
An icosahedral Au13 nanocluster with three adjacent chlorides on opposite poles catalyses hydroamination of phenylacetylene
Shen
Wu
,
Shinjiro
Takano
 * and
Tatsuya
Tsukuda
*
* and
Tatsuya
Tsukuda
*
Department of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Japan. E-mail: stakano@chem.s.u-tokyo.ac.jp; tsukuda@chem.s.u-tokyo.ac.jp
First published on 11th November 2025
Abstract
A novel anionic gold nanocluster, [Au13(dppeF)3Cl6]−, bearing six chlorides, has been synthesized using a bulky diphosphine, dppeF (1,2-bis[bis(3,5-ditrifluoromethylphenyl)phosphino]ethane) as a protecting ligand. Single-crystal X-ray diffraction analysis revealed that this nanocluster has an icosahedral Au13 core, which is protected by each of three Cl ligands on opposite poles and by three dppeF ligands in an equatorial plane. This nanocluster acted as a homogeneous catalyst for hydroamination between phenylacetylene and aniline, facilitated by the rapid exchange of the Cl− ligands with phenyl acetylides.
Introduction
In recent decades, the development of ligand-protected gold nanoclusters (AuNCs) has emerged as a significant area of research, due to their unique properties, which can be finely adjusted through the precise control of their size, shape, and surface modifications.1–3 Phosphines are some of the most common types of ligands used to protect AuNCs.4–9 Among numerous phosphine-protected AuNCs reported, icosahedral Au13 NCs protected by alkyl-bridged diphosphines (L = Ph2P-(CH2)n-PPh2; n = 1–5) provide an ideal opportunity to demonstrate how the ligand structure can precisely adjust the geometric structure, stability, and physicochemical properties of AuNCs. The chemical formulas in Table 1 show that the L/Cl ratio in the Au13 NC depends on the n value of L.10–12 The observed trend suggests that one bulkier L is replaced by two less bulky Cl ligands. This exchange decreases the total charge by two, keeping the total number of valence electrons at eight. Based on the sequence of chemical formulas in Table 1, we can predict that a negatively charged NC, such as [Au13L3Cl6]−, will form when L is bulkier than dpppe.| Formula | L | Ref. |
|---|---|---|
| (ArF = 3,5-bis(trifluoromethyl)phenyl). | ||
| [Au13L6]5+ | Ph2P-(CH2)1-PPh2 (dppm) | 10 |
| [Au13L5Cl2]3+ | Ph2P-(CH2)2-PPh2 (dppe) | 11 |
| [Au13L4Cl4]+ | Ph2P-(CH2)3-PPh2 (dppp) | 12 |
| Ph2P-(CH2)4-PPh2 (dppb) | ||
| Ph2P-(CH2)5-PPh2 (dpppe) | ||
| [Au13L3Cl6]− | (ArF)2P-(CH2)2-P(ArF)2 (dppeF) | This study |
The structure of L affects not only the L/Cl ratio but also the structure and optical properties of the Au13 core.13–17 The Au13 core of [Au13(dppe)5Cl2]3+ (Au13:dppe) is a nearly perfect icosahedron, whereas that of [Au13(dppm)6]5+ (Au13:dppm) is significantly distorted into D3 symmetry, probably due to the mechanical stress imposed on the two P sites of dppm. Au13:dppe exhibits photoluminescence (PL) at 766 nm with a quantum yield (QY) of 8%.11 The PLQY drops to 0.8% for [Au13(dppe)4Cl4]+ (Au13:dppp) and is below the detection limit for Au13:dppm.
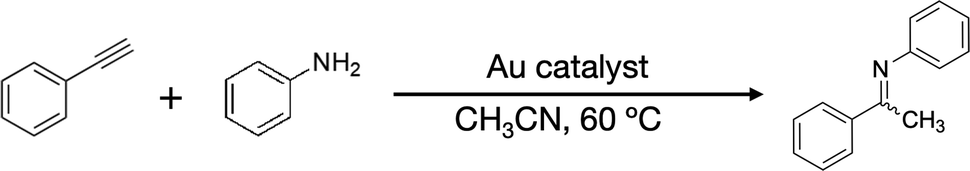
This work aimed at developing atomically precise AuNC catalysts with a Cl-rich surface. The idea comes from a report that the hydrochlorination of phenylacetylene (PA-H) was catalysed by a halide-rich AuNC: [Au13(NHC)6Br6]− (NHC = N-heterocyclic carbene).14 We attempted to synthesize a Cl-rich Au13 NC using (ArF)2PC2H4P(ArF)2 (ArF = 3,5-bis(trifluoromethyl)phenyl; dppeF) as a ligand, which is bulkier than dppe due to the presence of multiple CF3 groups within an otherwise identical molecular framework. We also expect that the resulting Au13 core will be stabilized electronically by the CF3 groups via an inductive mechanism.18 We successfully obtained [Au13(dppeF)3Cl6]− (Au13:dppeF) (Table 1) and determined its structure using single-crystal X-ray diffraction (SCXRD) analysis. Au13:dppeF has an icosahedral Au13 core that is surrounded by three dppeF ligands at the equator and six Cl ligands at the axial Au3 triangles on opposite poles. The superatomic nature of the Au13 core was confirmed by density functional theory (DFT) calculations. Au13:dppeF exhibited higher catalytic activity than Au13:dppe for the hydroamination of PA-H using aniline as the nucleophile.19 The reaction pathways were probed using in situ electrospray ionization mass spectrometry (ESI-MS) by taking advantage of homogeneous catalytic conditions.20–27 Based on the reaction intermediates detected by in situ ESI-MS, we propose that the reaction starts with the activation of PA-H via ligand exchange with Cl, followed by attack by a nucleophile (aniline) to produce the corresponding enamine, which releases imine as a final product.
Results and discussion
Synthesis and structural characterization of Au13:dppeF
A tetraphenylphosphonium (PPh4+) salt of Au13:dppeF was synthesized using a method analogous to the one used to prepare Au13:dppe.11 The synthetic details are provided in the Supporting Information (SI). Briefly, the (AuCl)2dppeF complex in tetrahydrofuran was reduced by NaBH4 at 253 K. The resultant polydispersed mixture of dppeF-protected AuNCs was subjected to an HCl etching process to focus the AuNC sizes. Au13:dppeF was recrystallized from dichloromethane and hexane after alumina column chromatography and repeated reprecipitation, yielding black crystals. The yield was 35% based on Au.Fig. 1a presents a typical negative-mode ESI mass spectrum of (PPh4+)·Au13:dppeF. The intense mass peak was assigned to the formula of [Au13(dppeF)3Cl6]−, as confirmed by comparing the experimental and the calculated isotope patterns. To the best of our knowledge, this is the first example of a negatively charged Au13 NC protected by phosphines. Fig. 1b shows the ultraviolet-visible (UV-Vis) absorption spectra of acetonitrile solutions of Au13:dppeF and Au13:dppe, which were prepared by dissolving crystals of (PPh4+)·Au13:dppeF and Au13:dppe·(PF6−)3, respectively. Both spectra exhibit characteristic peaks that reflect the quantized electronic structures. The spectral profiles of Au13:dppeF and Au13:dppe are similar, but do not match due to their structural differences. The energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of Au13:dppeF is estimated to be 2.0 eV from a spectral onset of the UV-Vis absorption spectrum. This value is comparable to that of Au13:dppe (∼1.9 eV).8,11,16 By combining with the nuclear magnetic resonance (NMR) data of (PPh4+)·Au13:dppeF (Fig. S1), we concluded that the target Au13 NC with six chlorides was successfully obtained.
 | ||
| Fig. 1 (a) Negative-mode ESI mass spectrum of (PPh4+)·Au13:dppeF. The inset compares the isotope patterns experimentally observed (red) and calculated (black) for Au13:dppeF. (b) UV-Vis absorption spectra of (PPh4+)·Au13:dppeF (red) and Au13:dppe·(PF6−)3 (black) in acetonitrile. | ||
Next, we examined the geometric structure of Au13:dppeF using SCXRD analysis. (PPh4+)·Au13:dppeF crystallized in the triclinic crystal system with the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group (Table S1). Fig. 2a shows the determined structure of Au13:dppeF and reveals that it has an icosahedral Au13 core. The continuous symmetry measure (CSM),28 which quantifies deformation from an ideal icosahedron (CSM = 0), was calculated to be 0.029 for the Au13 core in Au13:dppeF. This value is closer to 0 than that of Au13:dppe (CSM = 0.036)8 and indicates that the Au13 core of Au13:dppeF is a nearly perfect icosahedron. Fig. 2b shows the coordination geometry of the ligands around the Au13 core. Three dppeF ligands coordinate with the Au6 ring in a chair configuration on the equator of the Au13 core. The remaining axial Au3 triangles at the top and bottom are fully capped by three chlorides each, giving pseudo-C3 symmetry along the axial direction. Halide-accumulated facets were also observed in [Au13(NHC)6Br6]−, but they were divided into asymmetric sites of Br4 and Br2.14 These results imply that halides tend to segregate on the AuNC surface. However, the limited examples of polyhalogenated Au13 NCs make it difficult to conclude whether the segregated behavior of halides is a general feature.
space group (Table S1). Fig. 2a shows the determined structure of Au13:dppeF and reveals that it has an icosahedral Au13 core. The continuous symmetry measure (CSM),28 which quantifies deformation from an ideal icosahedron (CSM = 0), was calculated to be 0.029 for the Au13 core in Au13:dppeF. This value is closer to 0 than that of Au13:dppe (CSM = 0.036)8 and indicates that the Au13 core of Au13:dppeF is a nearly perfect icosahedron. Fig. 2b shows the coordination geometry of the ligands around the Au13 core. Three dppeF ligands coordinate with the Au6 ring in a chair configuration on the equator of the Au13 core. The remaining axial Au3 triangles at the top and bottom are fully capped by three chlorides each, giving pseudo-C3 symmetry along the axial direction. Halide-accumulated facets were also observed in [Au13(NHC)6Br6]−, but they were divided into asymmetric sites of Br4 and Br2.14 These results imply that halides tend to segregate on the AuNC surface. However, the limited examples of polyhalogenated Au13 NCs make it difficult to conclude whether the segregated behavior of halides is a general feature.
 | ||
| Fig. 2 (a) The entire structure of Au13:dppeF, as determined by SCXRD analysis. (b) Top and side views of Au13:dppeF. Hydrogen atoms, ArF moieties, a solvent molecule, and the PPh4+ ion are omitted for clarity. Color code: Au, yellow; P, orange; Cl, green; C, gray; F, yellow-green. | ||
The electronic structure of Au13:dppeF was investigated using DFT calculations with a full ligand model. Structural optimization was conducted using the crystal structure of Au13:dppeF as the initial structure. The optimized structure, shown in Fig. S2, reproduced the crystal structure shown in Fig. 2. Fig. 3 shows the energy diagrams and the corresponding Kohn–Sham (KS) orbitals of Au13:dppeF. As expected, given the eight valence electrons calculated for Au13:dppeF, the HOMO, HOMO−1, and HOMO−2 are assigned to three 1P superatomic orbitals. This confirms the electron configuration of (1S)2(1P)6. Despite the nearly perfect icosahedral Au13 core, the 1P orbitals are split into two subgroups, 1Pz and 1Px/1Py. The 1Pz orbital is more stable than the 1Px/1Py orbitals due to electrostatic stabilization by the six electron-withdrawing chlorides located along the z-axis. This also explains why the 1Dz2 orbital distributed along the z axis corresponds to the LUMO. The HOMO–LUMO gap was calculated to be 2.6 eV, which is larger than 2.0 eV determined by the optical method. A similar overestimation was observed in the case of Au13:dppe.13
 | ||
| Fig. 3 Energy diagrams and KS orbitals of Au13:dppeF (isovalue = 0.02). The top-right figure shows the orientation of the Au13:dppeF NC without the ArF moieties and H atoms, which have been removed for clarity. | ||
Catalytic application of Au13:dppeF to hydroamination reaction
Fig. 4 compares the space-filling models of Au13:dppeF and Au13:dppe. The Au13 core surface of Au13:dppeF is clearly visible through the gap between the three Cl ligands. In contrast, the Au13 core of Au13:dppe is barely visible. This remarkable difference in the steric environment suggests that reactants can readily access the Au13 core of Au13:dppeF than that of Au13:dppe. The catalytic application of well-defined AuNCs can provide useful information for establishing a correlation between structure and catalysis.29–32 Inspired by the hydrochlorination of PA-H by the halide-rich [Au13(NHC)6Br6]−,14 we compared the catalytic activity of Au13:dppeF and Au13:dppe for the hydroamination of terminal alkynes by amines under homogeneous conditions. PA-H and aniline were selected as the model terminal alkyne and amine, respectively.19 As shown in Table 2, Au13:dppeF catalysed the reaction (entry 1): the conversion after 24 h was 29% (Fig. S3), increasing up to ∼95% after two weeks. Conversion after 24 h increased with the amount of Au13:dppeF loading and reached up to 82% at 0.040 mol% (entries 2–4). The gradual decrease in TON values with increasing catalyst loading (entries 2–4) is due to the decreasing substrate concentration. In contrast, the conversions after 2 h were nearly proportional to the catalyst concentration, and TON values remained constant (Table S2). These results suggest that the active sites of the catalyst are uniform, despite the presence of multiple halide sites. The blank test (entry 10) and the poor conversion by the (AuCl)2-dppeF complex (entry 8) indicate that the higher conversion is associated with Au13:dppeF. Conversion by Au13:dppeF was higher than that by Au13:dppe (entry 9). This result supports our hypothesis that the less crowded chlorinated area provides the active site. | ||
| Fig. 4 Space-filling models of Au13:dppeF (left) and Au13:dppe (right) around the Cl coordination sites. Color code: Au, yellow; P, orange; Cl, green; C, gray; F, yellow-green. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | mol%b | Conversion (%) | TONc |
| a General conditions: PA-H (1.0 M), aniline (5.0 M), 500 rpm, 24 h. Conversion was estimated by 1H NMR analysis using 1,3,5-trimethoxybenzene as an internal standard. b Molecular base. c TON = Conversion (%)/mol%. d Not determined. | ||||
| 1 | Au13:dppeF | 0.010 | 29 | 2.9 × 103 |
| 2 | Au13:dppeF | 0.015 | 41 | 2.7 × 103 |
| 3 | Au13:dppeF | 0.027 | 67 | 2.4 × 103 |
| 4 | Au13:dppeF | 0.040 | 82 | 2.1 × 103 |
| 5 | Au13:dppeF/Br | 0.0098 | 6.3 | 6.3 × 102 |
| 6 | Au13:dppeF/I | 0.010 | <1 | n.d.d |
| 7 | Au13:dppeF/PA | 0.011 | 32 | 3.2 × 103 |
| 8 | (AuCl)2-dppeF | 0.065 | 9.1 | 1.4 × 102 |
| 9 | Au13:dppe | 0.010 | 4.8 | 4.8 × 102 |
| 10 | blank | — | <1 | n.d.d |
It has been reported that the Cl ligands in Au13:dppe can be replaced by PA−.13 To confirm that the same ligand exchange occurs in Au13:dppeF, we measured the ESI mass spectra of a mixed solution of Au13:dppeF and 50 equivalents of PA-H in the presence of the Brønsted base triethylamine. As expected, the dominant species detected was the fully PA-exchanged AuNCs [Au13(dppeF)3(PA)6]− (Au13:dppeF/PA) (Fig. S4). This observation led us to hypothesize that Au13:dppeF undergoes ligand exchange from Cl to PA, and that the resultant PA-functionalized Au13 NCs are involved in the catalytic reactions. To investigate this possibility, we characterized the AuNCs after a 24-hour catalytic reaction of Au13:dppeF using ESI-MS. As shown in Fig. S5, the main species detected was the fully PA-exchanged AuNCs Au13:dppeF/PA. We also studied the catalytic activity of the pre-synthesized Au13:dppeF/PA. As shown in entry 7 of Table 2, the catalytic activity of Au13:dppeF/PA was slightly higher than that of Au13:dppeF (entry 1, Table 2). These results suggest that the PA ligands introduced onto the Au13 NCs are key to hydroamination by aniline.
To further support the above suggestions, we examined the impact of halogens on activity. To this end, we synthesized the halide-exchanged analogues [Au13(dppeF)3Br6]− (Au13:dppeF/Br) and [Au13(dppeF)3I6]− (Au13:dppeF/I) by mixing Au13:dppeF with KBr and KI, respectively.15 The synthetic details and characterization results are provided in the SI (Fig. S6–S9). SCXRD analysis of the halide-exchanged products revealed the same coordination geometry (Fig. S10 and Table S1), indicating that these NCs can provide direct insight into the effect of halides on the catalytic activity. As shown in entries 1, 5, and 6 of Table 2, the reactivity decreases in the following order: Au13:dppeF > Au13:dppeF/Br ≫ Au13:dppeF/I. This trend reveals that the nature of the halides significantly affects activity. To elucidate halide-dependent catalytic activity, we compared the ligand exchange activity of Au13:dppeF, Au13:dppeF/Br, and Au13:dppeF/I with the PA− ligands using ESI-MS. We added 10 equivalents of PA-H to solutions of Au13:dppeF, Au13:dppeF/Br, and Au13:dppeF/I, in the presence of 10 equivalents of triethylamine, to promote the deprotonation of the terminal alkynyl proton. Fig. S11 shows the SI mass spectra of the mixed solutions after 24 h. Partially or fully PA-exchanged AuNCs [Au13(dppeF)3(PA)nX6−n]− (X: Cl, Br, or I) were detected similarly. The average number of PA ligands introduced (n) was ∼1.6, ∼0.3, and <0.1 for Au13:dppeF, Au13:dppeF/Br, and Au13:dppeF/I, respectively. These numbers increased to 5.9, 4.1, and 0.3, respectively, after 24 h of the catalytic reaction, where an excess of PA-H is present (Fig. S12). These results indicate that reactivity toward PA exchange decreased in the following order: Au13:dppeF > Au13:dppeF/Br ≫ Au13:dppeF/I. This is consistent with the activity order shown in entries 1, 5 and 6 of Table 2. Fig. S13 shows the time-course results for the catalysts of Au13:dppeF, Au13:dppeF/Br, Au13:dppeF/I and Au13:dppeF/PA (entries 1 and 5–7, Table 2). These results suggest that activating the PA ligands on Au13via ligand exchange is a critical step in the reaction.
The catalytic hydroamination reaction has been studied using an Au complex,33 an Au surface,34 Au nanoparticles (AuNPs),35–37 and AuNCs.19 All of these Au catalysts, except for the AuNCs, activate PA-H via a π-bond on an Au atom while leaving the terminal hydrogen atoms intact, as shown in Fig. 5a. In the case of [Au16(bi-NHC)5(PA)3Br2]3+ (bi-NHC = bidentate NHC ligand),19 although PA forms novel surface units of (bi-NHC)–Au(I)–PA on the icosahedral Au13 core, it is π-bonded to the Au13 core. Contrary to these examples, we propose σ-bonding of PA to the Au13 core of Au13:dppeF (Fig. 5b), based on the SCXRD result of a similar system, [Au13(dppe)5(PA)2]3+.13 To further support this hypothesis, DFT structural optimization was conducted for Au13:dppeF/PA by replacing six Cl ligands of Au13:dppeF with σ-ligated PA ligands. The optimized structure, shown in Fig. S14, supports σ-bonding of PA to the Au13 NC. The difference in the PA bonding mode is likely due to the difference in the steric environment at the adsorption site: σ-bonding is preferred at sterically crowded sites, while π-bonding is possible only at sterically open sites. The σ-bonding of PA to the Au13 NC alters the charge distribution of the PA molecule via electronic coupling between the 1P superatomic orbital of the Au13 core and the π orbital of the PA molecule, as theoretically demonstrated by Konishi.13 According to the natural bond orbital (NBO) analysis of our DFT-optimized structures of Au13:dppeF/PA and free PA−, the electronic charge transfers from PA to the Au13 NC. The NBO charges of the terminal C (Cα) and the adjacent C (Cβ) atoms of the PA ligand suggest that Cα becomes electron-rich and Cβ becomes electron-deficient upon ligation (Table S3).
 | ||
| Fig. 5 Schematic representation of (a) π-ligation of PA-H to an Au surface and (b) σ-bonding of PA to Au NC. (c) Proposed reaction mechanism of hydroamination of PA-H on Au13 NCs. | ||
Based on the above discussions, we propose the following catalytic mechanism for Au13:dppeF, as depicted schematically in Fig. 5c. The reaction begins with ligand exchange with PA-H in the presence of excess aniline, which acts as a Brønsted base (step i). Next, the σ-bonded PA is attacked by the nucleophile aniline. The nucleophilic attack by aniline occurs at the electron-deficient Cβ, to produce an enamine according to the Markovnikov rule (step ii). This nucleophilic attack of aniline on the σ-bonded PA substrate has also been proposed as a key step in the catalysis of Au16 NC.19 Finally, enamine tautomerizes to the imine (step iii), and then protodeauration and ligand exchange with a new PA occur (step iv).
Finally, we evaluated the stability of Au13:dppeF during the catalytic reaction. After 24 h of the reaction, we collected the catalysts by evaporating the solvent, followed by washing with a toluene–hexane mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) to remove the remaining reactants and the products. The precipitate exhibited a similar UV-Vis absorption profile to that of (PPh4+)·Au13:dppeF/PA (Fig. 6), which is consistent with the ESI-MS results. The catalysts after the 1st and 2nd usage exhibited similar spectral profiles (Fig. 6). These results suggested that the NC retained its Au13:dppeF/PA structure, indicating the stability of Au NCs during the catalysis. We also compared catalytic activity during recycled usage. The conversion by 0.01 mol% catalyst decreased from 32% to 19% and 11%, respectively, in the 2nd and 3rd cycles (Table 3). The gradual reduction in conversion with reuse is due to the reduced recovery of Au13:dppeF/PA NCs by washing with a toluene–hexane mixture. The weight of the recovered catalyst was reduced to ∼60% of the original value by washing (Table 3). Assuming the recovered precipitate was pure Au13:dppeF/PA NCs, the TON at the 2nd and 3rd cycles was calculated to be 3.1 × 103 and 3.0 × 103, respectively (Table 3). These values closely match the TON for Au13:dppeF/PA (3.2 × 103, entry 7, Table 2), which reinforces the high durability of this NC.
:3) to remove the remaining reactants and the products. The precipitate exhibited a similar UV-Vis absorption profile to that of (PPh4+)·Au13:dppeF/PA (Fig. 6), which is consistent with the ESI-MS results. The catalysts after the 1st and 2nd usage exhibited similar spectral profiles (Fig. 6). These results suggested that the NC retained its Au13:dppeF/PA structure, indicating the stability of Au NCs during the catalysis. We also compared catalytic activity during recycled usage. The conversion by 0.01 mol% catalyst decreased from 32% to 19% and 11%, respectively, in the 2nd and 3rd cycles (Table 3). The gradual reduction in conversion with reuse is due to the reduced recovery of Au13:dppeF/PA NCs by washing with a toluene–hexane mixture. The weight of the recovered catalyst was reduced to ∼60% of the original value by washing (Table 3). Assuming the recovered precipitate was pure Au13:dppeF/PA NCs, the TON at the 2nd and 3rd cycles was calculated to be 3.1 × 103 and 3.0 × 103, respectively (Table 3). These values closely match the TON for Au13:dppeF/PA (3.2 × 103, entry 7, Table 2), which reinforces the high durability of this NC.
 | ||
| Fig. 6 UV-Vis absorption spectra of initial Au13:dppeF (black) and Au13:dppeF after cycles 1 (dark brown) and 2 (brown), along with the spectrum of Au13:dppeF/PA (red) as a reference. | ||
| Cycle | Weight (mg)b | mol%c | Conversion (%) | TONd |
|---|---|---|---|---|
| a General conditions: PA-H (1.0 M), aniline (5.0 M), 500 rpm, 24 h. Conversion was estimated by 1H NMR analysis using 1,3,5-trimethoxybenzene as an internal standard. b Weight of the loading catalyst in each run. c Molecular base. d TON = Conversion (%)/mol%. e Calculated as Au13:dppeF/PA. | ||||
| 1 | 6.0 | 0.010 | 29 | 2.9 × 103 |
| 2 | 3.9 | 0.0061e | 19 | 3.1 × 103 |
| 3 | 2.3 | 0.0036e | 11 | 3.0 × 103 |
Conclusions
We successfully synthesized an anionic Au13 NC with six Cl ligands by using bulky diphosphines as protecting ligands. Single-crystal X-ray diffraction analysis revealed that the six chlorides coordinate with the two Au3 facets located at opposite poles of the icosahedral Au13 core. The Au13 NC was then used to catalyse the hydroamination reaction between PA-H and aniline to form an enamine. Based on the reaction intermediates detected by in situ ESI-MS, we propose that the reaction begins with the dissociative adsorption of PA-H, while releasing a Cl ligand. Then, a nucleophile aniline attacks at the electron-deficient Cβ of the adsorbed PA, producing the corresponding enamine. The efficient introduction of PA was attributed to the less crowded environment of the Cl-accumulated Au3 facets.Author contributions
S. Wu: investigation, visualization, writing – original draft, writing – review & editing. S. Takano: conceptualization, investigation, funding acquisition, supervision, validation, writing – original draft, writing – review & editing. T. Tsukuda: conceptualization, funding acquisition, resources, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5cp03444b.CCDC 2481066, 2484473 and 2484474 contain the supplementary crystallographic data for this paper.38a–c
Acknowledgements
This work was financially supported by JST CREST (grant number: JPMJCR20B2) and JSPS KAKENHI grants (no. JP23H00284 and JP23K26610).Notes and references
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208 CrossRef CAS PubMed.
- X. Zou, X. Kang and M. Zhu, Chem. Soc. Rev., 2023, 52, 5892 RSC.
- B. K. Teo and H. Zheng, Coord. Chem. Rev., 1995, 143, 611 CrossRef CAS.
- K. Konishi, Struct. Bonding, 2014, 161, 49 CrossRef.
- D. M. P. Mingos, Dalton Trans., 2015, 44, 6680 RSC.
- M. R. Narouz, K. M. Osten, P. J. Unsworth, R. W. Y. Man, K. Salorinne, S. Takano, R. Tomihara, S. Kaappa, S. Malola, C.-T. Dinh, J. D. Padmos, K. Ayoo, P. J. Garrett, M. Nambo, J. H. Horton, E. H. Sargent, H. Häkkinen, T. Tsukuda and C. M. Crudden, Nat. Chem., 2019, 11, 419 CrossRef CAS PubMed.
- S. Takano and T. Tsukuda, J. Am. Chem. Soc., 2021, 143, 1683 CrossRef CAS PubMed.
- Y. Niihori, S. Miyajima, A. Ikeda, T. Kosaka and Y. Negishi, Small Sci., 2023, 3, 2300024 CrossRef CAS PubMed.
- S.-S. Zhang, L. Feng, R. D. Senanayake, C. M. Aikens, X.-P. Wang, Q.-Q. Zhao, C.-H. Tung and D. Sun, Chem. Sci., 2018, 9, 1251 RSC.
- Y. Shichibu and K. Konishi, Small, 2010, 6, 1216 CrossRef CAS PubMed.
- Y. Shichibu, K. Suzuki and K. Konishi, Nanoscale, 2012, 4, 4125 RSC.
- M. Sugiuchi, Y. Shichibu, T. Nakanishi, Y. Hasegawa and K. Konishi, Chem. Commun., 2015, 51, 13519 RSC.
- H. Shen, S. Xiang, Z. Xu, C. Liu, X. Li, C. Sun, S. Lin, B. K. Teo and N. Zheng, Nano Res., 2020, 13, 1908 CrossRef CAS.
- Z.-H. Gao, J. Dong, Q.-F. Zhang and L.-S. Wang, Nanoscale Adv., 2020, 2, 4902 RSC.
- H. Hirai, S. Takano, T. Nakashima, T. Iwasa, T. Taketsugu and T. Tsukuda, Angew. Chem., Int. Ed., 2022, 61, e202207290 CrossRef CAS PubMed.
- E. L. Albright, T. I. Levchenko, V. K. Kulkarni, A. I. Sullivan, J. F. DeJesus, S. Malola, S. Takano, M. Nambo, K. Stamplecoskie, H. Häkkinen, T. Tsukuda and C. M. Crudden, J. Am. Chem. Soc., 2024, 146, 5759 CrossRef CAS.
- S. Ito, K. Koyasu, S. Takano and T. Tsukuda, J. Phys. Chem. Lett., 2021, 12, 10417 CrossRef CAS.
- H. Shen, Q. Wu, S. Malola, Y.-Z. Han, Z. Xu, R. Qin, X. Tang, Y.-B. Chen, B. K. Teo, H. Häkkinen and N. Zheng, J. Am. Chem. Soc., 2022, 144, 10844 CrossRef CAS PubMed.
- S.-F. Yuan, Z. Lei, Z.-J. Guan and Q.-M. Wang, Angew. Chem., Int. Ed., 2021, 60, 5225 CrossRef CAS.
- K. Isozaki, R. Ueno, K. Ishibashi, G. Nakano, H. Yin, K. Iseri, M. Sakamoto, H. Takaya, T. Teranishi and M. Nakamura, ACS Catal., 2021, 11, 13180 CrossRef CAS.
- S. Wang, L. Tang, B. Cai, Z. Yin, Y. Li, L. Xiong, X. Kang, J. Xuan, Y. Pei and M. Zhu, J. Am. Chem. Soc., 2022, 144, 3787 CrossRef CAS.
- J. Dong, J. R. Robinson, Z.-H. Gao and L.-S. Wang, J. Am. Chem. Soc., 2022, 144, 12501 CrossRef CAS PubMed.
- K. Isozaki, K. Iseri, R. Saito, K. Ueda and M. Nakamura, Angew. Chem., Int. Ed., 2023, 63, e202312135 CrossRef.
- C. Zhu, Z.-L. Chen, H. Li, L. Lu, X. Kang, J. Xuan and M. Zhu, J. Am. Chem. Soc., 2024, 136, 23212 CrossRef.
- K. Ueda, R. Saito, K. Iseri, S. Sekiya, M. Nakamura and K. Isozaki, ACS Catal., 2025, 15, 12260 CrossRef CAS.
- L.-J. Liu, X. Lei, J. Guo, X. Mo, Y. Lao, S. Zhuang, H. Zeng, S. Yang, Y. Zhao, W. W. Xu and J. He, J. Am. Chem. Soc., 2025, 147, 27981 CrossRef CAS PubMed.
- J. Echeverría, D. Casanova, M. Llunell, P. Alemany and S. Alvarez, Chem. Commun., 2008, 2717 RSC.
- P. Liu, R. Qin, G. Fu and N. Zheng, J. Am. Chem. Soc., 2017, 139, 2122 CrossRef CAS PubMed.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526 CrossRef CAS PubMed.
- R. Jin, G. Li, S. Sharma, Y. Li and X. Du, Chem. Rev., 2021, 121, 567 CrossRef CAS.
- S. Masuda, K. Sakamoto and T. Tsukuda, Nanoscale, 2024, 16, 4514 RSC.
- T. Müller, K. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795 CrossRef PubMed.
- Y. Lu, Y. Wang, H. Li, P. Li, X. Feng, Y. Yamamoto, M. Bao and J. Liu, RSC Adv., 2023, 13, 3371 RSC.
- A. D. Litta, A. Buonerba, A. Casu, A. Falqui, C. Capacchione, A. Franconetti, H. Garcia and A. Grassi, J. Catal., 2021, 400, 71 CrossRef.
- V. I. Isaeva, K. Papathanasiou, V. V. Chernyshev, L. Glukhov, G. Deyko, K. K. Bisht, O. P. Tkachenko, S. V. Savilov, N. A. Davshan and L. M. Kustov, ACS Appl. Mater. Interfaces, 2021, 13, 59803 CrossRef CAS.
- M. Boundor, N. Katir, S. Royer and A. El Kadib, ACS Appl. Nano Mater., 2025, 8, 639 CrossRef CAS.
- (a) CCDC 2481066: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2p8rd4; (b) CCDC 2484473: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2pd99q; (c) CCDC 2484474: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2pd9br.
| This journal is © the Owner Societies 2025 |