
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent highlights in mixed-coordinate oligophosphorus chemistry
Maximilian
Donath†
,
Felix
Hennersdorf†
and
Jan J.
Weigand
*
Department of Chemistry and Food Chemistry, TU Dresden, 01062 Dresden, Germany. E-mail: jan.weigand@tu-dresden.de
First published on 8th February 2016
Abstract
This review aims to highlight and comprehensively summarize recent developments in the field of mixed-coordinate phosphorus chemistry. Particular attention is focused on the synthetic approaches to compounds containing at least two directly bonded phosphorus atoms in different coordination environments and their unexpected properties that are derived from spectroscopic and crystallographic data. Novel substance classes are discussed in order to supplement previous reviews about mixed-coordinate phosphorus compounds.
 Maximilian Donath | Maximilian Donath studied at WWU Münster (Germany) with a research stay in Palermo (Italy). He received his diploma degree in 2011 and is currently a PhD student in the Weigand group. After starting his PhD thesis at the WWU Münster supported by a scholarship of the Fonds der Chemischen Industrie (FCI) he continued it at TU Dresden (Germany) since 2013. His research interest is focused on multinuclear NMR spectroscopy, the synthesis of cationic polyphosphorus frameworks and the activation of white phosphorus with p-block compounds. |
 Felix Hennersdorf | Felix Hennersdorf studied at Leipzig University (Germany) with research stays in Stellenbosch (South Africa), Singapore and Melbourne (Australia). He received his MSc degree in 2013 before he joined the Weigand group as a PhD student and now holds scholarships of the FCI and the Studienstiftung des Deutschen Volkes. Besides his interest in crystallography his research is focused on the synthesis of polyphosphorus compounds by activation of white phosphorus. |
 Jan J. Weigand | Jan J. Weigand obtained his diploma in chemistry in 2002 and his Dr. rer. nat. in 2005 from the LMU in Munich (Germany). In 2005 he was awarded the Bavarian culture prize and obtained a Lynen Scholarship from the AvH foundation for postdoctoral research at Dalhousie University in Halifax (Canada). He returned to Germany with a Lynen Return Fellowship and started his habilitation at the WWU Münster at the end of 2007 under the supervision of Prof. Hahn. He was awarded shortly after the Liebig scholarship of the FCI which allowed him to start his independent career in 2008. In April 2010 he became a fellow of the Emmy Noether research program awarded by the DFG and recently obtained the Wöhler research award for young scientists. In 2012, he obtained an ERC starting grant from the European Council. Since 2013 he has been Professor at the TU University Dresden. |
Introduction
Phosphorus is an intriguing element with several different bonding modes. The combination of two phosphorus atoms connected to each other multiplies the possibilities to gain compounds with different P–P bonds displaying a great variety of bonding modes. Such compounds have always attracted much attention and a first review in 1965 by Cowley covered the three basic types of substances containing phosphorus–phosphorus single bonds that are derived from classical diphosphanes and their oxidation products.1 In the report of Dillon et al., which appeared in 1995, the field of P–P bonded compounds was re-reviewed. A tremendous development was reported and a great variety of combinations of bonding modes including multiple bonds was discussed.2 However, charged and zwitterionic compounds were only considered very briefly. This review is aiming to give an overview of selected main-group compounds containing at least two phosphorus atoms bonded to each other. Those compounds possess different coordination numbers, valence states and/or formal charges. Consciously excluding the wide field of metal-coordinated compounds, this report focuses on certain intriguing examples for most of the substance classes in order to enable recognition of cross relations, recurring trends or properties. To keep the style of an overview, certain known substance classes or specific compounds which have been discussed extensively in the past, will only be commented on briefly. Thus, the more detailed discussions focus on cationic mixed-coordinate phosphorus compounds for which, in the last two decades, the most rapid development with respect to neutral or anionic compounds was spotted.Dillon et al. identified 170 hypothetical modes of P–P bonding which exceeds by far the scope of this review. Thus, the P–P bonding of the compounds in this article is classified only by the number of σ-bonds (vide infra). This allows to overcome the problem of categorizing molecules differently due to distinct resonance structures. Secondary consideration of substituent effects often reveals parallels and differences among a class, thus additional subdivisions are made.
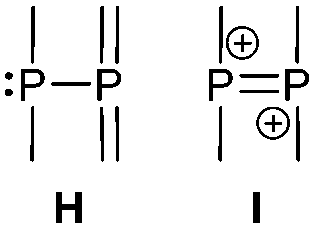
The term valency is only used if unambiguous. As an example, the IUPAC definition3 gives no clear instruction how to correctly entitle the valence state of an R4P+ cation that can be obtained by either protonation of the parent λ3-phosphane (phosphonium) or by hydride abstraction from the parent λ5-phosphane/phosphorane (λ5-phosphanylium).4 The nomenclature of catenated phosphorus compounds following the IUPAC rules is thoroughly described in literature.5 Crystal structure analysis permits a precise determination of coordination environments and P⋯P distances allowing a statement on bonding interactions. Appropriate computational calculations often give further insights into the strength of the respective bond. The P–P distance of 2.2 Å6 in P4 is often used as a benchmark in order to classify the bonding situation in the title compounds. Although the sum of the van der Waals radii of the P–P bonded framework strongly depends on the oxidation states of the corresponding phosphorus atoms (e.g. 3.6 Å),7 a value beyond 3 Å is considered as very weak interaction, if not repulsion, in cases where a bridging ligand prevents a longer distance. 31P NMR chemical shifts and 31P–31P-coupling constants are often indicative for a certain bonding situation and coordination numbers of the respective phosphorus atoms, but appeared to be of no ultimate proof in certain cases.8Table 1 displays selected P–P bonded frameworks with non-symmetrical bonding motifs A–H, J–K and M–O. In contrast to the well-known symmetrical combinations σ2–σ2 (diphosphenes)9 and σ3–σ3 (diphosphanes),1 rare examples exhibiting the symmetrical bonding situation I, L and P were included for reasons of completeness. The overall charge of the resulting systems strongly depends on the nature of the substituents attached to the P-atoms. For clarification reasons, a short comment about the σ-notation and the drawing of structures in this review is required. There are several ways to indicate the bonding situation of a P–P bonded system which might seem ambiguous. However, they require certain consideration such as following the octet rule and minimizing the number of formal charges or extended coordination numbers that are exceptions from the octet rule. For most of the depicted systems we decided to draw the simplest Lewis-type structure which primarily follows the octet rule in combination with formal charges (bonding modes: A–F, I, J and L). For a better recognition of lone pairs of electrons they are depicted (as two dots) only where relevant. We are aware that in many cases, these formal drawings do not display the actual electronic structures of the systems but indicate the actual number of bonding partners as denoted with the σ-notation. The number of bonding partners at phosphorus can exceed four in certain cases, thus representing hyper-coordinated bonding situations (bonding modes: G, K, and M–P). As Kilian et al. stated: “Hyper-coordination in phosphorus chemistry is not unusual, penta- and hexa-coordinate compounds are stable when highly electronegative substituents are attached to the phosphorus atom (as e.g. in PF5). On the other hand, hyper-coordination with less electronegative atoms around the central phosphorus is less common and more sophisticated approaches (resonance stabilization, favorable ring size formation) are needed to stabilize such species”.10
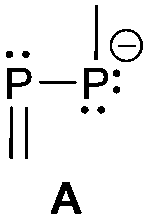
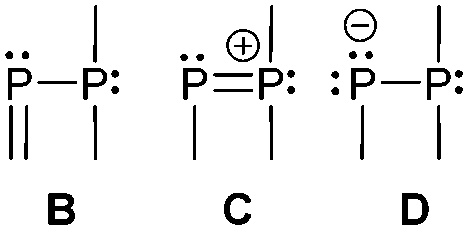
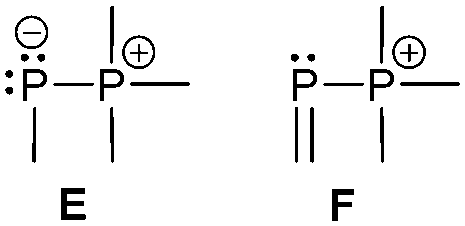
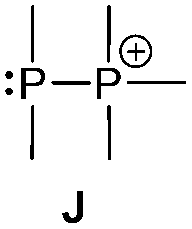

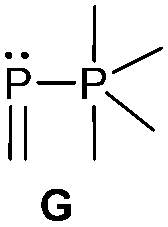
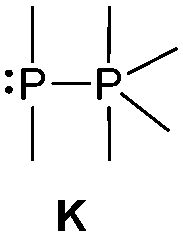
The discussion in the following section is divided in the combination of the σ-matrix as depicted in Table 1. Only for the combination of σ2–σ3, σ2–σ4, σ3–σ3 and σ4–σ4 a subdivision into classes of related compounds has been included. In general, for compound classes that are derived from related reactions, only one distinct synthetic approach is discussed. Although some of the related compounds have been reviewed in previous articles, we wish to emphasize on certain reactivity patterns, structural features or physico-chemical properties on the base of their connectivity. All drawings of the crystal structures in this contribution were generated from cif-files obtained exclusively from the CSD-Database or Supporting Information provided by the publisher. The ORTEP software was used for illustration.11 All ellipsoids are displayed at 50% probability.
σ2–σ2
The parent motif of σ2–σ2 phosphorus compounds is the well-known diphosphene unit. These compounds are typically intensively orange to red coloured, were first reported by Yoshifuji et al.12 and extensively reviewed elsewhere.9 However, diphosphenes bearing a phosphanido substituent are closely related, but comparable rare and representative examples of the bonding motif A (Table 1) are found in triphosphaallyl derivatives.
Bonding motif A
Selected examples of anionic (1−,132−,143−,154− (ref. 16)) and cationic (5+,176+ (ref. 18)) derivatives are summarized in Fig. 1. In these compounds the negative charge of the phosphanido moiety experiences resonance stabilization similar to the allyl and pentadienyl systems in carbon chemistry.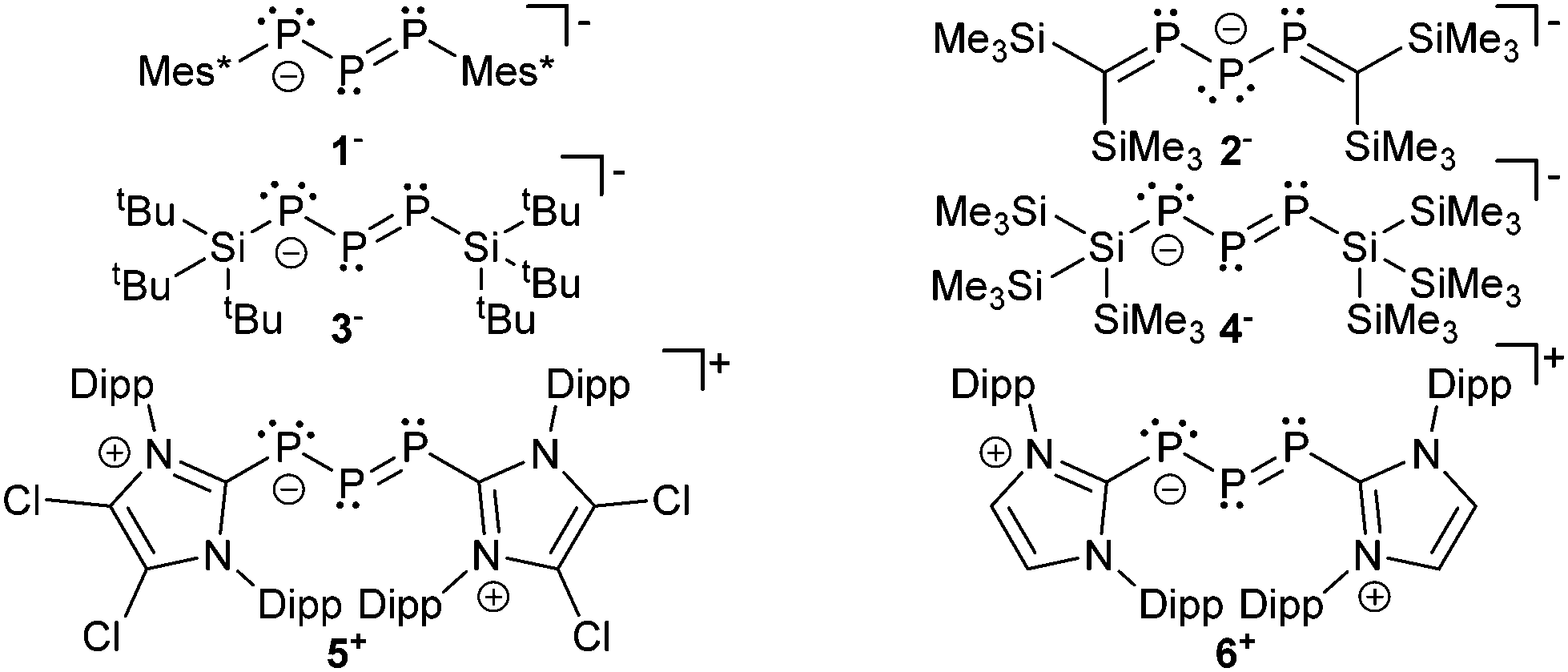 | ||
| Fig. 1 Selected examples of anionic and cationic derivatives containing the triphosphaallyl and triphosphapentadienyl motif; only one representative resonance structure is shown. | ||
The bonding principle in those compounds strongly depends on the substituents of the adjacent phosphorus atoms. Thus, examples 1−, 3−, 4−, 5+ and 6+ are best described by resonance structure I or II, whereas anion 2− should rather be described by III which resembles a 1,3-dimethylenetriphosphan-2-ide with formal charges on the central phosphorus or on the carbon atoms (Scheme 1). The difference in the electronic structures of these compounds is also reflected in the 31P chemical shifts (Table 2). The chemical shift of the central, dicoordinate phosphorus atom of ions 1−, 3−, 4−, 5+ and 6+ is typically observed in the region ranging between 500 and 750 ppm significantly low-field shifted compared to the adjacent two phosphorus atoms of which the chemical shift is observed at around 200 ppm (Table 2). Consistent with resonances structure I and II the central di-coordinate phosphorus atom is obviously involved in a normal conjugated allylic system and uses its p-type orbitals for both π- and σ-bonding. The s-type electron lone pair of the central phosphorus atom is therefore not available for electrophiles but the adjacent ones are. Thus, the addition of HOTf to a deep green solution of 5[GaCl4] instantly yields an orange oil19 and the 31P{1H} NMR spectrum reveals resonances of an AMX spin system which is assigned to the cyclo-triphosphanediium cation 72+ (Scheme 2).
| Li[1]a | [Li(dme)3][2] | [K(thf)2][3] | [Li(triaz)2][4] | [5][GaCl4] | [6][Cl] | |
|---|---|---|---|---|---|---|
| a No structural parameter available. | ||||||
| Ref. | 13 | 14 | 15c | 16b | 17 | 18 |
| Colour | Red violet | Deep red | Purple | Violet | Green | Green |
| δ(31P)adjacent in ppm | 208 | 494.1 | 232.1 | 199.8 | 199.4 | 190.6 |
| δ(31P)central in ppm | 548 | 295.5 | 730.0 | 717.2 | 597.9 | 591.9 |
| 1 J PP in Hz | 524 | 430 | 552.3 | 541 | 508.3 | 505.9 |
| d(P–P) in Å | 2.137(1) | 2.072(2) | 2.0730(8); 2.0774(8) | 2.093(1); 2.091(1) | 2.090(1); 2.097(1) | |
| d(P–C/Si) in Å | 1.687(3) | 2.252(2); 2.260(2) | 2.2459(8); 2.2454(9) | 1.815(2); 1.814(3) | 1.812(1); 1.811(1) | |
| Angle P–P–P in ° | 88.3(1) | 105.83(7) | 105.52(3) | 87.2(4) | 92.76(2) | |
 | ||
| Scheme 1 Resonance structures of triphosphaallyl and triphosphapentadienyl compounds. | ||
 | ||
| Scheme 2 Reversible protonation/deprotonation reaction between 5+ and 72+. | ||
The protonation of 5+ is reversible and the addition of a base to a solution of 72+ instantly forms a deep green solution of 5+.19 Upon protonation the phosphaallyl anion [(tBu3Si)2P3]− (3−) rearranges quickly to give the related cyclo-triphosphane derivative 8.15a The assignment of the resonances of cation 72+ is achieved by comparison to the 31P NMR parameter of related derivatives 815a and 920 (Fig. 2 and Table 3). The chemical shifts of 72+ are observed in the characteristic range for cyclo-triphosphanes.21 The resonance at highest field is assigned to the hydrogen-substituted P atom consistent with the additional splitting in the 31P NMR spectrum due to the 1J(PH) coupling (72+: 1J(PAH) = 155.7 Hz; compare 9: 1J(PAH) = 137.1 Hz). The substituents on the other two P atoms are in a transoid arrangement. A larger absolute value of the 1J(PP) coupling constants between the hydrogen-substituted P atom and the phosphorus atom with the cis-arranged substituent (72+: 1J(PAPM) = −158.8 Hz; compare 8: 1J(PAPX) = −224.2 Hz, 9: 1J(PAPM) = −226.6 Hz) contrasts the smaller coupling constant for the trans-arrangement involving the hydrogen-substituted P atom (72+: 1J(PAPX) = −130.4 Hz; compare 8: 1J(PAPX) = −141.3 Hz, 9: 1J(PAPM) = −144.1 Hz). A similar trend is observed for 2J(PH) coupling constants. Large values indicate a cis-arrangement of the hydrogen atom at PA and the lone pair of electrons at the adjacent P atom (72+: 2J(PXH) = 34.2 Hz, 8: 2J(PXH) = 16.6 Hz), whereas small values indicate a trans-arrangement (72+: 2J(PMH) = 17.8 Hz, 8: 2J(PMH) = 6.9 Hz).
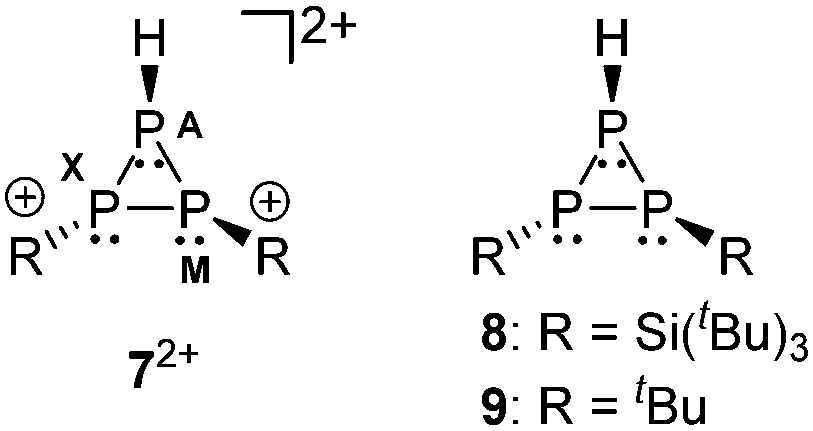 | ||
| Fig. 2 Substituent arrangement in cyclo-triphosphanes 72+, 8 and 9. | ||
| 31P | 72+ | 8 | 9 |
|---|---|---|---|
| a Coupling constants are not reported. | |||
| Ref. | 19 | 13a | 20 |
| Spin system | AMX | AMX | AMX |
| δ(PA) | −202.3 | −260.1 | −270.8 |
| δ(PM) | −182.6 | −259.5 | −149.2 |
| δ(PX) | −156.8 | −240.9 | −136.3 |
| 1 J(PAPM) | −158.8 | −224.2 | −144.1 |
| 1 J(PAPX) | −130.4 | −141.3 | −226.6 |
| 1 J(PMPX) | −203.0 | −188.0 | −223.7 |
| 1 J(PAH) | 155.7 | 137.1 | —a |
| 2 J(PMH) | 17.8 | 6.9 | —a |
| 2 J(PXH) | 34.2 | 16.6 | —a |
The fact that the protonation of 1− leads to the formation of the open-chain phosphanyldiphosphene Mes*PH–P = P–Mes* instead of a cyclo-triphosphane underlines the great dependence of the substituents on the electronic structure of triphosphaallyl compounds.13a A totally different reaction outcome is found upon protonation of anion 2− which is in agreement with the different resonance stabilization. According to III (Scheme 1) protonation should occur at the central phosphorus or the carbon atoms. Thermolysis of 2− in DME in the presence of traces of moisture led to the formation of the rearranged reaction product 10 suggesting the intermediate 11 with a protonated carbon atom (Scheme 3).14
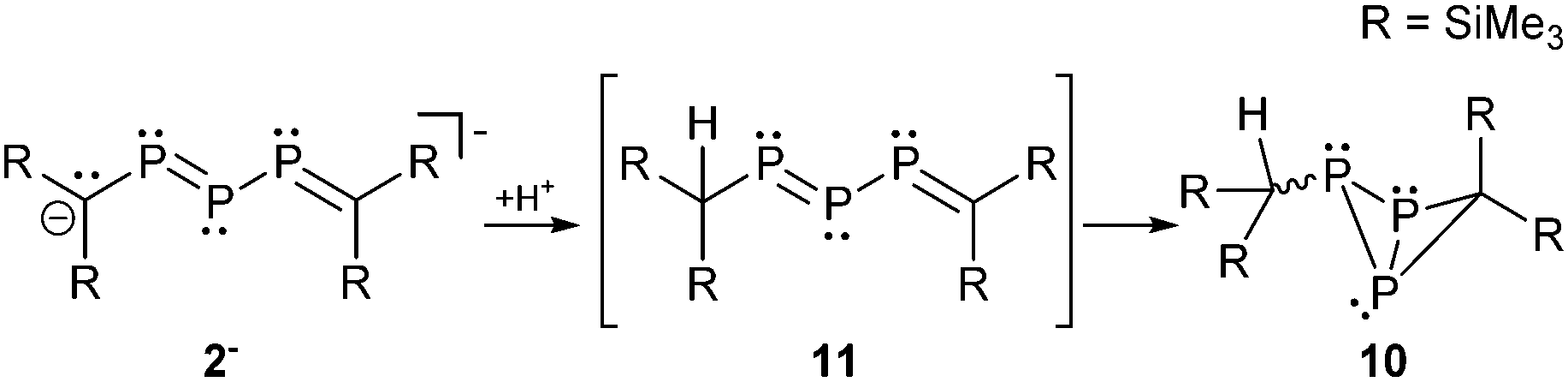 | ||
| Scheme 3 Rearrangement reaction of 2− upon protonation. | ||
The difference in the electronic structure of 2− is also evident in the reversed 31P NMR data. Thus, the more shielded central phosphorus atom is now observed at 295.5 ppm and the resonance for the adjacent phosphorus atoms is observed at significantly lower field at 494.1 ppm. The 1J(PP) coupling constant of 430 Hz is only slightly affected but in accordance with a significant structural change of the P3 bonded system. For triphosphaallyl ions 1−, 3−, 4−, 5+ and 6+ a significantly shortened P–P bond length (<2.1 Å) is observed indicating partial double-bond character as expected for an allyl system. In comparison, the P–P bond length is significantly elongated (2.137(1) Å) in the case of anion 2−. The other bonding parameters display the expected values for those compounds and the large deviations of the P–P–P angle ranging from 87–105° are best explained by the very soft bending potential of the P3-fragment. Unfortunately, for most of the compounds UV/VIS spectroscopic data is not available and only the observed colour in solution is reported (Table 2). The UV/VIS spectrum of the deep-green compound 5[GaCl4] in C6H5F shows two absorption bands of which the strongest band (Imax) is observed at 696 nm corresponding to an n → π* transition. The second absorption maximum at 443 nm can be assigned to a π → π* transition. The related cation 6+, dissolved in THF, shows the corresponding absorption bands at 666 nm and 434 nm, respectively. The reduction of 6+ into its corresponding neutral radical species 12 was achieved by reacting the cation with an excess of magnesium (Scheme 4).18
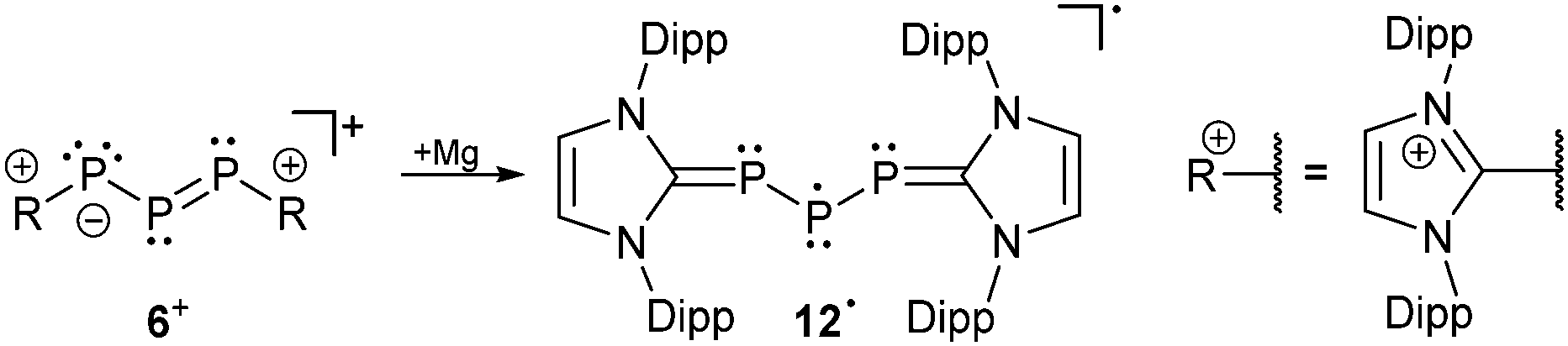 | ||
| Scheme 4 Reduction of 6+ to neutral radical species 12˙. | ||
Closely related to these triphosphaallyl systems are the cyclopentadienide related 1,2,3-triphospholides. Although first described already in 1990,22 the isolation of the non-coordinate parent compound [Pn3C2R2]− (Pn = P, As; 13−) and its arsa-analogue was achieved only recently by Goicoechea et al. Anions 13− are obtained from the reaction of heptapnictide trianions [Pn7]3− (Pn = P, As)23 or the monoprotonated derivatives [HPn7]2− with alkynes in dimethylformamide in the presence of a cation sequestering ligand like 2,2,2-crypt or 18-crown-6 (Scheme 5).24
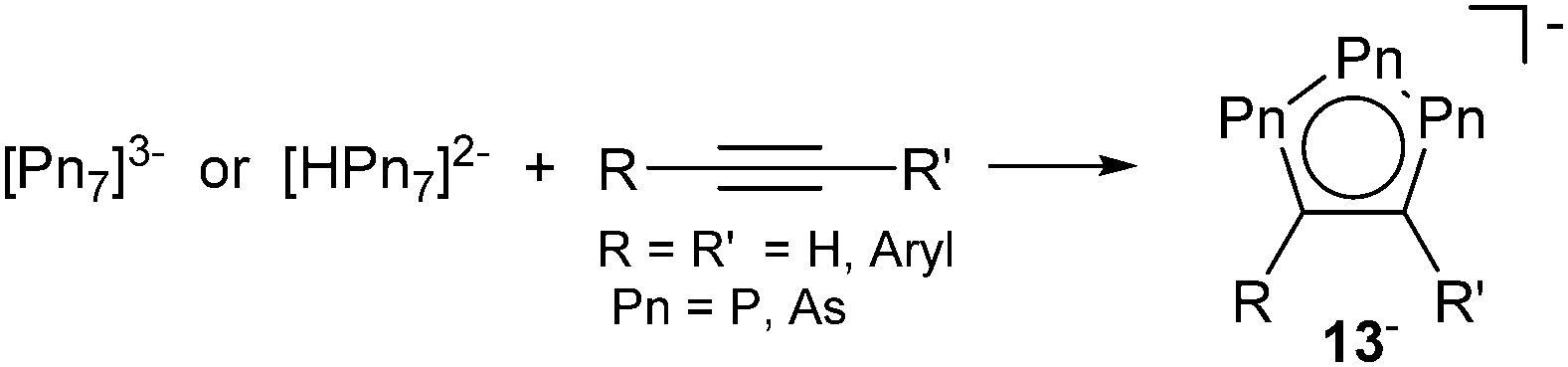 | ||
| Scheme 5 1,2,3-Tripnictoolides 13− formed by the reaction of heptapnictides and alkynes. | ||
Noteworthy is that the electronic structures of these “carbon-copy” ligands possess notable differences in comparison to cyclopentadienide ligands and derivatives thereof. They generally possess greater π-acceptor character than [Cp]− ligands which allows for the stabilization of complexes of electron-rich metal centres. The presence of Lewis basic sites on the phospholide ring allows for multiple coordination modes and the formation of multimetallic molecules and supramolecular systems.25
catena-Tetraphosphene dianions of the general formula [R–PPPP–R]2− may be described as doubly phosphanido substituted diphosphenes and are therefore related to the triphosphaallyl ions. The supersilyl-bearing (R = tBu3Si) alkali salts26 as well as the terphenyl substituted thallium analogue27 (R = C6H3-2,6-(C6H2-2,6-iPr2)2) were isolated and characterized. The bond lengths compare well to [Li(dme)3][2] (Fig. 1). All 31P chemical shifts are shifted to higher field. The alkali salts show highfield shifted resonances at 390–420 and −53 to 19 ppm for the inner and outer phosphorus atoms, respectively. The 31P NMR shifts of the thallium complex (285/180 ppm) could not be assigned.
Phosphanyl-substituted diphosphenes such as 14 represent another example related to bonding motif A and were first reported by Romanenko et al.28 The treatment of amino-substituted diphosphene 15 with triflic acid (Scheme 6) eliminates [iPr2NH2][OTf] accompanied by the in situ formation of the highly reactive phosphorus-analogue of a diazonium ion 16+ which, due to its high reactivity, is not stable even at −78 °C and forms bicyclo[1.1.0]tetraphosphanes besides unidentified products.
 | ||
| Scheme 6 Protonation of 15 in the presence of phosphanes leading to σ2–σ2–σ4-(17+) and σ2–σ2–σ4-compounds (14) formally via16+. Scheme simplified for clarity reason. | ||
In the presence of a tert-phosphane such as PPh3 the adduct 17a+ (R = R′ = Ph) is formed with a σ2–σ2–σ4 connectivity of the P3-backbone representing an example of bonding motif F (vide infra). The formation of 17a+ was confirmed by X-ray structure analysis and the obtained P–P bonding parameters of 2.025(1) Å and 2.206(1) Å are in the range of a double and a single bond, respectively. Employing a sec-phosphane such as tBu2PH in the reaction with 16+, the initially formed cation 17b+ slowly liberates HOTf to give the first example of a σ2–σ2–σ3 triphosphene 14 as an example of bonding motif B (vide infra).28,29
Recently, Grützmacher et al. reported on the synthesis of three polarized, cationic diphosphenes 18a–c+.30 Independently, a fourth derivative was also reported by our group.31 The synthesis of the precursor compound 19a–d, which belongs to bonding motif B, was achieved by two distinctly different synthetic strategies (Scheme 7). The group of Grützmacher followed the approach of DABCO (1,4-diaza[2.2.2]bicyclooctane) assisted condensation of the parent phosphinidene carbene adduct 20 with chlorophosphanes to synthesize 19a–c. Our group used the NHC induced [3+2] fragmentation reaction of the P5-cage cation 21a+ (bonding motif J, vide infra) yielding the triphosphaallyl compound 5+ as P3-fragment (vide supra) besides the P2-fragment 19d.17 The subsequent chloride abstraction from all four derivatives 19a–d by GaCl3 leads to the formation of the polarized, cationic diphosphenes 18a–d+. The derivatives 18b,d+ with sterically more demanding substituents could be isolated and structurally characterized. The P–P bond lengths are in the expected range for polarized double bonds (18b+: 2.061(1) Å, 18d+: 2.038(1) Å).
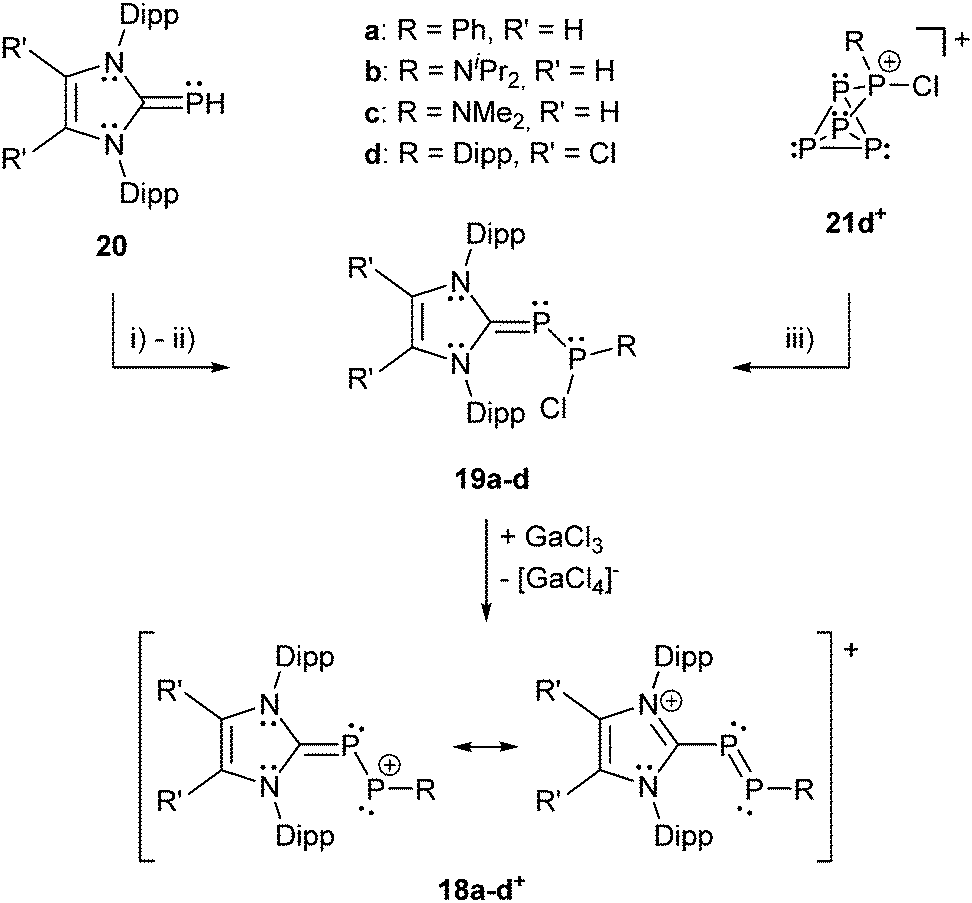 | ||
| Scheme 7 Two different approaches for the synthesis of NHC stabilized chlorodiphosphanes 19a–d comprising bonding motif B. For 19a,b: (i) +RPCl2, +DABCO, −DABCO*HCl; for 19c: (ii) +2 PCl(NMe2)2, +DABCO, −DABCO*HCl, −P(NMe2)3; for 19d: (iii) +3 1,3-bis(2,6-diisopropylphenyl)-4,5-dichloro-imidazole-2-ylidene, -5+. Subsequent chloride abstraction yields polarized cationic diphosphenes 18a–d+. Only two resonance structures that indicate the polarization of 18a–d+ are shown. | ||
The derivatives 18a,c+ with smaller residues on the phosphenium-type side prevent their isolation but enable dimerization to form dicationic cyclo-tetraphosphanes 22a,c2+ (Scheme 8, left) demonstrating the diphosphene-type reactivity. In contrast, the electrophilicity of the R-substituted P atom can be demonstrated by treatment of 18d+ with PMe3 yielding the unique σ2–σ3–σ4 species 23+ (Scheme 8, right) incorporating both bonding motif B and J (vide infra). The product may be depicted in another resonance structure including a phosphaalkene moiety, but a dihedral N–C–P–P angle of 50.5(3)° suggests a preference of the phosphanide structure. This is supported by quantum chemical calculations indicating a negative charge on the phosphanide P atom. The distance between the di- and tetracoordinate phosphorus atom is at 3.478(1) Å slightly below the edge of the sum of the van der Waals radii.
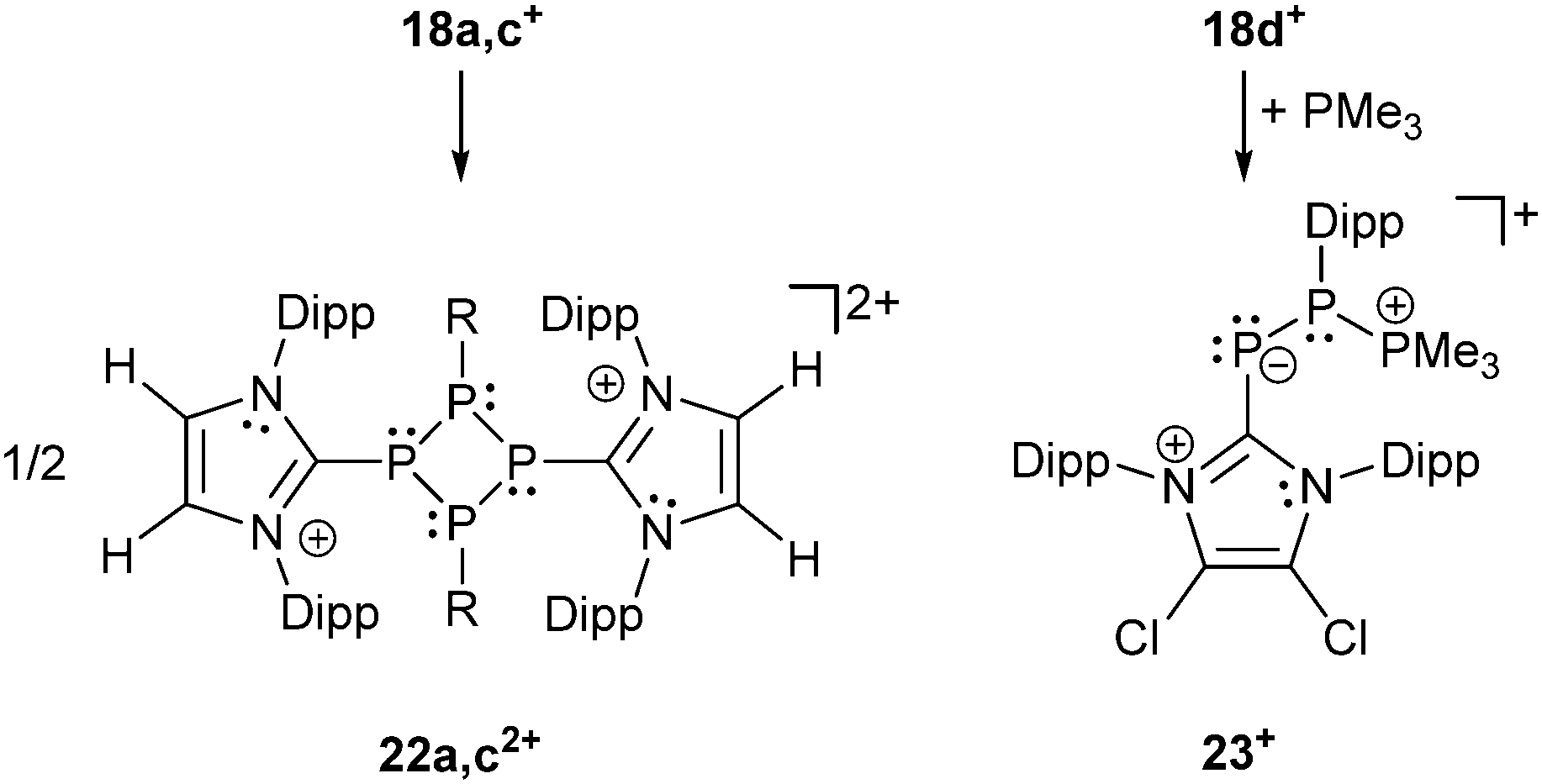 | ||
| Scheme 8 [2+2] dimerization reaction of diphosphenes 18a,c+ (left) and PMe3 attacking the more electrophilic P atom of diphosphene 18d+ forming σ2–σ3–σ4 compound 23+ (right). | ||
σ2–σ3
Three bonding motifs can be described connecting a di- and a tricoordinate phosphorus atom of which examples are already reported. They comprise alkylidene diphosphanes or phosphanylphosphaalkenes and related heteroatom-substituted derivatives of the general bonding motif B as well as diphosphen-1-ium C and diphosphan-1-ide D ions.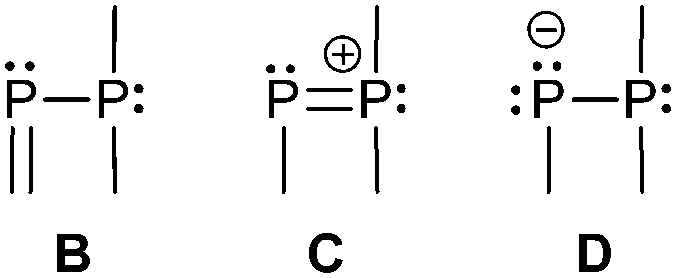
Bonding motif B
As compounds with bonding motif B are known for more than two to three decades32 and have been reviewed earlier2 we only want to comment shortly on selected examples depicted in Fig. 3.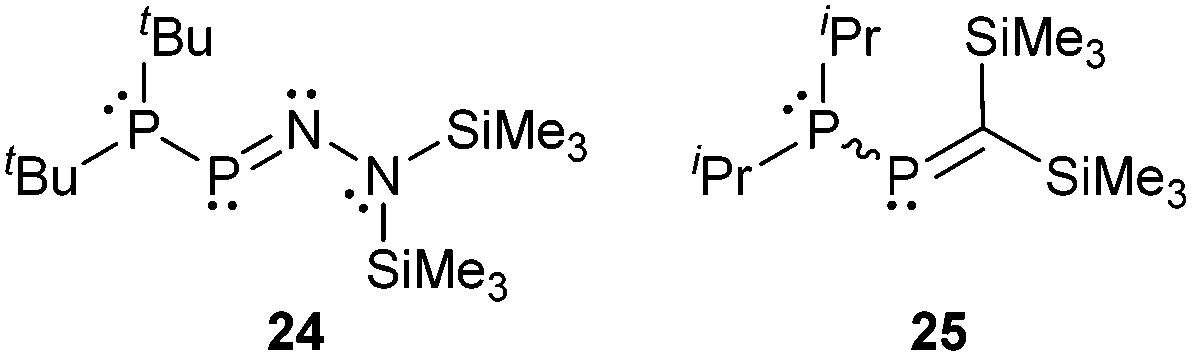 | ||
| Fig. 3 Selected examples of compounds comprising bonding motif B. | ||
In 1990 Niecke et al. reported on the 1-phosphanyldiazaphosphene 24 produced by a base-catalysed elimination of Me3SiCl from the corresponding phosphane.33 The amino group at the nitrogen atom lengthens the P–N double bond, whereas the phosphanyl group at phosphorus itself exerts only negligible changes on the P–N bond. In this example the phosphanyl group acts as a σ-donor and the amino group simultaneously as a σ-acceptor and a π-donor. As a consequence, both substituents at the P–N double bond refer to the case of σ-push pull substitution and enforce enhanced lengthening of the P–N double bond.
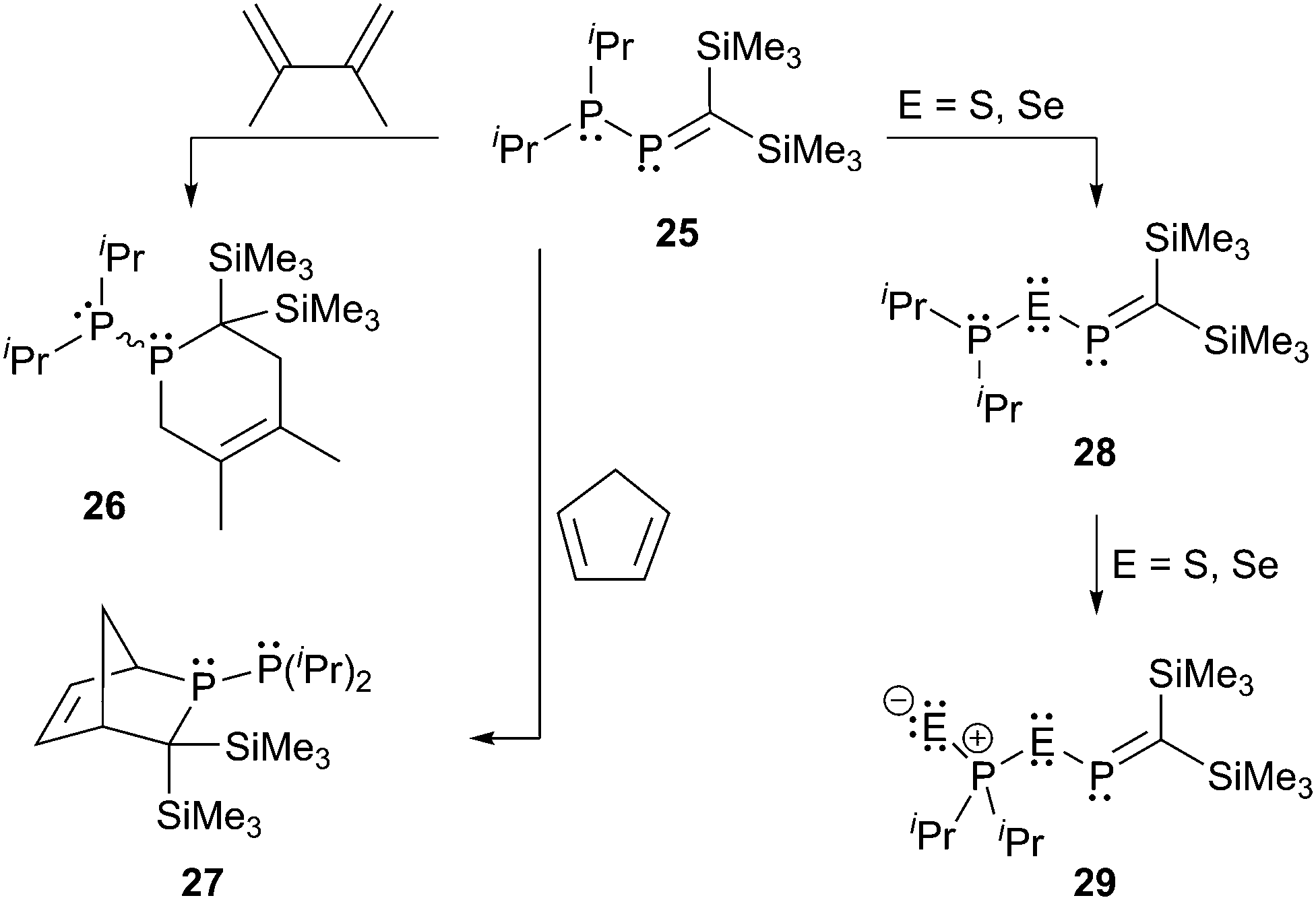
Du Mont et al. investigated the reactions of the P-phosphanyl phosphaalkene 25 at its P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and P–P bonds (Scheme 9).34 The reactions at the PC bond with 2,3-dimethylbutadiene or cyclopentadiene lead to the respective [4+2] cycloaddition products 26 and 27 which is a typical reactivity shown by phosphaalkenes. In the case of 27 also the Retro–Diels–Alder reaction could be observed at elevated temperatures. They reported that the main reaction in the oxidation of 25 with chalcogens E (E = S, Se) is the cleavage of the P–P bond to give 28 which is subsequently oxidized to 29.34a
C and P–P bonds (Scheme 9).34 The reactions at the PC bond with 2,3-dimethylbutadiene or cyclopentadiene lead to the respective [4+2] cycloaddition products 26 and 27 which is a typical reactivity shown by phosphaalkenes. In the case of 27 also the Retro–Diels–Alder reaction could be observed at elevated temperatures. They reported that the main reaction in the oxidation of 25 with chalcogens E (E = S, Se) is the cleavage of the P–P bond to give 28 which is subsequently oxidized to 29.34a
 | ||
| Scheme 9
P-Phosphanylphosphaalkene 25 undergoing [4+2] cycloaddition reactions at the PC bond (left) and oxidation of 25 under P–P cleavage (right). | ||
In the reaction of 25 with trichlorosilyltrimethylgermane (Me3GeSiCl3) they observed an unusual PC bond cleavage accompanied by a double dichlorosilylene (SiCl2) transfer, which represented an important contribution to the chemistry of stable Group 14 carbene analogues. The trichlorosilyltrimethylgermane represents a versatile reagent for the transfer of SiCl2 moieties to P-phosphanyl phosphaalkenes under very mild conditions.34b,c
Bonding motif C
The first phosphanylphosphenium ion 30a+ was obtained as triflate salt by Grützmacher et al. from the reaction of diphosphene 31a (R = tBu) with a 35-fold excess of MeOTf in CH2Cl2 as solvent (Scheme 10).35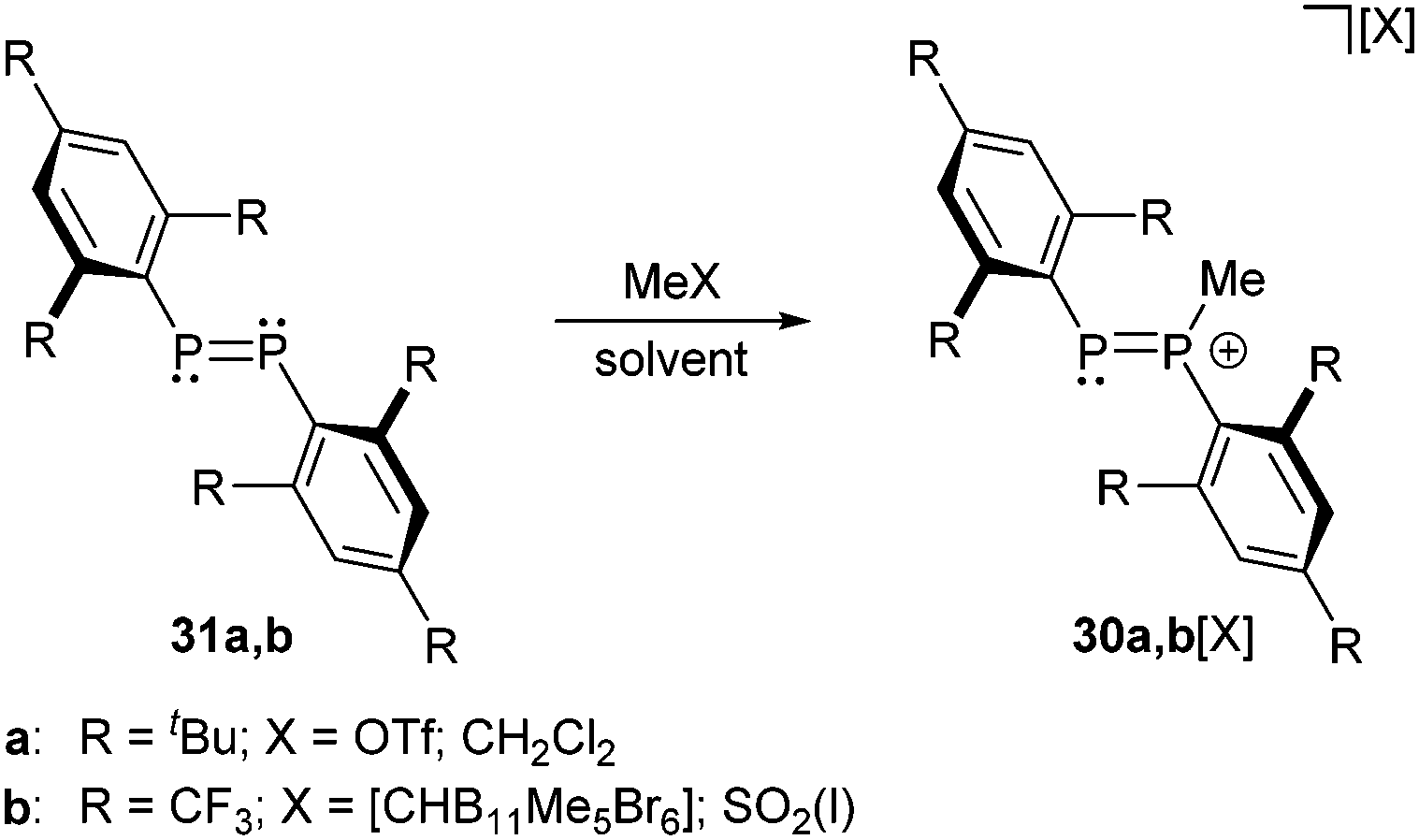 | ||
| Scheme 10 Diphosphenes 31a,b can only be methylated by very strong methylation agents to form diphosphenium cations 30a,b+. | ||
The same reaction with the very electron deficient diphosphene 31b (R = CF3) does not form the related cation 30b+ (R = CF3) even in boiling, neat methyl triflate. However, the methylation succeeds in liquid SO2(l) with very strong methylation reagents of the type R[CHB11Me5X6] (R = Me, Et, iPr; X = Br) introduced by Reed et al. (Scheme 10).36 This class of potent electrophilic “R+” alkylating agents uses the weakly nucleophilic carborane anions [CHB11Me5X6]− (X = Cl, Br) as leaving groups. In this way the Reed group managed to transform alkanes into carbenium ions via hydride abstraction below room temperature or to methylate electron deficient phosphorus compounds that are otherwise inert to conventional alkylating agents such as methyl triflate. The mixture of Me[CHB11Me5Br6] and diphosphene 31b (R = CF3) in SO2(l) was studied by NMR-spectroscopy revealing an AX spin system of 30b+ with chemical shifts at 260 and 279 ppm, typical for the tri- and dicoordinate phosphorus atoms, respectively and significantly upfield shifted compared to diphosphenes that typically display resonances at very low field (e.g.31a (R = tBu): δ = 495 ppm). The large coupling constant of 1J(PP) = 610 Hz confirms the direct connectivity. Similar values have been obtained for 30a+. Although 30a[OTf] is extremely sensitive in nonpolar solvents and decomposes readily in solution, they succeeded to obtain crystals suitable for X-ray diffraction. The methylation of 31a (P–P bond length: 2.034(2) Å) does not significantly affect the P–P bond length in cation 30a+ (2.024(2) Å). Attempts to deprotonate cation 30a+ with Et2NH as a base did not yield the expected ylide but resulted in the formation of the diphosphirane 32 (Scheme 11).35 The targeted product would have been well comparable to both bis(alkylidene)phosphoranes and bis(imino)phosphoranes. For those compound classes a thermally induced, conrotatory electrocyclic reaction according to the Woodward–Hoffmann rules has been suggested earlier on the base of theoretical and experimental results.37
 | ||
| Scheme 11 Formation of diphosphirane 32 by deprotonation of 30a[OTf]. | ||
Bonding motif D
Phosphanidophosphanes or diphosphan-1-ide ions belong to the bonding motif D and are the anionic congeners in the σ2–σ3 category of P–P bonded phosphorus compounds. Surprisingly little is known about this class of compounds, unless considering phosphorus rich oligo- and polyphosphides. This might be due to their high tendency towards condensation reactions. Baudler et al. extensively described the reaction of P2H4 in liquid ammonia. They stated that the initial step represents the deprotonation by an amide anion.38 The resulting P2H3− anion in turn again attacks diphosphane and initiates a cascade of disproportionation and aggregation reactions. Under evolution of PH3 gas the reaction ultimately leads to the formation of phosphorus rich anions which are only slightly nucleophilic and do not tend to incorporate further diphosphane. Interestingly, the formation of a large number of different Zintl type anions such as [P7]3−, [P14]4−, [P16]2−, [P19]3−, [P21]3−, [P22]4− and [P26]4− is observed.23 The reaction of P2H4 in ammonia in the presence of solvents such as THF yields hydrogen phosphides of which the selected examples [H3P3]2−,39 [H8P7]−, [H4P7]−, [H5P8]−,40 [HP7]2− (ref. 41) and [H2P7]− (ref. 42) are depicted in Fig. 4.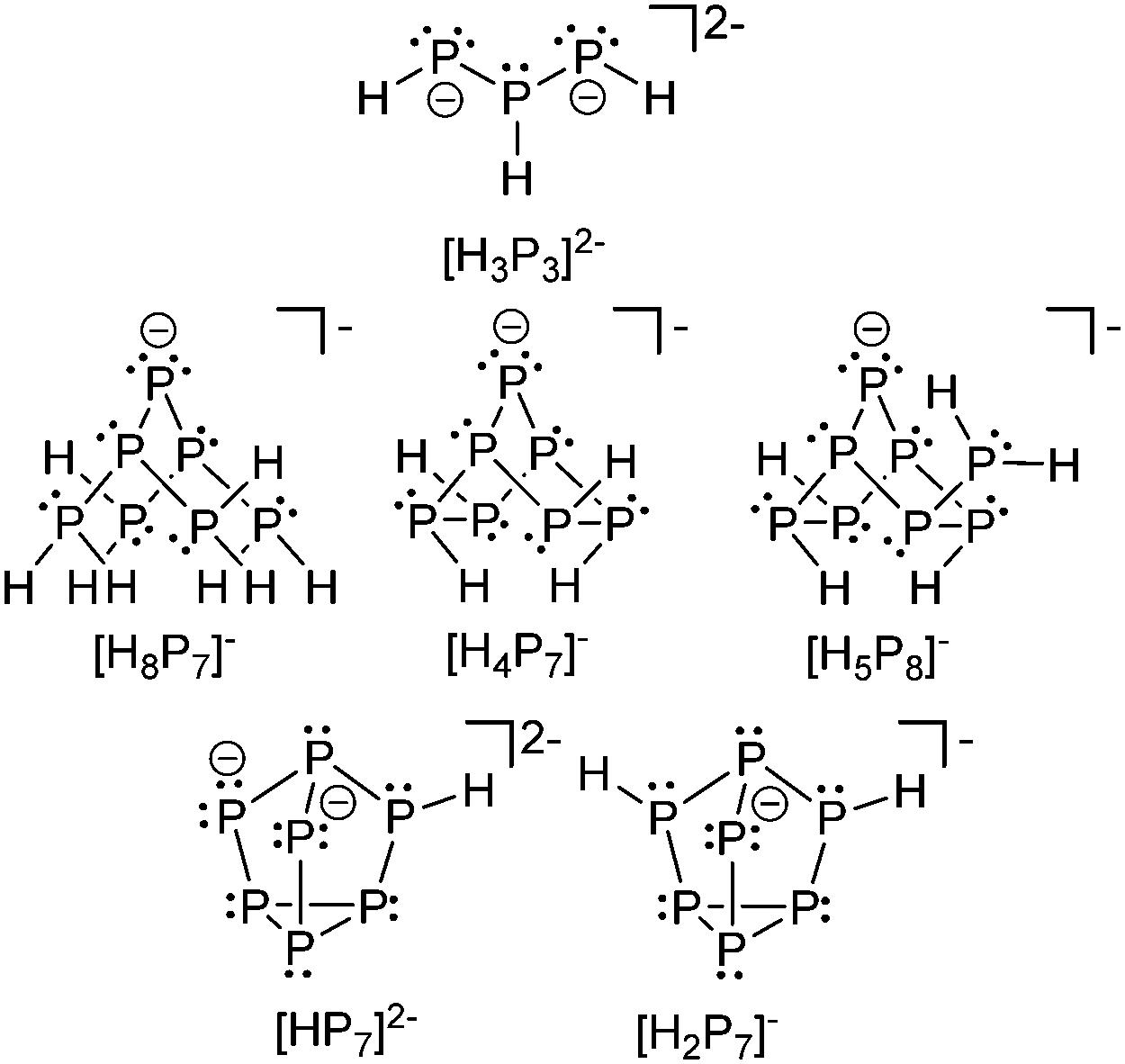 | ||
| Fig. 4 Selected examples of protonated oligophosphides. | ||
The degradation and aggregation pathways of white phosphorus in the presence of nucleophiles are of unquestionable complexity. Therefore, the underlying mechanisms, in which the phosphanylphosphanide motif also plays a key role, continue to attract attention which has led to numerous publications in the last decades. Parts of the interrelations between substituted oligophosphides, their reactivity and the mechanisms of their formation have been investigated employing e.g. bulky silanides and have been discussed elsewhere.43
Fritz et al. synthesized a series of bis-phosphanyl substituted lithium phosphanides 33a–d by treatment of the parent silyl phosphanes 34a–d with n-butyl lithium (Scheme 12).44 Interestingly, in the solid state, the lithium ion is not coordinated to the negatively charged phosphorus atom itself, but chelated by the two adjacent phosphane moieties instead. Several related acyclic and cyclic anions of the form [P(PR2)2]− are known and comprehensively discussed in the review by Macdonald et al.9
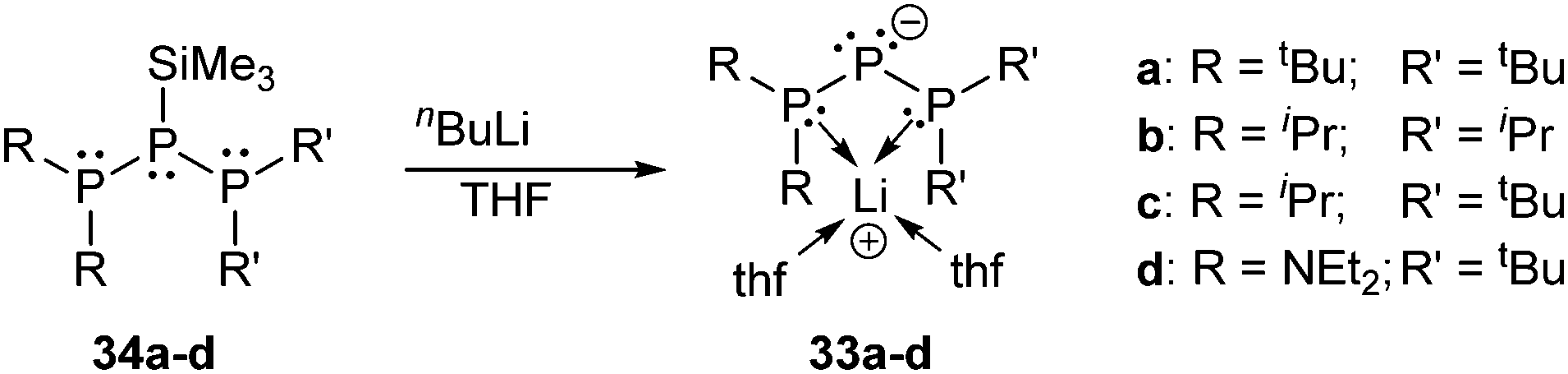 | ||
| Scheme 12 Upon treatment with nBuLi the silylated triphosphanes 34a–d form bis-phosphanyl substituted lithium phosphanides 33a–d. | ||
Tetraphosphane-1,4-diides represent another interesting substance class described by bonding motif D. Their solid state structures usually comprise mono- or dimeric ion-contact complexes.45Fig. 5 depicts the tetraphosphane-1,4-diide [Na(solv)x]2[34] as representative example, showing the characteristic M2P2 arrangement found in solid state structures of bis(alkali metal)-catena-oligophosphane-α,ω-diides [M2(solv)x(RnPn)] (M = Li, Na, K; n = 2, 3, 4; R = Ph, Mes, tBu; solv = solvent molecule or ligand). The formation of such bicyclobutane-shaped ion triples is found frequently for dianions as a result of a minimized net electrostatic energy. Four attractive Coulombic interactions (r+,−) are opposed by only two repulsive ones (r+,+ and r−,−).
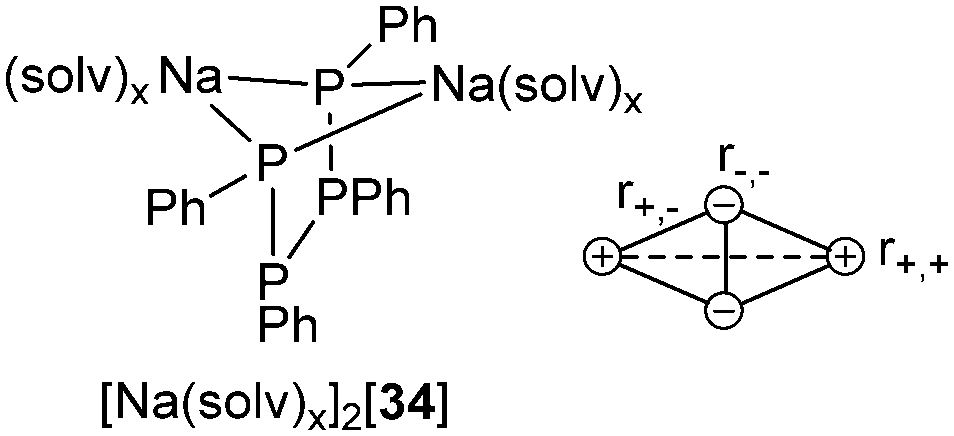 | ||
| Fig. 5 Schematic view of the tetraphosphane-1,4-diide [Na(solv)x]2[34] showing the characteristic bicyclobutane-shaped M2P2 arrangement (left); illustration of point charges and coulombic interactions (right). | ||
The group of Grützmacher reported an unexpected result from the removal of this charge-stabilizing cation anion contacts by the reaction of [Na(solv)x]2[34] with the cation sequestering ligand 2,2,2-crypt (Scheme 13).45c,d The yellow solution immediately turns to red-orange upon addition of 2,2,2-crypt accompanied by precipitation of a red, quickly crystallizing oil giving red (meso-diastereomer) and yellow crystals (rac-diastereomer) of [Na(2,2,2-crypt)]2[34].
 | ||
| Scheme 13 The sodium ions of [Na(solv)x]2[34] can be sequestrated by 2,2,2-crypt. In solution the product splits into two radical anions [35]˙−. Only one resonance structure of [35]˙− shown. | ||
As may be expected, the P4 chains in both diastereomers adopt a 1,4-anti-conformation due to electrostatic repulsion. Interestingly, the dissolution of the slightly soluble crystals yields an orange solution which contains free diphenyldiphosphene radical anions [35]˙− resulting from the homolytic cleavage of the central P–P bond in [34]2−. The radical anions exhibit a strong EPR signal (g = 2.0089) with a triplet splitting (aiso[P] = 115 MHz) resulting from two identical phosphorus nuclei. Further hyperfinecoupling to two non-equivalent ortho-protons (aiso[H] = 8.5 MHz, aiso[H′] = 4.0 MHz) indicates a hindered rotation of the phenyl groups on the EPR time scale along with a certain degree of π-type delocalization. Furthermore, the group of Grützmacher could show a complete reversibility of both reactions. Upon concentration of solutions of [35]˙− again crystals of [Na(2,2,2-crypt)]2[34] are obtained and the addition of [Na(2,2,2-crypt)]2[34] to a solution of NaOTf in THF reforms the ion triple [Na(solv)x]2[34]. Since the P–P bond lengths of the unstabilized [Ph4P4]2−-chains (central P–P: 2.224(2) Å, terminal P–P: 2.178(1) Å) in crystals of [Na(2,2,2-crypt)]2[meso-34] refer to normal single bonds they reasoned that the dissociation process into radical anions must originate from the Coulombic repulsion. Similarly, the P–P bond cleavage, leading to [P2Mes2]˙− radical anions, was also reported by another group to occur in concentrated THF solutions of the singly protonated species [K(18-crown-6)][P4HMes4]. In contrast to solutions of the dianions, [P4HMes4]˙− was found to undergo redistribution reactions forming complex mixtures of neutral and anionic species.46
σ2–σ4
The two bonding motifs E and F describe the connectivity of di- and tetracoordinate phosphorus atoms.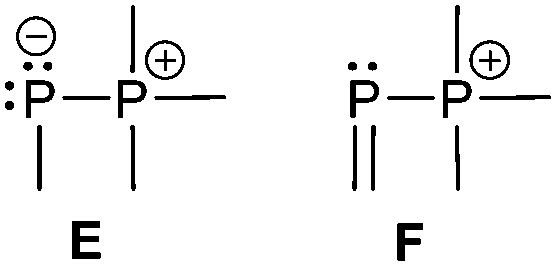
Bonding motif E
Bonding motif E comprises a large variety of zwitterionic phosphoniophosphanides. They represent the phosphorus analogues of alkylidene-σ4-phosphoranes (R2C−–P+R3) or Wittig reagents and are thus called phosphanylidene-σ4-phosphoranes or Phospha–Wittig reagents in this context. Compounds of type E have already been known for more than a half century and were first described as the donor stabilized Me3PPCF3 adduct (36a) resulting from the reaction of PMe3 and cyclic (PCF3)n (n = 4, 5; Scheme 14).47 Only very few examples are known (e.g.36b,c (b: R = Mes, c: R = Mes*),4837,493850 and 3951) and synthetically accessible in their free form (Fig. 6). They are mainly obtained as transition metal-stabilized compounds (LnM–P(R)–PR3), which are employed for PC bond formation alternatively to terminal phosphinidene complexes R–P = MLn (for reviews see ref. 25b, c and 52a–f; for a recent example of reversible phosphinidene transfer to triarylphosphane see ref. 52g).
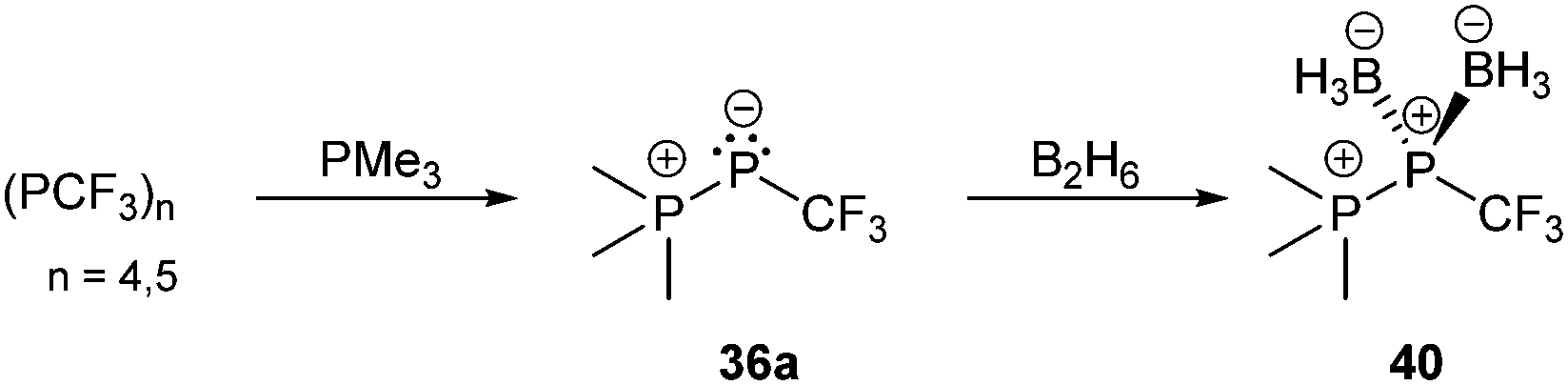 | ||
| Scheme 14 Deoligomerization of cyclo-phosphanes to phosphanylidene-σ4-phosphorane 36a and subsequent bis-borane adduct formation. | ||
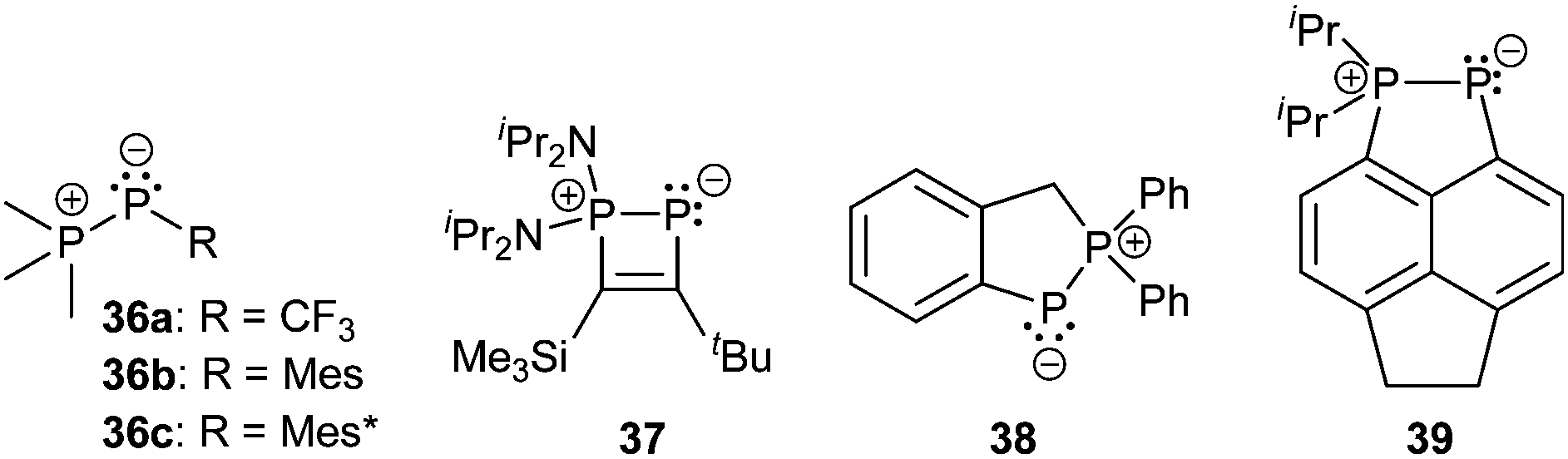 | ||
| Fig. 6 Examples of zwitterionic phosphoniophosphanides comprising bonding motif E. | ||
The known free phosphanylidene-σ4-phosphoranes have received only little attention so far, due to their high reactivity. Their isolation requires sterically demanding or electron withdrawing substituents at the σ2-phosphorus moiety in order to stabilize them kinetically and prevent di- or oligomerization.48,53 The peri-substituted compound 39 is sterically less encumbered than previous examples. Similar to the BH3 adduct 40, which is obtained from the reaction of 36a with B2H6 in Et2O (Scheme 14), the bis-borane adduct 39·2BH3 was isolated and fully characterized. It has been stated that steric rather than electronic properties are the limiting factor for the accessibility of the second lone-pair of electrons for coordination.51 A follow-up study on the coordination behaviour of 39 towards several transition metals, as well as on the oxidation products obtained by treatment with chalcogens is described elsewhere.54
Phosphanides bearing two adjacent phosphonio moieties are related to carbodiphosphoranes and cover triphosphenium cations and phospha-derivatives of cyclo-phosphazenes as their cyclic congeners. Cyclic triphosphenium cations 41a,b+ with 5- or 6-membered rings are typically obtained from the reaction of an appropriate chelating bisphosphane ligand (e.g. a: dppe; b: dppp) and PX3 (X = Cl, Br, I; Scheme 15).9,55
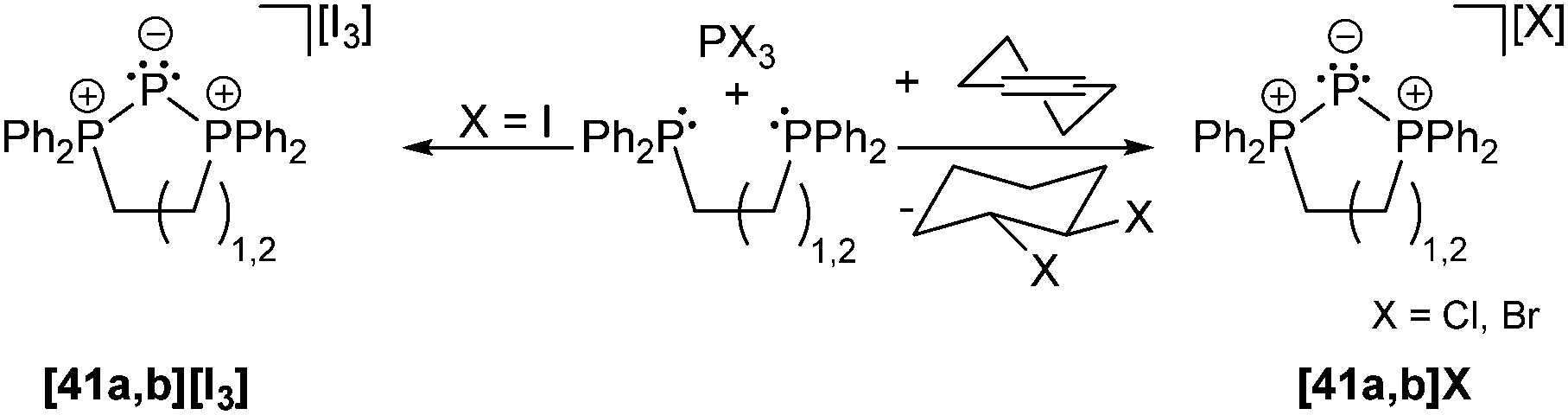 | ||
| Scheme 15 Synthesis of triphosphenium ions [41a,b]+ from PX3 and bidentate phosphanes. | ||
Although they were first reported by Fluck, extensive contributions to this field have been made by Schmidpeter, Dillon, Ragogna, Macdonald and others.9,56 Currently, triphosphenium ions are a topic of great interest as they serve as both, potential two- or four-electron donor ligands in transition metal complexes57 and as source of P+. Especially the latter allows for the synthesis of fused tricyclic 2-phosphaallylic cations such as 42+ which are derived from a convenient one-pot reaction of chelating bis-NHC (R = Me, Bn, nBu) and [41a]Br (Scheme 16). Calculations confirm that these species are best considered as carbene-ligated P(I) ions.58
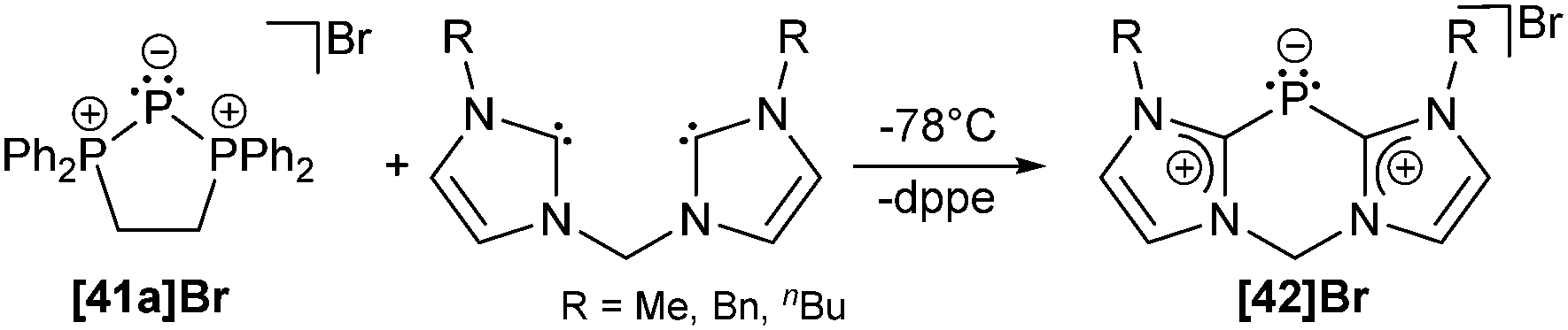 | ||
| Scheme 16 Transfer of P+ from [41a]+ onto bis-NHC to give [42a]+. | ||
Cyclic triphosphenium cations are very poor ligands and there are several arguments explaining why there is only a moderate number of metal complexes.57 Besides the positive charge lowering the frontier orbital energy, the accompanying anion may interfere in the complex formation. Furthermore, π-backbonding from the central phosphorus atom to the adjacent phosphonio moieties lowers the HOMO energy and therefore restrains coordination. Although metal complexes with cationic triphosphenium ions have been detected in solution, their low stability has so far precluded their isolation.57a,59 Ragogna et al. succeeded to synthesize appropriate ligands by introducing a bridging borate moiety into the backbone to give zwitterionic derivatives 43a,b (a: R = Ph, b: R = iPr). These ligands can undergo coordination to several transition metals as either two- or four-electron bridging donor to give stable and soluble neutral complexes (e.g. [AuCl(43a)], [{AuCl}2(μ-43a)], [{Co(CO)3}2 (μ-43a)]; Fig. 7).57a
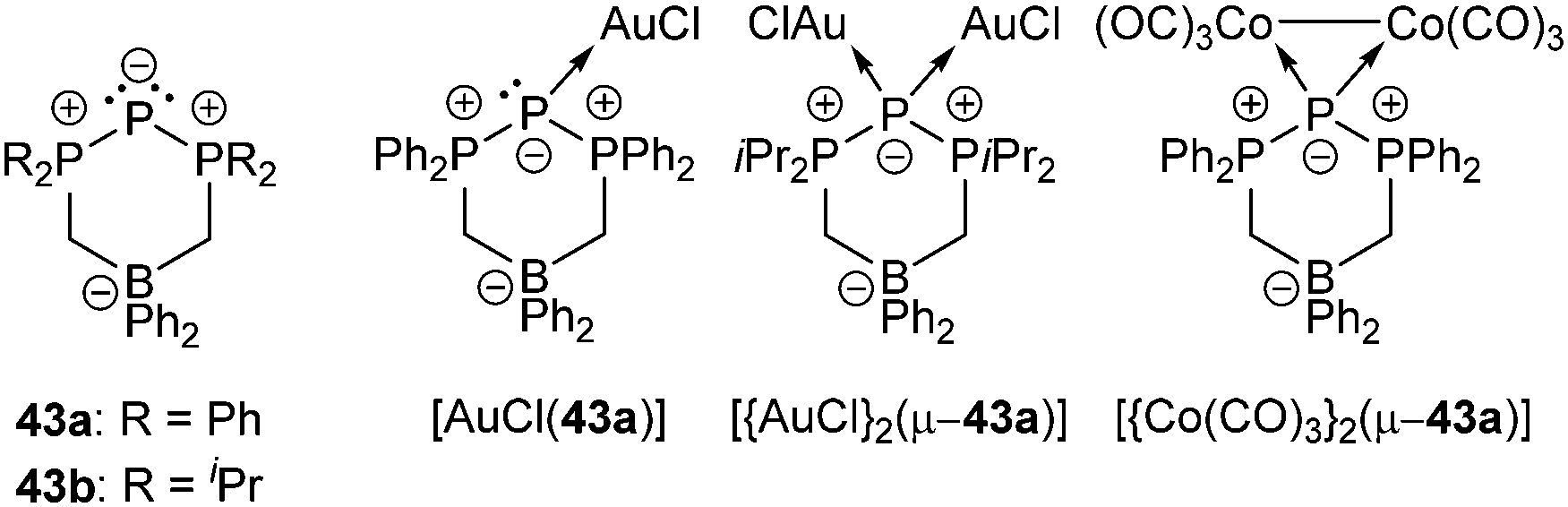 | ||
| Fig. 7 Zwitterionic triphosphenium derivatives 43a,b (a: R = Ph, b: R = iPr) and selected coordination complexes with AuCl and [Co(CO)3]2. | ||
The bromide salts proved to be suitable precursors for metathesis reactions. Accordingly, formation of phosphorus-rich oligomers by P+-transfer is observed when triphosphenium cation 41a+ is treated with LiN(PPh2)2. The release of dppe accompanies the formation of the known compound 44,60 however, in a much better yield.61 Reacting 41a[Br] with NaN(PiPr2)2 gives compound 45 in which two five-membered rings are linked via two phosphorus atoms. The mechanism of formation is unclear for both cases, however, their structural arrangement is confirmed by crystal structure analysis (Scheme 17).
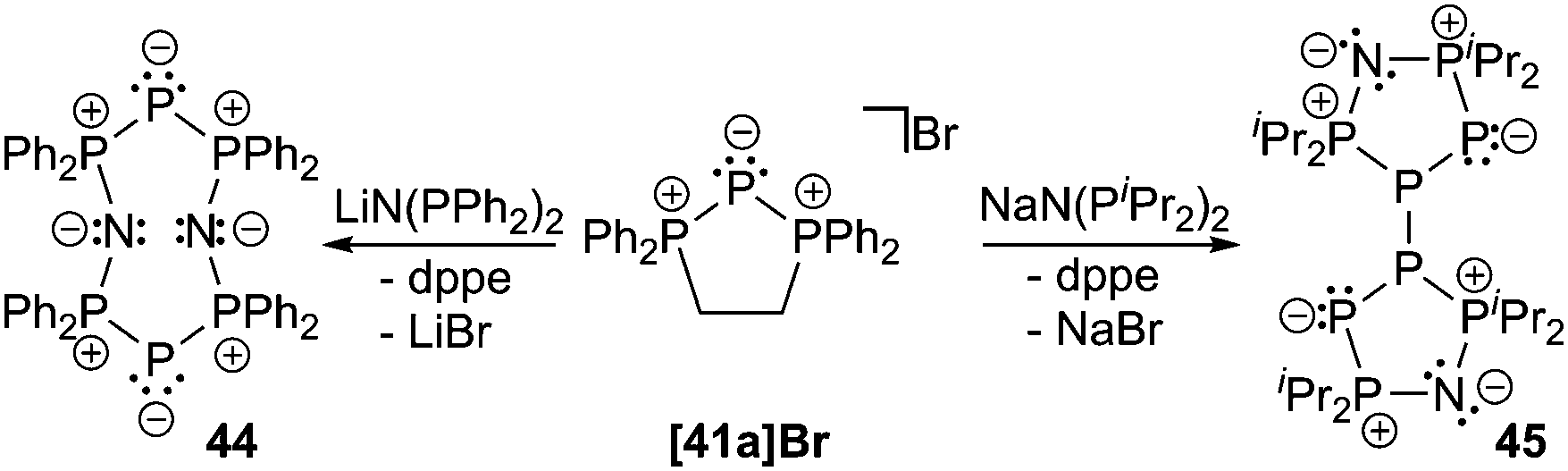 | ||
| Scheme 17 Formation of phosphorus-rich oligomers 44 and 45 by P+-transfer from [41a]+ onto bis(phosphanyl)amides. | ||
The readily available Janus-head type diphosphorus compound 46[OTf] (pyr = 3,5-dimethylpyrazolyl) is an attractive precursor for the construction of cationic ring and cage systems.8b,62 The stepwise reaction of 46[OTf] with Cy2PH gives the two novel cationic polyphosphorus frameworks [Cy4P4pyr]+ (47+) and [Cy6P7]+ (48+) as well as 49[OTf] as one of the isolated side-products (Scheme 18).63 Cations 47+ and 48+ (Fig. 8) feature di-, tri- and tetracoordinate phosphorus atoms derived from P1-synthons via a one-pot multiple P–P bond formation and represent the first examples of a σ2–σ4–σ3 bonding motif. In this reaction eight P–P bonds are formed by a combination of substitution and unprecedented base-induced reductive coupling steps to yield unusual cationic polyphosphorus compounds. The synthesis of these cations is an example of the distinct reactivity of phosphorus-centred cations compared to neutral and anionic phosphorus compounds.62a,b
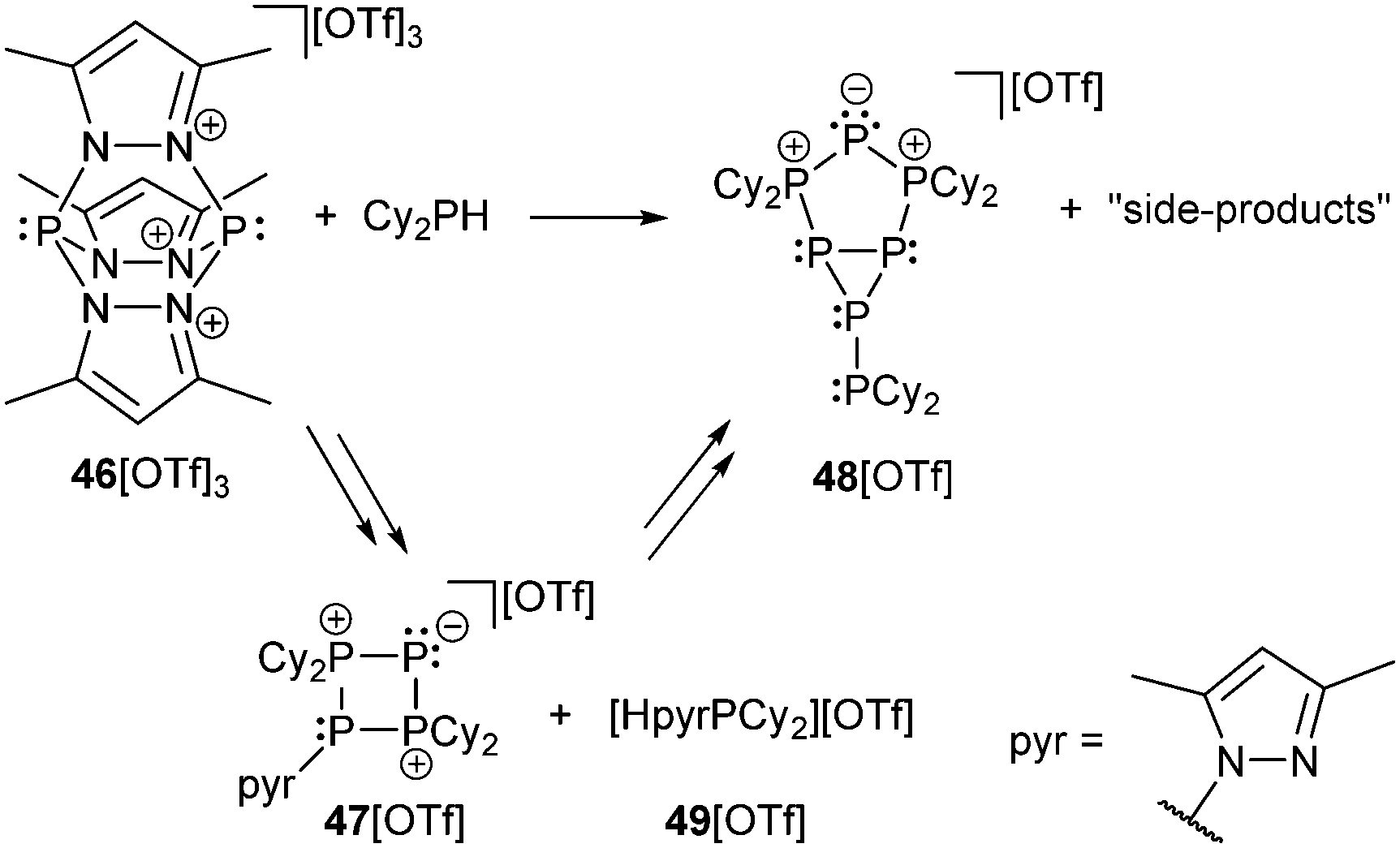 | ||
| Scheme 18 Formation of cyclic (47+) and bicyclic (48+) cationic polyphosphorus frameworks comprising the triphosphenium motif. Equation is not balanced. | ||
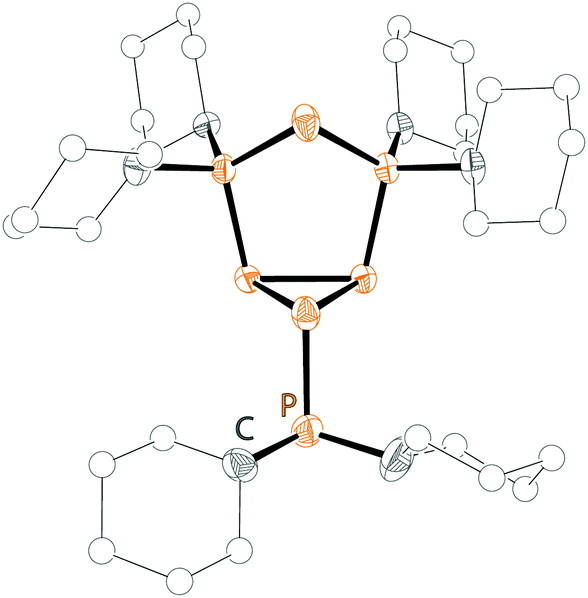 | ||
| Fig. 8 Molecular structure of cation 48+ in 48[OTf]; hydrogen atoms and anion are omitted for clarity. | ||
Bonding motif F
The main difference between bonding motif E and F is the π-acceptor ability of the substituent X relative to the phosphonium moiety (Fig. 9). This results in a different bond order or degree of π-(back)bonding of the dicoordinate phosphorus atom to the two substituents. From our point of view there is a continuous transition of π-(back)bonding predominantly to the phosphonium moiety (e.g. phosphanylidene-σ4-phosphoranes) to equal π-(back)bonding to both moieties (e.g. triphosphenium-compounds) (both bonding motif E). In compounds described by bonding motif F the π-(back)bonding is predominantly to the other substituent X and can mainly be described as genuine double bond.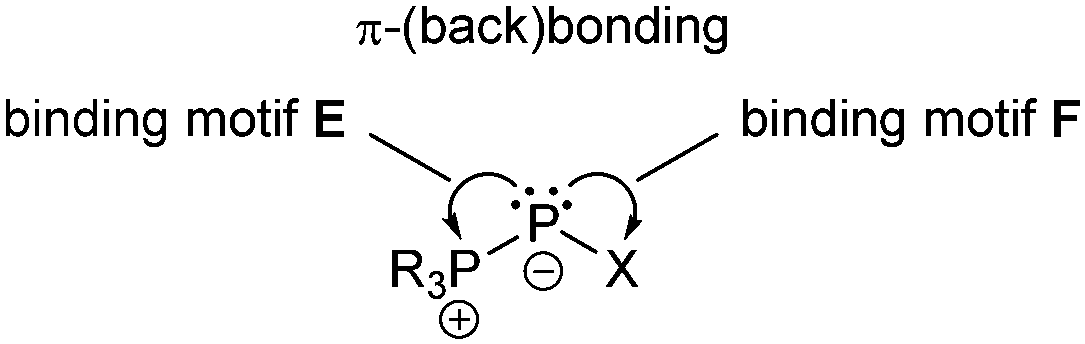 | ||
| Fig. 9 Illustration of π-(back)bonding in σ2–σ4 phosphorus compounds. | ||
A typical representative of bonding motif F was synthesized by Niecke et al. already in 1994.64 The phosphonio-phosphaalkene or triphenylphosphane stabilized methylenediylphosphenium ion 50+ as tetrachloroaluminate salt 50[AlCl4] remains unchanged in solution, whereas 50[OTf] decomposes quantitatively into phosphaalkyne 51 under elimination of Me3SiOTf and PPh3 (Scheme 19). A reaction proceeding in the reverse manner has been reported in which the phosphonio-phosphaalkene 52+ is formed by a 1,2-addition reaction of [HPPh3][OTf] to the bulky adamantyl-substituted phosphaalkyne 53.65 Further reactions of phosphaalkynes leading to comparable derivatives comprising bonding motif F are described elsewhere.66
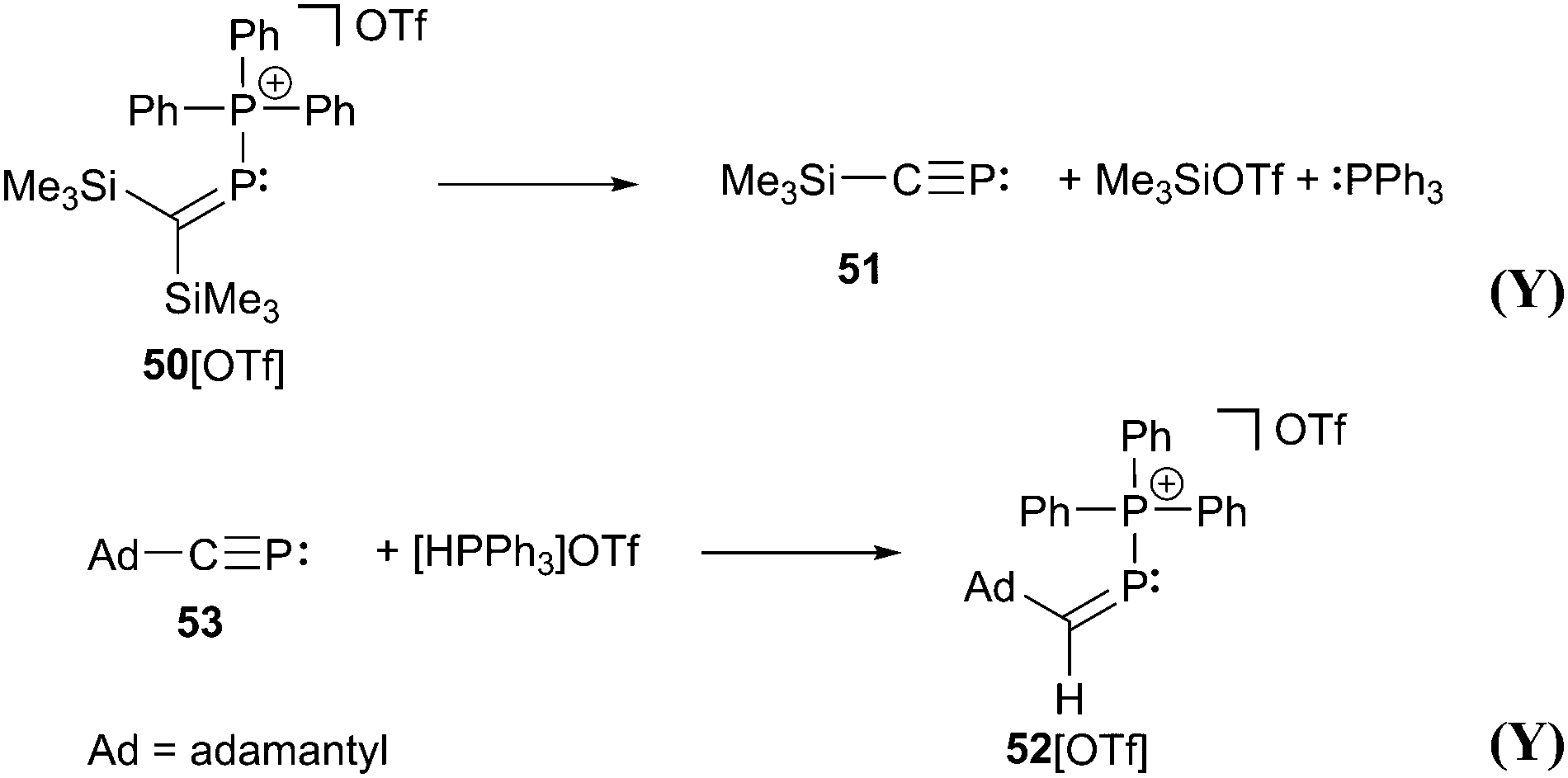 | ||
| Scheme 19 The phosphonio-phosphaalkene 50+ decomposes giving phosphaalkyne 51. In a reverse reaction 53 and triphenylphosphonium triflate form phosphonio-phosphaalkene 52[OTf]. | ||
The cation 17a+ which also comprises bonding motif F was already discussed in the context of bonding motif B.28 The regular P–P double (2.025(1) Å) and single bond (2.206(1) Å) of 17a+ allow a clear categorization. A formal substitution of the carbon bonded phosphorus atom by nitrogen leads to the cation 54+ which was described as the triphenylphosphane adduct of an iminophosphenium or phosphadiazonium ion. This compound is especially intriguing, since a classification of the bonding motif is not straightforward. In fact, on the one hand, 54+ is valence isoelectronic to cations 17a+ and 52+ which, due to a regular P–C or P–P double bond, clearly belong to bonding motif F. On the other hand, the P–N π-interaction to the Mes*N-moiety as X-substituent exceeds a bond order of two and is therefore much stronger than in examples belonging to bonding motif E or F.
In order to understand the bonding situation in 54+ we first want to discuss the phosphadiazonium ion (Mes*PN)+ which can be described by a set of resonance structures I–III (Scheme 20). Although it was frequently described as a cation with a σ1 phosphorus atom (I), this is certainly only the case with a weakly coordinating anion and in the absence of donors. Actually, the bonding environment of the phosphorus atom in Mes*NPX derivatives shows a great flexibility depending on the substituent X. This is achieved by a possible rehybridization of the nitrogen atom from sp2 in well-stabilized to sp in poorly stabilized (Mes*NP)+ derivatives. Mes*NPCl is best described as covalent chloroiminophosphane (II) with a σ2 P atom and a slightly elongated P–Cl single bond (2.142(4) Å). The P–N distance is rather short (1.495(4) Å) and the P–N–C angle (154.8(4)°) is much larger than expected for an sp2-imine, thus indicating a starting transition to an iminophosphenium ion.67 If X is a weakly coordinating anion such as [AlCl4]− the P–N distance (1.475(8) Å) is only slightly shortened, but the P–N–C angle (177.0(7)°) is widened to almost 180° suggesting sp-hybridization of nitrogen. Additionally, two P–Cl contacts (3.16 and 3.27 Å) shorter than the sum of the van der Waals radii (∼3.55 Å)7 are observed.67 Also the triflate derivative 55[OTf] shows a short P–N bond length (1.467(4) Å) and an almost linear environment at the nitrogen atom (P–N–C: 176.4(3)°). Despite a P–O distance of 1.923(3) Å that is much closer to a P–O single bond (∼1.6 Å) than to the sum of the van der Waals radii (∼3.3 Å),7 the large P–N–C angle reflects an efficient stabilization of the phosphorus atom by the Mes*N-moiety (II).68
 | ||
| Scheme 20 Representative resonance structures I–III illustrating the bonding situation in phosphadiazonium ions (Mes*NP)+. L can be a neutral ligand/donor or a counterion. | ||
Returning to compound 54[OTf], which is obtained by addition of PPh3 to 55[OTf], the crystal structure comprises a central σ3 phosphorus atom which adopts a distorted trigonal pyramidal geometry (Scheme 21 and Fig. 10).69 The P–P bond length (2.625 Å) corresponds to a long single bond and the P–O distance to the coordinate triflate anion (2.298 Å) is roughly 0.4 Å longer than in 55[OTf] but still closer to a P–O single bond than to the sum of the van der Waals radii. The P–N bond (1.486(4) Å) is slightly longer and the P–N–C angle (169.5(4)°) more acute compared to 55[OTf]. This may be rationalized by a more effective stabilization of the central phosphorus atom by PPh3 as additional donor ligand. Therefore, the solid state structure of 54[OTf] might best be described by resonance structures II and III. Interestingly, in solution the P–P bond is obviously not preserved as there was found no evidence of P–P coupling in 54[OTf]. In contrast, the weaker coordinating anion [AlCl4]− in 54[AlCl4] effects a stronger fixation of the PPh3 substituent and allows the observation of two doublets in the 31P NMR spectrum (δ(NP) = 87 ppm, δ(PPh3) = 22 ppm, 1J(PP) = 340 Hz).70
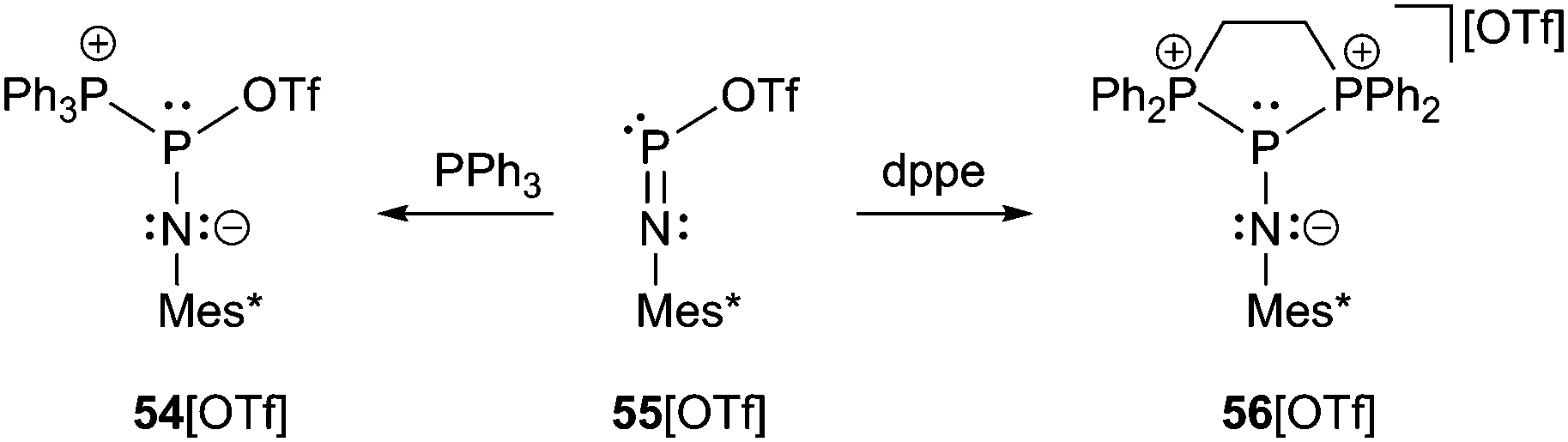 | ||
| Scheme 21 Reaction of lewis acidic 55[OTf] with PPh3 and dppe to yield P–P bonded compounds 54[OTf] and 56[OTf]. | ||
 | ||
| Fig. 10 Molecular structure of 54[OTf]; hydrogen atoms are omitted for clarity. | ||
A similar compound, in which the P–P bonds are maintained in solution is obtained by the addition of the bidentate ligand dppe to 55[OTf] yielding 56[OTf] (Scheme 21 and Fig. 11).71 The triflate anion is liberated and, in this case, non-coordinating, whereas two long P–P bonds are formed to the central σ3 P atom (2.5708(9) Å and 2.5392(9) Å). A low temperature NMR study at −80 °C revealed that the P–P bonds also remain unequal in solution (δ(NP) = 36 ppm, δ(dppe1) = 10.2 ppm, 1J(PP) = 492 Hz, δ(dppe2) = 10.3 ppm, 1J(PP) = 419 Hz). The coordination environment at the distorted trigonal pyramidal P atom may be rationalized by the occupied πx- and πy-orbitals of nitrogen overlapping the σ*-orbitals of the P–P bonds, thus elongating them, but shortening the P–N bond (1.489(1) Å) and facilitating an effective sp-hybridization of the nitrogen atom (P–N–C: 179.3(7)°) (Fig. 11, right).
 | ||
| Fig. 11 Molecular structure of 54[OTf]; hydrogen atoms are omitted for clarity (left); illustration of negative hyperconjugation in 56[OTf]. | ||
From our point of view 54+ and 56+ mark the transition zone to phosphane stabilized phosphenium or phosphanylphosphonium ions which belong to bonding motif J (vide infra).
σ2–σ5
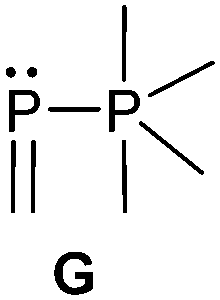
Bonding motif G
Examples of a dicoordinate phosphorus atom adjacent to a pentacoordinate phosphorus atom are very rare. We believe that up to date only two examples are reported of which one was proven its structural connectivity by X-ray diffraction analysis. In order to stabilize such an unusual bonding situation the dicoordinate phosphorus atom is required to be comparably electron poor. Regitz et al. treated the tetracyclic phosphanylphosphaalkene cage compound 57 with o-chloranil. Instead of the expected [4+2] cycloaddition reaction with the PC double bond, they observed the oxidative addition to the σ3 phosphorus atom to give compound 58 (Scheme 22). The phosphaalkene moiety occupies an equatorial position and the P–P bond length of 2.239(1) Å clearly indicates a regular P–P single bond (Fig. 12).72
 | ||
| Scheme 22 Oxidative addition of o-chloranil to phosphanylphosphaalkene 57 yielding a rare example (58) comprising bonding motif G. | ||
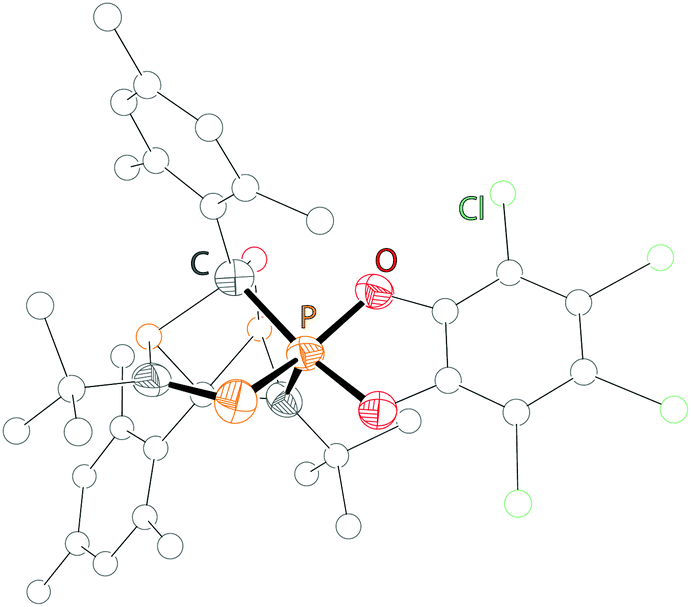 | ||
| Fig. 12 Molecular structure of compound 58; hydrogen atoms are omitted for clarity. | ||
Treatment of silylated phosphaalkene 59 with the chloro-bis(catecholato)phosphorane 60 yields another example of a compound (61) featuring a σ2–σ5 bonding motif. The reaction is accompanied by the formation of two eq. of Me3SiCl and the product can be obtained in 80% yield after recrystallization (Scheme 23). Characterization of this compound included NMR, IR and elemental analysis.73 Evidence of the structural arrangement was recently supported by a computational study of the same group on the mechanism of formation. They suggest a three step mechanism for the formation of the first experimentally observable intermediate, in which the P–P bond is already established, but the Si–O bond is still intact. The PC bond is found to be in slightly disfavoured (8.9 kJ mol−1) E-configuration.74
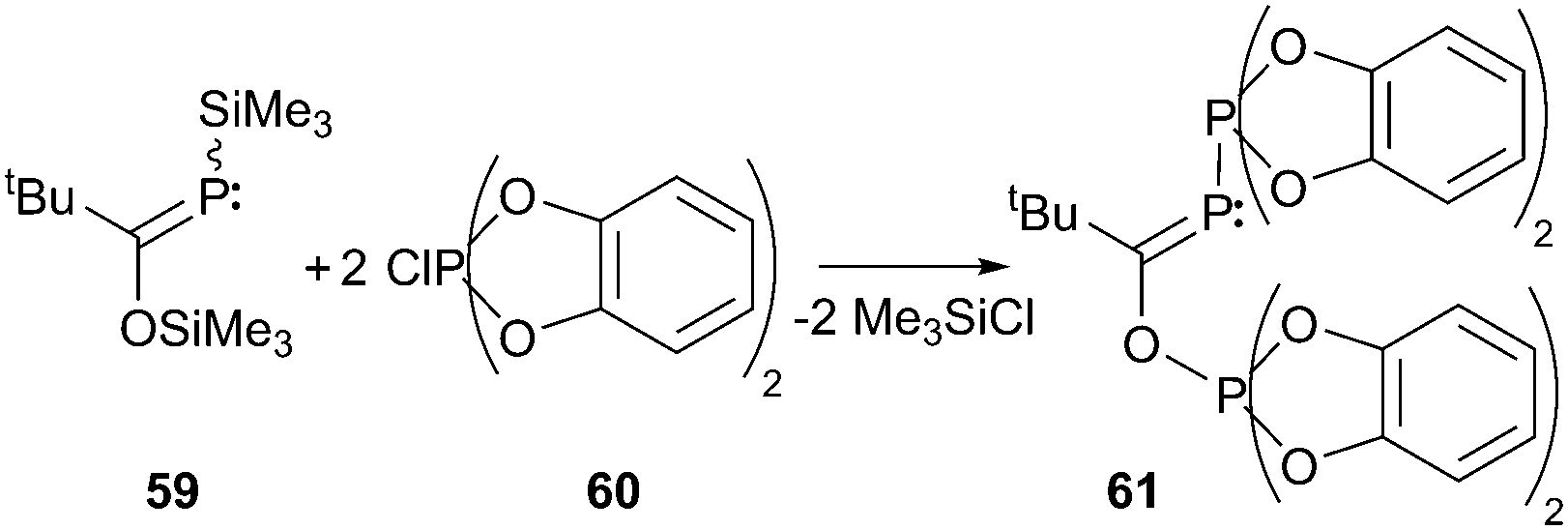 | ||
| Scheme 23 Condensation of silylated phosphaalkene 59 and chlorophosphorane 60 giving 61, which features the rare σ2–σ5 bonding motif G. | ||
σ3–σ3
Apart from diphosphanes, which are the most prominent representatives of σ3–σ3 phosphorus compounds, two other possibilities to connect two σ3 phosphorus atoms can be described by bonding modes H and I.
Bonding motif H
This substance class contains compounds in which a phosphanyl moiety is linked to a σ3 phosphorane. Predominantly in the early 1990's phosphanyl-bis(imino)phosphoranes and phosphanyl-bis(alkylidene)phosphoranes have been studied and discussed elsewhere.37a,b,75Bonding motif I
To the best of our knowledge, only once such a dication has been synthesized and characterized.76 Recognizing the relationship to the aforementioned σ2–σ3 phosphanylphosphenium ions (bonding motif C) and the difficulties in accessing them, the necessity to employ a weakly coordinating anion such as [Al(ORF)4]− (RF = C(CF3)3) seems obvious. The red dication 622+ was obtained from the parent diphosphane 63 by two single electron oxidation steps with Ag[Al(ORF)4] via the radical monocation (trip = 2,4,6-triisopropylphenyl; Scheme 24). The 31P NMR resonance at 168.8 ppm is shifted to even higher field than that of the related phosphanylphosphenium ions (30+). The P–P bond length of 2.021(2) Å and the planar geometry in the crystal structure indicate a regular double bond (Fig. 13). | ||
| Scheme 24 Oxidation of very bulky diphosphane 63 with silver salt of a very weakly coordinating anion yielding 622+, the only reported example of a diphosphenium dication. | ||
 | ||
| Fig. 13 Molecular structure of the diphosphenium dication 622+; hydrogen atoms are omitted for clarity. | ||
σ3–σ4
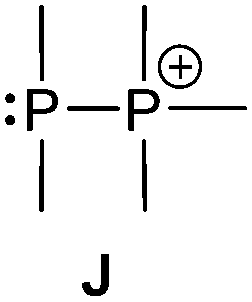
Bonding motif J
The category of the σ3–σ4 bonding motif J comprises neutral derivatives and a large variety of cations. The group of neutral derivatives (Fig. 14) includes the diphosphane monochalcogenides 64a (E = O, S). Although not many examples have appeared in the literature so far, they are of interest as flame retardants.77 The applicability of the well-known diphosphane monooxides R2P(O)–PR2 (64a; E = O)1,2 is somehow restricted by their lability towards moisture and oxygen. The tautomer of these oxides are di(phosphanyl) oxides or anhydrides of phosphinous acids, R2P–O–PR2 (64b; E = O) which are in most cases less stable than the corresponding diphosphane monoxides (46a). For compounds with alkoxy- (OR) and aminosubstituents (NR2) or mixtures of both equilibria between 64a and 64b are observed in certain cases depending also on steric demand.78 However, it has been shown that the introduction of strongly electron-withdrawing substitutents such as fluoride,79 trifluoromethyl- (CF3)80 or 2,4-bis(trifluoromethyl)phenyl- (2,4-(CF3)2C6H3) groups stabilize the anhydrides of the phosphinous acids.81 The bis(bis(trifluoromethyl)phosphanyl) oxide, (CF3)2P–O–P(CF3)2, is stable with respect to its corresponding phosphane oxide tautomer, however, it deflagrates readily on contact with air. Hoge et al. succeeded to synthesize and crystallographically characterize the (2,4-(CF3)2C6H3)2P–O–P(2,4-(CF3)2C6H3)2 derivative (δ = 105.6 ppm, 31P NMR in CDCl3) but also observed small quantities of the isomeric diphosphane monoxide (2,4-(CF3)2C6H3)2P(O)–P(2,4-(CF3)2C6H3)2 in the reaction, identified by its characteristic resonance in the 31P NMR spectrum at 39 ppm for the oxygen-bonded phosphorus atom and −32.5 ppm for the trivalent phosphorus atom (1J(PP) = 247 Hz).81 The stability of 64a is significantly improved for the heavier congeners with E = S or Se, however, large scale syntheses of these compounds in high purity are currently not available. Reported syntheses that are not suitable on an industrial scale involve desulfurization of diphosphane disulfides,82 oxidation reactions of diphosphanes with elemental sulfur,83 or comproportionation reactions of diphosphanes and diphosphane disulfides.83a,84 The reaction of sodium thiophosphinites with R2PCl has also been applied for the formation of compounds of type 64a.85 All these approaches are either not very selective, low-yielding or require harsh conditions. Recently, Morris et al. presented a convenient approach for the preparation of derivatives of 64a (R = Ph, Cy; E = S) by reacting R2PCl with Li2S. In a subsequent isomerization reaction the rarely reported di(phosphanyl) sulfides (64b; E = S) were obtained and used as ligands in ruthenium coordination complexes.86 To the best of our knowledge, synthetic protocols for diphosphane monoselenides (64a; E = Se) and -tellurides (64a; E = Te) are unreported. Only very few derivatives (R = tBu, R2N) of di(phosphanyl) selenides (64b; E = Se) and even fewer tellurides (64b; E = Te) are reported and prepared in a similar manner to the sulfur derivatives or via persistent phosphanyl radicals.87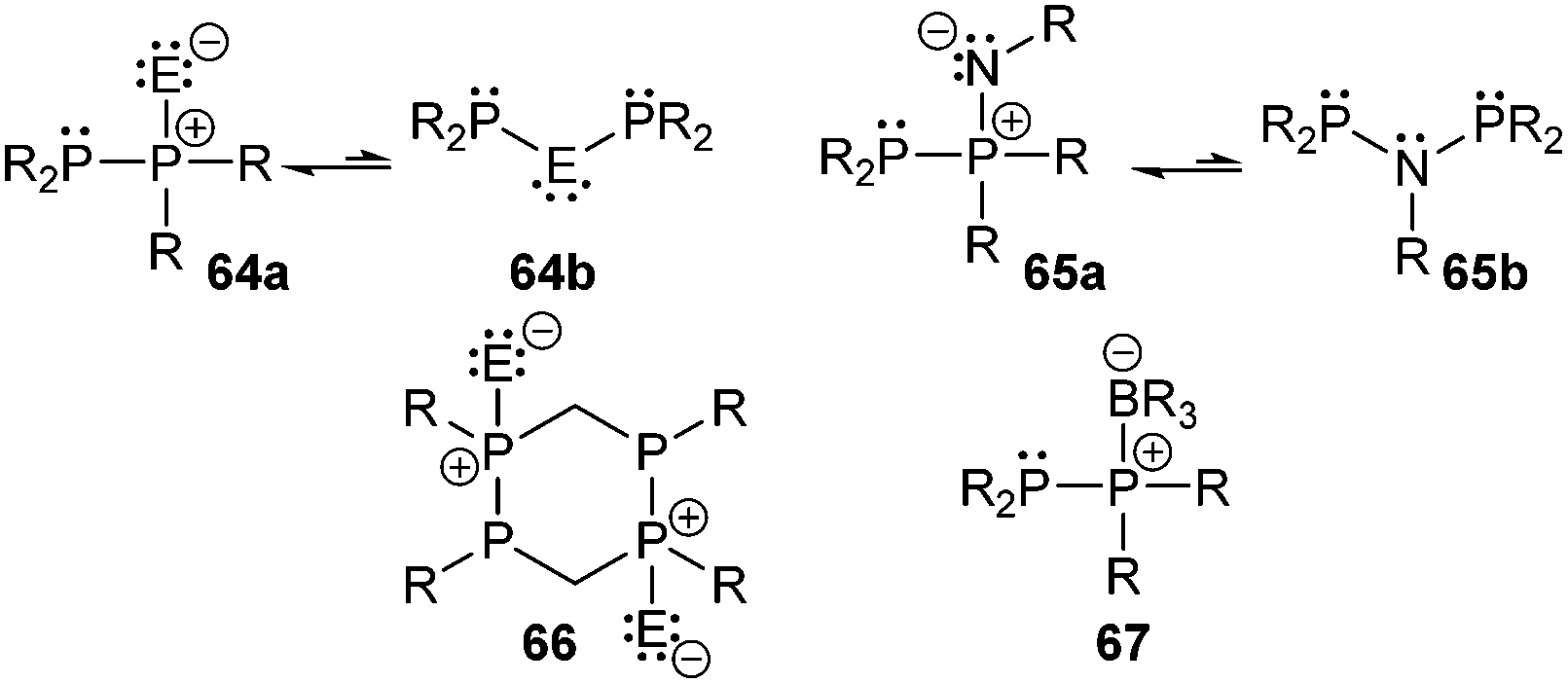 | ||
| Fig. 14 Selected examples of neutral derivatives 46–49 comprising bonding motif J. | ||
Iminophosphoranes88 have found applications in organic synthesis89 or as ligands in transition metal complexes.90 However, the chemistry of the related iminodiphosphanes of the general structure 65a is only scarcely developed (Fig. 14).91 They are isoelectronic to the diphosphane monochalcogenides 64a and are also tautomers of the more commonly encountered di(phosphanyl)amines 65b with an R2P–NR–PR2 skeleton.92 Similar to the rearrangement of 64a to 64b, a transformation of compounds of type 65a to 65b has been shown to take place upon coordination to transition metals.93
The oxidation of cyclo-phosphanes with chalcogens94 leads to the formation of product mixtures, as exemplified in the reaction of (PPh)5 with a deficiency of selenium in which one or two [PPh] units of the cyclo-phosphane are formally replaced by Se atoms.95 The oxidation of cyclo-1,4-(CH2)2(PtBu)4 with chalcogens showed that the two antipodal CH2 units of the starting material provide structural braces that allow dichalcogenation to occur without disruption of the six-membered C2P4 ring giving dichalcogenated derivatives such as the 2,5-isomers 66 (Fig. 14, R = tBu; E = S, Se).96 Density functional theory (DFT) calculations for the 2,5-chalcogenated derivatives 66 (E = S, Se) and the corresponding radical cations and dications predict significant structural changes of the six-membered ring upon oxidation. The formation of a transannular P–P single bond (ca. 2.25 Å) in the cyclic dications is indicated by geometry and consideration of the frontier orbitals.97 Boron adducts of diphosphanes of type 67 comprising bonding motif J are very scarce and only very few reports are known.98 However, borane complexes of cyclo-oligophosphanes are interesting objects of study since their structures give insight into the reactivity of cyclo-oligophosphanes and particularly into the relative nucleophilicity of the coordinated and uncoordinated phosphorus atoms. First reports by Cowley and Pinell on the treatment of cyclo-oligophosphanes cyclo-(PR)n (R = Et, nPr, nBu, Ph)99 with boron trihalides were recently complemented by Hey-Hawkins et al. They investigated the adduct formation of cyclo-oligophosphanes cyclo-(PPh)5 (68a) and cyclo-(P4Ph4CH2) (68b) in the reaction with BH3(SMe2) to form complexes 69a,b (Scheme 25). The latter complex 69b is of particular interest since they reasoned that the coordination of the borane facilitates the formation of the respective tetraphosphacyclopentanide anion.100 In their findings they concluded that the complexes 69a,b are readily formed from the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 reaction of 68a,b with BH3. The formation of adducts with a higher ratio of BH3 per molecule appears to be disfavoured. The position of the BH3 moieties in the cyclo-phosphane rings indicates a comparable high nucleophilicity of the respective P atoms in both cases. In solution, derivative 68a forms a mixture of two diastereomers which are related to each other by inversion of the tricoordinate P atoms, while a complex mixture of isomers is observed for 68b.
:2 reaction of 68a,b with BH3. The formation of adducts with a higher ratio of BH3 per molecule appears to be disfavoured. The position of the BH3 moieties in the cyclo-phosphane rings indicates a comparable high nucleophilicity of the respective P atoms in both cases. In solution, derivative 68a forms a mixture of two diastereomers which are related to each other by inversion of the tricoordinate P atoms, while a complex mixture of isomers is observed for 68b.
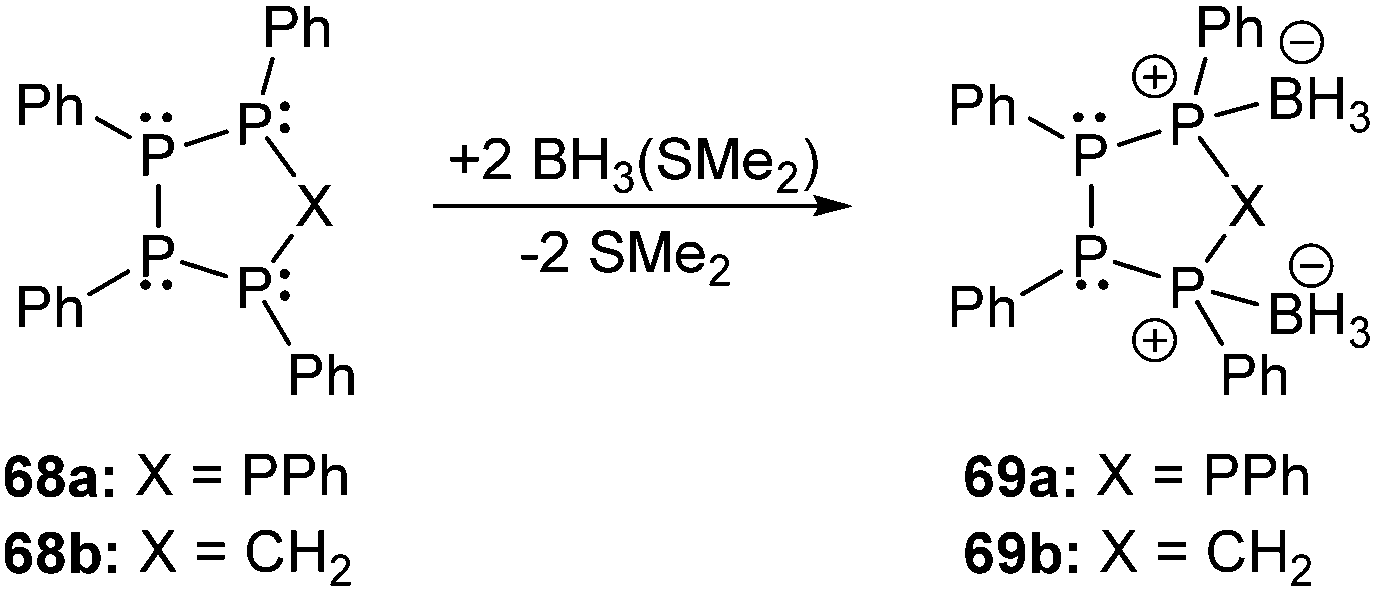 | ||
| Scheme 25 Formation of bis(borane) adducts of cyclo-phosphanes. | ||
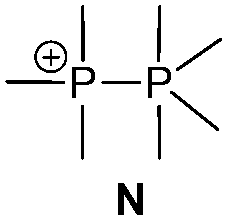
There is a large number of cationic species comprising bonding motif J. Phosphanylphosphonium ions consist of a tricoordinate phosphanyl moiety and a phosphonium centre which is analogous to an ammonium centre and conventionally defined as a tetracoordinate phosphorus center bearing a formal positive charge (iii, Scheme 26). According to another resonance structure, they are also termed phosphane stabilized phosphenium ions (ii). The question whether the σ3–σ4 P–P bond possesses rather a covalent or a dative character was theoretically investigated by Pietschnig.101 Calculations (MP2/6-311G(d)) on a model system (R = Me, X = Me or NH2; Scheme 26) revealed that according to Wiberg bond indices (0.867 for X = Me, 0.807 for X = NH2) the P–P bond in both derivatives are best described as single bonds. However, the heterolytic cleavage was only found to be slightly preferred over the homolytic P–P cleavage in case of the π-donating amino substituent. As already discussed in the context of phosphadiazonium ions (bonding motif F, Scheme 20) an efficient π-stabilization diminishes the necessity for stabilization by additional substituents/donors and may thus lead to a lower coordination number at the P atom. It is therefore not surprising that, to the best of our knowledge, no examples of isolated σ2 phosphenium ions without a π-donating substituent have been reported.
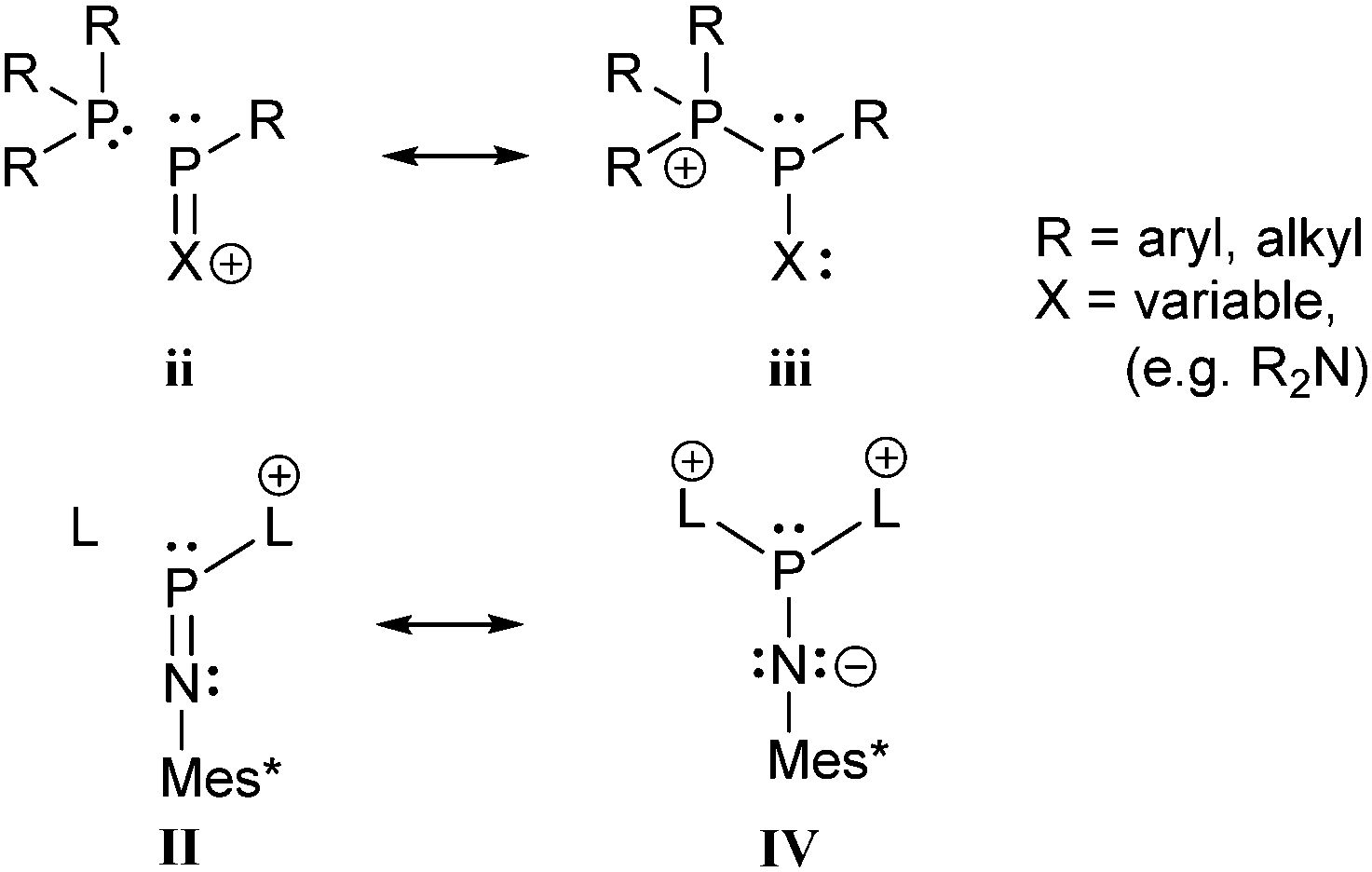 | ||
| Scheme 26 Resonance structures describing a phosphane stabilized phosphenium ion (ii) and a phosphanylphosphonium ion (iii). Resonance structures II and IV from Scheme 20 are illustrated for reasons of comparison. | ||
Basically independent of the nature of the P–P bond in a wide range of phosphanylphosphonium ions, the σ4 P atom represents a good nucleofuge and is therefore easily substituted by stronger Lewis bases. This inherent high reactivity makes cationic species containing polyphosphorus frameworks prominent synthetic targets.102 Catenated and cyclic polyphosphanylphosphonium salts are most intensely studied within this diverse group of cations.103 Typically phosphanylphosphonium cations can be accessed from either the reaction of a chlorophosphane with a halide abstracting agent (Abs) (Scheme 27, top), the reaction of a chlorophosphane, a phosphane and a halide abstracting agent (Scheme 27, middle) or by protonation or alkylation of a diphosphane (Scheme 27, bottom).
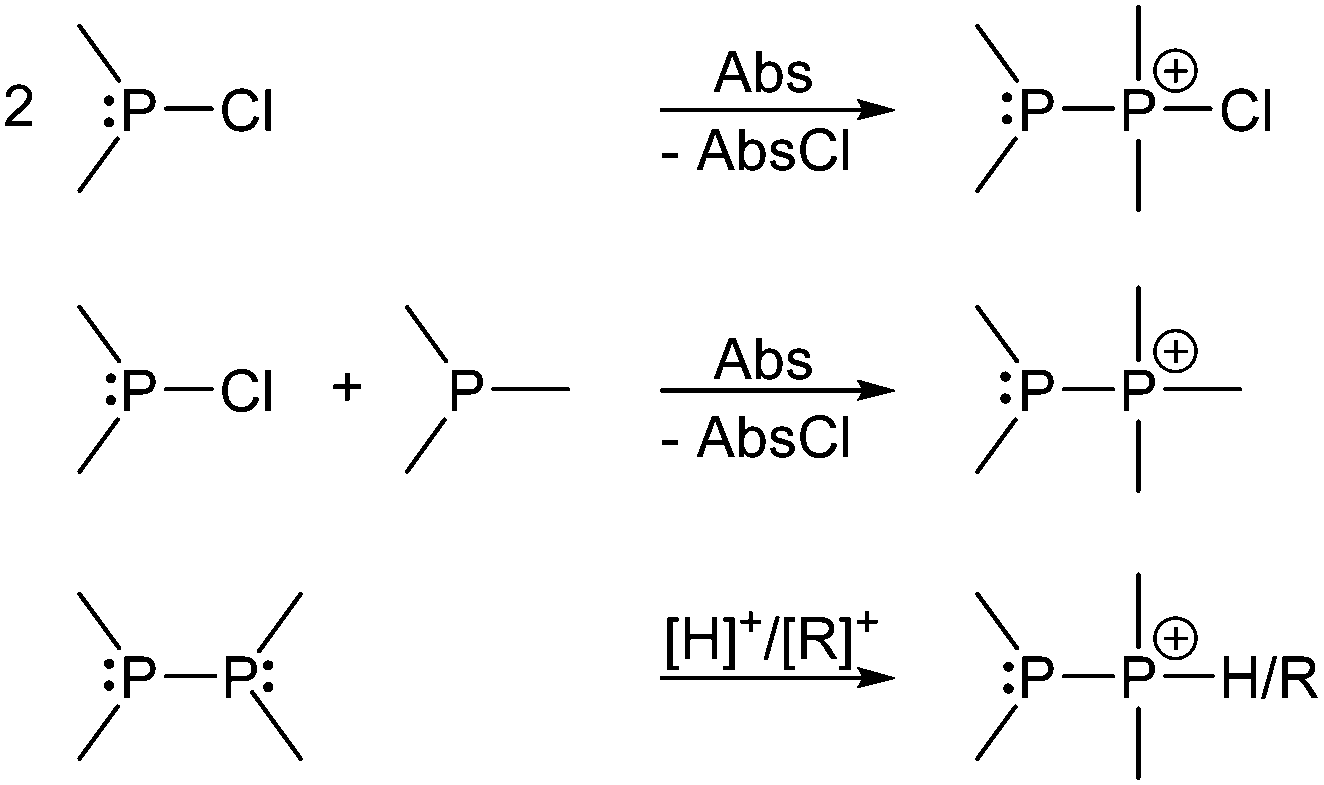 | ||
| Scheme 27 Synthesis of phosphanylphosphonium ion by P–P bond formation in the presence of a halide abstracting agent (top, middle) or protonation/alkylation of diphosphanes (bottom). | ||
A library of crystallographic data for several derivatives104 is available and shows consistent P–P distances that are close to 2.2 Å for most cases. The most important of the many reported catena- and cyclo-phosphorus cations comprising bonding motif J are depicted in Fig. 15.102c,105 They offer new synthetic approaches, not only for P–P bond formation, but also for other aspects in synthesis.106 Phosphanylphosphonium cations with two phosphorus atoms typically exhibit two well separated doublets in 31P NMR spectra with the high field shifted resonance corresponding to the tricoordinate phosphorus atom and the low field shifted to the tetracoordinate phosphorus atom. 1J(PP) coupling constants are normally observed in the range of 250–450 Hz. In the solid state phosphanylphosphonium cations contain a slightly distorted tetrahedral environment for the tetracoordinate and a pyramidal geometry for the tricoordinate phosphorus atom that is typical of a phosphane. As expected, the distortion can be more intense in more complex and cyclic systems as a result of ring strain or steric demand of the substituents.
The prototypical phosphanylphosphonium framework can be decorated with halo-substituents (X = Cl, Br, I) at either the phosphane or the phosphonium centres to yield an array of acyclic and cyclic halo-functionalized cations (Fig. 15). In this context, the preparation of diphosphanes or cyclo-polyphosphanes from the reduction of respective chlorophosphanes can be applied to access catenated cationic polyphopshorus frameworks.105d,e Cation 70+ readily reacts with Lewis bases such as 4-(dimethylamino)pyridine (dmap) and phosphanes (R3P), providing approaches to new open-chain and cyclic phosphorus frameworks (Scheme 28). Upon reaction with R3P (R = Me or nPr) or dmap the three-membered ring is opened to yield the adducts [R3P–PtBu–PtBu–P(Me)tBu][OTf] (R = Me (71a+), nPr (71b+)) and [(dmap)–PtBu–PtBu–P(Me)tBu][OTf] (72+). The complicated 31P{1H} NMR spectra of the three compounds were simulated, evidencing the presence of two diastereomeric forms of 71a+, and a single diastereomer of 71b+. This ring-opening reactivity of the cation 70+ parallels the reactivity of isolobal epoxides with nucleophiles under acidic conditions. The reaction with Me2PCl and Me3SiOTf resulted in the unexpected formation of dication 732+, which is postulated to result from two consecutive ring-opening and ring-closing steps. In contrast, employing MePCl2 in the reaction, cation 74+ is formed from a formal insertion of a “MeP” moiety into the cationic phosphorus framework of 70+.107
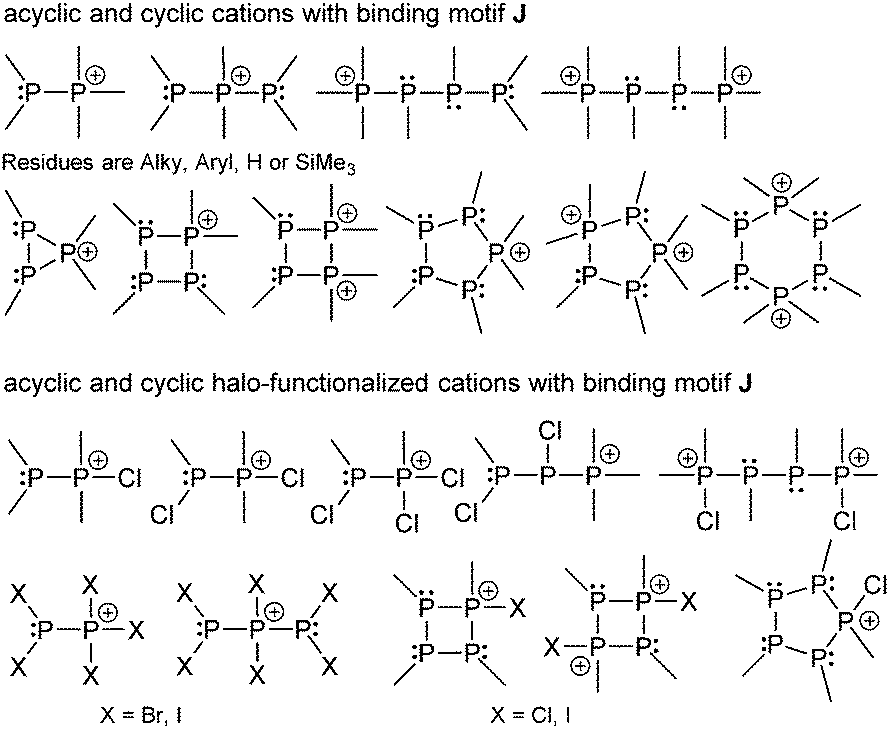 | ||
| Fig. 15 Selected examples of acyclic and cyclic phosphanylphosphonium cations comprising bonding motif J. | ||
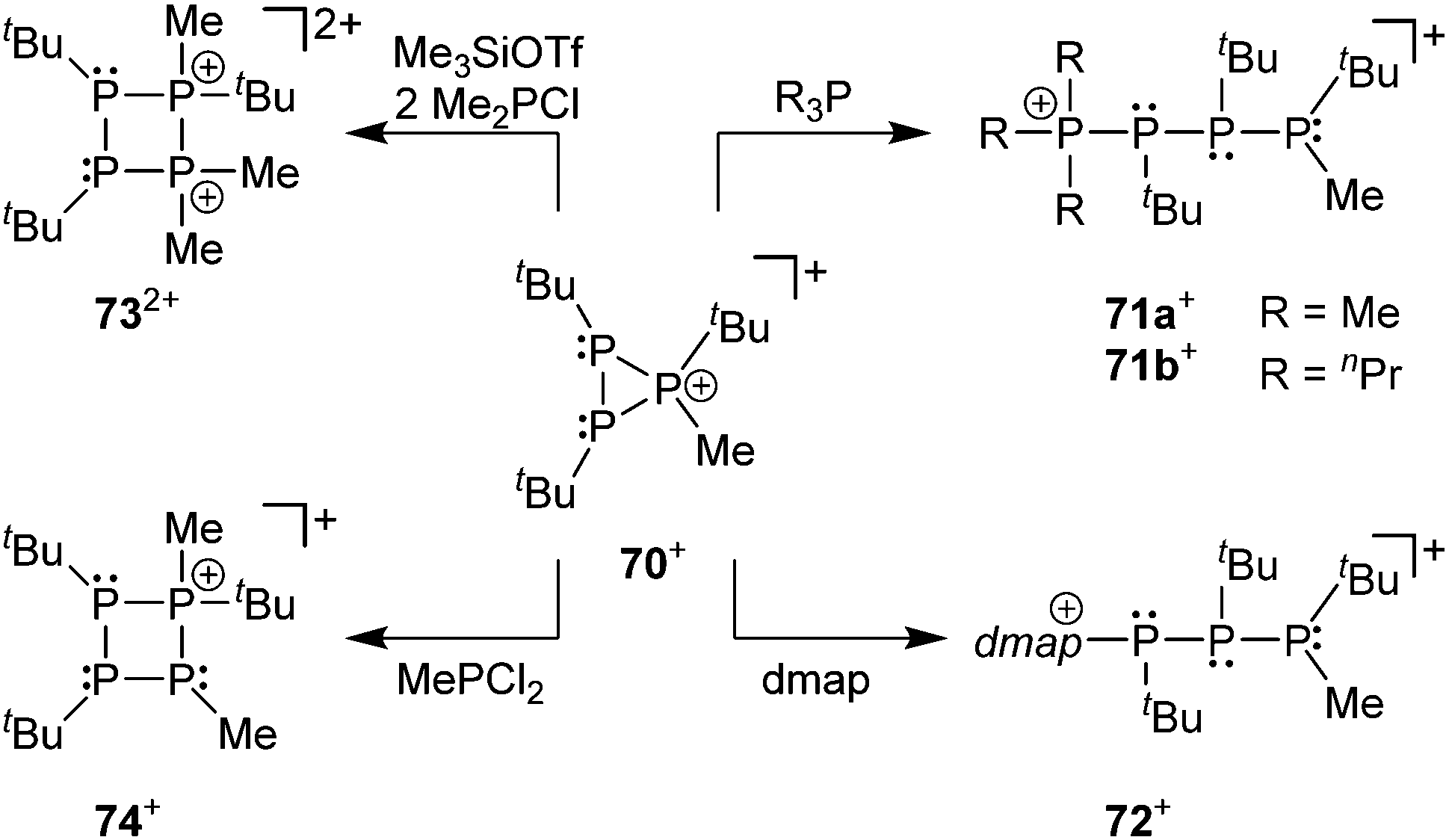 | ||
| Scheme 28 Diverse reactivity of cation 52+ in the reaction with a series of nucleophiles such as Me2PCl, MePCl2, R3P and dmap (4-(dimethylamino)pyridine). | ||
Related to this chemistry is the chlorination of cyclo-tetraphosphane 75 by PCl5 in the presence of the Lewis acid GaCl3 which provides a stepwise approach to salts of the first cyclo-phosphanylchlorophosphonium cations [Cy4P4Cl]+ and [Cy4P4Cl2]2+ (762+, (Scheme 29)).106,108 Reactions of the dication 762+ with dmpe effect a dissociation of the cyclic framework resulting in the formation of the cyclic cations 772+ and 782+ with an extended ring size. The new cations represent phosphane complexes that are formed from 762+ which dissociates formally via a retro [2+2] or [1+1+1′+1′] process releasing cationic [PCy]2+ and [P2Cy2]2+ fragments, demonstrating the partially coordinative nature of the σ3–σ4 P–P bonds in cyclo-phosphanylhalophosphonium cations.106
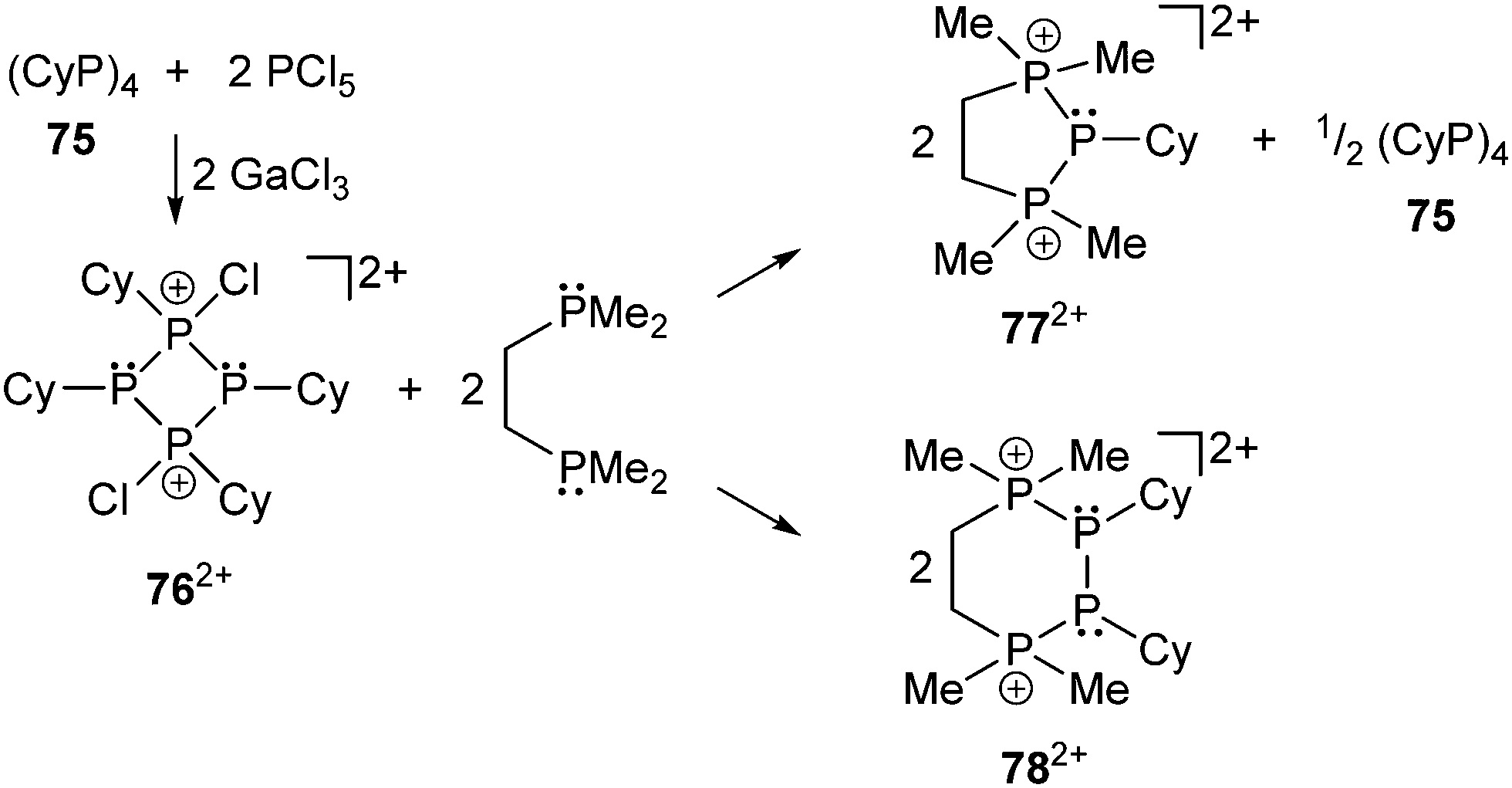 | ||
| Scheme 29 Synthesis of cyclo-phosphanylchlorophosphonium cation 762+ (left) and release of [PCy]2+ (top right) or [P2Cy2]2+ fragments (bottom right). | ||
The methylation of the diphosphirane 79 with MeOTf allows the formation of the P-methylated diphosphiranium salt 80[OTf] (Scheme 30, top), which has a related structure to cation 70+. This cation is only stable at low temperature in the presence of excess MeOTf.109 Following another approach, a diphosphiranium salt is obtained via the cationic cyclodimerization of a phosphaalkene 81 upon addition of HOTf (Scheme 30, bottom). In contrast to 80[OTf] the diphosphiranium triflate 82[OTf] showed sufficient stability in solution to be isolated and crystallographically characterized. The P–P bond length of 2.1637(5) Å corresponds to a slightly shortened single bond and the strain in the P2C ring is reflected by the small internal ring angles (σ3–P: 52.69(4)°, σ4–P: 55.82(5)°, C: 71.49(5)°).110
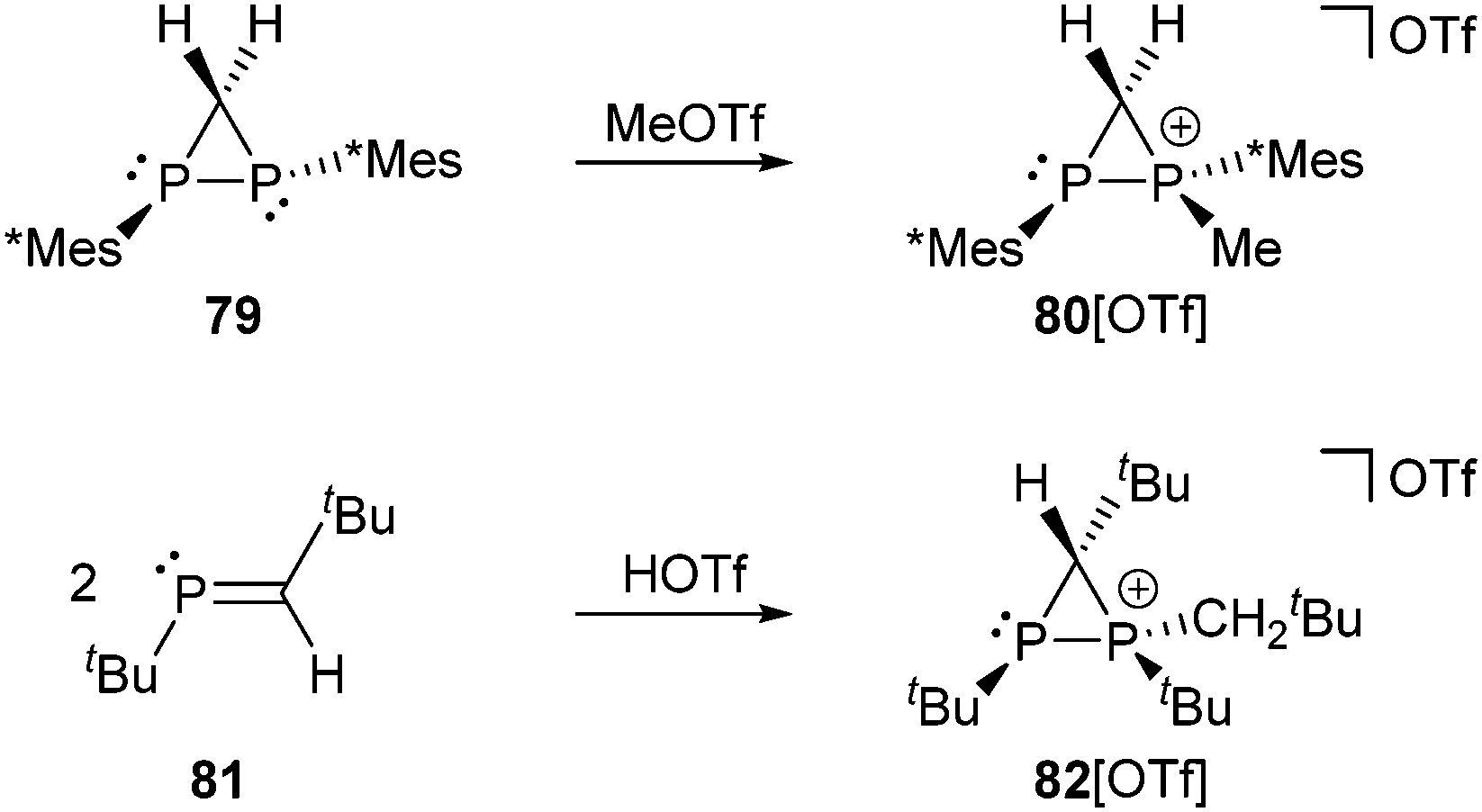 | ||
| Scheme 30 Synthesis of diphosphiranium cations 80+ by methylation of diphosphirane 79 (top) and 82+ by acid-induced dimerization of phosphaalkene 81 (bottom). | ||
The investigation of homoleptic polyphosphorus cations was limited to mass spectroscopy111 and quantum chemical calculation112 in the gas phase for decades. In general, Pn+ cations that feature an even number n of P atoms are paramagnetic. They are less stable than the respective diamagnetic Pn+ cations composed of an odd number of P atoms. A detailed discussion on polyphosphorus cations has been published.43a,113 The most stable cation according to quantum chemical calculations is the P9+-cage 83+, which is composed of two C2v-symmetric P5-cages fused by a common phosphonium moiety.112b Krossing et al. investigated the oxidation of P4 with [NO][A] (A = Al(OC(CF3)3)4−) which yields [P4NO]+-cage compound 84[A] (Scheme 31) via insertion of the nitrosonium cation into a P–P bond.114 A two-step mechanism was suggested on the basis of quantum chemical calculations indicating the HOMO of P4 and a π*-type LUMO at [NO]+ as the interacting frontier orbitals.114 The reaction of [P4NO]+-cage compound 84[A] with additional 1.5 eq. of P4 yields the P9+-cage compound 83[A] (Scheme 31).115 They suggest that cation 83+ forms via extrusion of 1/n (PNO)n and intermediary formation of a P3+-species. The 31P NMR spectrum of cation 83+ shows a characteristic A2A2′BC2C2′ spin system and confirms a D2d symmetric Zintl-type structure. Despite the electron precise Lewis formula of eight neutral, tricoordinate and one cationic, tetracoordinate P atom the charge is almost evenly distributed over all nine atoms according to quantum chemical calculations.115
 | ||
| Scheme 31 Synthesis of the first homoleptic polyphosphorus cation 83+ from P4 and 84+. | ||
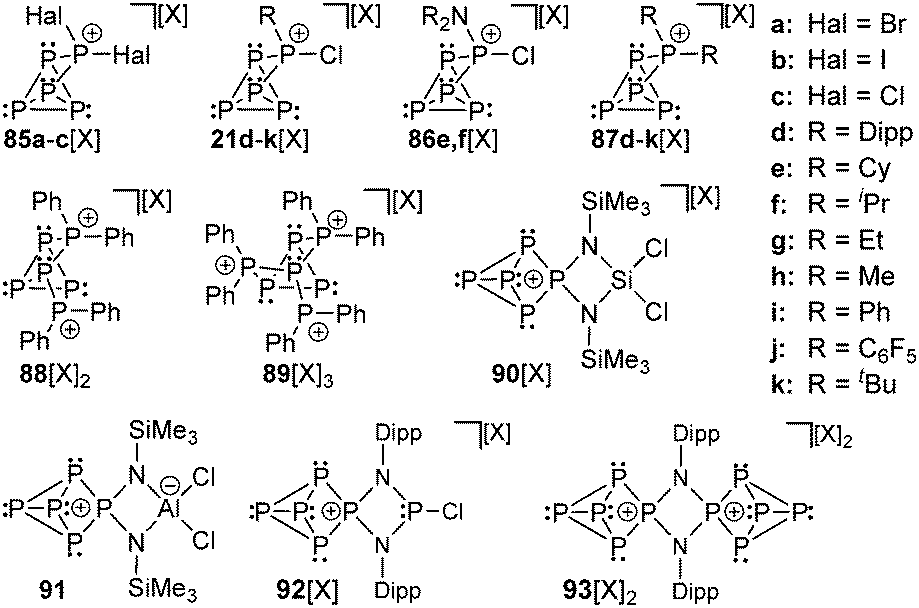
The same group also reported on the first cationic polyphosphorus cages featuring halogen substituents. They obtained the salts of cage cations 85a,b+ from the reaction of the silver salt [Ag(CH2Cl2)][X] (X = Al(OC(CF3)3)4) with PHal3 and P4 (Hal = Br, I; Fig. 16).116,104d Utilizing PCl3, the formation of the respective cation 85c+ was only observed in trace amounts.117 This reaction suffered from decomposition of the weakly coordinating anion Al(OC(CF3)3)4−. The structural motif of the [P5Hal2]+-cage is unprecedented and was not observed previously as part of the many known polyphosphides and organo-polyphosphanes. The 1:1 mixtures of RPCl2 and a Lewis acid ECl3 (E = Al, Ga) in fluorobenzene are potent sources of reactive equivalents of phosphenium ions [RPCl]+, which formally insert into P–P bonds of P4.118 Dissolution of P4 in these mixtures yields white to yellowish precipitates of the corresponding [RP5Cl]+-cage salts for a large range of distinct alkyl- and aryl-substituents R (21d–k[GaCl4]; Fig. 16; [X] = [GaCl4]). This approach can also be extended to R2N-subsituted derivatives 86e,f[GaCl4].119 The cationic [R2P5]+-cage compounds 87d–k[GaCl4] are synthesized via the stoichiometric reaction of R2PCl, GaCl3, and P4. The reaction conditions depend on the substituent R. Alkyl-substituted derivatives (87d–h[GaCl4]) are best synthesized using a solvent free melt reaction, whereas aryl-substituted derivatives (87i–k[GaCl4]) are formed in C6H5F solution (Fig. 16; [X] = [GaCl4]). All compounds can be prepared on a multi-gram scale in good to excellent yields and are fully characterized with an emphasis on 31P NMR spectroscopy in solution and single crystal structure determination. By addition of more eq. of GaCl3, the melt approach can be extended to dication [Ph4P6]2+ (88[X]2; [X] = [GaCl4]) and trication [Ph6P7]3+ (89[X]3; [X] = [Ga2Cl7]) via the consecutive insertion of up to three [Ph2P]+ fragments into the P–P bonds of the P4 tetrahedron.120 This approach has been extended to highly functionalized cation 90+ ([X] = [AlCl4]) and the zwitterionic P5-cage 91, which are formed by the insertion of the corresponding phosphenium cations that are derived from four-membered phosphorus–nitrogen–metal heterocycles.121 Similarly, the reaction of P4 with the cyclo-diphosphadiazane [DippNPCl]2 (Dipp = 2,6-disopropylphenyl) and GaCl3 in fluorobenzene yielded compound 92[GaCl4]. The addition of an excess of GaCl3 and a second eq. of P4 to a solution of 92[GaCl4] afforded the dicationic species 932+ as [Ga2Cl7]− salt.122
 | ||
| Fig. 16 Selected cationic cage compounds comprising bonding motif J. | ||
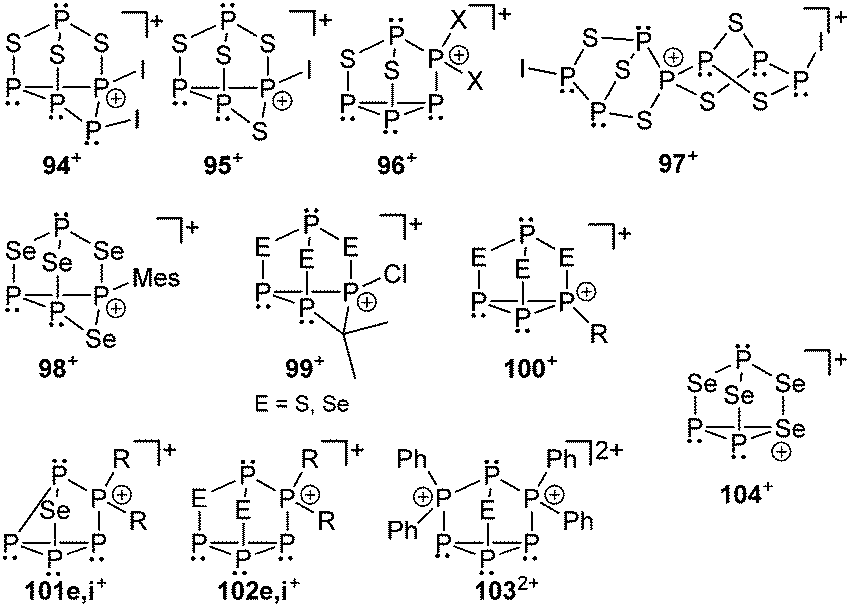
Also cationic polyphosphorus–chalcogen cages are rarely described in the literature. Until recently only two synthetic procedures were reported for their preparation. In the first approach P4S3 reacts with in situ generated [PI2]+-phosphenium ions yielding cation 94+ which disproportionates to cations 95+ and 96+ (Fig. 17).123 The second approach is based on halide abstraction from α-P4S3I2 using Krossing's silver salt Ag[Al(OC(CF3)3)4] giving the spiro-cyclic cage cation 97+.124 The formation of cation 98+ is observed in the protonation reaction of cyclic P–E-heterocycles (MesPSe)4 or reaction of P3Se4 with MesPCl2 in the presence of AlCl3 as Lewis acid.125 Reacting P4E3 with Me2CCl2 in the presence of AlCl3 the formation of cations 99+ (E = S, Se) is observed. The cations are only formed in small amounts but their connectivity was confirmed by extensive NMR investigation and X-ray analysis of suitable single crystals.126 Basal alkylation of P4E3 is observed in the reaction with secondary alkyhalides (sec-RX) to give cations of the type 100+ (E = S, Se).126
 | ||
| Fig. 17 Selected cationic polyphosphorus–chalcogen cages 94+–1032+ (E = S, Se) comprising bonding motif J; cation 104+ has been included since it represents the first binary SeP–cation and is related to the other cations. | ||
Another approach to generate cationic chalcogen–phosphorus cages (E = S, Se) is based on cationic polyphosphorus cages as starting materials. They constitute potentially versatile reagents due to the multitude of distinctly substituted derivates which are all conveniently obtained in one step procedures from white phosphorus.127 Chalcogenation reactions of [R2P5]+-cage compounds 87e,i[GaCl4] with elemental grey selenium yield the corresponding polyphosphorus-chalcogen cages 102[GaCl4] (E = Se, R = Cy, Ph; Fig. 17). Both cations are formed upon insertion of two selenium atoms into two P–P bonds adjacent to the phosphonium moieties in 87e,i+. Their structural motif resembles that of nortricyclane, with a basal P3-ring, the tetracoordinate P atom and the selenium atoms occupying the bridging positions, and one P atom defining the apex of the cage. This class of compound features interesting 31P and 77Se NMR characteristics. They reveal an AM2OX spin system for the isotopologues without a 77Se nucleus. These resonances are superimposed by the C1-symmetric isotopologues featuring one 77Se atom in one of the bridging positions. This gives rise to an AMNOXZ spin system which is strongly influenced by higher order effects. However, in the case of 102e+ (R = Cy), both spin systems were successfully simulated allowing for the exact determination of chemical shifts and coupling constants. A series of experiments employing variable temperatures, reaction times and stoichiometries gave meaningful insights into the mechanism of the chalcogenation. These experiments indicate that the insertion of Se atoms into P–P bonds of 87e,i+ proceeds in a stepwise manner via the intermediates 101e,i+.127 The targeted preparation of 103+ as [GaCl4]− salt was achieved by utilizing a 2:1 stoichiometry of 48+ and grey selenium (E = Se). Another synthetic approach for the preparation of 103+ (E = S, Se) is the targeted substitution of one [Ph2P]+-moiety in the tricationic cage 893+. This was achieved by reacting 893+ with grey selenium or sulfur under solvent-free conditions.127 Dication 1032+ was comprehensively characterized by X-ray crystallography as well as 31P and 77Se NMR spectroscopy (E = Se). The polyphosphorus cation 893+ and cationic polyphosphorus–chalcogen cages 1032+ and 102i+ are formally derived by stepwise isolobal exchange of [E] atoms by [Ph2P]+ units in the bridging positions of the nortricyclane type structure of P4E3 derivatives (E = S, Se).
Despite the fairly large number of binary group 15/16 element cations that have been reported, an example involving phosphorus in combination with a group 16 element has not been synthesized and characterized until very recently. Although cation [P3Se4]+ (104+) does not belong to bonding motif J it complements the series of polyphosphorus–selenium cages. Three distinct synthetic routes for salts of the nortricyclane type cation 104+ were independently discovered. Cation 104+ is obtained as metallate salt 104[MX4] (M = Al, Ga; X = Cl, Br), either via a melt approach from elemental red or white phosphorus, grey selenium and selenium tetrachloride in the Lewis-acidic ionic liquid BMImCl/AlCl3 (M = Al; X = Cl, Scheme 32, top) or an arylation reaction of P4Se3 with pentamethylbromobenzene (Me5C6Br) in the presence of AlCl3 in CH2X2 (M = Al; X = Cl, Br, Scheme 32, middle) or the addition of (Me3Si)2Se to a solution of PX3 and MX3 in CH2X2 at ambient temperature (M = Al, Ga; X = Cl, Br, Scheme 32, bottom).128 The materials have been structurally characterized by single-crystal X-ray diffraction, Raman, in solution and solid state NMR and quantum chemical calculations. Remarkably, the tricoordinate Se atom in the basal plane of the adamantane-like P3Se4+ cation presents a 1J(31P77Se) coupling constant near zero Hz, in agreement with results from quantum chemical calculations. The dynamic disordering phenomena suggested by the results from X-ray crystallography were further investigated by detailed solid state NMR spectroscopy. These studies indicate an interesting dynamic heterogeneity phenomenon, where one of the two cations in the asymmetric unit shows slow reorientation dynamics on the NMR timescale, while the other one can be considered rigid.128
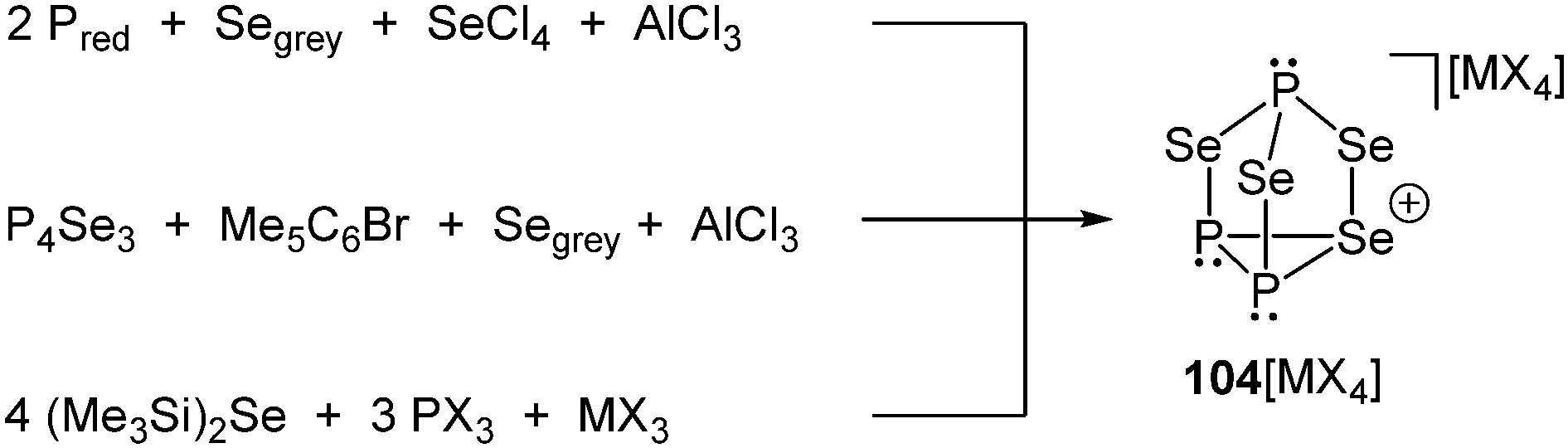 | ||
| Scheme 32 Three different approaches for the synthesis of the first binary phosphorus chalcogen cation P3Se4+ (104+) as metallate salt. | ||
The symmetrical butterfly-shaped P6-diaction 1052+ is obtained by substitution of the AsPh3 moieties of 1062+ by PPh3 (Scheme 33). The experimental and iteratively fitted 31P{1H} NMR spectrum reveals an A2MM′XX′ spin system (Fig. 18). The large 31P–31P coupling constants observed in 1052+ (2J(MM′) = 339 Hz, 3J(MX′) = −139 Hz and 4J(XX′) = 39 Hz) indicate a substantial degree of trough space coupling between the cis-orientated free-lone pairs located at each “butterfly-wing”.129
 | ||
| Scheme 33 AsPh3 can be substituted by PPh3 in the bicyclic compound 1062+ to give the analogous dicationic polyphosphorus framework 1052+. | ||
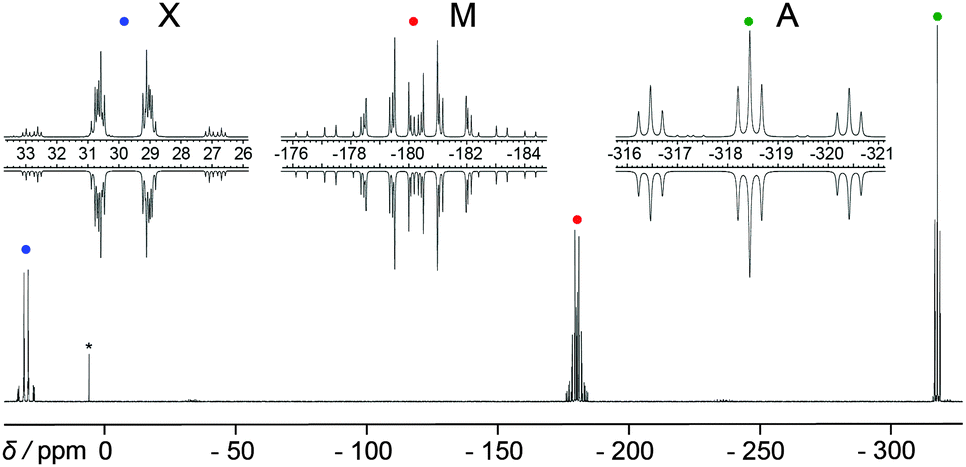 | ||
| Fig. 18 31P{1H} NMR spectrum of the butterfly-shaped diaction 1052+ (upwards) and simulation (downwards). This figure has been reproduced from Angewandte Chemie with permission from John Wiley and Sons.129 | ||
Bertrand et al. reported on the P5N cage compound 107 obtained from the insertion reaction of a room temperature stable phosphanylnitrene130108 into one edge of a P4 tetrahedron (Scheme 34). The zwitterionic cage compound 107 reveals an A2CMX spin system with 31P chemical shifts in the expected ranges for endo,endo-bicyclo[1.1.0]tetraphosphane derivatives but for one exception (δ(PA) = −255.2, δ(PC) = −154.4, δ(PM) = 57.6, δ(PX) = 68.2 ppm). The PM atom adjacent to the nitrogen atom is significantly shifted to lower field with respect to the related PC atom which is connected with the PX phosphonium atom. This peculiar bonding environment of a P atom incorporated into the rigid bicyclo[1.1.0]tetraphosphane framework is also reflected by the P–P bond lengths found in the crystal structure of 107. The distances between PA and PM (2.288(3) and 2.275(3) Å) are significantly longer than those between PA and PC (2.217(2) and 2.194(3) Å). This finding may be reasoned by the negative hyperconjugation of the nitrogen lone-pairs into the σ*(PX)–(N–PX 1.575(5) Å) as well as into the σ*(PM)-orbitals (N–PM 1.614(4) Å) therefore loosening the PA–PM bonds.
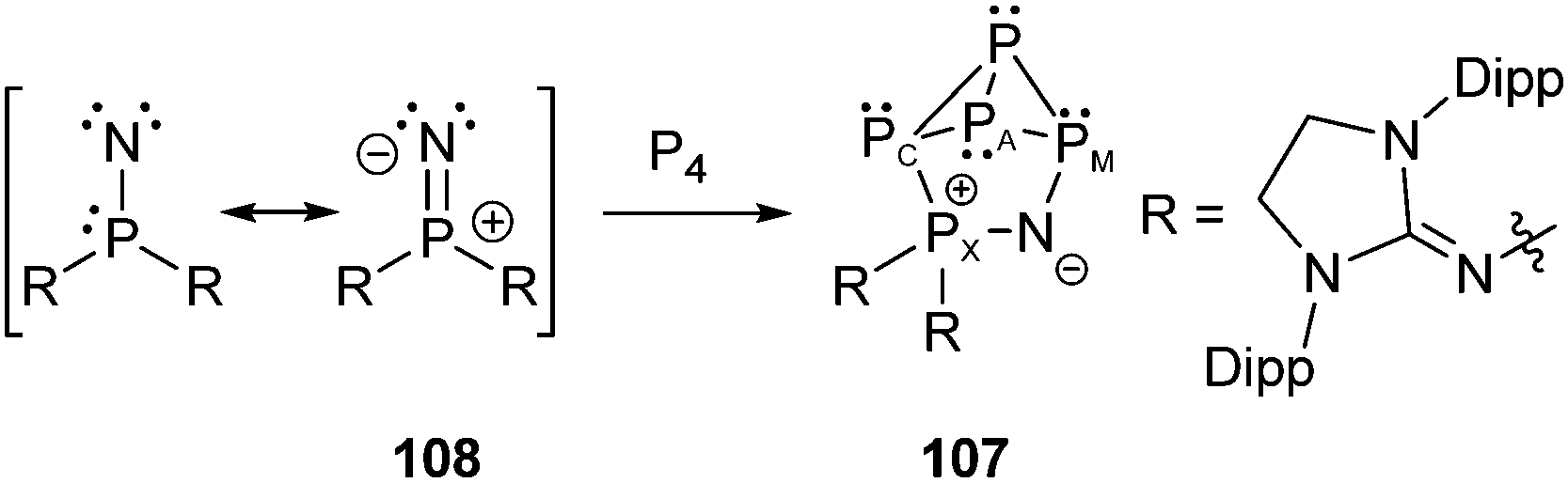 | ||
| Scheme 34 Insertion of a phosphanylnitrene 108 into one edge of the P4 tetrahedron giving 107. | ||
σ3–σ5
The class of σ3–σ5 diphosphorus compounds comprises basically only phosphanylphosphoranes (bonding motif K), as no example of the combination of a σ3λ5 phosphorus atom adjacent to a σ5 phosphorane has been reported.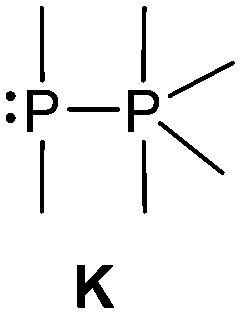
Bonding motif K
As this class of compounds was reviewed earlier,2 we refrain from a repetition and only want to discuss two particular examples instead.Compound 109 was obtained by reduction of the corresponding phosphane oxide 110 (bonding motif N) with chloroform (Scheme 35).131 The 31P NMR spectrum displays a complex A3B coupling pattern with the σ5 phosphorus resonance (A-part) high field shifted with respect to the σ3 phosphorus resonance (B-part) (81 MHz, CDCl3, δ(PA) = −28.2, δ(PB) = 40.9 ppm, 1J(PP) = 341.3 Hz). The crystal structure of 109 (Fig. 19) comprises a central σ3 phosphorus atom bearing three σ5 phosphoranyl moieties. The P–P bonds are in the expected range of single bonds (2.244 Å) and occupy the equatorial positions of the trigonal bipyramidal coordinate σ5 phosphorus atom. Since normal phosphane oxides (e.g. Ph3PO or nBu3PO) are not reduced to the corresponding phosphanes by chloroform, the reduction of 110 to 109 is quite remarkable though the mechanism is unknown. The authors ascribe the unusual reactivity to the three P–P bonded phosphoranyl substituents. Decreased π-backbonding of the oxygen lone pairs into P based σ*-orbitals would render the oxygen atom more nucleophilic and susceptible for the attack of a weak electrophile like chloroform.132
 | ||
| Scheme 35 Synthesis of 110 and reduction with CHCl3 to triphosphoranylphosphane 109. | ||
 | ||
| Fig. 19 Molecular structure of triphosphoranyl phosphane 109; hydrogen atoms are omitted for clarity. | ||
As the urea-bridged phosphoniophosphoranide 111 (bonding motif M) and the phosphanyl phosphorane 112 (bonding motif K) have been reviewed earlier,133 we only want to mention one interesting aspect which points out how closely related the two bonding motifs K (σ3–σ5) and M (σ4–σ4) are. When 111 is treated with sodium fluoride in acetonitrile solution the fluoride does not simply substitute the chloride moiety at the same P atom. Both coordination environments are altered as fluoride is binding to the amino-substituted phosphonium P atom (Scheme 36).133 Thus, the σ3–σ5 diphosphorus compound 112 is obtained with the fluoro-substituent and phosphanyl moiety located in axial positions. This transformation also involves an elongation of the P–P bond length from 2.195 Å in 111 to 2.267 Å in 112.134 While the initial apical P–Cl bond length (2.816 Å) is much longer compared to literature known examples (∼2.3 Å)135 and comparable to the [PCl4]− anion,136 the P–F distance (1.628 Å) is only slightly longer than those observed in PF5 (1.577(5) Å).137
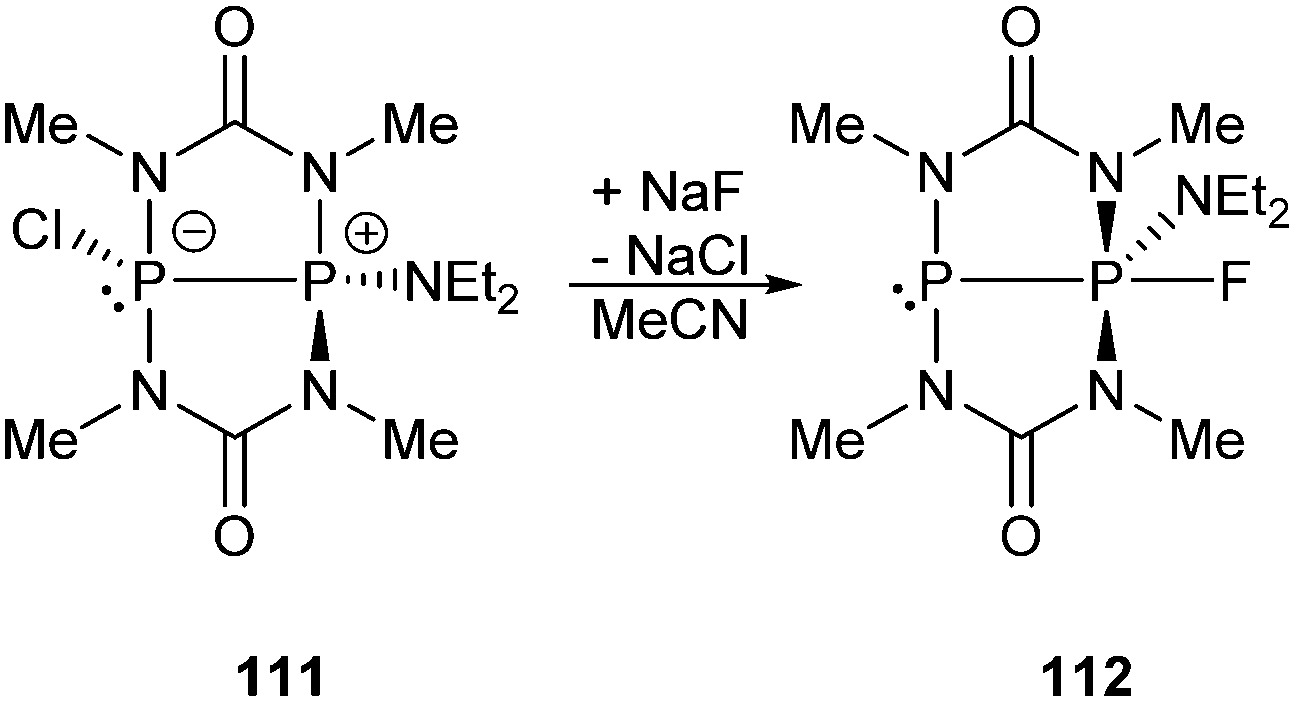 | ||
| Scheme 36 Transformation of 111 (bonding motif M) into 112 (bonding motif K) by halide exchange. | ||
σ4–σ4
The two bonding motifs L and M describe the connectivity of two tetracoordinate phosphorus atoms that can be linked either as two phosphonium units giving a dication (L) or as the formally neutral phosphoniophosphoranide (M).
Bonding motif L
The fundamentally important parent or benchmark P–P bonded diphosphonium unit is present in a few structurally characterized compounds that involve bulky amino substituents (1132+),138 unsymmetrically substituted alkyl-derivatives (1142+)139 or a polycyclic framework (1152+,1401162+ (vide infra),1411172+ (vide infra);142Fig. 20). Hexaalkyldiphosphonium triiodide salts were speculated as the products of reactions of red phosphorus with alkyl iodides or from trialkylphosphanes with iodine,143 and [Me3PPMe3][PF6]2 (962+, R = R′ = Me) has first been assigned on the basis of elemental analysis data and IR spectroscopy.144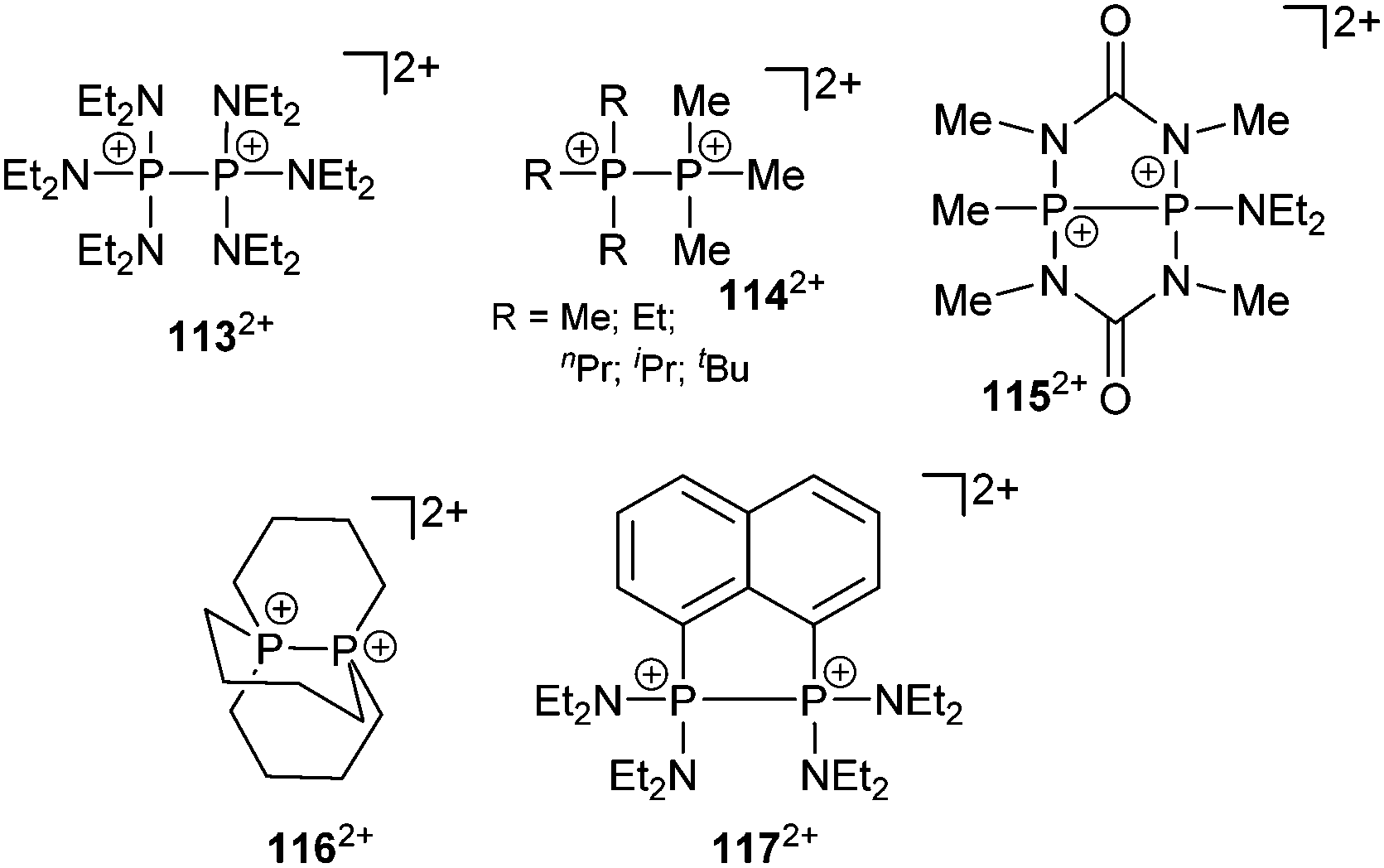 | ||
| Fig. 20 Selected examples of diphosphonium dications comprising bonding motif L. | ||
Polyphosphane adducts of electrophilic antimony,145 thallium,144 copper,144 or iron146 acceptors are known to spontaneously eliminate diphosphonium dications [R3PPR3]2+. Dialkylation of diphosphines, reductive elimation processes or methylation of P–P phosphanylphosphonium cations are versatile synthetic approaches to fundamentally important prototypical examples of diphosphonium dications (Fig. 20) that define the origin of a potentially extensive and diverse catena-phosphorus chemistry, paralleling catena-carbon chemistry. The quantitative nature of the reactions bodes well for the development of polyphosphonium chemistry. Very recently diphosphonium dications were shown to activate B–H, Si–H, C–H and H–H bonds while being part of a frustrated Lewis pair.147
Bonding motif M
Phosphoniophosphoranides may in principal be obtained as Lewis adducts of two phosphanes with one acting as a donor and the other one as an acceptor. The resulting mixed-coordinate P–P compounds display a phosphonium P atom with a tetrahedral and phosphoranide P atom with a disphenoidal geometry.Reports of such intermolecular Lewis adducts between simple phosphanes go back to the 1950's.148 The lack of structural proof lasted for almost a half century due to the low stability of this bonding motif in solution. Müller et al. obtained the phosphoniophosphoranide 118 as crystalline solid by slow diffusion of PMe3 into a solution of PBr3 in toluene (Fig. 21).149 Compound 118 shows a great susceptibility towards decomposition reactions. Keeping the solid below −40 °C without solvent allows storage over several days. In contrast, the decomposition of 118 in solution is markedly accelerated making an immediate NMR measurement at low temperatures necessary. In comparison to the resonances of free PMe3 (δ = −61.9 ppm in C6D6) and PBr3 (228.5 ppm in CD2Cl2) the resonance of the donating PMe3 unit (Pdon) is shifted to lower field, whereas that of the accepting PBr3 unit (Pacc) is shifted to significantly higher field (δ(Pdon) = 29.5 ppm, δ(Pacc) = 78.5 ppm, 1J(PP) = 450.3 Hz). The crystal structure of 118 (Fig. 22) shows the typical disphenoidal geometry of the phosphoranide P atom with the phosphonio and one bromo substituent occupying the two equatorial positions. While these bond lengths (P–P 2.264(2) Å, P–Breq 2.250(2) Å) correspond to single bonds the two axially bonded bromo substituents display elongated P–Br distances (P–Brax: 2.424(2) and 2.677(2) Å). The most distant bromine atom forms a bridge (3.327(2) Å; dashed bonds) about an inversion centre towards another PBr3 unit resulting in two edge-sharing distorted square pyramids. Although the distances between two monomeric units of 118 are closer to the sum of the van der Waals radii (3.74 Å)7 than to a classical single bond, the formation of dimeric structures in the solid state was also observed for other derivatives.133,134
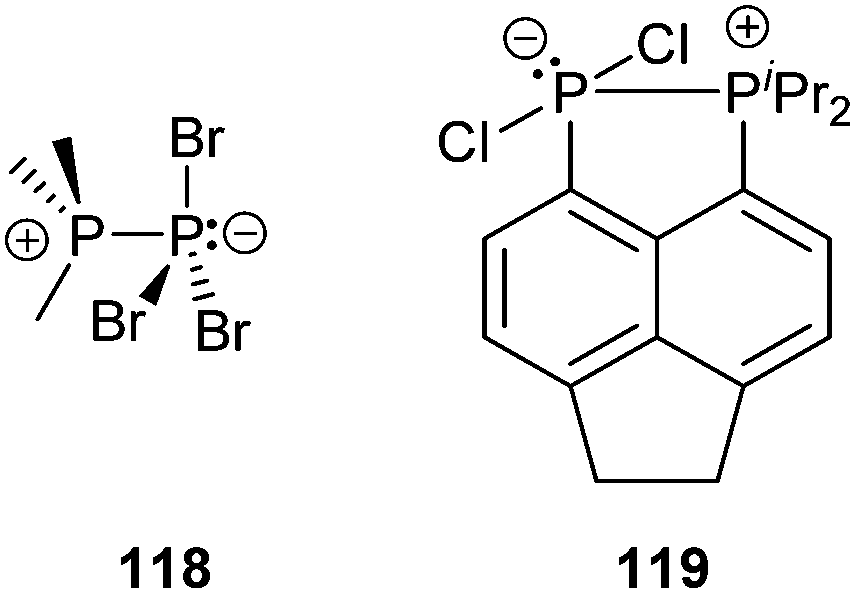 | ||
| Fig. 21 Selected examples of compounds comprising bonding motif M. | ||
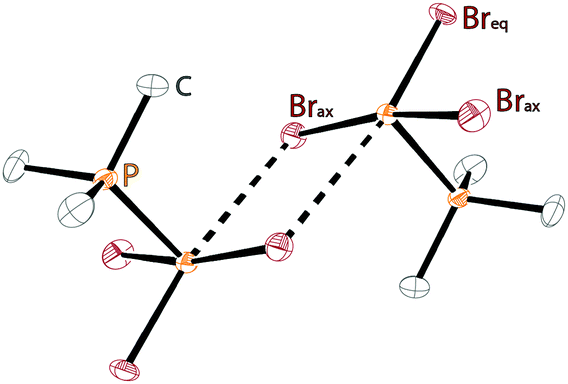 | ||
| Fig. 22 Molecular structure of 118 displaying two phosphoniophosphoranide units forming a dimeric bridge; hydrogen atoms are omitted for clarity. | ||
A more elaborate example of a phosphoniophosphoranide (119) features an electron accepting PCl2- and an electron donating iPr2P-moiety connected by the rigid acenaphthene backbone (Fig. 21). Compound 119 represents the first phosphoniophosphoranide which is stable at room temperature although it tends to decompose under reductive coupling in the presence of nucleophiles. The molecular structure reveals a covalent P–P single bond (2.257(2) Å) with the phosphonio substituent in the equatorial and the chlorine atoms in the axial positions. The resonances of solution 31P NMR spectra (PCl2 68.8 and PiPr2 40.4 ppm) and the observation of a 1J(PP) coupling constant of 363 Hz are consistent with the retention of the P–P bond upon redissolution.135d
The reaction of the anthracene derived triphosphane10120 with P2I4 in the presence of LiCl and chlorinated solvents leads to the formation of the diphosphoniophosphoranide 121 as a result of a formal two electron oxidation and substitution process (Scheme 37). The rather long P–P bond lengths are unequal in the solid state (2.382(5) and 2.528(5) Å). In contrast, the solution 31P NMR spectrum shows only two sharp signals at 193 K. A dynamic NMR investigation did not indicate a bond switching process (bell–clapper rearrangement). The chalcogenation of 120 after methanolysis leads to the formation of compound 122 (E = S, Se) which represents an unprecedented neutral example of bonding motif L (vide supra).
 | ||
| Scheme 37 Reactions of 120 giving 122 (bonding motif L) and diphosphoniophosphoranide 121 (bonding motif M). | ||
σ4–σ5
As already mentioned in the discussion of the phosphoniophosphoranide 118, the solid state structures of some phosphoniophosphoranides (bonding motif M) comprise bridged dimeric structures. From a geometrical point of view they may be considered as σ4–σ5 motif although interatomic distances suggest not more than attractive interactions.133,134,149 Therefore we do not consider them as a separate bonding motif.The connection of a phosphonio and a phosphorane moiety leads to bonding motif N.
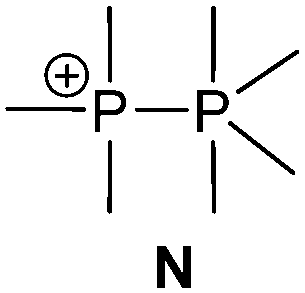
Bonding motif N
Alder et al. showed that the addition of nucleophiles to tricyclic propellane-type diphosphonium dications (bonding motif L) of different ring sizes gives phosphoniophosphoranes. Although some aspects of this work have already been summarized elsewhere,150 the different extent of transannular P–P bonding with respect to the applied nucleophiles and ring sizes is worthy to be mentioned here.151 Crystal structure analyses are available for the benzyl-, hydroxo- and hydrido-substituted derivatives indicating a different degree of pyramidalization of the σ5 phosphorane moiety. Different P–P-distances in the range from 2.514 to 2.813 Å are observed.There seems to be no obvious correlation between P–P distances and coupling constants in such systems. Deprotonation of the doubly protonated bis(phosphonium)dication 1232+ initially gives the monoprotonated bicyclic cation out,out-124+ which undergoes in,out-inversion of the free lone pair (Scheme 38).152 While out,out-124+ shows broad signals in the 31P NMR spectra, in,out-124+ shows two doublets with a 1J(PP) coupling constant of 253 Hz. The energy barrier of inversion in 124+ was determined to be only 70 kJ mol−1, an extraordinary low value for trialkylphosphanes. The phosphoniophosphorane was calculated to be 24 kJ mol−1 more stable.153
 | ||
| Scheme 38 Deprotonation of 1232+ leading to out,out-124+ followed by inversion of P(III) giving in,out-124+ which exhibits transannular P–P bonding. | ||
Schmutzler et al. reinvestigated the single protonation (Scheme 39, left) and methylation (Scheme 39, right) of 1,8-bis(diphenylphosphanyl)naphthalene 125 and reported the crystal structure of the cations 126+ and 127+ as triflate salts.8a Unexpectedly, the reaction with the electrophiles showed a pronounced increase in the P–P bond distances (126+: 3.211 Å; 127+: 3.265 Å) in contrast to the expected attractive interaction as observed for the Alder systems (vide supra). Very recently, Stephan et al. synthesized the phosphoniofluorophosphorane as perfluorinated borate salt 128[B(C6F5)4] by fluorination of 125 and subsequent fluoride ion abstraction from the difluorophosphorane 129 (Scheme 39, bottom).154 Both, the fluorine and the phosphonio moiety occupy apical positions with elongated P–P (2.530(1) Å) and P–F distance (1.637(2) Å) which can be explained by a 3c4e-bonding interaction.
 | ||
| Scheme 39 Derivatization of 125 to compounds with (128+) and without intramolecular P–P bonding (126+, 127+). | ||
Kilian et al. reported on the isomeric diphosphoniumdications meso-1302+ and rac-1302+ that are formed by two consecutive methylation steps of the diphosphaacenaphthene 131via a phosphanylphosphonium intermediate 132+ (Scheme 40, top). The formation of the respective diastereomer strongly depends on the methylation agent used. An interesting interconversion process of both stereoisomers is facilitated by the presence of fluoride ions which was supported by quantum chemical calculations and 31P-EXSY NMR investigations indicating that the inversion occurs via phosphoniophosphorane intermediates 133a,b+.155
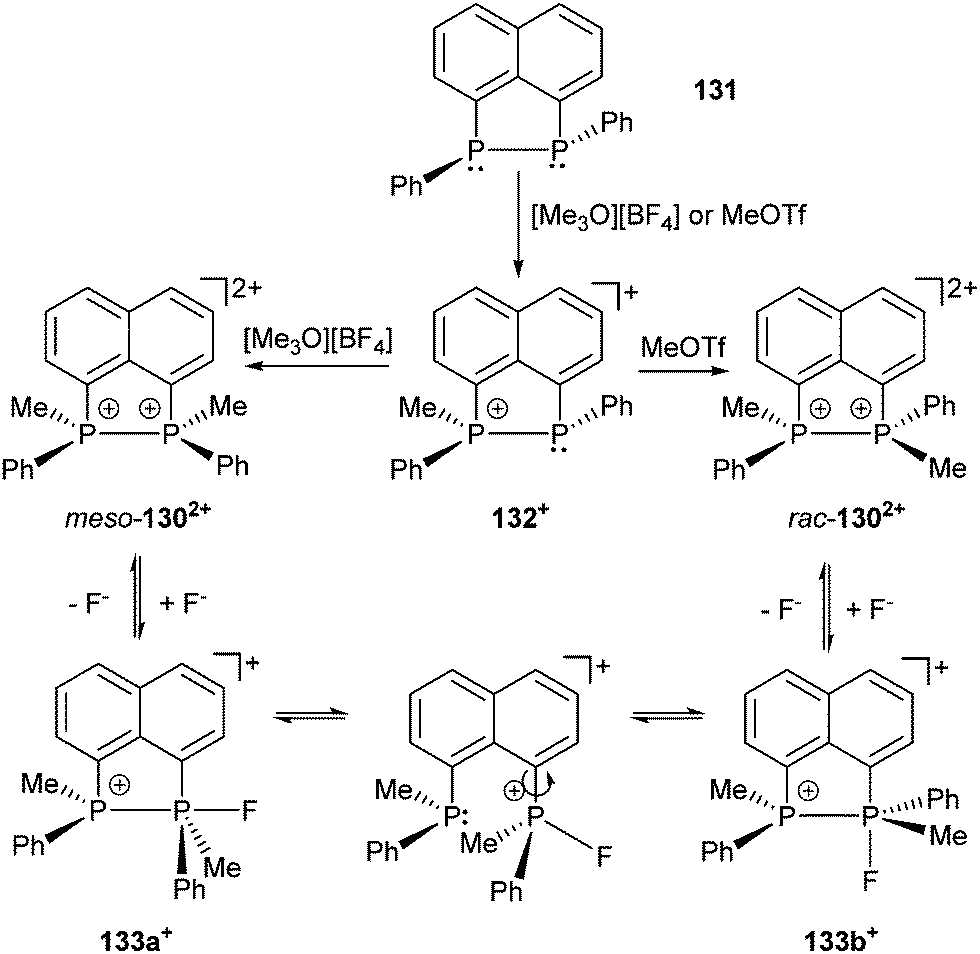 | ||
| Scheme 40 Single and double methylation of 1,8-naphthalene bridged diphosphane 131 to meso- or rac-1302+ (top) and interconversion in the presence of fluoride ions (bottom). | ||
These contrary results finally lead to the question which electronic conditions must be fulfilled for σ4–σ5 phosphoniophosphoranes to be stable towards dissociation to phosphanes and phosphonium cations and whether they can exist unless restrained by a bridging ligand forcing them to close contact. Besides charged phosphoniophosphoranes, phosphane oxides131,156 (see also 110) and sulfides133,157 adjacent to the σ5 phosphorane unit exhibit similar bonding motifs. Although they have been discussed earlier,2 we like to point out that they are sometimes surprisingly stable and some of them have been also structurally characterized. They feature the tetracoordinate phosphorus atom in an equatorial position. This is in sharp contrast to the aforementioned phosphoniophosphoranes, as the few which could be isolated so far, feature the tetracoordinate phosphorus in an apical position.158 These inherent differences might be explained by the fact that the Lewis adduct formation occurs between a cationic phosphonium ion and in one case a neutral phosphane and in the other case an anionic σ3–λ3 phosphane (phosphinite, R2PO− or phosphinothioite, R2PS−). In addition to an expectable coulombic attraction, the significantly enhanced donor strength of the latter ones should lead to a considerably stronger interaction between both phosphorus atoms in a σ4–σ5 environment.
σ4–σ6
The only phosphorus moiety bonded to a hexacoordinate phosphate forming stable products that have been observed so far is the phosphonium moiety. Phosphoniophosphates are described by bonding motif O.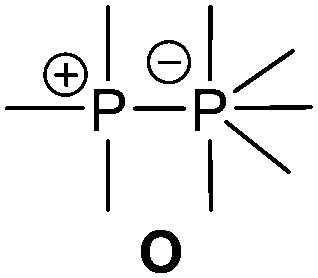
Bonding motif O
Only few reports on phosphoniophosphates have appeared in the literature. At first, adducts of PF5 and alkylphosphanes were studied by NMR spectroscopy.159The first isolated and structurally characterized σ4–σ6 P–P compound 134 from an electron deficient trifluoro-λ5-dioxophospholane was published by Röschenthaler et al.160 The molecular structure displays a tetrahedral phosphonio moiety bonded to an octahedral σ6-phosphate. The P–P bond length (2.234(5) Å) lies well in the expected range for a single bond. Other examples comprising bonding motif O (Fig. 23) display comparable bond lengths with those of 135161 and 136133 being in the upper and lower range of typical P–P single bonds, respectively. Although nearly all structurally characterized examples contain bridging ligands, prearranging a close contact of the phosphorus atoms, we would like to point out that in diphosphete 137,162 a relaxed σ3–σ5 phosphanylphosphorane without P–P-bond and a C–C–P angle of 120° at the sp2-hybridized carbon atoms would imply a much higher distance between the two P atoms. Therefore, the distance of 2.2962(6) Å clearly indicates a strong attractive interaction between both phosphorus atoms. In the oxidation product 136 of the phosphoniophosphoranide 111 (bonding motif M) by o-chloranil, the hexacoordinate phosphorus comprises a regular octahedral environment and a bond length of 2.165 Å to the phosphonium moiety. The tetracoordinate phosphorus atom is in a heavily distorted tetrahedral environment due to the urea ligand forcing the two P–P–N angles to be rather small at 97°.133 The structures of the cation 138+ (ref. 163) and the unsymmetrically chlorinated diphosphaacenaphthene 135161 exhibit P–P distances of 2.202(1) and 2.338(2) Å, respectively. The latter was suggested to undergo a tautomerization via a not isolable diphosphorane intermediate (σ5–σ5) similar to the equilibrium between molecular PCl5 and its ionic form [PCl4][PCl6].164 The optimized geometry of the calculated diphosphorane intermediate shows an axial-apical connection of the phosphorus moieties.
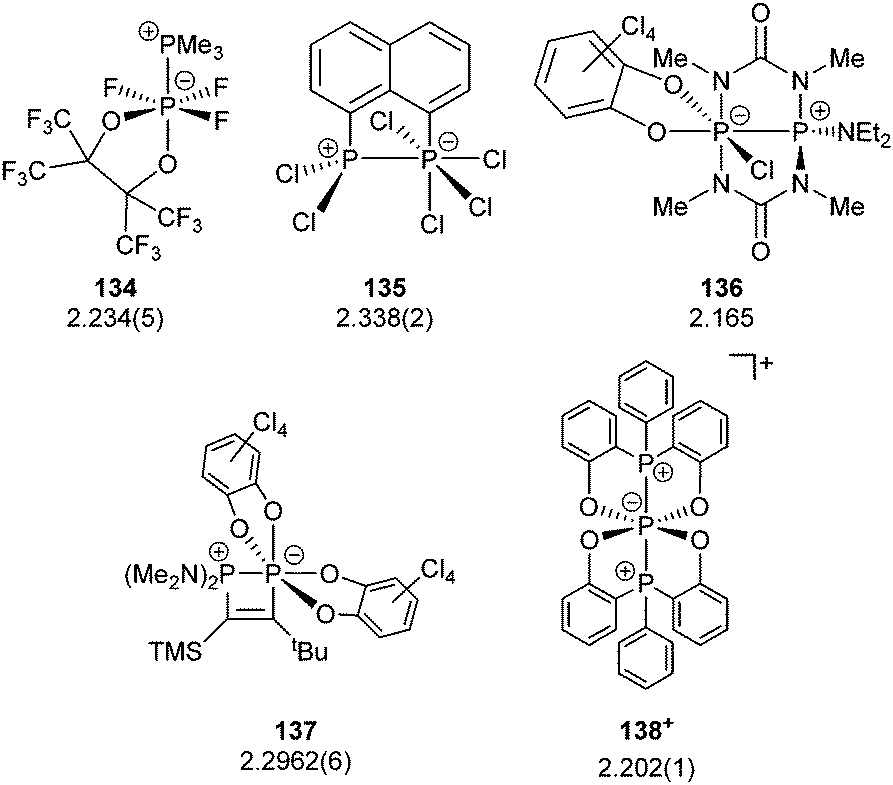 | ||
| Fig. 23 Selected derivatives comprising bonding motif O with respective P–P bond lengths in Å. | ||
σ5–σ5
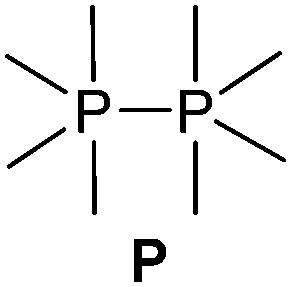
Bonding motif P
Bonding motif P is very rare and the very few known examples are already included in previous reviews.2 One of the two structurally characterized examples shows a Peq–Peq (2.264(1) Å)165 and the other one a Pax–Pax (2.255(1) Å)166 connection of both phosphorus moieties. However, compound 139 is of particular interest (Fig. 24), as both phosphorus atoms are in a different pentacoordinate environment which allows for the observation of a large 1J(PP) coupling constant of 749.6 Hz. Although no crystal structure is available, it was proposed that the P–P bond is in equatorial position for both pentacoordinate moieties which can also explain the large 1J(PP) value observed.167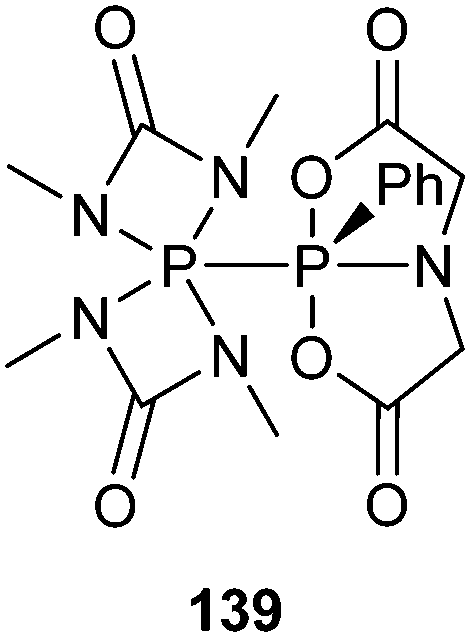 | ||
| Fig. 24 Compound 139 comprising the very rare example of bonding motif P. | ||
Abbreviations
| 18-crown-6 | 1,4,7,10,13,16-Hexaoxacyclooctadecane |
| 2,2,2-crypt | 4,7,13,16,21,24-Hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane |
| Ad | Adamantyl |
| Abs | Halide abstracting agent |
| Bu | Butyl |
| Cp | Cyclopentadienide |
| Cy | Cyclohexyl |
| DABCO | 1,4-Diaza[2.2.2]bicyclooctane |
| Dipp | 2,6-Diisopropylphenyl |
| dmpe | 1,2-Bis(dimethylphosphanyl)ethane |
| dppe | 1,2-Bis(diphenylphosphanyl)ethane |
| dppp | 1,3-Bis(diphenylphosphanyl)propane |
| Et | Ethyl |
| eq. | Equivalent(s) |
| i | Iso |
| Me | Methyl |
| Mes | 2,4,6-Trimethylphenyl |
| Mes* | 2,4,6-Tri(tert-butyl)phenyl |
| n | Normal/unbranched |
| NHC | N-heterocyclic carbene |
| OTf | Triflate/trifluoromethanesulfonate |
| Pr | Propyl |
| Ph | Phenyl |
| pyr | 3,5-Dimethylpyrazolyl |
| t | tert |
| triaz | 1,3,5-Trimethylhexahydro-1,3,5-triazine |
| trip | 2,4,6-Triisopropylphenyl |
| vide infra | See below |
| vide supra | See above |
Acknowledgements
This work was supported by the Fonds der Chemischen Industrie (FCI, scholarships for F. H. and M. D.) and the ERC (SynPhos 307616).References
- A. H. Cowley, Chem. Rev., 1965, 65, 617–634 CrossRef CAS.
- (a) L. Lamandé, K. Dillon and R. Wolf, Phosphorus, Sulfur Silicon Relat. Elem., 1995, 103, 1–24 CrossRef; (b) R. R. Holmes, Phosphorus, Sulfur Silicon Relat. Elem., 1996, 109, 1–42 CrossRef.
- IUPAC, Compendium of Chemical Terminology, 2nd edn. (the “Gold Book”), Compiled by A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford, 1997, XML on-line corrected version: http://goldbook.iupac.org (2006) created by M. Nic, J. Jirat, B. Kosata; updates compiled by A. Jenkins, ISBN 0-9678550-9-8, DOI: http://10.1351/goldbook.
- W. H. Powell, Pure Appl. Chem., 1993, 65, 1357–1455 CrossRef CAS.
- J. J. Weigand and N. Burford, in Comprehensive Inorganic Chemistry II, ed. J. R. Poeppelmeier, Elsevier, Amsterdam, 2nd edn, 2013, pp. 119–149 Search PubMed.
- (a) L. R. Maxwell, S. B. Hendricks and V. M. Mosley, J. Chem. Phys., 1935, 3, 699–709 CrossRef CAS; (b) N. J. Brassington, H. G. M. Edwards and D. A. Long, J. Raman Spectrosc., 1981, 11, 346–348 CrossRef CAS; (c) A. Simon, H. Borrmann and H. Craubner, Phosphorus Sulfur Relat. Elem., 1987, 30, 507–510 CrossRef CAS; (d) M. Haser and O. Treutler, J. Chem. Phys., 1995, 102, 3703–3711 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- (a) A. Karaçar, V. Klaukien, M. Freytag, H. Thönnessen, J. Omelanczuk, P. G. Jones, R. Bartsch and R. Schmutzler, Z. Anorg. Allg. Chem., 2001, 627, 2589–2603 CrossRef; (b) K.-O. Feldmann, Dissertation, Westfälische Wilhelms-Universität Münster, 2012 Search PubMed.
- (a) B. D. Ellis and C. L. B. Macdonald, Coord. Chem. Rev., 2007, 251, 936–973 CrossRef CAS; (b) L. Weber, Eur. J. Inorg. Chem., 2000, 2425–2441 CrossRef CAS.
- P. Kilian and A. M. Z. Slawin, Dalton Trans., 2007, 3289–3296 RSC.
- L. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- M. Yoshifuji, I. Shima, N. Inamoto, K. Hirotsu and T. Higuchi, J. Am. Chem. Soc., 1981, 103, 4587–4589 CrossRef CAS.
- (a) P. Jutzi and U. Meyer, Phosphorus Sulfur Relat. Elem., 1988, 40, 275–277 CrossRef CAS; (b) M. Scheer, C. Kuntz, M. Stubenhofer, M. Linseis, R. F. Winter and M. Sierka, Angew. Chem., Int. Ed., 2009, 48, 2600–2604 CrossRef CAS PubMed; (c) M. Scheer, C. Kuntz, M. Stubenhofer, M. Linseis, R. F. Winter and M. Sierka, Angew. Chem., 2009, 121, 2638–2642 CrossRef.
- (a) V. Thelen, D. Schmidt, M. Nieger, E. Niecke and W. W. Schoeller, Angew. Chem., 1996, 108, 354–356 CrossRef; (b) V. Thelen, D. Schmidt, M. Nieger, E. Niecke and W. W. Schoeller, Angew. Chem., Int. Ed. Engl., 1996, 35, 313–315 CrossRef CAS.
- (a) N. Wiberg, A. Wörner, H.-W. Lerner, K. Karaghiosoff, D. Fenske, G. Baum, A. Dransfeld and P. von Ragué Schleyer, Eur. J. Inorg. Chem., 1998, 833–841 CrossRef CAS; (b) H.-W. Lerner, I. Sanger, F. Schodel, A. Lorbach, M. Bolte and M. Wagner, Dalton Trans., 2008, 787–792 RSC; (c) H.-W. Lerner, M. Bolte, K. Karaghiosoff and M. Wagner, Organometallics, 2004, 23, 6073–6076 CrossRef CAS.
- (a) V. Cappello, J. Baumgartner, A. Dransfeld, M. Flock and K. Hassler, Eur. J. Inorg. Chem., 2006, 2393–2405 CrossRef CAS; (b) S. Yao, Dissertation, Johannes Gutenberg-University of Mainz, 2005 Search PubMed.
- (a) M. H. Holthausen, S. K. Surmiak, P. Jerabek, G. Frenking and J. J. Weigand, Angew. Chem., Int. Ed., 2013, 52, 11078–11082 CrossRef CAS PubMed; (b) M. H. Holthausen, S. K. Surmiak, P. Jerabek, G. Frenking and J. J. Weigand, Angew. Chem., 2013, 125, 11284–11288 CrossRef.
- A. M. Tondreau, Z. Benko, J. R. Harmer and H. Grützmacher, Chem. Sci., 2014, 5, 1545–1554 RSC.
- M. H. Holthausen, Dissertation, Westfälische Wilhelms-Universität Münster, 2013 Search PubMed.
- M. Baudler, B. Makowka and K. Langerbeins, Z. Naturforsch., B: J. Chem. Sci., 1985, 40, 1274–1276 Search PubMed.
- (a) M. Baudler, J. Hahn, H. Dietsch and G. Fuerstenberg, Z. Naturforsch., B: J. Chem. Sci., 1976, 31, 1305–1310 Search PubMed; (b) J. Hahn, M. Baudler, C. Krueger and Y. H. Tsay, Z. Naturforsch., B: J. Chem. Sci., 1982, 37, 797–805 Search PubMed.
- M. Baudler and J. Hahn, Z. Naturforsch., B: J. Chem. Sci., 1990, 45, 1139–1142 CAS.
- (a) S. Scharfe, F. Kraus, S. Stegmaier, A. Schier and T. F. Fässler, Angew. Chem., Int. Ed., 2011, 50, 3630–3670 CrossRef CAS PubMed; (b) S. Scharfe, F. Kraus, S. Stegmaier, A. Schier and T. F. Fässler, Angew. Chem., 2011, 123, 3712–3754 CrossRef.
- (a) R. S. P. Turbervill and J. M. Goicoechea, Chem. Commun., 2012, 48, 6100–6102 RSC; (b) R. S. P. Turbervill, A. R. Jupp, P. S. B. McCullough, D. Ergöçmen and J. M. Goicoechea, Organometallics, 2013, 32, 2234–2244 CrossRef CAS.
- (a) P. Le Floch and F. Mathey, Coord. Chem. Rev., 1998, 179–180, 771–791 CrossRef; (b) F. Mathey, Angew. Chem., Int. Ed., 2003, 42, 1578–1604 CrossRef CAS PubMed; (c) F. Mathey, Angew. Chem., 2003, 115, 1616–1643 CrossRef; (d) P. Le Floch, Coord. Chem. Rev., 2006, 250, 627–681 CrossRef CAS; (e) J. F. Nixon, Chem. Soc. Rev., 1995, 24, 319–328 RSC; (f) K. B. Dillon, F. Mathey and J. F. Nixon, Phosphorus: The Carbon Copy: From Organophosphorus to Phospha-organic Chemistry, Wiley, Chichester, Toronto, 1st edn, 1998 Search PubMed.
- (a) A. Lorbach, A. Nadj, S. Tüllmann, F. Dornhaus, F. Schödel, I. Sänger, G. Margraf, J. W. Bats, M. Bolte, M. C. Holthausen, M. Wagner and H.-W. Lerner, Inorg. Chem., 2009, 48, 1005–1017 CrossRef CAS PubMed.
- (a) A. R. Fox, R. J. Wright, E. Rivard and P. P. Power, Angew. Chem., Int. Ed., 2005, 44, 7729–7733 CrossRef CAS PubMed; (b) A. R. Fox, R. J. Wright, E. Rivard and P. P. Power, Angew. Chem., 2005, 117, 7907–7911 CrossRef.
- V. D. Romanenko, V. L. Rudzevich, E. B. Rusanov, A. N. Chernega, A. Senio, J.-M. Sotiropoulos, G. Pfister-Guillouzo and M. Sanchez, J. Chem. Soc., Chem. Commun., 1995, 1383–1385 RSC.
- L. N. Markovskii, V. D. Romanenko, M. I. Povolotskii, A. V. Ruban and E. O. Klebanskii, Zh. Obshch. Khim., 1986, 56, 2157–2158 CAS.
- A. Beil, R. J. Gilliard Jr. and H. Grützmacher, Dalton Trans., 2016, 45, 2044–2052 RSC.
- K. Schwedtmann, M. H. Holthausen, C. H. Sala, F. Hennersdorf, R. Fröhlich and J. J. Weigand, Chem. Commun., 2016, 52, 1409–1412 RSC.
- (a) R. Appel and U. Kündgen, Angew. Chem., Int. Ed. Engl., 1982, 21, 219–220 CrossRef; (b) R. Appel and U. Kündgen, Angew. Chem., 1982, 94, 227 CrossRef CAS.
- O. Altmeyer, E. Niecke, M. Nieger, T. Busch, W. W. Schoeller and D. Stalke, Heteroat. Chem., 1990, 1, 191–194 CrossRef CAS.
- (a) J. Mahnke, A. Zanin, W.-W. du Mont, F. Ruthe and P. G. Jones, Z. Anorg. Allg. Chem., 1998, 624, 1447–1454 CrossRef CAS; (b) W.-W. du Mont, T. Gust, E. Seppälä, C. Wismach, P. G. Jones, L. Ernst, J. Grunenberg and H. C. Marsmann, Angew. Chem., Int. Ed., 2002, 41, 3829–3832 CrossRef CAS; (c) W.-W. du Mont, T. Gust, E. Seppälä, C. Wismach, P. G. Jones, L. Ernst, J. Grunenberg and H. C. Marsmann, Angew. Chem., 2002, 114, 3977–3979 CrossRef.
- (a) S. Loss, C. Widauer and H. Grützmacher, Angew. Chem., Int. Ed., 1999, 38, 3329–3331 CrossRef CAS; (b) S. Loss, C. Widauer and H. Grützmacher, Angew. Chem., 1999, 111, 3546–3548 CrossRef.
- T. Kato, E. Stoyanov, J. Geier, H. Grützmacher and C. A. Reed, J. Am. Chem. Soc., 2004, 126, 12451–12457 CrossRef CAS PubMed.
- (a) P. Becker, H. Brombach, G. David, M. Leuer, H.-J. Metternich and E. Niecke, Chem. Ber., 1992, 125, 771–782 CrossRef CAS; (b) E. Niecke, V. von der Gönna and M. Nieger, Chem. Ber., 1990, 123, 2329–2333 CrossRef CAS; (c) W. W. Schoeller and T. Busch, Chem. Ber., 1992, 125, 1319–1323 CrossRef CAS; (d) R. Streubel and E. Niecke, Chem. Ber., 1990, 123, 1245–1251 CrossRef CAS; (e) R. Streubel, M. Nieger and E. Niecke, Chem. Ber., 1993, 126, 645–648 CrossRef CAS.
- (a) M. Baudler, Angew. Chem., 1982, 94, 520–539 CrossRef CAS; (b) M. Baudler, Angew. Chem., Int. Ed. Engl., 1982, 21, 492–512 CrossRef; (c) M. Baudler, Angew. Chem., 1987, 99, 429–451 CrossRef CAS; (d) M. Baudler, Angew. Chem., Int. Ed. Engl., 1987, 26, 419–441 CrossRef; (e) M. Baudler and K. Glinka, Chem. Rev., 1993, 93, 1623–1667 CrossRef CAS; (f) M. Baudler and K. Glinka, Chem. Rev., 1994, 94, 1273–1297 CrossRef CAS.
- N. Korber and J. Aschenbrenner, J. Chem. SocDalton Trans., 2001, 1165–1166 RSC.
- M. Baudler and P. Winzek, Z. Anorg. Allg. Chem., 1999, 625, 417–422 CrossRef CAS.
- (a) J. C. Aschenbrenner and N. Korber, Z. Anorg. Allg. Chem., 2004, 630, 31–32 CrossRef CAS; (b) F.-R. Dai and L. Xu, Inorg. Chim. Acta, 2006, 359, 4265–4273 CrossRef CAS.
- N. Korber and H.-G. von Schnering, J. Chem. SocChem. Commun., 1995, 1713–1714 RSC.
- (a) M. Scheer, G. B. Balázs and A. Seitz, Chem. Rev., 2010, 110, 4236–4256 CrossRef CAS PubMed; (b) S. Gómez-Ruiz and E. Hey-Hawkins, Coord. Chem. Rev., 2011, 255, 1360–1386 CrossRef; (c) W. T. K. Chan, F. García, A. D. Hopkins, L. C. Martin, M. McPartlin and D. S. Wright, Angew. Chem., 2007, 119, 3144–3146 CrossRef; (d) W. T. K. Chan, F. García, A. D. Hopkins, L. C. Martin, M. McPartlin and D. S. Wright, Angew. Chem., Int. Ed., 2007, 46, 3084–3086 CrossRef CAS PubMed.
- (a) I. Kovacs, E. Matern and G. Fritz, Z. Anorg. Allg. Chem., 1996, 622, 935–941 CrossRef CAS; (b) I. Kovacs, H. Krautscheid, E. Matern, E. Sattler, G. Fritz, W. Hönle, H. Borrmann and H. G. Von Schnering, Z. Anorg. Allg. Chem., 1996, 622, 1564–1572 CrossRef CAS.
- (a) R. Wolf, A. Schisler, P. Lönnecke, C. Jones and E. Hey-Hawkins, Eur. J. Inorg. Chem., 2004, 3277–3286 CrossRef CAS; (b) R. Wolf and E. Hey-Hawkins, Z. Anorg. Allg. Chem., 2006, 632, 727–734 CrossRef CAS; (c) J. Geier, J. Harmer and H. Grützmacher, Angew. Chem., Int. Ed., 2004, 43, 4093–4097 CrossRef CAS PubMed; (d) J. Geier, J. Harmer and H. Grützmacher, Angew. Chem., 2004, 116, 4185–4189 CrossRef.
- R. Wolf, S. Gomez-Ruiz, J. Reinhold, W. Böhlmann and E. Hey-Hawkins, Inorg. Chem., 2006, 45, 9107–9113 CrossRef CAS PubMed.
- A. B. Burg and W. Mahler, J. Am. Chem. Soc., 1961, 83, 2388–2389 CrossRef CAS.
- S. Shah, G. P. A. Yap and J. D. Protasiewicz, J. Organomet. Chem., 2000, 608, 12–20 CrossRef CAS.
- M. Sanchez, R. Réau, C. J. Marsden, M. Regitz and G. Bertrand, Chem. – Eur. J., 1999, 5, 274–279 CrossRef CAS.
- A. Schmidpeter and G. Burget, Z. Naturforsch., B: J. Chem. Sci., 1985, 40, 1306–1313 Search PubMed.
- (a) B. A. Surgenor, M. Bühl, A. M. Z. Slawin, J. D. Woollins and P. Kilian, Angew. Chem., Int. Ed., 2012, 51, 10150–10153 CrossRef CAS PubMed; (b) B. A. Surgenor, M. Bühl, A. M. Z. Slawin, J. D. Woollins and P. Kilian, Angew. Chem., 2012, 124, 10297–10300 CrossRef.
- (a) S. Shah and J. D. Protasiewicz, Coord. Chem. Rev., 2000, 210, 181–201 CrossRef CAS; (b) H. Aktaş, J. C. Slootweg and K. Lammertsma, Angew. Chem., Int. Ed., 2010, 49, 2102–2113 CrossRef PubMed; (c) H. Aktaş, J. C. Slootweg and K. Lammertsma, Angew. Chem., 2010, 122, 2148–2159 CrossRef; (d) A. Schmidpeter, in Multiple Bonds and Low Coordination in Phosphorus Chemistry, ed. M. Regitz, O. J. Scherer and R. Appel, Thieme, Stuttgart, 1990, pp. 338–351 Search PubMed; (e) F. Mathey, A. Marinetti, S. Bauer and P. Le Floch, Pure Appl. Chem., 1991, 63, 855–858 CrossRef CAS; (f) F. Mathey, N. H. T. Huy and A. Marinetti, Helv. Chim. Acta, 2001, 84, 2938–2957 CrossRef CAS; (g) N. A. Piro and C. C. Cummins, J. Am. Chem. Soc., 2009, 131, 8764–8765 CrossRef CAS PubMed.
- (a) S. Shah and J. D. Protasiewicz, Chem. Commun., 1998, 1585–1586 RSC; (b) S. Shah, M. C. Simpson, R. C. Smith and J. D. Protasiewicz, J. Am. Chem. Soc., 2001, 123, 6925–6926 CrossRef CAS PubMed; (c) D. V. Partyka, M. P. Washington, J. B. Updegraff, R. A. Woloszynek and J. D. Protasiewicz, Angew. Chem., Int. Ed., 2008, 47, 7489–7492 CrossRef CAS PubMed; (d) D. V. Partyka, M. P. Washington, J. B. Updegraff, R. A. Woloszynek and J. D. Protasiewicz, Angew. Chem., 2008, 120, 7599–7602 CrossRef; (e) U. J. Kilgore, H. Fan, M. Pink, E. Urnezius, J. D. Protasiewicz and D. J. Mindiola, Chem. Commun., 2009, 4521–4523 RSC.
- B. A. Surgenor, B. A. Chalmers, K. S. Athukorala Arachchige, A. M. Z. Slawin, J. D. Woollins, M. Bühl and P. Kilian, Inorg. Chem., 2014, 53, 6856–6866 CrossRef CAS PubMed.
- (a) E. L. Norton, K. L. S. Szekely, J. W. Dube, P. G. Bomben and C. L. B. Macdonald, Inorg. Chem., 2008, 47, 1196–1203 CrossRef CAS PubMed; (b) B. D. Ellis and C. L. B. Macdonald, Inorg. Chem., 2006, 45, 6864–6874 CrossRef CAS PubMed.
- (a) D. Weber, G. Heckmann and E. Fluck, Z. Naturforsch., B: J. Chem. Sci., 1976, 31, 81–84 Search PubMed; (b) A. Schmidpeter, S. Lochschmidt and W. S. Sheldrick, Angew. Chem., 1985, 97, 214–215 CrossRef CAS; (c) A. Schmidpeter, S. Lochschmidt and W. S. Sheldrick, Angew. Chem., Int. Ed. Engl., 1985, 24, 226–227 CrossRef; (d) K. B. Dillon and R. J. Olivey, Heteroat. Chem., 2004, 15, 150–154 CrossRef CAS; (e) J. D. Burton, R. M. K. Deng, K. B. Dillon, P. K. Monks and R. J. Olivey, Heteroat. Chem., 2005, 16, 447–452 CrossRef CAS.
- (a) J. W. Dube, C. L. B. Macdonald, B. D. Ellis and P. J. Ragogna, Inorg. Chem., 2013, 52, 11438–11449 CrossRef CAS PubMed; (b) J. W. Dube, C. L. B. Macdonald and P. J. Ragogna, Angew. Chem., Int. Ed., 2012, 51, 13026–13030 CrossRef CAS PubMed; (c) J. W. Dube, C. L. B. Macdonald and P. J. Ragogna, Angew. Chem., 2012, 124, 13203–13207 CrossRef.
- J. F. Binder, A. A. Swidan, M. Tang, J. H. Nguyen and C. L. B. Macdonald, Chem. Commun., 2015, 51, 7741–7744 RSC.
- P. K. Coffer, R. M. K. Deng, K. B. Dillon, M. A. Fox and R. J. Olivey, Inorg. Chem., 2012, 51, 9799–9808 CrossRef CAS PubMed.
- A. Schmidpeter, F. Steinmüller and W. S. Sheldrick, Z. Anorg. Allg. Chem., 1989, 579, 158–172 CrossRef CAS.
- S. C. Kosnik, G. J. Farrar, E. L. Norton, B. F. T. Cooper, B. D. Ellis and C. L. B. Macdonald, Inorg. Chem., 2014, 53, 13061–13069 CrossRef CAS PubMed.
- (a) J. J. Weigand, K.-O. Feldmann, A. K. C. Echterhoff, A. W. Ehlers and K. Lammertsma, Angew. Chem., 2010, 122, 6314–6317 CrossRef; (b) J. J. Weigand, K. O. Feldmann, A. K. C. Echterhoff, A. W. Ehlers and K. Lammertsma, Angew. Chem., Int. Ed., 2010, 49, 6178–6181 CrossRef CAS PubMed; (c) K.-O. Feldmann and J. J. Weigand, Angew. Chem., 2012, 124, 6670–6672 CrossRef; (d) K.-O. Feldmann and J. J. Weigand, Angew. Chem., Int. Ed., 2012, 51, 6566–6568 CrossRef CAS PubMed.
- (a) K.-O. Feldmann and J. J. Weigand, Angew. Chem., Int. Ed., 2012, 51, 7545–7549 CrossRef CAS PubMed; (b) K.-O. Feldmann and J. J. Weigand, Angew. Chem., 2012, 124, 7663–7667 CrossRef.
- G. David, E. Niecke, M. Nieger, J. Radseck and W. W. Schoeller, J. Am. Chem. Soc., 1994, 116, 2191–2192 CrossRef CAS.
- J. A. C. Clyburne and M. J. Schriver, Inorg. Chem., 1996, 35, 3062–3063 CrossRef CAS.
- (a) B. Breit, A. Hoffmann, U. Bergsträßer, L. Ricard, F. Mathey and M. Regitz, Angew. Chem., 1994, 106, 1541–1543 CrossRef CAS; (b) B. Breit, A. Hoffmann, U. Bergsträsser, L. Ricard, F. Mathey and M. Regitz, Angew. Chem., Int. Ed. Engl., 1994, 33, 1491–1493 CrossRef; (c) E. A. Ishmaeva, E. V. Popova, V. F. Mironov, R. M. Aminova, Y. A. Vereshchagina, V. I. Galkin, K. Moeller and R. Schmutzler, Russ. J. Org. Chem., 2004, 40, 1076–1079 CrossRef CAS.
- (a) E. Niecke, M. Nieger and F. Reichert, Angew. Chem., 1988, 100, 1781–1782 CrossRef CAS; (b) E. Niecke, M. Nieger and F. Reichert, Angew. Chem., Int. Ed. Engl., 1988, 27, 1715–1716 CrossRef.
- (a) N. Burford, H. A. Spinney, M. J. Ferguson and R. McDonald, Chem. Commun., 2004, 2696–2697 RSC; (b) E. Niecke, R. Detsch, M. Nieger, F. Reichert and W. W. Schoeller, Bull. Soc. Chim. Fr., 1993, 130, 25–31 CAS.
- N. Burford, T. S. Cameron, J. A. C. Clyburne, K. Eichele, K. N. Robertson, S. Sereda, R. E. Wasylishen and W. A. Whitla, Inorg. Chem., 1996, 35, 5460–5467 CrossRef CAS PubMed.
- E. Niecke, G. David, R. Detsch, B. Kramer, M. Nieger and P. Wenderoth, Phosphorus, Sulfur Silicon Relat. Elem., 1993, 76, 25–28 CrossRef.
- N. Burford, A. D. Phillips, H. A. Spinney, M. Lumsden, U. Werner-Zwanziger, M. J. Ferguson and R. McDonald, J. Am. Chem. Soc., 2005, 127, 3921–3927 CrossRef CAS PubMed.
- A. Mack, U. Bergsträßer, G. J. Reiß and M. Regitz, Eur. J. Org. Chem., 1999, 587–595 CrossRef CAS.
- E. V. Popova, V. F. Mironov, E. A. Ishmaeva and I. I. Patsanovskii, Russ. J. Gen. Chem., 1999, 69, 48–50 CAS.
- E. A. Ishmaeva, D. V. Chachkov, A. Z. Alimova and Y. A. Vereshchagina, Russ. J. Org. Chem., 2014, 50, 608–610 CrossRef CAS.
- D. Gudat, E. Niecke, A. Ruban and V. von der Gönna, Magn. Reson. Chem., 1996, 34, 799–806 CrossRef CAS.
- X. Pan, Y. Su, X. Chen, Y. Zhao, Y. Li, J. Zuo and X. Wang, J. Am. Chem. Soc., 2013, 135, 5561–5564 CrossRef CAS PubMed.
- (a) M. Staal and P. Charoensirisomboon, Germany Pat., WO2009121821A1, 2009 Search PubMed; (b) B. M. Balbo, J. Ferbitz, O. S. Henze, C. Fleckenstein and K. Massonne, Germany Pat., WO2011092232A1, 2011 Search PubMed.
- I. F. Lutsenko and V. L. Foss, Pure Appl. Chem., 1980, 52, 917–944 CrossRef CAS.
- R. W. Rudolph, R. C. Taylor and R. W. Parry, J. Am. Chem. Soc., 1966, 88, 3729–3734 CrossRef CAS PubMed.
- (a) J. E. Griffiths and A. B. Burg, J. Am. Chem. Soc., 1960, 82, 1507–1508 CrossRef CAS; (b) J. E. Griffiths and A. B. Burg, J. Am. Chem. Soc., 1962, 84, 3442–3450 CrossRef CAS.
- (a) B. Hoge and B. Kurscheid, Angew. Chem., 2008, 120, 6920–6922 CrossRef; (b) B. Hoge and B. Kurscheid, Angew. Chem., Int. Ed., 2008, 47, 6814–6816 CrossRef CAS PubMed.
- L. Maier, J. Inorg. Nucl. Chem., 1962, 24, 275–283 CrossRef CAS.
- (a) L. Maier, Helv. Chim. Acta, 1962, 45, 2381–2383 CrossRef CAS; (b) H. Matschiner, F. Krech and A. Steinert, Z. Anorg. Allg. Chem., 1969, 371, 256–262 CrossRef CAS.
- M. Gruber, P. G. Jones and R. Schmutzler, Chem. Ber., 1990, 123, 1313–1317 CrossRef CAS.
- V. L. Foss, Y. A. Veits, P. L. Kukhmisterov and I. F. Lutsenko, Zh. Obshch. Khim., 1977, 47, 478–479 CAS.
- P. E. Sues, A. J. Lough and R. H. Morris, Chem. Commun., 2014, 50, 4707–4710 RSC.
- (a) R. C. Dobbie and M. J. Hopkinson, J. Fluorine Chem., 1974, 3, 367–374 CrossRef; (b) W.-W. du Mont, R. Hensel, W. McFarlane, I. J. Colquhoun, M. L. Ziegler and O. Serhadli, Chem. Ber., 1989, 122, 37–41 CrossRef CAS; (c) H. Westerman, M. Nieger and E. Niecke, Chem. Ber., 1991, 124, 13–16 CrossRef; (d) N. J. Hill, G. Reeske and A. H. Cowley, Main Group Chem., 2010, 9, 5–10 CAS; (e) N. A. Giffin, A. D. Hendsbee, T. L. Roemmele, M. D. Lumsden, C. C. Pye and J. D. Masuda, Inorg. Chem., 2012, 51, 11837–11850 CrossRef CAS PubMed.
- E. Ciganek, J. Org. Chem., 1970, 35, 3631–3636 CrossRef CAS.
- (a) A. Sola, F. Oton, A. Espinosa, A. Tarraga and P. Molina, Dalton Trans., 2011, 40, 12548–12559 RSC; (b) Á. Lorenzo, E. Aller and P. Molina, Tetrahedron, 2009, 65, 1397–1401 CrossRef; (c) M. Alajarín, A. Vidal and R.-A. Orenes, Eur. J. Org. Chem., 2002, 4222–4227 CrossRef.
- (a) T. Q. Ly and J. D. Woollins, Coord. Chem. Rev., 1998, 176, 451–481 CrossRef CAS; (b) Z. Fei and P. J. Dyson, Coord. Chem. Rev., 2005, 249, 2056–2074 CrossRef CAS.
- (a) Z. Fei, E. Păunescu, W. H. Ang, R. Scopelliti and P. J. Dyson, Eur. J. Inorg. Chem., 2014, 1745–1750 CrossRef CAS; (b) H. Rossknecht, W. P. Lehmann and A. Schmidpeter, Phosphorus, 1975, 5, 195–201 CAS; (c) A. Schmidpeter and H. Rossknecht, Z. Naturforsch., B: J. Chem. Sci., 1971, 26, 81–82 CAS.
- (a) M. S. Balakrishna, V. S. Reddy, S. S. Krishnamurthy, J. F. Nixon and J. C. T. R. Burckett St. Laurent, Coord. Chem. Rev., 1994, 129, 1–90 CrossRef CAS; (b) T. Appleby and J. Derek Woollins, Coord. Chem. Rev., 2002, 235, 121–140 CrossRef CAS.
- (a) Z. Fei, R. Scopelliti and P. J. Dyson, Eur. J. Inorg. Chem., 2004, 530–537 CrossRef CAS; (b) Z. Fei, R. Scopelliti and P. J. Dyson, Dalton Trans., 2003, 2772–2779 RSC; (c) Z. Fei, N. Biricik, D. Zhao, R. Scopelliti and P. J. Dyson, Inorg. Chem., 2004, 43, 2228–2230 CrossRef CAS PubMed; (d) Z. Fei, W. H. Ang, D. Zhao, R. Scopelliti and P. J. Dyson, Inorg. Chim. Acta, 2006, 359, 2635–2643 CrossRef CAS.
- (a) P. Bhattacharyya, A. M. Z. Slawin and J. D. Woollins, Chem. – Eur. J., 2002, 8, 2705–2711 CrossRef CAS PubMed; (b) K. Karaghiosoff, K. Eckstein and R. Motzer, Phosphorus, Sulfur Silicon Relat. Elem., 1994, 93, 185–188 CrossRef; (c) G. Großmann, G. Ohms, K. Krüger, K. Karaghiosoff, K. Eckstein, J. Hahn, A. Hopp, O. L. Malkina and P. Hrobarik, Z. Anorg. Allg. Chem., 2001, 627, 1269–1278 CrossRef.
- (a) G. Hua and J. D. Woollins, Angew. Chem., 2009, 121, 1394–1403 CrossRef; (b) G. Hua and J. D. Woollins, Angew. Chem., Int. Ed., 2009, 48, 1368–1377 CrossRef CAS PubMed.
- P. J. W. Elder and T. Chivers, Inorg. Chem., 2013, 52, 7791–7804 CrossRef CAS PubMed.
- P. J. W. Elder, T. L. Roemmele, M. Taghavikish, T. A. Engesser, H. Scherer, I. Krossing, R. T. Boeré and T. Chivers, Heteroat. Chem., 2014, 25, 501–513 CrossRef CAS.
- (a) A. B. Burg, J. Inorg. Nucl. Chem., 1959, 11, 258 CrossRef CAS; (b) H. L. Carrell and J. Donohue, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1968, 24, 699–707 CrossRef CAS; (c) H. Schmidbaur, T. Wimmer, A. Grohmann, O. Steigelmann and G. Müller, Chem. Ber., 1989, 122, 1607–1612 CrossRef CAS.
- A. H. Cowley and R. P. Pinnell, Inorg. Chem., 1966, 5, 1463–1464 CrossRef CAS.
- R. Wolf, M. Finger, C. Limburg, A. C. Willis, S. B. Wild and E. Hey-Hawkins, Dalton Trans., 2006, 831–837 RSC.
- R. Pietschnig, J. Organomet. Chem., 2007, 692, 3363–3369 CrossRef CAS.
- (a) N. Burford, T. S. Cameron, D. J. LeBlanc, P. Losier, S. Sereda and G. Wu, Organometallics, 1997, 16, 4712–4717 CrossRef CAS; (b) N. Burford, T. S. Cameron, P. J. Ragogna, E. Ocando-Mavarez, M. Gee, R. McDonald and R. E. Wasylishen, J. Am. Chem. Soc., 2001, 123, 7947–7948 CrossRef CAS PubMed; (c) N. Burford, P. J. Ragogna, R. McDonald and M. J. Ferguson, J. Am. Chem. Soc., 2003, 125, 14404–14410 CrossRef CAS PubMed.
- (a) N. Burford, C. A. Dyker and A. Decken, Angew. Chem., Int. Ed., 2005, 44, 2364–2367 CrossRef CAS PubMed; (b) N. Burford, C. A. Dyker, M. Lumsden and A. Decken, Angew. Chem., 2005, 117, 6352–6355 CrossRef; (c) C. A. Dyker and N. Burford, Chem. – Asian J., 2008, 3, 28–36 CrossRef CAS PubMed.
- (a) M. B. Abrams, B. L. Scott and R. T. Baker, Organometallics, 2000, 19, 4944–4956 CrossRef CAS; (b) J. M. Slattery, C. Fish, M. Green, T. N. Hooper, J. C. Jeffery, R. J. Kilby, J. M. Lynam, J. E. McGrady, D. A. Pantazis, C. A. Russell and C. E. Willans, Chem. – Eur. J., 2007, 13, 6967–6974 CrossRef CAS PubMed; (c) M. Gee, R. E. Wasylishen, P. J. Ragogna, N. Burford and R. McDonald, Can. J. Chem., 2002, 80, 1488–1500 CrossRef CAS; (d) M. Gonsior, I. Krossing, L. Müller, I. Raabe, M. Jansen and L. van Wüllen, Chem. – Eur. J., 2002, 8, 4475–4492 CrossRef CAS PubMed; (e) N. Burford, D. E. Herbert, P. J. Ragogna, R. McDonald and M. J. Ferguson, J. Am. Chem. Soc., 2004, 126, 17067–17073 CrossRef CAS PubMed.
- (a) N. Burford, P. J. Ragogna, R. McDonald and M. J. Ferguson, Chem. Commun., 2003, 2066–2067 RSC; (b) C. A. Dyker, N. Burford, G. Menard, M. D. Lumsden and A. Decken, Inorg. Chem., 2007, 46, 4277–4285 CrossRef CAS PubMed; (c) C. A. Dyker, N. Burford, M. D. Lumsden and A. Decken, J. Am. Chem. Soc., 2006, 128, 9632–9633 CrossRef CAS PubMed; (d) Y.-y. Carpenter, N. Burford, M. D. Lumsden and R. McDonald, Inorg. Chem., 2011, 50, 3342–3353 CrossRef CAS PubMed; (e) Y.-y. Carpenter, C. A. Dyker, N. Burford, M. D. Lumsden and A. Decken, J. Am. Chem. Soc., 2008, 130, 15732–15741 CrossRef CAS PubMed.
- J. J. Weigand, N. Burford, R. J. Davidson, S. Cameron and P. Seelheim, J. Am. Chem. Soc., 2009, 131, 17943–17953 CrossRef CAS PubMed.
- A. P. M. Robertson, C. A. Dyker, P. A. Gray, B. O. Patrick, A. Decken and N. Burford, J. Am. Chem. Soc., 2014, 136, 14941–14950 CrossRef CAS PubMed.
- M. H. Holthausen, D. Knackstedt, N. Burford and J. J. Weigand, Aust. J. Chem., 2013, 66, 1155–1162 CAS.
- S. Loss, C. Widauer, H. Ruegger, U. Fleischer, C. M. Marchand, H. Grützmacher and G. Frenking, Dalton Trans., 2003, 85–91 RSC.
- J. I. Bates and D. P. Gates, J. Am. Chem. Soc., 2006, 128, 15998–15999 CrossRef CAS PubMed.
- T. P. Martin, Z. Phys. D: At., Mol. Clusters, 1986, 3, 211–217 CrossRef CAS.
- (a) M. D. Chen, R. B. Huang, L. S. Zheng, Q. E. Zhang and C. T. Au, Chem. Phys. Lett., 2000, 325, 22–28 CrossRef CAS; (b) T. Xue, J. Luo, S. Shen, F. Li and J. Zhao, Chem. Phys. Lett., 2010, 485, 26–30 CrossRef CAS.
- I. Krossing, in Molecular Clusters of the Main Group Elements, ed. M. Driess and H. Nöth, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2005, pp. 209–229 Search PubMed.
- (a) T. Köchner, S. Riedel, A. J. Lehner, H. Scherer, I. Raabe, T. A. Engesser, F. W. Scholz, U. Gellrich, P. Eiden, R. A. Paz Schmidt, D. A. Plattner and I. Krossing, Angew. Chem., Int. Ed., 2010, 49, 8139–8143 CrossRef PubMed; (b) T. Köchner, S. Riedel, A. J. Lehner, H. Scherer, I. Raabe, T. A. Engesser, F. W. Scholz, U. Gellrich, P. Eiden, R. A. Paz Schmidt, D. A. Plattner and I. Krossing, Angew. Chem., 2010, 122, 8316–8320 CrossRef.
- (a) T. Köchner, T. A. Engesser, H. Scherer, D. A. Plattner, A. Steffani and I. Krossing, Angew. Chem., Int. Ed., 2012, 51, 6529–6531 CrossRef PubMed; (b) T. Köchner, T. A. Engesser, H. Scherer, D. A. Plattner, A. Steffani and I. Krossing, Angew. Chem., 2012, 124, 6635–6637 CrossRef.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2001, 40, 4406–4409 CrossRef CAS.
- A. Bihlmeier, M. Gonsior, I. Raabe, N. Trapp and I. Krossing, Chem. – Eur. J., 2004, 10, 5041–5051 CrossRef CAS PubMed.
- M. H. Holthausen, K.-O. Feldmann, S. Schulz, A. Hepp and J. J. Weigand, Inorg. Chem., 2012, 51, 3374–3387 CrossRef CAS PubMed.
- M. H. Holthausen and J. J. Weigand, Z. Anorg. Allg. Chem., 2012, 638, 1103–1108 CrossRef CAS.
- (a) J. J. Weigand, M. Holthausen and R. Fröhlich, Angew. Chem., Int. Ed., 2009, 48, 295–298 CrossRef CAS PubMed; (b) J. J. Weigand, M. Holthausen and R. Fröhlich, Angew. Chem., 2009, 121, 301–304 CrossRef.
- M. H. Holthausen, C. Richter, A. Hepp and J. J. Weigand, Chem. Commun., 2010, 46, 6921–6923 RSC.
- M. H. Holthausen and J. J. Weigand, J. Am. Chem. Soc., 2009, 131, 14210–14211 CrossRef CAS PubMed.
- M. Gonsior, I. Krossing and E. Matern, Chem. – Eur. J., 2006, 12, 1703–1714 CrossRef CAS PubMed.
- (a) M. Gonsior, I. Krossing and E. Matern, Chem. – Eur. J., 2006, 12, 1986–1996 CrossRef CAS PubMed; (b) I. Krossing, Chem. – Eur. J., 2001, 7, 490–502 CrossRef CAS PubMed.
- (a) K. Eckstein, Dissertation, LMU München, 1997 Search PubMed; (b) C. Strauhal, Diploma thesis, LMU München, 1996 Search PubMed.
- O. Schön, Dissertation, LMU München, 2007 Search PubMed.
- M. H. Holthausen, A. Hepp and J. J. Weigand, Chem. – Eur. J., 2013, 19, 9895–9907 CrossRef CAS PubMed.
- K.-O. Feldmann, T. Wiegand, J. Ren, H. Eckert, J. Breternitz, M. F. Groh, U. Müller, M. Ruck, B. Maryasin, C. Ochsenfeld, O. Schön, K. Karaghiosoff and J. J. Weigand, Chem. – Eur. J., 2015, 21, 9697–9712 CrossRef CAS PubMed.
- (a) M. Donath, E. Conrad, P. Jerabek, G. Frenking, R. Fröhlich, N. Burford and J. J. Weigand, Angew. Chem., Int. Ed., 2012, 51, 2964–2967 CrossRef CAS PubMed; (b) M. Donath, E. Conrad, P. Jerabek, G. Frenking, R. Fröhlich, N. Burford and J. J. Weigand, Angew. Chem., 2012, 124, 3018–3021 CrossRef.
- F. Dielmann, O. Back, M. Henry-Ellinger, P. Jerabek, G. Frenking and G. Bertrand, Science, 2012, 337, 1526–1528 CrossRef CAS PubMed.
- W. Wang, R. Cao and L. Liu, J. Chem. Soc., Perkin Trans. 1, 2000, 1673–1675 RSC.
- W.-H. Wang, G.-Y. Xu, R.-Z. Cao and L.-Z. Liu, Heteroat. Chem., 2000, 11, 512–517 CrossRef CAS.
- G. Bettermann, H. Buhl, R. Schmutzler, D. Schomburg and U. Wermuth, Phosphorus Sulfur Relat. Elem., 1983, 18, 77–80 CrossRef CAS.
- D. Schomburg, Acta Crystallogr., Sect. A: Found. Crystallogr., 1984, 40, C265 Search PubMed.
- (a) K. Schwedtmann, M. H. Holthausen, K.-O. Feldmann and J. J. Weigand, Angew. Chem., 2013, 125, 14454–14458 CrossRef; (b) K. Schwedtmann, M. H. Holthausen, K.-O. Feldmann and J. J. Weigand, Angew. Chem., Int. Ed., 2013, 52, 14204–14208 CrossRef CAS PubMed; (c) T. Böttcher, B. S. Bassil, L. Zhechkov, T. Heine and G.-V. Röschenthaler, Chem. Sci., 2013, 4, 77–83 RSC; (d) P. Wawrzyniak, A. L. Fuller, A. M. Z. Slawin and P. Kilian, Inorg. Chem., 2009, 48, 2500–2506 CrossRef CAS PubMed.
- K. B. Dillon, A. W. G. Platt, A. Schmidpeter, F. Zwaschka and W. S. Sheldrick, Z. Anorg. Allg. Chem., 1982, 488, 7–26 CrossRef CAS.
- K. W. Hansen and L. S. Bartell, Inorg. Chem., 1965, 4, 1775–1776 CrossRef CAS.
- E. V. Nikitin, A. S. Romakhin, V. A. Zagumennov and Y. A. Babkin, Electrochim. Acta, 1997, 42, 2217–2224 CrossRef CAS.
- J. J. Weigand, S. D. Riegel, N. Burford and A. Decken, J. Am. Chem. Soc., 2007, 129, 7969–7976 CrossRef CAS PubMed.
- D. Schomburg, G. Bettermann, L. Ernst and R. Schmutzler, Angew. Chem., Int. Ed. Engl., 1985, 24, 975–976 CrossRef.
- R. W. Alder, C. Ganter, C. J. Harris and A. G. Orpen, J. Chem. Soc., Chem. Commun., 1992, 1170–1172 RSC.
- P. Kilian, A. M. Z. Slawin and J. D. Woollins, Dalton Trans., 2006, 2175–2183 RSC.
- (a) Y. P. Makovetskii, N. G. Feshchenko, V. V. Malovik, V. Y. Semenii, I. E. Boldeskul, V. A. Bondar and N. P. Chernukho, Zh. Obshch. Khim., 1980, 50, 1967 Search PubMed; (b) Y. P. Makovetskii, V. E. Lidkovskii, I. E. Boldeskul, N. G. Feshchenko and N. N. Kalibabchuk, Zh. Obshch. Khim., 1989, 52, 1989 Search PubMed.
- R. M. Siddique and J. M. Winfield, Can. J. Chem., 1989, 67, 1780–1784 CrossRef CAS.
- (a) S. S. Chitnis, Y.-Y. Carpenter, N. Burford, R. McDonald and M. J. Ferguson, Angew. Chem., Int. Ed., 2013, 52, 4863–4866 CrossRef CAS PubMed; (b) S. S. Chitnis, A. P. M. Robertson, N. Burford, J. J. Weigand and R. Fischer, Chem. Sci., 2015, 6, 2559–2574 RSC; (c) S. S. Chitnis, N. Burford, J. J. Weigand and R. McDonald, Angew. Chem., Int. Ed., 2015, 54, 7828–7832 CrossRef CAS PubMed; (d) S. S. Chitnis, N. Burford, J. J. Weigand and R. McDonald, Angew. Chem., 2015, 127, 7939–7943 CrossRef; (e) A. P. M. Robertson, N. Burford, R. McDonald and M. J. Ferguson, Angew. Chem., Int. Ed., 2014, 53, 3480–3483 CrossRef CAS PubMed; (f) A. P. M. Robertson, N. Burford, R. McDonald and M. J. Ferguson, Angew. Chem., 2014, 126, 3548–3551 CrossRef.
- G. Bellachioma, G. Cardaci, A. Macchioni, C. Venturi and C. Zuccaccia, J. Organomet. Chem., 2006, 691, 3881–3888 CrossRef CAS.
- M. H. Holthausen, J. M. Bayne, I. Mallov, R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 7298–7301 CrossRef CAS PubMed.
- R. R. Holmes and E. F. Bertaut, J. Am. Chem. Soc., 1958, 80, 2980–2983 CrossRef CAS.
- G. Müller, H.-J. Matheus and M. Winkler, Z. Naturforsch., B: J. Chem. Sci., 2001, 56, 1155–1162 Search PubMed.
- I. Bauer and W. D. Habicher, Collect. Czech. Chem. Commun., 2004, 69, 1195–1230 CrossRef CAS.
- R. W. Alder, D. D. Ellis, R. Gleiter, C. J. Harris, H. Lange, A. G. Orpen, D. Read and P. N. Taylor, J. Chem. Soc., Perkin Trans. 1, 1998, 1657–1668 RSC.
- R. W. Alder, C. P. Butts, A. G. Orpen, D. Read and J. M. Oliva, J. Chem. Soc., Perkin Trans. 2, 2001, 282–287 RSC.
- (a) R. W. Alder and D. Read, Angew. Chem., 2000, 112, 3001–3004 CrossRef; (b) R. W. Alder and D. Read, Angew. Chem., Int. Ed., 2000, 39, 2879–2882 CrossRef CAS.
- M. H. Holthausen, R. R. Hiranandani and D. W. Stephan, Chem. Sci., 2015, 6, 2016–2021 RSC.
- D. M. U. K. Somisara, M. Bühl, T. Lebl, N. V. Richardson, A. M. Z. Slawin, J. D. Woollins and P. Kilian, Chem. – Eur. J., 2011, 17, 2666–2677 CrossRef CAS PubMed.
- H. W. Roesky and H. Djarrah, Inorg. Chem., 1982, 21, 844–845 CrossRef CAS.
- (a) D. Schomburg, N. Weferling and R. Schmutzler, J. Chem. Soc., Chem. Commun., 1981, 609–610 RSC; (b) L. Lamande and A. Munoz, Phosphorus Sulfur Relat. Elem., 1987, 32, 1–17 CrossRef CAS.
- R. W. Alder, C. P. Butts, A. G. Orpen and D. Read, J. Chem. Soc., Perkin Trans. 2, 2001, 288–295 RSC.
- C. W. Schultz and R. W. Rudolph, J. Am. Chem. Soc., 1971, 93, 1898–1903 CrossRef.
- (a) J. A. Gibson, G.-V. Röschenthaler and R. Schmutzler, J. Chem. Soc., Dalton Trans., 1975, 918–924 RSC; (b) W. S. Sheldrick, J. A. Gibson and G.-V. Röschenthaler, Z. Naturforsch., B: J. Chem. Sci., 1978, 33, 1102–1105 Search PubMed.
- P. Kilian, D. Philp, A. M. Z. Slawin and J. D. Woollins, Eur. J. Inorg. Chem., 2003, 249–254 CrossRef CAS.
- (a) M. Sanchez, R. Réau, F. Dahan, M. Regitz and G. Bertrand, Angew. Chem., 1996, 108, 2386–2388 CrossRef; (b) M. Sanchez, R. Réau, F. Dahan, M. Regitz and G. Bertrand, Angew. Chem., Int. Ed. Engl., 1996, 35, 2228–2230 CrossRef CAS.
- (a) H. Luo, R. McDonald and R. G. Cavell, Angew. Chem., 1998, 110, 1172–1174 CrossRef; (b) H. Luo, R. McDonald and R. G. Cavell, Angew. Chem., Int. Ed., 1998, 37, 1098–1099 CrossRef CAS.
- (a) R. W. Suter, H. C. Knachel, V. P. Petro, J. H. Howatson and S. G. Shore, J. Am. Chem. Soc., 1973, 95, 1474–1479 CrossRef CAS; (b) K. B. Dillon and T. A. Straw, J. Chem. Soc., Chem. Commun., 1991, 234–235 RSC.
- J. E. Richman, R. O. Day and R. R. Holmes, J. Am. Chem. Soc., 1980, 102, 3955 CrossRef CAS.
- H. W. Roesky, D. Amirzadeh-Asl and W. S. Sheldrick, J. Am. Chem. Soc., 1982, 104, 2919–2920 CrossRef CAS.
- L. Lamande and A. Munoz, Tetrahedron, 1990, 46, 3527–3534 CrossRef CAS.
Footnote |
| † Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |