanoheliangolides, such as budlein (22), this rearrangement occurs with the participation of the vinylic methyl at the angelate ester and leads to the formation of the m/z 293 (22.1) product ion (Fig. 13).21
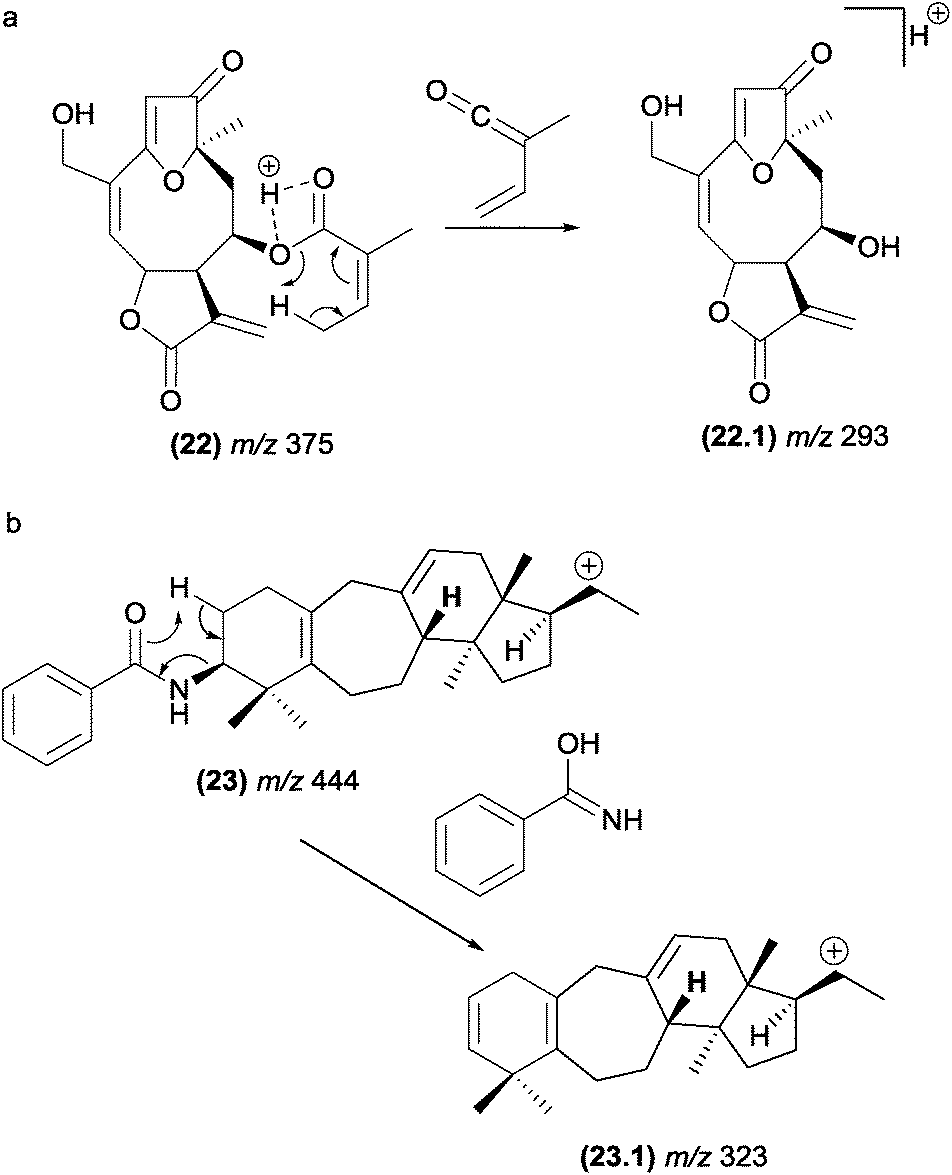 | ||
| Fig. 13 Retro-heteroene reactions in furanoheliangolides (a) benzoyl eliminations in steroidal alkaloids (b). | ||
Benzoyl groups attached to the basic skeleton of natural products are often described as being eliminated due to McLafferty-type rearrangements. For example, these rearrangements were reported to be involved in the formation of benzoyl amide (fragment ion, m/z 323, (23.1)) from steroidal alkaloids found in Buxus papillosa (Fig. 13).83
One of the most common fragmentation processes in glycosylated natural products is the elimination of the sugar moiety. When the aglycone unit has a carbonyl close to the sugar, a McLafferty-type rearrangement is expected to be involved in this elimination. Fig. 14a illustrates the sugar loss from chamaedryoside (24), a neo-clerodane diterpene isolated from Teucrium chamaedrys, which has sterically favourable conditions for the occurrence of a McLafferty-type rearrangement.48
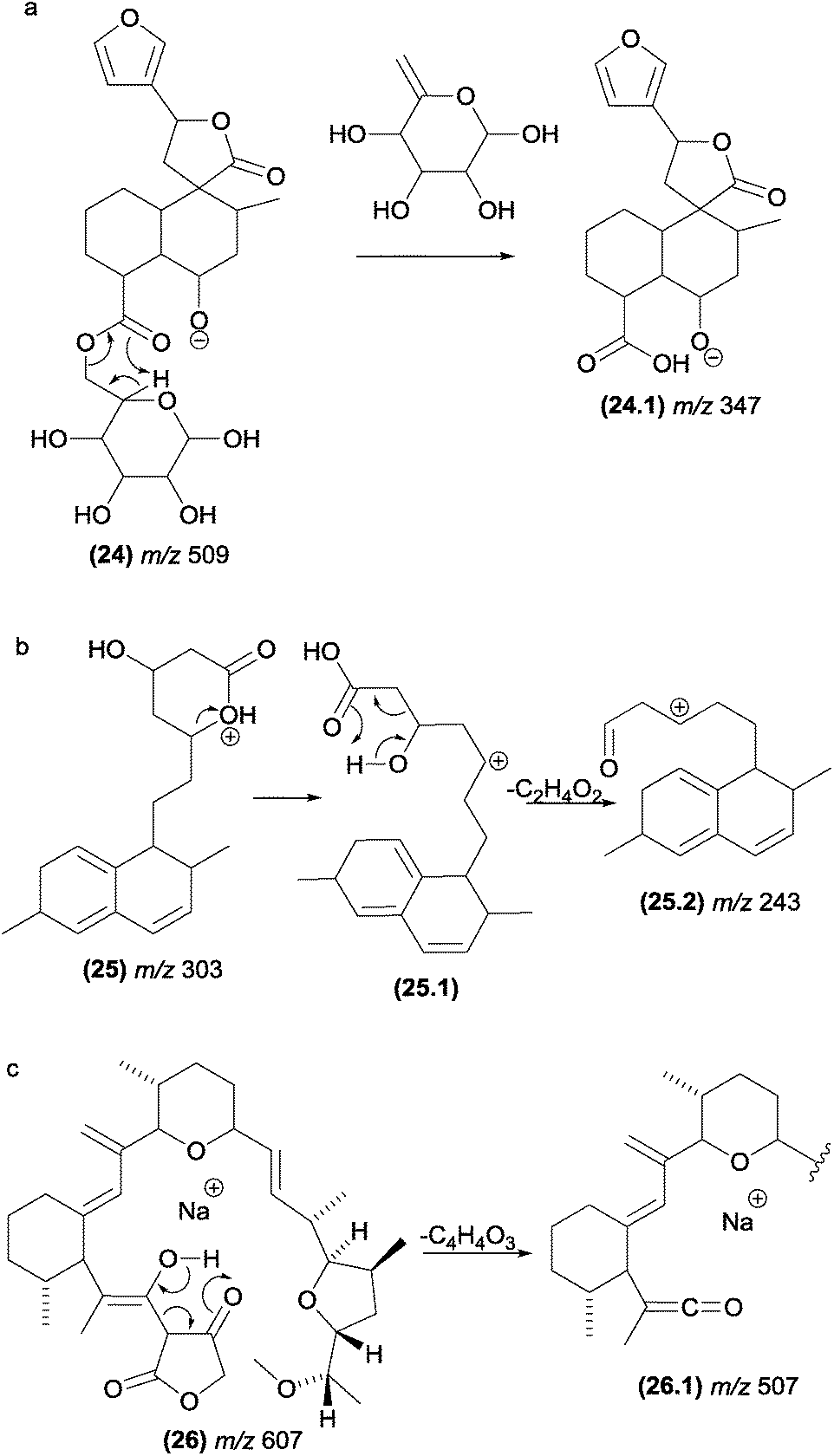 | ||
| Fig. 14 McLafferty-type rearrangements involved in the eliminations of sugar moieties in natural products (a) and retro-aldol fragmentation reactions in simvastatin/lovastatin (b) and tetrosin (c). | ||
Wang and co-workers studied the fragmentation of simvastatin (25) and lovastatin,97 two drugs responsible for lowering plasma cholesterol levels (Fig. 14b). The authors reported that the protonation occurred on the carbonyl oxygen of the lactone moiety. However, under CID conditions, the proton migrates to the ester oxygen, opening the ring. The protonation of the carbonyl oxygen and further proton migration to the ester oxygen with subsequent opening of lactone rings under CID conditions have also been supported by thermochemical data obtained through computational chemistry.98 The formation of the m/z 243 (25.2) product ion of simvastatin and lovastatin was proposed to occur from the m/z 303 fragment ion as a result of a retro-aldol fragmentation.
This class of retro-heteroene fragmentation reactions is often reported for β-hydroxy carbonyl compounds such as tetronasin (26), a polyether ionophore antibiotic isolated from Streptomyces longisporus (Fig. 14c).99
5.5 Charge remote fragmentations
Charge remote fragmentations typically occur in compounds possessing saturated long-chains, such as alkyl amines, carboxylic acid, fatty alcohols, acids, and esters.32,100 In the case of unsaturated fatty acids, allylic bonds are preferentially broken by charge remote fragmentations, providing information about the double bond position and substitution sites (Fig. 15a).37,101,102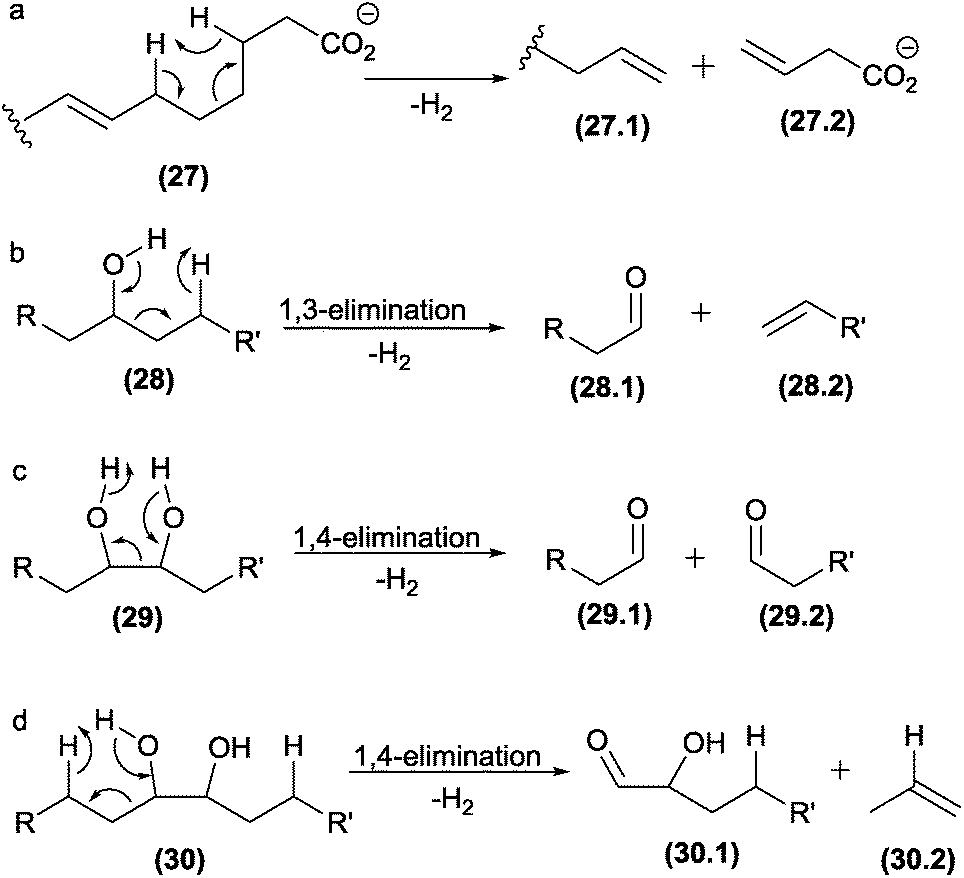 | ||
| Fig. 15 Charge-remote fragmentation reactions in fatty acids (a), and oxidized diacylphosphatidylcholines (b–d). | ||
Charge remote fragmentations were investigated in diacylphosphatidylcholines submitted in an oxidation process through a Fenton reaction.90 The oxidation introduced two hydroxy groups into the long chain, which were involved in C–C cleavages by different charge remote fragmentations, highlighting 1,3- and 1,4-eliminations (Fig. 15b–d).
5.6 Aromatic eliminations
Previous studies noted an unusual elimination of the aromatic ring from select polyenes, such as carotenoids;38 retinoids;38 mycotoxins103,104 and macrocyclic compounds.38 A systematic investigation of these reactions was performed by Guaratini and co-workers; the authors found that the aromatic elimination occurs in positive (protonated and sodiated molecules) and negative (deprotonated molecules) ionization modes.38 In addition, these elimination reactions were reported to occur with molecular ions (M+˙) generated by ESI and under CID conditions, thus confirming that a CRF reaction is involved in these processes.5The similarities between the chemical properties of conjugated cyclic compounds (with no continuous p-orbital overlap) and their acyclic counterparts are well established.12 Thus, the identical valence tautomerism observed for cyclo-octatetraene105 at room temperature can also occur for acyclic homologue structures.38 Finally, macrocyclic structures, such as amphotericin and nystatin, confirm the in source isomerization ability of cyclic structures to eliminate the identical neutral groups. Structures such as rapamycin (31), which has only three conjugate double bonds, can also obtain the minimum requirements (four double bonds) after methanol elimination (Fig. 16).38
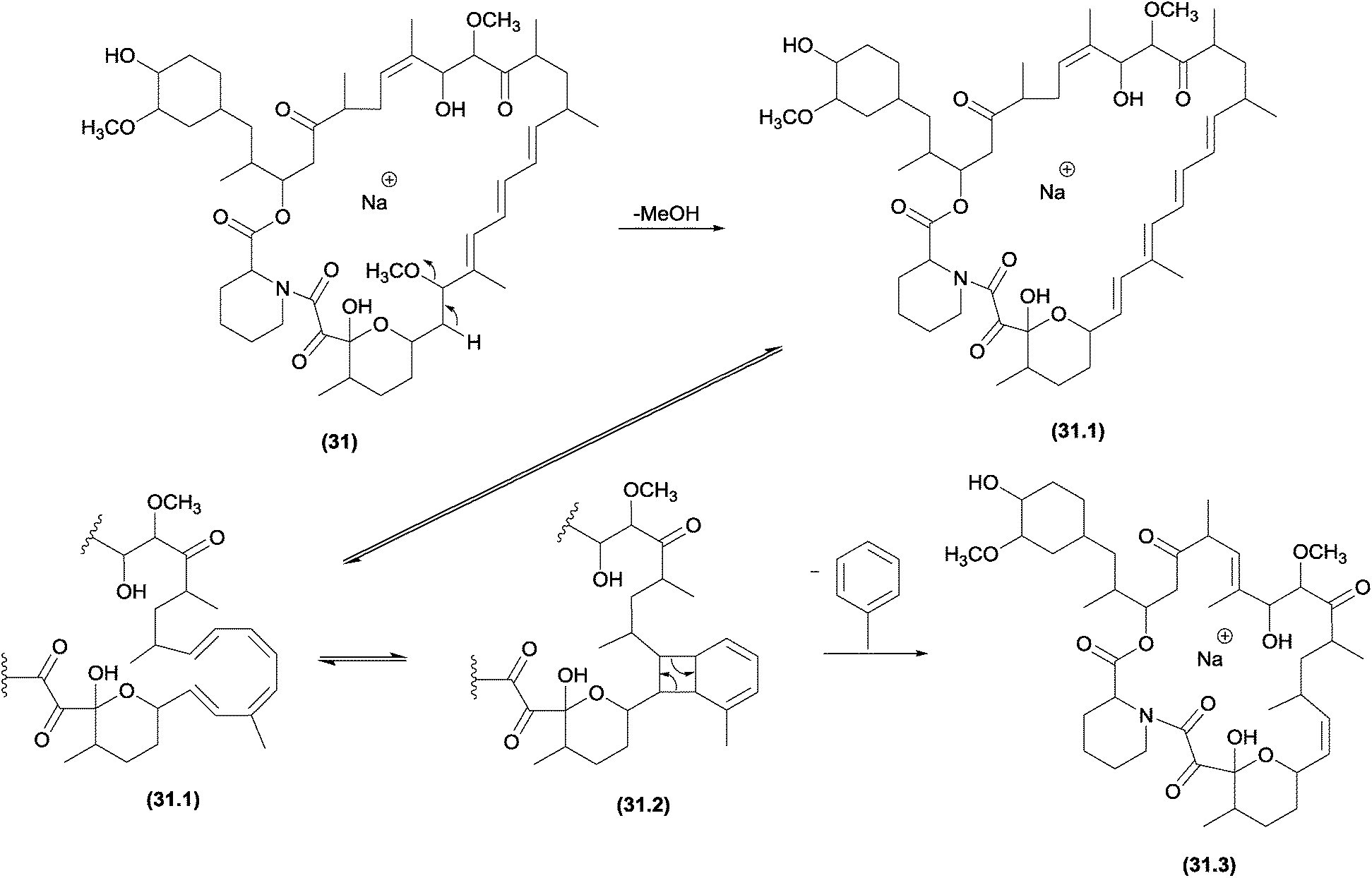 | ||
| Fig. 16 Fragmentation pathway of rapamycin, in which the initial intramolecular 1,2-elimination of MeOH yields the basic requirement for the pericyclic aromatic elimination. | ||
5.7 Other pericyclic processes
The first cycloreversion process that will be discussed is a fragmentation involving cyclohexane rings. Recently, cycloreversions in cyclohexane rings in CID experiments were reported for alkaloids (Fig. 17).49,106 In the case of diterpene alkaloids (Fig. 17b), this cycloreversion leads to the formation of the m/z 253 product ion. The elimination of C2H4 (28 Da) requires distinguishing accurate-mass measurements from CO elimination. Other examples may be found in molecules with no double bonds in cyclohexane rings.77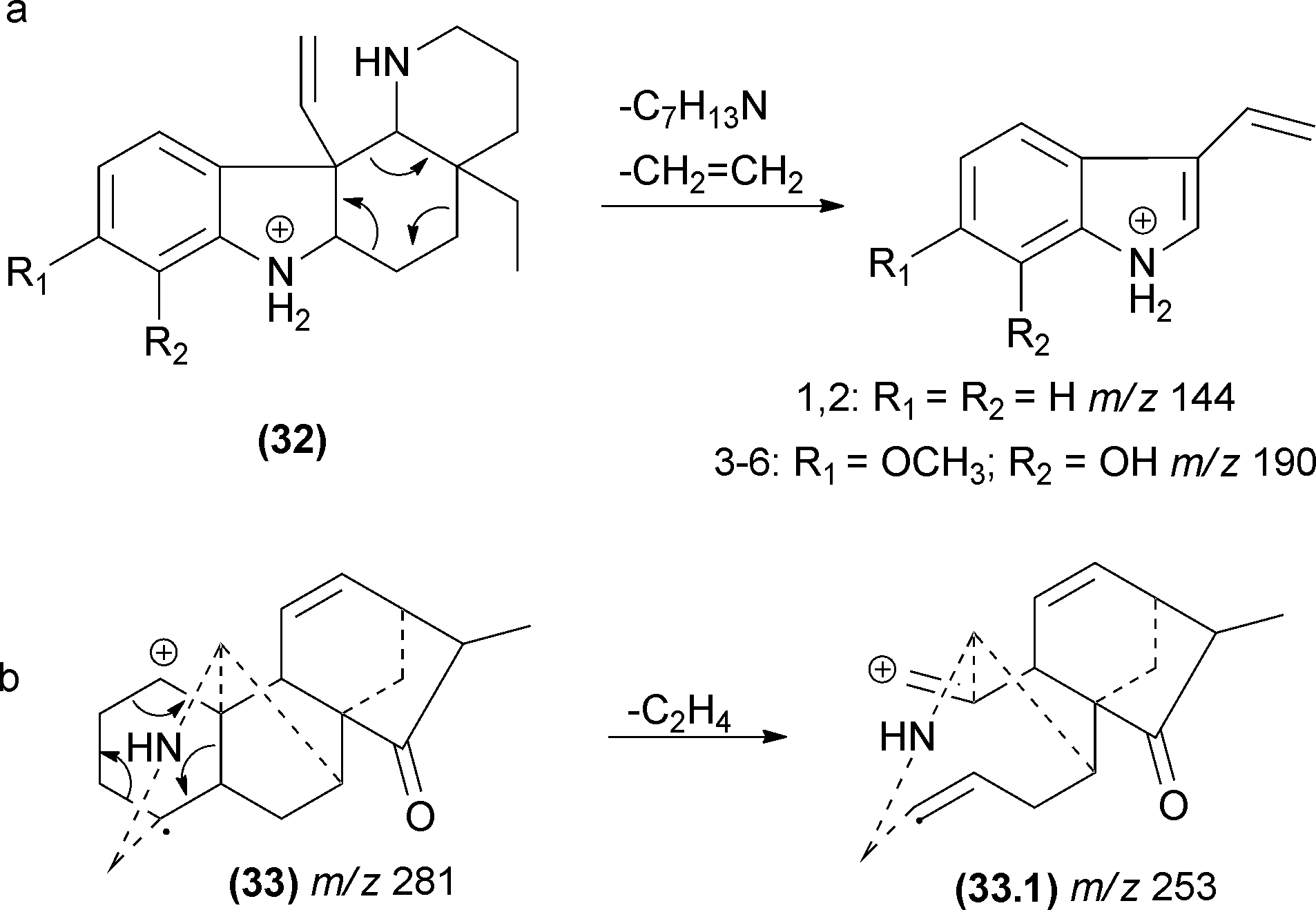 | ||
| Fig. 17 Pericyclic cycloreversions involving cyclohexane rings. | ||
Another important pericyclic process was recently reported by Pinto and co-workers.107 The occurrence of these hydrogen sigmatropic rearrangements was reported to occur during the fragmentation of Velosia diterpenes.107 Water elimination was reported from the first ion, and the only possibilities without a rearrangement are the formation of a cyclic alkene moiety or a vinylic carbocation. Both proposals require severe restrictions to occur; therefore, another process must be responsible for this water elimination. Detailed analyses of a diterpene series allowed the authors to confirm the 1,3-hydrogen shift by a sigmatropic rearrangement before the water elimination (Fig. 18). Elimination of water from the initial ion structure could not result in a stable ion structure. However, a 1,3-shift (sigmatropic rearrangement) alters the double bond position, thus allowing the ion to lose water by a remote hydrogen rearrangement.
 | ||
| Fig. 18 1,3-Shift rearrangements in diterpenes allowing sequential water elimination through a stable carbocation. | ||
5.8 Carbon monoxide eliminations from cyclic carbonyls compounds
Several natural products contain cyclic ketones, lactones and lactams moieties in their structures. Two elimination reactions may occur in this group: (a) water elimination after an internal addition or (b) CO elimination to afford a ring contraction product. Gates and co-workers prepared labelled samples of erythromycin containing 18O to confirm the water elimination mechanism after carbonyl protonation.108 This unexpected reaction normally does not compete with the CO elimination and occurs only under special conditions. However, the CO elimination due to a ring contraction is a common process in terpenoids,109 iridoids,61 alkaloids,78,110 quinones,111–113 and flavonoids,114 among other structures.Fabre and co-workers published a systematic investigation of flavonoids in ESI-MSn (negative mode) and organized the fragmentation of the unusual losses of CO, CO2 and C3O2, in addition to the retro Diels–Alder mechanism (RDA).115 Considering the CO elimination from the central ring, different influences are observed between flavanones and flavanonols (no double bond between carbons 2 and 3) and flavones and flavonols (with a double bond at carbons 2–3). This first group has the typical moiety expected for the general mechanism proposed in Fig. 4.8. In this case, we presented in Fig. 19 the possible initial equilibrium displayed in Fig. 4.8b, but both reactions can occur (35 and 36). Furthermore, for flavone and flavonols, the vinylic carbanion may restrict the CO elimination for this mechanism, suggesting a displacement reaction similar to that in Section 6.1.3.
 | ||
| Fig. 19 CO elimination in flavonoids. | ||
5.9 Radical fragmentation
In radical fragmentation, the most common process that converts an even-electron ion into an odd-electron ion is the elimination of ˙CH3 from flavonoids (in negative and positive modes),116 antraquinones117 and terpenoids.118 To clarify this process, Vessecchi and co-workers applied experimental and computational analysis to calculate the energies of the radical elimination of ˙H and ˙CH3 in isoflavonoids.43 Radical species form above an Elab of 20 eV, and the calculated enthalpies of the ˙CH3 elimination were approximately 46 kcal mol−1 for both of the methylated isoflavonoids analysed. These observations confirmed the radical elimination as a low energy process. For other non-methylated isoflavonoids, ˙H elimination from the deprotonated molecule [M − H − H]−˙ can also occur but may require higher energy than ˙CH3 elimination in methylated isoflavonoids.43 Additionally, after ˙CH3 or ˙H elimination, a second ˙H elimination may occur to reinstate the closed-shell system (Fig. 20).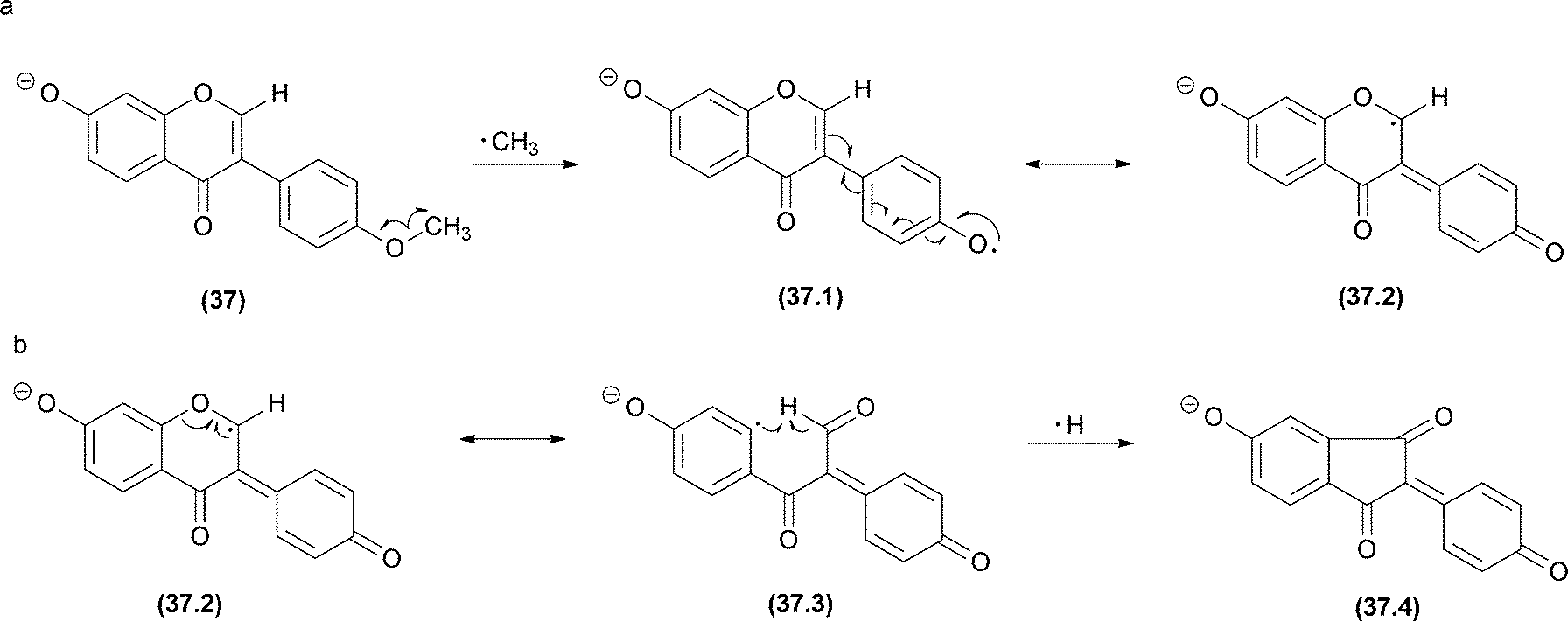 | ||
| Fig. 20 Biochanin A radical fragmentations. (a) The major pathway starts with ˙CH3 radical elimination. (b) The minor ion is produced by ˙H elimination. | ||
The identical reaction for ˙CH3 was also observed in non-phenolic compounds, such as mycosporine-like amino acids (MAA) (Fig. 21).119 Again, theoretical calculations in addition to high-resolution mass spectrometry allowed a rationalization of the gas phase mechanism. The authors applied natural bond orbital (NBO) and atoms in molecules (AIM) calculations to explain [(M + H) − 15]+˙ in a MAA moiety. The ion was generated by the cleavage of the ether bond through a reinforcement of the C(7; alkene)–O(14) bond and the consequent weakening of the O(14)–C(15; methyl) bond (Fig. 21). For MAA with an extra acid function, the characteristic methyl radical loss was not observed in the positive mode.120 This result can be easily explained because the homolytic cleavage was found to be dependent on the weakening of the O–C bond after the protonation and on the CO2 elimination to reduce the anion internal energy.
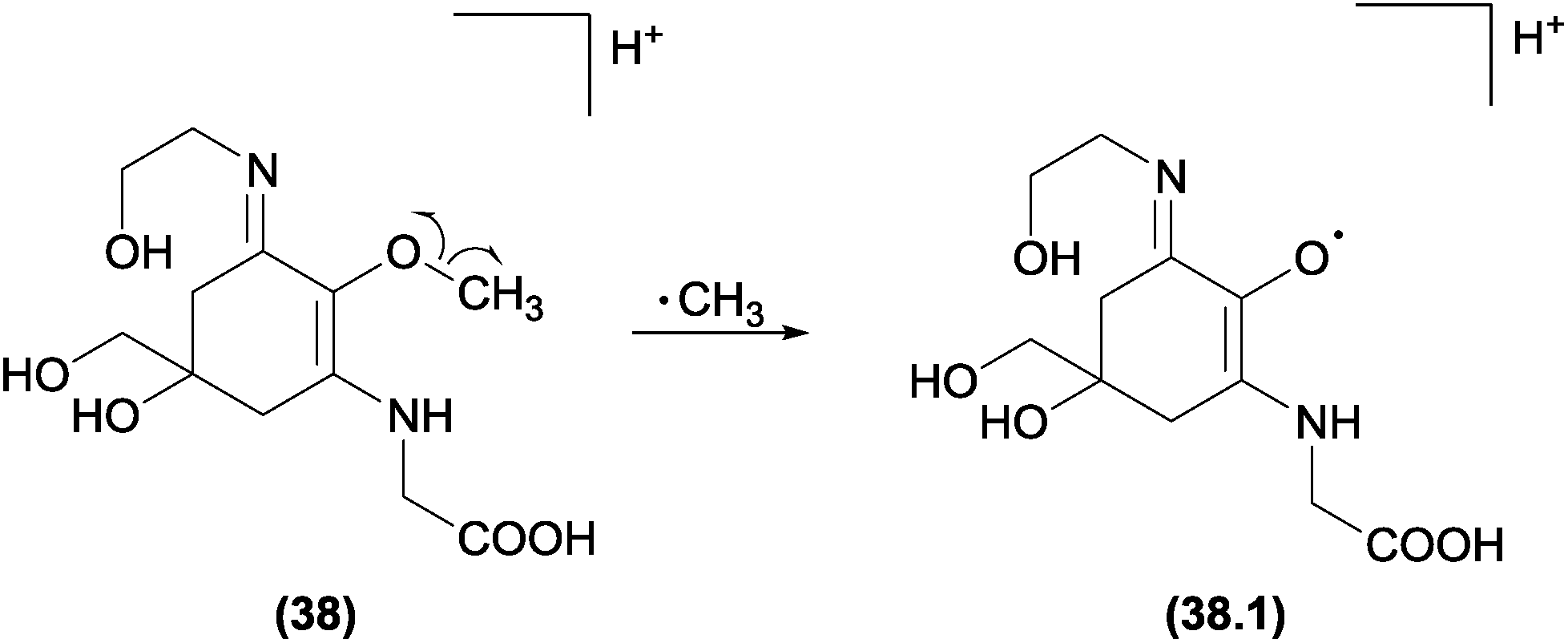 | ||
| Fig. 21 Asterin radical fragmentations. All sequential major fragments observed in the spectra were products from the ion afforded by ˙CH3 elimination. | ||
6 CMF examples
6.1 Charge migration fragmentations (CMF) in positive ions
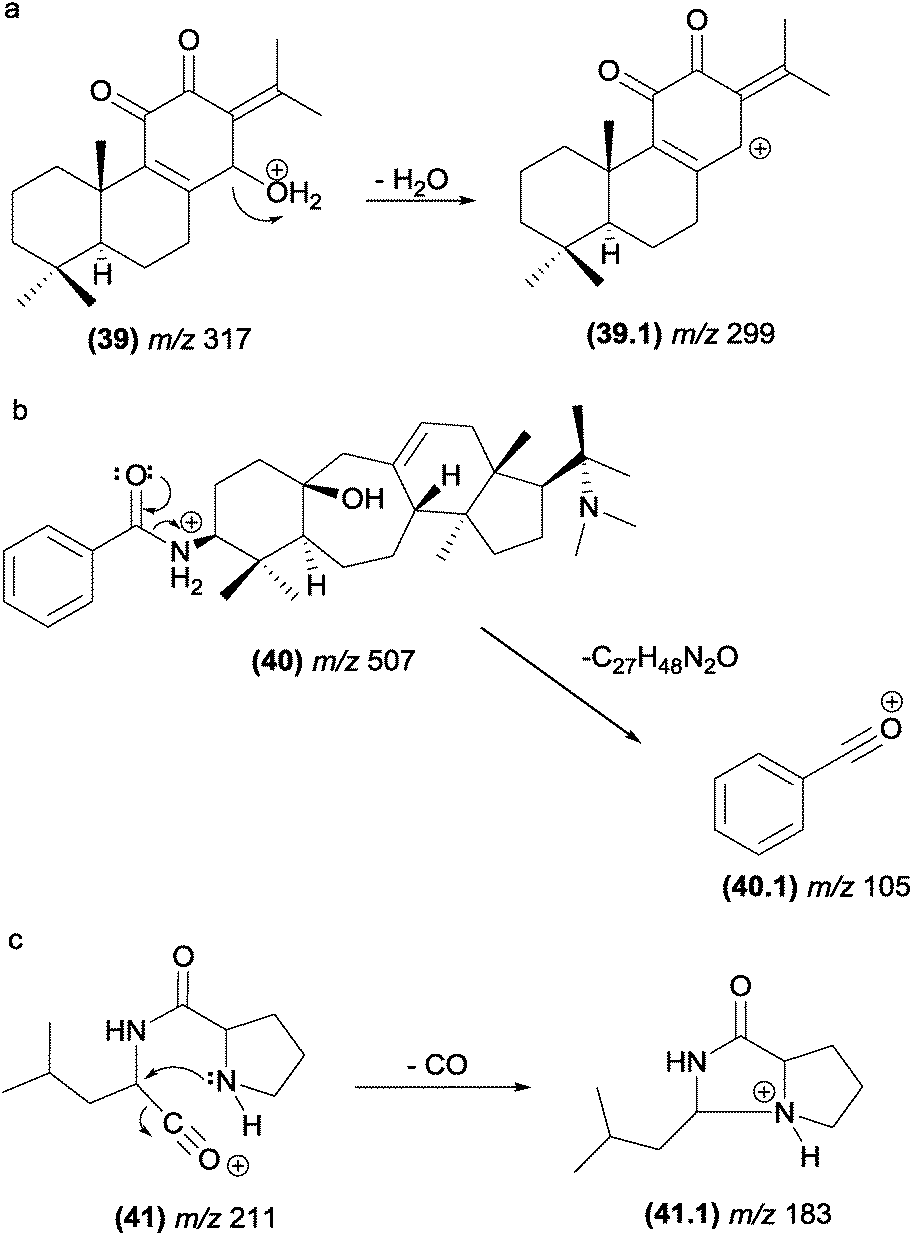 | ||
| Fig. 22 (a) Dehydration in diterpenes by simple inductive cleavage. (b) Eliminations assisted by adjacent heteroatoms in alkaloids and (c) elimination of CO from diketopiperazines through a displacement reaction. | ||
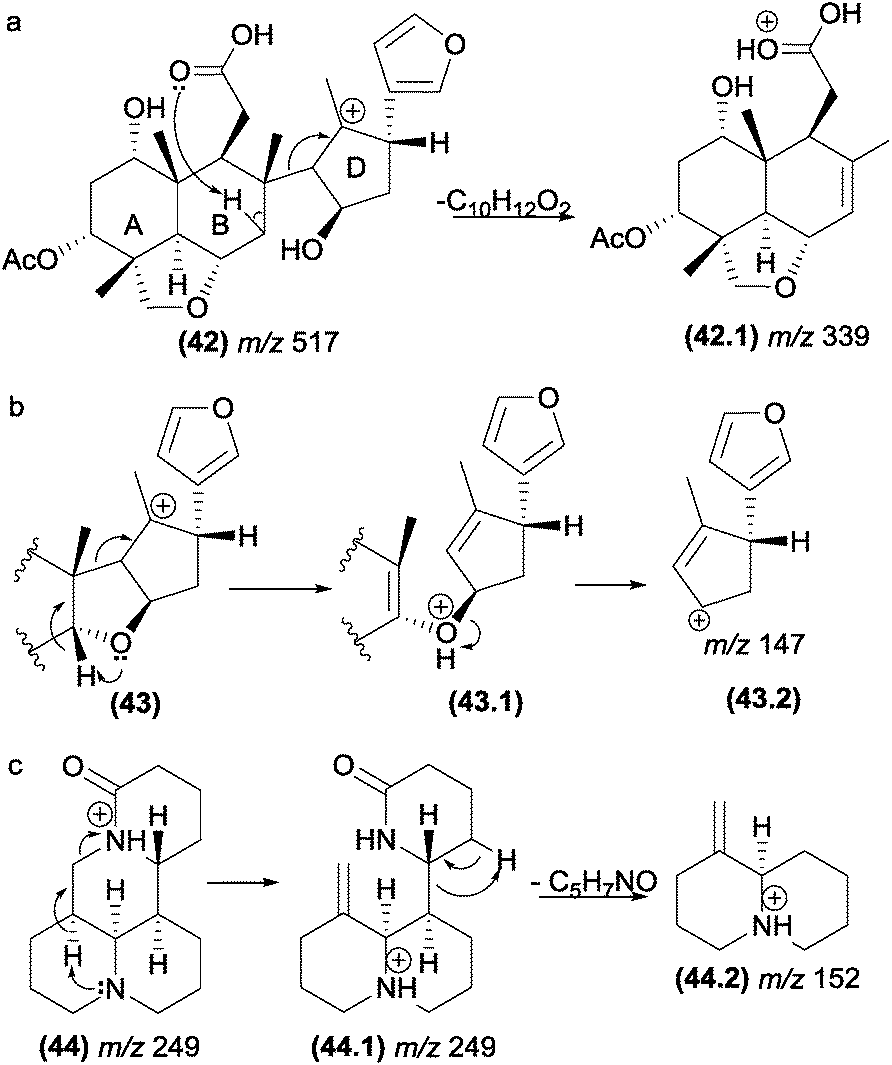 | ||
| Fig. 23 Inductive cleavages assisted by β-hydrogen removal in triterpenes (a and b) and alkaloids (c). | ||
This class of fragmentation reactions can also be involved in the formation of intermediate ion structures of salannin-like triterpenoids (Fig. 23b). In the first step of this reaction, an inductive cleavage assisted by β-hydrogen removal of the oxygen atom occurs to break a carbon–carbon bond. This facilitates the second step of the reaction, which is promoted by a simple inductive cleavage to generate the fragment m/z 147 (43.2).
In alkaloids such as sophoridine (44), the nitrogen acts as a basis for the removal of the β-hydrogen of the protonated nitrogen; this protonated nitrogen is in the detected fragment m/z 152 (44.2), which is formed in the second step by a remote hydrogen rearrangement.125
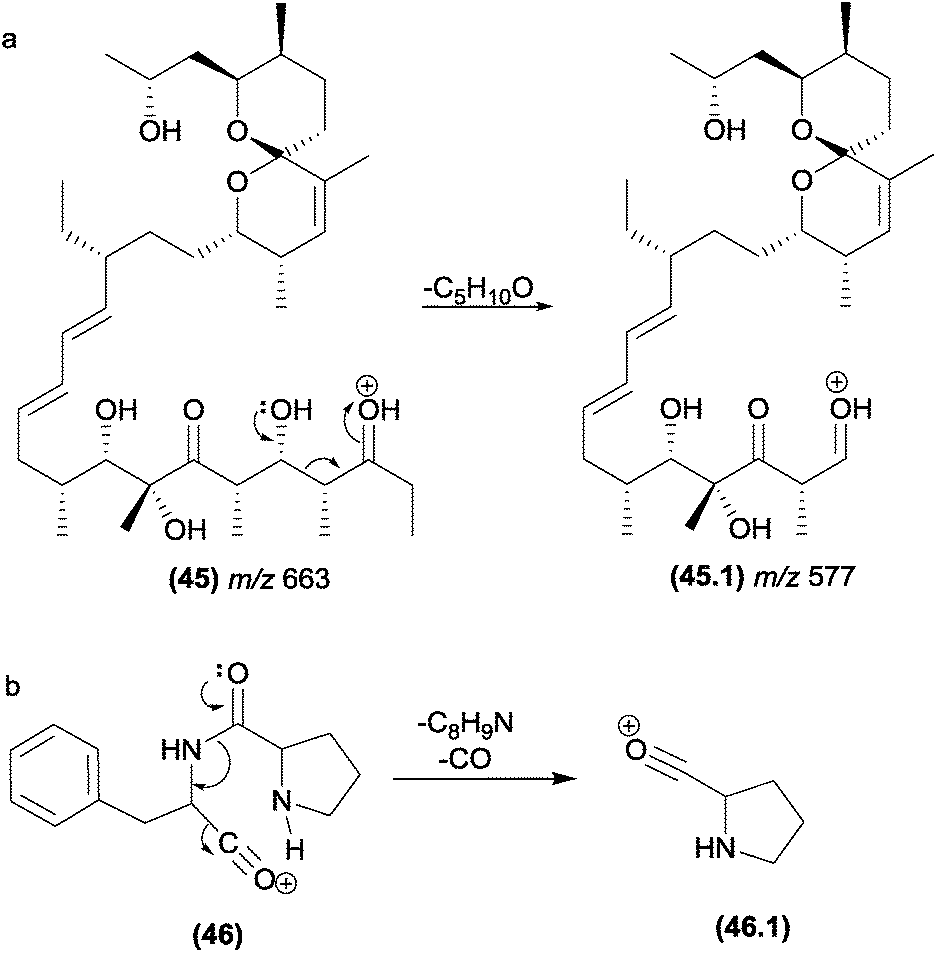 | ||
| Fig. 24 Grob–Wharton fragmentation in polyketide (a) and diketopiperazines (b). | ||
6.2 Charge migration fragmentations (CMF) in negative ions
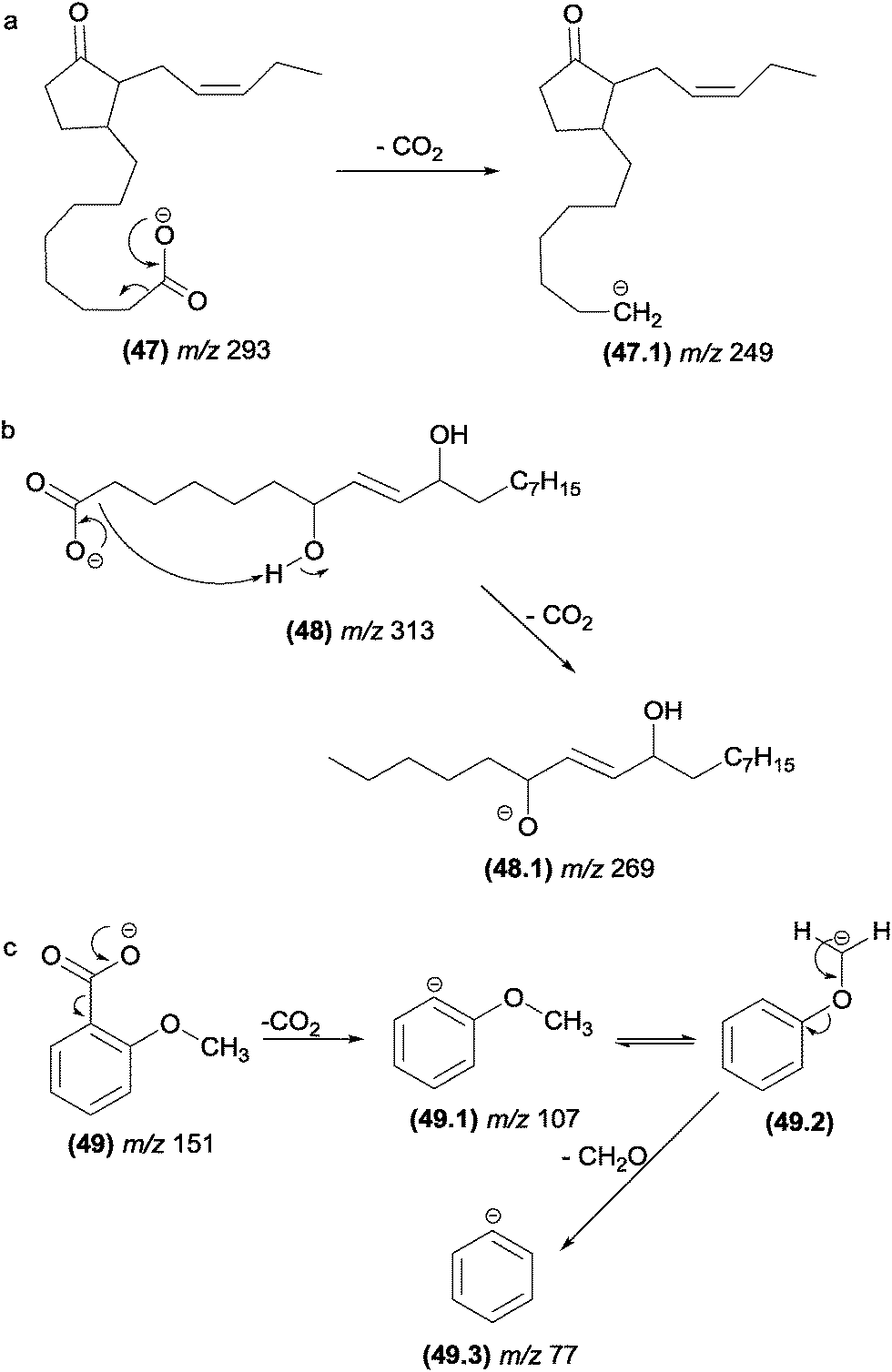 | ||
| Fig. 25 α-Eliminations in cyclopentenoneoxylipins (a), fatty acids (b) and methoxybenzoate anions (c). | ||
The elimination of formaldehyde from deprotonated compounds containing methoxyl groups attached to aromatic rings has been reported for many classes of natural products. Herath and co-workers129 investigated the fragmentation of deprotonated dimethoxybenzoic acid derivatives and proposed that the elimination of carbon dioxide and formaldehyde involves α-elimination (Fig. 25c). For natural products possessing functional groups more acidic than methoxyl, the elimination of formaldehyde is likely to be preceded by proton transfer under CID conditions.
The α-eliminations can be energetically favoured when the negative charge in the product ion is stabilized by resonance. In Fig. 26, the structure of the resonance stabilized enolate ion (m/z 675) resulting from an α-elimination is similar to those of products of retro-aldol reactions.94
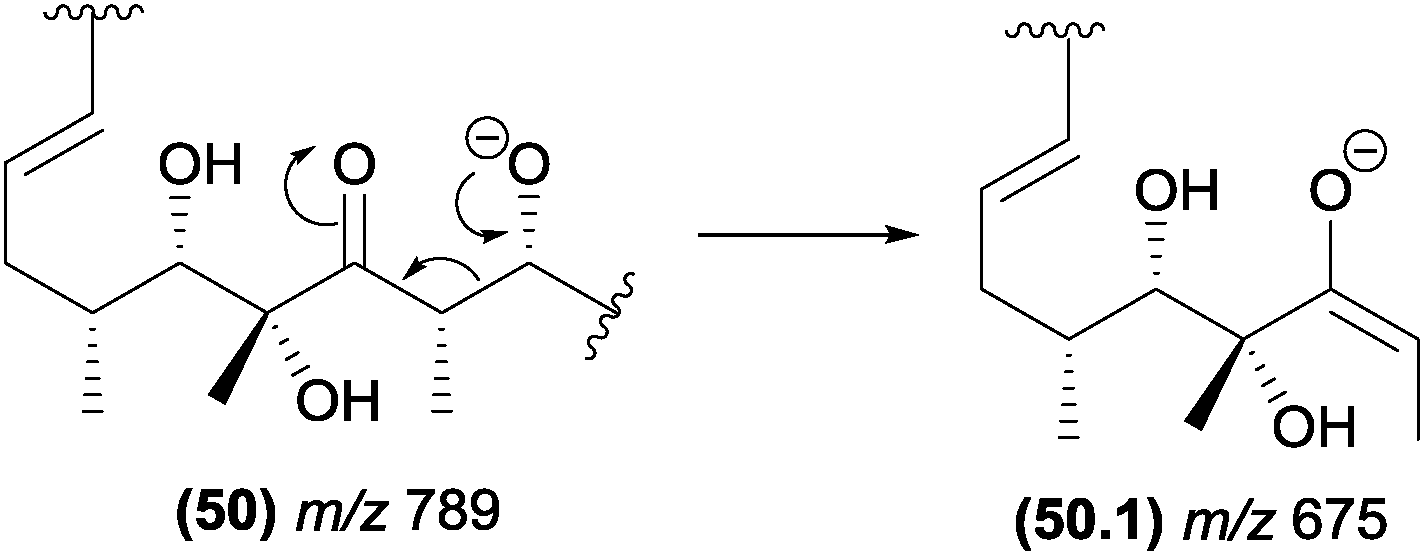 | ||
| Fig. 26 α-Elimination in polyketides leading to the formation of the resonance stabilized m/z 675 product ion. | ||
Eliminations of CO from negative ions can also result from α-elimination fragmentation reactions. In Fig. 27a, the simultaneous eliminations of CO (28 u) and C8H4O3 (148 u) from the flavonoid structure results in the m/z 109 product ion, which is stabilized by resonance.
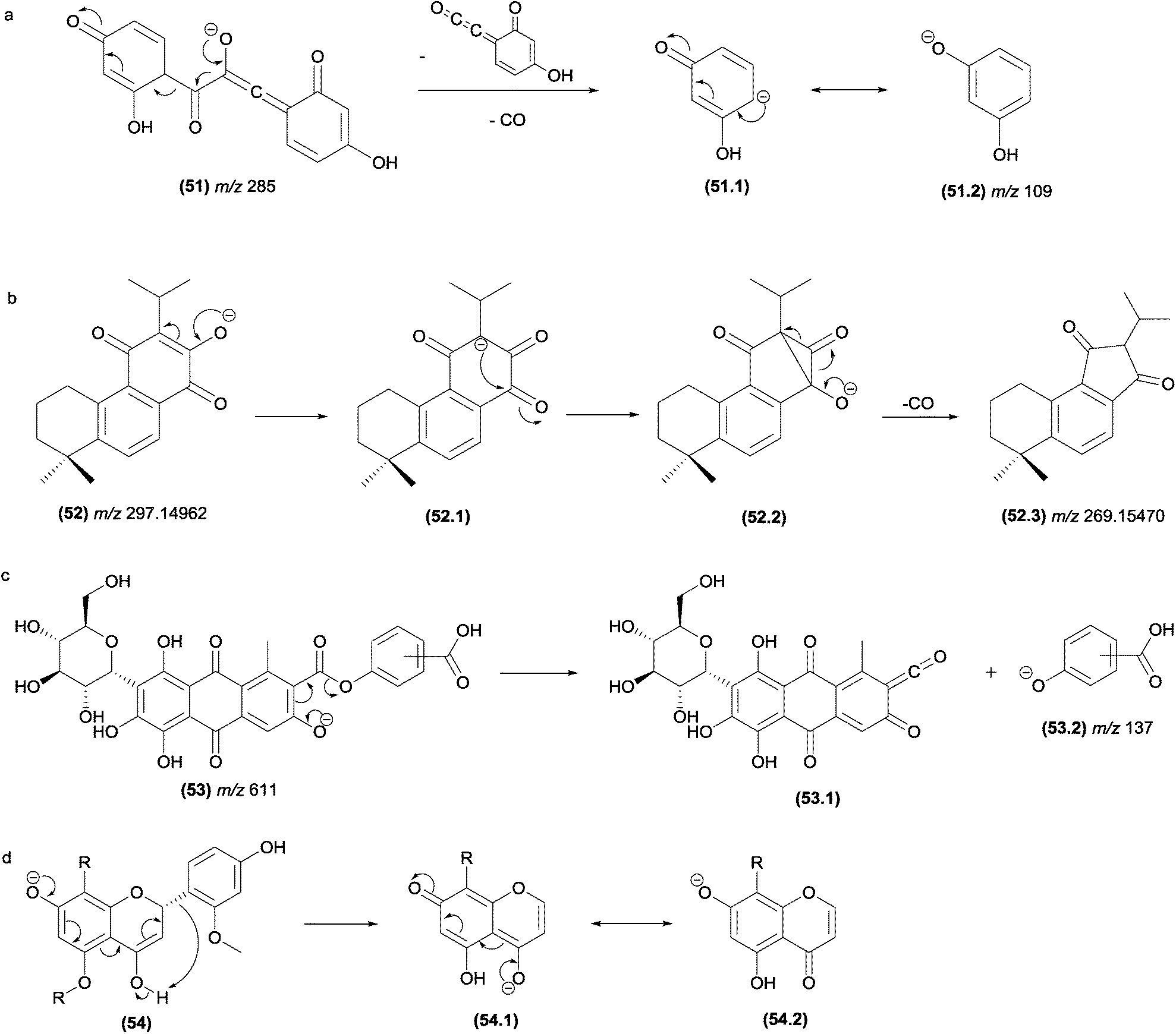 | ||
| Fig. 27 α-Eliminations resulting in the loss of CO in flavonoids (a) and abietane-type diterpenes. γ-Elimination in antraquinones (c) and ζ-elimination in flavonoids (d). | ||
Musharraf and co-workers investigated the fragmentation of abietane-type diterpenoid salvialeriafone and related compounds using high-resolution electrospray ionization tandem mass spectrometry (HR-ESI-MS/MS).121 The authors reported the formation of the m/z 269.15470 product ion from its m/z 297.1462 precursor ion involving the formation of a three-centred intermediate ion, from which the CO elimination occurs by α-elimination (Fig. 27b).
In flavonoids (Fig. 27d), the fragmentation in the C-ring resulting from a ε-elimination or ζ-elimination in addition to the typical RDA fragmentation allows the identification of substituents attached to the rings.131
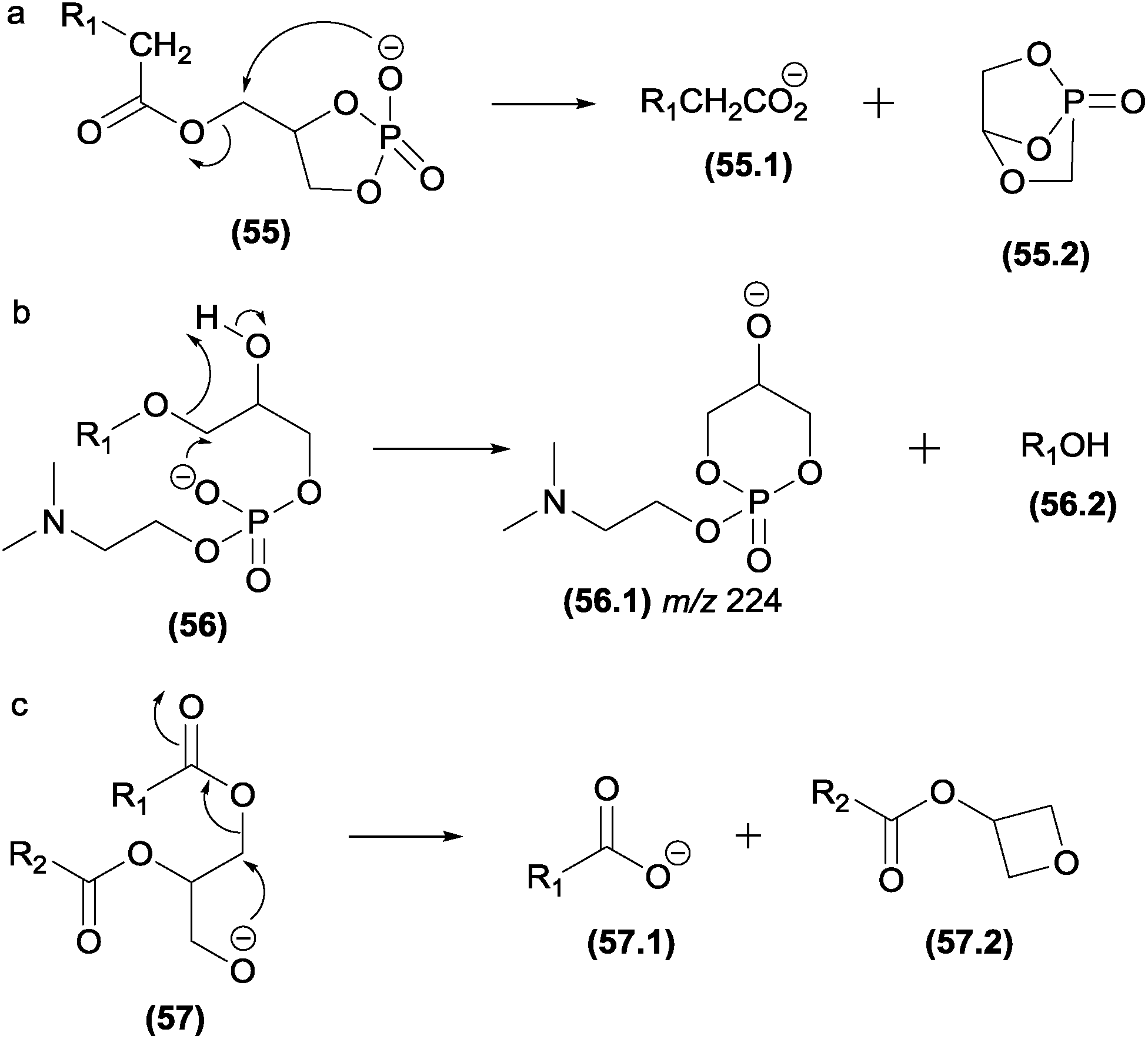 | ||
| Fig. 28 Displacement reactions in diacylglycerophosphaticid acid (a), glycerophosphocholines (b) and diacylglycerols (c). | ||
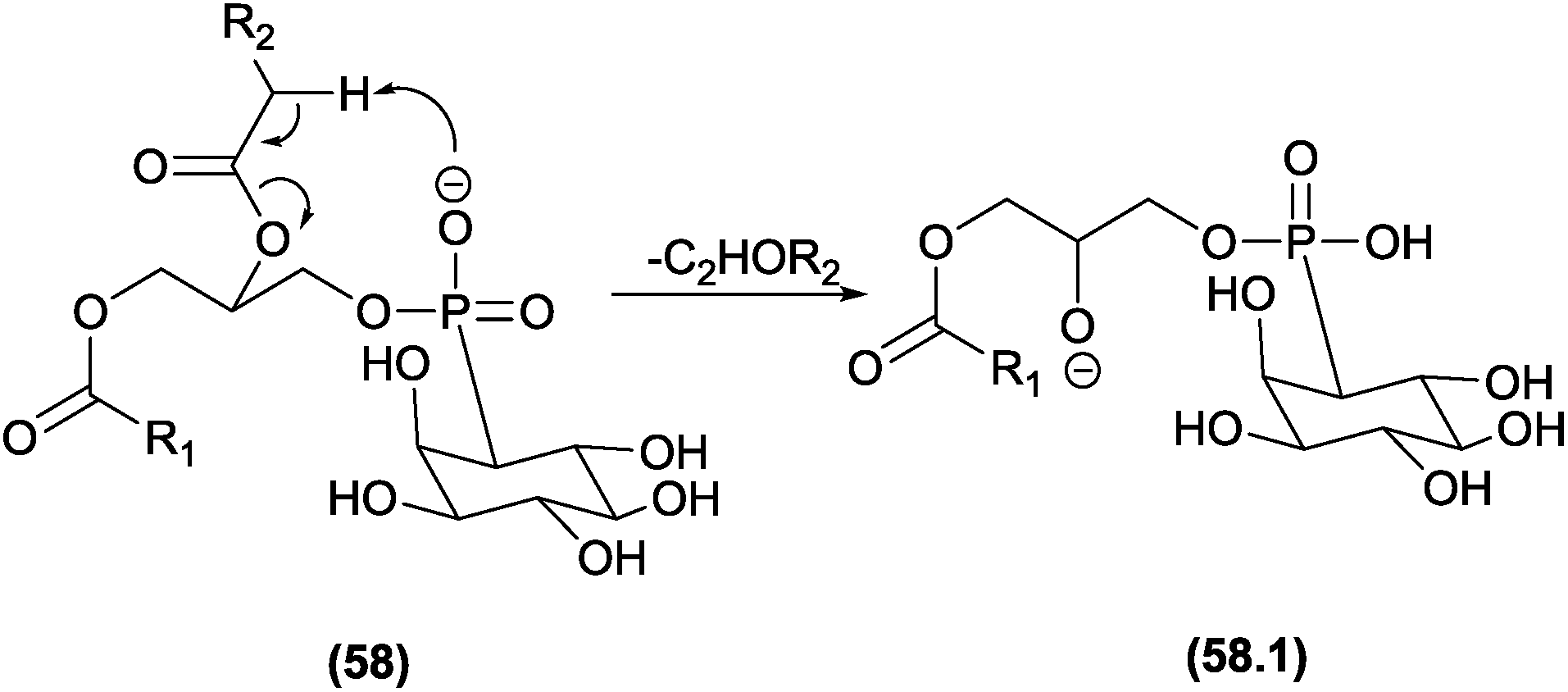 | ||
| Fig. 29 Eliminations assisted by β-hydrogen removal in phosphatidylinositol. | ||
7 CRF versus CMF
The competition between CRF and CMF reactions in the gas phase under CID conditions has been investigated for select classes of organic compounds, and the effects of this competition have been reported. In cases in which CRF and CMF originate from the identical fragment ion, experiments using isotopic exchange are mandatory. This strategy was used by Cydzik and co-workers to investigate the competition between CRF and CMF in peptides derivatized as quaternary ammonium salts.136 According to the MS/MS spectra obtained from peptides containing deuterium in place of all the exchangeable hydrogen atoms, the authors demonstrated that both CRF and CMF reactions occur in the fragmentation of these compounds.Hsu and Turk investigated the fragmentation pathways of diacylglycerophosphoethanolamine and the influence of steric effects on the competition between CRF and CMF reactions.132 In Fig. 30, the configuration of the double bond plays a key role in this competition: removal of the α-carbonyl hydrogen by the phosphate group follows the CMF route and produces the m/z 140 fragment ion, whereas for the CRF route, the carbonyl must remove a vinylic hydrogen through a McLafferty-type rearrangement. The E (trans) configuration favours the McLafferty-type rearrangement and does not allow removal of the α-carbonyl; therefore, the m/z 176 product ion is not observed. However, the Z (cis) configuration favours the α-carbonyl hydrogen removal and does not allow the McLafferty-type rearrangement.
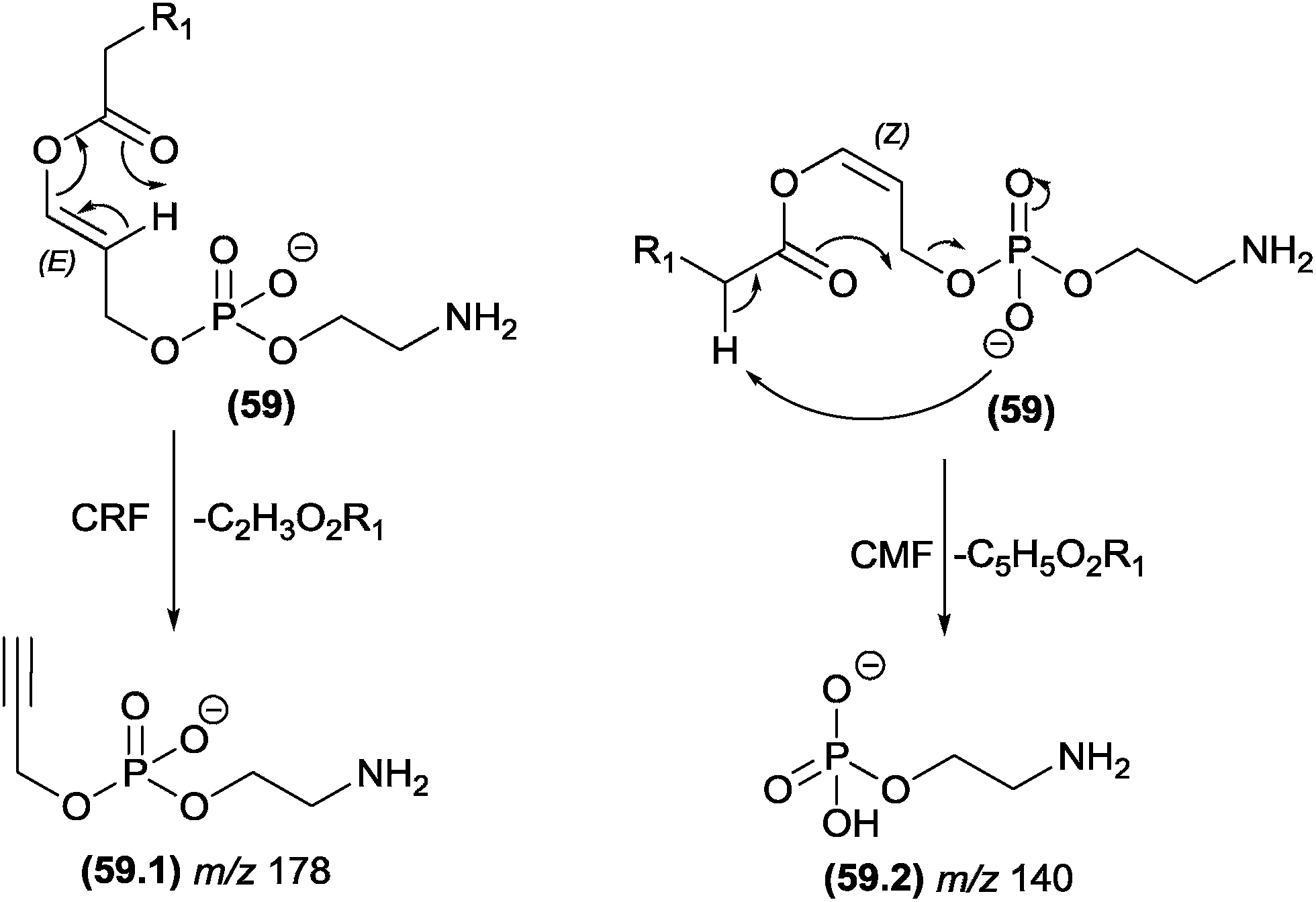 | ||
| Fig. 30 Competition between CRF and CMF in glycerophosphoethanolamine. | ||
Select authors have reported the effect of the structural stability of the product ion reaction pathways between CRF and CMF. Denekamp and co-workers137 investigated the fragmentation of triphenylphosphonium cations using molecules with different substituents at the aromatic ring. The authors reported that an electron withdrawing group (e.g., −NO2) in the para position favoured CRF reactions (Fig. 31a), whereas electron releasing groups (e.g., −CH3) at the identical position favoured the CMF reactions (Fig. 32b). The tendency of electron releasing groups to favour CMF reactions is related to their ability to stabilize the positive charge in the transition state involved in the cleavage of the benzylic bond.
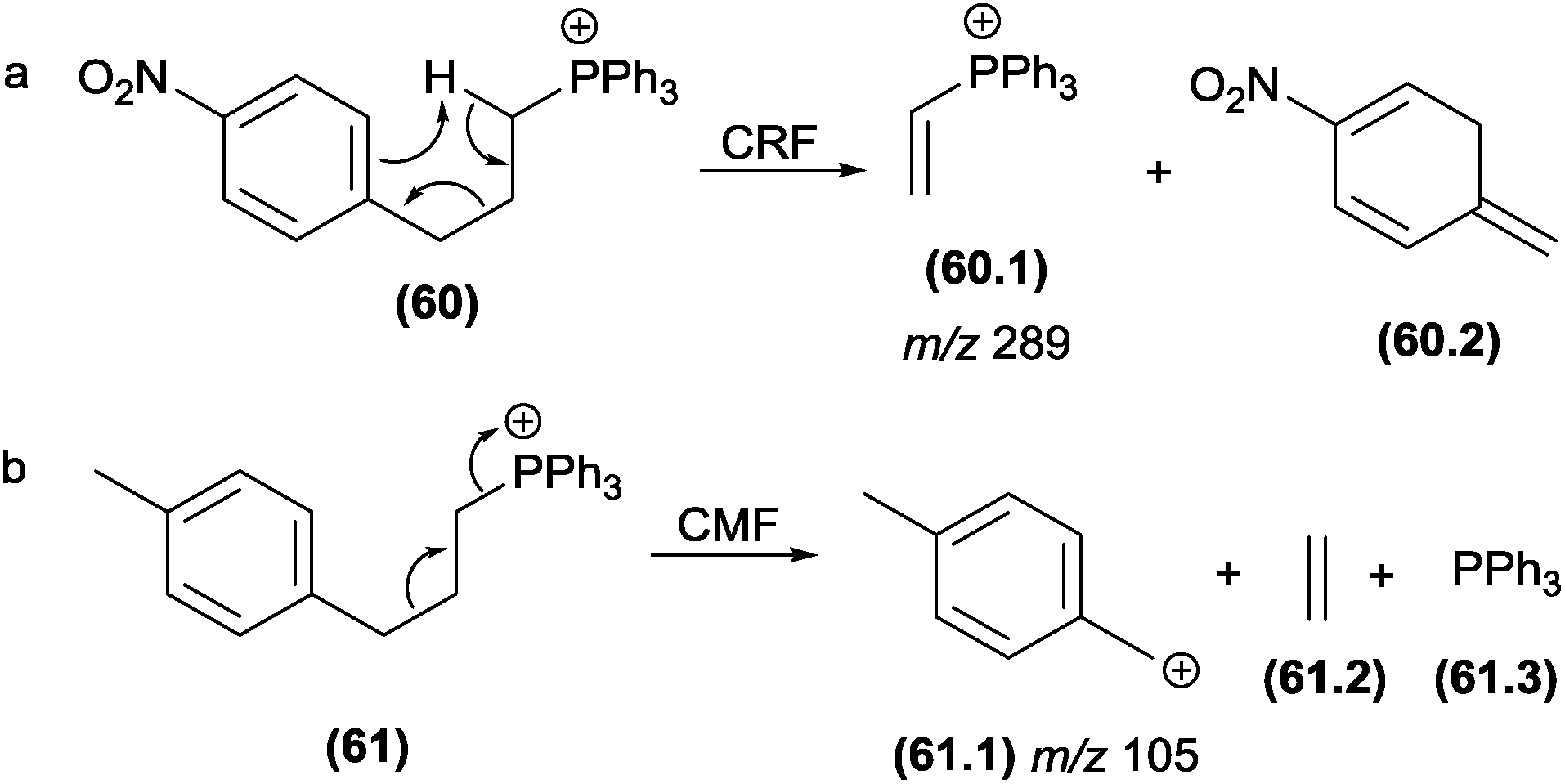 | ||
| Fig. 31 Competition between CRF and CMF in triphenylphosphonium cations. | ||
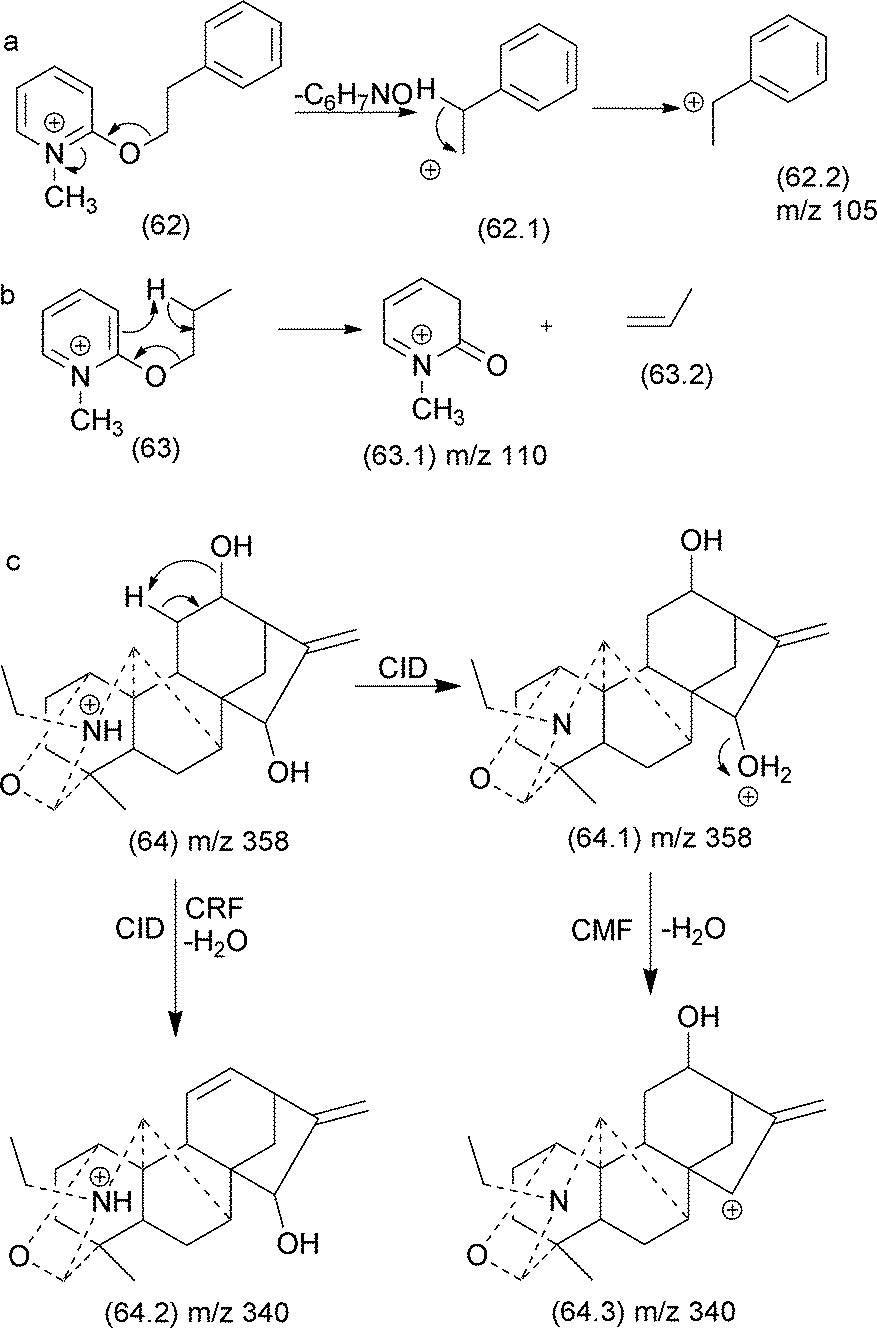 | ||
| Fig. 32 CMF and CRF reactions in alkyl 1-methylpydirinium ions (a and b) and competition between CRF and CMF in diterpenealkaloids (c). | ||
Quirke and van Berkel investigated the competition between CRF and CMF in alkyl 1-methylpyridinium ether derivatives of alcohols.138 The authors established a correlation between the occurrence of CRF/CMF and the nature of the substituent attached at the side chain. When the substituent at the side chain can generate a stable fragment ion, CMF reactions are the predominant fragmentation reactions (Fig. 32a). When the side chain exhibited a γ-hydrogen, a retro-ene reaction produced the m/z 110 product ion, which is the most intense in the MS/MS spectrum (Fig. 32b).
In natural products, a good example of the competition between CRF and CMF is the water elimination from protonated diterpene alkaloids.106 This elimination can result from a remote hydrogen rearrangement (CRF) or a single inductive cleavage (CMF) (Fig. 32c). When water is eliminated by a CMF reaction, a proton migration from the nitrogen atom to the oxygen of the hydroxyl group under CID conditions appears to be involved and results in a product ion stabilized by resonance.
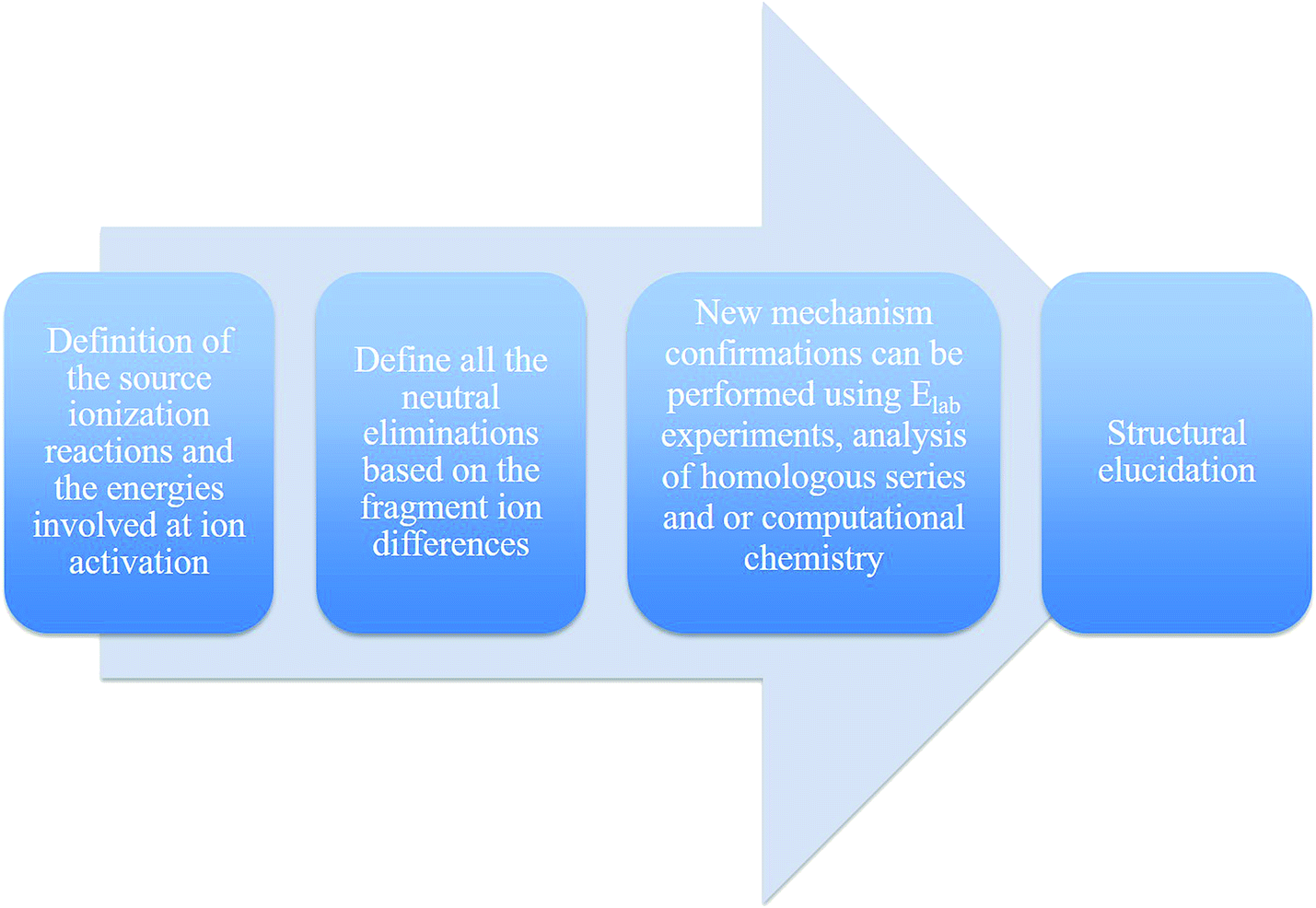 | ||
| Fig. 33 Suggested steps for structural elucidation using MS fragmentation. | ||
8 Concluding remarks
In this review, the aim was to furnish details of the possible gas phase fragmentation reactions involved in MS/MS experiments. Based on our compilation, it is important to understand that different collision gases and energy transfers, in addition to other parameters, may change the expected ion formation. Different classes of compounds can be analysed using electrospray mass spectrometry in the positive or negative ion modes, depending on their chemical structures. The fragmentation reactions observed are dependent on the structural core. It is important to reinforce the importance of working with the mass differences rather than attempting to define the reactions involved in each fragment formation.In this work, the readers can find general mechanisms and examples to understand the fragmentation reaction types that can occur in the gas phase. However, in the fragmentation rationalization processes depicted in Fig. 1, the understanding of the mechanism must be combined with the knowledge about the identity of the ion types that may be generated in the ionization source. Thus, the general mechanisms of charge migration fragmentation (CMF) and charge retention fragmentation (CRF) reactions (Fig. 4 and 7) proposed in this review can provide valuable information about the structural requirements the molecule must have to proceed through each reaction and can help the readers identify structural features in the analytes of interest.
Finding the description of fragmentation mechanisms to support identification in the literature remains challenging, and basic concepts in organic chemistry are not always considered when proposing the product ion structure from its precursor ion, resulting in erroneous interpretations. Therefore, we recommend that inexperienced users employ Fig. 33 to remember the important steps before defining a new structure by ESI-MS/MS. Finally, this review aims to help readers interested in the structure elucidation of synthetic and natural products using ESI-CID-MS/MS and LC-MS/MS to propose reasonable fragmentation mechanisms based on organic chemistry principles.
9 Acknowledgements
Daniel P. Demarque would like to thank FAPESP for the doctoral scholarship (2014/18052-0). The authors thank the Brazilian foundations FAPESP (2013/20094-0; 2014/23604-1; 2014/20302-4; 2014/50265-3), CNPq (442384/2014-9; 306385/2011-2; 467646/2014-7) and INCT-if. The open access was supported by Bruker Daltonics and Sinc do Brasil.10 Notes and references
- J. B. Fenn, Angew. Chem., Int. Ed., 2003, 42, 3871–3894 CrossRef CAS PubMed.
- J. B. Fenn, M. Mann, C. K. Meng, S. F. Wong and C. M. Whitehouse, Science, 1989, 246, 64–71 CAS.
- M. Ernst, D. B. Silva, R. R. Silva, R. Z. N. Vencio and N. P. Lopes, Nat. Prod. Rep., 2014, 31, 784–806 RSC.
- H. J. Dias, N. I. Melo and A. E. M. Crotti, in Tandem mass spectrometry - applications and principles, ed. J. K. Prasain, Intech, 2012, ch. 26, DOI:10.5772/32680.
- R. Vessecchi, A. E. M. Crotti, T. Guaratini, P. Colepicolo, S. E. Galembeck and N. P. Lopes, Mini-Rev. Org. Chem., 2007, 4, 75–87 CrossRef CAS.
- R. Vessecchi, Z. Naal, J. N. C. Lopes, S. E. Galembeck and N. P. Lopes, J. Phys. Chem. A, 2011, 115, 5453–5460 CrossRef CAS PubMed.
- L. Feketeova, A. L. Albright, B. S. Sorensen, M. R. Horsman, J. White, R. A. J. O'Hair and N. Bassler, Int. J. Mass Spectrom., 2014, 365, 56–63 CrossRef.
- K. Levsel, H. M. Schiebel, J. K. Terlouw, K. J. Jobst, M. Elend, A. Preib, H. Thiele and A. Ingendoh, J. Mass Spectrom., 2007, 42, 1024–1044 CrossRef PubMed.
- O. W. Hand, L. D. Detter, S. A. Lammert, R. G. Cooks and R. A. Walton, J. Am. Chem. Soc., 1989, 111, 5577–5583 CrossRef CAS.
- A. E. M. Crotti, C. A. Carollo, L. Gobbo-Neto, M. D. Santos, P. J. Gates and N. P. Lopes, in Modern Biotechnology in Medicinal Chemistry and Industry, ed. C. A. Taft, Research Signpost, Kerala, India, 2006 Search PubMed.
- E. Hoffmann and V. Stroobant, Mass spectrometry - principles and applications, John Wiley & Sons, Chichester, UK, 3rd edn, 2007 Search PubMed.
- R. B. Cole, Electrospray and MALDI Mass Spectrometry, Wiley, 2010 Search PubMed.
- F. W. McLafferty and F. Tureek, Interpretation of mass spectra, University Science Books, Sausalito, 4th edn, 1993 Search PubMed.
- F. A. Dorr, J. C. Tomaz, N. P. Lopes and E. Pinto, Rapid Commun. Mass Spectrom., 2008, 22, 2015–2020 CrossRef CAS PubMed.
- F. A. Dorr, B. Kovacevi, Z. B. Maksi, E. Pinto and D. A. Volmer, J. Am. Soc. Mass Spectrom., 2011, 22, 2011–2020 CrossRef CAS PubMed.
- N. P. Lopes, C. B. W. Stark, H. Hong, P. J. Gates and J. Staunton, Rapid Commun. Mass Spectrom., 2002, 16, 414–420 CrossRef CAS PubMed.
- A. Kholomeev, A. Makarov, E. Denisov, O. Lange, W. Balshun and S. Horning, presented in part at the 54th ASMS Conference on Mass Spectrometry and Applied Topics, Seattle, WA, 2006 Search PubMed.
- H. Muenster, K. Strupat, A. Makarov, O. Lange, S. R. Horning, M. R. Larsen and O. N. Jensen, presented in part at the 17th International Mass Spectrometry Conference, Prague, 2003 Search PubMed.
- M. R. Larsen, T. E. Thingholm, O. N. Jensen, P. Roepstorff and T. J. D. Jorgensen, Mol. Cell. Proteomics, 2005, 4, 873–886 CAS.
- Y. Huang, S. Liu, S. Miao and P. M. Jeanville, presented in part at the Application Note: 417, San Jose, CA, USA, 2008 Search PubMed.
- L. R. Sartori, R. Vessecchi, H. U. Humpf, F. B. Da Costa and N. P. Lopes, Rapid Commun. Mass Spectrom., 2014, 28, 723–730 CrossRef CAS PubMed.
- K. K. Murray, R. K. Boyd, M. N. Eberlin, G. J. Langley, L. Li and Y. Naito, Pure Appl. Chem., 2013, 85, 1515–1609 CrossRef CAS.
- F. A. Dorr, D. Oliveira-Silva, N. P. Lopes, J. Iglesias, D. A. Volmer and E. Pinto, Rapid Commun. Mass Spectrom., 2011, 25, 1981–1992 CrossRef PubMed.
- I. S. Kim, G. H. Nguyen, S. J. Kim and A. Jang, J. Ind. Eng. Chem., 2015, 29, 375–381 CrossRef CAS.
- K. Levsen and H. Schwarz, Mass Spectrom. Rev., 1983, 2, 77–148 CrossRef CAS.
- M. Holcapek, R. Jirasko and M. Lisa, J. Chromatogr. A, 2010, 1217, 3908–3921 CrossRef CAS PubMed.
- J. M. Zhang and J. S. Brodbelt, J. Mass Spectrom., 2003, 38, 555–572 CrossRef CAS PubMed.
- A. Schwarzenberg, H. Dossmann, R. B. Cole, X. Machuron-Mandard and J. C. Tabet, J. Mass Spectrom., 2014, 49, 1330–1337 CrossRef CAS PubMed.
- P. Miketova, K. H. Schram, J. Whitney, E. H. Kearns and B. N. Timmermann, J. Mass Spectrom., 1999, 34, 1240–1252 CrossRef CAS.
- K. B. Tomer and M. L. Gross, Biomed. Environ. Mass Spectrom., 1988, 15, 89–98 CrossRef CAS PubMed.
- C. F. Cheng and M. L. Gross, Mass Spectrom. Rev., 2000, 19, 398–420 CrossRef CAS PubMed.
- V. H. Wysocki and M. M. Ross, Int. J. Mass Spectrom. Ion Processes, 1991, 104, 179–211 CrossRef CAS.
- C. Denekamp, A. Weisz and A. Mandelbaum, J. Mass Spectrom., 1996, 31, 1028–1032 CrossRef CAS.
- Z. Wang, Comprehensive organic name reactions and reagents, John Wiley & Sons, Chichester, UK, 2010 Search PubMed.
- J. Adams and M. L. Gross, J. Am. Chem. Soc., 1986, 108, 6915–6921 CrossRef CAS.
- J. Adams and M. L. Gross, J. Am. Chem. Soc., 1989, 111, 435–440 CrossRef CAS.
- M. L. Gross, Int. J. Mass Spectrom. Ion Processes, 1992, 118, 137–165 CrossRef.
- T. Guaratini, N. P. Lopes, E. Pinto, P. Colepicolo and P. J. Gates, Chem. Commun., 2006, 4110–4112, 10.1039/b609672g.
- R. O. C. Norman and J. M. Coxon, Principles of Organic Synthesis, CRC Press, New York, NY, 1993 Search PubMed.
- N. J. A. Coughlan, B. D. Adamson, K. J. Catani, U. Wille and E. J. Bieske, J. Phys. Chem. Lett., 2014, 5, 3195–3199 CrossRef CAS PubMed.
- D. L. Pavia, G. M. Lampman and G. S. Kriz, Introduction to Spectroscopy, Cengage Learning, 2015 Search PubMed.
- S. Sankararaman and R. Hoffmann, Pericyclic Reactions - A Textbook: Reactions, Applications and Theory, Wiley, 2005 Search PubMed.
- R. Vessecchi, G. J. Zocolo, D. R. Gouvea, F. Hubner, B. Cramer, M. R. R. de Marchi, H. U. Humpf and N. P. Lopes, Rapid Commun. Mass Spectrom., 2011, 25, 2020–2026 CrossRef CAS PubMed.
- T. W. G. Solomons, C. B. Fryhle and S. A. Snyder, Organic Chemistry, Wiley, 11th edn, 2014 Search PubMed.
- C. A. Grob, Angew. Chem., Int. Ed., 1969, 8, 535–546 CrossRef CAS.
- C. A. Grob and W. Baumann, Helv. Chim. Acta, 1955, 38, 594–610 CrossRef CAS.
- Y. G. Xia, G. Y. Li, J. Liang, C. A. Ortori, B. Y. Yang, H. X. Kuang and D. A. Barrett, J. Pharm. Biomed. Anal., 2014, 100, 109–122 CrossRef CAS PubMed.
- A. Ricci, S. Piccolella, A. Fiorentino, F. Pepi, B. D'Abrosca and P. Monaco, Rapid Commun. Mass Spectrom., 2010, 24, 1543–1556 CrossRef CAS PubMed.
- G. P. Aguiar, K. A. L. Wakabayashi, G. F. Luz, V. B. Oliveira, L. Mathias, I. J. C. Vieira, R. Braz-Filho and A. E. M. Crotti, Rapid Commun. Mass Spectrom., 2010, 24, 295–308 CrossRef CAS PubMed.
- F. F. Hsu and J. Turk, J. Am. Soc. Mass Spectrom., 2001, 12, 1036–1043 CrossRef CAS.
- L. C. M. Ngoka and M. L. Gross, J. Mass Spectrom., 2000, 35, 265–276 CrossRef CAS.
- K. D. Roberts and G. E. Reid, J. Mass Spectrom., 2007, 42, 187–198 CrossRef CAS PubMed.
- C. G. Gu, G. Tsaprailis, L. Breci and V. H. Wysocki, Anal. Chem., 2000, 72, 5804–5813 CrossRef CAS PubMed.
- J. M. Froelich and G. E. Reid, J. Am. Soc. Mass Spectrom., 2007, 18, 1690–1705 CrossRef CAS PubMed.
- N. Sadagopan and J. T. Watson, J. Am. Soc. Mass Spectrom., 2001, 12, 399–409 CrossRef CAS PubMed.
- P. C. Liao, Z. H. Huang and J. Allison, J. Am. Soc. Mass Spectrom., 1997, 8, 501–509 CrossRef CAS.
- J. Ruzicka, C. Weisbecker and A. B. Attygalle, J. Mass Spectrom., 2011, 46, 12–23 CrossRef CAS PubMed.
- A. Delcambre and C. Saucier, J. Mass Spectrom., 2012, 47, 727–736 CrossRef CAS PubMed.
- H. Mcnab, E. S. B. Ferreira, A. N. Hulme and A. Quye, Int. J. Mass Spectrom., 2009, 284, 57–65 CrossRef CAS.
- J. Y. Zhang, N. Li, Y. Y. Che, Y. Zhang, S. X. Liang, M. B. Zhao, Y. Jiang and P. F. Tu, J. Pharm. Biomed. Anal., 2011, 56, 950–961 CrossRef CAS PubMed.
- L. W. Qi, C. Y. Chen and P. Li, Rapid Commun. Mass Spectrom., 2009, 23, 3227–3242 CrossRef CAS PubMed.
- C. Y. Ma, H. P. Lv, X. Z. Zhang, Z. M. Chen, J. Shi, M. L. Lu and Z. Lin, Anal. Chim. Acta, 2013, 795, 15–24 CrossRef CAS PubMed.
- G. C. Justino, C. M. Borges and M. H. Florencio, Rapid Commun. Mass Spectrom., 2009, 23, 237–248 CrossRef CAS PubMed.
- R. Simons, J. P. Vincken, M. C. Bohin, T. F. M. Kuijpers, M. A. Verbruggen and H. Gruppen, Rapid Commun. Mass Spectrom., 2011, 25, 55–65 CrossRef CAS PubMed.
- Y. Zhou, X. Liu, J. Yang, Q. B. Han, J. Z. Song, S. L. Li, C. F. Qiao, L. S. Ding and H. X. Xu, Anal. Chim. Acta, 2008, 629, 104–118 CrossRef CAS PubMed.
- J. Xu, D. W. Qian, S. Jiang, J. M. Guo, E. X. Shang, J. A. Duan and J. Yang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 944, 123–127 CrossRef CAS PubMed.
- K. Ablajan and A. Tuoheti, Rapid Commun. Mass Spectrom., 2013, 27, 451–460 CrossRef CAS PubMed.
- L. Zhang, L. Xu, S. S. Xiao, Q. F. Liao, Q. Li, J. Liang, X. H. Chen and K. S. Bi, J. Pharm. Biomed. Anal., 2007, 44, 1019–1028 CrossRef CAS PubMed.
- E. de Rijke, P. Out, W. M. A. Niessen, F. Ariese, C. Gooijer and U. A. T. Brinkman, J. Chromatogr. A, 2006, 1112, 31–63 CrossRef CAS PubMed.
- Y. L. Ma, I. Vedernikova, H. van den Heuvel and M. Claeys, J. Am. Soc. Mass Spectrom., 2000, 11, 136–144 CrossRef CAS PubMed.
- S. Su, Y. Wang, L. Bai, B. Xia, X. Li, Y. Tang, P. Xu and M. Xue, J. Pharm. Biomed. Anal., 2015, 104, 38–46 CrossRef CAS PubMed.
- H. J. Li and M. L. Deinzer, Anal. Chem., 2007, 79, 1739–1748 CrossRef CAS PubMed.
- W. Friedrich, A. Eberhardt and R. Galensa, Eur. Food Res. Technol., 2000, 211, 56–64 CrossRef CAS.
- R. Jaiswal, L. Jayasinghe and N. Kuhnert, J. Mass Spectrom., 2012, 47, 502–515 CrossRef CAS PubMed.
- H. Li, L. R. Wan, Y. Hashi and S. Z. Chen, Rapid Commun. Mass Spectrom., 2007, 21, 2497–2504 CrossRef CAS PubMed.
- W. Yang, D. M. Fang, H. P. He, X. J. Hao, Z. J. Wu and G. L. Zhang, Rapid Commun. Mass Spectrom., 2013, 27, 1203–1212 CrossRef CAS PubMed.
- Y. J. Li, H. L. Wei, L. W. Qi, J. Chen, M. T. Ren and P. Li, Rapid Commun. Mass Spectrom., 2010, 24, 2975–2985 CrossRef CAS PubMed.
- Z. X. Qing, P. Cheng, X. B. Liu, Y. S. Liu and J. G. Zeng, J. Pharm. Biomed. Anal., 2014, 103, 26–34 CrossRef PubMed.
- S. P. Wang, L. Liu, L. L. Wang, Y. H. Hu, W. D. Zhang and R. H. Liu, Molecules, 2012, 17, 10470–10493 CrossRef CAS PubMed.
- X. Y. Xian, B. H. Sun, X. T. Ye, G. Y. Zhang, P. Y. Hou and H. Y. Gao, J. Sep. Sci., 2014, 37, 1533–1545 CrossRef CAS PubMed.
- G. Bianco, S. Abate, C. Labella and T. R. I. Cataldi, Rapid Commun. Mass Spectrom., 2009, 23, 1065–1074 CrossRef CAS PubMed.
- Y. Zhou, Q. B. Han, J. Z. Song, C. F. Qiao and H. X. Xu, J. Chromatogr. A, 2008, 1206, 131–139 CrossRef CAS PubMed.
- J. C. J. M. D. S. Menezes, M. G. P. M. S. Neves, J. A. S. Cavaleiro, C. Barros, S. M. Santos, F. D. da Silva, V. F. Ferreira and M. R. M. Domingues, Int. J. Mass Spectrom., 2013, 343, 1–8 CrossRef.
- L. Sleno, M. J. Chalmers and D. A. Volmer, Anal. Bioanal. Chem., 2004, 378, 977–986 CrossRef CAS PubMed.
- L. Sleno, A. J. Windust and D. A. Volmer, Anal. Bioanal. Chem., 2004, 378, 969–976 CrossRef CAS PubMed.
- B. Carey, M. J. F. Saez, B. Hamilton, J. O'Halloran, F. N. A. M. van Pelt and K. J. James, Rapid Commun. Mass Spectrom., 2012, 26, 1793–1802 CrossRef CAS PubMed.
- B. Li, X. Zhang, J. Wang, L. Zhang, B. W. Gao, S. P. Shi, X. H. Wang, J. Li and P. F. Tu, Phytochem. Anal., 2014, 25, 229–240 CrossRef CAS PubMed.
- F. F. Hsu and J. Turk, J. Am. Soc. Mass Spectrom., 2010, 21, 657–669 CrossRef CAS PubMed.
- F. F. Hsu and J. Turk, J. Am. Soc. Mass Spectrom., 2008, 19, 1673–1680 CrossRef CAS PubMed.
- A. Reis, P. Domingues, A. J. V. Ferrer-Correia and M. R. M. Domingues, J. Mass Spectrom., 2004, 39, 1513–1522 CrossRef CAS PubMed.
- D. Ponomarev and V. Takhistov, J. Mol. Struct., 2006, 784, 198–213 CrossRef CAS.
- W. J. Griffiths, Y. Yang, J. A. Lindgren and J. Sjovall, Rapid Commun. Mass Spectrom., 1996, 10, 21–28 CrossRef CAS.
- E. J. Crevelin, A. E. M. Crotti, T. D. Zucchi, I. S. Melo and L. A. B. Moraes, J. Mass Spectrom., 2014, 49, 1117–1126 CrossRef CAS PubMed.
- A. Fredenhagen, C. Derrien and E. Gassmann, J. Nat. Prod., 2005, 68, 385–391 CrossRef CAS PubMed.
- N. P. Lopes, P. J. Gates, J. P. G. Wilkins and J. Staunton, Analyst, 2002, 127, 1224–1227 RSC.
- A. E. M. Crotti, J. L. C. Lopes and N. P. Lopes, J. Mass Spectrom., 2005, 40, 1030–1034 CrossRef CAS PubMed.
- H. Wang, Y. H. Wu and Z. X. Zhao, J. Mass Spectrom., 2001, 36, 58–70 CrossRef CAS PubMed.
- A. E. M. Crotti, T. Fonseca, H. Hong, J. Staunton, S. E. Galembeck, N. P. Lopes and P. J. Gates, Int. J. Mass Spectrom., 2004, 232, 271–276 CrossRef CAS.
- T. Fonseca, N. P. Lopes, P. J. Gates and J. Staunton, J. Am. Soc. Mass Spectrom., 2004, 15, 325–335 CrossRef CAS PubMed.
- W. Vetter, W. Meister and G. Oesterhelt, J. Mass Spectrom., 1998, 33, 461–472 CrossRef CAS.
- K. B. Tomer, F. W. Crow and M. L. Gross, J. Am. Chem. Soc., 1983, 105, 5487–5488 CrossRef CAS.
- M. L. Gross, Int. J. Mass Spectrom., 2000, 200, 611–624 CrossRef CAS.
- H. Hong, P. J. Gates, J. Staunton, T. Stinear, S. T. Cole, P. F. Leadlay and J. B. Spencer, Chem. Commun., 2003, 2822–2823, 10.1039/b308163j.
- K. Kleigrewe, F. Aydin, K. Hogrefe, P. Piecuch, K. Bergander, E. U. Wurthwein and H. U. Humpf, J. Agric. Food Chem., 2012, 60, 5497–5505 CrossRef CAS PubMed.
- J. McMurry, Organic Chemistry, Brooks/Cole Publishing Company, Belmont, CA, 1992 Search PubMed.
- R. Li, Z. J. Wu, F. Zhang and L. S. Ding, Rapid Commun. Mass Spectrom., 2006, 20, 157–170 CrossRef CAS PubMed.
- A. C. Pinto, R. Vessecchi, C. G. Silva, A. C. L. Amorim, H. M. S. Júnior, M. J. C. Rezende, C. M. Rezende and N. P. Lopes, Rapid Commun. Mass Spectrom., 2016, 30, 61–68 CrossRef PubMed.
- P. J. Gates, G. C. Kearney, R. Jones, P. F. Leadlay and J. Staunton, Rapid Commun. Mass Spectrom., 1999, 13, 242–246 CrossRef CAS PubMed.
- Y. F. Zhang, Q. R. Shi, P. Y. Shi, W. D. Zhang and Y. Y. Cheng, Rapid Commun. Mass Spectrom., 2006, 20, 2328–2342 CrossRef CAS PubMed.
- Y. Wang, Z. Q. Liu, F. R. Song and S. Y. Liu, Rapid Commun. Mass Spectrom., 2002, 16, 2075–2082 CrossRef CAS PubMed.
- R. Vessecchi, F. S. Emery, S. E. Galembeck and N. P. Lopes, J. Mass Spectrom., 2012, 47, 1648–1659 CrossRef CAS PubMed.
- R. Vessecchi, J. N. C. Lopes, N. P. Lopes and S. E. Galembeck, J. Phys. Chem. A, 2011, 115, 12780–12788 CrossRef CAS PubMed.
- R. Vessecchi, P. G. B. D. Nascimento, J. N. C. Lopes and N. P. Lopes, J. Mass Spectrom., 2006, 41, 1219–1225 CrossRef CAS PubMed.
- H. Wang and F. Feng, J. Pharm. Biomed. Anal., 2009, 49, 1157–1165 CrossRef CAS PubMed.
- N. Fabre, I. Rustan, E. de Hoffmann and J. Quetin-Leclercq, J. Am. Soc. Mass Spectrom., 2001, 12, 707–715 CrossRef CAS PubMed.
- U. Justesen, J. Mass Spectrom., 2001, 36, 169–178 CrossRef CAS PubMed.
- W. Jin, Y. F. Wang, R. L. Ge, H. M. Shi, C. Q. Jia and P. F. Tu, Rapid Commun. Mass Spectrom., 2007, 21, 2351–2360 CrossRef CAS PubMed.
- Y. A. Jeilani, B. H. Cardelino and V. Ibeanusi, J. Mass Spectrom., 2011, 46, 625–634 CrossRef CAS PubMed.
- K. H. M. Cardozo, V. M. Carvalho, E. Pinto and P. Colepicolo, Rapid Commun. Mass Spectrom., 2006, 20, 253–258 CrossRef CAS PubMed.
- K. H. M. Cardozo, R. Vessecchi, S. E. Galembeck, T. Guaratini, P. J. Gates, E. Pinto, N. P. Lopes and P. Colepicolo, J. Braz. Chem. Soc., 2009, 20, 1625–1631 CrossRef CAS.
- S. G. Musharraf, M. Goher, A. Hussain and M. I. Choudhary, Chem. Cent. J., 2012, 6, 120 CrossRef CAS PubMed.
- S. G. Musharraf, M. Goher, S. Shahnaz, M. I. Choudhary and A. U. Rahman, Rapid Commun. Mass Spectrom., 2013, 27, 169–178 CrossRef CAS PubMed.
- N. A. J. C. Furtado, R. Vessecchi, J. C. Tomaz, S. E. Galembeck, J. K. Bastos, N. P. Lopes and A. E. M. Crotti, J. Mass Spectrom., 2007, 42, 1279–1286 CrossRef CAS PubMed.
- S. Haldar, F. A. Mulani, T. Aarthy, D. S. Dandekar and H. V. Thulasiram, J. Chromatogr. A, 2014, 1366, 1–14 CrossRef CAS PubMed.
- Z. J. Wu, D. M. Sun, D. M. Fang, J. Z. Chen, P. Cheng and G. L. Zhang, Int. J. Mass Spectrom., 2013, 341, 28–33 CrossRef.
- J. N. Sousa, B. A. Rocha, M. D. Assis, A. P. F. Peti, L. A. B. Moraes, Y. Iamamoto, P. J. Gates, A. R. M. de Oliveira and N. P. Lopes, Rev. Bras. Farmacogn., 2013, 23, 621–629 CrossRef.
- J. Y. Bao, X. L. Gao and A. D. Jones, Rapid Commun. Mass Spectrom., 2014, 28, 457–464 CrossRef CAS PubMed.
- T. Nilsson, E. Martinez, A. Manresa and E. H. Oliw, Rapid Commun. Mass Spectrom., 2010, 24, 777–783 CrossRef CAS PubMed.
- K. B. Herath, C. S. Weisbecker, S. B. Singh and A. B. Attygalle, J. Org. Chem., 2014, 79, 4378–4389 CrossRef CAS PubMed.
- K. Lech, K. Witkoś, B. Wileńska and M. Jarosz, Anal. Bioanal. Chem., 2015, 855–867 CrossRef CAS PubMed.
- Y. F. Zhang, P. Zhang and Y. Y. Cheng, J. Mass Spectrom., 2008, 43, 1421–1431 CrossRef CAS PubMed.
- F. F. Hsu and J. Turk, J. Am. Soc. Mass Spectrom., 2000, 11, 892–899 CrossRef CAS PubMed.
- F. F. Hsu, I. J. Lodhi, J. Turk and C. F. Semenkovich, J. Am. Soc. Mass Spectrom., 2014, 25, 1412–1420 CrossRef CAS PubMed.
- C. Muller, B. Kanawati, T. M. Rock, S. Forcisi, F. Moritz and P. Schmitt-Kopplin, Rapid Commun. Mass Spectrom., 2014, 28, 1735–1744 CrossRef PubMed.
- F. F. Hsu and J. Turk, J. Am. Soc. Mass Spectrom., 2000, 11, 986–999 CrossRef CAS PubMed.
- M. Cydzik, M. Rudowska, P. Stefanowicz and Z. Szewczuk, J. Am. Soc. Mass Spectrom., 2011, 22, 2103–2107 CrossRef CAS PubMed.
- C. Denekamp, E. Tenetov and Y. Horev, J. Am. Soc. Mass Spectrom., 2003, 14, 790–801 CrossRef CAS PubMed.
- J. M. E. Quirke and G. J. van Berkel, J. Mass Spectrom., 2001, 36, 1294–1300 CrossRef CAS PubMed.
Footnote |
| † These authors equally contributed this work. |
| This journal is © The Royal Society of Chemistry 2016 |