
Silylative coupling of olefins with vinylsilanes in the synthesis of functionalized alkenes†
Justyna
Szudkowska-Frątczak
,
Grzegorz
Hreczycho
and
Piotr
Pawluć
*
Faculty of Chemistry, Adam Mickiewicz University in Poznan, Umultowska 89b, 61-614 Poznan, Poland. E-mail: piotrpaw@amu.edu.pl
First published on 2nd March 2015
Abstract
The development of highly selective methods for the synthesis of functionalized olefins, based on sequential catalytic reactions of organometallic reagents, has been the subject of extensive study because of their versatile applications in organic synthesis and materials science. The silylative coupling of olefins with vinyl-substituted organosilicon compounds (discovered in our group) represents one of the most efficient and straightforward methods for the synthesis of stereodefined alkenylsilanes and bis(silyl)alkenes, which are particularly attractive scaffolds for further transformations including palladium-catalyzed cross-coupling with organic halides (Hiyama coupling) or electrophile-induced desilylation. The article highlights the recent developments and covers the literature mainly from the last decade in the sequential (also one-pot) synthetic strategies including ruthenium-catalyzed silylative coupling followed by desilylative cross-coupling, acylation and halogenation, leading to stereodefined organic derivatives such as (E)-alkenyl halides, (E)-α,β-unsaturated ketones or arylene-(E)-vinylene derivatives which are widely applied as fine chemicals, functional materials or building blocks in organic synthesis.
 Justyna Szudkowska-Frątczak | Justyna Szudkowska-Frątczak was born in 1986 in Konin (Poland). In 2010, she joined Professor Bogdan Marciniec's research group at the Organometallic Chemistry Department, Faculty of Chemistry AMU, as a graduate student and performed her Ph.D. work in organometallic chemistry. Her doctoral studies cover the area of new catalytic methods for the synthesis of organometalloid (Si, Ge, and B) compounds and their application in organic synthesis. |
 Grzegorz Hreczycho | Grzegorz Hreczycho was born in Zielona Góra (Poland) in 1977. He obtained his Ph.D. in 2007 from Adam Mickiewicz University in Poznan (Poland) under the supervision of Professor Bogdan Marciniec. His research interests cover novel applications of unsaturated silicon and germanium compounds in addition and cross-coupling reactions catalyzed by transition metal complexes. |
 Piotr Pawluć | Piotr Pawluć received a Ph.D. (2004) and habilitation (2012) in chemistry from Adam Mickiewicz University in Poznan (Poland). He was a postdoctoral fellow with Professor Andre Mortreux at the Lille University (France). His research interests involve developing catalytic methodologies for the preparation of organosilicon building blocks and their application to organic synthesis. More than 50 research publications, patents, and book chapters document his activity in the fields of organometallic chemistry, homogeneous catalysis and organic synthesis. |
1. Introduction
The use of main group elements as reagents in stereoselective organic synthesis has brought for the last 30 years many new and selective methods for the syntheses of functionalized organic and organometallic compounds of fundamental importance for development of organic chemistry and chemical technology.1,2 Spectacular examples of selective reactions of organometallic compounds with organic electrophiles that have been permanently accepted in the canon of modern organic synthesis, are the catalytic coupling processes: Suzuki–Miyaura (boron compounds),3 Stille (tin compounds),4 Negishi (zinc compounds)5 and Hiyama (silicon compounds).6 The significance of these discoveries has been emphasized by honoring of Professors A. Suzuki, E.-i. Negishi and R. Heck, who are the pioneers in investigation of organometallic or organic reagents’ coupling with organic halides, with the Nobel Award in chemistry in 2010. The interest in the area is evidenced by a great increase in the number of papers and monographs on the above subjects, published in the top ranked scientific journals in the last decade.The particular importance of silicon from among the main group elements follows from the fact that (in contrast to the other p-block elements) the majority of its compounds are relatively cheap and commercially available, they show high stability, insignificant toxicity and have highly selective reactivity towards electrophiles. Chemical properties of organosilicon compounds, in particular vinylsilanes and their derivatives, make them highly valuable in the reactions leading to functionalized organic compounds.7,8
Unsaturated organosilicon compounds have been for many years successfully applied in stereoselective organic synthesis.7–9 Particularly useful tools in these processes are the reactions of electrophilic-induced desilylation of vinylsilanes and their derivatives as they permit the selective introduction of a given substituent (e.g. acyl, halogen, nitro groups) to replace a silyl group (ipso-substitution), usually ensuring the retention of geometry.8,9
Of the available methods for the preparation of stereodefined alkenylsilanes, the transition metal-catalyzed silylative coupling of functionalized olefins with vinylsilanes (called also trans-silylation or Marciniec coupling) is one of the most direct, flexible and powerful approaches (Scheme 1).10 The silylative coupling reaction, is an example of a catalytic process taking place as a result of C–H bond cleavage at the α and β carbon atom from the vinyl group and activation of the Cvinyl–Si bond in the vinylsilane molecule with the evolution of the ethylene molecule. The catalysts of this process are complexes of transition metals (M = Ru, Rh, Co, Ir), containing or capable of in situ generation of M–H and M–Si bonds.10
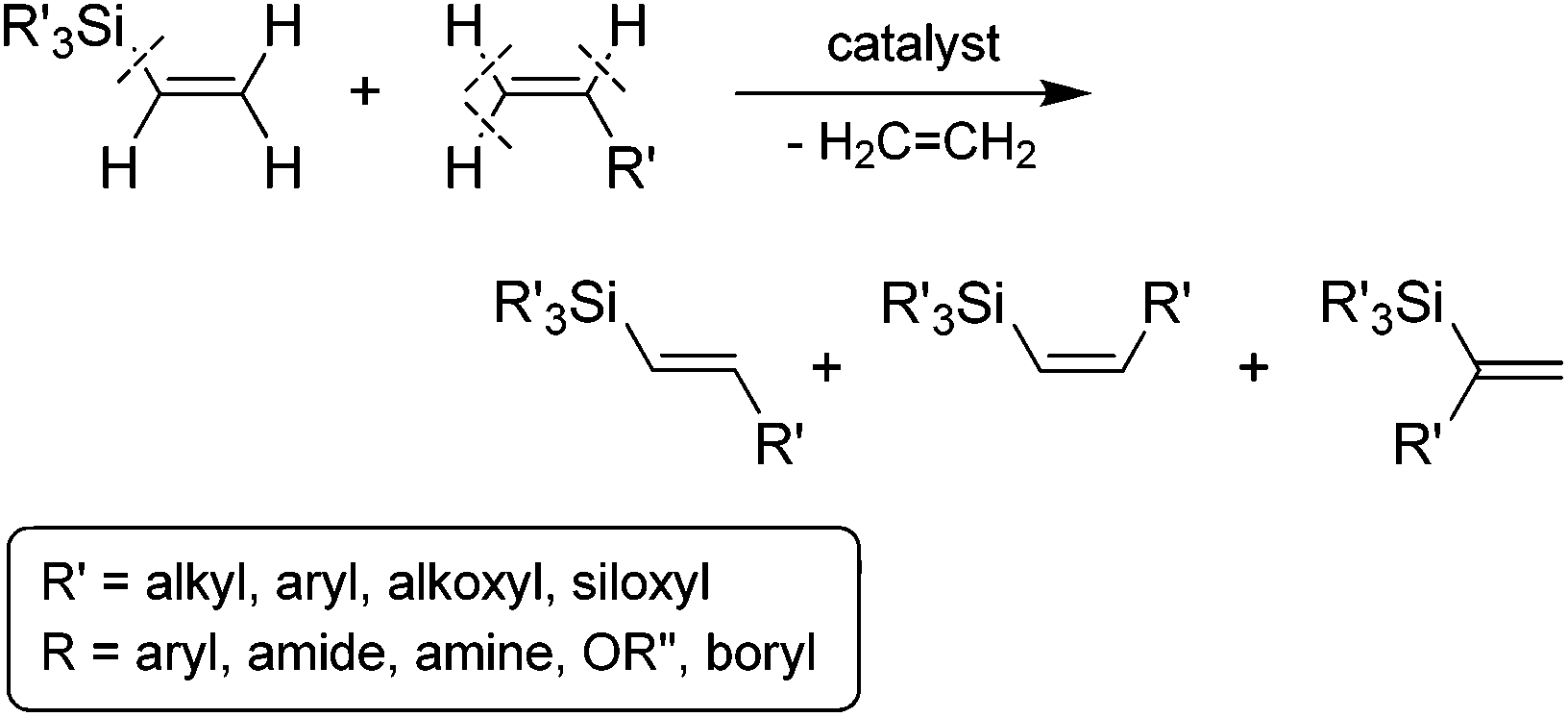 | ||
| Scheme 1 Silylative coupling of olefins with vinylsilanes. | ||
The silylative coupling was discovered by professor Bogdan Marciniec in 1984.11 The mechanism of the process proposed for the Ru-complexes by Wakatsuki12 and corrected by the Marciniec group,13 and for other metal complexes such as Rh,14 Co15 and Ir16 proceeds via the insertion of vinylsilane into the M–H bond and β-Si transfer to the metal with the elimination of ethylene to generate M–Si species followed by the insertion of alkene and β-H transfer to the metal with the elimination of substituted vinylsilane (Scheme 2).
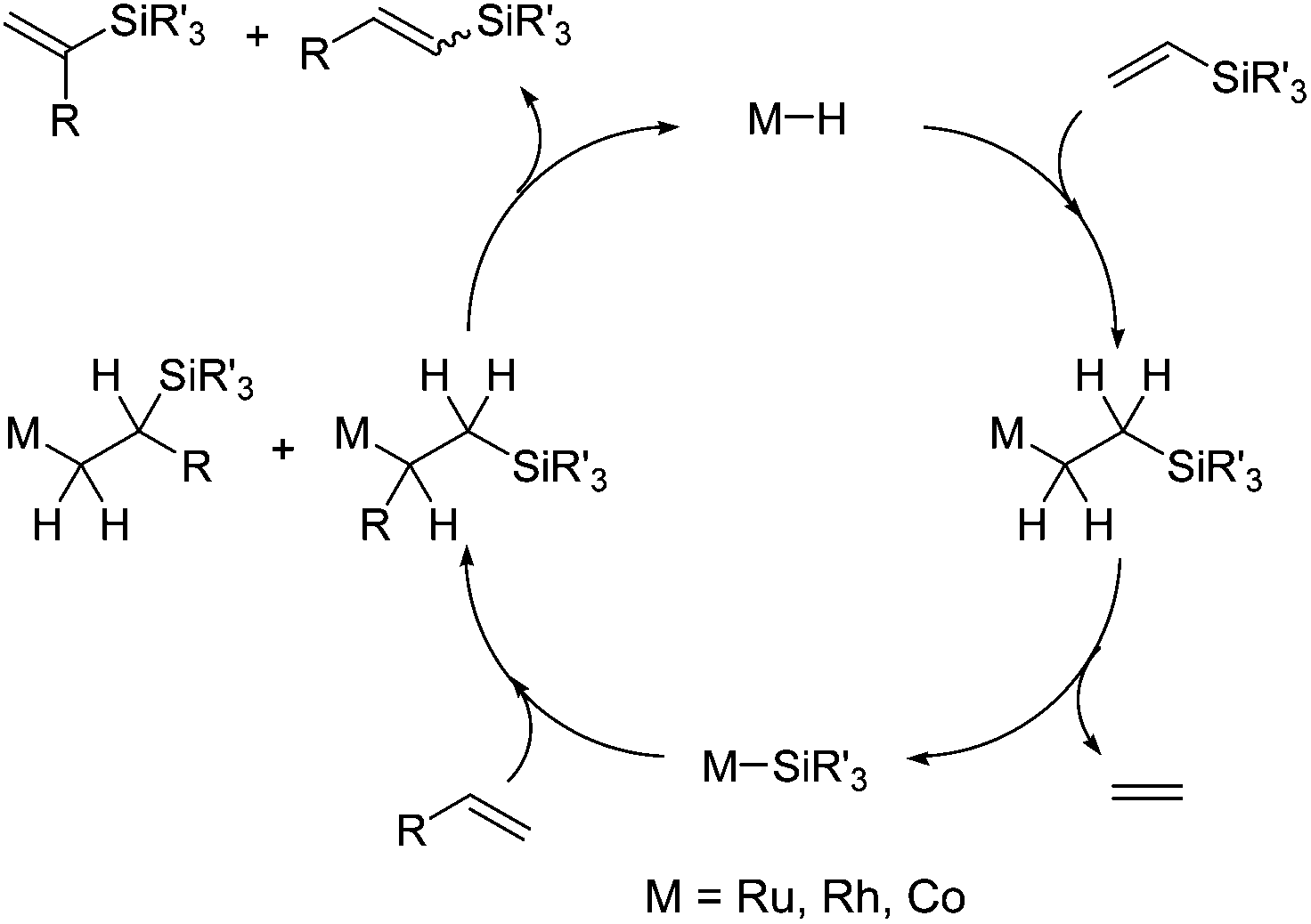 | ||
| Scheme 2 Silylative coupling mechanism. | ||
Intensive research work by the Poznan group from the beginning of 1990s has brought about the development of selective methods of synthesis of β-functionalized vinyl-substituted silicon compounds by catalytic silylative coupling reaction (mainly in the presence of ruthenium and rhodium complexes) of a wide gamut of olefins, e.g. substituted styrenes,17N-vinylamides,18N-vinylcarbazole19 and vinyl ethers20 with alkyl-, aryl- and alkoxy-substituted vinylsilanes and vinylsiloxanes. The selectivity of this process depends on the structure of the substrate and the catalyst used. The most active and selective catalysts for this process are ruthenium(II)-hydride complexes containing phosphines as ligands i.e. [Ru(H)(Cl)(CO)(PPh3)3] and [Ru(H)(Cl)(CO)(PCy3)2]. The reaction proceeds most effectively and selectively with functionalized olefins containing aryl and nitrogen-containing groups (e.g. substituted styrenes, N-vinylamides, N-vinylcarbazole). Reactions involving terminal 1-alkenes such as 1-hexene in the presence of Ru complexes occur with lower E/Z selectivity and are accompanied by competitive isomerization of olefins. Silylative coupling in the presence of ruthenium complexes has also been successfully applied to the functionalization of multivinyl-substituted organosilicon compounds such as cyclosiloxanes and cyclosilazanes,21,22 silsesquioxanes and spherosilicates23,24 as well as to the modification of vinyl-substituted polysiloxanes.25 Functionalized polyhedral silsesquioxanes (POSS) and spherosilicates have attracted widespread attention as precursors and components of a variety of inorganic/organic hybrid materials. The vinyl-functionalized silsesquioxanes are of particular interest as the vinyl substituents are reactive in various transformations leading to hybrid materials, for example, chromophore–POSS systems which recently have been studied for OLEDs. Reactivities of divinyl-substituted organosilicon compounds in the competitive processes of catalytic polycondensation26–29 and intramolecular cyclization30–34 by silylative coupling in the presence of ruthenium(II) or rhodium(I) complexes have also been studied.
The silylative coupling, in combination with subsequent desilylation reactions such as Hiyama cross-coupling and halodesilylation, appears to be a valuable step to provide functionalized unsaturated organic compounds.35 The article highlights the recent developments in the sequential synthetic strategies including ruthenium-catalyzed silylative coupling followed by desilylative cross-coupling, acylation and halogenation, leading to stereodefined organic derivatives containing arylene–vinylene units which are widely applied as fine chemicals, functional materials or unsaturated building blocks in organic synthesis.
2. Results and discussion
2.1 Synthesis and applications of (E)-alkenyl halides
The combination of the ruthenium-catalyzed silylative coupling with electrophilic halodesilylation reaction has been used for the stereoselective preparation of synthetically useful (E)-alkenyl halides from terminal olefins.36–39 The study has been concentrated on the search for new direct and selective methods for the syntheses of functionalized (E)-alkenyl halides from terminal alkenes with the use of certain products of sequential reactions in selected catalytic processes (e.g. Suzuki–Miyaura and Sonogashira coupling reactions) in order to obtain more complex functionalized organic compounds containing π-conjugated systems of double or triple bonds.[Ru(H)(Cl)(CO)(PPh3)3]-catalyzed E-selective silylative coupling of styrenes with vinyltrimethylsilane followed by N-iodosuccinimide-mediated iododesilylation has been reported as a valuable synthetic method for the one-pot conversion of easily available and relatively inexpensive styrenes into (E)-β-aryl vinyl iodides,36 which are widely applied as useful building blocks in transition metal-catalyzed organic transformations and natural product synthesis. Both reactions are not air-sensitive and can be performed with commercially available reagents and solvents without further purification. Under the optimal conditions, substituted styrenes bearing functional groups such as –Me, –Ph, –OMe, –Cl, –Br and –F, reacted successfully to give the (E)-β-aryl vinyl iodides in high yields via the corresponding (E)-styrylsilane intermediates, irrespective of the substituent electronic character and position on the aromatic ring (Scheme 3). The application of the one-pot ruthenium-catalyzed silylative coupling/N-bromosuccinimide-mediated bromodesilylation strategy for the synthesis of (E)-β-aryl vinyl bromides has also been reported (Scheme 3).36 Using this procedure, (E)-β-aryl vinyl bromides have been prepared from styrenes containing both electron-donating and electron-withdrawing groups with over 98% E-selectivity and high yield. The above sequential reactions have been the first among the literature examples of one-pot halogenation of alkenes leading selectively to (E)-alkenyl halides.
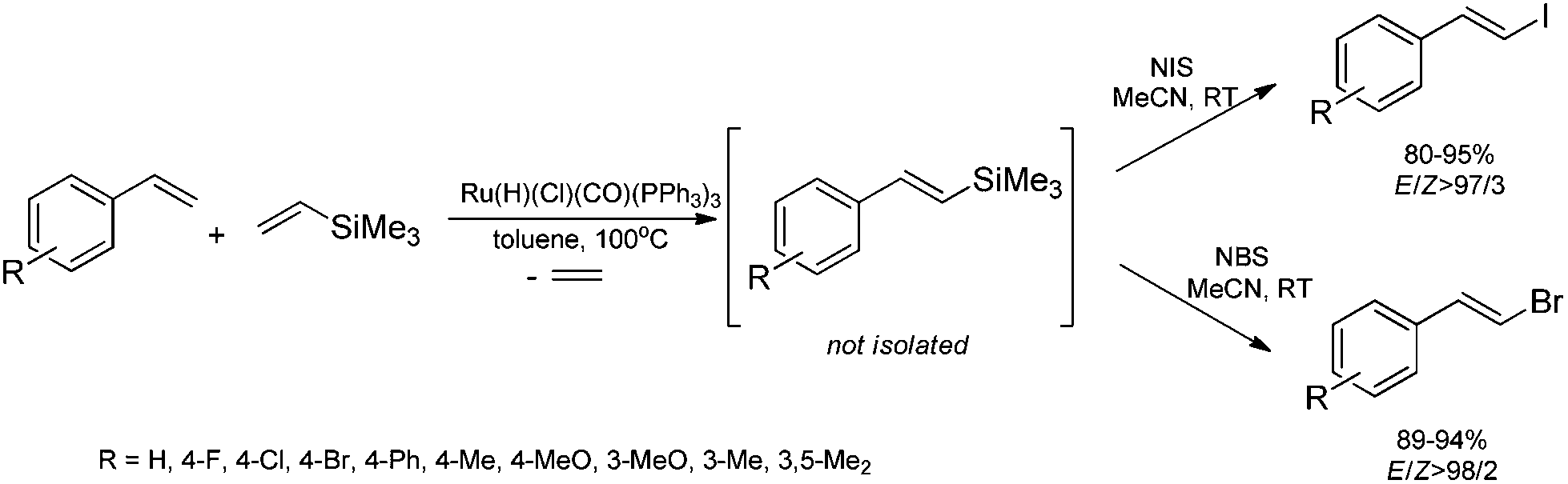 | ||
| Scheme 3 Synthesis of (E)-β-aryl vinyl halides. | ||
The discovery of the new method for the synthesis of (E)-styryl halides from styrenes has encouraged the search for other substrates of sequential reactions of silylative coupling and halodesilylation. Investigation of the sequential silylative coupling and iododesilylation of N-vinylcarbazole has brought about a method for the synthesis of (E)-9-(2-iodovinyl)-9H-carbazole that has proved to be a valuable reagent in the sequential reactions of carbon–carbon bond coupling.37 The silylative coupling of N-vinylcarbazole with vinyltrimethylsilane has been performed in the presence of the [Ru(H)(Cl)(CO)(PCy3)2] complex, while the iododesilylation of (E)-N-2-(trimethylsilyl)vinylcarbazole has been the most effective in the presence of N-iodosuccinimide (NIS) or molecular iodine. The conditions permitting effective realization of silylative coupling and iododesilylation in one-pot were established to give (E)-9-(2-iodovinyl)-9H-carbazole in 76% yield and high selectivity (Scheme 4). It has been first described in the literature example of an effective stereospecific halodesilylation of functionalized nitrogen-containing vinylsilanes.
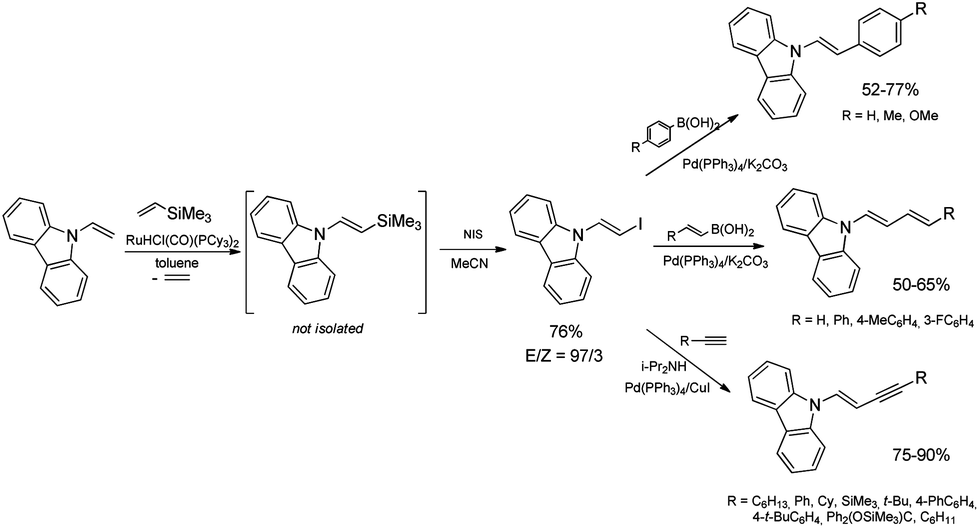 | ||
| Scheme 4 Synthesis and application of (E)-9-(2-iodovinyl)-9H-carbazole. | ||
The use of (E)-9-(2-iodovinyl)-9H-carbazole as a building block in catalytic coupling reactions permitted obtaining a series of new unsaturated organic compounds containing substituted (E)-N-vinylcarbazole groups having many interesting potential applications.37 In particular, (E)-9-(2-iodovinyl)-9H-carbazole has been shown to undergo palladium catalyzed Sonogashira and Suzuki–Miyaura coupling reactions to yield carbazole-containing (E)-but-1-en-3-ynes and substituted (E,E)-9-(buta-1,3-dien-1-yl)carbazoles in good yields (Scheme 4). N-Substituted π-conjugated carbazole derivatives have found many applications in organic electronics, nonlinear optics, chemistry of sensors, and are building blocks of a large group of pharmaceuticals an alkaloids.
Ruthenium-catalyzed E-selective silylative coupling of easily available N-vinylamides or imides with vinyltrimethylsilane followed by halodesilylation has been found to be a valuable and general synthetic method for the conversion of N-vinylamides into (E)-N-(2-halovinyl)amides or their imide analogues.38,39 A series of (E)-β-iodoenamides and (E)-β-iodoenimides has been easily obtained from N-vinyl derivatives (N-vinylamides and N-vinylimides) by stereoselective [Ru(H)(Cl)(CO)(PCy3)2]-catalyzed silylative coupling with vinyltrimethylsilane and subsequent stereospecific silicon–iodine exchange mediated by N-iodosuccinimide or molecular iodine under mild conditions (Scheme 5).38,39
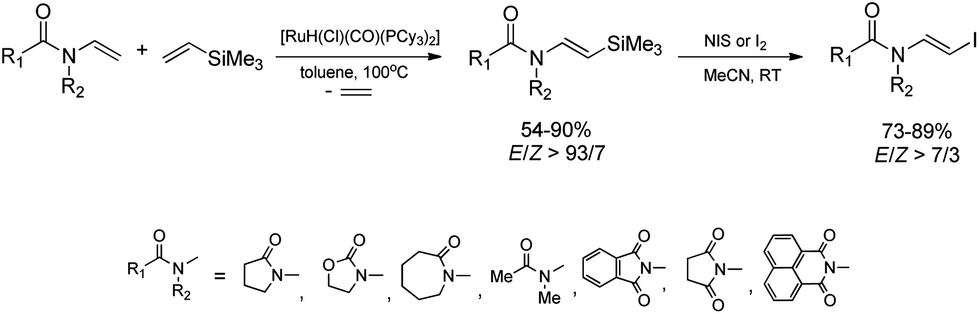 | ||
| Scheme 5 Synthesis of (E)-β-iodoenamides and (E)-β-iodoenimides. | ||
Bromodesilylation of (E)-β-silylenimides affords (E)-β-bromoenimides, while the analogous reactions involving (E)-β-silylenamides led to decomposition of substrates.38 The ruthenium-catalyzed silylation/halodesilylation sequence can be also performed in a one-pot procedure without the isolation of silylenamide derivatives to give (E)-β-haloenamides in moderate to good yields (68–88%).38
β-Haloenamides are synthetically versatile variants of enamides, offering rapid access to β-metalated enamides that can serve as acyl anion equivalents or can be involved in transition metal-catalyzed reactions. In addition, stereodefined haloenamides are excellent candidates for the use as coupling partners in Pd-catalyzed cross-coupling reactions allowing regio- and stereoselective construction of complex enamide and dienamide derivatives.40 In this context, (E)-N-2-iodovinylphthalimide has been successfully applied as a new building block in catalytic reactions involving carbon–carbon bond formation i.e. Suzuki–Miyaura and Sonogashira coupling reactions catalyzed by palladium complexes (Scheme 6).39 The catalytic Suzuki–Miyaura coupling reaction allowed selective synthesis of a series of (E)-N-(2-arylvinyl)phthalimides (reactions with aryl boronic acids) and (E,E)-N-(buta-1,3-dienyl)phthalimides (reactions with (E)-arylvinyl boronic acids or esters) in good yields (60–90%). Sonogashira coupling of (E)-N-iodovinylphthalimide with terminal alkynes containing aryl, (cyclo)alkyl and silyl substituents taking place in the presence of a catalytic system PdCl2(PPh3)2/CuI gave selectively (E)-N-(but-1-en-3-yn-1-yl)phthalimides with good yields (60–84%) and selectivity.39
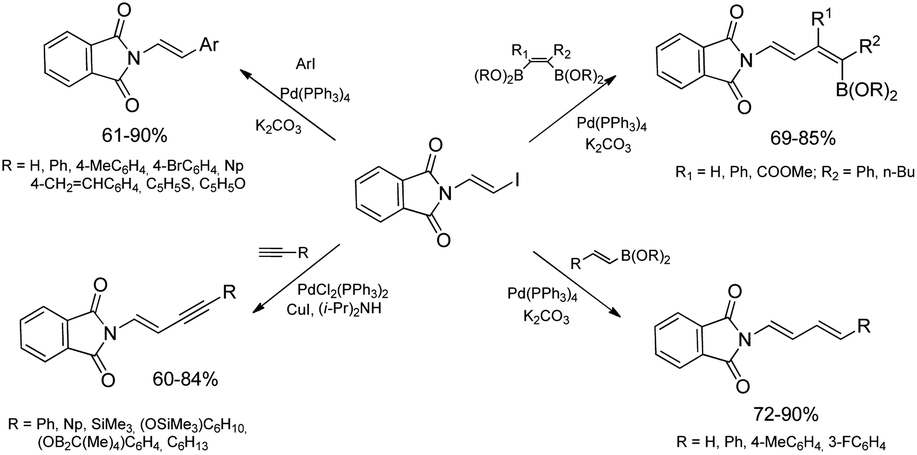 | ||
| Scheme 6 Synthetic application of (E)-N-iodovinylphthalimide. | ||
N-Substituted phthalimides are key structural units in a variety of biologically important compounds. Several fungicides, metabolic drugs and functional materials contain alkenyl-substituted phthalimides as key structural elements.41
2.2 Synthesis of (E)-α,β-unsaturated ketones
One recently-reported method for the synthesis of (E)-styryl ketones is based on one-pot sequential silylative coupling of substituted styrenes with vinyltrimethylsilane in the presence of [Ru(H)(Cl)(CO)(PPh3)3] and desilylative acylation of (E)-trimethylsilylstyrenes with acid anhydrides catalyzed by [Rh(Cl)(CO)2]2 (Narasaka coupling).42 Under the optimal conditions, substituted styrenes bearing both electron withdrawing and electron donating functional groups, reacted successfully to give the (E)-styryl ketones in high yields via the corresponding (E)-styrylsilane intermediates, irrespective of the substituent electronic character and position on the aromatic ring (Scheme 7). Anhydrides of aliphatic, aromatic and unsaturated carboxylic acids can be applied in this process.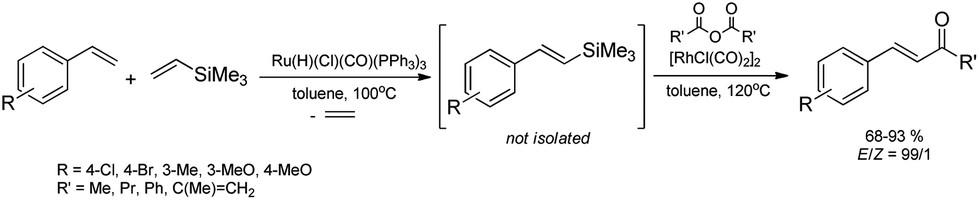 | ||
| Scheme 7 Synthesis of (E)-styryl ketones. | ||
A combination of silylative homo-coupling of vinylsilanes (catalyzed by [Ru(H)(Cl)(CO)(PCy3)2] or [Ru(Cl)2(CO)3]2). and rhodium-catalyzed desilylative acylation of (E)-1,2-bis(silyl)ethenes with acid anhydrides leads to the selective formation of (E)-β-silylvinyl ketones with the retention of olefin configuration (Scheme 8).43 Products of sequential reactions of silylative coupling and acylation, i.e. (E)-styryl and (E)-β-silylvinyl ketones are valuable reagents in organic synthesis (nucleophilic addition, Michael condensation, Diels–Alder process, etc.).44
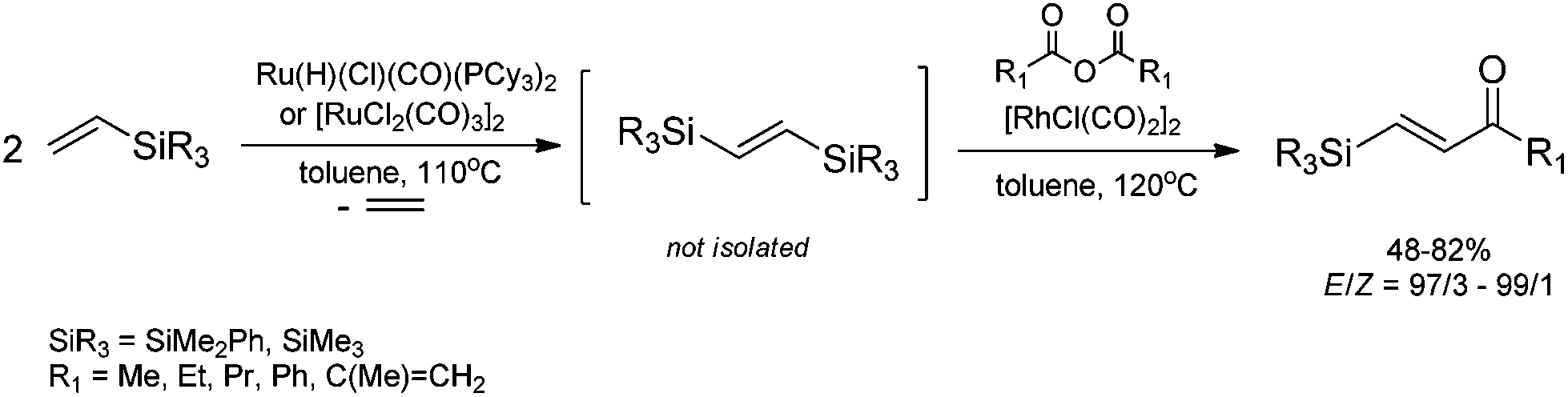 | ||
| Scheme 8 Synthesis of (E)-β-silylvinyl ketones. | ||
2.3 Synthesis of aryl–vinyl derivatives
Functionalization of alkoxy- or siloxy-substituted vinylsilanes via silylative coupling with olefins can be successfully combined with palladium-catalyzed Hiyama coupling to yield a wide variety of aryl–vinyl derivatives. The sequential silylative coupling–Hiyama coupling strategy has been successfully used for stereoselective synthesis of (E)-stilbenes,45 (E)-9-styrylcarbazoles,46 bis[(E)-styryl]arenes,47 stilbenoid dendrimers48 and arylene-(E)-vinylene polymers.49 Stilbenoid compounds with extended π-electron systems show interesting photophysical and photochemical properties and are therefore suitable for various applications in materials science.Sequential [Ru(H)(Cl)(CO)(PPh3)3]-catalyzed silylative coupling of substituted styrenes with alkoxy- or siloxy-substituted vinylsilanes and [Pd2(dba)3]-catalyzed Hiyama coupling of such obtained (E)-styrylsilanes with iodoarenes has been applied in stereoselective synthesis of substituted (E)-stilbenes (Scheme 9).45 Substituted (E)-4-chlorostilbenes have also been successfully obtained in one-pot procedures without isolation of organosilicon intermediates.45 Application of 1,4-diiodobenzene or other aryl dihalides as coupling reagents in sequential one-pot silylative coupling–Hiyama coupling reaction led to bis[(E)-4-halostyryl]arenes with good yield and high stereoselectivity (Scheme 9).47 Multistyryl-substituted arenes with polyconjugated branches can be also synthesized via one-pot silylative coupling/Hiyama coupling protocol (Scheme 9). Ruthenium hydride-catalyzed silylative coupling of 1,3-divinyltetramethyldisiloxane with 4-halostyrenes followed by palladium-catalyzed cross-coupling of distyryl-siloxanes with tri- or tetrahaloarenes led to 1,3,5-tris((E)-4-chlorostyryl)benzene or 1,2,4,5-tetrakis((E)-4-halostyryl)benzenes respectively, in high yields (72–95%) and stereoselectivity (>99%).48
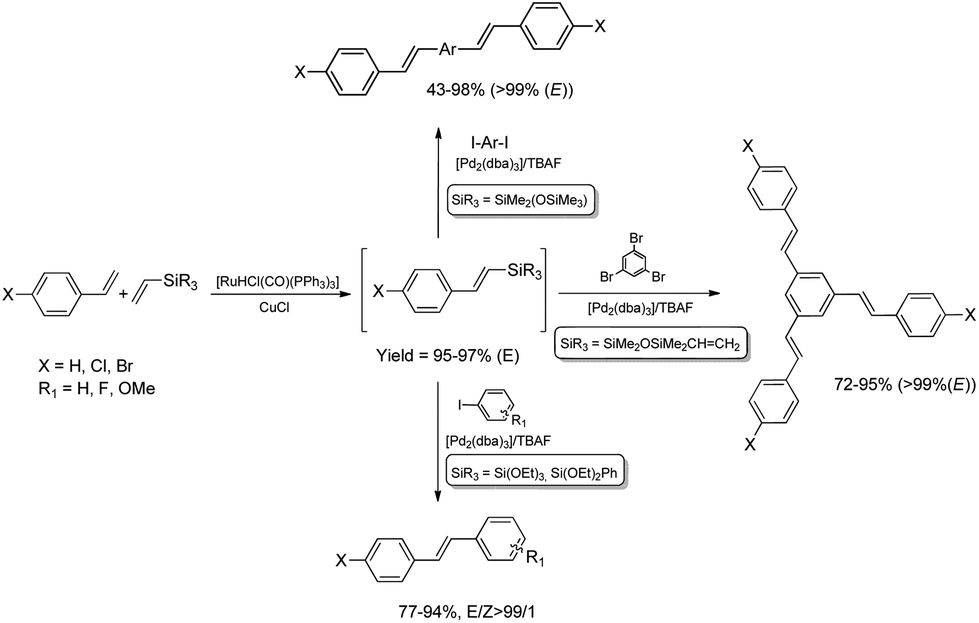 | ||
| Scheme 9 Synthesis of (E)-stilbenes, bis[(E)-styryl]arenes and stilbenoid dendrimers. | ||
A highly stereoselective, one-pot synthetic methodology for the construction of (E)-poly(arylenevinylene)s based on sequential silylative homo-coupling–Hiyama cross-coupling of isopropoxy-dimethylvinylsilane with aryl dihalides has been reported (Scheme 10). A double bond of isomeric bis(silyl)ethene intermediates can thus be very efficiently grafted into the aromatic structure, offering the potential of constructing (E)-stilbene and polyene derivatives.49
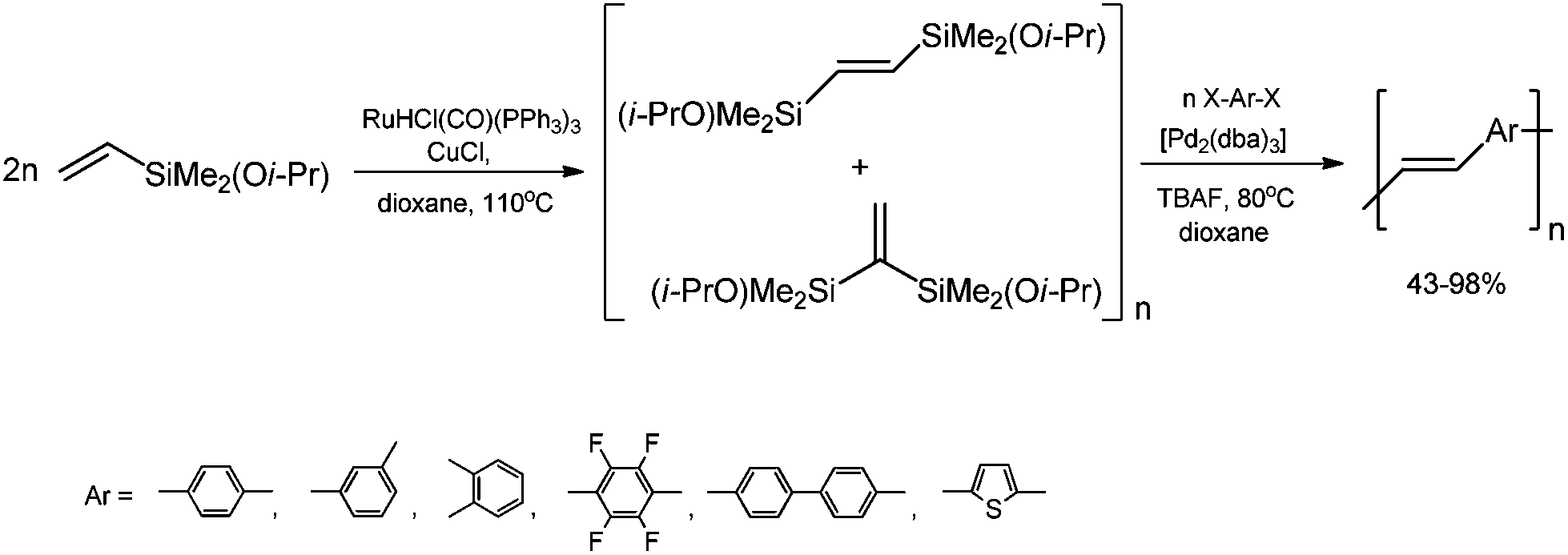 | ||
| Scheme 10 Synthesis of (E)-poly(arylenevinylene)s. | ||
A combination of the ruthenium-catalyzed silylative coupling and palladium-catalyzed Hiyama coupling processes, using vinylcyclosiloxanes as supporting reagents leads to stereoselective synthesis of (E)-styryl derivatives with high yield. Silylative coupling of tetravinylcyclotetrasiloxane with 4-bromostyrene followed by cross-coupling with p-substituted aryl iodides yielded unsymmetrical (E)-4-bromostilbenes (Scheme 11).50
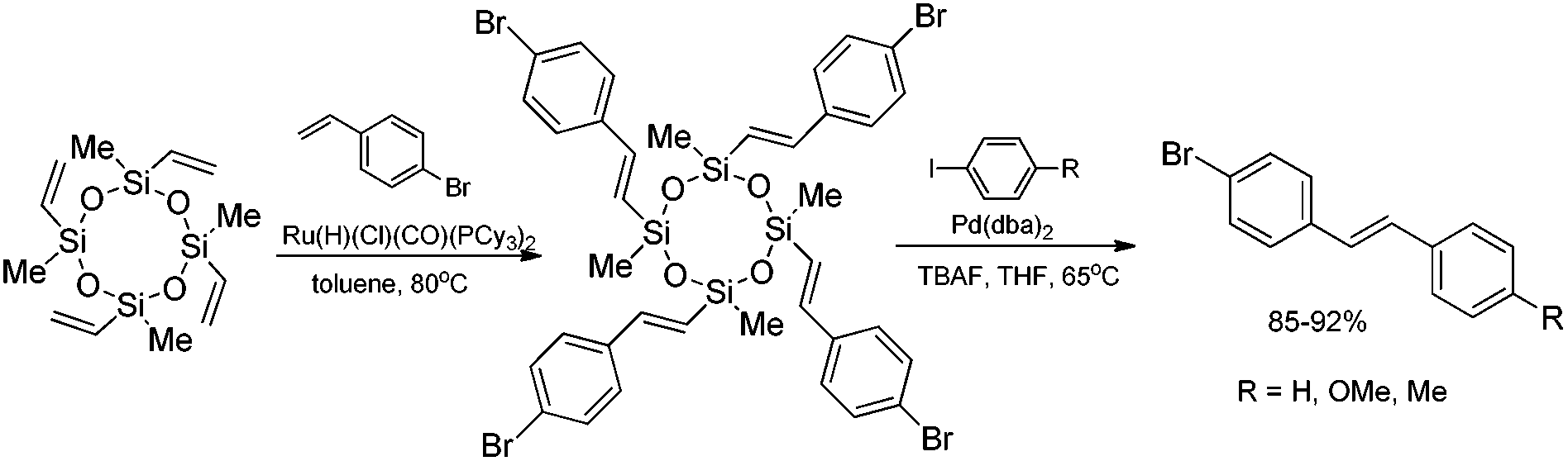 | ||
| Scheme 11 Synthesis of (E)-4-bromostilbenes. | ||
Hiyama coupling of the β-n-butoxyvinyl-substituted cyclotrisiloxane (synthesized by its silylative coupling with vinyl n-butyl ether) with iodobenzene has been successfully applied for the synthesis of β-n-butoxystyrenes (Scheme 12) which are difficult to synthesize using other methods.21
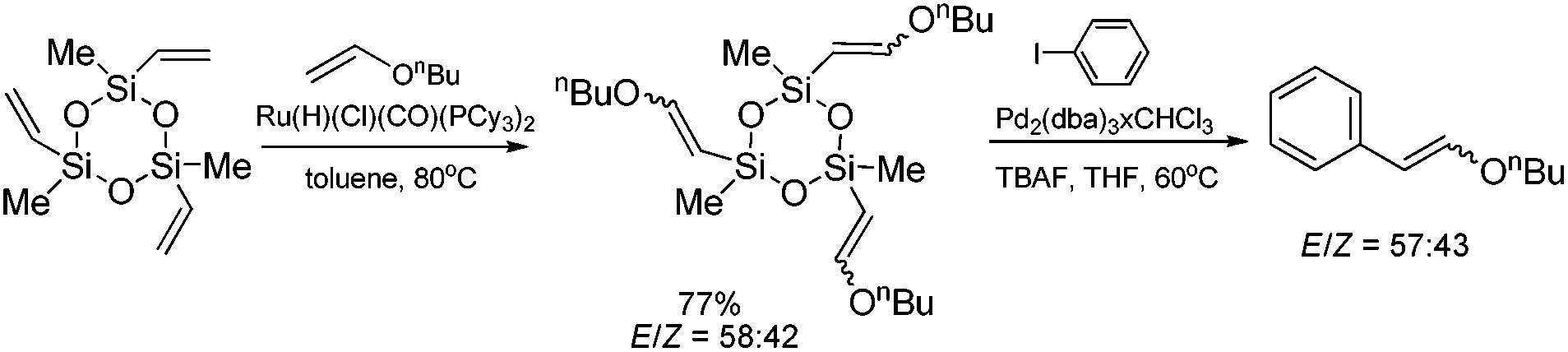 | ||
| Scheme 12 Synthesis of β-n-butoxystyrenes. | ||
Stereoselective β-arylation of N-vinylcarbazole can be performed via sequential silylative coupling/Hiyama coupling. The (E)-9-[2-(triethoxysilyl)ethenyl]-9H-carbazole, obtained via silylative-coupling undergoes the cross-coupling (Hiyama coupling) reactions in the presence of [Pd2(dba)3] catalyst, giving exclusively, N-(E)-arylvinylcarbazole derivatives (Scheme 13).19,46
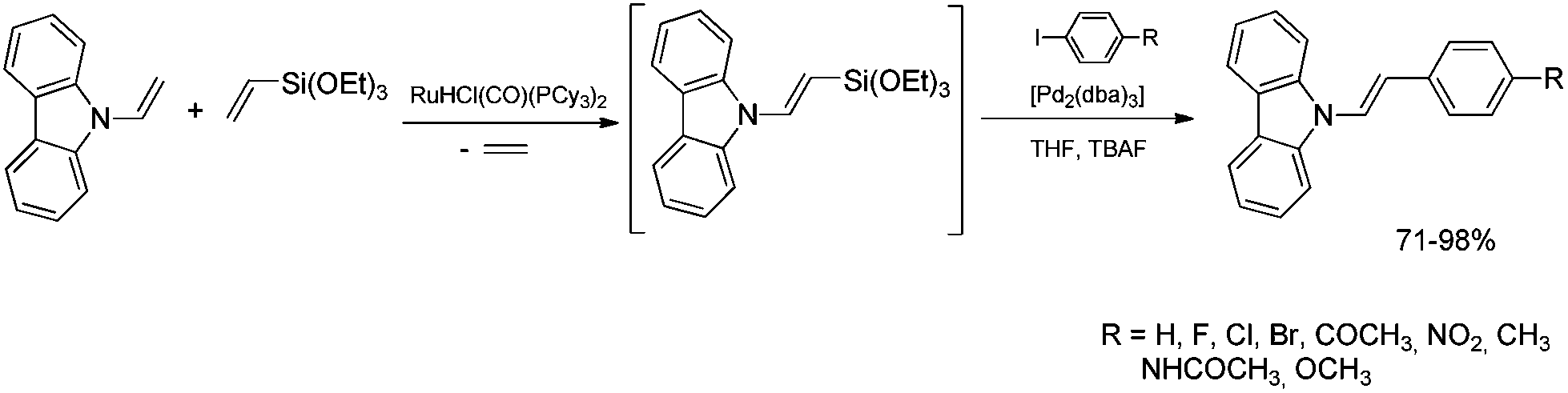 | ||
| Scheme 13 Synthesis of (E)-9-styrylcarbazoles. | ||
A combination of the ruthenium-catalyzed silylative coupling of p-substituted styrenes with vinyltrimethylsilane and InCl3-catalyzed coupling of styrylsilanes with benzyl alcohol derivatives51 leads to the synthesis of disubstituted alkenes containing aromatic groups with a moderate yield and selectivity (Scheme 14). The application of 1,2-dichloroethane as a solvent enabled us to carry out both processes in a one-pot fashion.
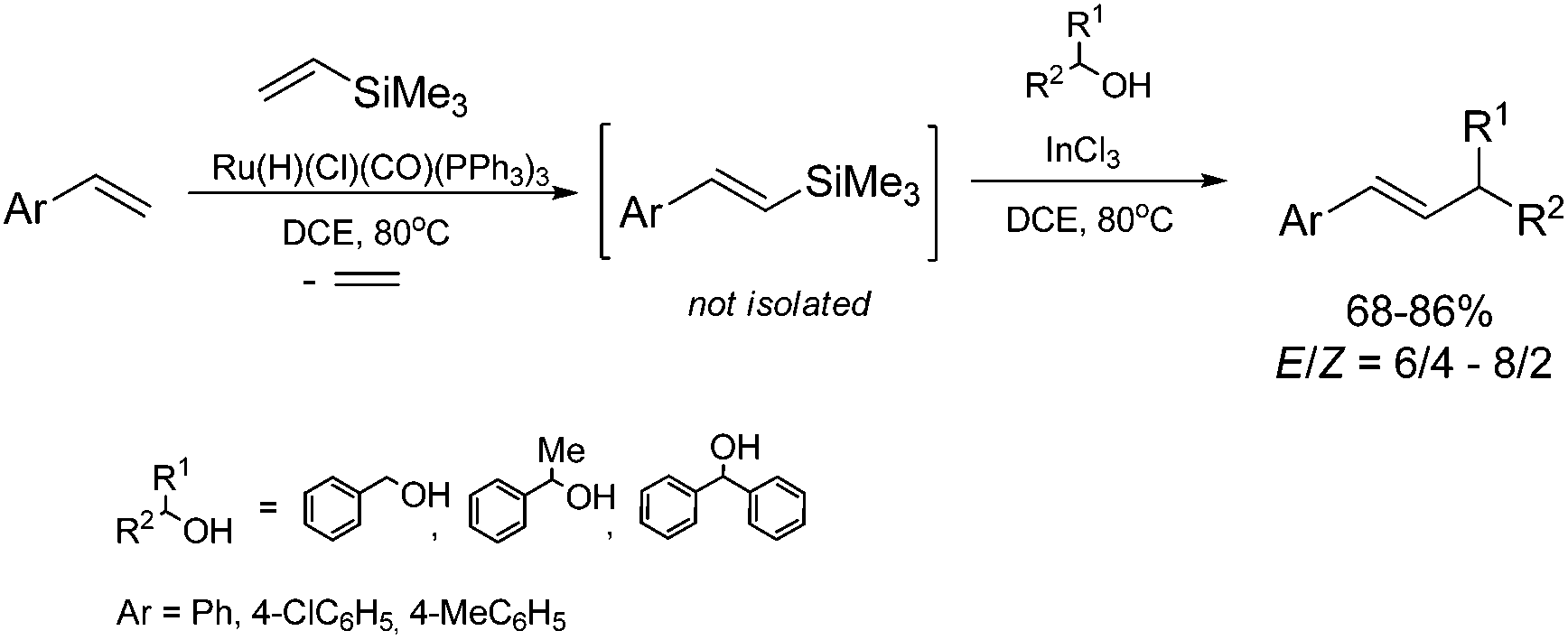 | ||
| Scheme 14 Synthesis of disubstituted alkenes containing aromatic groups. | ||
2.4 Synthesis of trisubstituted olefins
The unique feature of the silylative coupling reaction, distinguishing this process from cross-metathesis is the formation of 1,1-bis(silyl)ethene fragment under given conditions. Although, 1,1-bis(silyl)ethenes cannot be selectively obtained through direct silylative homo-coupling (disproportionation) of vinylsilanes, new efficient protocols for their synthesis using ruthenium-catalyzed silylative coupling exo-cyclization of divinyl-substituted monomers, followed by the reaction with Grignard reagents or alcohols have been reported.31,52 The resulting 1,1-bis(silyl)ethenes have been efficiently coupled in the presence of silver nitrate and palladium acetate with aryl iodides to give the corresponding 1,1-bis(silyl)-2-arylethenes with high yield. Two complementary three-step approaches based on sequential procedures have been reported, in which the first step is either one-pot silylative coupling cyclization or Grignard reagent treatment, the second is Heck coupling or silylative coupling cyclization, and the last is Heck coupling or Grignard reagent treatment (Scheme 15).53,54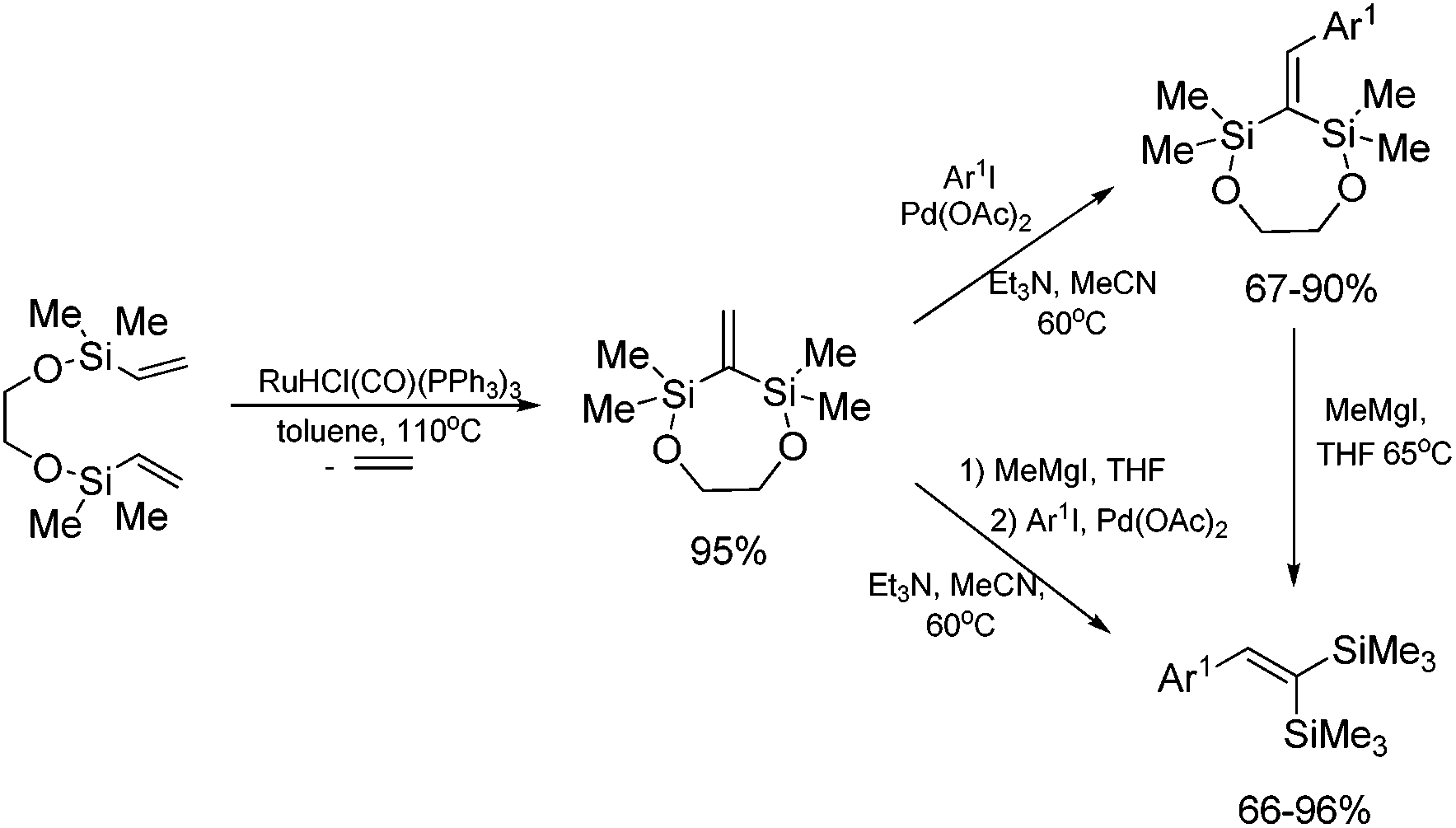 | ||
| Scheme 15 Synthetic routes towards 1,1-bis(silyl)-2-arylethenes. | ||
1,1-Bis(silyl)-2-arylethenes have been found as attractive precursors for electrophilic substitution to give functionalized trisubstituted olefins. Bromodesilylation of the resulting 1,1-bis(trimethylsilyl)-2-arylethenes in the presence of N-bromosuccinimide (NBS) led to the discovery of a new method for the synthesis of 1,1-dibromo-2-arylethenes.55 The selective double bromodesilylation of 1,1-bis(trimethylsilyl)-2-arylethenes takes place under mild conditions for a wide gamut of organosilicon substrates containing electron acceptor or donor substituents in the aromatic ring, and is an attractive alternative to the hitherto used methods of synthesis of the above compounds (Scheme 16). The products of bromodesilylation are important precursors in the syntheses of 1-bromo-1-alkynes, internal and terminal alkynes, aryl–methyl ketones and (E)- or (Z)-1-bromoalkenes.55 Geminal dibromoalkenes are also used as reagents in many coupling reactions catalyzed with palladium complexes leading to a wide gamut of functionalized alkenes and alkynes, e.g. (Z)-1-aryl-1-bromo-1-alkenes, (Z)-2-bromo-1,3-butadienes, triarylethenes, 2-aryl-1,1-dialkynylethenes, (E)-3-methyl-3-en-1-ynes, and symmetric or asymmetric 1,3-diynes.56
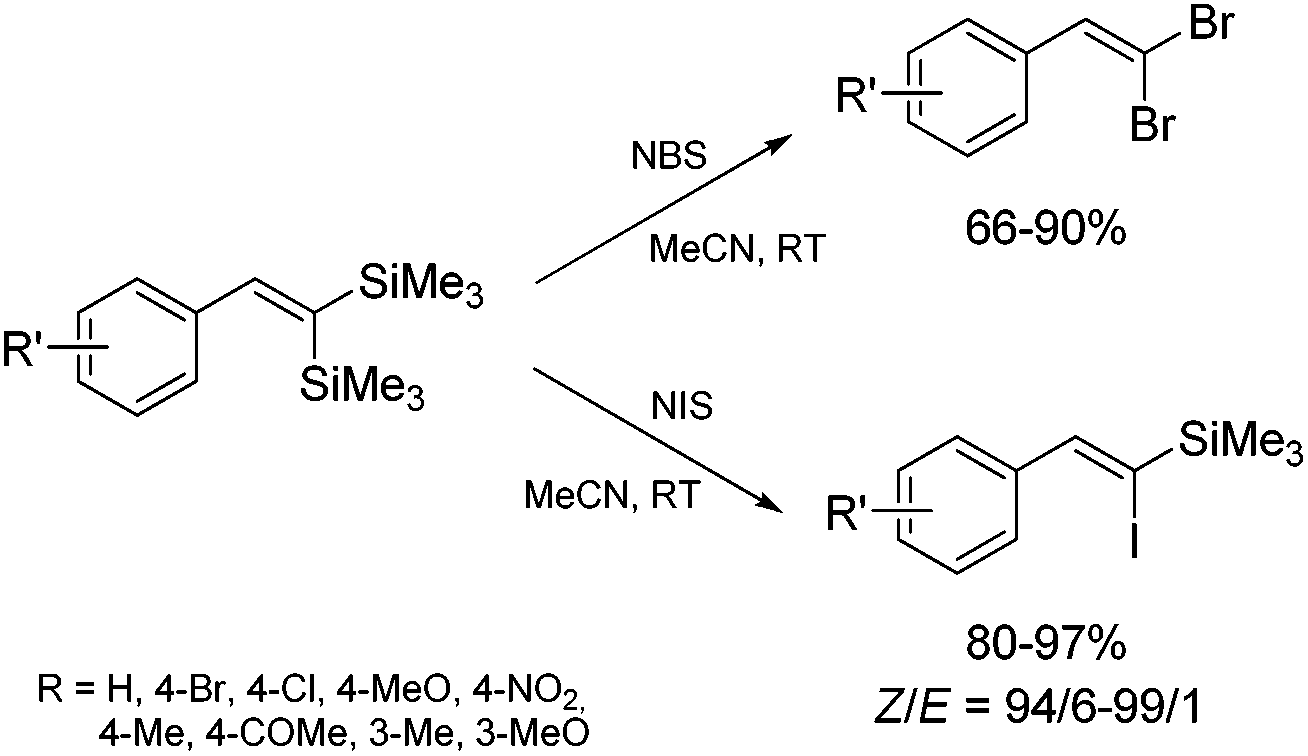 | ||
| Scheme 16 Halodesilylation of 1,1-bis(trimethylsilyl)-2-arylethenes. | ||
The studies of iododesilylation of 1,1-bis(trimethylsilyl)-2-arylethenes in the presence of iodinating agents (N-iodosuccinimide and iodopyridinium tetrafluoroborate) have shown that the process takes place under mild conditions with a high stereoselectivity to yield mono-substitution products (despite the use of excess of iodinating agents) – (Z)-1-iodo-1-silyl-2-arylethenes (Scheme 16).57
Stereodefined α-iodovinylsilanes can serve as precursors for the stereoselective preparation of 1-lithio-1-silylalkenes, trisubstituted olefins, secondary amides as well as a variety of important organosilicon intermediates such as silyl-substituted alkenes, (Z,E)-1,3- and 1,4-dienes or α-bromovinylsilanes.58
2,2,4,4-Tetramethyl-1,5-dioxa-3-methylene-2,4-disilacycloheptane (easily obtained via ruthenium-catalyzed silylative coupling cyclization of dimethylvinylsilyl-substituted ethylene glycol) in the reaction with aryl iodides, under standard cross-coupling conditions forms exclusively (instead of expected 1,1-diarylethenes) cine-substitution products with perfect stereoselectivity and almost quantitative yield.49 A double bond of bis(silyl)ethene can thus be efficiently grafted into the aromatic structure, offering the potential of constructing symmetrical (E)-stilbene derivatives. The extension of this silicon-assisted protocol for aryl dihalides has led to stereoselective synthesis of (E)-poly(arylenevinylene)s (Scheme 17).49
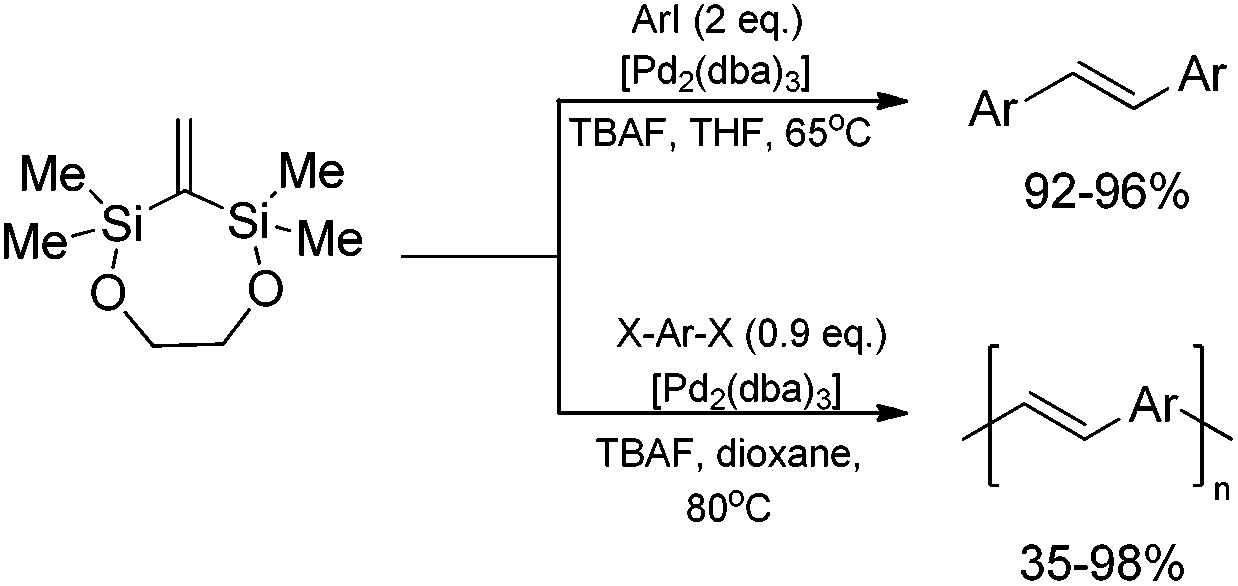 | ||
| Scheme 17 Synthesis of symmetrical (E)-stilbene derivatives and (E)-poly(arylenevinylene)s using cyclic 1,1-bis(silyl)ethene as a platform. | ||
Cyclic 1,1-bis(silyl)-2-arylethenes (obtained via Heck arylation of 2,2,4,4-tetramethyl-1,5-dioxa-3-methylene-2,4-disilacycloheptane) have been applied as precursors for the palladium-catalyzed synthesis of unsymmetrically substituted (E)-stilbenes and (E,E)-1,4-diarylbuta-1,3-dienes (Scheme 18).59
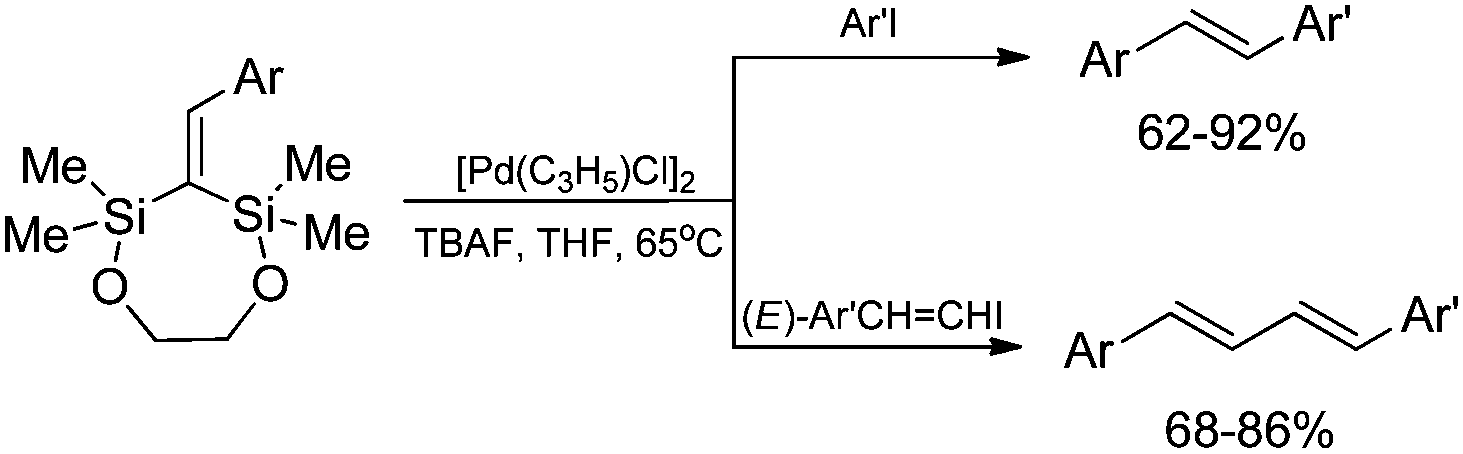 | ||
| Scheme 18 Synthetic application of cyclic 1,1-bis(silyl)alkenes. | ||
3. Conclusions
The silylative coupling of olefins with vinyl-substituted silicon compounds, in combination with subsequent desilylation reactions such as Hiyama cross-coupling, desilylative acylation and halodesilylation, appears to be a valuable step to provide functionalized unsaturated organic compounds.The main idea behind the methodologies developed is the use, at the first stage, of silylative coupling of olefins with vinylsilanes (in inter- or intra-molecular version) in the presence of ruthenium complexes and then the use of the organosilicon products (β-substituted vinylsilanes or isomeric bis(silyl)ethenes) as intermediates in the sequential stoichiometric (bromo- and iododesilylation) or catalytic (acylation and Hiyama coupling) desilylation reactions.
The unique feature of these methodologies is that the stereochemistry of the processes can be controlled during the initial step as the subsequent desilylation proceeds with the retention of the configuration at the carbon atom and allows the formation of stereodefined products. The outcome of the research presented is the development of new alternative methods for the syntheses of functionalized di- and tri-substituted alkenes for potential use in materials chemistry (π-conjugated derivatives of stilbenes, phthalimides, carbazoles) and in organic synthesis (new precursors of coupling reactions, alkenyl halides with imide and carbazole groups, alkenyl dihalides etc.) leading to the application of selected organic products as new building blocks in the synthesis of a wide gamut of substituted dienes and enynes.
Acknowledgements
Financial support from the National Science Centre (Poland); grant no. 2011/03/B/ST5/01034 is gratefully acknowledged.Notes and references
- Main Group Metals in Organic Synthesis, ed. H. Yamamoto and K. Oshima, Wiley-VCH, Weinheim, 2004 Search PubMed.
- Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, 2004 Search PubMed.
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (b) A. Molnar, Chem. Rev., 2011, 111, 2251 CrossRef CAS PubMed; (c) C. C. J. Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062 CrossRef PubMed.
- (a) P. Espinet and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 4704 CAS; (b) V. Farina, V. Krishnamurthy and W. J. Scott, The Stille Reaction, Wiley, New York, 1998 Search PubMed.
- S. Xu, H. Kamada, E. H. Kim, A. Oda and E.-i. Negishi, Pd-Catalyzed Cross-Coupling with Organometals Containing Zn, Al, Zr, and so on – The Negishi Coupling and Its Recent Advances, in Metal-Catalyzed Cross-Coupling Reactions and More, ed. A. de Meijere, S. Bräse and M. Oestreich, Wiley-VCH, Weinheim, Germany, 2014 Search PubMed.
- (a) Y. Nakao and T. Hiyama, Chem. Soc. Rev., 2011, 40, 4893 RSC; (b) H. F. Sore, W. R. J. D. Galloway and D. R. Spring, Chem. Soc. Rev., 2012, 41, 1845 RSC.
- I. Fleming, A. Barbero and D. Walter, Chem. Rev., 1997, 97, 2063 CrossRef CAS PubMed.
- M. J. Curtis-Long and Y. Aye, Chem. – Eur. J., 2009, 15, 5402 CrossRef CAS PubMed.
- M. A. Brook, Silicon in Organic, Organometallic and Polymer Chemistry, John Wiley & Sons, Chichester, 2000 Search PubMed.
- For review on silylative coupling of olefins with vinylsilicon compounds see: (a) B. Marciniec, Acc. Chem. Res., 2007, 40, 943 CrossRef CAS PubMed; (b) B. Marciniec, Coord. Chem. Rev., 2005, 249, 2374 CrossRef CAS PubMed.
- B. Marciniec and J. Guliński, J. Organomet. Chem., 1984, 266, C19 CrossRef CAS.
- Y. Wakatsuki, H. Yamazaki, N. Nakano and Y. Yamamoto, J. Chem. Soc., Chem. Commun., 1991, 703–704 RSC.
- (a) B. Marciniec and C. Pietraszuk, J. Chem. Soc., Chem. Commun., 1995, 2003 RSC; (b) B. Marciniec and C. Pietraszuk, Organometallics, 1997, 16, 4320 CrossRef CAS.
- B. Marciniec, E. Walczuk-Guściora and P. Błażejewska-Chadyniak, J. Mol. Catal., 2000, 160, 165 CrossRef CAS.
- B. Marciniec, I. Kownacki and D. Chadyniak, Inorg. Chem. Commun., 1999, 2, 581 CrossRef CAS.
- B. Marciniec, I. Kownacki and M. Kubicki, Organometallics, 2002, 21, 3263 CrossRef CAS.
- B. Marciniec and C. Pietraszuk, Organometallics, 1997, 16, 4320 CrossRef CAS.
- B. Marciniec, D. Chadyniak and S. Krompiec, Tetrahedron Lett., 2004, 45, 4065 CrossRef CAS PubMed.
- B. Marciniec, M. Majchrzak, W. Prukała, M. Kubicki and D. Chadyniak, J. Org. Chem., 2005, 70, 8550 CrossRef CAS PubMed.
- M. Kujawa and C. Pietraszuk, Organometallics, 2000, 19, 1677 CrossRef.
- Y. Itami, B. Marciniec and M. Kubicki, Organometallics, 2003, 22, 3717 CrossRef CAS.
- B. Marciniec, J. Waehner, P. Pawluc and M. Kubicki, J. Mol. Catal., 2007, 265, 25 CrossRef CAS PubMed.
- J. Waehner, B. Marciniec and P. Pawluć, Eur. J. Inorg. Chem., 2007, 2975 CrossRef CAS.
- (a) Y. Itami, B. Marciniec and M. Kubicki, Chem. – Eur. J., 2004, 10, 1239 CrossRef CAS PubMed; (b) P. Żak, C. Pietraszuk, B. Marciniec, G. Spólnik and W. Danikiewicz, Adv. Synth. Catal., 2009, 351, 2675 CrossRef; (c) P. Żak, B. Dudziec, M. Kubicki and B. Marciniec, Chem. – Eur. J., 2014, 20, 9387 CrossRef PubMed.
- P. Żak, M. Skrobanska, C. Pietraszuk and B. Marciniec, J. Organomet. Chem., 2009, 694, 1903 CrossRef PubMed.
- M. Majchrzak, Y. Itami, B. Marciniec and P. Pawluć, Tetrahedron Lett., 2000, 41, 10303 CrossRef CAS.
- M. Majchrzak, B. Marciniec and Y. Itami, Adv. Synth. Catal., 2005, 347, 1285 CrossRef CAS.
- B. Marciniec, E. Małecka and M. Ścibiorek, Macromolecules, 2003, 36, 5545 CrossRef CAS.
- M. Majchrzak, M. Ludwiczak, M. Bayda, B. Marciniak and B. Marciniec, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 127 CrossRef CAS.
- B. Marciniec and M. Lewandowski, Tetrahedron Lett., 1997, 38, 3777 CrossRef CAS.
- P. Pawluć, B. Marciniec, G. Hreczycho, B. Gaczewska and Y. Itami, J. Org. Chem., 2005, 70, 370 CrossRef PubMed.
- P. Pawluć, G. Hreczycho and B. Marciniec, Synlett, 2005, 1105 Search PubMed.
- P. Pawluć, B. Marciniec, B. Dudziec, G. Hreczycho and M. Kubicki, Synthesis, 2006, 3739 Search PubMed.
- T. Mise, Y. Takaguchi, T. Umemiya, S. Shimizu and Y. Wakatsuki, Chem. Commun., 1998, 699 RSC.
- P. Pawluc, W. Prukala and B. Marciniec, Eur. J. Org. Chem., 2010, 219 CrossRef CAS.
- P. Pawluc, G. Hreczycho, J. Szudkowska, M. Kubicki and B. Marciniec, Org. Lett., 2009, 11, 3390 CrossRef CAS PubMed.
- P. Pawluc, A. Franczyk, J. Walkowiak, G. Hreczycho, M. Kubicki and B. Marciniec, Org. Lett., 2011, 13, 1976 CrossRef CAS PubMed.
- J. Szudkowska-Frątczak, M. Zaranek, G. Hreczycho, M. Kubicki, T. Grabarkiewicz and P. Pawluć, Appl. Organomet. Chem., 2015 DOI:10.1002/aoc.3284.
- P. Pawluc, A. Franczyk, J. Walkowiak, G. Hreczycho, M. Kubicki and B. Marciniec, Tetrahedron, 2012, 68, 3545 CrossRef CAS PubMed.
- (a) A. E. Pasqua, L. L. Thomas, J. J. Crawford and R. Marquez, Tetrahedron, 2011, 67, 7611 CrossRef CAS PubMed; (b) A. B. Smith III, M. O. Duffey, K. Basu, S. P. Walsh, H. W. Suennemann and M. Frohn, J. Am. Chem. Soc., 2008, 130, 422 CrossRef PubMed; (c) H. Liang, Z. Ren, Y. Wang and Z. Guan, Chem. – Eur. J., 2013, 19, 9789 CrossRef CAS PubMed.
- (a) H. Uhr, C. Boie, H. Rieck, B. Krueger, U. Heinemann, R. Market, M. Vaupel, M. Kluger, K. Stenzel, U. Wachendorff-Neumann, A. Mauler-Mchnick, K. Kuck, P. Loesel and S. Narabu, DE Patent19918294, 2000 Search PubMed; Chem. Abstr. 2000 133 309910 Search PubMed; (b) A. D. Brown, M. E. Bunnage, C. A. L. Lane, R. A. Lewthwaite, P. A. Glossop, K. James and D. A. Price, US 20050215590, 2005; Chem. Abstr. 2005 143 346910 Search PubMed; (c) T. Ogawa and H. Yamada, JP 08176107, 1996; Chem. Abstr. 1996 125 221571 Search PubMed.
- P. Pawluc, J. Szudkowska, G. Hreczycho and B. Marciniec, J. Org. Chem., 2011, 76, 6438 CrossRef CAS PubMed.
- P. Pawluc, Catal. Commun., 2012, 23, 10 CrossRef CAS PubMed.
- (a) M. P. Sibi and S. Manyem, Tetrahedron, 2000, 56, 8033 CrossRef CAS; (b) N. Krause and A. Hoffmann-Roder, Synthesis, 2001, 171 CrossRef CAS PubMed; (c) D. Almasi, D. A. Alonso and C. Najera, Tetrahedron: Asymmetry, 2007, 18, 299 CrossRef CAS PubMed; (d) M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520 CrossRef CAS PubMed.
- (a) W. Prukala, M. Majchrzak, C. Pietraszuk and B. Marciniec, J. Mol. Catal. A: Chem., 2006, 254, 58 CrossRef CAS PubMed; (b) A. Skarzynska, M. Majchrzak, A. M. Trzeciak and B. Marciniec, J. Mol. Catal. A: Chem., 2011, 351, 128 CrossRef CAS PubMed.
- W. Prukała, B. Marciniec, M. Majchrzak and M. Kubicki, Tetrahedron, 2007, 63, 1107 CrossRef PubMed.
- W. Prukala, M. Majchrzak, K. Posala and B. Marciniec, Synthesis, 2008, 3047 CAS.
- W. Prukala, Synlett, 2008, 3026 CrossRef CAS PubMed.
- W. Prukała, P. Pawluć, K. Posała and B. Marciniec, Synlett, 2008, 41 Search PubMed.
- B. Marciniec, J. Waehner, P. Pawluc and M. Kubicki, J. Mol. Catal. A: Chem., 2007, 265, 25 CrossRef CAS PubMed.
- Y. Nishimoto, M. Kajioka, T. Saito, M. Yasuda and A. Baba, Chem. Commun., 2008, 6396 RSC.
- P. Pawluc, G. Hreczycho and B. Marciniec, Synlett, 2005, 1105 CAS.
- P. Pawluc, G. Hreczycho and B. Marciniec, J. Org. Chem., 2006, 71, 8676 CrossRef CAS PubMed.
- P. Pawluc, G. Hreczycho, M. Madalska, J. Szudkowska, M. Kubicki and B. Marciniec, Synthesis, 2009, 3843 CrossRef CAS PubMed.
- P. Pawluc, G. Hreczycho, J. Walkowiak and B. Marciniec, Synlett, 2007, 2061 CAS.
- For review see: (a) J. R. Wang and K. Manabe, Synthesis, 2009, 1405 Search PubMed; (b) G. Chelucci, Chem. Rev., 2012, 112, 1344 CrossRef CAS PubMed.
- P. P. Pawluc, M. Madalska, G. Hreczycho and B. Marciniec, Synthesis, 2008, 3687 CAS.
- (a) G. Zweifel, R. E. Murray and H. P. On, J. Org. Chem., 1981, 46, 1292 CrossRef CAS; (b) E.-i. Negishi and T. Takahashi, J. Am. Chem. Soc., 1986, 108, 3402 CrossRef CAS; (c) A. Arefolov, N. F. Langille and J. S. Panek, Org. Lett., 2001, 3, 3281 CrossRef CAS PubMed; (d) J. C. Anderson and R. H. Munday, J. Org. Chem., 2004, 69, 8971 CrossRef CAS PubMed; (e) A. Inoue, K. Kitagawa, H. Shinokubo and K. Oshima, J. Org. Chem., 2001, 66, 4333 CrossRef CAS PubMed.
- P. Pawluc, G. Hreczycho, A. Suchecki, M. Kubicki and B. Marciniec, Tetrahedron, 2009, 65, 5497 CrossRef CAS PubMed.
Footnote |
| † Dedicated to Prof. Ei-ichi Negishi on the occasion of his 80th birthday. |
| This journal is © the Partner Organisations 2015 |