
Self-healing, antibiofouling and anticorrosion properties enabled by designing polymers with dynamic covalent bonds and responsive linkages†
Jenpob
Sokjorhor
a,
Tiwa
Yimyai
b,
Raweewan
Thiramanas
 c and
Daniel
Crespy
*a
c and
Daniel
Crespy
*a
aDepartment of Materials Science and Engineering, School of Molecular Science and Engineering, Vidyasirimedhi Institute of Science and Technology (VISTEC), Rayong 21210, Thailand. E-mail: daniel.crespy@vistec.ac.th
bDepartment of Chemical and Bimolecular Engineering, School of Energy Science and Engineering, Vidyasirimedhi Institute of Science and Technology (VISTEC), Rayong 21210, Thailand
cNational Nanotechnology Center (NANOTEC), National Science and Technology Development Agency (NSTDA), Pathum Thani 12120, Thailand
First published on 7th June 2024
Abstract
Coating metal structures with a protective material is a popular strategy to prevent their deterioration due to corrosion. However, maintaining the barrier properties of coatings after their mechanical damage is challenging. Herein, we prepared multifunctional coatings with self-healing ability to conserve their anticorrosion performance after damage. The coating was formed by blending synthesized redox-responsive copolymers with the ability to release a corrosion inhibitor upon the onset of corrosion with synthesized self-healing polyurethanes containing disulfide bonds. The corrosion rate of steel substrates coated with a blend is approximately 24 times lower than that of steel coated with only self-healing polyurethane. An exceptional healing efficiency, as high as 95%, is obtained after mechanical damage. The antibiofouling property against bacterial and microalgal attachments on coatings is facilitated by the repellent characteristic of fluorinated segments and the biocidal activity of the inhibitor moieties in the copolymer.
Introduction
The degradation of metallic materials due to corrosion can be accelerated by the presence of corrosive media, which leads to a dramatic reduction in the lifespan of metallic materials.1 Corrosion affects human safety, leads to environmental pollution, and creates huge financial losses, amounting to 2.5 trillion dollars annually worldwide and representing 3.4% of the global gross domestic product.2 Coatings can be applied on metal substrates for hindering their corrosion by providing a barrier film between exposed metal surfaces and their environments. Mechanical damage to coatings leads to a reduction in their anticorrosion properties.3,4 Herein, we designed a coating displaying simultaneous anticorrosion and self-healing properties for alleviating this issue.Several strategies have been developed for increasing the lifetime of coatings.5–7 A first approach relies on the release of corrosion inhibitors in the defect regions on coatings, where they form a protective layer against corrosive media. Corrosion inhibitors can be encapsulated in containers, which are designed to allow for the selective release of inhibitors when triggered by a mechanical force;8–10 irradiation of light;11,12 change in temperature;13,14 or factors related to corrosion such as change in redox potential,15,16 pH,17,18 or ion concentration.19,20 The second strategy relies on the release of healing agents from the capsules or microvascular networks, which can fill up the defects and then polymerize.5,21–24 Finally, the healing of damages can also be achieved by introducing intrinsic self-healing polymers with either physical or chemical healing processes into coatings.25–28 Examples of physical processes are shape memory effects,29,30 interchain diffusion,31 or phase-separated morphologies,32 whereas chemical processes involve dynamic covalent chemistry (e.g., disulfide bonds,33,34 Diels–Alder reactions,35,36 or imine bonds37,38) or non-covalent chemistries (e.g., hydrogen bonding,39–41 metal–ligand coordination,42,43 or guest–host interactions44,45).
However, healing strategies based on the release of corrosion inhibitors or healing agents have some limitations related to their finite amounts in coatings, including a restricted number of healing cycles.5,21,46–48 To overcome these limitations, intrinsic self-healing materials have been developed because they allow for a virtually unlimited number of healing cycles.49–53 Intrinsic self-healing can be carried out with external interventions, such as heat,54 light,55,56 or electricity,57 which promote the recovery of dynamic bonds and chain diffusion in polymer networks. Reversible systems based on bonds formed via Diels–Alder reactions led to an improvement of polylactide filaments compared with the same polymers without Diels–Alder bonds.58 Cured natural rubber containing furan-maleimide adducts allowed healing by heating to regenerate dienes and dienophiles that could further reform covalent bonds.59 The cured rubber could recover 80% of the original tensile strength after the healing of scratched samples (scratch depth ∼38 μm, width ∼114 μm) for 4 h at a temperature of 130 °C. However, the self-healing based on Diels–Alder chemistry required high temperatures (exceeding 120 °C) to heal damages.60–62 Dynamic disulfide bonds were incorporated in the materials for healing damaged materials under moderate temperatures (25–90 °C)63–66 due to the low bond dissociation energy (60 kcal mol−1) of the metathesis reaction.34 A healing efficiency of ∼97% could be achieved by heating scratched samples of a polyurethane containing disulfide bonds (scratch depth ∼1.3 μm, width ∼12.8 μm) at 80 °C for 1 h.67 Additionally, the barrier properties of coatings were improved by introducing carbon nanotubes68 or graphene oxide69,70 in intrinsic self-healing polymers. Thus, the self-healing coatings of an acrylic resin containing fluorinated carbon nanotubes displayed a larger impedance modulus of acrylic composite coatings (|Z|0.01 = 2.4 × 109 Ω cm2), compared with the same coatings without carbon nanotubes (|Z|0.01 = 1.4 × 107 Ω cm2) after 10 days of immersion in a saline solution.68 Furthermore, a waterborne composite coating was prepared by Diels–Alder reaction between nanofillers with a sandwich structure (polydopamine/graphene oxide/polyaniline) containing furan groups and polyurethane terminated with maleimide groups.69 Polyurethane composite coatings could recover 81% of their original impedance moduli at a frequency of 0.01 Hz after healing of the scratched samples for 0.5 h at 130 °C, followed by 24 h at 65 °C. Anticorrosion properties were imparted by the formation of an oxide layer by the reaction between polyaniline and steel and the increase in the diffusion pathways for corrosive species upon the introduction of the nanofillers. Additionally, antibiofouling and anticorrosion functions were added to intrinsic self-healing polyurethane coatings. The coatings were prepared by a reaction between isocyanate-terminated prepolymer, 2-hydroxyethyl disulfide and graphene oxide functionalized with isocyanate groups in the presence of 2-methyl-4-isothiazoline-3-ketone as the antifouling agent.70 The healing of scratched coatings was activated by near-infrared irradiation while the presence of 5 wt% antifouling agent and 3 wt% graphene oxide in the coatings led to the highest impedance modulus (|Z|0.01 = 3.3 × 108 Ω cm2) after immersion for 15 days in saline solution. However, the presence of fillers in coatings may provide new penetration pathways for corrosive substances. Furthermore, intrinsic self-healing is typically relatively slow, which is a limitation for anticorrosion applications.
Therefore, polymers with multiple healing mechanisms were designed so that corrosion reactions are hindered at an early stage. Some healing strategies rely on the presence of organic corrosion inhibitors, which suppress the onset of electrochemical corrosion by releasing inhibitors at the site of damages.71–73 Coatings with self-healing and anticorrosion properties were formed by incorporating benzotriazole or silica nanoparticles loaded with benzotriazole,74 polyurethane nanocapsules loading with 2-mercaptobenzimidazole,75 or nanorod of attapulgite loaded with 2-undecylimidazoline as a corrosion inhibitor and anti-biofouling agent76 in an intrinsic self-healing silicone-based polymer. The release of corrosion inhibitors improved the inhibition efficiency of the coating by the formation of a protective thin film on the surface of steel. Additionally, an anticorrosive composite coating was prepared by adding graphene oxide modified with TiO2 nanocapsules loaded with benzothiazole to a waterborne self-healing polyurethane containing disulfide bonds.77 However, an uncontrolled leaching of the inhibitor from coatings can occur, leading to a reduction of their service life. A possibility for controlling the release of inhibitors from the anticorrosion coatings is to conjugate them to polymer chains via linkages that are responsive to consequences of corrosion. Thus, a coating with the abilities of repairing mechanical damages and releasing conjugated corrosion inhibitors was prepared. The self-healing coating with dual antifouling and anticorrosion properties was prepared by conjugating 2-aminobenzothiazole with the silicone-based polymer.78 However, silicone-based polymers have too low surface energy, which enables low adhesion and easy detachment from the substrates.79–81 Additionally, the dual-function was ensured by pH-responsive copolymer containing 8-hydroxyquinoline units, which coordinated cerium ions.82 The corrosion process was hindered upon the release of 8-hydroxyquinoline and cerium ions, which can form an insoluble layer at damaged sites, while healing relied on the regeneration of the coordinated bonds. However, cerium ions can be released during corrosion, hence reducing the healing ability of the coatings in long-term service. Therefore, a challenge is to combine the two healing strategies into the same coatings for simultaneously healing damages and releasing inhibitors upon corrosion, enhancing adhesion strength, and preserving the healing ability of the coating in the long term.
Herein, we prepared coatings with the ability of simultaneous self-healing and reducing the corrosion rate. The coatings were designed to heal mechanical damages while releasing a corrosion inhibitor. Both healing and release functions were implemented using the reversibility of disulfide linkages. The coatings were designed by blending a polyurethane providing self-healing ability and a redox-responsive copolymer with the ability to release a corrosion inhibitor. The anticorrosion performance and the self-healing properties of coatings prepared with these polymers were investigated.
Results and discussion
In order to achieve self-healing and anticorrosion properties in the same coating, we blended a polyurethane providing self-healing ability and a redox-responsive copolymer with the ability to release a corrosion inhibitor. The functions of the polymers could be activated by disulfide exchange reactions upon heating.The polyurethane containing disulfide bonds (PUDS) was synthesized by condensation polymerization between 2-hydroxyethyl disulfide and an isocyanate-terminated prepolymer, which was synthesized by reacting polytetrahydrofuran with dicyclohexylmethane 4,4′-diisocyanate (Fig. 1(a)). Disulfide bonds were introduced into the polyurethane main chain to enable a self-healing property,63,65,83 which can be activated by increasing the temperature above the glass-transition temperature (Tg).84,85 A redox-responsive copolymer, P(TFEM-co-MBTS2MA) (PTM), was synthesized by free-radical copolymerization between 2,2,2-trifluoroethyl methacrylate and 2-(benzo[d]thiazol-2-yldisulfaneyl)ethyl methacrylate (Fig. 1(b)). 2-Mercaptobenzothiazole (MBT) can be released from the copolymer by disulfide exchange reaction activated by heating (Fig. 2(a)). Heteroatoms in MBT allowed its adsorption on metal substrates to form a hydrophobic layer.86–88
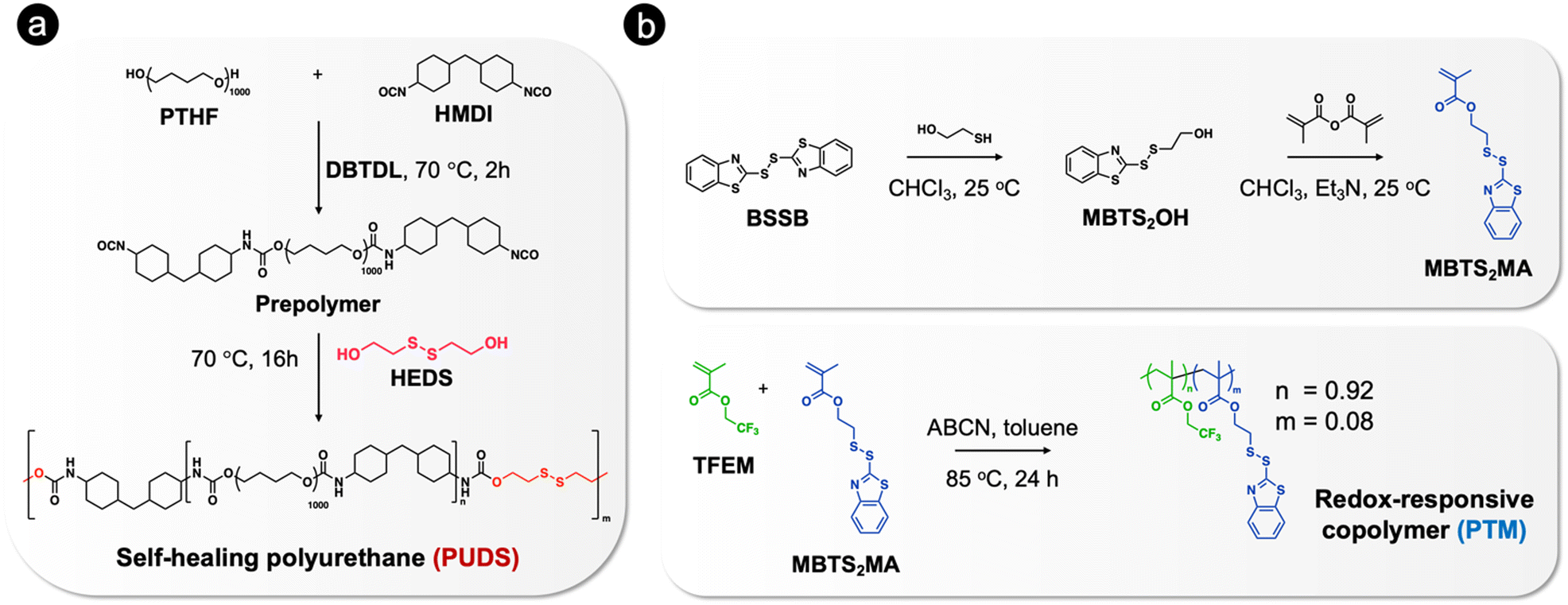 | ||
| Fig. 1 Synthetic routes for the preparation of (a) self-healing polyurethane containing disulfide bonds (PUDS) and (b) redox-responsive copolymer (PTM). | ||
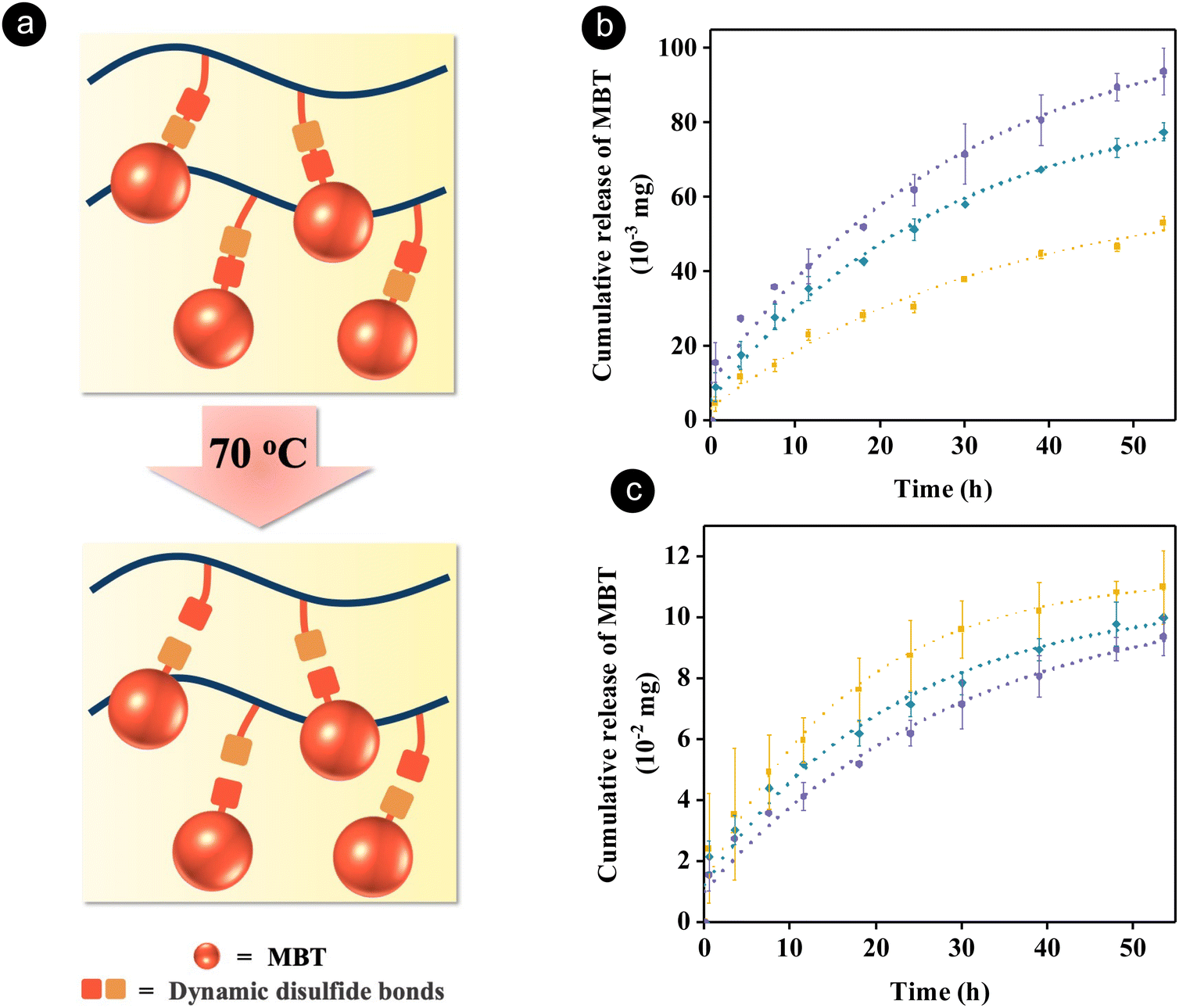 | ||
Fig. 2 (a) Schematics showing the release process of a corrosion inhibitor from a polymer via the dynamic disulfide bond exchange reaction upon heating. Release of 2-mercaptobenzothiazole (MBT) from the redox-responsive copolymer (PTM) determined using 1H NMR spectroscopy in a mixture of DMSO-d6 and CDCl3 (80/20 v/v) at 70 °C while keeping the concentration of polymer blends constant (b) or the concentration of PTM constant (c) ( , 10 wt% PTM/PUDS; , 10 wt% PTM/PUDS;  , 20 wt% PTM/PUDS; , 20 wt% PTM/PUDS;  , 30 wt% PTM/PUDS). , 30 wt% PTM/PUDS). | ||
To verify that MBT can be released from the blend of PTM and PUDS at the healing temperature of PUDS (70 °C), the amount of released MBT in solutions containing various ratios of the two polymers was measured by 1H NMR spectroscopy (Fig. S1, ESI†). The release of MBT was attributed to the thermal dissociation of disulfide bonds upon heating, leading to the formation of sulfur-centered radicals attacking other neighboring disulfide bonds.64,66 Furthermore, the healing temperature was selected to be above the glass transition temperature of PUDS, hence allowing the migration of polymer chains, which facilitated the disulfide exchange reaction.63 The amount of released MBT increased with increasing ratios of PTM at a constant concentration of the polymer blend (Fig. 2(b)). In another experiment, the release of MBT from PTM present in a solution containing various amounts of PUDS was measured by keeping the concentration of PTM constant. Interestingly, the amount of released MBT increased with increasing ratios of PUDS (Fig. 2(c)), which was attributed to the presence of more disulfide bonds in the system. Indeed, the concentration of disulfide bonds increased from 0.15 to 0.51 wt% with increasing ratios of PUDS from 70 to 90 wt% (Table S1, ESI†).
The mechanical properties of the steel substrates coated with PUDS and blends of PTM and PUDS were investigated by pencil hardness and pull-off adhesion tests. The pencil hardness of the coatings of PUDS or blends of PTM and PUDS were similar (B level), as shown in Table 1. The adhesion strength of the coatings decreased with increasing ratios of PTM, as shown in Table 1, due to the fluorinated molecules in the 2,2,2-trifluoroethyl methacrylate units in PTM.73,89,90 The adhesion strengths of the coatings were in the same range as for other polyurethane coatings on steel substrate for anticorrosion (Table S2, ESI†).
| Entry | Pencil hardness | Adhesion strength (MPa) |
|---|---|---|
| PUDS | 3B | 6.5 ± 1.3 |
| 10 wt% PTM/PUDS | 4B | 4.5 ± 0.5 |
| 20 wt% PTM/PUDS | 4B | 4.0 ± 0.8 |
| 30 wt% PTM/PUDS | 5B | 3.7 ± 0.9 |
Potentiodynamic polarization measurements were carried out to investigate the anticorrosion performance of coatings in the presence of artificial seawater (3.5 wt% NaCl aqueous solution). The measurements were performed on steel substrates and steel coated with a ∼40 μm layer off PUDS or blends of PTM and PUDS. The corrosion rates of steel coated with blends containing 10, 20, and 30 wt% PTM were 24, 41, and 59 times lower than that for PUDS, respectively (Fig. 3(a) and Table S3, ESI†). The anticorrosion properties were attributed to the adsorption of MBT on steel surface,86 which formed a monolayer, preventing the surface from interacting with corrosive media. The anodic metal dissolution and cathodic hydrogen evolution decreased when increasing the ratio of PTM in the coatings (Fig. S2, ESI†). The anticorrosion performance of the blends of PTM and PUDS coatings was found to exceed other reported anticorrosion coatings based on polyurethane on steel substrates (Table S4, ESI†).
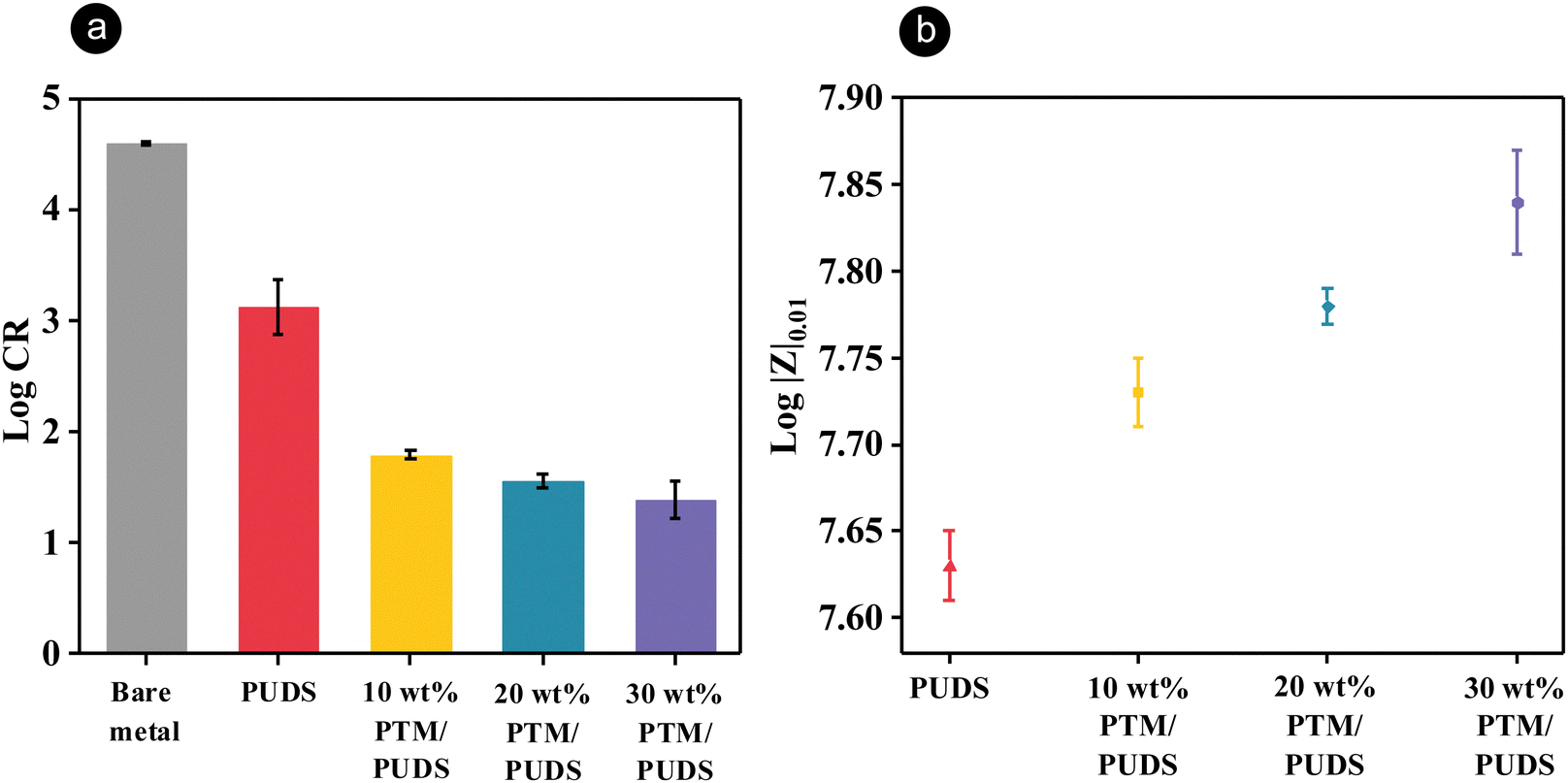 | ||
| Fig. 3 (a) Corrosion rate of steel substrates and steel substrates coated with PUDS or blends of PTM and PUDS in a 3.5 wt% NaCl aqueous solution. (b) Bode impedance modulus at 0.01 Hz (|Z|0.01) of the steel substrates coated with PUDS or blends of PTM and PUDS in a 3.5 wt% NaCl aqueous solution. | ||
Electrochemical impedance spectroscopy (EIS) measurements were carried out to investigate the barrier properties of coatings (∼40 μm) in the presence of artificial seawater. The coating resistance and charge transfer resistance for the blends of both the polymers increased with an increasing ratio of PTM from 10 to 30 wt% (Table S5, ESI†), suggesting that the blends containing more PTM in the coatings displayed better barrier properties. High values of |Z|0.01 correspond to the high anticorrosion performance of the coatings (Table S6, ESI†).71,72,91 |Z|0.01 of the steel substrates coated with blends of PTM and PUDS increased in the presence of more PTM (Fig. 3(b) and Fig. S4, ESI†).
In order to characterize the self-healing property of the coatings, scratches on the coatings were applied using a fresh razor blade, followed by healing at 70 °C, above the glass transition temperature of PUDS. The self-healing ability of the coatings was then monitored with an optical microscope overtime (see Fig. S5, ESI†). The scratch areas on the coating were healed after being at 70 °C for 2 days, while the scratch on the steel surface remained visible. The healing mechanism of coatings was attributed to dynamic disulfide bonds exchange. After the scratched coatings were heated, the polymer chains were mobile and the disulfide bonds were reformed.92 Furthermore, the healing ability of the coatings was evaluated by comparing the Bode plots of the impedance of the pristine coating to the coatings after scratching (width of 0.4 mm) and subsequent healing at 70 °C for 48 h (see Fig. S6, ESI†). The healing efficiency of the coatings was calculated by dividing the integral area of the Bode plot measured after damage/healing to the integral area for the pristine coating (Fig. 4(a)). The healing efficiencies for PUDS, 10 wt% PTM/PUDS, 20 wt% PTM/PUDS, and 30 wt% PTM/PUDS decreased from 100% to 95, 90 and 83% because the ratio of total disulfide bonds compared with the total amount of polymers in the coatings decreased from 6.2 wt% to 5.9, 5.6 and 5.2 wt%, respectively. This result indicated that the healing efficiency decreased when the ratio of PTM in the coating increased (Fig. 4(b)) due to the lower concentration of total disulfide bonds. The healing efficiency of the blends of PTM and PUDS was better than other reported self-healing coatings based on polyurethane for steel substrates (Table S7, ESI†). This is especially remarkable given the fact that our coatings were thinner (∼40 μm) than previously reported coatings. Indeed, thick coatings can heal better than thin coatings.83,93 The flow recovery of the polymer matrix after damage is driven by surface tension, and the wetting energy is hence dependent on the exposed surface at the damage area.29 Hence, a strategy relying on the combination of the self-healing ability of PUDS and the presence of corrosion organic inhibitors in PTM is promising for anticorrosion applications.
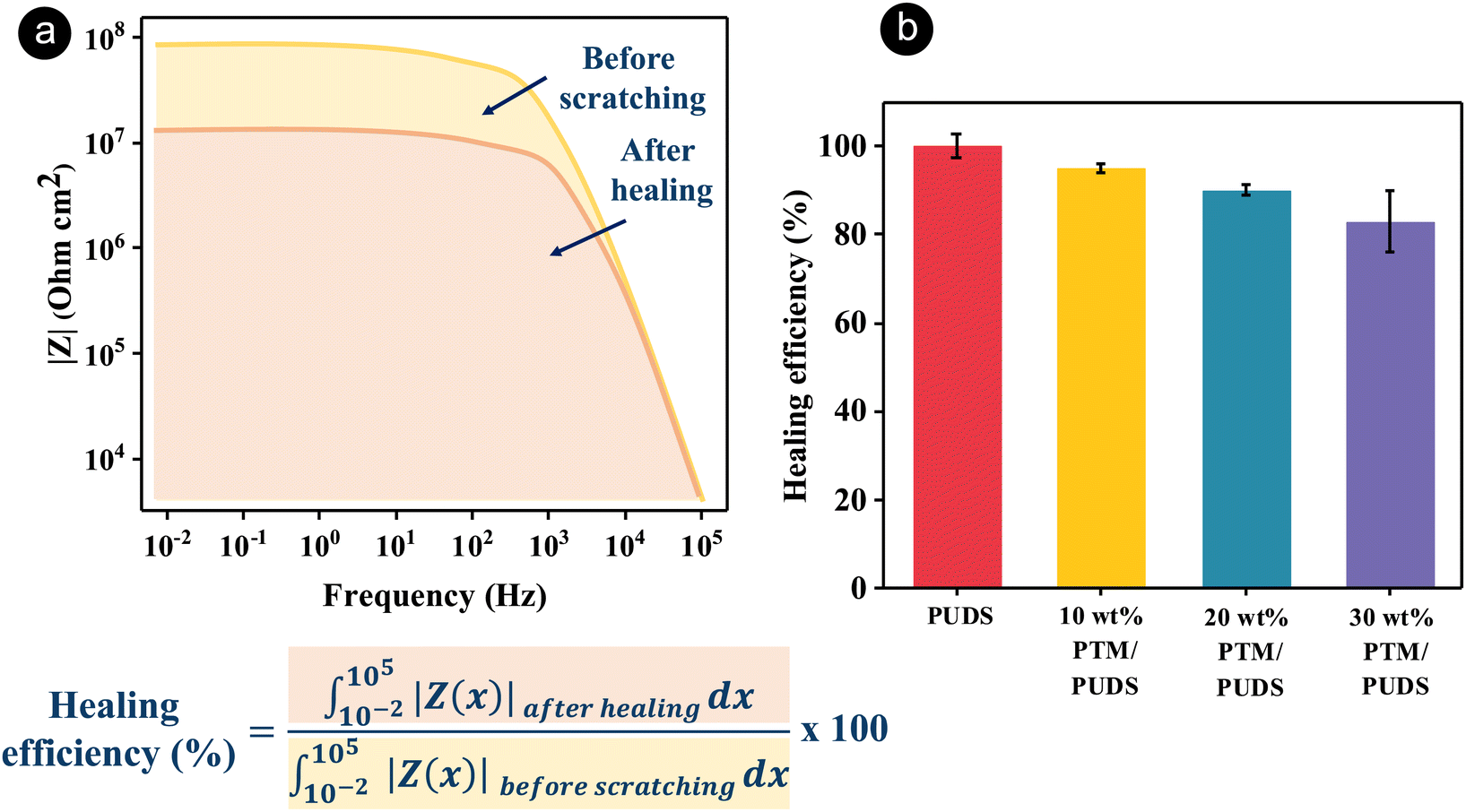 | ||
| Fig. 4 (a) Schematics showing the calculation of healing efficiency by comparing the Bode plots of the impedance of the pristine coating to the scratched coatings after healing at 70 °C. (b) Healing efficiency of the steel substrate coated with PUDS or blends of PTM and PUDS measured by electrochemical impedance spectroscopy (EIS) measurements. | ||
The long-term anticorrosion performance of pristine coatings and coatings that were healed after damage was investigated by salt spray tests. During the test, an aqueous solution of 5 wt% NaCl was sprayed at 35 °C onto the coatings to accelerate the corrosion. The RGB color threshold method was applied to all pixels of photographs of steel and coated steel substrates after being exposed to the salt spray test at various intervals of time (see Fig. 5(a)). The bare steel substrate was almost completely corroded (94% of the total surface area) after 16 h (see Fig. 5(b) and (c)). In contrast, the corroded surface area after 16 h decreased from ∼31% to 2% with increasing ratios of PTM in the coatings from 0 to 30 wt%. For the coated substrates, the ratio of corroded to total area for PUDS, 10 wt% PTM/PUDS, 20 wt% PTM/PUDS, and 30 wt% PTM/PUDS were 88, 23, 13, and 8% after 48 h, respectively. The salt spray tests hence confirmed that the coatings with a larger amount of PTM in the coatings displayed a better anticorrosion performance compared with coatings without PTM. To verify the hypothesis that blends of PTM and PUDS could heal damages and recover anticorrosion properties, corroded areas of coated steel substrates after scratching and after scratching and healing were monitored upon their exposure to salt spray tests. The corroded areas of healed coatings with 10, 20, and 30 wt% PTM/PUDS were 25, 16, and 11%, respectively, after 48 h (see Fig. S7a and b, ESI†). These values were smaller than the corroded areas of scratched coatings (42, 34, and 25% for 10, 20, and 30 wt% PTM/PUDS coatings). Salt spray tests confirmed that the strategies relying on the combination of intrinsic self-healing ability via reversible disulfide bonds and the presence of polymer-corrosion inhibitors conjugates is promising for anticorrosion applications.
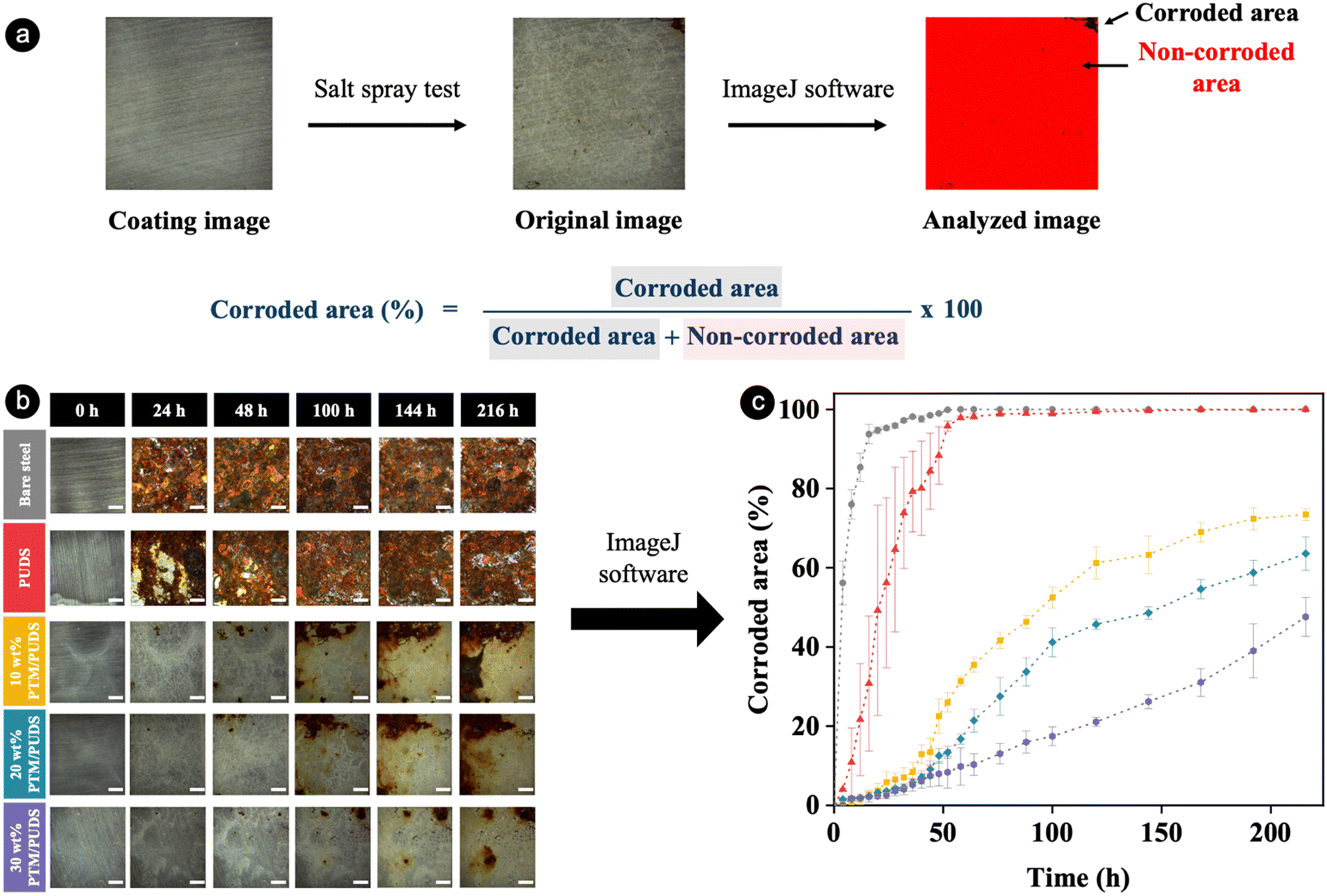 | ||
| Fig. 5 (a) Schematics showing the calculation of corroded area using the ImageJ software for a photograph of steel substrate coated with PUDS or blends of PTM and PUDS after being exposed to the salt spray test at 35 °C. (b) Photographs of steel substrates and steel substrates coated with PUDS or blends of PTM and PUDS at different times during the salt spray test at 35 °C. Scale bars are 2 mm. (c) Temporal evolution of the corroded area of steel substrates coated with PUDS or blends of PTM and PUDS during the salt spray test at 35 °C. | ||
To support the hypothesis that MBT was released upon corrosion, non-scratched and scratched coatings of 30 wt% PTM/PUDS on steel substrates were exposed to air or a 3.5 wt% NaCl aqueous solution for 24 h. The S/F atomic ratio on the surface of the coatings was then investigated by energy-dispersive X-ray spectroscopy (EDS) to investigate a possible change in the chemical composition due to the release of MBT from the copolymer. The S/F atomic ratio was monitored because fluorine is only present in the copolymer while sulfur atoms are present in released MBT, in the side-chains of copolymer, and in the self-healing polyurethane. Therefore, a change in the S/F ratio indicates a migration of released MBT. For non-scratched coatings, S/F ∼0.3, similar to the theoretical value of 0.3 predicted from the composition of the polymer blend between PTM and PUDS. Interestingly, S/F increased to ∼0.4 in the scratched coating exposed to the saline solution. This increase was attributed to the migration of released MBT to the scratches (see Fig. S8, ESI†). The release mechanism of MBT is attributed to the change in the electrochemical potential, associated with redox reactions involving metal oxidation and oxygen reduction upon corrosion, as shown previously.73,94 MBT can also be released upon chemical reduction, as demonstrated earlier.73 During the redox reaction, MBT is released upon electrochemical reduction by cleaving the redox-labile disulfide bonds at the surfaces of the coating on the damage site. After MBT was released from the copolymer due to electrochemical reduction, the released MBT at the damage site migrates to the exposed surface of the metal substrate via diffusion in the saline solution. The inhibitor MBT, containing sulfur, oxygen, and a nitrogen heterocyclic structure, can form protective passive layers at the corroded area of the coating via physical adsorption and chemical adsorption on metal surfaces through electrostatic interactions and coordination complexes (see Fig. S9, ESI†).95
The formation of biofilms consisting of proteins, bacteria, or marine organisms on coating is an undesirable process that increases the corrosion of immersed metallic structures.96–98 To evaluate the antibiofouling performance of the coatings, coatings of epoxy (control sample), PUDS, or blends of PTM and PUDS were immersed in a solution containing the unicellular microalgae, Chlorella ellipsoidea for 57 days. The algae from the genus Chlorella are extensively used for studying the antibiofouling performance of coatings due to their fast growth.73,99–102 As shown in Fig. 6(a) and Fig. S10 (ESI†), the coatings after incubation displayed a fluorescence characteristic of the presence of chlorophyll in the algal cells. The surface coverages with algae on epoxy and PUDS (control) coatings calculated from the analysis of fluorescence microscope images were 55% and 51%, respectively (see Fig. 6(b)). The surface coverages on the coatings were reduced by 11, 20, and 31% for 10, 20, and 30 wt% PTM/PUDS, respectively, compared with the PUDS coating. This enhanced performance was attributed to the presence of fluorinated moieties in PTM.89,90,103,104 Furthermore, we also evaluated the concentration of chlorophyll of the adhered algae, which is proportional to their amount (see Fig. 6(c) and Fig. S11, ESI†). The concentration of chlorophyll on the epoxy coating was larger than other polyurethane coatings. The amount of algae adhered on the surface of 10, 20, and 30 wt% PTM/PUDS coatings were reduced by 4, 8, and 29%, respectively, compared with the PUDS coating. In another experiment, we determined the bacterial adhesion on the coatings of epoxy, PUDS, or blends of PTM and PUDS on glass substrates. The coatings were immersed in a dispersion of Gram-negative bacterium Escherichia coli (E. coli) and Gram-positive bacterium Staphylococcus aureus (S. aureus) at 37 °C. After 24 h of incubation, bacterial cells on the surface of the coatings were then extracted using a crystal violet solution (0.1 vol% in deionized water), as shown in Fig. S12 (ESI†), and the absorbance of extracted crystal violet solution was measured at 590 nm by UV spectrophotometry. As shown in Fig. 5(d) and (e), there was less adhesion of E. coli and S. aureus on the coating for PUDS, 10 wt% PTM/PUDS, 20 wt% PTM/PUDS, and 30 wt% PTM/PUDS than for the epoxy coating (control). Furthermore, the amounts of E. coli adhered on the coatings were reduced by 40, 48, and 58% for 10, 20, and 30 wt% PTM/PUDS compared with the PUDS coating, while S. aureus adhesion was reduced by 27, 33, and 66% for 10, 20, and 30 wt% PTM/PUDS compared with the PUDS coating, respectively. This reduced bacterial adhesion on the coating of blends was attributed to the biocidal activity of MBT,73,105 suggesting that the blends containing more PTM in the coatings displayed better antibiofouling and antibacterial properties.
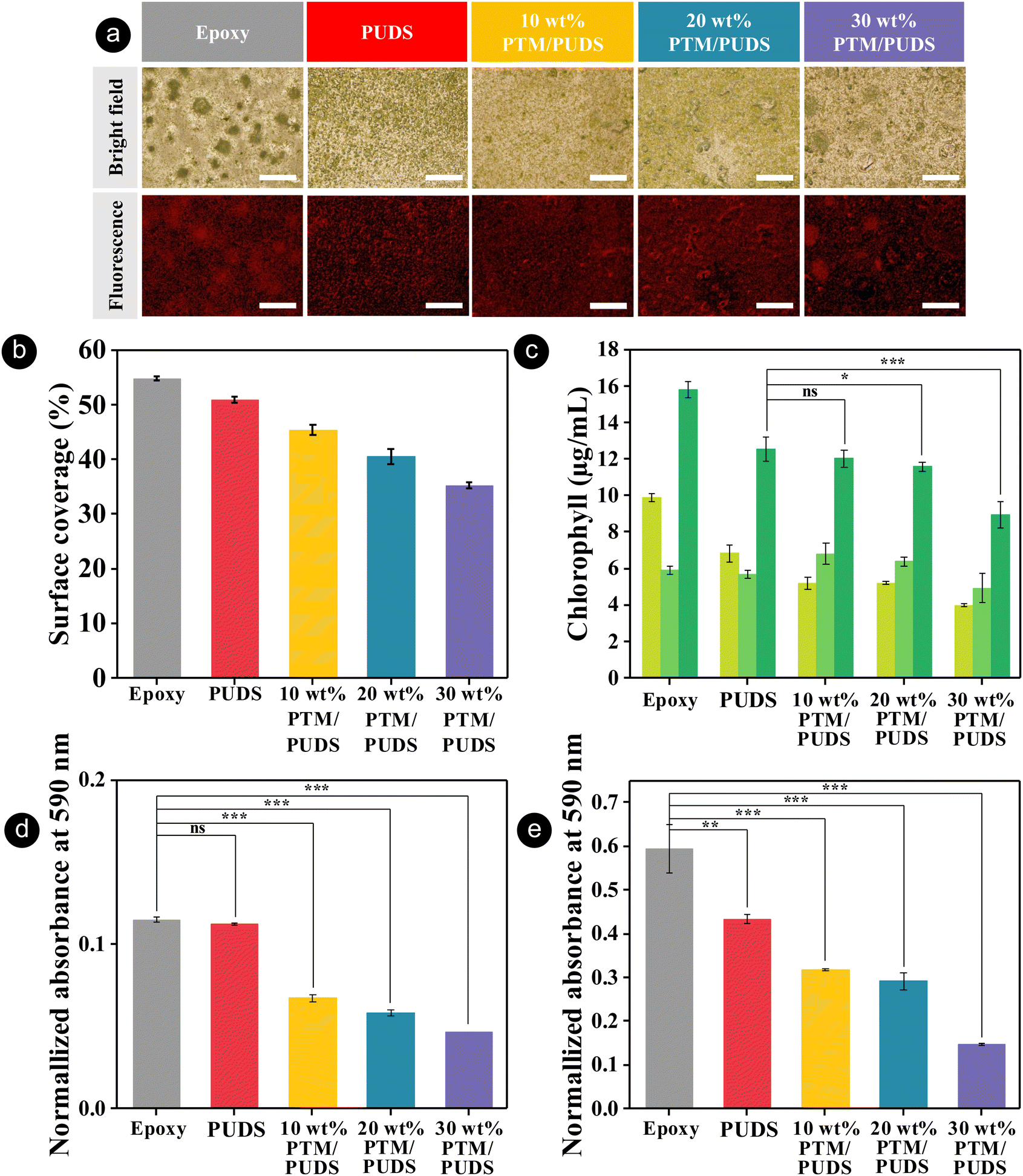 | ||
Fig. 6 (a) Photographs of the algae adhered on epoxy, PUDS, or blends of PTM and PUDS coatings on glass substrates taken with a bright field and fluorescence microscope. The red color is produced by the chlorophyll autofluorescence of algae cells. Scale bars are 500 μm. (b) Surface coverage of algae on the coatings measured from the image analysis of fluorescence images obtained by fluorescence microscopy. (c) Concentration of chlorophyll extracted ( , chlorophyll a; , chlorophyll a;  , chlorophyll b; , chlorophyll b;  , total chlorophyll) from algae adhering on the coatings (ns, not significant; *, significant; ***, highly significant). (d) Normalized absorbance at 590 nm of epoxy, PUDS, or blends of PTM and PUDS coatings on glass substrates incubated in Escherichia coli and (e) Staphylococcus aureus solution at 37 °C for 24 h. , total chlorophyll) from algae adhering on the coatings (ns, not significant; *, significant; ***, highly significant). (d) Normalized absorbance at 590 nm of epoxy, PUDS, or blends of PTM and PUDS coatings on glass substrates incubated in Escherichia coli and (e) Staphylococcus aureus solution at 37 °C for 24 h. | ||
Conclusion
Dual-functional coatings consisting of a redox-responsive copolymer containing a corrosion inhibitor conjugated via disulfide linkages blended with a polyurethane containing disulfide bonds were synthesized. The polymer blend could release the corrosion inhibitor upon heating at moderate temperatures. The coatings fabricated from the polymer blend exhibited an excellent anticorrosion performance due to the controlled release of the inhibitor on steel substrates. Moreover, the coatings displayed a healing efficiency as high as ∼95%, calculated by comparing the anticorrosion properties of the healed coating after damage to one of the pristine coatings before damage. The healing was attributed to exchange reaction between dynamic disulfide bonds in the coating. Moreover, the coatings displayed repellent properties against the microalgae Chlorella ellipsoidea, Escherichia coli, and Staphylococcus aureus bacteria due to the existence of fluorinated moieties and the biocidal activity of MBT moieties. Thus, the concept demonstrated in this study could be potentially applied for automotive, aerospace, packaging and/or electronics applications. In the future, self-healing chemistry relying on dynamic bonds and the release of conjugated inhibitors via responsive linkages, such as disulfide, β-thiopropionate, or hydrazone bonds, could be combined in the same polymer structure.Materials and methods
Materials
2,2′-Dibenzothiazolyl disulfide (BSSB, >96.0%, TCI Chemicals), 2-mercaptoethanol (99%, Acros Organics), methacrylic anhydride (MAAn, 94%, Sigma-Aldrich), hydroquinone (HQ, >99.0%, TCI Chemicals), triethylamine (≥99.0%, Merck), 1,1′-azobis(cyclohexanecarbonitrile) (ABCN, 98%, Sigma-Aldrich), chloroform (≥99%, Carlo Erba), toluene extra dry (99.85%, Acros Organics), dichloromethane (DCM, 99.9%, Honeywell Burdick & Jackson), n-hexane (95%, Carlo Erba), chloroform-D (99.8%, Cambridge Isotope Laboratories Inc.), dimethyl sulfoxide-D6 (99.8%, Merck), 2-hydroxyethyl disulfide (HEDS, ≥85.0%, Sigma-Aldrich), 1,6-hexanediol (HDO, >97.0%, TCI Chemicals), dibutyltin dilaurate (DBTDL, 95%, Sigma-Aldrich), dicyclohexylmethane 4,4′-diisocyanate (HMDI, >90.0%, TCI Chemicals), tetrahydrofuran (THF, ≥99.9%, Carlo Erba), N,N-dimethylacetamide (DMAc, >99.0%, TCI Chemicals), and 1,4-dioxane (99.8%, Carlo Erba) were used as received. 2,2,2-Trifluoroethyl methacrylate (TFEM, >99.0%, TCI Chemicals) was purified by vacuum distillation (120 mbar) at 59 °C prior to use. Polytetrahydrofuran (PTHF, Mn ∼ 1000 g mol−1, Sigma-Aldrich) was dried at 120 °C under vacuum for 2 h before use.Synthesis of the functional monomer MBTS2MA
The synthesis was carried out following the procedures that were previously reported.73 A thiol-disulfide exchange reaction between BSSB and 2-mercaptoethanol yielded 2-benzothiazolyl-2′-hydroxyethyl disulfide (MBTS2OH, 56% yield) after purification by recrystallization with hexane. MBTS2OH (2.00 g, 8.23 mmol), MAAn (3.68 mL, 24.7 mmol), and HQ (0.027 g, 0.25 mmol) were dissolved in DCM (70 mL) under nitrogen atmosphere. The mixture was stirred in an ice-water bath, followed by the dropwise addition of Et3N (3.44 mL, 24.7 mmol). The mixture was stirred for 10 min in a cooling bath for 24 h at 25 °C. The crude product was extracted with DI water (5 × 45 mL) and dried over Na2SO4. The solvent was then removed with a rotary evaporator. The product (MBTS2MA) was purified by column chromatography on silica gel with 8 vol% ethyl acetate in hexane (62% yield). HRMS: calculated for C13H13NO2S3 312.0181 [M + H]+, found 312.0143.Synthesis of P(TFEM-co-MBTS2MA)
TFEM (3.18 g, 18.89 mmol), MBTS2MA (0.59 g, 1.9 mmol), and ABCN (52 mg, 0.21 mmol) were dissolved in anhydrous toluene (9.6 mL). The mixture was bubbled with nitrogen gas for 30 min and subsequently stirred at 85 °C for 24 h. The reaction mixture was diluted with DCM (3 mL), precipitated in excess hexane (100 mL) 3 times, and dried under vacuum at 25 °C to provide the copolymer P(TFEM-co-MBTS2MA) with a yield of 96%. The apparent number-average molecular weight (Mn) and the molecular weight distribution (MWD) of the copolymer were 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 g mol−1 and 3.71, respectively. The copolymer composition was determined by 1H-NMR spectroscopy. The proton signals of methylene in the –CH2CF3 of the TFEM units and the –OCH2– of the MBTS2MA units of copolymers in CDCl3 were detected at δ = 4.34 ppm for two monomer units, and the proton signal of methylene in the –CH2S– of the MBTS2MA units was detected at δ = 3.20 ppm for MBTS2MA.
400 g mol−1 and 3.71, respectively. The copolymer composition was determined by 1H-NMR spectroscopy. The proton signals of methylene in the –CH2CF3 of the TFEM units and the –OCH2– of the MBTS2MA units of copolymers in CDCl3 were detected at δ = 4.34 ppm for two monomer units, and the proton signal of methylene in the –CH2S– of the MBTS2MA units was detected at δ = 3.20 ppm for MBTS2MA.
Synthesis of polyurethane containing disulfide bonds (PUDS)
PTHF (20.1 g, 20.1 mmol) was first melted in a 250 mL round bottom flask at 70 °C. HMDI (15.2 mL, 62.3 mmol) was then added into the flask and stirred to give a well-mixed mixture, followed by the addition of DBTDL (38.00 μL, 0.06 mmol). After 2 h reaction, a viscous prepolymer solution was obtained and diluted with DMAc (80 mL). The flask was then connected to a digital overhead stirrer (RW 20, IKA) with a polytetrafluoroethylene-coated propeller for stirring the viscous polymer solution. HEDS (5.2 mL, 41.7 mmol) was then charged into the solution and the reaction was allowed to proceed at 70 °C for 16 h to obtain PUDS. After reaction, the solution of PUDS was diluted with DMAc (55 mL) and THF (20 mL). The apparent number-average molecular weight (Mn) and the molecular weight distribution (MWD) of PUDS were 34000 g mol−1 and 2.28, respectively.
Preparation of the coatings
Steel SPCC (TCRSS, Thailand), with the chemical composition (wt%) Mn (0.500), C (0.120), S (0.045), P (0.040), and Fe (remainder), density of 7.8 g cm−3 and equivalent weight of 27.88 g mol−1 was used as the substrate. The metal substrates were cut with an area of 2.0 cm × 2.0 cm and thickness of 0.1 cm. The metal substrates were polished with 100 to 1000 grit SiC abrasive papers. Then, the metal substrates were cleaned with deionized water and sonicated with ethanol (10 mL) in an ultrasound bath for 5 min and dried under vacuum at 25 °C in a desiccator for 2 h. Then, the metal substrates were covered with a tape (851 greenback printed circuit board tape, 3M) to provide an open surface area of 1.2 cm × 1.2 cm. The solution containing 5 mg PUDS, the mixture of 4.5 mg PUDS and 0.5 mg PTM, the mixture of 4.0 mg PUDS and 1.0 mg PTM, or the mixture of 3.5 mg PUDS and 1.5 mg PTM in the mixture solvent of 1,4-dioxane (35 μL) and THF (15 μL) was spin-coated (SPIN150i, SPS) onto an open surface area of metal substrates with a rotational speed of 180 rpm and acceleration of 90 rpm s−1 for 100 s. Finally, the coated substrates were kept under vacuum at 60 °C for 24 h to obtain coatings with a thickness of ∼40 μm and the coated surface areas were fixed to 1.0 cm × 1.0 cm by covering with a tape (851 Greenback Printed Circuit Board Tape, 3M).Anticorrosion property of the coatings
Electrochemical impedance spectroscopy (EIS) and potentiodynamic polarization measurements were performed at 25 °C with a potentiostat (Autolab PGSTAT302N, Metrohm) and the software Nova version 2.1.4. The coated substrate was used as the working electrode, Ag/AgCl as the reference electrode, and platinum rod as the counter electrode. The EIS measurements were performed at 25 °C in the frequency range of 105–10−2 Hz, perturbation voltage of 10 mV, and in a 3.5 wt% aqueous solution of NaCl as the electrolyte. Potentiodynamic polarization was performed at 25 °C with a scan rate of 1 mV s−1 and potentials ranging from −1 to 1 V. The corrosion rate was calculated following the equation below.106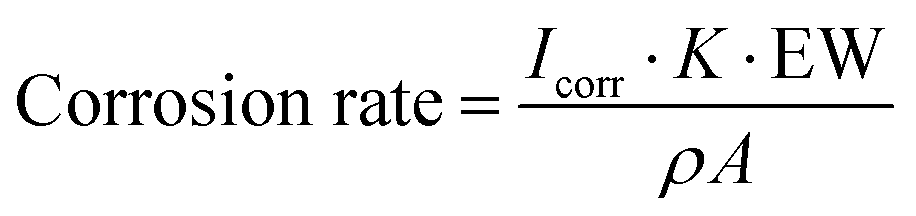 | (1) |
Self-healing property of the coatings
The coated samples were scratched (length = 5.0 mm, width = 0.4 mm) using a razor blade, and the scratched samples were heated in a fan oven at 70 °C for 2 days. The self-healing efficiency of the coatings was calculated by the following equation.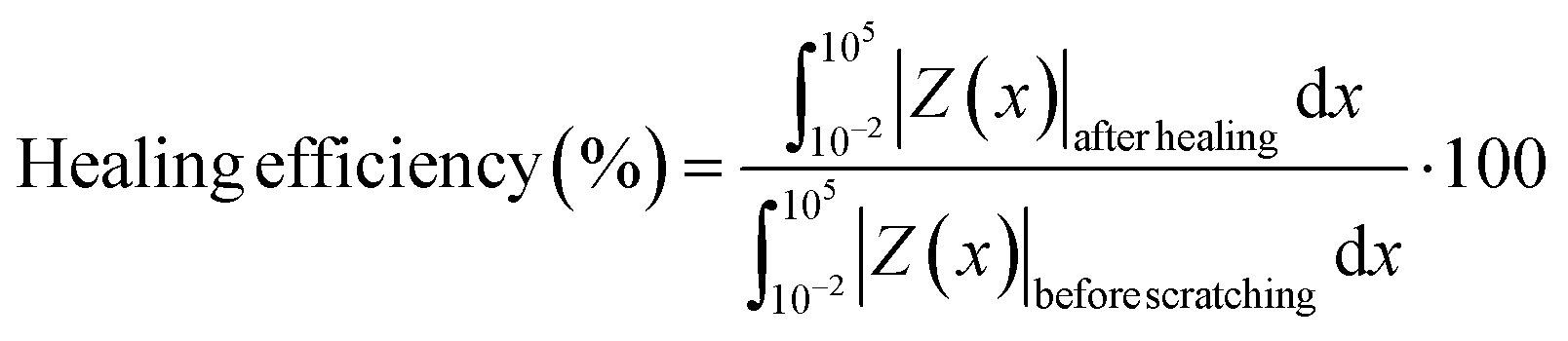 | (2) |
healing| and Z(x)beforecutting| are the impedance (|Z|) from the Bode plot of the samples after healing and before scratching, respectively.
Investigation of Microalgal attachment
Chlorella ellipsoidea TISTR 8260 and BG-11 medium were purchased from the Thailand Institute of Scientific and Technological Research. The limnetic microalgae (Chlorella ellipsoidea) were cultured in a conical flask containing a sterilized BG-11 growth medium at 28 °C under cool-white fluorescent lamps until the stock culture reached a density of ∼1 × 107 cells mL−1. The algae dispersion of the stock culture was then diluted with fresh BG-11 medium at a ratio of 1/5 (v/v). Glass substrates coated with epoxy, PUDS, 10 wt% PTM/PUDS, 20 wt% PTM/PUDS, or 30 wt% PTM/PUDS with an exposed surface area of 2.0 cm × 2.0 cm were immersed into 90 mm-Petri dishes containing 30 mL of the algae solution under the culture condition (16:8 light–dark cycles at 25 °C). After 57 days, the coatings were then gently dip-rinsed with sterile distilled water to remove the non-adhered algae. Fluorescence images of the coatings were taken with a fluorescence microscope (Olympus IX71) using a digital camera to observe and quantify the algae cells. The surface coverage of algae adhered on the coatings was estimated from the red channel of the fluorescence images using the ImageJ software.The coatings were dried at 25 °C for 5 days. 100 μL of dimethylformamide (DMF) was then dropped on the coatings to extract chlorophyll from the adhered algae cells, following the procedure of Schumann et al.107 The extraction of chlorophyll with DMF was repeated two times. Then, the collected DMF were combined and incubated in the dark at 4 °C for 24 h. The UV-vis absorbance of the incubated solution was measured using a microplate reader (PowerWave XS2, BioTek). The amount of chlorophyll was calculated using the following equation:108 Chlorophyll a (μg mL−1) = 12A664 − 3.11A647, chlorophyll b (μg mL−1) = 20.78A647 − 4.88A664, and total chlorophyll = 17.67A647 + 7.12A664, where A664 is the absorbance at 664 nm after removing the sample absorbance at 750 nm, and A647 is the absorbance at 647 nm after removing the sample absorbance at 750 nm. Student's t-tests were performed, and the calculated p values were considered to be significant for *p < 0.05, **p < 0.01 and ***p < 0.001.
Bacteria adhesion assay
Escherichia coli (E. coli) ATCC 25922 and Staphylococcus aureus (S. aureus) ATCC 6538, representatives of Gram-negative and Gram-positive bacteria, respectively, were grown in Mueller Hinton broth (MHB) with 150 rpm shaking at 37 °C for 24 h. The samples were placed separately into 35 mm-Petri dishes containing 4 mL of the bacterial suspension (107 colony forming unit (CFU) mL−1). After incubation at 37 °C for 24 h, the samples were gently dip-rinsed with deionized water to remove non-adherent bacteria. The crystal violet staining method was applied to quantitatively measure the adherent bacteria on the polymer coating surface.109 Crystal violet solution (0.1 mL, 0.1%) was first dropped onto the coated area. The samples were then left at room temperature (28 °C) for 1 h and then gently dip-rinsed with deionized water for 4 times to remove the excess dye. Afterward, the coated crystal violet was extracted from the surface by adding an acetic acid solution (0.24 mL, 10% in water) and incubated for 10 min. Finally, the absorbance of the dissolved crystal violet solution was measured at 590 nm by a microplate reader (PowerWave XS2, BioTek).Analytical methods
H1 NMR spectra were recorded with a Bruker 600 MHz NMR spectrometer at 25 °C. To investigate the release of 2-mercaptobenzothiazole (MBT) from P(TFEM-co-MBTS2MA) (PTM), a mixture of 62.99 mg PUDS and 7.00 mg PTM, a mixture of 28.00 mg PUDS and 7.00 mg PTM, or a mixture of 16.33 mg PUDS and 7.00 mg PTM were dissolved in a solution containing chloroform-D (130 μL) and dimethyl sulfoxide-D6 (520 μL) in NMR tubes. In another experiment, a mixture of 21.00 mg PUDS and 2.33 mg PTM, a mixture of 18.66 mg PUDS and 4.67 mg PTM, or a mixture of 16.33 mg PUDS and 7.00 mg PTM were dissolved in a solution containing chloroform-D (130 μL) and dimethyl sulfoxide-D6 (520 μL) in NMR tubes. The tubes were then heated at 70 °C in an oven. Tubes were withdrawn for NMR measurements after 30 min, 3.5 h, 7.5 h, 11.5 h, 18 h, 24 h, 30 h, 39 h, 48 h, and 53.5 h. The amount of released MBT was measured by comparing the characteristic peaks of aromatic protons in MBT (δ = 7.62–7.59) with the characteristic peaks of aromatic protons of conjugated MBT in the PTM (8.05–7.75 ppm). The apparent weight- and number-average molecular weights of the polymers were measured by gel permeation chromatography (GPC, Viscotek TDAmax, Malvern) with a refractive index detector and three single-pore GPC/size exclusion chromatography columns (6, 7, and 10 μm particle size, linear M). Polymer solutions with a concentration of 3 mg mL−1 in tetrahydrofuran (THF) were filtrated through a polytetrafluoroethylene (PTFE) membrane filter (0.45 mm pore size) and injected at a flow rate of 1 mL min−1 and measured at 35 °C. The hardness of the polymer coating was measured with a pencil hardness tester (VF2378, TQC) according to ASTM D3363. The coated samples were prepared by the spin-coating of a mixture of 1,4-dioxane (140 mL) and THF (60 mL) containing 20.0 mg PUDS (control sample), 2.0 mg PTM and 18.0 mg PUDS (10 wt% PTM/PUDS), 4.0 mg PTM and 16.0 mg PUDS (20 wt% PTM/PUDS), or 6.0 mg PTM and 14.0 mg PUDS (30 wt% PTM/PUDS) onto the metal substrates with a surface area of 2.4 cm × 2.4 cm. Afterwards, the coated substrates were kept under vacuum at 60 °C for 24 h to obtain coatings with a thickness of ∼40 μm. The coated surface area was then reduced to 2.0 cm × 2.0 cm by covering the edges of the substrates with a tape (851 Greenback Printed Circuit Board Tape, 3M). The adhesion strength of the polymer coating was determined using a pull-off adhesion tester (PosiTest, DEFELSKO) according to ASTM D4541. The coatings for the pull-off adhesion measurements were prepared similarly to the coatings for the hardness test, except the amounts of solvents and polymers were increased 4-fold to cover the substrates with a surface area of 4.4 cm × 4.4 cm.Author contributions
Jenpob Sokjorhor: conceptualization, methodology, formal analysis, writing – original draft, validation, investigation, Tiwa Yimyai: conceptualization, validation, investigation, Raweewan Thiramanas: validation, investigation, Daniel Crespy: conceptualization, supervision, writing – review & editing, validation, funding acquisition.Data availability
The data supporting this article have been included as part of ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research has received funding support from the NSRF (National Science, Research and Innovation Fund) via the Program Management Unit for Human Resources & Institutional Development, Research and Innovation (grant number B05F640208).References
- X.-m Zhang, Z.-y Chen, H.-f Luo, T. Zhou, Y.-l Zhao and Z.-c Ling, Trans. Nonferrous Met. Soc. China, 2022, 32, 377–410 CrossRef CAS.
- B. Hou, X. Li, X. Ma, C. Du, D. Zhang, M. Zheng, W. Xu, D. Lu and F. Ma, NPJ Mater. Degrad., 2017, 1, 4 CrossRef.
- W. Fan, Y. Zhang, W. Li, W. Wang, X. Zhao and L. Song, J. Chem. Eng., 2019, 368, 1033–1044 CrossRef CAS.
- F. Zhang, P. Ju, M. Pan, D. Zhang, Y. Huang, G. Li and X. Li, Corros. Sci., 2018, 144, 74–88 CrossRef CAS.
- G. Cui, Z. Bi, S. Wang, J. Liu, X. Xing, Z. Li and B. Wang, Prog. Org. Coat., 2020, 148, 105821 CrossRef CAS.
- L. Wang, S. Li and J. Fu, Prog. Org. Coat., 2023, 175, 107381 CrossRef CAS.
- Z. Chen, N. Scharnagl, M. L. Zheludkevich, H. Ying and W. Yang, J. Chem. Eng., 2023, 451, 138582 CrossRef CAS.
- K. Thongchaivetcharat, S. Salaluk, D. Crespy, H. Thérien-Aubin and K. Landfester, ACS Appl. Mater. Interfaces, 2020, 12, 42129–42139 CrossRef CAS PubMed.
- A. Khan, M. H. Sliem, A. Arif, M. A. Salih, R. A. Shakoor, M. F. Montemor, R. Kahraman, S. Mansour, A. M. Abdullah and A. Hasan, Prog. Org. Coat., 2019, 137, 105319 CrossRef CAS.
- Y. Huang, L. Deng, P. Ju, L. Huang, H. Qian, D. Zhang, X. Li, H. A. Terryn and J. M. C. Mol, ACS Appl. Mater. Interfaces, 2018, 10, 23369–23379 CrossRef CAS PubMed.
- E. V. Skorb, A. G. Skirtach, D. V. Sviridov, D. G. Shchukin and H. Möhwald, ACS Nano, 2009, 3, 1753–1760 CrossRef CAS PubMed.
- T. Chen, R. Chen, Z. Jin and J. Liu, J. Mater. Chem. A, 2015, 3, 9510–9516 RSC.
- G. L. Li, Z. Zheng, H. Möhwald and D. G. Shchukin, ACS Nano, 2013, 7, 2470–2478 CrossRef CAS PubMed.
- P. Yang, S. Gai and J. Lin, Chem. Soc. Rev., 2012, 41, 3679–3698 RSC.
- S. Sun, X. Zhao, M. Cheng, Y. Wang, C. Li and S. Hu, Prog. Org. Coat., 2019, 136, 105302 CrossRef CAS.
- T. Wang, L. Tan, C. Ding, M. Wang, J. Xu and J. Fu, J. Mater. Chem. A, 2017, 5, 1756–1768 RSC.
- D. Borisova, D. Akçakayıran, M. Schenderlein, H. Möhwald and D. G. Shchukin, Adv. Funct. Mater., 2013, 23, 3799–3812 CrossRef CAS.
- Z. Zheng, X. Huang, M. Schenderlein, D. Borisova, R. Cao, H. Möhwald and D. Shchukin, Adv. Funct. Mater., 2013, 23, 3307–3314 CrossRef CAS.
- F. Maia, J. Tedim, A. D. Lisenkov, A. N. Salak, M. L. Zheludkevich and M. G. S. Ferreira, Nanoscale, 2012, 4, 1287–1298 RSC.
- C. Ding, Y. Liu, M. Wang, T. Wang and J. Fu, J. Mater. Chem. A, 2016, 4, 8041–8052 RSC.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS PubMed.
- S. An, M. W. Lee, A. L. Yarin and S. S. Yoon, J. Chem. Eng., 2018, 344, 206–220 CrossRef CAS.
- S. H. Cho, S. R. White and P. V. Braun, Adv. Mater., 2009, 21, 645–649 CrossRef CAS.
- D. G. Bekas, K. Tsirka, D. Baltzis and A. S. Paipetis, Composites, Part B, 2016, 87, 92–119 CrossRef CAS.
- S. Wang and M. W. Urban, Nat. Rev. Mater., 2020, 5, 562–583 CrossRef CAS.
- S. Utrera-Barrios, R. Verdejo, M. A. López-Manchado and M. Hernández Santana, Mater. Horiz., 2020, 7, 2882–2902 RSC.
- M. Goyal, S. N. Agarwal and N. Bhatnagar, J. Appl. Polym. Sci., 2022, 139, e52816 CrossRef CAS.
- F. Sun, L. Liu, J. Xu and J. Fu, Mater. Chem. Front., 2023, 7, 3494–3523 RSC.
- C. C. Hornat and M. W. Urban, Nat. Commun., 2020, 11, 1028 CrossRef CAS PubMed.
- J. Chen, Z. Wang, B. Yao, Y. Geng, C. Wang, J. Xu, T. Chen, J. Jing and J. Fu, Adv. Mater., 2024, 2401178, DOI:10.1002/adma.202401178.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC.
- Y. Chen, A. M. Kushner, G. A. Williams and Z. Guan, Nat. Chem., 2012, 4, 467–472 CrossRef CAS PubMed.
- T. Yimyai, T. Phakkeeree and D. Crespy, Adv. Sci., 2020, 7, 1903785 CrossRef CAS PubMed.
- H. Guo, Y. Han, W. Zhao, J. Yang and L. Zhang, Nat. Commun., 2020, 11, 2037 CrossRef CAS PubMed.
- C. M. Madl and S. C. Heilshorn, Chem. Mater., 2019, 31, 8035–8043 CrossRef CAS PubMed.
- K. K. Oehlenschlaeger, J. O. Mueller, J. Brandt, S. Hilf, A. Lederer, M. Wilhelm, R. Graf, M. L. Coote, F. G. Schmidt and C. Barner-Kowollik, Adv. Mater., 2014, 26, 3561–3566 CrossRef CAS PubMed.
- X. Cao, P. Zhang, N. Guo, Y. Tong, Q. Xu, D. Zhou and Z. Feng, RSC Adv., 2021, 11, 2985–2994 RSC.
- J. Hu, R. Mo, X. Sheng and X. Zhang, Polym. Chem., 2020, 11, 2585–2594 RSC.
- Z. Xie, B.-L. Hu, R.-W. Li and Q. Zhang, ACS Omega, 2021, 6, 9319–9333 CrossRef CAS PubMed.
- Y. Eom, S.-M. Kim, M. Lee, H. Jeon, J. Park, E. S. Lee, S. Y. Hwang, J. Park and D. X. Oh, Nat. Commun., 2021, 12, 621 CrossRef CAS PubMed.
- J. Xu, J. Chen, Y. Zhang, T. Liu and J. Fu, Angew. Chem., Int. Ed., 2021, 60, 7947–7955 CrossRef CAS PubMed.
- Y.-L. Rao, A. Chortos, R. Pfattner, F. Lissel, Y.-C. Chiu, V. Feig, J. Xu, T. Kurosawa, X. Gu, C. Wang, M. He, J. W. Chung and Z. Bao, J. Am. Chem. Soc., 2016, 138, 6020–6027 CrossRef CAS PubMed.
- Z. Li, Y. Shan, X. Wang, H. Li, K. Yang and Y. Cui, J. Chem. Eng., 2020, 394, 124932 CrossRef CAS.
- J.-B. Hou, X.-Q. Zhang, D. Wu, J.-F. Feng, D. Ke, B.-J. Li and S. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 12105–12113 CrossRef CAS PubMed.
- G. Sinawang, M. Osaki, Y. Takashima, H. Yamaguchi and A. Harada, Chem. Commun., 2020, 56, 4381–4395 RSC.
- K. Urdl, A. Kandelbauer, W. Kern, U. Müller, M. Thebault and E. Zikulnig-Rusch, Prog. Org. Coat., 2017, 104, 232–249 CrossRef CAS.
- C. E. Diesendruck, N. R. Sottos, J. S. Moore and S. R. White, Angew. Chem., Int. Ed., 2015, 54, 10428–10447 CrossRef CAS PubMed.
- Z. Jin, H. Liu, Z. Wang, W. Zhang, Y. Chen, T. Zhao, G. Meng, H. Liu and H. Liu, Prog. Org. Coat., 2022, 172, 107121 CrossRef CAS.
- L. F. Fan, M. Z. Rong, M. Q. Zhang and X. D. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 38538–38546 CrossRef CAS PubMed.
- M. Q. Zhang and M. Z. Rong, Polym. Chem., 2013, 4, 4878–4884 RSC.
- S. D. Bergman and F. Wudl, J. Mater. Chem., 2008, 18, 41–62 RSC.
- H. Yue, Z. Wang and Y. Zhen, ACS Omega, 2022, 7, 18197–18205 CrossRef CAS PubMed.
- I. Lee Hia, E.-S. Chan, S.-P. Chai and P. Pasbakhsh, J. Mater. Chem. A, 2018, 6, 8470–8478 RSC.
- Y. Xu and D. Chen, Macromol. Chem. Phys., 2016, 217, 1191–1196 CrossRef CAS.
- D. Habault, H. Zhang and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7244–7256 RSC.
- F. Sun, L. Liu, T. Liu, X. Wang, Q. Qi, Z. Hang, K. Chen, J. Xu and J. Fu, Nat. Commun., 2023, 14, 130 CrossRef CAS PubMed.
- W. Pu, D. Fu, Z. Wang, X. Gan, X. Lu, L. Yang and H. Xia, Adv. Sci., 2018, 5, 1800101 CrossRef PubMed.
- J. R. Davidson, G. A. Appuhamillage, C. M. Thompson, W. Voit and R. A. Smaldone, ACS Appl. Mater. Interfaces, 2016, 8, 16961–16966 CrossRef CAS PubMed.
- P. Tanasi, M. Hernández Santana, J. Carretero-González, R. Verdejo and M. A. López-Manchado, Polymer, 2019, 175, 15–24 CrossRef CAS.
- B. Willocq, F. Khelifa, J. Brancart, G. Van Assche, P. Dubois and J. M. Raquez, RSC Adv., 2017, 7, 48047–48053 RSC.
- B. Strachota, J. Hodan, J. Dybal and L. Matějka, Macromol. Mater. Eng., 2021, 306, 2000474 CrossRef CAS.
- L. Feng, Z. Yu, Y. Bian, J. Lu, X. Shi and C. Chai, Polymer, 2017, 124, 48–59 CrossRef CAS.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541 CrossRef CAS.
- Y. Lai, X. Kuang, P. Zhu, M. Huang, X. Dong and D. Wang, Adv. Mater., 2018, 30, 1802556 CrossRef PubMed.
- Z. Q. Lei, H. P. Xiang, Y. J. Yuan, M. Z. Rong and M. Q. Zhang, Chem. Mater., 2014, 26, 2038–2046 CrossRef CAS.
- X. Wu, J. Li, G. Li, L. Ling, G. Zhang, R. Sun and C.-P. Wong, J. Appl. Polym. Sci., 2018, 135, 46532 CrossRef.
- Y. Xu and D. Chen, J. Mater. Sci., 2018, 53, 10582–10592 CrossRef CAS.
- A. Zhou, H. Yu, J. Tang, B. Zhang, F. Qi, Y. Zhou, N. Zhao and X. Ouyang, Prog. Org. Coat., 2023, 180, 107589 CrossRef CAS.
- J. Xu, F. Gao, H. Wang, R. Dai, S. Dong and H. Wang, Prog. Org. Coat., 2023, 174, 107244 CrossRef CAS.
- W. Tian, S. Wang, Z. Guo, H. Yu and L. Tian, J. Chem. Eng., 2023, 462, 142346 CrossRef CAS.
- K. Mantala, T. Phakkeeree and D. Crespy, J. Ind. Eng. Chem., 2021, 97, 500–505 CrossRef CAS.
- K. Auepattana-Aumrung, T. Phakkeeree and D. Crespy, J. Appl. Polym. Sci., 2022, 139, 51730 CrossRef CAS.
- T. Yimyai, R. Thiramanas, T. Phakkeeree, S. Iamsaard and D. Crespy, Adv. Funct. Mater., 2021, 31, 2102568 CrossRef CAS.
- G. Chen, S. Wen, J. Ma, Z. Sun, C. Lin, Z. Yue, J. M. C. Mol and M. Liu, Surf. Coat. Technol., 2021, 421, 127388 CrossRef CAS.
- O. Razaghi Kashani, S. Amiri and M. Hosseini-zori, J. Nanostruct., 2022, 12, 726–737 Search PubMed.
- Y. Wu, J. Wei, X. Shi and W. Zhao, J. Mater. Sci. Technol., 2023, 148, 222–234 CrossRef CAS.
- J. Zhou, H. Liu, Y. Sun and K. Chen, Adv. Mater. Interfaces, 2022, 9, 2102001 CrossRef CAS.
- J. Sun, J. Duan, X. Liu, X. Dong, Y. Zhang, C. Liu and B. Hou, Appl. Mater. Today., 2022, 28, 101551 CrossRef.
- Z. Sun, J. Wen, W. Wang, H. Fan, Y. Chen, J. Yan and J. Xiang, Prog. Org. Coat., 2020, 146, 105744 CrossRef CAS.
- Y. Liu, C. Leng, B. Chisholm, S. Stafslien, P. Majumdar and Z. Chen, Langmuir, 2013, 29, 2897–2905 CrossRef CAS PubMed.
- S. Cha and C. Kim, ACS Appl. Mater. Interfaces, 2018, 10, 24003–24012 CrossRef CAS PubMed.
- K. Auepattana-Aumrung and D. Crespy, J. Chem. Eng., 2023, 452, 139055 CrossRef CAS.
- J. A. Yoon, J. Kamada, K. Koynov, J. Mohin, R. Nicolaÿ, Y. Zhang, A. C. Balazs, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2012, 45, 142–149 CrossRef CAS.
- S.-M. Kim, H. Jeon, S.-H. Shin, S.-A. Park, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Adv. Mater., 2018, 30, 1705145 CrossRef PubMed.
- K. Imato, A. Takahara and H. Otsuka, Macromolecules, 2015, 48, 5632–5639 CrossRef CAS.
- M. Gholami, I. Danaee, M. H. Maddahy and M. RashvandAvei, Ind. Eng. Chem. Res., 2013, 52, 14875–14889 CrossRef CAS.
- D. A. Winkler, M. Breedon, A. E. Hughes, F. R. Burden, A. S. Barnard, T. G. Harvey and I. Cole, Green Chem., 2014, 16, 3349–3357 RSC.
- I. A. Kartsonakis, S. G. Stanciu, A. A. Matei, R. Hristu, A. Karantonis and C. A. Charitidis, Corros. Sci., 2016, 112, 289–307 CrossRef CAS.
- B. Xu, Y. Liu, X. Sun, J. Hu, P. Shi and X. Huang, ACS Appl. Mater. Interfaces, 2017, 9, 16517–16523 CrossRef CAS PubMed.
- A. M. C. Maan, A. H. Hofman, W. M. de Vos and M. Kamperman, Adv. Funct. Mater., 2020, 30, 2000936 CrossRef CAS.
- S. Salaluk, K. Auepattana-Aumrung, K. Thongchaivetcharat, L.-P. Lv, Y. Wang, E. Viyanit and D. Crespy, Adv. Mater. Interfaces, 2020, 7, 2001073 CrossRef CAS.
- T. Yimyai, D. Crespy and A. Pena-Francesch, Adv. Funct. Mater., 2023, 33, 2213717 CrossRef CAS.
- C. Zhu, Y. Fu, C. Liu, Y. Liu, L. Hu, J. Liu, I. Bello, H. Li, N. Liu, S. Guo, H. Huang, Y. Lifshitz, S.-T. Lee and Z. Kang, Adv. Mater., 2017, 29, 1701399 CrossRef PubMed.
- A. Vimalanandan, L.-P. Lv, T. H. Tran, K. Landfester, D. Crespy and M. Rohwerder, Adv. Mater., 2013, 25, 6980–6984 CrossRef CAS PubMed.
- T. Yimyai, D. Crespy and M. Rohwerder, Adv. Mater., 2023, 35, 2300101 CrossRef CAS PubMed.
- A. M. C. Maan, A. H. Hofman, W. M. de Vos and M. Kamperman, Adv. Funct. Mater., 2020, 30, 2000936 CrossRef CAS.
- I. Banerjee, R. C. Pangule and R. S. Kane, Adv. Mater., 2011, 23, 690–718 CrossRef CAS PubMed.
- R. Chen, Y. Zhang, Q. Xie, Z. Chen, C. Ma and G. Zhang, Adv. Funct. Mater., 2021, 31, 2011145 CrossRef CAS.
- S. Ma, Q. Ye, X. Pei, D. Wang and F. Zhou, Adv. Mater. Interfaces, 2015, 2, 1500257 CrossRef.
- L. K. Roepke, D. Brefeld, U. Soltmann, C. J. Randall, A. P. Negri and A. Kunzmann, Sci. Rep., 2022, 12, 15935 CrossRef CAS PubMed.
- S. Natarajan, D. S. Lakshmi, M. Bhuvaneshwari, V. Iswarya, P. Mrudula, N. Chandrasekaran and A. Mukherjee, RSC Adv., 2017, 7, 27645–27655 RSC.
- S. Bi, K. Xu, G. Shao, K. Yang and J. Tian, J. Mater. Sci. Technol., 2023, 159, 125–137 CrossRef CAS.
- C. M. Grozea and G. C. Walker, Soft Matter, 2009, 5, 4088–4100 RSC.
- S. Krishnan, Y.-J. Kwark and C. K. Ober, Chem. Rec., 2004, 4, 315–330 CrossRef CAS PubMed.
- F. Maia, A. P. Silva, S. Fernandes, A. Cunha, A. Almeida, J. Tedim, M. L. Zheludkevich and M. G. S. Ferreira, J. Chem. Eng., 2015, 270, 150–157 CrossRef CAS.
- D. Prasai, J. C. Tuberquia, R. R. Harl, G. K. Jennings and K. I. Bolotin, ACS Nano, 2012, 6, 1102–1108 CrossRef CAS PubMed.
- R. Schumann, N. Häubner, S. Klausch and U. Karsten, Int. Biodeterior. Biodegrad., 2005, 55, 213–222 CrossRef CAS.
- R. J. Porra, Photosynth. Res., 2002, 73, 149–156 CrossRef CAS PubMed.
- D. Zhao, X.-d Xu, S.-s Yuan, S.-j Yan, X.-h Wang, S.-f Luan and J.-h Yin, Chin. J. Polym. Sci., 2017, 35, 887–896 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb00736k |
| This journal is © The Royal Society of Chemistry 2024 |