
THE DISCOVERY OF SERS: an idiosyncratic account from a vibrational spectroscopist
Patrick
Hendra
Prof. Emeritus in Chemistry, University of Southampton
Introduction
Let's start by explaining what SERS is and the experiment where it was first demonstrated. If you know all about SERS please skip the Introduction.Let's assume that you have a set electrochemical kit so that you can apply a known potential to an electrochemical cell and a cell suitable for Raman spectroscopy – and oh yes a Raman instrument! The cell must contain the electrolyte and a working electrode surrounded by a secondary electrode (a ring of Pt wire) and that is connected by a capillary full of electrolyte, to a reference electrode. The working electrode is mounted behind a window and the whole cell is mounted in a Raman instrument. You can see the arrangement in Fig. 1.
 | ||
| Fig. 1 Photograph of the Raman spectroelectrochemistry cell, in the sample compartment of a Cary 82 Raman spectrometer illuminated by 514.5 nm argon laser exciting light. The glass cell filled with electrolyte has a plane glass window to the left. The reference electrode is poking out upwards. The working electrode is inclined at 45 degrees with its roughened surface inclined downwards. The laser beam comes up from below. The photographs were taken in the Chemistry Department, Southampton University in 1975. Jim McQuillan collection. | ||
The working electrode is silver, the electrolyte contains chloride ions and pyridine. The first step is to repeatedly sweep the working electrode from positive to negative. After about 10 sweeps the surface will seem to be cream in colour. What you have done is to etch the surface and then plate it repeatedly, thus roughening it's surface. The surface area will have increased by about 10×. At this stage, the cell is aligned in the instrument sample area resulting in a strong spectrum being observed, see Fig. 2. The spectrum is that of pyridine but somewhat modified. It is then possible to alter the potential on the silver electrode in steps of 0.1 V producing a set of spectra as seen in Fig. 3.
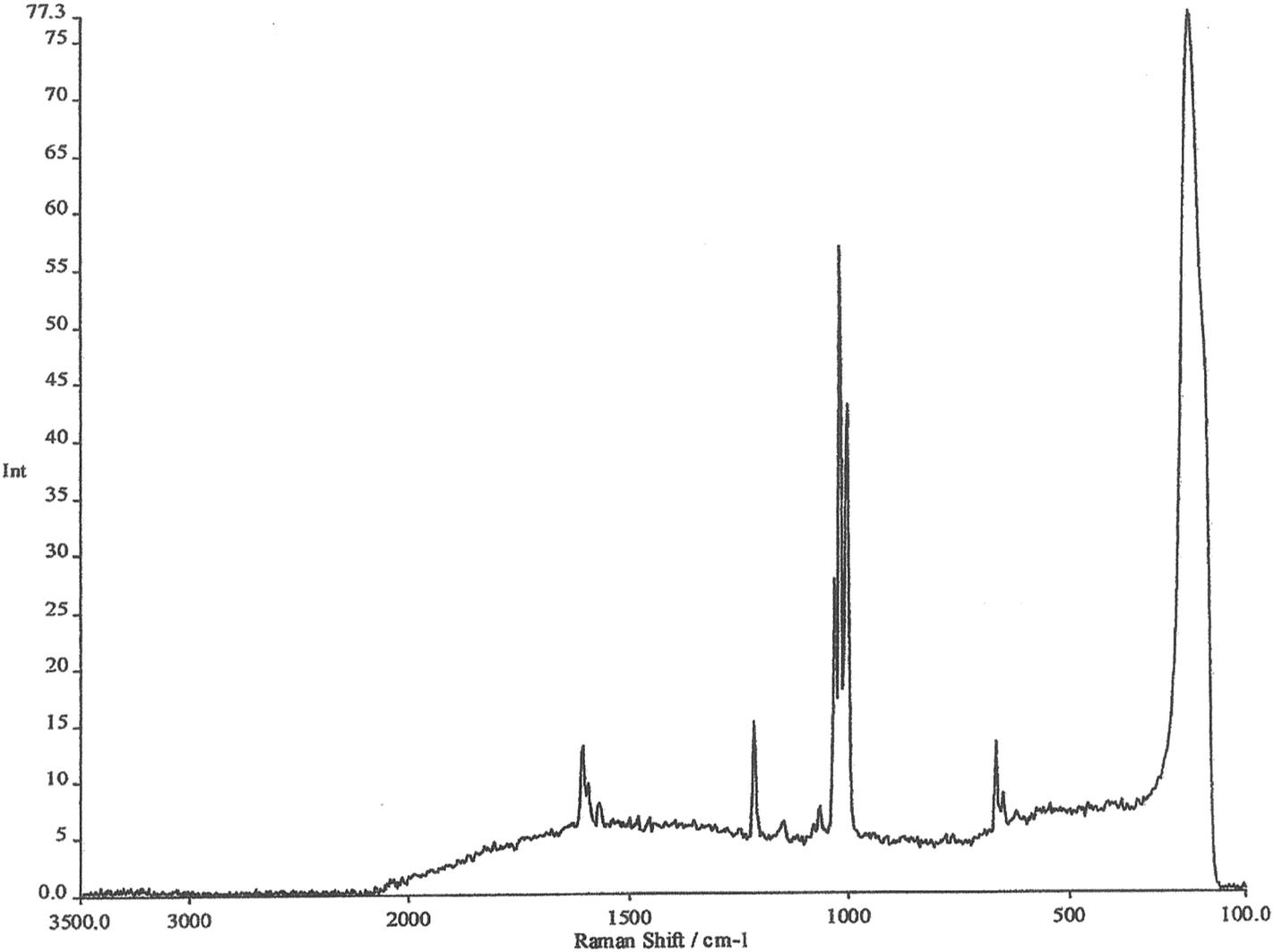 | ||
| Fig. 2 FT-Raman spectrum of pyridine on a roughened silver electrode. | ||
 | ||
| Fig. 3 Surface-enhanced Raman spectra recorded at different potentials on a roughened silver electrode using an electrochemical Raman cell. | ||
Experience of Raman spectra of the adsorption of pyridine over silica acid catalysts enables us to interpret these spectra with confidence. The region between 1000 and 1300 cm−1 enables us to identify bands due to pyridine hydrogen bonded to water, pyridine chemisorbed to silver atoms on the surface and pyridinium cations. What we are seeing is the spectrum of pyridine species lying within the electrode/electrolyte interface as they respond to the huge potential gradients in the ‘electrical double layer’.
The remarkable thing about these spectra is not just the detail available (which was totally unique at the time) but their INTENSITY. They are at least a million times as intense as one might normally expect, thus the name Surface Enhanced Raman Scattering. I will stop there by citing the paper that we wrote back in 1974 describing the experiment.1
If you want a really first class detailed account please pick up Jim McQuillan's recollection in Notes and Records of the Royal Society. It describes the origin of the SERS enhancement process and includes extracts from his lab notes and is to be highly recommended.2
Background
Martin Fleischmann and I were appointed to Southampton – he in 1968 and myself two years earlier. Unbeknown to either of us, our appointer, Graham Hills, decided that my interest in Raman could be compounded with Fleischmann's considerable skills in electrochemistry to yield information on the chemical reactions occurring at electrode electrolyte interfaces for the first time. A word of explanation is essential – I was no electrochemist – the sum total of my retained knowledge of electrochemistry was to swear when the car battery went flat. However, I learned fast.In a nutshell; detailed and precise electrochemical measurements on a cell were routine at the time and included static measurements of potential and current. Cyclic measurements were also very precise and all could be made over a range of electrolyte contents and concentrations. Some kinetic measurements were possible. However, the nature of the reactions close to the interface had to be deduced.
It was possible to run a cell, measure its characteristics, switch off, clean and dry the electrode and then examine it by surface chemical techniques under high vacuum. Sort of baby minus the bathwater almost literally! Graham Hills was well aware that very few measurements then available were likely to probe the interface especially when the cell was actually operating, i.e., in situ. The only methods potentially available were reflection (ATR) infrared and maybe Raman. ATR could minimise the path thickness and thus hopefully pull out the contribution made by the interface to the total spectrum but this would be very difficult particularly in aqueous systems. And Oh Yes – Raman works well in aqueous solutions – or so it is said!
When Martin came to Southampton from Newcastle, he brought Allan Bewick with him and Allan gamely took on the IR challenge. To a considerable degree he had success and demonstrated beautiful results using reflection methods. I was intended to be the Raman Guinea Pig. Fortunately, no-one told me about the Master Plan when I was appointed in 1966. Mind you, there were occasional questions around ‘have you considered looking at aqueous solutions?’ but I ignored these and I started out on another tack (more anon).
In the early 1970s Graham engineered a head bashing between Martin and I (Martin was one of the best electrochemists in the business with a reputation to match, I was a young lowly over-ambitious lecturer and at that juncture I hardly knew Martin at all).
We discussed the Raman spectroscopic problem if it was to be applied to working electrodes, I emphasised that to succeed the absolute maximum amount of the Raman scatterer must fall onto the patch of the electrode surface viewed by the spectrometer (the patch is the imaginary image of the entrance slit at the electrode surface) and illuminated by the laser at the highest available brightness. What experiment might work?
As a spoiled inorganic chemist (my PhD was in co-ordination chemistry) I pointed out that calomel-Hg2Cl2 was one of the strongest Raman scatterers known to man (and woman, of course!), so this was an obvious starter because electrochemists habitually paddle in the stuff. At this crucial moment Jim McQuillan arrived as a post-doc, Fig. 4.
 | ||
| Fig. 4 Martin Fleischmann, Pat Hendra and Jim McQuillan. | ||
Jim was/is a superb experimenter – very thoughtful, thorough and kept excellent notes. Martin and Jim volunteered to generate a surface coated with micro globular mercury (this increases the surface area to 10 times that of a smooth surface). After some difficulty it worked, as we were able to record the spectrum of Hg2Cl2 over the mercury surface and confirmed the experiment by using an electrolyte containing Br− to generate a spectrum of Hg2Br2. It was certain however that the calomel layer was multilayer.
Encouraged, Martin and Jim then searched a range of possibilities. Martin's Group came up with many roughened surfaces – platinum, palladium, iron, lead, you name it – and studied them in electrodes containing good Raman scattering prospects such as CN− and CNS− and organics but to be honest, progress was a bit slowwww!
Among numerous ideas, good, and more frequently bad, it was decided to try silver using an electrolyte containing pyridine. Why did this almost bizarre combination make it? Reasons – desperation and tiny bit of further background and luck.
In the early 60s, I did a one year postdoc at the Royal Holloway College in traditional surface chemistry – vacuum systems, BET isotherms etc. – so I learned something of the appropriate arts. My PhD had been in IR of inorganics and especially co-ordination compounds of the Pt group elements, and also those of Cu, Ag and Au. I became interested in Raman from 1964 or so and tried various methods to record spectra from deeply coloured co-ordination compounds. In 1966, I was lucky enough to be in the right place at the right time and recorded a huge number of laser Raman spectra of inorganic solids (including calomel) in Lippincott's lab in Maryland on the prototype Cary 81L laser Raman spectrometer. All the specimens were powders and many were deeply coloured (Raman of coloured powders – you must, at the time be joking!).
Once at Southampton, I was able to use equipment owned by Ian Beattie† and started a programme to demonstrate to sceptical infrared spectroscopists that Raman was worth trying. We expanded from inorganics, including many more deeply coloured ones like the Group 16 mono- and di-halides and even KMnO4, on to organics and then with Harry Willis into polymers and then in the early 70s we tried surfaces.
One of the classic infrared measurements in the study of high area oxide catalysts was to examine them as incredibly thin pellets and to adsorb pyridine and then follow the spectrum as the pyridine coverage was pumped off. Bands in the IR absorption spectrum could be reliably and quantitatively related to Lewis, Brönsted and hydrogen bonding sites on the surface and gave a unique insight into this form of catalytic chemistry.
We demonstrated that this so-called ‘Perry method for distinguishing surface sites’ could also be exploited in the Raman. Clear bands could be seen down to coverage as low as 0.5 monolayers, BUT only if the surface area of the catalyst was large enough (typically 500 m2 per gram) and the fluorescence could be tamed. Norman Sheppard was involved in similar studies at that time and to an extent did much better than we did in controlling the nuisance of high levels of fluorescence.
So, trying the combination of a silver electrode and pyridine as a tell-tale molecule wasn't as bizarre as it sounds. Well everyone interested in SERS, as the experiment was later dubbed, knows that the measurements were extended to Cu and Au and then a long term effort was made to understand the origin of the phenomenon. I played no part in all of this because I had other fish to fry. See Ref. 1 for some more details on SERS and its origin.
My interest was always centred on the lack of interest in Raman compared with infrared.‡
Why don't vibrational spectroscopists do their work in STEREO? So, I let the electrochemists get on with SERS and expanded Raman into polymer morphology and even into combustion chemistry (yes, you can record the Raman spectrum of the reacting gases in a flame – oddly the combustion people were never impressed). And then along came FT Raman and I thought we had cracked the problem, buy the right instrument and no-one had any excuse, you could do the IR and the Raman on the same instrument and display the results using the same software. Hmmm!
Dare I suggest that by 2016 Raman has usurped IR's premier position as an analytical tool, or has it?
Acknowledgements
It is impossible to thank everyone who made all this happen, there are dozens of them, but I must thank Jim McQuillan who supplied the photo in Fig. 1 and checked the manuscript for me. Fig. 2 and 3 are modern images. They were kindly recorded by Dr Sumeet Mahajan of the University of Southampton on an FT Instrument. They are typical of what could be recorded in the 1970s.Finally, I must mention my debt to Martin Fleischmann who sadly has passed away. Few of us have the opportunity to work with true genius – I did.
References
- M. Fleischmann, P. Hendra and J. McQuillan, Chem. Phys. Lett., 1974, 26, 163 CrossRef CAS.
- A. J. Mcquillan, Notes Rec. Roy. Soc., 2009, 63, 105–109 CrossRef.
Footnotes |
| † I. R. Beattie, Prof. of Inorganic Chemistry, supported me through thick and thin, night and day. His group used the instrument in the day and my lot took the nights! An incredibly generous offer to a young upstart who must have appeared as a competitor. Thanks Ian. |
| ‡ It's worth remembering that for many years the IRDG was the Infrared Discussion Group. Apart from the 1930s, Raman was little more than an academic plaything until relatively recently. |
| This journal is © The Royal Society of Chemistry 2016 |