
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfrared photoinduced electrochemiluminescence microscopy of single cells†
Julie
Descamps
 a,
Yiran
Zhao
b,
Bertrand
Goudeau
a,
Dragan
Manojlovic
c,
Gabriel
Loget
*bd and
Neso
Sojic
*a
a,
Yiran
Zhao
b,
Bertrand
Goudeau
a,
Dragan
Manojlovic
c,
Gabriel
Loget
*bd and
Neso
Sojic
*a
aUniv. Bordeaux, CNRS UMR 5255, Bordeaux INP, Site ENSMAC, 33607 Pessac, France. E-mail: sojic@u-bordeaux.fr
bUniv. Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes)-UMR6226, Rennes F-35000, France. E-mail: gabriel.loget@cnrs.fr
cUniversity of Belgrade, Faculty of Chemistry, 11000 Belgrade, Serbia
dInstitute of Energy and Climate Research, Fundamental Electrochemistry (IEK-9), Forschungszentrum Jülich GmbH, Jülich, 52425, Germany
First published on 8th December 2023
Abstract
Electrochemiluminescence (ECL) is evolving rapidly from a purely analytical technique into a powerful microscopy. Herein, we report the imaging of single cells by photoinduced ECL (PECL; λem = 620 nm) stimulated by an incident near-infrared light (λexc = 1050 nm). The cells were grown on a metal–insulator–semiconductor (MIS) n-Si/SiOx/Ir photoanode that exhibited stable and bright PECL emission. The large anti-Stokes shift allowed for the recording of well-resolved images of cells with high sensitivity. PECL microscopy is demonstrated at a remarkably low onset potential of 0.8 V; this contrasts with classic ECL, which is blind at this potential. Two imaging modes are reported: (i) photoinduced positive ECL (PECL+), showing the cell membranes labeled with the [Ru(bpy)3]2+ complex; and (ii) photoinduced shadow label-free ECL (PECL−) of cell morphology, with the luminophore in the solution. Finally, by adding a new dimension with the near-infrared light stimulus, PECL microscopy should find promising applications to image and study single photoactive nanoparticles and biological entities.
Introduction
Electrochemiluminescence (ECL) is a well-known phenomenon of light emission generated by an electrochemical reaction in the vicinity of the electrode surface.1 In an aqueous environment, it involves a highly exergonic electron-transfer reaction between a luminophore and the electrogenerated radicals of a sacrificial co-reactant, populating the excited state of the luminophore that relaxes to the ground state by emitting a photon.2 In simple terms, in ECL technologies, an electrical signal is converted into an optical signal. Thanks to its high sensitivity, spatial and temporal resolution, and near-zero background, ECL has become an important tool in medical diagnosis or clinical immunoassays.3–6 The most efficient and most used ECL system comprises the tris(2,2′-bipyridine)ruthenium(II) complex ([Ru(bpy)3]2+) and tri-n-propylamine (TPrA), which act as the luminophore and the co-reactant, respectively.7–10 With this tandem system, bright ECL emission is generated during oxidation in a neutral aqueous solution (pH = 7.4).11,12 Since ECL provides an optical readout, in the last decade, it has evolved into a powerful technique for microscopy applications, allowing for the study of micro/nanoparticles and biological entities such as cells or organelles.13–23 Two main imaging approaches have been developed: (i) positive ECL (ECL+) with a light-emitting object, which directly generates ECL or is labeled with an ECL tag, on a dark background; and (ii) shadow or negative label-free ECL (ECL−), which is based on a non-ECL emissive object hindering the ECL reagents diffusion and thus shadowing the ECL background.18,24 Both provide complementary information about the imaged object. In the latter configuration, Su and co-workers described the ECL imaging of fingerprints and cell motion.25–28 To pursue the quest for higher sensitivity, single photons and single biomolecules were imaged at the level of cells, organelles or bacteria by developing original approaches and specifically tailored nanomaterials.29–31 Feng and co-workers reported a direct optical method for imaging single ECL photons generated by individual electron-transfer reaction events in solution.32 They extended their original approach to image cells and nanoparticles with remarkable optical resolution.33–35 Liu and co-workers reported the imaging of single biomolecules at the cell membrane level with ECL nano-emitters and their motion tracking.36–38A different ECL approach has been developed first in organic solvents39,40 and more recently in water41–45 by photo-stimulating the ECL emission at depleted semiconductors (SCs). It can be considered counter-intuitive because, contrary to classical ECL, where the experiments are performed in the dark, this approach, named photoinduced electrochemiluminescence (PECL), requires an incident light (λexc).46–48 The photogenerated charges react with the ECL reagents dissolved in the solution and produce in fine the ECL emission. This means that the SC working electrode acts as a light absorber (λexc) and a light emitter (λem).49 More precisely, the absorption of a photon with an energy higher than the bandgap of the SC results in the creation of electron/hole (e−/h+) pairs.50 The e− is promoted to the CB, and the electronic vacancy h+ is left in the depletion region of the VB and driven to the SC/electrolyte interface, generating a photovoltage and decreasing the required potential to trigger the anodic ECL reactions. According to the choice of the SC material and of the ECL system, Stokes and anti-Stokes PECL can be triggered. The first can be compared to photoluminescence (PL), where a photon of higher energy induces the emission of a photon of lower energy with a Stokes shift (λexc < λem).45 Conceptually, it is reminiscent of a light downconversion process, even if it requires electrochemical reactions to trigger ECL. The latter mechanism induces the light upconversion with an anti-Stokes shift (λexc > λem)41,47 and has the benefit of avoiding employing harmful incident light such as UV light for microscopy applications. Overall, both types of PECL conversion need the imposition of a potential on the SC. Generally, ECL requires a relatively high onset potential, above 1 V for the [Ru(bpy)3]2+–TPrA system (all potentials in this article are vs. Ag/AgCl). Thanks to the photovoltage generated inside the SC, PECL also has the advantage of decreasing the potential required to emit ECL.41,47 The photovoltage is a consequence of a Fermi level mismatch between the n-doped SC and its contacting phase, resulting in a depletion region and a built-in potential on the SC surface. Under illumination, the SC absorbs light, which creates e−/h+ pairs. Due to the built-in potential in the depletion region, the concentration of h+ increases at the solid/liquid interface. The photogenerated minority carriers (h+) are transferred at the SC/electrolyte interface and can trigger the ECL reaction.49 PECL has been used to photoaddress charge transfer at a local level and therefore to partially activate the electrode, either with the incident light or with the use of a heterogeneous electrode.47,51 Recently, Xu and co-workers imaged the activity of single gold nanoparticles on a TiO2 surface using PECL,52 and our group investigated the local reactivity of Ir microbands on Si/SiOx using PECL microscopy.51 To the best of our knowledge, PECL microscopy has not been reported so far for biological samples.
Here, we report the development of PECL microscopy for visualizing single Chinese hamster ovary cells (CHO-K1) using the model [Ru(bpy)3]2+–TPrA system. A red ECL emission (λem = 620 nm) is achieved by irradiating an n-type silicon (Si) photoanode with a near-infrared incident light (λexc = 1050 nm) in back-illumination. In this work, we selected the near-infrared excitation light at 1050 nm because it allows for a high penetration depth inside Si and a very important anti-Stokes shift of −430 nm (versus λem = 620 nm), making it easy to separate both wavelengths for cell imaging. PECL microscopy was demonstrated in both positive and shadow imaging modes (Fig. 1). Combining PECL with bio-imaging opens new routes for photoinduced and localized microscopy of single biological entities.
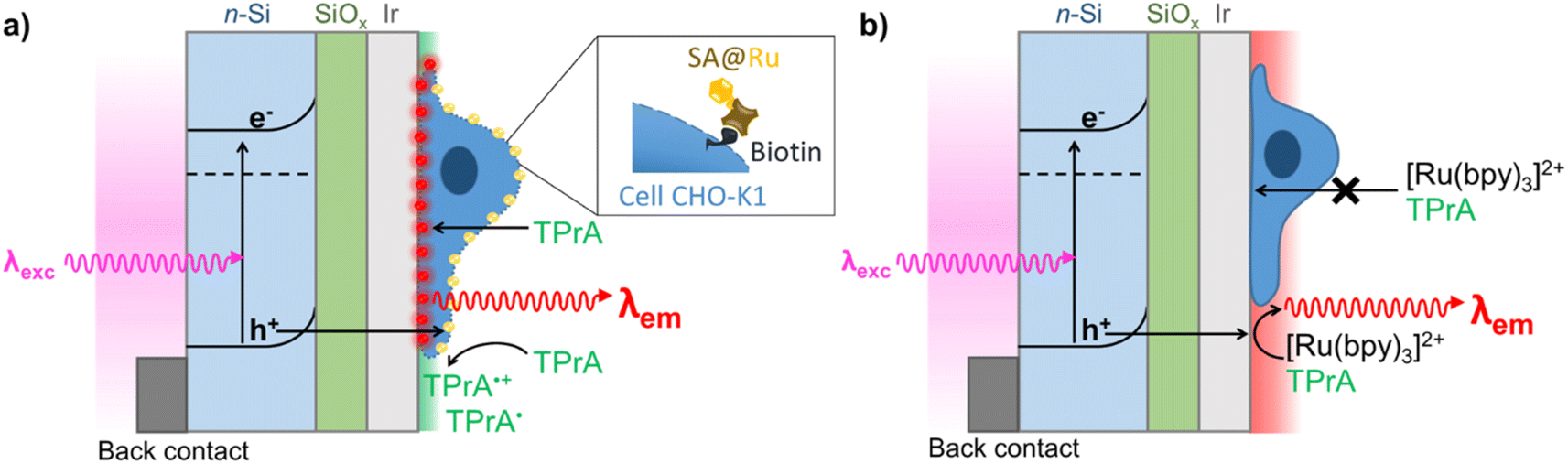 | ||
| Fig. 1 Schematic representation of two PECL microscopy modes for imaging single cells with back-illumination configuration. (a) Direct imaging by PECL+ of cell membrane labeled with the streptavidin-modified [Ru(bpy)3]2+ luminophore (SA@Ru) in TPrA solution; and (b) label-free imaging of cells by PECL− with cells immersed in a PBS solution containing both [Ru(bpy)3]2+ and TPrA. | ||
Results and discussion
Si was chosen for PECL microscopy because of its abundance and its narrow bandgap (Eg = 1.12 eV, i.e., 1107 nm),53–56 which enables large anti-Stokes shifts and thus easy separation of excitation (λexc = 1050 nm) and PECL wavelength (λem = 620 nm). This is crucial for the present application because excitation light should not affect the output optical signal during bio-imaging experiments. This near-infrared excitation wavelength was also selected because it ensures deep photon penetration within Si (α−1 ≈ 0.6 mm),57 which is important to favor the presence of photogenerated carriers at the photoanode frontside (in the depletion region) and, thus, the participation of photogenerated holes in the interfacial ECL reactions. To avoid photo-passivation of Si in water, it was protected by a 1.5 nm-thick SiOx layer (made by chemical oxidation) and a 2 nm-thick (sputtered) Ir thin film to form a metal–insulator–semiconductor (MIS) n-Si/SiOx/Ir junction,41,47 as shown in Fig. 1. This structure has proved to provide the longest PECL stability with a bright ECL emission43 and cell growth comparable to carbon or glass substrates.58 The preparation of n-Si/SiOx/Ir has been fully detailed in our previous reports.41,47To check that the detected light was generated through the interfacial transfer of photogenerated holes, we investigated the electrochemical and ECL properties of a moderately doped (photoactive) n-Si/SiOx/Ir SC anode dedicated to PECL and compared it with a highly doped degenerated (non-photoactive) p++-Si/SiOx/Ir anode active in the dark, as for classical ECL. The electrochemical and ECL behaviors of the electrodes were investigated by cyclic voltammetry (Fig. 2). Experiments were performed in a PBS (0.65 M, pH 7.4) solution containing 30 μM [Ru(bpy)3]2+ and 0.1 M TPrA. As expected, neither a current nor an ECL signal were detected for n-Si/SiOx/Ir in the dark, indicating its rectifying nature. In contrast, under 1050 nm illumination, the n-Si/SiOx/Ir electrode produced a photocurrent, meaning that the electrochemical reactions were exclusively induced by photogenerated charges. It is interesting to note that oxidation reactions on n-Si/SiOx/Ir started at 0.4 V, whereas ECL was emitted only for potentials higher than 0.65 V. This means that TPrA was first oxidized, but ECL emission occurred only with the concomitant oxidation of [Ru(bpy)3]2+, as shown in a previous study of PECL with these electrodes.47 Similar behavior was observed for the p++-Si/SiOx/Ir electrode. In this case, the current density and the ECL intensity are higher than in the PECL configuration because, in the latter case, they depend on the incident photon density and the photoconversion efficiency of Si at λexc = 1050 nm. The plateau observed at n-Si/SiOx/Ir indicates that, in these conditions, the reaction is limited by the density of absorbed photons. Conversely, the p++-Si/SiOx/Ir electrode does not show a plateau as seen on the on n-Si/SiOx/Ir curve because the reactions are not light-limited. The photoactivity is confirmed by the onset potential for ECL generation that is shifted by −296 mV compared to the p++-Si/SiOx/Ir electrode, as was the case in our previous report.41,47
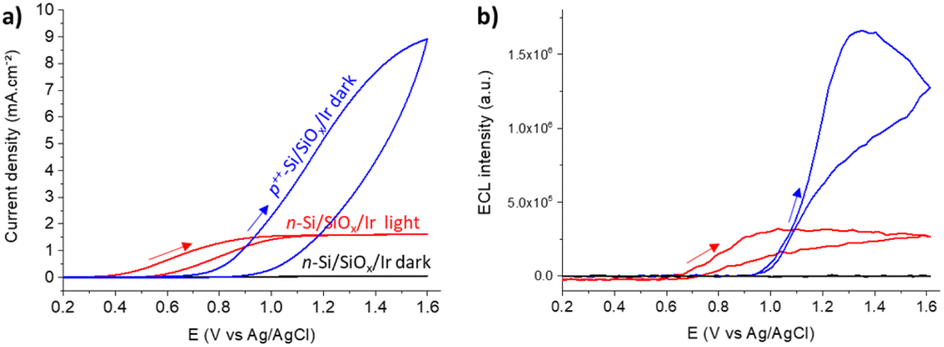 | ||
| Fig. 2 (a) Cyclic voltammograms and (b) corresponding ECL signals of n-Si/SiOx/Ir in the dark (black curve) or under 1050 nm infrared illumination (red curve) compared to the p++-Si/SiOx/Ir electrode in the dark (blue curve) in a PBS solution (pH 7.4) containing 30 μM [Ru(bpy)3]2+ and 0.1 M TPrA. Scan rate: 50 mV s−1. | ||
These results show that potentials higher than 0.7 V are sufficient to generate PECL emission. Thus, we selected a potential of 0.8 V for further PECL microscopy experiments on cells.
This data demonstrates that the excitation stimulus at λexc = 1050 nm triggers PECL emission (λem = 620 nm) at n-Si/SiOx/Ir. The anti-Stokes shift of −430 nm is large enough to allow for efficient filtering of the near-infrared excitation wavelength, thus easily discriminating the PECL signal from the incident near-infrared light (Fig. 3). Therefore, it enables microscopy experiments on cells using the PECL emission from [Ru(bpy)3]2+–TPrA. Moreover, the stability of these electrodes demonstrated in a recent report, with a bright PECL emission for at least 35 h, makes them suitable for microscopy applications.47 Next, the n-Si/SiOx/Ir photoanodes were tested for imaging CHO-K1 cells using both PECL+ and PECL− and compared to the PL and ECL images recorded with their corresponding but non-photoactive p++-Si/SiOx/Ir anodes.
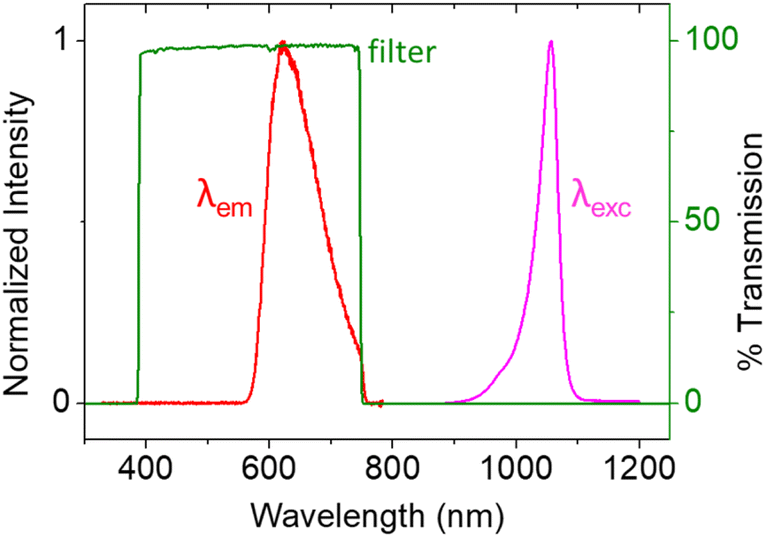 | ||
| Fig. 3 Spectra of the 1050 nm LED excitation (purple curve), the resulting PECL emission (red curve), and the filter transmission (green curve). | ||
CHO-K1 cells were grown on both n-Si/SiOx/Ir and p++-Si/SiOx/Ir photoanodes. Cell growth on the sputtered Ir thin film was comparable to a glassy carbon electrode or a glass substrate, in good agreement with previously reported results.58 The cells were imaged on an inverted epifluorescence microscope in two ways: PECL+ and PECL−, with a three-electrode set-up mounted on a commercially available chamber (Idylle tech transfer platform, Fig. S1†). For the first set of imaging experiments, we tested the PECL+ mode (Fig. 1a) with cells that were labeled using the [Ru(bpy)3]2+ luminophore bearing a streptavidin (SA@Ru).59 The cells were fixed and permeabilized with Triton X-100. This step enables the TPrA to diffuse through the membranes and to initiate the electrochemical steps over the whole cell and not only on the cell periphery. As reported previously, the entire cell became visible by ECL with this procedure.60 Then, the cells were incubated with biotin X, which reacts with the primary amino groups of proteins. Biotinylation is a classical method to label cell proteins.61 After incubation with SA@Ru, the cell membranes were decorated with the ECL emitter. PECL+ microscopy was performed in a commercially available ProCell buffer solution, which contains 0.18 M TPrA (Fig. 4). The direct oxidation of TPrA at the electrode surface generates the cation radical TPrA˙+. This is followed by its fast deprotonation reaction, which gives the neutral radical TPrA˙.62 The SA@Ru labels located on the cell membranes react with both radicals, as demonstrated previously, and generate the excited state [Ru(bpy)3]2+*, which decays by emitting ECL locally.
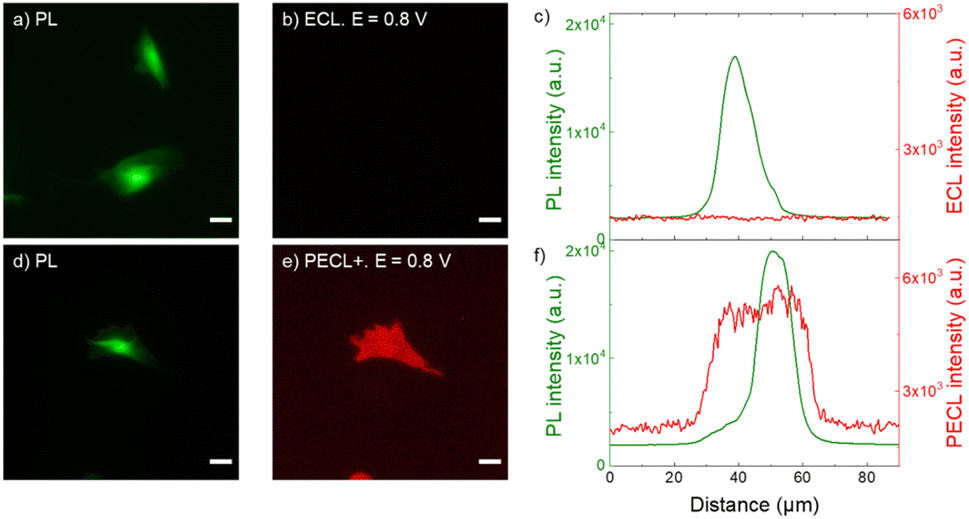 | ||
| Fig. 4 (a and d) PL micrographs (green color) of CHO-K1 cells labeled with SA@Ru. (b) ECL and (e) PECL+ images (red color) of the corresponding labeled cells recorded in ProCell under near-infrared (λexc = 1050 nm) back-illumination at 0.8 V on (b) p++-Si/SiOx/Ir and (e) n-Si/SiOx/Ir electrodes. (c and f) Comparison of the luminescence intensity profiles of the cells in PL, (c) ECL and (f) PECL+. The axis along which the profiles were extracted is shown in Fig. S4.† Green and red are false colors coding the luminescence intensity. Cells were grown on the Ir surfaces, fixed, permeabilized with Triton X-100 and then labeled with SA@Ru. Scale bar: 20 μm. | ||
Fig. 4 shows the PECL+ study of the cells labeled with SA@Ru (Fig. 1a). The microscopic images (green false color; Fig. 4a and d) correspond to the PL of the [Ru(bpy)3]2+ and so highlight the SA@Ru label localization. The entire cell is visible using PL. The pictures on the right (red false color; Fig. 4b and e) are the ECL and PECL+ images recorded at 0.8 V. Green and red false colors have been selected to code the PL and ECL intensities of the images, respectively, but the same wavelength (i.e., 620 nm) is emitted by the SA@Ru in the PL, ECL or PECL modes. At this potential, the cells are not visible by classic ECL because TPrA and SA@Ru are not oxidized on the p++-Si/SiOx/Ir electrode (Fig. 2). The labeled cells become visible using ECL only at more anodic potentials, typically 1.2 V (Fig. S2†). In the PECL+ mode, one can see the whole cell on the n-Si/SiOx/Ir electrode at 0.8 V under near-infrared illumination (Fig. 4e). On the one hand, this highlights an important advantage of the reported PECL+ approach, since it shows that lower potentials are required in comparison to classic ECL experiments. On the other hand, illumination of the biological samples is required. This is not an issue in the reported conditions since the cells were fixed, but potential phototoxicity effects should be considered in future research on PECL imaging, even if the illumination wavelength is in the near-infrared range. The control experiments of the n-type electrode recorded without near-infrared light at 0.8 V and under near-infrared light at open circuit potential did not show any PECL+ images in these conditions (Fig. S3†). The cells remain invisible in these experimental conditions. This demonstrates that the PECL+ images correspond to photogenerated h+ that oxidize TPrA and populate the excited state of the SA@Ru labels. Indeed, there is no PECL emission without the synergetic effect of incident near-infrared illumination and the imposition of a 0.8 V potential, meaning that both stimuli are required to produce PECL+ images. This is confirmed by examining the PL, ECL (Fig. 4c) and PECL+ (Fig. 4f) intensity profiles that were extracted along the axis depicted in Fig. S4.† The luminescence intensity profiles plotted in green for PL and in red for ECL (Fig. 4c) and PECL+ (Fig. 4f) confirm the sharp contrast between ECL and PECL+ for these conditions. As observed previously on a glassy carbon electrode in classic ECL mode, Fig. 4d and e shows that a significantly larger spatial extension of the cell membrane is visible by PECL+ when compared to PL. This confirms the surface-confined features of the PECL+ microscopy, which depends on the concentrations of both ECL reagents and their respective reactivity.62,68,69 This thin emitting layer improves the contrast in comparison to PL images. Finally, this set of microscopy experiments also proved the possibility of back-illumination with an MIS structure to record PECL+ images of the cells.
Finally, to complete this PECL study, we tested the second ECL imaging mode, i.e., the shadow PECL (PECL−) imaging of the cells on the n-Si/SiOx/Ir electrode (Fig. 1b). The negative or shadow ECL mode is a label-free imaging method where ECL reagents (i.e., [Ru(bpy)3]2+ and TPrA) are not covalently bonded to the cell and are dissolved in solution. This allows imaging of the morphology of the objects under investigation and the transport properties of the ECL reagents through it.60,63–65 The object, which is captured on the electrode surface, locally hinders the diffusion of both the luminophore and/or the co-reactant and thus partially or totally inhibits the initial electrochemical step and, thus, the eventual ECL generation. Therefore, the object is visible by a negative optical contrast: it appears dark on a bright luminescent background because stronger ECL intensity is produced at the bare electrode.
Herein, the cells were labeled with the calcein-AM dye. This is a standard procedure to discriminate between live and dead cells.66 Indeed, the hydrophobic AM (acetomethoxy) group makes it enter viable cells. Then, the acetomethoxy group is removed by the intracellular esterases, and the resulting green dye is trapped within the cell and fluoresces strongly. This labeling step simplifies the imaging process since it enables us to see them first by fluorescence (FL) and to compare the FL and the PECL− images. In addition, calcein-AM (λabs = 494 nm, λem = 517 nm) did not interfere with the electrochemical reactions involved in ECL or with the near-infrared excitation and ECL wavelengths. The fixed and calcein-labeled cells were immersed in the PBS solution containing the freely diffusing [Ru(bpy)3]2+ and TPrA (Fig. 1b). FL images of the calcein-AM labels define the cells' shapes (Fig. 5a and d). The images depicted in Fig. 5b and e correspond to the ECL and PECL− signals, respectively. Here again, no ECL light was emitted at 0.8 V, showing that the potential was too low to enable ECL generation at the p++-type anode. In contrast, PECL occurred at 0.8 V on a n-Si/SiOx/Ir back-illuminated electrode, and it revealed the cells in a negative (or shadow) contrast. The intensity profiles of the cells (Fig. S6†) were extracted from the FL and ECL images (Fig. 5c) and PECL− (Fig. 5f). There was no signal in ECL, whereas a local decrease in intensity was observed in the PECL− configuration. The correlation between the positive FL and the negative PECL− information is better emphasized on the 3D images. This is shown by comparing Fig. 5g with Fig. 5h, which demonstrates that the 3D FL images of a typical single cell have the same but inverted shape as the PECL intensity. As in the PECL+ mode, the control experiment of the n-type electrode without near-infrared back-illumination at 0.8 V and under near-infrared light at open circuit potential did not show any PECL in these conditions (Fig. S7†). However, the cells' morphology remains visible by classical ECL on a p++-type electrode at 1.2 V (Fig. S7†). This highlights the interest in the PECL approach, which requires a lower potential. In addition, since ECL intensity decreases when recording successive ECL images of cells or other entities in TPrA solution due to a progressive lower TPrA oxidation current,67 local photo-addressing different regions of the electrode may constitute an efficient and highly beneficial approach to avoid this general drawback in ECL microscopy.
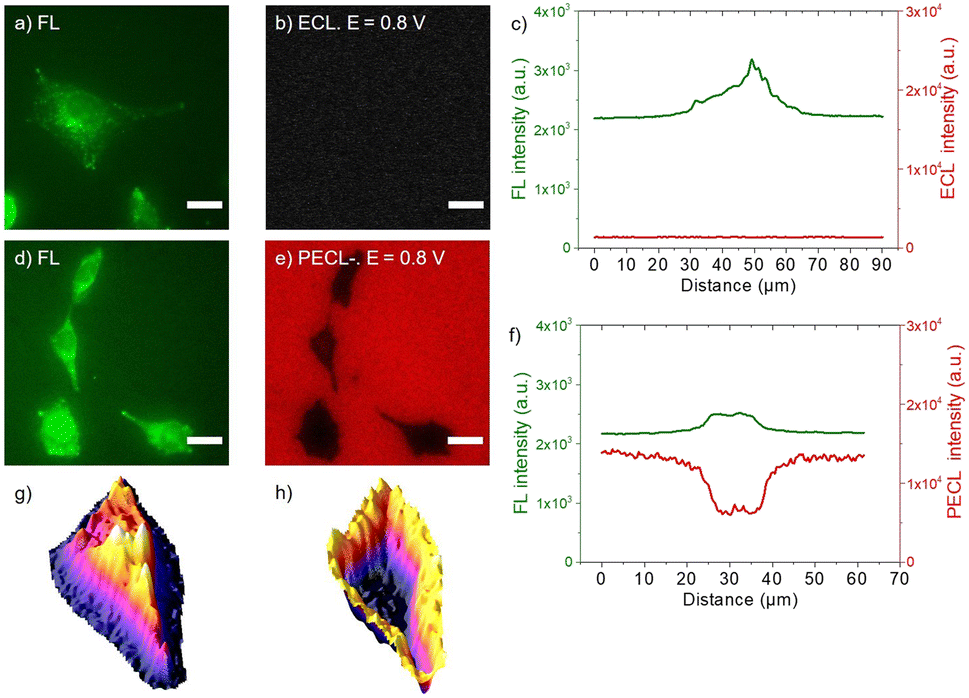 | ||
| Fig. 5 (a and d) Fluorescence micrographs (green color) of CHO-K1 cells labeled with calcein-AM. (b) ECL and (e) PECL− images (red color) of the same calcein-labeled cells recorded in PBS with 30 μM [Ru(bpy)3]2+ and 0.1 M TPrA under near-infrared (λexc = 1050 nm) back-illumination at 0.8 V on (b) p++-Si/SiOx/Ir and (e) n-Si/SiOx/Ir electrodes. (c and f) Comparison of the luminescence intensity profiles of the same cells in FL, (c) ECL and in (f) PECL−. (g and h) 3D imaging of the same cell in (g) PL and (h) PECL− modes. The axis along which the profiles were extracted is shown in Fig. S6.† Green and red are false colors coding the luminescence intensity. Cells were grown on the Ir surfaces, labeled with calcein-AM and then fixed. Scale bar: 20 μm. | ||
Conclusions
Herein, we reported the PECL microscopy of single cells with the anodic [Ru(bpy)3]2+/TPrA system, which generates a visible emission at 620 nm under an incident non-visible light at 1050 nm in back illumination. This corresponds to an anti-Stokes shift of −430 nm that allows easy separation of both wavelengths and thus makes the approach suitable for microscopy applications. The strategy is based on an n-type Si semiconductor coated by Ir into a MIS structure, n-Si/SiOx/Ir, which provides high stability and bright PECL emission. CHO-K1 cells were grown on the Ir surface of the photoanode and were imaged by PECL, exploiting the photogenerated charges. PECL microscopy was demonstrated at a remarkably low potential of 0.8 V, where cells cannot be visualized in classic ECL with a non-photoactive p++-type anode. Positive PECL (PECL+) of the cells was reported first by labeling the plasma membrane with the [Ru(bpy)3]2+ luminophore. Then, the cell morphology was imaged by an alternative label-free shadow PECL (PCL−) by immersing n-Si/SiOx/Ir in the electrolyte containing free diffusing luminophore and co-reactant. The local control of imaging with the incident near-infrared light has a strong potential in the microscopy field considering the large penetration depth of these wavelengths in biological samples. Finally, PECL microscopy adds a new dimension to the ECL toolbox with the tunable incident illumination, opening new possibilities for local photo-addressing of the electrode and for imaging biological samples.Data availability
Additional data can be obtained from the corresponding author upon request.Author contributions
J. Descamps: investigation, methodology, analysis, writing – original draft. Y. Zhao: investigation, methodology, writing – original draft. B. Goudeau: investigation, methodology, analysis. D. Manojlovic: conceptualization, methodology, writing – review & editing. G. Loget: conceptualization, supervision, writing – review & editing. N. Sojic: conceptualization, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Agence Nationale de la Recherche (LiCORN, ANR-20-CE29-0006 and ELISE, ANR-21-CE42-001) and the PHC “Pavle Savic”.Notes and references
- Z. Liu, W. Qi and G. Xu, Chem. Soc. Rev., 2015, 44, 3117–3142 RSC.
- M. Hesari and Z. Ding, J. Electrochem. Soc., 2016, 163, H3116–H3131 CrossRef CAS.
- H. Qi and C. Zhang, Anal. Chem., 2020, 92, 524–534 CrossRef CAS PubMed.
- C. Ma, Y. Cao, X. Gou and J.-J. Zhu, Anal. Chem., 2020, 92, 431–454 CrossRef CAS PubMed.
- M. Guo, D. Du, J. Wang, Y. Ma, D. Yang, M. A. Haghighatbin, J. Shu, W. Nie, R. Zhang, Z. Bian, L. Wang, Z. J. Smith and H. Cui, Chem. Biomed. Imaging, 2023, 1, 179–185 CrossRef CAS.
- X. Yang, J. Hang, W. Qu, Y. Wang, L. Wang, P. Zhou, H. Ding, B. Su, J. Lei, W. Guo and Z. Dai, J. Am. Chem. Soc., 2023, 145, 16026–16036 CrossRef CAS PubMed.
- Y. Yuan, S. Han, L. Hu, S. Parveen and G. Xu, Electrochim. Acta, 2012, 82, 484–492 CrossRef CAS.
- E. Kerr, E. H. Doeven, D. J. D. Wilson, C. F. Hogan and P. S. Francis, Analyst, 2016, 141, 62–69 RSC.
- A. Zanut, A. Fiorani, S. Canola, T. Saito, N. Ziebart, S. Rapino, S. Rebeccani, A. Barbon, T. Irie, H.-P. Josel, F. Negri, M. Marcaccio, M. Windfuhr, K. Imai, G. Valenti and F. Paolucci, Nat. Commun., 2020, 11, 2668 CrossRef CAS PubMed.
- A. Fiorani, D. Han, D. Jiang, D. Fang, F. Paolucci, N. Sojic and G. Valenti, Chem. Sci., 2020, 11, 10496–10500 RSC.
- J. Ding, P. Zhou and B. Su, ChemElectroChem, 2022, 9, e202200236 CrossRef CAS.
- Y. Wang, J. Ding, P. Zhou, J. Liu, Z. Qiao, K. Yu, J. Jiang and B. Su, Angew. Chem., Int. Ed., 2023, 62, e202216525 CrossRef CAS PubMed.
- W. Zhao, H.-Y. Chen and J.-J. Xu, Chem. Sci., 2021, 12, 5720–5736 RSC.
- X. Gou, Z. Xing, C. Ma and J.-J. Zhu, Chem. Biomed. Imaging, 2023, 1, 414–433 CrossRef CAS.
- S. Rebeccani, A. Zanut, C. I. Santo, G. Valenti and F. Paolucci, Anal. Chem., 2021, 94, 336–348 CrossRef PubMed.
- Y. B. Vogel, C. W. Evans, M. Belotti, L. Xu, I. C. Russell, L.-J. Yu, A. K. K. Fung, N. S. Hill, N. Darwish, V. R. Gonçales, M. L. Coote, K. Swaminathan Iyer and S. Ciampi, Nat. Commun., 2020, 11, 6323 CrossRef CAS PubMed.
- M. Sentic, F. Virgilio, A. Zanut, D. Manojlovic, S. Arbault, M. Tormen, N. Sojic and P. Ugo, Anal. Bioanal. Chem., 2016, 408, 7085–7094 CrossRef CAS PubMed.
- A. Zanut, A. Fiorani, S. Rebeccani, S. Kesarkar and G. Valenti, Anal. Bioanal. Chem., 2019, 411, 4375–4382 CrossRef CAS PubMed.
- C. Cui, R. Jin, D. Jiang, J. Zhang and J.-J. Zhu, Anal. Chem., 2020, 92, 578–582 CrossRef CAS PubMed.
- J. Zhang, R. Jin, D. Jiang and H.-Y. Chen, J. Am. Chem. Soc., 2019, 141, 10294–10299 CrossRef CAS PubMed.
- X. Gou, Y. Zhang, Z. Xing, C. Ma, C. Mao and J.-J. Zhu, Chem. Sci., 2023, 14, 9074–9085 RSC.
- A. Chovin, P. Garrigue and N. Sojic, Electrochim. Acta, 2004, 49, 3751–3757 CrossRef CAS.
- C. Ma, X. Gou, Z. Xing, M.-X. Wang, W. Zhu, Q. Xu, D. Jiang and J.-J. Zhu, Research, 2023, 6, 0257 CrossRef CAS.
- J. Zhang, S. Arbault, N. Sojic and D. Jiang, Annu. Rev. Anal. Chem., 2019, 12, 275–295 CrossRef CAS PubMed.
- H. Ding, P. Zhou, W. Fu, L. Ding, W. Guo and B. Su, Angew. Chem., Int. Ed., 2021, 60, 11769–11773 CrossRef CAS PubMed.
- H. Ding, W. Guo and B. Su, Angew. Chem., Int. Ed., 2020, 59, 449–456 CrossRef CAS PubMed.
- L. Xu, Y. Li, S. Wu, X. Liu and B. Su, Angew. Chem., Int. Ed., 2012, 51, 8068–8072 CrossRef CAS PubMed.
- L. Ding, P. Zhou, Y. Yan and B. Su, Chem. Biomed. Imaging, 2023, 1, 558–565 CrossRef CAS.
- J. Feng, Curr. Opin. Electrochem., 2022, 34, 101000 CrossRef CAS.
- J. Dong and J. Feng, Anal. Chem., 2023, 95, 374–387 CrossRef CAS PubMed.
- S. Knezevic, L. Bouffier, B. Liu, D. Jiang and N. Sojic, Curr. Opin. Electrochem., 2022, 35, 101096 CrossRef CAS.
- J. Dong, Y. Lu, Y. Xu, F. Chen, J. Yang, Y. Chen and J. Feng, Nature, 2021, 596, 244–249 CrossRef CAS PubMed.
- Y. Zhou, J. Dong, P. Zhao, J. Zhang, M. Zheng and J. Feng, J. Am. Chem. Soc., 2023, 145, 8947–8953 CrossRef CAS PubMed.
- W. Zhu, J. Dong, G. Ruan, Y. Zhou and J. Feng, Angew. Chem., Int. Ed., 2023, 62, e202214419 CrossRef CAS PubMed.
- J. Dong, Y. Xu, Z. Zhang and J. Feng, Angew. Chem., Int. Ed., 2022, 61, e202200187 CrossRef CAS PubMed.
- B. Li, X. Huang, Y. Lu, Z. Fan, B. Li, D. Jiang, N. Sojic and B. Liu, Adv. Sci., 2022, 9, 2204715 CrossRef CAS PubMed.
- Y. Liu, H. Zhang, B. Li, J. Liu, D. Jiang, B. Liu and N. Sojic, J. Am. Chem. Soc., 2021, 143, 17910–17914 CrossRef CAS PubMed.
- Y. Lu, X. Huang, S. Wang, B. Li and B. Liu, ACS Nano, 2023, 17, 3809–3817 CrossRef CAS PubMed.
- D. Laser and A. J. Bard, Chem. Phys. Lett., 1975, 34, 605–610 CrossRef CAS.
- J. D. Luttmer and A. J. Bard, J. Electrochem. Soc., 1979, 126, 414–419 CrossRef CAS.
- Y. Zhao, J. Yu, G. Xu, N. Sojic and G. Loget, J. Am. Chem. Soc., 2019, 141, 13013–13016 CrossRef CAS PubMed.
- Y. B. Vogel, N. Darwish and S. Ciampi, Cell Rep. Phys. Sci., 2020, 1, 100107 CrossRef.
- Y. Zhao, J. Descamps, Y. Léger, L. Santinacci, S. Zanna, N. Sojic and G. Loget, Electrochim. Acta, 2023, 444, 142013 CrossRef CAS.
- J. Yu, H. Saada, N. Sojic and G. Loget, Electrochim. Acta, 2021, 381, 138238 CrossRef CAS.
- J. Yu, H. Saada, R. Abdallah, G. Loget and N. Sojic, Angew. Chem., Int. Ed., 2020, 59, 15157–15160 CrossRef CAS PubMed.
- J. Descamps, Y. Zhao, J. Yu, G. Xu, Y. Léger, G. Loget and N. Sojic, Chem. Commun., 2022, 58, 6686–6688 RSC.
- Y. Zhao, J. Descamps, S. Ababou-Girard, J.-F. Bergamini, L. Santinacci, Y. Léger, N. Sojic and G. Loget, Angew. Chem., Int. Ed., 2022, 61, e202201865 CrossRef CAS PubMed.
- Y. Zhao, J. Descamps, B. Le Corre, Y. Léger, A. Kuhn, N. Sojic and G. Loget, J. Phys. Chem. Lett., 2022, 13, 5538–5544 CrossRef CAS PubMed.
- Y. Zhao, L. Bouffier, G. Xu, G. Loget and N. Sojic, Chem. Sci., 2022, 13, 2528–2550 RSC.
- Y. B. Vogel, J. J. Gooding and S. Ciampi, Chem. Soc. Rev., 2019, 48, 3723–3739 RSC.
- J. Descamps, Y. Zhao, J. Le-Pouliquen, B. Goudeau, P. Garrigue, K. Tavernier, Y. Léger, G. Loget and N. Sojic, Chem. Commun., 2023, 59, 12262–12265 RSC.
- J.-W. Xue, C.-H. Xu, W. Zhao, H.-Y. Chen and J.-J. Xu, Nano Lett., 2023, 23, 4572–4578 CrossRef CAS PubMed.
- M. J. Kenney, M. Gong, Y. Li, J. Z. Wu, J. Feng, M. Lanza and H. Dai, Science, 2013, 342, 836–840 CrossRef CAS PubMed.
- B. Fabre and G. Loget, Acc. Mater. Res., 2023, 4, 133–142 CrossRef CAS.
- K. Sun, S. Shen, Y. Liang, P. E. Burrows, S. S. Mao and D. Wang, Chem. Rev., 2014, 114, 8662–8719 CrossRef CAS PubMed.
- S. Hu, M. R. Shaner, J. A. Beardslee, M. Lichterman, B. S. Brunschwig and N. S. Lewis, Science, 2014, 344, 1005–1009 CrossRef CAS PubMed.
- M. Saritaş and H. D. McKell, J. Appl. Phys., 1987, 61, 4923–4925 CrossRef.
- K. Göbbels, T. Kuenzel, A. van Ooyen, W. Baumgartner, U. Schnakenberg and P. Bräunig, Biomaterials, 2010, 31, 1055–1067 CrossRef.
- G. Valenti, S. Scarabino, B. Goudeau, A. Lesch, M. Jović, E. Villani, M. Sentic, S. Rapino, S. Arbault, F. Paolucci and N. Sojic, J. Am. Chem. Soc., 2017, 139, 16830–16837 CrossRef CAS PubMed.
- S. Voci, B. Goudeau, G. Valenti, A. Lesch, M. Jović, S. Rapino, F. Paolucci, S. Arbault and N. Sojic, J. Am. Chem. Soc., 2018, 140, 14753–14760 CrossRef CAS PubMed.
- M. Howarth and A. Y. Ting, Nat. Protoc., 2008, 3, 534–545 CrossRef CAS PubMed.
- W. Miao, J.-P. Choi and A. J. Bard, J. Am. Chem. Soc., 2002, 124, 14478–14485 CrossRef CAS PubMed.
- S. Knežević, E. Kerr, B. Goudeau, G. Valenti, F. Paolucci, P. S. Francis, F. Kanoufi and N. Sojic, Anal. Chem., 2023, 95, 7372–7378 CrossRef PubMed.
- C. Ma, S. Wu, Y. Zhou, H.-F. Wei, J. Zhang, Z. Chen, J.-J. Zhu, Y. Lin and W. Zhu, Angew. Chem., Int. Ed., 2021, 60, 4907–4914 CrossRef CAS PubMed.
- X. Hu, S. Yu, C. Wang, X. Zhang, J. Pan and H. Ju, Anal. Chem., 2023, 95, 4496–4502 CrossRef CAS PubMed.
- L. Gutiérrez, G. Stepien, L. Gutiérrez, M. Pérez-Hernández, J. Pardo, J. Pardo, V. Grazú and J. M. de la Fuente, in Comprehensive Medicinal Chemistry III, ed. S. Chackalamannil, D. Rotella and S. E. Ward, Elsevier, Oxford, 2017, pp. 264–295 Search PubMed.
- D. Han, B. Goudeau, D. Jiang, D. Fang and N. Sojic, Anal. Chem., 2021, 93, 1652–1657 CrossRef CAS PubMed.
- W. Guo, P. Zhou, L. Sun, H. Ding and B. Su, Angew. Chem., Int. Ed., 2021, 60, 2089–2093 CrossRef CAS.
- Y. Wang, W. Guo, Q. Yang and B. Su, J. Am. Chem. Soc., 2020, 142, 1222–1226 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05983a |
| This journal is © The Royal Society of Chemistry 2024 |