
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper-catalyzed four-component borocarbonylative allylation of vinyl arenes to β-boryl enones
Sufang
Shao
ab and
Xiao-Feng
Wu
 *ab
*ab
aDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 116023 Dalian, Liaoning, China. E-mail: xwu2020@dicp.ac.cn
bLeibniz-Institut für Katalyse e.V., 18059 Rostock, Germany
First published on 30th October 2025
Abstract
We present here a novel copper-catalyzed four-component borocarbonylative allylation of aryl olefins. Utilizing carbon monoxide as the carbonyl source under mild reaction conditions, this protocol employs readily available aryl olefins, bis(pinacolato)diboron, and allylic phosphates as the substrates to enable the simultaneous introduction of boron, carbonyl, and allyl groups.
The carboboration of alkenes is a process that has been proven to be an efficient approach to the construction of complex molecular architectures from simple olefin precursors. This stems from the unique reactivity of carbon-boron bonds, which can be efficiently converted into carbon–carbon, carbon-nitrogen and carbon–oxygen bonds.1 This confers significant scientific and applied value to the development of carboboration systems under mild conditions. Several catalytic systems have been developed for alkene carboboration. In transition metal catalysis, copper or nickel-catalyzed systems have been used for alkylboration,2 hydroxyboration-alkylation,3 carboxyboration of alkenes,4 and so on. For introducing an aryl functional group, Cu/Pd co-catalysis,5 Cu/Ni co-catalysis,6 Ni-catalysis7 and Pd8 monometallic catalysis have shown exceptional reactivity and selectivity (Fig. 1a).
 | ||
| Fig. 1 Metal-catalyzed carboboration of alkenes. | ||
β-Boryl carbonyl compounds serve as pivotal intermediates in organic synthesis, enabling efficient transformations into diverse organoboron derivatives. However, the concurrent installation of carbonyl and organoboron functionalities in simple molecular frameworks remains a synthetic challenge. Inspired by alkene borofunctionalization, significant advances have been made in their synthesis via the simultaneous introduction of carbonyl and boryl groups across C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, enabling the efficient construction of complex molecular frameworks. The investigations demonstrated that transition metal catalytic systems, such as those based on palladium or copper, exhibited exceptional reactivity towards such transformations.9 Notably, in 2021 we reported a Pd/Cu co-catalyzed borocarbonylation with reversed selectivity, achieving fully regioselective synthesis of β-boryl ketones (Fig. 1b).10 Building on this, the present study realized the one-step multifunctionalization of alkenes with successful allyl group introduction. This work reports a novel copper-catalyzed four-component borocarbonylative allylation of aryl olefins. Using carbon monoxide (CO) as the carbonyl source,11 this method uses aryl olefins, bis(pinacolato)diboron and allylic phosphates under mild conditions to construct boryl, carbonyl and allyl groups simultaneously, demonstrating broad substrate scope and functional group compatibility (Fig. 1c).
C bonds, enabling the efficient construction of complex molecular frameworks. The investigations demonstrated that transition metal catalytic systems, such as those based on palladium or copper, exhibited exceptional reactivity towards such transformations.9 Notably, in 2021 we reported a Pd/Cu co-catalyzed borocarbonylation with reversed selectivity, achieving fully regioselective synthesis of β-boryl ketones (Fig. 1b).10 Building on this, the present study realized the one-step multifunctionalization of alkenes with successful allyl group introduction. This work reports a novel copper-catalyzed four-component borocarbonylative allylation of aryl olefins. Using carbon monoxide (CO) as the carbonyl source,11 this method uses aryl olefins, bis(pinacolato)diboron and allylic phosphates under mild conditions to construct boryl, carbonyl and allyl groups simultaneously, demonstrating broad substrate scope and functional group compatibility (Fig. 1c).
In the preliminary investigation of this study, the catalytic reaction of vinylarene (1f) with 2-methylallyl diphenyl phosphate (2f) was systematically examined using B2pin2 as the boron source in DMAc solvent at 50 °C under 10 bar CO atmosphere (Table 1). Initially, a screening experiment was conducted in which a variety of ligands were examined under the reaction conditions with lithium methoxide employed as the base (Table 1, entries 1–8). However, experimental data demonstrated that utilization of ligand L2 resulted in an enantiomeric purity (ee) value of the target product of only 26%, with a reaction yield of 49% (Table 1, entry 2). While the use of ligand L6 led to a significant decline in ee value to 14%, accompanied by a yield as low as 24% (Table 1, entry 6). It was observed that reactions with other chiral ligands resulted in the production of racemic products. By increasing the reaction temperature to 50 °C, the yield of 4f can be improved to 80% with 71% isolated yield (Table 1, entry 9). In order to enhance the synthesis efficiency of racemic product, the feeding ratio of reaction substrates 1f and 2f was further optimized (Table 1, entries 1, 10–13). However, no better yield of the target product could be obtained. The enantioselectivity of the desired product could not be improved by further testing with chiral ligands which might due to the high acidity of the tertiary benzylic C–H bond under strong basic conditions. Additionally, similar result was obtained with racemic Ph-BPE ligand. Due to the low stability of alkylborane reagent and also for easier purification, the generated borocarbonylation product was oxidized into the corresponding alcohol 4f.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 1f (eq.) | 2f (eq.) | Ligand | T (°C) | Yield (%) | ee |
| Reaction conditions: 1f (0.1 mmol), 2f (0.3 mmol), CuCl (10 mol%), ligand (10 mol%), LiOMe (2.0 equiv) and DMAc (1 ml) at 50 °C for 24 h under CO (10 bar). Yields were determined by GC-FID analysis using n-dodecane as internal standard.a Yield of isolated product. | ||||||
| 1 | 1 | 3 | L1 | 20 | 63 | 0 |
| 2 | 1 | 3 | L2 | 20 | 49 | 26 |
| 3 | 1 | 3 | L3 | 20 | 0 | — |
| 4 | 1 | 3 | L4 | 20 | 57 | 0 |
| 5 | 1 | 3 | L5 | 20 | 38 | 0 |
| 6 | 1 | 3 | L6 | 20 | 24 | 14 |
| 7 | 1 | 3 | L7 | 20 | 8 | — |
| 8 | 1 | 3 | L8 | 20 | 17 | — |
| 9 | 1 | 3 | L1 | 50 | 80(71a) | 0 |
| 10 | 1 | 2 | L1 | 50 | 60 | 0 |
| 11 | 1 | 1 | L1 | 50 | 34 | 0 |
| 12 | 2 | 1 | L1 | 50 | 24 | 0 |
| 13 | 3 | 1 | L1 | 50 | 30 | 0 |
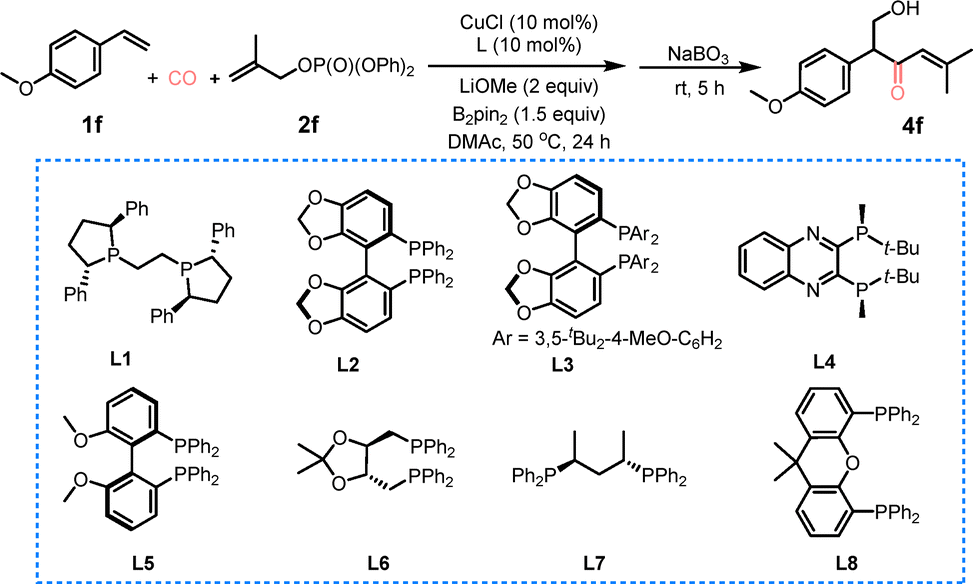
Following the optimization of the reaction conditions, a systematic investigation was conducted into the substrate scope of alkenes (Scheme 1). In order to facilitate the isolation process, the Bpin moiety was oxidized to a hydroxyl group, thereby yielding product 4. As illustrated in Scheme 1, a variety of aryl alkenes underwent the copper-catalyzed four-component borocarbonylative allylation, yielding the corresponding products in moderate yields (up to 76%). It is noteworthy that the positional effect of alkyl substituents on the aromatic ring was negligible (4a–4d, 66–72% yields). Substrates bearing electron-donating groups (4f, 4g) exhibited minimal yield variation, whereas the halogen-substituted alkene (4e) afforded the target product in a slightly reduced yield (55%). The N-containing alkene (4j) exhibited notable reactivity, yielding the desired product in a 59% yield. It is important to note that natural product-derived alkenes, including furfuryl alcohol-, nerol-, and DL-menthol-derived substrates (4l–4n), were efficiently transformed into the desired products in good yields. The scope of electrophiles was further explored as well. Allylic phosphates bearing ortho-, meta-, or para-substituted aromatic rings (o-tolyl, m-tolyl, p-methoxyphenyl) at the 2-position were found to engage in the reaction smoothly, furnishing the corresponding borocarbonylative allylation products (4p–4r) in 50–58% yields.
 | ||
| Scheme 1 Substrate scope. Reaction conditions: 1 (0.1 mmol), 2 (0.3 mmol), CuCl (10 mol%), L1 (10 mol%), LiOMe (2.0 eq.) and DMAc (1 ml) at 50 °C for 24 h under CO (10 bar), isolated yields. | ||
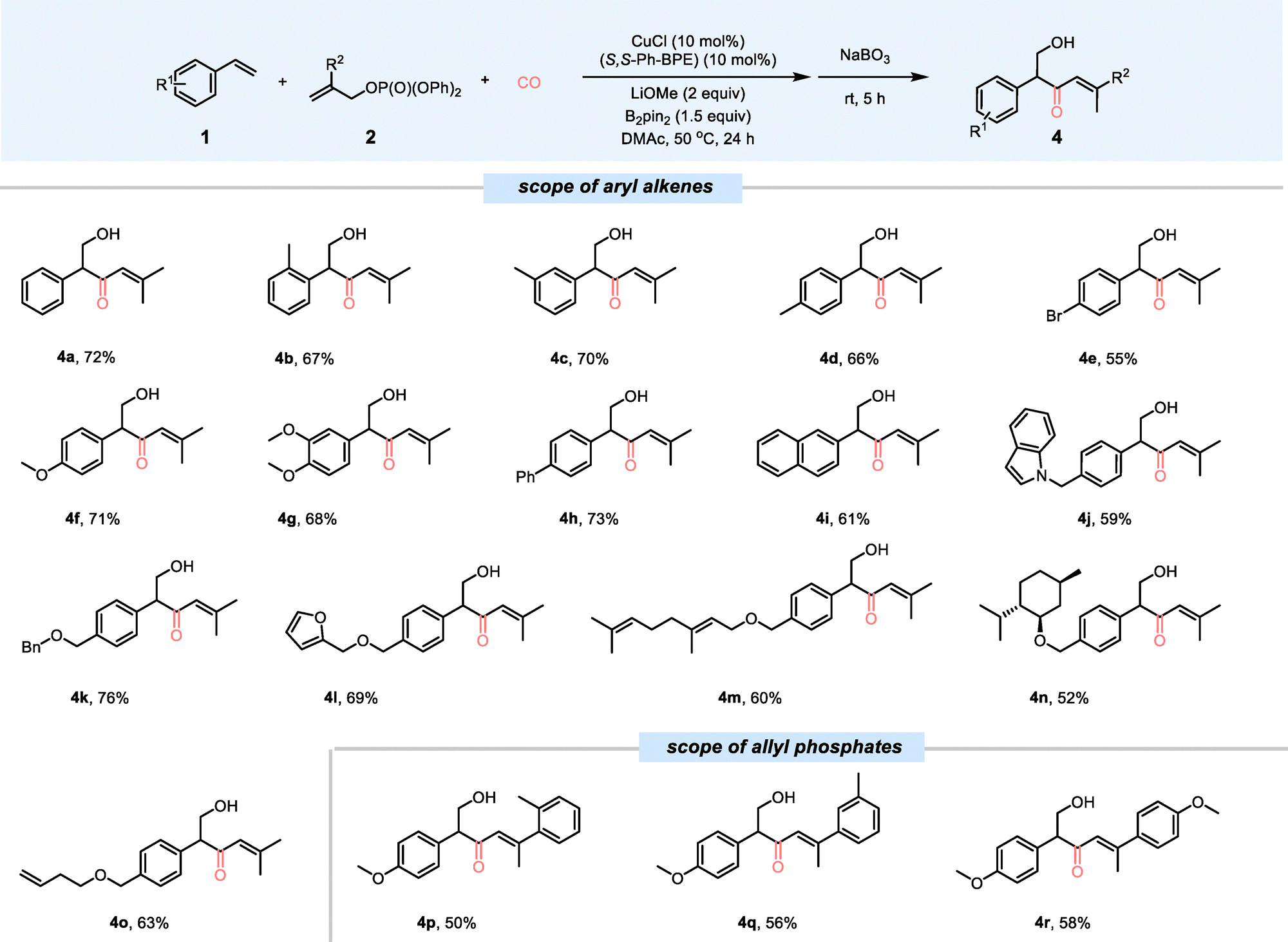
Under the optimized reaction system, it is possible to scale up this transformation to a 1 mmol scale and afforded product 4a in a 60% isolated yield (Scheme 2a). In order to expand the synthetic utility of this method further, diverse derivatizations of product 3a were conducted. The oxidation of 3a with NaBO3 resulted in the production of 4a, with an overall yield of 80% (Scheme 2b). When 3a was employed as a nucleophilic component in the Suzuki–Miyaura cross-coupling reaction, it reacted with bromobenzene to yield the desired product 5a in a 68% yield (Scheme 2c).
 | ||
| Scheme 2 Scale-up reaction and derivatizations. | ||
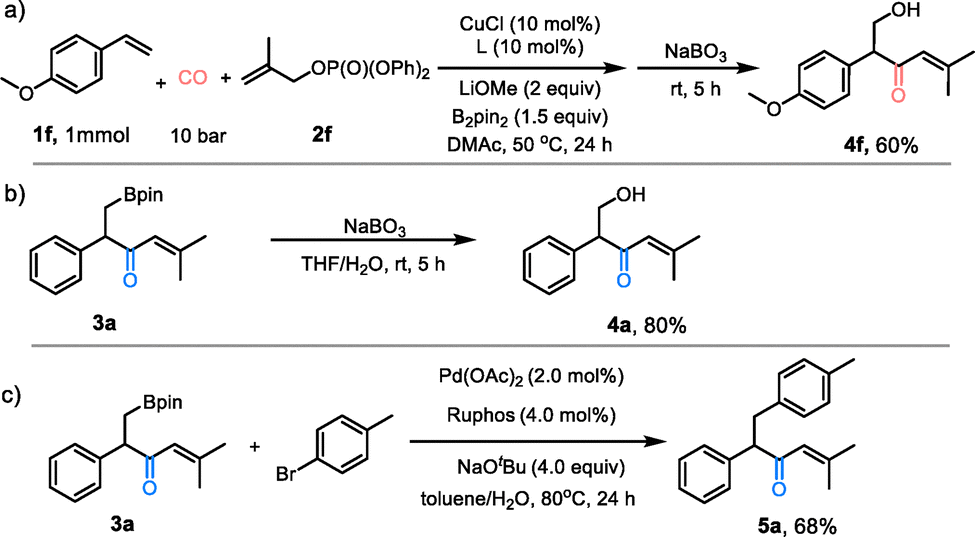
In order to elucidate the reaction mechanism, control experiments were performed (Scheme 3). Initially, the incorporation of three equivalents of TEMPO yielded the isolation of product 4a in a yield of 34% (Scheme 3a). This outcome suggests that the reaction proceeds without significant radical involvement. Subsequently, a radical clock experiment was performed using (1-cyclopropylvinyl)benzene as a diagnostic substrate. The formation of product 4a in a 50% yield, accompanied by the complete retention of the cyclopropyl moiety which excludes the intermediacy of free radicals (Scheme 3b). Utilizing these experimental insights, a plausible catalytic cycle is proposed (Scheme 3c). The transformative sequence is initiated by migratory insertion of the olefin into the Cu–B bond of the LCuBpin complex (Int-I), generating alkylcopper intermediate Int-II. The sequential coordination of CO and the allylic phosphate to Int-II forms Int-III. The subsequent steps involved CO insertion, allylic substitution, and reductive elimination, leading to the formation of product A. Product A then underwent isomerization to afford the final borocarbonylative allylation product. Finally, the LCuBpin catalyst was subjected to a process of regeneration by the action of base and B2pin2, thereby bringing the catalytic cycle to a conclusion.
 | ||
| Scheme 3 Mechanistic investigations and proposed reaction pathway. | ||
In summary, a novel copper-catalyzed four-component borylation-carbonylation-allylation reaction of aryl olefins has been developed. In the context of mild reaction conditions, this method has been demonstrated to be efficacious in the synthesis of β-boryl unsaturated ketones. The reaction demonstrates a substantial substrate scope and functional group compatibility, thus establishing a novel synthetic platform for the expeditious construction of complex molecules.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: general comments, general procedure, analytic data and NMR spectra. See DOI: https://doi.org/10.1039/d5cc05832e.Notes and references
- R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef PubMed.
- (a) H. Ito, Y. Kosaka, K. Nonoyama, Y. Sasaki and M. Sawamura, Angew. Chem., Int. Ed., 2008, 47, 7424–7427 CrossRef PubMed; (b) K. Kubota, E. Yamamoto and H. Ito, J. Am. Chem. Soc., 2013, 135, 2635–2640 CrossRef PubMed.
- (a) S. Crotti, F. Bertolini, F. Macchia and M. Pinesch, Org. Lett., 2009, 11, 3762–3765 CrossRef; (b) H. Y. Cho and J. P. Morke, J. Am. Chem. Soc., 2008, 130, 16140–16141 CrossRef PubMed; (c) J. C. Green, M. V. Joannou, S. A. Murray, J. M. Zanghi and S. J. Meek, ACS Catal., 2017, 7, 4441–4445 CrossRef.
- T. W. Butcher, E. J. McClain, T. G. Hamilton, T. M. Perrone, K. M. Kroner, G. C. Donohoe, N. G. Akhmedov, J. L. Petersen and B. V. Popp, Org. Lett., 2016, 18, 6428–6431 CrossRef PubMed.
- (a) T. Jia, P. Cao, B. Wang, Y. Lou, X. Yin, M. Wang and J. Liao, J. Am. Chem. Soc., 2015, 137, 13760–13763 CrossRef; (b) K. M. Logan, K. B. Smith and M. K. Brown, Angew. Chem., Int. Ed., 2015, 54, 5228–5231 CrossRef; (c) S. R. Sardini and M. K. Brown, J. Am. Chem. Soc., 2017, 139, 9823–9826 CrossRef PubMed.
- K. Semba, Y. Ohtagaki and Y. Nakao, Org. Lett., 2016, 18, 3956–3959 CrossRef PubMed.
- K. M. Logan, S. R. Sardini, S. D. White and M. K. Brown, J. Am. Chem. Soc., 2017, 140, 159–162 CrossRef.
- K. Yang and Q. Song, J. Org. Chem., 2016, 81, 1000–1005 CrossRef.
- (a) Y. Yuan, F. P. Wu, J. X. Xu and X. F. Wu, Angew. Chem., Int. Ed., 2020, 59, 17055–17061 CrossRef; (b) Y. Yuan, J.-X. Xu and X.-F. Wu, Chem. Commun., 2022, 58, 12110–12113 RSC; (c) Y. Yuan, F. Ge, T. Yang and X.-F. Wu, J. Catal., 2025, 450, 116280 CrossRef.
- F.-P. Wu and X.-F. Wu, Chem. Sci., 2021, 12, 10341–10346 RSC.
- B. Gabriele, Carbon Monoxide in Organic Synthesis-Carbonylation Chemistry, Wiley, 2022 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |