A novel phase-switching protecting group for multi-step parallel solution phase synthesis
Xin Lia, Chris Abellb, Miles S. Congreve†a, Brian H. Warringtona and Mark Ladlow*a
aGlaxoSmithKline Cambridge Technology Centre, University Chemical Laboratory, Lensfield Road, Cambridge, UK CB2 1EW. E-mail: Mark.2.Ladlow@gsk.com; Fax: +44 (0)1223 331532; Tel: +44 (0)1223 336349
bUniversity of Cambridge, University Chemical Laboratory, Lensfield Road, Cambridge, UK CB2 1EW
First published on 25th February 2004
Abstract
A new phase-tag 1 which facilitates the parallel solution phase synthesis of carboxylic acids, esters, and carboxamides is reported. The new phase tag assists compound purification by enabling the selective resin capture of reaction products in either a reversible pH dependent manner (solid-phase extraction), or irreversibly in a Diels–Alder reaction.
Introduction
The high-throughput screening of synthetic compound libraries has emerged as a key strategy for the identification of starting points in new medicinal chemistry programs.1 This has created an increased demand for large numbers of new compounds. Towards this end, solid-phase synthesis has enjoyed much success in allowing the high-throughput synthesis of a wide-range of compound types.2 However, this approach often suffers from long development times, together with the inability to adequately monitor chemistries and separate by-products prior to cleavage from the resin. This has prompted a re-evaluation of solution phase strategies, whereby reactions are easily monitored using well-established techniques.3 Moreover, the introduction of an increasingly wide variety of polymer-supported reagents and scavenger resins has greatly facilitated reaction work-up and often allows for the isolation of the desired products in high purity without the need for subsequent column chromatography.4Both of these strategies confine the substrate to either the solid or solution phase respectively, and relegate other reactants to the alternative phase. An attractive complementary approach, which combines the inherent advantages of both solid- and solution phase methodologies, uses the concept of ‘phase-switching’. Here, the substrate is moved between the solid and solution phases as required to assist both synthesis and compound isolation. In practice, this may be achieved by solid-phase extraction (SPE) or resin ‘capture and release’ protocols, whereby the substrate is temporarily ‘immobilised’ via an activated linkage to a functionalised support.5 This linkage is subsequently cleaved by introducing a co-reactant which derivatises and concomitantly releases the modified substrate back into solution.
Alternatively, where suitable ‘affinity’ groups are not present, phase-switching during synthesis may be performed by temporarily ‘tagging’ the substrate with a removable soluble ‘phase-switching’ tag. Several methods including the introduction of anthracenyl,6 polyaromatic,7 crown ether affinity,8 ionisable,9 precipitation-enabling,10 Cu-ion chelating,11 and fluorous phase-tags12 have been described to allow substrate, or reagent, sequestration from solution.
Herein, we report the preparation and use of a new bifunctional anthracenyl-tagged protecting group 1 that aids the solution phase synthesis and purification of carboxylic acids, esters and carboxamides (Fig. 1).
 | ||
| Fig. 1 Tagged protecting groups for resin ‘capture and release’ phase-switching applications. | ||
The new ‘phase-switching’ protecting group benefits from several key features. Firstly, the presence of a UV chromophoric anthracenyl moiety both enables convenient substrate visualisation by UV detection (ε254
= 36![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 000) and facilitates quantitative reaction monitoring by UV detection at 386 nm (ε386
= 9000), a region of the UV spectrum that is typically free from competing absorptions.13 Secondly, in contrast to our recently reported anthracenyl tagging group 2,14 which undergoes an effectively irreversible15
‘phase-switch’ in a Diels–Alder reaction with polystyrene-supported maleimide, the new tagging group 1 may alternatively be reversibly attached to a solid support in an orthogonal manner. This latter process is achieved by the incorporation of a tertiary amino functionality which enables pH dependent reversible solid phase extraction by salt formation with an acidic solid support. This affords a particularly versatile bifunctional phase-tag. In addition, the incorporation of Fukuyama's anilino safety-catch methodology16 to robustly attach the tagging group 1 to the substrate ensures a high resistance to premature cleavage during compound synthesis and renders the tagged adduct 3 compatible with a wide variety of chemistries.
000) and facilitates quantitative reaction monitoring by UV detection at 386 nm (ε386
= 9000), a region of the UV spectrum that is typically free from competing absorptions.13 Secondly, in contrast to our recently reported anthracenyl tagging group 2,14 which undergoes an effectively irreversible15
‘phase-switch’ in a Diels–Alder reaction with polystyrene-supported maleimide, the new tagging group 1 may alternatively be reversibly attached to a solid support in an orthogonal manner. This latter process is achieved by the incorporation of a tertiary amino functionality which enables pH dependent reversible solid phase extraction by salt formation with an acidic solid support. This affords a particularly versatile bifunctional phase-tag. In addition, the incorporation of Fukuyama's anilino safety-catch methodology16 to robustly attach the tagging group 1 to the substrate ensures a high resistance to premature cleavage during compound synthesis and renders the tagged adduct 3 compatible with a wide variety of chemistries.
Results and discussion
The bifunctional tagged protecting group 1 was prepared starting from 3-methyl-4-nitrophenol 4 which was protected as the benzyl ether 5 prior to treatment with N,N-dimethylformamide dimethyl acetal and subsequent de-benzylation to afford the phenyl acetaldehyde 7 (Scheme 1). This was converted to the acetal 8 upon exposure to 1,3-propanediol followed by O-alkylation with ethyl iodoacetate to provide the ester 9. The ester was converted to the corresponding methyl carboxamide 10 and then reduced with NaBH4–I217 to afford the secondary amine 11 in good yield. The anthracenyl group was then introduced to give the carboxamide 12. Reduction of the nitro group proceeded smoothly in the presence of NaBH4–Cu(acac)218 affording the aniline 13 and, finally, exposure of 13 to lithium aluminium hydride in THF gave gram quantities of the desired tertiary amine phase-tag 1 in 40% overall yield from 4.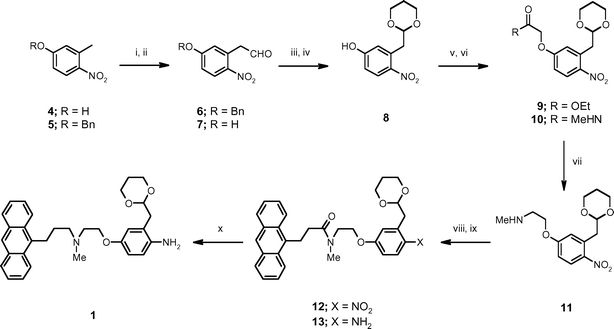 | ||
| Scheme 1 Reagents and conditions: i. benzyl bromide, K2CO3, DMF, 90 °C, 91%; ii. a) dimethylformamide dimethyl acetal, DMF, reflux; b) 10% aqueous HCl, diethyl ether, reflux, 82%; iii. TFA, 93%; iv. 1,3-propanediol, Amberlyst H-15, CH2Cl2, reflux, 92%; v. iodoacetic acid ethyl ester, DMF, PS-BEMP, 98%; vi. methylamine, Amberlyst H-15, THF, 98%; vii. a) NaBH4, I2, THF, reflux; b) diethylamine, THF, 55 °C, 89%; viii. 3-anthracen-9-yl-propionyl chloride, PS-DIPEA, CH2Cl2, 93%; ix. NaBH4, Cu(acac)2, CH2Cl2/MeOH/iPrOH, reflux, 89%; x. LiAlH4, THF, 90%. | ||
To illustrate the use of 1 in assisting parallel multi-step solution phase synthesis, we targeted the preparation of a representative selection of carboxylic acid derivatives displaying the well-known biaryl pharmacophoric motif.19 Routinely, purification of intermediates was performed by a standard protocol which involved resin capture onto an acidic Isolute SCX-220 cartridge, followed by a wash step to remove contaminants, and then release of the desired substrate by elution with 2 M methanolic ammonia solution. Importantly, the use of the acidic SCX-2 support was not observed to give rise to any by-products associated with premature loss of the acetal protecting group. However, clearly when both a reagent and the desired product contain basic functionality, then purification is not possible simply by SPE with an acidic support. In these instances, an alternative work-up and purification procedure was invoked whereby the anthracenyl phase-tag participates in a Diels–Alder reaction with a polymer-supported maleimide resin to immobilise the desired tagged substrate and facilitate its separation from all soluble reagents and impurities (Fig. 2).
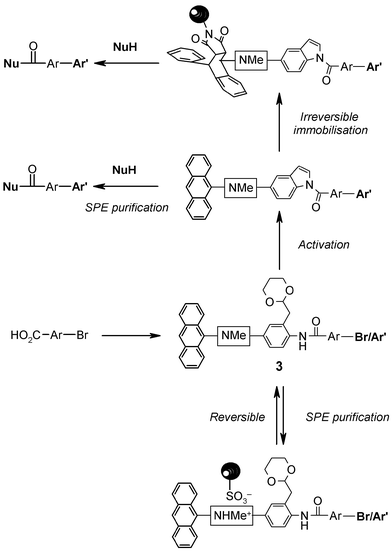 | ||
| Fig. 2 Schematic application of bifunctional phase-tags to mediate both reversible and irreversible phase-switching during solution phase synthesis and compound manipulation. | ||
The phase tag 1 was first introduced as the anilide of a representative carboxylic acid. For example, 4-bromobenzoic acid was derivatised with 1 in the presence of diisopropylcarbodiimide (DIC) and 1-hydroxybenzotriazole (HOBt) to afford the corresponding carboxamide 3a (Scheme 2) which was readily purified by the standard SCX ‘capture and release’ protocol previously described, as shown in Fig. 3. Alternatively, this amidation could also be performed by treating 1 with 4-bromobenzoyl chloride in the presence of polymer-supported diisopropylamine to afford 3a in 92% yield and >99% purity according to LC-MS. The nicotinamide 3b was prepared in a similar way using the DIC/HOBt protocol. In this case, the introduction of aminomethyl polystyrene resin was necessary to remove excess 5-bromonicotinic acid prior to ‘catch and release’ purification with an SCX-2 SPE cartridge.
 | ||
| Scheme 2 Reagents & conditions: i. a) 4-bromobenzoic acid, DIC, HOBt, DMF, 84% or 4-bromobenzoyl chloride, PS-DIPEA, DCE, 92%; b) SCX-2 SPE; ii. a) 5-bromonicotinic acid, DIC, HOBt, DMF, 89%; b) PS-NH2; c) SCX-2 SPE, 89%; iii a) 14a–c, Pd(Ph3P)4, Cs2CO3, DME, 80 °C; b) SCX-2 SPE; iv. a) N-methylbenzylamine, PS-BH(OAc)3, CH2Cl2, rt, 4 h; b) PS-NCO; c) SCX-2 SPE, 88%. | ||
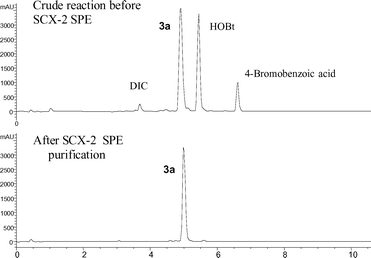 | ||
| Fig. 3 Purification of tagged intermediate 3a by solid-phase extraction with an acidic SCX-2 SPE cartridge. | ||
Next we investigated arylation of the tagged aryl bromide 3a (Scheme 2). Under typical Suzuki coupling conditions (Pd(PPh3)4, Cs2CO3, DME, 80 °C), reaction with a series of boronic acids 14a–c (Fig. 4) all proceeded rapidly in solution within 0.5 h to afford the corresponding tagged biaryls 15a–c in good yields. Again, all products were isolated with excellent purities (>91% by LC-MS), following purification by SPE using SCX-2 cartridges. In a similar way, the nicotinamide 3b was converted into the biaryl 15d which after SCX-2 SPE purification was obtained in quantitative yield.
 | ||
| Fig. 4 Boronic acid set for biaryl couplings. | ||
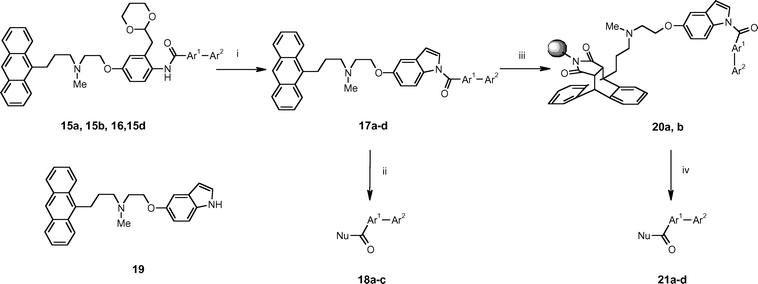
To demonstrate the compatibility of tagged substrates with the use of polymer-supported reagents, the tagged aldehyde 15c was subjected to reductive amination with N-methylbenzylamine in the presence of polymer-supported triacetoxyborohydride (PS-BH(OAc)3) to afford the tertiary amine 16. Removal of excess N-methylbenzylamine was readily achieved by the introduction of the scavenger resin PS-NCO, which does not react with the tertiary amine in the tag. Subsequently, purification by SCX-2 SPE gave the desired amine 16 in 88% yield and 86% purity according to LC-MS analysis.
At this stage, we turned our attention to the removal of the phase-tag by nucleophilic displacement. After some experimentation, we found that treatment of the tagged anilide intermediates 15a, 15b, 15d and 16 with 20% trifluoroacetic acid in aqueous acetone at 58 °C for 6 h reliably led to transacetalisation and cyclisation to form the corresponding N-acyl indoles 17a–d (Table 1). These reactions could be conveniently monitored by HPLC at 386 nm and, following purification by SCX-2 SPE, the desired compounds were all obtained in high yields (>85%) and purities (>85% by LC-MS). Two complementary nucleophilic cleavage protocols were then envisaged depending upon the presence or absence of basic functionality in the desired product.
Reagents and conditions: i. a) (1 : 1 : 3) TFA : H2O : Me2CO, 58 °C, 6 h, b) SCX-2 SPE; ii. NuH, THF, rt−80 °C; iii. PS-maleimide, PhMe, 100 °C, 16 h; iv. NuH, THF, rt−80 °C
| Starting material | Product | Ar1 | Ar2 | NuH | Yield (%)a | HPLC purity (%)b |
|---|---|---|---|---|---|---|
| a Isolated yield.b Estimated from HPLC peak areas 220–290 nm. | ||||||
| 15a | 17a |  |  | — | 96 | 98 |
| 15b | 17b |  |  | — | 100 | 97 |
| 16 | 17c |  |  | — | 100 | >99 |
| 15d | 17d |  |  | — | 86 | >99 |
| 17c | 20a |  |  | — | Quant. | N/A |
| 17d | 20b |  |  | — | Quant. | N/A |
| 17a | 18a |  |  | MeNH2 | 96 | >99 |
| 17a | 18b |  |  | MeOH | 76 | >99 |
| 17b | 18c |  |  | Piperidine | 75 | 97 |
| 20a | 21a |  |  | n-PrNH2 | 82 | 98 |
| 20a | 21b |  |  | Piperidine | 79 | 96 |
| 20b | 21c |  |  | MeNH2 | 79 | 98 |
| 20b | 21d |  |  | H2O, CsOH·H2O | 62 | >99 |
For target molecules lacking a basic group, the N-acyl indoles were treated with an appropriate nucleophile and the cleaved phase-tag 19 was subsequently captured by SCX-2 SPE, leaving only the desired product in solution. Thus, 17a was treated with excess methylamine at ambient temperature to afford the methylamide 18a, and with methanol in the presence of 4-dimethylaminopyridine (DMAP) to afford the methyl ester 18b. Similarly, treatment of 17b with piperidine gave the tert-carboxamide 18c. In all of these cases, purification of the products was readily achieved using the standard SCX-2 SPE protocol to remove the cleaved phase-tag 19.
Conversely, when the desired cleavage product contained basic functionality, it was necessary to first invoke the orthogonal Diels–Alder separation strategy. Thus, the tagged-amines 17c and 17d were heated at reflux with polystyrene-supported maleimide21 in toluene to afford the immobilised Diels–Alder adducts 20a and 20b respectively. Under these conditions, no anthracenyl containing material was left in solution according to HPLC analysis. The resin 20b was then treated with either excess methylamine in tetrahydrofuran or aqueous caesium hydroxide to afford the methylamide 21c and the carboxylic acid 21d respectively. In principle, further purification of the products could have been performed by SCX-2 SPE, but this was not necessary since both compounds were obtained in good yield and excellent purity.
In a similar way, treatment of the resin 20a with propylamine in tetrahydrofuran gave the carboxamide 21a in high yield and purity after simple evaporation, and treatment of 20a with piperidine at 65 °C gave the tert-carboxamide 21b. In this latter case, removal of traces of residual piperidine from the product was performed by incubation with PS-NCO scavenger resin. All the adducts 21a–d were obtained in good yields (62–82%) and with high purities (>96% according to LC-MS and NMR). The 1H NMR spectrum of a representative analogue 21a is shown in Fig. 5.
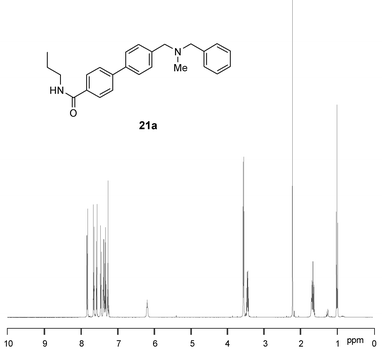 | ||
| Fig. 5 400 MHz 1H NMR spectrum of 21a. | ||
Conclusions
In summary, we have described the preparation of the novel bifunctional phase-tag 1, and demonstrated its use to facilitate the convenient solution phase preparation of carboxylic acids, esters and carboxamides in good yields and excellent purities, without the need for conventional column chromatographic purification. The incorporation of both anthracenyl and tert-amino functionalities in the phase-tagging group allows for either irreversible solid-phase capture of the tagged substrate in a Diels–Alder reaction with PS-maleimide resin, or reversible ‘capture and release’ by an acidic solid-support, such as a sulfonic acid derivatised silica. The judicious combination of these orthogonal phase-switching processes allows for great versatility in product purification and manipulation, and in particular, is compatible with the incorporation of polar basic functionalities in reaction products. Where RP-HPLC retention time under standard conditions (tR) is taken as representative of lipophilicity,22 it is notable that the biaryl derivatives compounds 18a–c and 21a–d prepared displayed a wide range of values (tR = 2.58–5.90 min23). The ability to prepare such compounds in a single process is an attractive attribute of this phase-tagging approach. Indeed, the incorporation of compounds displaying diverse structural and physical characteristics is a desirable objective in preparing compound libraries for drug discovery applications. We believe that the present study clearly illustrates the advantages inherent in the use of bifunctional tagging strategies to facilitate the parallel solution phase multi-step synthesis of compound arrays. The additional effort required to introduce the phase-tag in the first instance is offset by both simplified downstream work-up procedures, and the ability to obtain reaction products in high purity without the need for conventional chromatographic purification.Experimental
All moisture-sensitive reactions were carried out under a nitrogen atmosphere in oven-dried glassware. All solvents and reagents were used as supplied unless otherwise stated. Analytical thin layer chromatography (TLC) was carried out on Polygram™ SIL G/UV254 plates. Analytical high pressure liquid chromatography (HPLC) was performed using a Hewlett Packard Series 1050 instrument. Column: Supelcosil™ ABZ+PLUS 3.3 cm × 4.6 mm, 3 μm. Eluent A: water, 0.1% TFA, B: acetonitrile 95%, water 5%, TFA 0.05%. Flow rate: 1 cm3 min−1. Detection: UV (diode array). Method: gradient 10–95% B in A over 7 min. Infrared spectra were collected on a Perkin-Elmer Spectrum One ATR FT-IR instrument or by DRIFTS using a BioRad FT-IR. Liquid chromatography-mass spectra (LC-MS) were recorded on a Waters Alliance HT/Micromass ZQ under electrospray positive and negative ionisation conditions. Column: Supelcosil™ ABZ+PLUS 3.3 cm × 4.6 mm, 3 μm. Eluent A: 10 mM solution of ammonium acetate in water, 0.1% formic acid, B: acetonitrile 95%, water 5%, formic acid 0.05%. Flow rate: 1 mL min−1. Detection: UV (diode array: 215, 230, 254 nm). Method: gradient 0–100% B in A over 3.5 min. Accurate mass spectra were recorded on a VG Autospec mass spectrometer in positive electrospray mode. NMR spectra were recorded in the indicated solvent. Chemical shifts (δ) are reported in parts per million (ppm) relative to tetramethylsilane as an internal standard; the following abbreviations are used for multiplicities: s = singlet; d = doublet; t = triplet; m = multiplet; dd = doublet of doublets; br = broad; and coupling constant J-values are quoted in Hz. 1H NMR spectra were recorded on a Bruker AM 400 spectrometer at 400 MHz. 13C NMR spectra were recorded on a Bruker AM 400 at 100 MHz with proton decoupling.4-Benzyloxy-2-methyl-1-nitrobenzene (5)
To a solution of 3-methyl-4-nitro-phenol 4 (16.8 g, 110 mmol) in anhydrous DMF (100 ml), was added anhydrous potassium carbonate (15.0 g, 152 mmol) and benzyl bromide (12.0 ml, 100 mmol). The resulting suspension was stirred at 90 °C for 2 h then concentrated in vacuo, and the residue diluted with 2 M aq. NaOH (200 ml). The mixture was extracted with ethyl acetate (3 × 150 ml) and the combined organic extracts were dried (Na2SO4) and concentrated in vacuo to afford 5 as a yellow solid (22.2 g, 91% yield). HPLC: (254 nm): tR = 6.03 min (100%); IR: νmax/cm−1 1578 and 1333; 1H NMR (400 MHz, CDCl3): δ 2.62 (3H, s), 5.12 (2H, s), 6.86 (2H, m), 7.38 (5H, m), 8.07 (1H, d, J = 9.6); 13C NMR (100 MHz, CDCl3): δ 161.5, 141.6, 136.5, 135.0, 128.1 (2C), 127.8, 126.9, 126.9 (2C), 117.7, 111.9, 69.8, and 21.1; LC-MS (ESI): tR = 4.57 min (242.4 [M − H]−). HRMS (ESI): m/z calcd (C14H13NO3Na) 266.0793, found 266.0790 [M + Na]+.(5-Benzyloxy-2-nitrophenyl)-acetaldehyde (6)
To a solution of benzyl ether 5 (18.7 g, 77.0 mmol) in DMF (100 ml), was added N,N-dimethylformamide dimethyl acetal (20 ml, 150 mmol). The resulting mixture was heated at reflux under nitrogen for 24 h and then concentrated in vacuo. The residue was dissolved in diethyl ether (100 ml), treated with 10% aq. HCl (25 ml), and the resulting mixture was heated at reflux for 2 h. The separated organic layer was washed with saturated brine, dried (Na2SO4), and concentrated in vacuo until a precipitate was formed (∼20 ml). After cooling at 4 °C for 2 h, the precipitate was collected by filtration and washed with a mixture of ethyl acetate and hexane (1 : 1) to give a solid which was recrystallised from diethyl ether to afford 6 as an off-white solid (17.2 g, 82%). HPLC: (254 nm): tR = 5.22 min (100%); IR: νmax/cm−1 1724, 1579 and 1334; 1H NMR (400 MHz, CDCl3): δ 4.08 (2H, s), 5.14 (2H, s), 6.84 (1H, d, J = 2.8), 6.98 (1H, dd, J = 2.8 and 9.2), 7.38 (5H, m), 8.20 (1H, d, J = 9.2) and 9.83 (1H, s); 13C NMR (100 MHz, CDCl3): δ 196.1, 162.1, 141.2, 134.7, 130.9, 128.2 (2C), 127.9, 127.5, 126.9 (2C), 118.9, 113.3, 70.1, and 48.4; LC-MS (ESI): tR = 4.20 min (272.3 [M + H]+). HRMS (ESI): m/z calcd (C15H13NO4Na) 294.0742, found 294.0739 [M + Na]+.(5-Hydroxy-2-nitrophenyl)-acetaldehyde (7)
Benzyl ether 6 (7.48 g, 27.6 mmol) was dissolved in trifluoroacetic acid (50 ml) and the resulting solution was stirred at room temperature for 24 h. The reaction mixture was concentrated in vacuo, and the residue was purified by column chromatography to give phenol 7 as white solid (4.67 g, 93%). HPLC (254 nm): tR = 4.70 min (100%); IR: νmax/cm−1 3360, 1719, 1582 and 1324; 1H NMR (400 MHz, CDCl3): δ 4.08 (2H, s), 6.24 (1H, br), 6.69 (1H, d, J = 2.4), 6.84 (1H, dd, J = 2.4 and 8.8), 8.15 (1H, d, J = 8.8), 9.84 (1H, s); 13C NMR (100 MHz, CDCl3): δ 198.5, 162.4, 140.0, 132.5, 127.8, 119.7, 114.7, and 48.1; LC-MS (ESI): tR = 3.31 min (180.4 [M − H]−); HRMS (ESI): m/z calcd (C8H7NO4Na) 204.0273, found 204.0265 [M + Na]+.3-[1,3]Dioxan-2-ylmethyl-4-nitrophenol (8)
To a solution of acetaldehyde 7 (3.19 g, 17.6 mmol) in anhydrous CH2Cl2 (100 ml) was added 1,3-propanediol (10 ml) and Amberlyst H-15 resin (500 mg). The resulting suspension was stirred at room temperature for 16 h then filtered and the resin thoroughly washed with CH2Cl2 (3 × 15 ml). The combined filtrates were washed with saturated brine, dried (Na2SO4) and concentrated in vacuo to afford the acetal 8 as an oil (3.87 g, 92%). HPLC (254 nm): tR = 3.81 min (100%); IR: νmax/cm−1 3286, 1581 and 1332; 1H NMR (400 MHz, CDCl3): δ 1.33 (1H, m), 2.08 (1H, m), 3.24 (2H, d, J = 5.2), 3.74 (2H, m), 4.07 (2H, m), 4.84 (1H, t, J = 5.2), 6.02 (1H, s), 6.77 (1H, dd, J = 2.8 and 8.8), 6.80 (1H, d, J = 2.8), 7.98 (1H, d, J = 8.8); 13C NMR (100 MHz, CDCl3): δ 158.8, 142.0, 133.9, 127.1, 119.6, 113.8, 100.3, 66.3 (2C), 38.9, and 25.0; LC-MS (ESI): tR = 3.56 min (240.3 [M + H]+). HRMS (ESI): m/z calcd (C11H13NO5Na) 262.0691, found 262.0678 [M + Na]+.(3-[1,3]Dioxan-2-ylmethyl-4-nitrophenoxy)-acetic acid ethyl ester (9)
To a solution of phenol 8 (3.00 g, 12.6 mmol) in DMF (100 ml), was added anhydrous potassium carbonate (5.00 g, 36.2 mmol) and ethyl iodoacetate (3.00 ml, 25.3 mmol). The resulting suspension was stirred at 90 °C for 2 h, then concentrated in vacuo, and the residue partitioned between 2 M aq. NaOH (50 ml) and diethyl ether (50 ml). The organic layer was separated, washed with saturated brine, and dried (Na2SO4). The solvent was evaporated in vacuo, and the residue was purified by column chromatography eluting with a mixture of ethyl acetate and hexane (1 : 4) to afford the ethyl ester 9 as light yellow solid (4.00 g, 98%). HPLC (254 nm): tR = 4.68 min (100%); IR: νmax/cm−1 1755, 1581 and 1339; 1H NMR (400 MHz, CDCl3): δ 1.31 (4H, m), 2.05 (1H, m), 3.22 (2H, d, J = 5.2), 3.70 (2H, m), 4.04 (2H, m), 4.27 (2H, q, J = 7.2), 4.67 (2H, s), 4.78 (1H, t, J = 5.2), 6.82 (1H, dd, J = 2.8 and 9.2), 6.88 (1H, d, J = 2.8), 7.98 (1H, d, J = 9.2); 13C NMR (100 MHz, CDCl3): δ 167.2, 159.9, 142.9, 133.8, 126.5, 118.7, 112.6, 100.0, 66.2 (2C), 64.7, 61.0, 38.8, 25.0, and 13.5; LC-MS (ESI): tR = 3.94 min (326.3 [M + H]+); HRMS (ESI): m/z calcd (C15H19NO7Na) 348.1059, found 348.1046 [M + Na]+.2-(3-[1,3]Dioxan-2-ylmethyl-4-nitrophenoxy)-N-methyl-acetamide (10)
The ethyl ester 9 (2.88 g, 8.86 mmol) was dissolved in a solution of methylamine in THF (1 M × 75 ml) and the resulting solution was treated with Amberlyst H-15 resin (100 mg) at room temperature for 24 h. The reaction mixture was filtered and the resin was thoroughly washed with THF (3 × 20 ml). The combined organic filtrates were concentrated in vacuo and the residue was recrystallised from ethyl acetate to give the carboxamide 10 as a yellow solid (2.70 g, 98%). HPLC (254 nm): tR = 3.47 min (100%); IR: νmax/cm−1 3332, 1669, 1581 and 1338; 1H NMR (400 MHz, CDCl3): δ 1.32 (1H, m), 2.05 (1H, m), 2.91 (3H, d, J = 4.8), 3.24 (2H, d, J = 5.0), 3.71 (2H, m), 4.05 (2H, m), 4.54 (2H, s), 4.80 (1H, t, J = 5.0), 6.49 (1H, brs), 6.85 (1H, dd, J = 2.8 and 9.2), 6.93 (1H, d, J = 2.8), 8.00 (1H, d, J = 9.2); 13C NMR (100 MHz, CDCl3): δ 166.8, 159.1, 143.3, 134.0, 126.7, 119.0, 112.4, 99.9, 66.8, 66.3 (2C), 38.7, 25.2, and 25.0; LC-MS (ESI): tR = 3.31 min (311.3 [M + H]+); HRMS (ESI): m/z calcd (C14H18N2O6Na) 333.1063, found 333.1077 [M + Na]+.[2-(3-[1,3]Dioxan-2-ylmethyl-4-nitrophenoxy)-ethyl]-methyl-amine (11)
To a suspension of amide 10 (1.50 g, 4.80 mmol) and NaBH4 (0.60 g, 15.9 mmol) in anhydrous THF (30 ml), was slowly added (30 min) a solution of iodine (1.22 g, 4.80 mmol) in THF (10 ml) with ice–water bath cooling. The resulting suspension was heated at reflux for 4 h, then cooled to 0 °C and quenched by carefully adding 10% aqueous HCl solution. When gas evolution ceased, the mixture was neutralised using 2 M aq. NaOH and extracted with diethyl ether (3 × 50 ml). The combined organic extracts were washed with saturated brine, dried (Na2SO4), and the solvent was evaporated in vacuo. The residue was dissolved in anhydrous THF (50 ml), and the resulting solution was treated with diethylamine (5 ml) at 55 °C for 2 h to liberate the free amine. The reaction mixture was evaporated in vacuo and the residue was purified by SCX-2 SPE to give the amine 11 as yellow solid (1.27 g, 89%). HPLC: (254 nm): tR = 2.43 min (100%); IR: νmax/cm−1 1579 and 1336; 1H NMR (400 MHz, CDCl3): δ 1.30 (1H, m), 1.60 (1H, brs), 2.05 (1H, m), 2.50 (3H, s), 2.98 (2H, t, J = 5.2), 3.24 (2H, d, J = 5.2), 3.71 (2H, m), 4.06 (2H, m), 4.12 (2H, t, J = 5.2), 4.79 (1H, t, J = 5.2), 6.82 (1H, dd, J = 2.8 and 9.2), 6.87 (1H, d, J = 2.8), 7.99 (1H, d, J = 9.2); 13C NMR (100 MHz, CDCl3): δ 161.3, 142.1, 133.8, 126.8, 118.6, 112.4, 100.1, 67.2, 66.3 (2C), 49.8, 39.0, 35.7, and 25.0; LC-MS (ESI): tR = 2.65 min (297.3 [M + H]+); HRMS: (ESI): m/z calcd (C14H21N2O5) 297.1450, found 297.1451 [M + H]+.3-Anthracen-9-yl-N-[2-(3-[1,3]dioxan-2-ylmethyl-4-nitrophenoxy)-ethyl]-N-methyl-propionamide (12)
To a suspension of 3-anthracen-9-yl-propionic acid (1.18 g, 4.70 mmol) in anhydrous CH2Cl2 (100 ml), was added a solution of 2 M oxalyl chloride in CH2Cl2 (10 ml) with ice–water bath cooling. The resulting suspension was warmed to ambient temperature, and heated at reflux for 2 h. The reaction mixture was concentrated in vacuo to dryness and the residue was re-dissolved in CH2Cl2 (50 ml). The resulting solution was treated with triethylamine (5 ml), and then a solution of amine 11 (1.03 g, 3.48 mmol) in CH2Cl2 (5 ml) was added. The mixture was stirred at room temperature for 2 h and then concentrated in vacuo. The residue was diluted with saturated brine (50 ml) and extracted with ethyl acetate (3 × 50 ml). The combined organic extracts were dried (Na2SO4), concentrated in vacuo, and the residue was purified by column chromatography eluting with a mixture of ethyl acetate and hexane (2 : 1) to give the carboxamide 12 (1.72 g, 93%). HPLC (254 nm): tR = 6.28 min (100%); IR: νmax/cm−1 1640, 1579 and 1335; 1H NMR (400 MHz, DMSO, 140 °C): δ 1.27 (1H, m), 1.85 (1H, m), 2.79 (2H, t, J = 7.8), 2.89 (3H, s), 3.13 (2H, d, J = 5.0), 3.63 (4H, m), 3.92 (4H, m), 4.15 (2H, t, J = 5.7), 4.71 (1H, t, J = 5.0), 6.89 (2H, m), 7.49 (4H, m), 7.84 (1H, d, J = 8.9), 8.03 (2H, d, J = 8.0), 8.29 (2H, d, J = 8.6), 8.41 (1H, s); 13C NMR (100 MHz, CDCl3): δ 172.0, 161.5, 143.7, 134.1, 133.9, 131.7 (2C), 129.6 (2C), 129.2 (2C), 126.8, 126.0, 125.9 (2C), 125.1 (2C), 124.4 (2C), 119.3, 113.6, 100.7, 66.8, 66.4 (2C), 40.2 (2C), 38.3, 34.1, 25.5 and 23.3; LC-MS (ESI): tR = 4.68 min (529.4 [M + H]+); HRMS (ESI): m/z calcd (C31H32N2O6Na) 551.2158, found 551.2145 [M + Na]+.N-[2-(4-Amino-3-[1,3]dioxan-2-ylmethyl-phenoxy)-ethyl]-3-anthracen-9-yl-N-methyl-propionamide (13)
To a solution of the nitrobenzene 12 (1.45 g, 2.75 mmol) in a mixture of CH2Cl2 and isopropanol (50 ml/5 ml) was added copper acetylacetonate (0.72 g, 2.75 mmol) and NaBH4(0.32 g, 8.25 mmol) with ice–water bath cooling. The resulting mixture was stirred at room temperature for 5 min, then methanol (5 ml) was slowly added and the mixture was heated at reflux for 30 min. The reaction was quenched by slowly adding water (∼5 ml). When gas evolution ceased, the mixture was filtered through a Celite pad. The filtrate was concentrated in vacuo and the residue was diluted with saturated brine (50 ml) and extracted with ethyl acetate (3 × 50 ml). The combined, dried (Na2SO4) organic extracts were concentrated in vacuo and the crude product was loaded onto a pre-washed SCX-2 cartridge. The cartridge was thoroughly washed with methanol, and then with 2 M ammonia in methanol. The eluate was concentrated in vacuo to give the amine 13 as yellow solid (1.21 g, 89%). HPLC (254 nm): tR = 4.21 min (100%); IR: νmax/cm−1 3417, 3346 and 1634; 1H NMR (400 MHz, DMSO, 120 °C): δ 1.29 (1H, m), 1.89 (1H, m), 2.65 (2H, d, J = 5.0), 2.78 (2H, t, J = 7.9), 2.89 (3H, s), 3.54 (2H, t, J = 5.5), 3.66 (2H, m), 3.94 (6H, m), 4.11 (2H, brs), 4.68 (1H, t, J = 5.0), 6.54 (3, m), 7.48 (4H, m), 8.02 (2H, d, J = 8.0), 8.28 (2H, d, J = 8.6), 8.42 (1H, s); 13C NMR (100 MHz, CDCl3): δ 172.2, 151.0, 141.2, 134.4, 131.9 (2C), 129.9 (2C), 129.4 (2C), 126.2 (2C), 126.1, 125.3 (2C), 124.6 (2C), 123.1, 118.6, 116.9, 114.5, 102.2, 67.1, 66.6 (2C), 40.2 (2C), 37.9, 34.4, 25.8 and 23.7; LC-MS (ESI): tR = 4.04 min (499.4 [M + H]+); HRMS (ESI): m/z calcd (C31H34N2O4Na) 521.2416, found 521.2403 [M + Na]+.4-{2-[(3-Anthracen-9-yl-propyl)-methylamino]-ethoxy}-2-[1,3]dioxan-2-ylmethyl-phenylamine (1)
To a solution of the amide 13 (1.16 g, 2.33 mmol) in THF (50 ml), was added a solution of LiAlH4 in diethyl ether (1 M × 7.5 ml, 7.50 mmol) with ice–water bath cooling. The resulting suspension was stirred at room temperature for 16 h, then the reaction was quenched by carefully adding methanol (∼10 ml). When gas evolution ceased, the mixture was washed with water (100 ml) and the organic layer was separated. The aqueous layer was extracted with diethyl ether (3 × 50 ml) and the combined organic extracts were dried (Na2SO4). The solvent was evaporated in vacuo and the residue was purified by column chromatography eluting with a mixture of methanol and CH2Cl2 (1 : 15) to give the phase-tag 1 (1.02 g, 90%) as an oil. HPLC (254 nm): tR = 3.47 min (100%); IR: νmax/cm−1 3425, 3347 and 1623; 1H NMR (400 MHz, CDCl3): δ 1.29 (1H, m), 2.03 (3H, m), 2.38 (3H, s), 2.69 (2H, t, J = 7.0), 2.81 (4H, m), 3.69 (4H, m), 4.07 (4H, m), 4.67 (1H, t, J = 5.2), 6.60 (1H, d, J = 8.4), 6.69 (2H, m), 7.47 (4H, m), 7.99 (2H, d, J = 8.2), 8.32 (3H, m); 13C NMR (100 MHz, CDCl3): δ 151.2, 139.2, 134.3, 131.0 (2C), 129.0 (2C), 128.5 (2C), 125.0, 124.8 (2C), 124.1 (2C), 123.9 (2C), 122.9, 117.5, 116.5, 113.3, 102.7, 66.2 (2C), 66.1, 57.5, 55.7, 42.2, 37.7, 28.2, 24.9 (2C); LC-MS (ESI): tR = 3.31 min (485.4 [M + H]+); HRMS (ESI): m/z calcd (C31H37N2O3) 485.2804, found 485.2813 [M + H]+.N-(4-{2-[(3-Anthracen-9-yl-propyl)-methylamino]-ethoxy}-2-[1,3]dioxan-2-ylmethyl-phenyl)-4-bromobenzamide (3a)
Method A: To a suspension of 4-bromobenzoyl chloride (439 mg, 2.00 mmol) and PS-DIPEA (2.00 g, 3.83 mmol g−1) in 1,2-dichloroethane (DCE; 16 ml), was added a solution of the phase-tag 1 (484 mg, 1.00 mmol) in DCE (4 ml). The resulting mixture was stirred at room temperature for 30 min, then filtered and the resin washed with CH2Cl2 (3 × 3 ml). The combined organic extracts were evaporated, and the residue was loaded onto an SCX-2 cartridge. The cartridge was thoroughly washed with methanol, and finally with 2 M ammonia solution in methanol to release the carboxamide 3a which was isolated as a yellow solid (614 mg, 92%).Method B: To a solution of 4-bromobenzoic acid (80 mg, 0.40 mmol) and HOBt (20 mg, 0.15 mmol) in DMF (2 ml), was added DIC (100 µl, 0.64 mmol). After 15 min, a solution of the phase-tag 1 (400 µl × 0.25 M, 0.10 mmol) in DMF (1 ml) was added and the resulting mixture was stirred at room temperature for 2 h before loading onto an SCX-2 SPE cartridge. This was eluted as in method A to afford the carboxamide 3a as yellow solid (56.1 mg, 84%). Mp: 89–90 °C; HPLC (254 nm): tR = 5.29 min (94%); IR: νmax/cm−1 3344 and 1668; 1H NMR (400 MHz, CDCl3): δ 1.37 (1H, m), 2.04 (3H, m), 2.39 (3H, s), 2.69 (2H, t, J = 7.2), 2.84 (2H, t, J = 5.8), 2.91 (2H, d, J = 4.8), 3.67 (2H, t, J = 8.0), 3.76 (2H, m), 4.11 (4H, m), 4.71 (1H, t, J = 4.8), 6.79 (1H, d, J = 2.8), 6.88 (1H, dd, J = 2.8 and 8.8), 7.47 (4H, m), 7.61 (2H, d, J = 8.8), 7.80 (2H, d, J = 8.4), 7.88 (1H, d, J = 8.8), 7.99 (2H, d, J = 8.4), 8.32 (3H, m), 9.43 (1H, s); 13C NMR (100 MHz, CDCl3): δ 163.6, 155.2, 134.3, 133.5, 131.1 (2C), 131.0 (2C), 129.5, 129.0 (2C), 128.7, 128.5 (2C), 128.1 (2C), 125.4, 125.0, 124.8 (2C), 124.5, 124.2 (2C), 123.8 (2C), 117.3, 112.5, 102.6, 66.4 (2C), 65.8, 57.5, 55.6, 42.2, 38.1, 28.2, 24.9 (2C); LC-MS (ESI): tR = 3.93 min (669.3 [M + H]+); HRMS (ESI): m/z calcd (C38H40N2O4Br) 667.2171, found 667.2166 [M + H]+.
N-(4-{2-[(3-Anthracen-9-yl-propyl)-methylamino]-ethoxy}-2-[1,3]dioxan-2-ylmethyl-phenyl)-5-bromonicotinamide (3b)
To a solution of 5-bromonicotinyl acid (81 mg, 0.40 mmol) and HOBt (20 mg, 0.15 mmol) in DMF (2 ml), was added DIC (100 µl, 0.64 mmol). After 15 min, a solution of aniline 1 (400 µl × 0.25 M 0.10 mmol) in DMF (1 ml) was added followed, after 2 h, by aminomethyl polystyrene resin (200 mg, 4.5 mmol g−1, 0.90 mmol). The suspension was agitated for 16 h, then filtered and the resin washed with CH2Cl2 (3 × 3 ml). The combined organic filtrates were loaded onto a pre-washed SCX-2 cartridge and the cartridge was thoroughly washed with methanol. Elution with 2 M ammonia in methanol afforded the nicotinamide 3b which was isolated as a yellow solid (59 mg, 89%). Mp 159–160 °C; HPLC (254 nm): tR = 4.79 min (>99%); IR: νmax/cm−1 3335 and 1675; 1H NMR (400 MHz, CDCl3): δ 1.39 (1H, m), 2.02 (2H, m), 2.16 (1H, m), 2.39 (3H, s), 2.69 (2H, t, J = 7.0), 2.84 (2H, t, J = 5.8), 2.92 (2H, d, J = 4.8), 3.67 (2H, t, J = 8.0), 3.77 (2H, m), 4.14 (4H, m), 4.74 (1H, t, J = 4.8), 6.80 (1H, d, J = 2.8), 6.89 (1H, dd, J = 2.8 and 8.8), 7.47 (4H, m), 7.87 (1H, d, J = 8.8), 7.99 (2H, d, J = 8.4), 8.32 (3H, m), 8.40 (1H, s), 8.81 (1H, s), 9.04 (1H, s), 9.44 (1H, s); 13C NMR (100 MHz, CDCl3): δ 161.0, 155.5, 152.5, 145.7, 137.2 (2C), 134.3, 131.5, 131.0 (2C), 129.0 (2C), 128.7, 128.5 (2C), 125.0, 124.8 (2C), 124.4, 124.2 (2C), 123.8 (2C), 120.3, 117.4, 112.6, 102.5, 66.5 (2C), 65.8, 57.4, 55.5, 42.2, 38.2, 28.3, 24.9, 24.8; LC-MS (ESI): tR = 3.74 min (670.4 [M + H]+); HRMS (ESI): m/z calcd (C37H39N3O4Br) 668.2124, found 668.2104 [M + H]+.General Suzuki coupling procedure
A solution of the tagged bromide 3a or 3b (approx. 0.10 mmol) in DME (1 ml) was added to a reaction tube containing a suspension of Cs2CO3 (8 eq.), Pd(PPh3)4 (0.1 eq.) and a boronic acid 14a–c (2 eq.) in DME (3 ml) and water (1 ml). The resulting mixtures were stirred at 80 °C for 0.5 h, then diluted with saturated brine and extracted with ethyl acetate (3 × 3 ml). The combined organic extracts were loaded onto a pre-washed SCX-2 cartridge and the cartridge was thoroughly washed with methanol. Elution with 2 M ammonia in methanol gave the biaryls 15a (84%), 15b (59%), 15c (100%) and 15d (98%).2′-Fluoro-biphenyl-4-carboxylic acid N-(4-{2-[(3-anthracen-9-yl-propyl)-methylamino]-ethoxy}-2-[1,3]dioxan-2-ylmethyl-phenyl)amide (15a)
Mp 92–93 °C; HPLC (254 nm): tR = 5.44 min (98%); IR: νmax/cm−1 3345 and 1664; 1H NMR (400 MHz, CDCl3): δ 1.37 (1H, m), 2.02 (2H, m), 2.12 (1H, m), 2.40 (3H, s), 2.70 (2H, t, J =7.2), 2.86 (2H, t, J = 5.8), 2.95 (2H, d, J = 4.8), 3.68 (2H, t, J = 8.0), 3.77 (2H, m), 4.13 (4H, m), 4.74 (1H, t, J = 4.8), 6.81 (1H, d, J = 2.8), 6.90 (1H, dd, J = 2.8 and 8.8), 7.19 (1H, m), 7.25 (1H, m), 7.37 (1H, m), 7.47 (5H, m), 7.67 (2H, d, J = 8.2), 7.93 (1H, d, J = 8.8), 8.01 (4H, m), 8.32 (3H, m), 9.50 (1H, s); 13C NMR (100 MHz, CDCl3): δ 164.2, 159.1 (1C, d, 1JC,F = 246.8), 155.1, 138.3, 134.3, 133.7, 131.0 (2C), 130.0, 129.7, 129.0 (3C), 128.7, 128.5 (4C), 127.4, 126.6 (2C), 125.0, 124.8 (2C), 124.6, 124.2 (2C), 123.9 (3C), 117.2, 115.6 (1C, d, 2JC,F = 22.3), 112.5, 102.6, 66.4 (2C), 65.8, 57.5, 55.6, 42.2, 38.1, 28.2 and 24.9 (2C); LC-MS (ESI): tR = 4.07 min (683.4 [M + H]+); HRMS (ESI): m/z calcd (C44H44N2O4F) 683.3285, found 683.3281 [M + H]+.N-(4-{2-[(3-Anthracen-9-yl-propyl)-methylamino]-ethoxy}-2-[1,3]dioxan-2-ylmethyl-phenyl)-4-thiophen-2-yl-benzamide (15b)
Mp 79–80 °C; HPLC (386 nm): tR = 5.55 min (91%); IR: νmax/cm−1 3345 and 1663; 1H NMR (400 MHz, CDCl3): δ 1.38 (1H, m), 2.02 (2H, m), 2.14 (1H, m), 2.40 (3H, s), 2.69 (2H, t, J = 7.2), 2.85 (2H, t, J = 5.8), 2.93 (2H, d, J = 4.4), 3.67 (2H, t, J = 7.8), 3.77 (2H, m), 4.13 (4H, m), 4.74 (1H, t, J = 4.4), 6.80 (1H, d, J = 2.8), 6.89 (1H, dd, J = 2.8 and 9.0), 7.11 (1H, dd, J = 3.6 and 5.0), 7.34 (1H, dd, J = 0.8 and 5.0), 7.41 (1H, dd, J = 0.8 and 3.6), 7.47 (4H, m), 7.71 (2H, d, J = 8.4), 7.98 (5H, m), 8.33 (3H, m), 9.46 (1H, s); 13C NMR (100 MHz, CDCl3): δ 164.0, 155.1, 142.6, 136.7, 134.2, 133.2, 131.0 (2C), 129.8, 129.0 (2C), 128.7, 128.5 (2C), 127.6, 127.2 (2C), 125.3, 125.1 (2C), 125.0, 124.8 (2C), 124.6, 124.2 (2C), 123.8 (2C), 123.5, 117.2, 112.5, 102.6, 66.4 (2C), 65.6, 57.4, 55.5, 42.1, 38.1, 28.1, 24.9 (2C); LC-MS (ESI): tR = 4.01 min (671.5 [M + H]+); HRMS (ESI): m/z calcd (C42H43N2O4S) 671.2943, found 671.2951 [M + H]+.4′-Formyl-biphenyl-4-carboxylic acid N-(4-{2-[(3-anthracen-9-yl-propyl)-methylamino]-ethoxy}-2-[1,3]dioxan-2-ylmethyl-phenyl)amide (15c)
Mp 60–61 °C; HPLC (386 nm): tR = 5.22 min (92%); IR: νmax/cm−1 3339, 1700 and 1670. 1H NMR (400 MHz, CDCl3): δ 1.39 (1H, m), 2.02 (2H, m), 2.13 (1H, m), 2.40 (3H, s), 2.70 (2H, t, J = 7.0), 2.86 (2H, t, J = 5.8), 2.94 (2H, d, J = 4.8), 3.67 (2H, t, J = 8.0), 3.78 (2H, m), 4.14 (4H, m), 4.74 (1H, t, J = 4.8), 6.80 (1H, d, J = 2.8), 6.90 (1H, dd, J = 2.8 and 9.0), 7.47 (4H, m), 7.74 (2H, d, J = 8.4), 7.80 (2H, d, J = 8.0), 7.93 (1H, d, J = 9.2), 7.99 (4H, m), 8.06 (2H, d, J = 8.0), 8.32 (3H, m), 9.50 (1H, s), 10.09 (1H, s); 13C NMR (100 MHz, CDCl3): δ 191.1, 185.4, 164.0, 155.2, 145.4, 142.0, 135.0, 134.4, 134.3, 131.0 (2C), 129.7 (2C), 129.0 (2C), 128.7, 128.5 (2C), 127.23 (2C), 127.18 (2C), 126.9 (2C), 125.0, 124.8 (2C), 124.6, 124.2 (2C), 123.9 (2C), 117.3, 112.5, 102.6, 66.6 (2C), 65.8, 57.5, 55.6, 42.2, 38.1, 28.2, 24.9 (2C); LC-MS (ESI): tR = 3.96 min (693.5 [M + H]+); HRMS (ESI): m/z calcd (C45H45N2O5) 693.3328, found 693.3348 [M + H]+.N-(4-{2-[(3-Anthracen-9-yl-propyl)-methylamino]-ethoxy}-2-[1,3]dioxan-2-ylmethyl-phenyl)-5-(2-fluorophenyl)-nicotinamide (15d)
Mp 104–105 °C; HPLC (254 nm): tR = 4.89 min (>99%); IR: νmax/cm−1 3336 and 1672; 1H NMR (400 MHz, CDCl3): δ 1.33 (1H, m), 2.05 (3H, m), 2.40 (3H, s), 2.69 (2H, t, J = 7.0), 2.85 (2H, t, J = 5.8), 2.94 (2H, d, J = 4.8), 3.67 (2H, t, J = 8.0), 3.75 (2H, m), 4.12 (4H, m), 4.73 (1H, t, J = 4.8), 6.81 (1H, d, J = 2.8), 6.90 (1H, dd, J = 2.8 and 9.0), 7.25 (2H, m), 7.46 (6H, m), 7.90 (1H, d, J = 8.8), 7.99 (2H, d, J = 8.2), 8.32 (3H, m), 8.43 (1H, s), 8.93 (1H, s), 9.14 (1H, s), 9.62 (1H, s); 13C NMR (100 MHz, CDCl3): δ 162.4, 159.2 (1C, d, 1JC,F = 247.6), 155.4, 151.3, 146.6, 134.7, 134.3, 131.0, 130.98 (2C), 129.99, 129.92, 129.86, 129.77, 129.2, 128.99 (2C), 128.88, 128.5 (2C), 124.98, 124.80 (2C), 124.6, 124.2 (3C), 123.9 (2C), 117.3, 115.7 (1C, d, 2JC,F = 22.0), 112.6, 102.6, 66.5 (2C), 65.8, 57.5, 55.6, 42.2, 38.1, 28.3, 24.9 and 24.7; LC-MS (ESI): tR = 3.88 min (684.5 [M + H]+); HRMS (ESI): m/z calcd (C43H43N3O4F) 684.3237, found 684.3223 [M + H]+.4′-[(Benzyl-methylamino)-methyl]-biphenyl-4-carboxylic acid N-(4-{2-[(3-anthracen-9-yl-propyl)-methylamino]-ethoxy}-2-[1,3]dioxan-2-ylmethyl-phenyl)amide (16)
To a solution of aldehyde 15c (83.5 mg, 0.12 mmol) in anhydrous CH2Cl2 (5 ml), was added N-benzylmethylamine (47 µl, 0.36 mmol). The mixture was stirred at room temperature for 1 h when PS-BH(OAc)3 (300 mg, 2.1 mmol g−1, 0.63 mmol) was added. The resulting suspension was shaken at room temperature for 3 h, then filtered and the resin washed with CH2Cl2 (3 × 3 ml). The combined filtrates were treated with PS-isocyanate resin (500 mg, 1.6 mmol g−1, 0.80 mmol) for 16 h, then filtered and the resin washed with CH2Cl2 (3 × 3 ml). The combined filtrates were loaded onto a pre-washed SCX-2 cartridge. The cartridge was thoroughly washed with methanol and then the amine 16 was eluted with 2 M ammonia in methanol (84 mg, 88%). HPLC (386 nm): tR = 4.35 min (86%); IR: νmax/cm−1 3336 and 1666; 1H NMR (400 MHz, CDCl3): δ 1.37 (1H, m), 2.03 (2H, m), 2.13 (1H, m), 2.23 (3H, s), 2.40 (3H, s), 2.70 (2H, t, J = 7.0), 2.86 (2H, t, J = 5.8), 2.94 (2H, d, J = 4.8), 3.56 (2H, s), 3.58 (2H, s), 3.68 (2H, t, J = 7.8), 3.78 (2H, m), 4.14 (4H, m), 4.74 (1H, t, J = 4.8), 6.81 (1H, d, J = 2.8), 6.90 (1H, dd, J = 2.8 and 9.0), 7.25 (1H, m), 7.33 (2H, m), 7.38 (2H, m), 7.47 (6H, m), 7.61 (2H, d, J = 8.4), 7.70 (2H, d, J = 8.4), 7.93 (1H, d, J = 9.2), 8.00 (4H, m), 8.32 (3H, m), 9.48 (1H, s); 13C NMR (100 MHz, CDCl3): δ 164.4, 155.1, 143.4, 138.7, 138.6, 138.1, 134.3, 133.1, 131.0 (2C), 129.8, 129.0 (2C), 128.8 (2C), 128.7, 128.5 (2C), 128.3 (2C), 127.6 (2C), 127.0 (2C), 126.4 (4C), 126.3, 125.0, 124.8 (2C), 124.6, 124.2 (2C), 123.9 (2C), 117.2, 112.5, 102.6, 66.4 (2C), 65.7, 61.2, 60.8, 57.4, 55.6, 42.2, 41.7, 38.1, 28.2, 24.9 (2C); LC-MS (ESI): tR = 3.78 min (798.5 [M + H]+ and 400.0 [M + 2H]2+); HRMS (ESI): m/z calcd (C53H56N3O4) 798.4271, found 798.4294 [M + H]+.General procedure for activation of the safety-catch
The acetals 15a (82 mg, 120 µmol), 15b (57 mg, 85 µmol), 16 (98 mg, 123 µmol), and 15d (145 mg, 213 µmol) were separately treated with a mixture of TFA, acetone and water (1 : 3 : 1, ∼40 µmol ml−1) at 58 °C for 6 h. The reaction mixtures were evaporated in vacuo and the residues loaded onto pre-washed SCX-2 cartridges. The cartridges were thoroughly washed with ethyl acetate, and then eluted with a mixture of triethylamine and ethyl acetate (1 : 1) to afford the indoles 17a (70 mg, 96%), 17b (50 mg, 100%), 17c (92 mg, 100%), and 17d (114 mg, 86%) respectively.(5-{2-[(3-Anthracen-9-yl-propyl)-methylamino]-ethoxy}-indol-1-yl)-(2′-fluorobiphenyl-4-yl)-methanone (17a)
HPLC (220–290 nm): tR = 5.88 min (98%); IR: νmax/cm−1 1678; 1H NMR (400 MHz, CDCl3): δ 2.04 (2H, m), 2.42 (3H, s), 2.72 (2H, t, J = 7.0), 2.90 (2H, t, J = 5.9), 3.68 (2H, t, J = 8.0), 4.19 (2H, d, J = 5.9), 6.54 (1H, d, J = 3.8), 7.02 (1H, dd, J = 2.8 and 9.0), 7.09 (1H, d, J = 2.8), 7.23 (2H, m), 7.32 (1H d, J = 3.8), 7.38 (1H, m), 7.47 (5H, m), 7.70 (2H, d, J = 8.0), 7.80 (2H, d, J = 8.0), 7.99 (2H, d, J = 8.0), 8.33 (4H, m); 13C NMR (100 MHz, CDCl3): δ 167.4, 159.1 (1C, d, 1JC,F = 246.9), 155.4, 138.7, 134.3, 133.0, 131.1, 131.0 (2C), 130.1, 130.0, 129.3 (1C, d, 4JC,F = 8.0), 129.0 (2C), 128.7 (2C), 128.5 (4C), 127.6 (2C), 127.1 (1C, d, 2JC,F = 13.1), 125.0, 124.8 (2C), 124.2 (2C), 123.9, 123.8, 116.6, 115.7 (1C, d, 2JC,F = 22.4), 113.4, 108.0, 104.0, 66.1, 57.5, 55.6, 42.3, 28.2 and 24.9; LC-MS (ESI): tR = 4.28 min (607.5 [M + H]+); HRMS (ESI): m/z calcd (C41H36N2O2F) 607.2761, found 607.2763 [M + H]+.(5-{2-[(3-Anthracen-9-yl-propyl)-methylamino]-ethoxy}-indol-1-yl)-(4-thiophen-2-yl-phenyl)-methanone (17b)
HPLC (220–290 nm): tR = 5.87 min (97%); IR: νmax/cm−1 1677; 1H NMR (400 MHz, CDCl3): δ 2.03 (2H, m), 2.42 (3H, s), 2.72 (2H, t, J = 7.0), 2.89 (2H, t, J = 5.9), 3.68 (2H, t, J = 8.0), 4.18 (2H, d, J = 5.9), 6.53 (1H, d, J = 3.6), 7.01 (1H, dd, J = 2.6 and 8.8), 7.08 (1H, d, J = 2.8), 7.13 (1H, dd, J = 3.6 and 5.0), 7.30 (1H, d, J = 3.6), 7.38 (1H, dd, J = 0.8 and 5.0), 7.47 (5H, m), 7.74 (4H, s), 7.99 (2H, d, J = 8.0), 8.29 (1H, d, J = 8.8), 8.33 (3H, m); 13C NMR (100 MHz, CDCl3): δ 167.2, 155.3, 142.1, 137.1, 134.2, 132.4, 131.1, 131.0 (2C), 130.1, 129.4 (2C), 129.0 (2C), 128.5 (2C), 127.7, 127.5, 125.7, 125.0 (3C), 124.8 (2C), 124.2 (2C), 123.8 (3C), 116.5, 113.4, 107.9, 104.0, 66.1, 57.5, 55.6, 42.3, 28.2 and 24.9; LC-MS (ESI): tR = 4.32 min (595.5 [M + H]+); HRMS (ESI): m/z calcd (C39H35N2O2S) 595.2419, found 595.2418 [M + H]+.(5-{2-[3-Anthracen-9-yl-propyl)-methylamino]-ethoxy}-indol-1-yl)-{4′-[(benzyl-methylamino)-methyl]-biphenyl-4-yl}-methanone (17c)
HPLC (220–290 nm): tR = 4.65 min (>99%); IR: νmax/cm−1 1679; 1H NMR (400 MHz, CDCl3): δ 2.06 (2H, m), 2.23 (3H, s), 2.44 (3H, s), 2.75 (2H, t, J = 7.0), 2.93 (2H, t, J = 5.9), 3.56 (2H, s), 3.59 (2H, s), 3.69 (2H, t, J = 8.0), 4.20 (2H, t, J = 5.9), 6.54 (1H, d, J = 3.6), 7.00 (1H, dd, J = 2.6 and 9.0), 7.08 (1H, d, J = 2.6), 7.26 (1H, m), 7.36 (5H, m), 7.47 (6H, m), 7.60 (2H, d, J = 8.0), 7.72 (2H, d, J = 8.4), 7.79 (1H, d, J = 8.4), 7.99 (2H, d, J = 8.0), 8.32 (4H, m); 13C NMR (100 MHz, CDCl3): δ 167.6, 155.2, 143.9, 138.9, 138.4, 137.8, 134.0, 132.3, 131.1, 131.0 (2C), 130.2, 129.2 (2C), 128.9 (4C), 128.5 (2C), 128.3 (2C), 127.6 (3C), 126.5 (2C), 126.4 (3C), 125.1, 124.9 (2C), 124.2 (2C), 123.8 (2C), 116.5, 113.3, 107.9, 103.9, 65.8, 61.2, 60.7, 57.4, 55.4, 42.1, 41.7, 27.9 and 24.9; LC-MS (ESI): tR = 3.94 min (722.6 [M + H]+ and 362.2 [M + 2H]2+); HRMS (ESI): m/z calcd (C50H48N3O2) 722.3746, found 722.3747 [M + H]+.(5-{2-[(3-Anthracen-9-yl-propyl)-methylamino]-ethoxy}-indol-1-yl)-[5-(2-fluorophenyl)-pyridin-3-yl]-methanone (17d)
HPLC (220–290 nm): tR = 5.20 min (>99%); IR: νmax/cm−1 1684; 1H NMR (400 MHz, CDCl3): δ 2.03 (2H, m), 2.42 (3H, s), 2.72 (2H, t, J = 7.0), 2.89 (2H, t, J = 5.9), 3.68 (2H, t, J = 8.0), 4.19 (2H, d, J = 5.9), 6.59 (1H, d, J = 3.8), 7.03 (1H, dd, J = 2.6 and 9.0), 7.09 (1H, d, J = 2.6), 7.25 (3H, m), 7.46 (6H, m), 7.99 (2H, d, J = 8.0), 8.21 (1H, m), 8.32 (4H, m), 8.94 (1H, d, J = 2.2), 8.99 (1H, t, J = 1.8); 13C NMR (100 MHz, CDCl3): δ 165.1, 155.7, 151.4, 147.8, 136.0, 134.2, 131.2, 131.0 (3C), 130.2, 130.1, 130.0, 129.7, 129.0 (2C), 128.5 (2C), 126.8, 125.0, 124.8 (3C), 124.3, 124.1 (3C), 123.8 (2C), 116.6, 115.8 (1C, d, 2JC,F = 22.2), 113.7, 109.1, 104.1, 66.1, 57.5, 55.6, 42.3, 28.2 and 24.9; LC-MS (ESI): tR = 3.97 min (608.6 [M + H]+); HRMS (ESI): m/z calcd (C40H35N3O2F) 608.2713, found 608.2709 [M + H]+.General procedure for Diels–Alder cycloaddition with PS-maleimide resin
To a reaction tube (30 ml) containing swollen maleimide resin (1.5 eq. ∼1.2 mmol g−1) in toluene (3 ml), was added a solution of anthracene 17c (68 mg, 95 µmol) or 17d (114 mg, 188 µmol) in toluene (3 ml). The mixtures were stirred at 100 °C for 16 h and then the resins were thoroughly washed with CH2Cl2, and dried under high vacuum to give the Diels–Alder adducts brown beads 20a (308 mg, quant.) and 20b (355 mg, quant.) as brown beads. Under these conditions, no starting materials remained in solution according to HPLC analysis.2′-Fluoro-biphenyl-4-carboxylic acid methylamide (18a)
Indolyl amide 17a (26 mg, 43 µmol) was dissolved in 1 M methylamine solution in THF (4 ml). The resulting solution was stirred at room temperature for 16 h and then concentrated in vacuo. The residue was loaded onto a pre-washed SCX-2 cartridge (500 mg), and the cartridge was eluted with methanol (3 ml). The eluant was concentrated in vacuo to give the methylamide 18a as a solid (9.6 mg, 96%). HPLC (254 nm): tR = 4.46 min (>99%); 1H NMR (400 MHz, CDCl3): δ 3.03 (3H, d, J = 5.2), 6.24 (1H, brs), 7.18 (1H, m), 7.34 (1H, m), 7.43 (1H, m), 7.61 (2H, d J = 8.2), 7.81 (2H, d, J = 8.2); 13C NMR (100 MHz, CDCl3): δ 167.2, 159.0 (1C, d, 1JC,F = 277.1), 138.2, 133.0, 130.0 (1C, d, 3JC,F = 2.8), 129.0 (1C, d, 4JC,F = 8.3), 128.5 (2C), 127.3 (1C, d, 2JC,F = 13), 126.3 (2C), 123.8 (1C, d, 3JC,F = 3.3), 115.6 (1C, d, 2JC,F = 22.5), and 26.3; LC-MS (ESI): tR = 3.85 min (230.4 [M + H]+); HRMS (EI): m/z calcd (C14H12NOF) 229.0903, found 229.0903 [M]+.2′-Fluoro-biphenyl-4-carboxylic acid methyl ester (18b)
To a solution of indolyl amide 17a (26 mg, 43 µmol) in a mixture of THF and methanol (2 ml/2 ml), was added DMAP (50 mg, 0.41 mmol). The resulting mixture was refluxed for 48 h and then loaded onto a pre-washed SCX-2 cartridge (2 g). The cartridge was eluted with methanol (8 ml), and the eluate was concentrated in vacuo to give methyl ester 18b as a white solid (7.6 mg, 76%). HPLC (254 nm): tR = 5.90 min (>99%); 1H NMR (400 MHz, CDCl3): δ 3.94 (3H, s), 7.19 (1H, m), 7.35 (1H, m), 7.45 (1H, m), 7.62 (2H, d J = 8.2), 8.11 (2H, d, J = 8.2); 13C NMR (100 MHz, CDCl3): δ 166.3, 159.1 (1C, d, 1JC,F = 247.1), 139.7, 130.0 (1C, d, 3JC,F = 2.7), 129.2 (1C, d, 4JC,F = 8.3), 129.0 (2C), 128.6, 128.3 (2C), 127.3 (1C, d, 2JC,F = 13.2), 123.9 (1C, d, 3JC,F = 3.2), 115.6 (1C, d, 2JC,F = 22.5), and 51.5; LC-MS (ESI): tR = 4.55 min (231.4 [M + H]+); HRMS (EI): m/z calcd (C14H11O2F) 230.0743, found 230.0736 [M]+.Piperidin-1-yl-(4-thiophen-2-yl-phenyl)-methanone (18c)
Indolyl amide 17b (40 mg, 68 µmol) was dissolved in 20% piperidine solution in acetonitrile (2.5 ml). The resulting solution was stirred at 80 °C for 16 h, and then concentrated in vacuo. The residue was loaded onto a pre-washed SCX-2 cartridge (500 mg), the cartridge was eluted with methanol, and the eluate was concentrated in vacuo to give piperidinyl amide 18c (13.8 mg, 75%). HPLC (254 nm): tR = 5.24 min (97%); 1H NMR (400 MHz, CDCl3): δ 1.60 (6H, brs), 3.38 (2H, brs), 3.70 (2H, brs), 7.08 (1H, dd, J = 3.6 and 5.2), 7.31 (1H, dd, J = 1.2 and 5.2), 7.34 (1H, dd, J = 1.2 and 3.6), 7.40 (2H, d J = 8.0), 7.62 (2H, d, J = 8.0);. 13C NMR (100 MHz, CDCl3): δ 169.3, 142.8, 134.8, 134.6, 127.5, 127.0 (2C), 125.1 (2C), 124.8, 123.0, 48.2, 42.6, 25.9, 25.0, and 23.9; LC-MS (ESI): tR = 4.23 min (272.4 [M + H]+); HRMS (ESI): m/z calcd (C16H17NOSNa) 294.0929, found 294.0917 [M + Na]+.4′-[(Benzyl-methylamino)-methyl]-biphenyl-4-carboxylic acid propylamide (21a)
A suspension of indolyl amide resin 20a (130 mg, 0.31 mmol g−1, 40 μmol) in 20% propylamine solution in THF (2 ml) was shaken at room temperature for 16 h. The mixture was filtered, and the resin was washed with THF (3×). The combined filtrates were concentrated in vacuo to give propylamide 21a as a white solid (12.3 mg, 82%). HPLC (254 nm): tR = 3.44 min (98%); 1H NMR (400 MHz, CDCl3): δ 0.98 (3H, t, J = 7.6), 1.66 (2H, tq, J = 6.7 and 7.6), 2.21 (3H, s), 3.44 (2H, q, J = 6.7), 3.54 (2H, s), 3.56 (2H, s), 6.18 (1H, brs), 7.25 (1H, m), 7.34 (4H, m), 7.45 (2H, d, J = 8.0), 7.56 (2H, d, J = 8.0), 7.64 (2H, d, J = 8.0), 7.82 (2H, d, J = 8.0); 13C NMR (100 MHz, CDCl3): δ 166.6, 143.3, 138.7, 138.6, 138.0, 132.7, 128.8 (2C), 128.3 (2C), 127.6 (2C), 126.7 (2C), 126.4 (2C), 126.4 (2C), 126.3, 61.2, 60.8, 41.7, 41.1, 22.3 and 10.8;. LC-MS (ESI): tR = 3.12 min (373.6 [M + H]+); HRMS (EI): m/z calcd (C25H28N2O) 372.2202, found 372.2198 [M]+.{4′-[(Benzyl-methylamino)-methyl]-biphenyl-4-yl}-piperidin-1-yl-methanone (21b)
A suspension of indolyl amide resin 20a (130 mg, ∼0.31 mmol g−1, 40 μmol) in 20% piperidine solution in THF (2 ml) was heated at 65 °C for 48 h. The mixture was filtered, and the resin was washed with THF (3 × 2 ml). The combined filtrates were concentrated in vacuo to give piperidinylamide 21b (12.7 mg, 79%). HPLC (254 nm): tR = 3.70 min (96%); 1H NMR (400 MHz, CDCl3): δ 1.62 (6H, brs), 2.21 (3H, s), 3.40 (2H, br), 3.54 (2H, s), 3.56 (2H, s), 3.72 (2H, brs), 7.25 (1H, m), 7.35 (4H, m), 7.45 (4H, m), 7.54 (2H, d, J = 8.0), 7.60 (2H, d, J = 8.0); 13C NMR (100 MHz, CDCl3): δ 169.5, 141.5, 138.6, 138.3 (2C), 134.4, 128.8 (2C), 128.3 (2C), 127.6 (2C), 126.7 (2C), 126.3 (5C), 61.2, 60.8, 48.2, 42.5, 41.7, 25.9, 25.0 and 24.0; LC-MS (ESI): tR = 3.21 min (399.6 [M + H]+); HRMS (EI): m/z calcd (C27H30N2O) 398.2358, found 398.2354 [M]+.5-(2-Fluorophenyl)-N-methyl-nicotinamide (21c)
A suspension of indolyl amide resin 20b (150 mg, 0.52 mmol g−1, 78 μmol) in 2 M methylamine solution in THF (2 ml) was shaken at room temperature for 16 h. The suspension was filtered, and the resin was washed with THF (3×). The combined filtrates were concentrated in vacuo to give methylamide 21c as a white solid (14.1 mg, 79%). HPLC (254 nm): tR = 2.58 min (98%); 1H NMR (400 MHz, CDCl3): δ 3.04 (3H, d, J = 4.8), 6.50 (1H, brs), 7.22 (2H, m), 7.42 (2H, m), 8.27 (1H, s), 8.87 (1H, s), 8.94 (1H, s); 13C NMR (100 MHz, CDCl3): δ 165.5, 159.1 (1C, d, 1JC,F = 247.5), 151.2 (1C, d, 3JC,F = 3.4), 146.0 (2C), 134.6, 131.0, 129.9 (1C, d, 4JC,F = 8.2), 129.4, 124.2 (1C, d, 3JC,F = 3.4), 123.9 (1C, d, 2JC,F = 13.2), 115.7 (1C, d, 2JC,F = 22.1) and 26.3; LC-MS (ESI): tR = 3.33 min (231.4 [M + H]+); HRMS (EI): m/z calcd (C13H11N2OF) 230.0855, found 230.0861 [M]+.5-(2-Fluorophenyl)-nicotinic acid (21d)
Caesium hydroxide (5 mg, 30 μmol) was added to a suspension of indolyl amide resin 20b (150 mg, 0.52 mmol g−1, 78 μmol) in a mixture of THF and water (1.9 ml/0.1 ml). The resulting mixture was shaken at room temperature for 16 h when saturated brine (2 ml) was added. The mixture was extracted with ethyl acetate (3 × 2 ml), and the combined organic layer was concentrated in vacuo to give the carboxylic acid 17g as a white solid. HPLC (254 nm): tR = 3.16 min (>99%); 1H NMR (400 MHz, DMSO): δ 7.36 (2H, m), 7.49 (1H, m), 7.66 (1H, m), 8.36 (1H, s), 8.96 (1H, s), 9.06 (1H, s); 13C NMR (100 MHz, CDCl3): δ 165.8, 159.1 (1C, d, 1JC,F = 245.2), 152.5, 149.1 (2C), 136.4 (1C, d, 3JC,F = 2.8), 130.7 (1C, d, 4JC,F = 8.3), 130.6, 126.3, 125.1 (1C, d, 3JC,F = 3.0), 123.8 (1C, d, 2JC,F = 12.9), and 116.0 (1C, d, 2JC,F = 22.1); LC-MS (ESI): tR = 3.85 min (218.4 [M + H]+); HRMS (EI): m/z calcd (C12H8NO2F) 217.0539, found 217.0537 [M]+.Acknowledgements
We thank GlaxoSmithKline for financial support of this work.References
- (a) E. M. Gordon, M. A. Gallop and D. V. Patel, Acc. Chem. Res., 1996, 29, 144 CrossRef CAS; (b) J. Li, C. W. Murray, B. Waszkowycz and S. C. Young, Drug Discovery Today, 1998, 3, 105 CrossRef CAS; (c) B. A. Kenny, M. Bushfield, D. J. Parry-Smith, S. Fogarty and J. M. Treherne, Prog. Drug Res., 1998, 51, 245 Search PubMed; (d) A. Golebiowski, S. R. Klopfenstein and D. E. Portlock, Curr. Opin. Chem. Biol., 2001, 5, 273 CrossRef.
- (a) J. Yoshida and K. Itami, Chem. Rev., 2002, 102, 3693 CrossRef CAS; (b) A. Ganesan, Drug Discovery Today, 2002, 7, 47 CrossRef CAS; (c) F. Z. Dörwald, Organic Synthesis on Solid Phase, Wiley-VCH, Weinheim, 2000 Search PubMed; (d) D. Obrecht and J. M. Villalgordo, Solid-Supported Combinatorial and Parallel Synthesis of Small-Molecular-Weight Compound Libraries, Pergamon, Oxford, 1998 Search PubMed.
- (a) N. Bailey, A. W. J. Cooper, M. J. Deal, A. W. Dean, A. L. Gore, M. C. Hawes, D. B. Judd, A. T. Merritt, R. Storer, S. Travers and S. P. Watson, Chimia, 1997, 51, 832 CAS; (b) I. Hughes and D. Hunter, Curr. Opin. Chem. Biol., 2001, 5(3), 243 CrossRef CAS.
- (a) D. H. Drewry, D. M. Coe and S. Poon, Med. Res. Rev., 1999, 19, 97 CrossRef; (b) S. V. Ley, I. R. Baxendale, R. N. Bream, P. S. Jackson, A. G. Leach, D. A. Longbottom, M. Nesi, J. S. Scott, R. I. Storer and S. J. Taylor, Chem. Soc., Perkin Trans. 1, 2000, 3815 RSC; (c) A. Kirschning, H. Monenschein and R. Wittenberg, Angew. Chem., Int. Ed., 2001, 40, 650 CrossRef CAS; (d) D. L. Flynn, S. C. Berk and G. M. Makara, Curr. Opin. Drug Discovery Dev., 2002, 5, 580 Search PubMed; (e) S. V. Ley and I. R. Baxendale, Nat. Rev. Drug Discovery, 2002, 1, 573 Search PubMed.
- (a) L. A. Thompson, Curr. Opin. Chem. Biol., 2000, 4, 324 CrossRef CAS; (b) D. L. Flynn, Med. Res. Rev., 1999, 19, 408 CrossRef CAS.
- (a) P. Lan, J. A. Porco Jr., M. S. South and J. J. Parlow, J. Comb. Chem., 2003, 5, 660 CrossRef CAS; (b) X. Wang, J. J. Parlow and J. A. Porco Jr., Org. Lett., 2001, 2, 3509 CrossRef.
- (a) A. M. Hay, S. Hobbs-Dewitt, A. A. MacDonald and R. Ramage, Synthesis, 1999, 1979 CrossRef CAS; (b) J. S. Warmus and M. I. da Silva, Org. Lett., 2000, 2, 1807 CrossRef CAS.
- K. Itami, T. Nokami and J. Yoshida, Angew. Chem., Int. Ed., 2001, 40, 1074 CrossRef CAS.
- S. Zhang, K. Fukase and S. Kusumoto, Tetrahedron Lett., 1999, 40, 7479 CrossRef CAS.
- (a) H. Perrier and M. Labelle, J. Org. Chem., 1999, 64, 2110 CrossRef CAS; (b) D. J. Gravert and K. D. Janda, Chem. Rev., 1997, 97, 489 CrossRef CAS.
- S. V. Ley, A. Massi, F. Rodriguez, D. C. Horwell, R. A. Lewthwaite, M. C. Pritchard and A. M. Reid, Angew. Chem., Int. Ed., 2001, 40, 1053 CrossRef CAS.
- (a) Review: W. Zhang, Tetrahedron, 2003, 59, 4475 Search PubMed; (b) FluoroFlash: D. P. Curran, Synlett, 2001, 1488 Search PubMed; (c) D. P. Curran, D. P. Curran and Y. Oderaotoshi, Tetrahedron, 2001, 57, 5243 CrossRef CAS; (d) D. Schwinn and W. Bannwarth, Helv. Chim. Acta, 2002, 85, 255 CrossRef CAS; (e) D. Schwinn, H. Glatz and W. Bannwarth, Helv. Chim. Acta, 2003, 86, 188 CrossRef CAS.
- S. P. Andrews and M. Ladlow, J. Org. Chem., 2003, 68, 5525 CrossRef CAS.
- X. Li, C. Abell and M. Ladlow, J. Org. Chem., 2003, 68, 4189 CrossRef CAS.
- (a) M. E. Bunnage and K. C. Nicolaou, Angew. Chem., Int. Ed., 1996, 35, 1110 CAS; (b) M. E. Bunnage and K. C. Nicolaou, Chem. Eur. J., 1997, 3, 187 CrossRef CAS.
- (a) E. Arai, H. Tokuyama, M. S. Linsell and T. Fukuyama, Tetrahedron Lett., 1998, 39, 71 CrossRef CAS; (b) M. H. Todd, S. F. Oliver and C. Abell, Org. Lett., 1999, 1, 1149 CrossRef CAS.
- A. S. B. Prasad, J. V. B. Kanth and M. Periasamy, Tetrahedron, 1992, 48, 4623 CrossRef CAS.
- K. Hanaya, T. Muramatsu, H. Kudo and Y. L. Chow, J. Chem Soc., Perkin Trans. 1, 1979, 10, 2409 RSC.
- For example: (a) S. Peukert, J. Brendel, B. Pirard, A. Brüggemann, P. Below, H.-W. Kleemann, H. Hemmerle and W. Schmidt, J. Med. Chem., 2003, 46, 486 CrossRef CAS; (b) D. Lesuisse, E. Albert, F. Bouchoux, E. Cérède, J.-M. Lefrançois, M.-O. Levif, S. Tessier, B. Tric and G. Teutsch, Bioorg. Med. Chem. Lett., 2001, 11, 1713 CrossRef CAS; (c) S. Takekawa, A. Asami, Y. Ishihara, J. Terauchi, K. Kato, Y. Shimomura, M. Mori, H. Murakoshi, K. Kato, N. Suzuki, O. Nishimura and M. Fujino, Eur. J. Pharmacol., 2002, 438, 129 CrossRef CAS.
- Isolute™ SCX-2 (alkylsulfonic acid) cartridges were obtained from Jones Chromatography (e.g. Cat. No. 532-0100-C)..
- The polymer-supported maleimide resin was prepared on a 1–2% cross-linked polystyrene and was determined to have a loading of approximately 1.2 mmol g−1 according to N combustion analysis..
- K. Valko, C. M. Du, C. Bevan, D. P. Reynolds and M. H. Abraham, Curr. Med. Chem., 2001, 8, 1137 Search PubMed.
- HPLC retention times tR were measured using a wide polarity reverse phase method with an 7 min total gradient time as described in the Experimental section.
Footnote |
| † Current address: Astex Technology Ltd, 436 Cambridge Science Park, Milton Road, Cambridge, UK CB4 0QA. |
| This journal is © The Royal Society of Chemistry 2004 |