
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Semisynthetic ferritin-based nanoparticles with high magnetic anisotropy for spatial magnetic manipulation and inductive heating†
Andreas
Neusch
 a,
Ulf
Wiedwald
b,
Iuliia P.
Novoselova‡
a,
Daniel A.
Kuckla
a,
Nikolaos
Tetos
b,
Sarah
Sadik§
a,
Philipp
Hagemann
a,
Michael
Farle
b and
Cornelia
Monzel
*a
a,
Ulf
Wiedwald
b,
Iuliia P.
Novoselova‡
a,
Daniel A.
Kuckla
a,
Nikolaos
Tetos
b,
Sarah
Sadik§
a,
Philipp
Hagemann
a,
Michael
Farle
b and
Cornelia
Monzel
*a
aExperimental Medical Physics, Heinrich-Heine University Düsseldorf, 40225 Düsseldorf, Germany. E-mail: Cornelia.Monzel@hhu.de
bFaculty of Physics and Center for Nanointegration (CENIDE), University of Duisburg-Essen, 47057 Duisburg, Germany
First published on 11th July 2024
Abstract
The human iron storage protein ferritin represents an appealing template to obtain a semisynthetic magnetic nanoparticle (MNP) for spatial manipulation or inductive heating applications on a nanoscale. Ferritin consists of a protein cage of well-defined size (12 nm), which is genetically modifiable and biocompatible, and into which a magnetic core is synthesised. Here, we probed the magnetic response and hence the MNP's suitability for (bio-)nanotechnological or nanomedical applications when the core is doped with 7% cobalt or 7% zinc in comparison with the undoped iron oxide MNP. The samples exhibit almost identical core and hydrodynamic sizes, along with their tunable magnetic core characteristics as verified by structural and magnetic characterisation. Cobalt doping significantly increased the MNP's anisotropy and hence the heating power in comparison with other magnetic cores with potential application as a mild heat mediator. Spatial magnetic manipulation was performed with MNPs inside droplets, the cell cytoplasm, or the cell nucleus, where the MNP surface conjugation with mEGFP and poly(ethylene glycol) gave rise to excellent intracellular stability and traceability within the complex biological environment. A magnetic stimulus (smaller than fN forces) results in the quick and reversible redistribution of the MNPs. The obtained data suggest that semisynthetic ferritin MNPs are highly versatile nanoagents and promising candidates for theranostic or (bio-)nanotechnological applications.
Introduction
Over the last few decades, magnetic nanoparticles (MNPs) have become indispensable tools in nanomedicine for delivery,1,2 imaging,3–6 therapy,5,7,8 or combined approaches. More recently, (bio)nanotechnology-based approaches have used the specific coupling of MNPs to biomolecules: (1) to inductively heat them to create nanoscale hot spots9–11 (2) to spatially redistribute molecules,12,13 since the spatiotemporal distribution of molecules plays a key role in biological signalling processes,14,15 or (3) to apply forces to molecules for their controlled conformational change. The particular advantage of combining such nanoparticle-based approaches with a remote magnetic stimulus is to actively change biomolecular states in a contact-free and controlled manner. This is in contrast to what is hitherto possible by only observing the system's passive response.The active stimulation provided by cases (1)–(3) is highly relevant for biomaterial development, subcellular signalling or biomechanical studies,16–19 as well as for applying a stimulus in a whole organism. The aforementioned nanomedical approaches always aim at an in vivo application. The use of MNPs in such a biological context, however, requires their biocompatibility, to endow them with specific targeting properties, and a magnetic core which is sufficiently magnetisable and tuneable with regard to its magnetic anisotropy. In addition, the overall particle size should be below 100 nm.20 As a result, partly contradictory demands need to be reconciled, which generally is a difficult task.
Here, we synthesised ferritin-based MNPs, commonly referred to as magnetoferritin (MFt), which have attracted considerable attention due to their semi-synthetic and multifunctional nature.21–26 We show how MFts meet most of the abovementioned demands and scrutinise their suitability for spatial magnetic manipulation and inductive heating approaches, the latter also as a response to a recent discussion in the field.27,28
MFts are based on the human iron storage protein ferritin, which forms a well-defined hollow sphere with an internal and external diameter of 8 nm and 12 nm, respectively.29,30 As such, the protein serves as a template, into which magnetic nanocrystals of high homogeneity, in terms of their shape and size, are synthesised. This semi-synthetic design using a protein shell as a template has several advantages for theranostic or (bio-)nanotechnological applications, which include excellent solubility and stability in physiological solutions as well as a low toxicity. Moreover, due to their well-defined, small size, they remain mobile in various biological environments.9,17,31 Furthermore, MFts can be easily modified by genetic engineering or chemical reactions involving one of the primary amines, carboxylates or thiols exposed to their exterior. Thus, fluorescent labels or tags for site-specific targeting can be attached.32–34 Previously, MFts were shown to couple to other biomolecules without apparent alteration of their natural function.12,35,36 Finally, ferritin exhibits an exceptional stability over a wide range of temperatures (up to ∼80 °C) and pH (3–10).
Ferritin is the most abundant iron-storage protein and its globular, hollow structure is conserved in most organisms. It is responsible for mediating iron homeostasis and to prevent oxidative stress.22 Human ferritin forms a 24-mer protein37 composed of two subunits: heavy chain ferritin (HCF) and light chain ferritin (LCF).22,38 Due to their structural similarities, cages can be formed by any numerical combination of the two subunits. Although LCF and HCF are almost identical in structure, only HCF is equipped with a ferroxidase domain capable of oxidising toxic, soluble Fe(II) to insoluble and less harmful Fe(III) upon intake. The oxidised iron atoms sediment inside the cage and can fill it up to approximately 4500 iron atoms.39 Naturally occurring ferritin cores are ferrihydrites and were reported to exhibit paramagnetic or superparamagnetic properties above 12 K.40
With regard to the core, a wide choice of inorganic materials have so far been mineralised inside the ferritin cage, such as Fe3O4,41–44 Co3O4,45,46 Mn3O4,47 CoPt,48 Pd,49 Ag,50 CdS,51 CdSe,52 and ZnSe.53 Here, the inner cage diameter of 8 nm naturally confines the core and gives rise to a narrow size distribution. The synthesised MNPs are generally superparamagnetic at ambient or higher temperature, as they exhibit a magnetocrystalline anisotropy with an energy density that cannot block the particle's magnetic dipole moment at an upper size limit of 8 nm given by the ferritin cage. Superparamagnetism is particularly advantageous for many applications, since the nanoparticles do not show significant particle–particle (magnetic dipole–dipole) interactions. As a result, they do not form chains or aggregates in suspension and are more stable over time. On the other hand, the constraint to the growth of the inorganic core imposed by the ferritin size represents an important limitation to its magnetic properties. In particular, the applicability of ferritin for the hyperthermic heating of malignant cells or for the switching of thermally sensitive molecules was recently questioned.27 Theoretical and experimental studies have indeed demonstrated that the maximum efficiency of hyperthermic heating is achieved for magnetite MNPs of 15–18 nm diameter and that it drops off sharply upon MNP size reduction.19,54–57
It is hence important to show that the MFts’ properties can be tuned to maximise their efficiency for hyperthermic heating and their suitability for spatial magnetic remote manipulation. A viable route is to increase the magnetic anisotropy of the inorganic core, which can be simply realised through the replacement of ferrous ions in Fe oxides with dopants.11 A prominent example is cobalt-doped ferrite nanoparticles, whose magnetocrystalline anisotropy can be up to 20 times larger than that of the undoped ferrite, retaining a good heat dissipation property down to a size of about 8 nm.58 Interestingly, the highest values of magnetic anisotropy in ferritin were previously obtained using cobalt (Co) doping at a small percentage.9,59,60 When ferrite nanoparticles were doped with a small percentage of zinc (Zn), the saturation magnetisation was shown to increase slightly.61 Additional changes in the magnetic behaviour of the particles may arise, due to the inverse spinel structure of Co-doped ferrites (with Co2+ ions in octahedral sites and Fe3+ ions equally distributed between tetrahedral and octahedral sites) and the normal spinel structure of Zn-doped ferrites (with Zn2+ ions in tetrahedral and Fe3+ in octahedral sites).62 On the basis of these findings, MFt is a highly interesting nanoagent for spatial redistribution studies in external magnetic fields and has been controversially discussed as a candidate to mediate inductive heating processes.9,27,63
In this work, we probed the characteristics of MFt and doped the magnetic core to tailor its magnetic properties (i.e., to increase the magnetic anisotropy energy density or saturation magnetisation) for these applications. Former studies showed a strong improvement concerning heat dissipation in hyperthermia assays of MFt between no doping and 5% of Co doping, while 10% led to less heat dissipation.9 Accordingly, the effective anisotropy of γ-Fe2O3 MNPs reaches a plateau at doping levels >5% of Co2+.64 Hence, we chose to investigate the effect of doping MFt cores with 7% Co or 7% Zn with respect to Fe. The manuscript is structured as follows: first, the structural and magnetic properties of pure and doped MFts were determined. Thereafter, an example to increase the MFts’ inductive heating response is presented. The spatial manipulation of MFts applying magnetic field gradients is then shown for simple droplets and inside living cells. In the cell cytoplasm and in the cell nucleus, a repetitive and reversible tuning of MFts’ spatial distributions was achieved. In cellular environments, the protein shell of MFts allows the MNPs to remain biocompatible despite the presence of Co2+, as demonstrated previously in the absence and presence of external magnetic fields.9,31 Overall, the comparison of different MFts and their applications provides important guidelines for their use as biomedical or nanotechnological agents as a possible strategy for improved spatial manipulation and magnetic hyperthermia to fully exploit the multiple advantages that ferritin MNPs offer.
Materials and methods
Ferritin purification
A semi-synthetic ferritin nanoprobe was prepared as previously described.31 In short, the ferritin cage was genetically engineered so that each subunit of the 24-homomer consisted of the heavy chain ferritin subunit with mEGFP fused to its N-terminus. The cDNA of this fused subunit was inserted into the pET21a(+) vector (reporter gene: ampR, see Fig. S1†). The construct was a generous gift from the Coppey/Hajj laboratory at the Laboratoire Physico-Chimie, Institut Curie, Paris, France and the Piehler laboratory at the University of Osnabrück, Germany.The engineered protein was expressed in BL21-CodonPlus (DE3)-RIPL competent cells (Agilent, Santa Clara, CA, USA). Bacteria were transformed via heat shock and plated on LB-Agar containing ampicillin (0.1 μg mL−1). A single colony was chosen and used to grow a preculture and later the main culture in 2×YT medium (containing 0.1 μg mL−1 ampicillin). Bacteria were grown at 37 °C and 230 rpm in an orbital shaking incubator up to an OD600 of 0.6 to 0.8. At this point, expression was induced by the addition of the lac operon activator IPTG (isopropyl β-D-1-thiogalactopyranoside) at a final concentration of 1 mM. Cultures were incubated for 16 h at 16 °C and 160 rpm in a shaking incubator. Bacteria were then harvested at 8000g for 10 min, washed in PBS, collected at 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 5 min and finally resuspended in HEPES-2 buffer (50 mM HEPES, 150 mM NaCl, pH 8.0, filtered 0.2 μm). To prevent unwanted protein degradation, protease inhibitors were added to the bacteria according to the manufacturer's advice (cOmplete EDTA-free protease inhibitor cocktail, Roche, Basel, Switzerland). To that, 0.2 mg mL−1 of both DNAse I (Roche) and lysozyme (PanReac AppliChem, Darmstadt, Germany) were added. Bacteria were homogenised using a microfluidiser (M110P microfluidizer, Microfluidics, Westwood, MA, USA). After cell disruption, PMSF (phenylmethylsulfonyl fluoride, Merck, Darmstadt, Germany) was added to the solution at a final concentration of 5 mM. The lysate was then centrifuged for 40 min at 19000g and 4 °C. The released proteins were further purified by heat denaturation in a water bath at 70 °C for 15 min and ammonium sulphate precipitation at 30% and 70% at 4 °C. After desalting via dialysis (20 kDa cut-off) against buffer HEPES-1 (20 mM HEPES, 100 mM NaCl, pH 8.0, filtered 0.2 μm), the protein solution was separated using a size exclusion column (HiPrep Sephacryl S-400 HR, Cytiva, Marlborough, MA, USA) in a FPLC system (NGC, Bio-Rad, Hercules, CA, USA). The purified ferritin cages were then PEGylated using O-[(N-succinimidyl)succinyl-aminoethyl]-O′-methylpolyethylene glycol 2′000 (Sigma Aldrich, St Louis, MO, USA) in DMSO at a molar excess of 3500 PEG units per ferritin cage. The mixture was left to react at room temperature for 2 h on a rotary shaker and cleaned afterwards using a PD-10 desalting column (Cytiva).
000g for 5 min and finally resuspended in HEPES-2 buffer (50 mM HEPES, 150 mM NaCl, pH 8.0, filtered 0.2 μm). To prevent unwanted protein degradation, protease inhibitors were added to the bacteria according to the manufacturer's advice (cOmplete EDTA-free protease inhibitor cocktail, Roche, Basel, Switzerland). To that, 0.2 mg mL−1 of both DNAse I (Roche) and lysozyme (PanReac AppliChem, Darmstadt, Germany) were added. Bacteria were homogenised using a microfluidiser (M110P microfluidizer, Microfluidics, Westwood, MA, USA). After cell disruption, PMSF (phenylmethylsulfonyl fluoride, Merck, Darmstadt, Germany) was added to the solution at a final concentration of 5 mM. The lysate was then centrifuged for 40 min at 19000g and 4 °C. The released proteins were further purified by heat denaturation in a water bath at 70 °C for 15 min and ammonium sulphate precipitation at 30% and 70% at 4 °C. After desalting via dialysis (20 kDa cut-off) against buffer HEPES-1 (20 mM HEPES, 100 mM NaCl, pH 8.0, filtered 0.2 μm), the protein solution was separated using a size exclusion column (HiPrep Sephacryl S-400 HR, Cytiva, Marlborough, MA, USA) in a FPLC system (NGC, Bio-Rad, Hercules, CA, USA). The purified ferritin cages were then PEGylated using O-[(N-succinimidyl)succinyl-aminoethyl]-O′-methylpolyethylene glycol 2′000 (Sigma Aldrich, St Louis, MO, USA) in DMSO at a molar excess of 3500 PEG units per ferritin cage. The mixture was left to react at room temperature for 2 h on a rotary shaker and cleaned afterwards using a PD-10 desalting column (Cytiva).
Magnetic core synthesis
Ferritin was loaded with a magnetic core consisting of ferrihydrite including cobalt or zinc as dopants. For the synthesis, 25 mL of PEGylated ferritin was heated to 65 °C in 100 mM NaCl at a ferritin concentration of 50 nM. The pH was kept constant at 8.5 by the addition of 100 mM NaOH using an autotitrator (Titration Excellence T5, Mettler-Toledo, Columbus, OH, USA). The whole reaction was performed under a constant stream of N2 to suppress unwanted oxidation.To produce magnetic cores, both the ferrous precursor (iron(II) sulphate hexahydrate, Honeywell, Charlotte, NC, USA; see also Table 1) and H2O2 (5.5 mM, Merck) were dissolved in 2 mL of 100 mM NaCl and filtered through a 0.2 μm syringe filter. These two reagents were then added to the reaction vessel at a rate of 200 μL min−1 over a course of 10 min. In this setup, the soluble Fe2+ diffuses into the core and can be oxidised to the insoluble form in a controlled manner via the HCF's ferroxidase centre. The oxidised Fe3+ ions sediment inside the protein cage to form the MFt core, leading to brown colouration of the solution. In order to dope MFt cores, parts of the ferrous precursor were replaced with precursors of either Co2+ (CoCl2, Honeywell) or Zn2+ (ZnCl2, Acros Organics, Waltham, MA, USA; for detailed compositions see Table 1). This method enabled the implementation of doping ions into the core structure during its formation.9,65
| Sample | Dopant | Molarity of the precursor | Final % | |
|---|---|---|---|---|
| Fe (μM) | Dopant (μM) | |||
| Pure | — | 1034 | — | — |
| Zn7 | Zn | 983 | 52 | 6.7 ± 1.3 |
| Co7 | Co | 983 | 116 | 6.4 ± 2.2 |
After complete addition of the precursors and upon completion of the reaction, the reaction was quenched with trisodium citrate (Sigma Aldrich) at a final concentration of 2 mM. The fresh product was centrifuged at 19000g at 4 °C for 30 min, then filtered through a 0.2 μm syringe filter and concentrated using Amicon Ultra centrifugal filters (100 kDa cut-off, Merck).
For further characterisation of the MNPs (X-ray diffraction and vibrating sample magnetometry), the samples were dialysed against low concentration HEPES buffer (0.2 mM HEPES, 1 mM NaCl, pH 8.0) to reduce the amount of salt and buffering agent in the sample. The samples were then plunge frozen in liquid nitrogen and lyophilised. For all other characterisation studies, the samples were kept in HEPES-1.
Transmission electron microscopy
Protein cages and magnetic cores were imaged by transmission electron microscopy (TEM). Cages and cores were recorded separately, since organic materials such as proteins are hardly visible by TEM. Protein cages were hence stained with uranyl acetate, while magnetic cores were recorded without additional staining.For TEM measurements, freshly prepared samples of both ferritin and differently doped magnetoferritin (MFt) were diluted to 0.05 μM. A droplet was placed on a TEM grid (Ni grid, Formvar carbon film, Plano GmbH, Wetzlar, Germany) and left to sediment for 1 min. Excess solution was removed using filter paper. Unstained samples were left to dry under ambient conditions. For uranyl acetate staining, the grid was dipped into a drop of 2% uranyl acetate for 3 s after sample sedimentation and the remaining liquid was removed using filter paper. Afterwards, the grid was placed onto a second drop of 2% uranyl acetate and left for 30 s. Excess solution was removed using filter paper and the grid was left to dry in air.
Stained images were taken with a JEOL JEM-2100Plus (Akishima, Tokyo, Japan) with an acceleration voltage of 80 kV. Unstained bright-field (BF) high-resolution TEM (HRTEM) and scanning TEM (STEM) images were acquired with a JEOL 2200FS transmission electron microscope at an acceleration voltage of 200 kV using a 2k × 2k GATAN UltraScan1000XP CCD camera. The local chemical composition was determined using EDX in STEM mode with an Oxford windowless 80 mm2 SDD X-MaxN 80 TLE detector with a 0.21 sr solid angle. HRTEM and EDX data were analysed using Gatan Micrograph Suite and Oxford's Aztec software.
X-ray diffraction
X-ray diffraction (XRD) patterns were recorded using a PANalytical X'Pert PRO diffractometer equipped with a spinner for powders using Cu-Kα radiation and an X'Celerator detector. The background of the XRD patterns was subtracted and further data processing was performed using OriginPro. A 50-point moving average smoothing was applied to the diffractograms. Due to the small step width, the MFt peak width remained unchanged while the sharp NaCl peaks exhibited slightly smaller intensities.Vibrating sample magnetometry
The magnetic properties of all samples were studied using Vibrating Sample Magnetometry (VSM) with a PPMS DynaCool system (Quantum Design, San Diego, CA, USA). The measurements were taken on powder samples within the field range of ±9 T at various temperatures ranging from 5 K to 330 K and normalised to the total sample mass.Dynamic light scattering and electrophoretic light scattering
For characterisation of the nanoparticles in solution and, hence, under biological conditions, magnetoferritin was analysed using dynamic light scattering (DLS) and electrophoretic light scattering (ELS). Prior to measuring, the samples were diluted to 1 μM in HEPES-1 buffer (pH 8.0, see the Ferritin purification section) and filtered through a 0.2 μm filter to remove agglomerates. All measurements were performed using a Zetasizer Nano ZS (Malvern Panalytical Ltd, Malvern, UK). Each sample was characterised by averaging over 75 sub-runs (5 subsequent runs, each containing 15 sub-runs) with 10 s measurements per sub-run. A log–normal fit was applied to the measured number distribution to calculate the hydrodynamic diameter DH, representing the diameter of a sphere with the same diffusion properties as the analysed particle.ELS measurements were performed at 40 V to account for the high conductivity of saline buffers and to prevent damage of the measuring cell's electrodes. For each sample, 300 sub-runs (10 subsequent runs, each containing 30 sub-runs) were recorded.
Alternating magnetic field-induced heating
Magnetic hyperthermia measurements were performed in a custom-made alternating magnetic field (AMF) setup described previously.66 In short, 200 μL of magnetoferritin solution was placed in a single-use cuvette and subjected to an AC magnetic field (45 mT induction amplitude at 93.75 kHz or 104.5 kHz). Thermal changes were monitored by recording the solution surface with a thermal imager (VarioCam HD, InfraTec GmbH, Dresden, Germany) with an IR 1.0/30 JENOPTIK objective (InfraTec GmbH). Measurements were performed every 10 s over a duration of 30 min. The magnetic field was switched on directly after the first image and switched off after 15 min (91 images). To obtain the sample's temperature, the average temperature of the sample surface in the cuvette (approximately 5 × 10 mm2 or 300 pixels) was determined. The measurement was repeated three times for all samples. Images were evaluated using the software IRBIS 3 plus (InfraTec GmbH).Droplet assay for quantification of magnetic forces
In order to characterise the forces that can be applied to MFt, a droplet assay on coverslips was performed. In a first step, coverslips were coated with a PDMS layer (SYLGARD 184 Silicone Elastomer Kit, Dow Europe GmbH, Wiesbaden, Germany) to reduce unspecific binding of MFt to the glass surface. For this, a 1:10 mixture (curing agent to elastomer by weight) of PDMS was diluted in twice the amount (by weight) of hexane.67 A coverslip (22 × 22 mm2, thickness: 170 ± 5 μm) was sonicated in an ultrasonic bath for 10 min in isopropanol, followed by another sonication step in ultrapure water. In between treatments, the coverslip was thoroughly flushed with ultrapure water. Afterwards, the coverslip was blown dry using pressurised N2. The coverslip was then mounted onto a spin coater and 100 μL of the PDMS mixture was placed on top. The spin coater was run for 150 s at 6000 rpm with an acceleration of 100 rpm s−1. After spin coating, the coverslip was cured at 110 °C for 20 min.
For the droplet assay, MFt was diluted to 0.1 μM in HEPES-1 with 25 V% of glycerol. A droplet of 1 μL was placed onto a PDMS-coated coverslip and a magnetic tip was brought close to it. The magnetic tip consisted of two magnets (cube: NdFeB, side length: 5 mm, gold-plated, product number: W-05-G; cuboid: NdFeB, 10 × 4 × 1.2 mm3, gold-plated, product number: Q-10-4-1.2-G; both from supermagnete.de by Webcraft GmbH, Gottmadingen, Germany) and a steel wire (diameter: 0.4 mm, product number: 1416, Röslau Stahldraht, Röslau, Germany) attached to the magnets.31 The attraction of the fluorescent MFt was observed using an inverted epifluorescence microscope (IX83 by Olympus, Shinjuku, Tokyo, Japan) equipped with a 60× oil-objective with a numerical aperture N.A. = 1.25 and phase contrast PH3 (Olympus UPLFLN60XOIPH/1,25, Olympus). Data analysis was performed using Matlab (R2023a, The Mathworks Inc., Natick, MA, USA). A horizontal line scan (thickness: 1 μm) over the intensity in the fluorescence channel was performed starting from the position of the magnetic tip. Position 0 was placed at the edge of the drop with the inside of the drop in the positive direction. A linear background (y = mx + c) was fitted to intensities at higher positions and subtracted from the line scan to obtain the final intensity curve.
Spatial magnetic manipulation
For imaging and manipulation, Cos7 cells were plated on sterilised glass coverslips in 35 mm cell-culture dishes at about 50% confluency. Prior to experiments, the cells were washed with PBS buffer and re-incubated in preheated Leibovitz medium (L15, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% foetal calf serum (FCS) and 1% PenStrep (Thermo Fisher Scientific). Imaging was performed in a heating chamber (TempController 2000-2, PeCon GmbH, Erbach, Germany) set to 37 °C and mounted on an inverted microscope (IX83 from Olympus) equipped with a 60× oil-objective with N.A. = 1.25 and phase contrast PH3 (Olympus UPLFLN60XOIPH/1,25, Olympus). MFts were centrifuged (10000g for 10 min) and the supernatant was used. MNPs were injected into the cells using a micromanipulation system (InjectMan 4 and FemtoJet 4i, Eppendorf, Hamburg, Germany) with microinjection capillaries (Femtotip II, inner diameter of 500 nm, Eppendorf). For injection, MFt with a concentration of 1 μM was used. After microinjection, the sample was washed with the previously mentioned Leibovitz medium to remove the remaining MFt in the medium. Thereafter, a home-built magnetic tip (described in the Droplet assay for quantification of magnetic forces section and in the study by Novoselova et al.31) was approached to the cell using InjectMan 4 of the micromanipulation system. Videos of MFt redistribution in the fluorescence channel were acquired typically every 10 s over 30–45 min at a 200 ms camera exposure time (Prime BSI, Photometrics, Tucson, AZ, USA, see also Videos S1–S3†). Phase contrast and fluorescence image recordings were analysed using in-house developed analysis routines written in Matlab (R2023a, The Mathworks Inc.) and ImageJ (version 1.49v, U. S. National Institutes of Health, Bethesda, MD, USA).68 A background subtraction was applied to the fluorescence videos.
Cell viability assay
For evaluation of the toxicity of MFt on the cells, standardised CellTiter-Blue (CTB) assay from Promega (Fitchburg, WI, USA) was performed as described previously.31 Cos7 cells were seeded in a 96-well plate (25 000 cells per well) one day in advance. The next day, they were incubated with varying concentrations of MFt for 1 h in a mixture of 100 μL of both Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal calf serum and 1% PenStrep (all from Thermo Fisher Scientific) and HEPES-1. As the negative control, the cells were treated with 0.1% of Triton X-100 (PanReac AppliChem) for 30 min, and the positive control was treated only with DMEM. All conditions were used in triplicate. After incubation, cells were washed and incubated in 190 μL of a mixture of CTB solution and DMEM (1 : 9 by volume) for 4 h. Afterwards, 100 μL of each cell supernatant was transferred to a fresh well and the plate was analysed using an Infinite M Plex plate reader (Tecan, Männedorf, Switzerland). Each well was excited at 560 nm and fluorescence was detected at 590 nm over 25 flashes and 5 repetitions. The measured intensity is proportional to the number of living cells inside the corresponding well.Results and discussion
Synthesis and structural characterisation
A set of three MFts was prepared using a ferritin cage based on only HCF subunits fused N-terminally to mEGFP. For the magnetic core synthesis, either pure Fe2+ or Fe2+ doped with a final doping ratio of 7% (mol mol−1) Co2+ or doped with 7% (mol mol−1) Zn2+ with respect to the total metal amount was used (see Fig. 1, 2 and Table 1). The dopant molarity used during the synthesis differed by small percentage (see the Materials and methods section), but effectively gave rise to the 7% doping reported here (see also Fig. 3). The biomineralisation reaction was carried out inside the purified and PEGylated ferritin. The obtained MFts were first characterised by transmission electron microscopy (see Fig. 1 and 2) and dynamic light scattering to probe the protein and core size, respectively.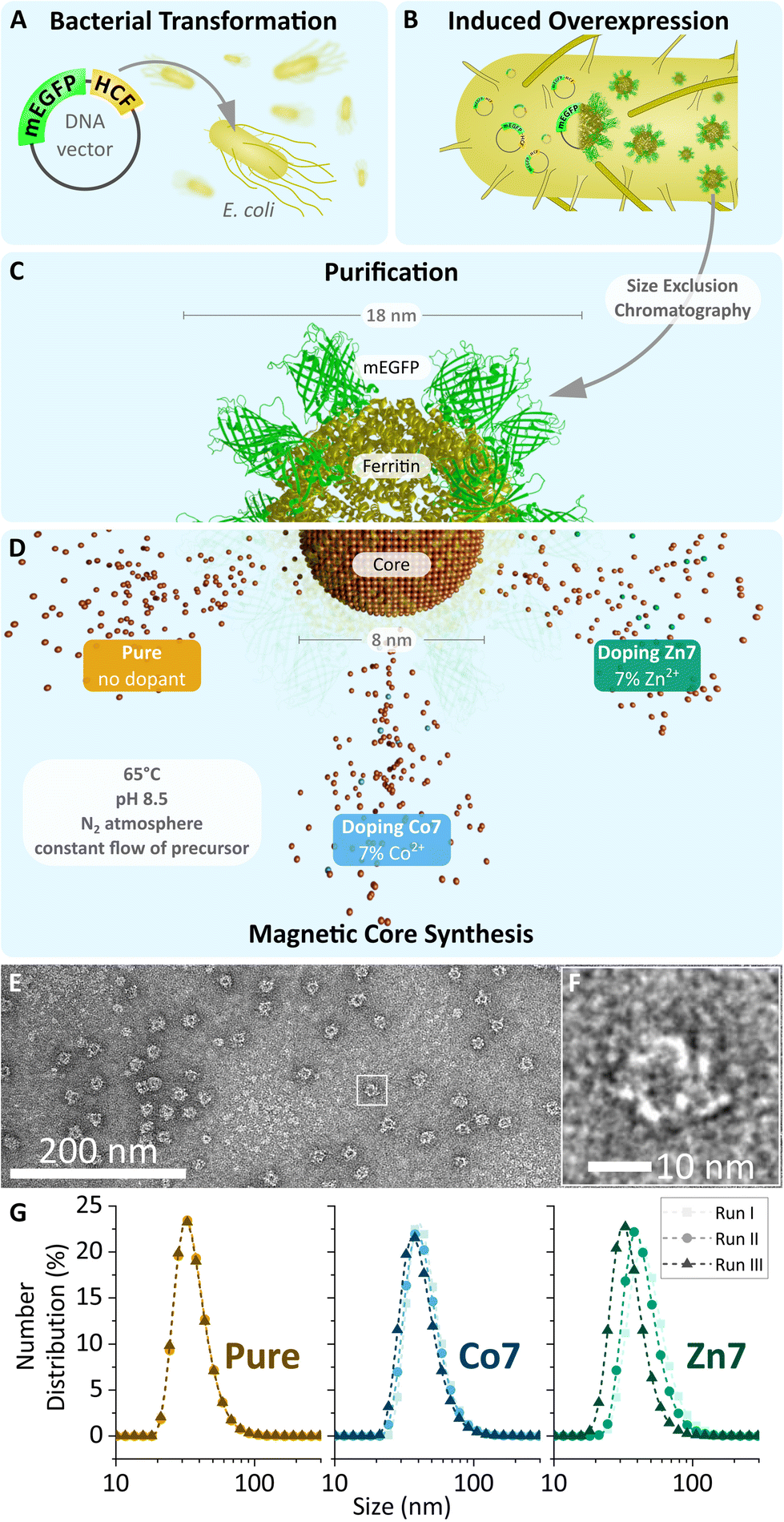 | ||
| Fig. 1 (A–D) Schematic overview of MFt production. Apoferritin (yellow, PDB code: 1FHA), genetically fused to mEGFP (green, PDB code: 2Y0G, created with Avogadro v. 1.2.090 and blender v.2.93.2, Blender Foundation, Amsterdam, Netherlands), is expressed in transformed E. coli and then purified by size exclusion chromatography. Empty cages are filled with their respective metal oxide core (brown) during magnetic core synthesis. (C and D) Schematic illustration of the semi-synthetic MFt cage. The three different cores used in this study are indicated. (E) Transmission electron microscopy image of pure MFt's protein shell stained with 2% uranyl acetate. (F) Magnified image of (E) displaying one single ferritin cage. (G) Hydrodynamic diameter obtained using dynamic light scattering for MFt with three differently doped cores over three independent synthesis runs. | ||
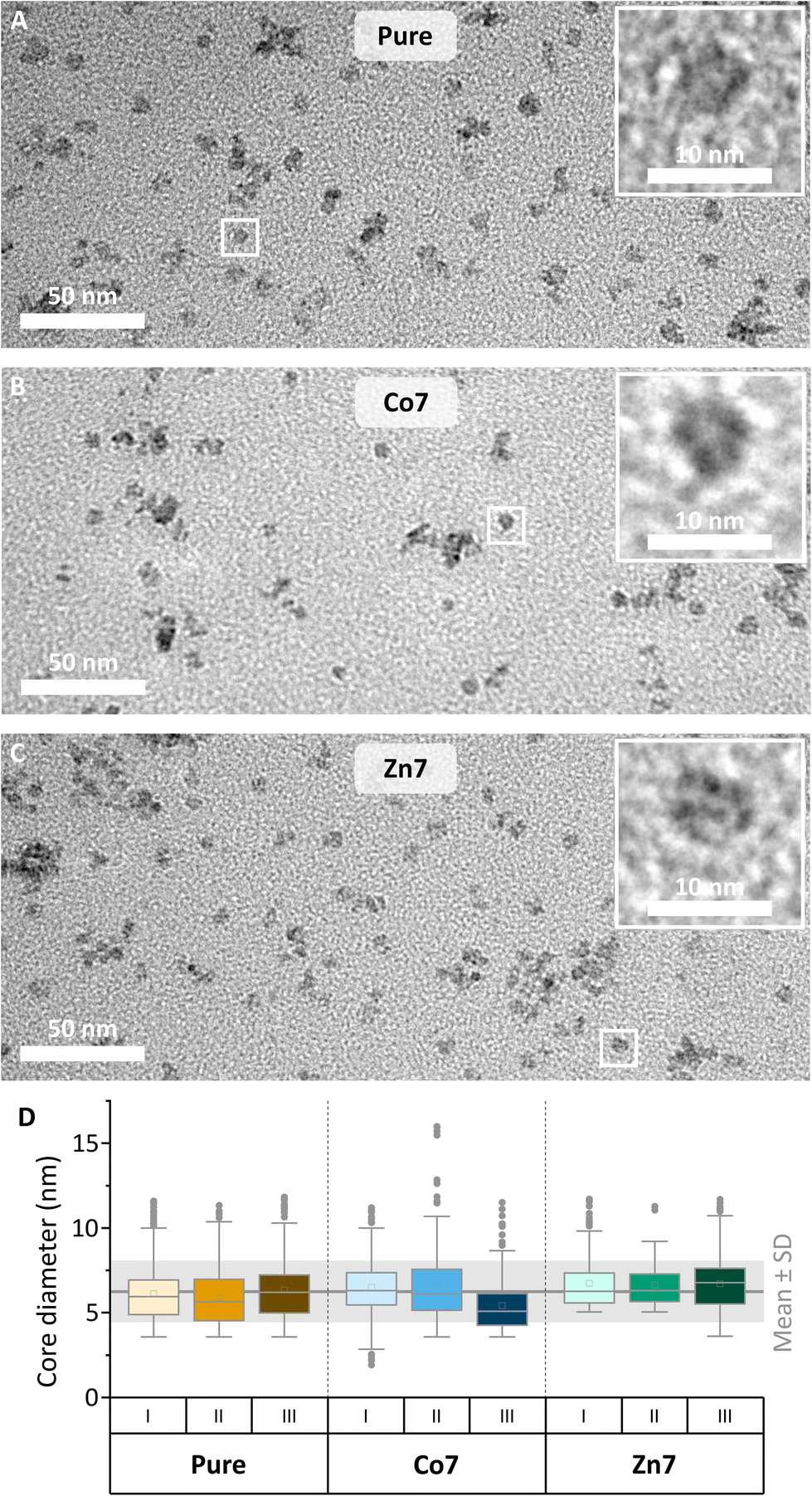 | ||
| Fig. 2 (A–C) Transmission electron microscopy images of magnetoferritin. Images of all doping conditions were taken without staining to visualise metallic cores. (D) Box plots showing the diameters of cores from three independent synthesis runs (I, II and III) of all three doping conditions (each N ≥ 93; mean ± SD = 6.3 ± 1.8 nm). Mean values are shown as boxes and median values are shown as horizontal lines. | ||
 | ||
| Fig. 3 Representative EDX-STEM images of MFt (pure) and doped (Co7 and Zn7) samples and elemental maps of indicated elements Fe, Co, and Zn (scale bar: 50 nm). The graph shows the dopant concentration relative to Fe of individual runs I–III. | ||
The mean diameter of all MFt cages measured with TEM was 13.9 ± 2.2 nm (see Fig. S2†), whereas the mean hydrodynamic diameters of MFts measured with DLS were 36.7 ± 1.9 nm for pure MFt, 44.7 ± 3.3 nm for Co2+-doped MFt, and 43.4 ± 7.1 nm for Zn2+-doped MFt (Fig. 1 and Table 2). The polydispersity index was ≤0.17 for all MFts, confirming the expected small size distribution within the sample (Table 2). The diameter values are in accordance with previous reports,31 where the TEM size of the protein structure matches the theoretically calculated mEGFP-decorated protein diameter (12 + 2 × 2.3 = 16.6 nm).69 The larger hydrodynamic size in comparison with the physical size was typically observed after surface PEGylation and core synthesis, giving rise to an increased hydrophilicity. This effective hydrodynamic diameter is particularly suitable for subcellular applications, since cytoplasmic non-specific interactions between a nanoprobe and the proteins/fibres in the cytoplasm dramatically increase above 50 nm, as previously reported.13,70 In addition, the measured negative ζ-potential of around −5 mV (Table 2) prevents non-specific interactions within the primarily negatively charged biological environment.
| Sample | D H (nm) | PdI | ζ-Potential (mV) |
|---|---|---|---|
| Pure | 36.7 ± 1.9 | 0.16 ± 0.01 | −5.1 ± 0.7 |
| Co7 | 44.7 ± 3.3 | 0.17 ± 0.02 | −4.7 ± 1.1 |
| Zn7 | 43.4 ± 7.1 | 0.17 ± 0.01 | −4.8 ± 0.8 |
The mean diameter of all cores was 6.3 ± 1.8 nm (Fig. 2), which is in accordance with previously reported values of synthetically loaded ferritin cages.17,31 There were no systematic differences in size after loading the cages with Fe or together with Co or Zn as dopants. The reproducibility of the magnetic core synthesis was evaluated by three separate syntheses (called runs in the following) and analysed separately, unless stated otherwise.
The doping levels of Co and Zn were determined by energy dispersive X-ray spectroscopy in scanning transmission electron microscopy (EDX-STEM). Fig. 3 shows high-resolution STEM images in the upper row, along with Fe, Co, and Zn elemental maps in the middle and lower rows. High-resolution STEM images appear blurry, due to the organics surrounding the magnetic cores. Under the focused electron beam, significant amounts of amorphous carbon grew, thus limiting the resolution and acquisition time for the elemental maps of Fe, Co, and Zn. Nonetheless, individual cores are visible in the pure MFt samples, while core accumulations yielded larger EDX signals. The elemental maps in Fig. 3 clearly show the colocalisation of Co or Zn with Fe. For the quantification of Co and Zn doping levels with respect to Fe, more than ten EDX spectra, each over small areas, were measured including about 20–30 magnetic cores. The results of each run are summarised in the graph in Fig. 3. The desired low doping levels of 6.8 ± 1.3 at% for Co and 6.4 ± 1.9 at% for Zn relative to the Fe intensity in the EDX spectra were obtained, with error bars indicating their standard deviation. We are the first to measure this in the case of MFts and find that the effective dopant molarities of 6.8 (Co) and 6.4 (Zn) are slightly different from the amount applied during the synthesis (5% for Zn and 11% for Co). This deviation shows that it is necessary to determine the exact doping state in the core after the synthesis. All three individual runs of Co-doped (from now on called Co7) or Zn-doped (Zn7) samples contained only cores with doping elements, excluding the presence of pure iron or cobalt and zinc oxide. At this point, we can state that Co and Zn doping of MFt cores is possible and reproducible with a fairly homogeneous distribution of dopants from core to core.
To further investigate the crystal structure of MFts, we used X-ray diffraction. Three individual samples for each dopant (runs I–III) were pooled to increase the signal intensity. Since the small size of MFts and the large amount of organic material gave rise to a background signal, Fig. 4 presents the X-ray diffractogram using Cu-Kα radiation after background removal and smoothing.
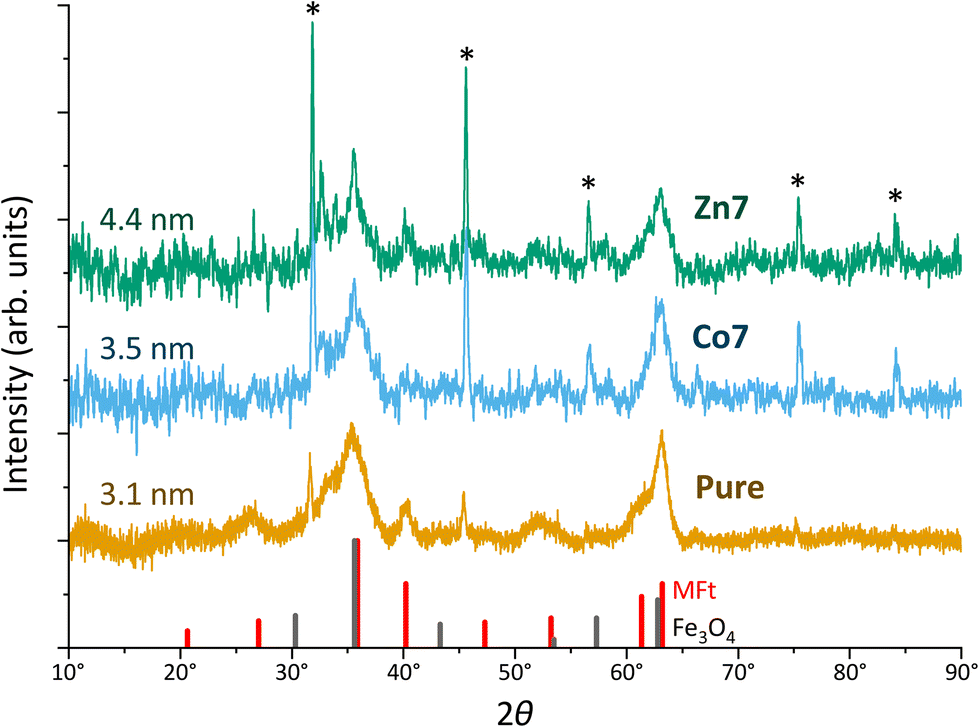 | ||
| Fig. 4 X-ray diffraction patterns of magnetoferritin (pure) and doped (Co7 and Zn7) samples using Cu-Kα radiation. Runs I–III were pooled. Sharp peaks indicated by asterisks originate from residual NaCl after drying. Crystallite sizes obtained from the Scherrer analysis are given on the left. Sticks in black and red indicate the angles and intensities for Fe3O4 and MFt powder samples, respectively. Diffractograms are shifted vertically for better visibility. | ||
Overall, the diffractograms of pure and doped MFt samples are similar and consist of two sets of sharp and broader peaks. The sharp peaks (indicated by asterisks) originate from NaCl crystallising during the drying process. However, more interesting are the broader peaks of the diffractogram of few-nm crystallites, i.e., the pure MFt and Co7- and Zn7-doped samples. The crystallite sizes of 3–4 nm given in Fig. 3 were evaluated from the two largest peaks of each diffractogram using Scherrer's approach.71 Considering the average core diameter of 6.3 nm and 3–4 nm crystallite sizes, it is estimated that each MFt consists of 4–9 crystallites regardless of doping. These numbers are realistic in light of the number of the HCF crystallisation points in a cage.72 The diffractograms have a close relationship to those of 6-line ferrihydrite73,74 while ferrites including the often-observed ferrimagnetic magnetite (Fe3O4) or maghemite (Fe2O3) can be excluded from the peak positions and relative intensities. For this comparison, powder-like samples75,76 were assumed and the patterns of standard bulk cobalt ferrite (PDF-00-0221086), magnetite (PDF-00-019-0629) and maghemite (PDF-00-039-1346) were used. While different structures of ferrihydrite have been suggested over the decades, it is clear from previous studies that defects and relative site occupancies slightly modify the XRD response.
Magnetic characterisation
The structural characterisation of MFts in terms of their size and core composition showed a high homogeneity and similar properties between the three different samples. Any change in terms of their suitability for spatial manipulation and inductive heating approaches should thus depend on their magnetic properties. In Fig. 5A, the zero field-cooling (ZFC) and field-cooling (FC) magnetisation curves at B = 5 mT are displayed. For direct comparison of the blocking temperature, all curves are normalised to the FC value at T = 5 K. For clarity, we show only one curve for the pure MFt and Co7- and Zn7-doped samples as indicated by the run number while the values given below are accompanied by the statistics of the three independent runs. All individual runs confirm the trends. It is obvious from Fig. 5A that both the ZFC/FC maximum and the irreversibility point shift with doping towards higher temperatures, reflecting an increased magnetic anisotropy energy density (MAE) at a constant volume given by the MFt nanocompartments (cf.Fig. 2D). Taking the ZFC maxima as a measure of the blocking temperature TB, we obtained 35 ± 5 K for pure MFt, 130 ± 37 K for Co7 and 74 ± 14 K for Zn7. The statistics hereby also reflect the individual doping levels achieved (cf. graph in Fig. 3). For pure MFt in our semisynthetic approach, TB = 35 K is about 3 times higher than that usually observed for horse spleen ferritin (TB = 10 K)6 likely signalling different crystallisation mechanisms. While Zn doping doubles TB as compared to pure MFt, it is striking that a low Co doping of 7% significantly increases the blocking temperatures to 130 K and thus the effective MAE Keff correspondingly; TB can be translated in Keff for single-domain nanoparticles using the following equation:where kB is the Boltzmann constant and V is the particle volume. The prefactor A = ln(τ/τ0) accounts for the measurement time (for VSM 1 s) with respect to the inverse of the intrinsic attempt frequency (typically 10−9 s). Using a prefactor A of 21 for VSM, Keff values are 0.8 × 105 J m−3 for pure MFt, 2.9 × 105 J m−3 for Co7 and 1.6 × 105 J m−3 for Zn7 assuming a constant volume of V = 131 nm3. Such 3.5 times larger TB is very attractive for inductive heating since the increased anisotropy may lead to blocking effects at frequencies of 100 kHz at constant particle volume and thus to higher heat losses via Néel relaxation processes. A rough estimate can be drawn when comparing the prefactors AVSM = 21 for VSM and Aind = 9 for inductive heating at 100 kHz. Then, we compare AVSM· TB = Aind· TB,ind and expect a TB,ind = 21/9· 130 K ≈ 303 K for Co7 in inductive heating experiments. Thus, hysteresis losses can be expected at 100 kHz, leading to better heating in magnetic hyperthermia. Note that this estimate assumes a frequency-independent magnetic susceptibility which is generally not the case.
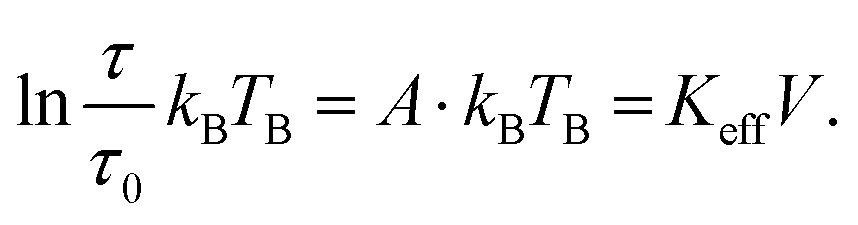
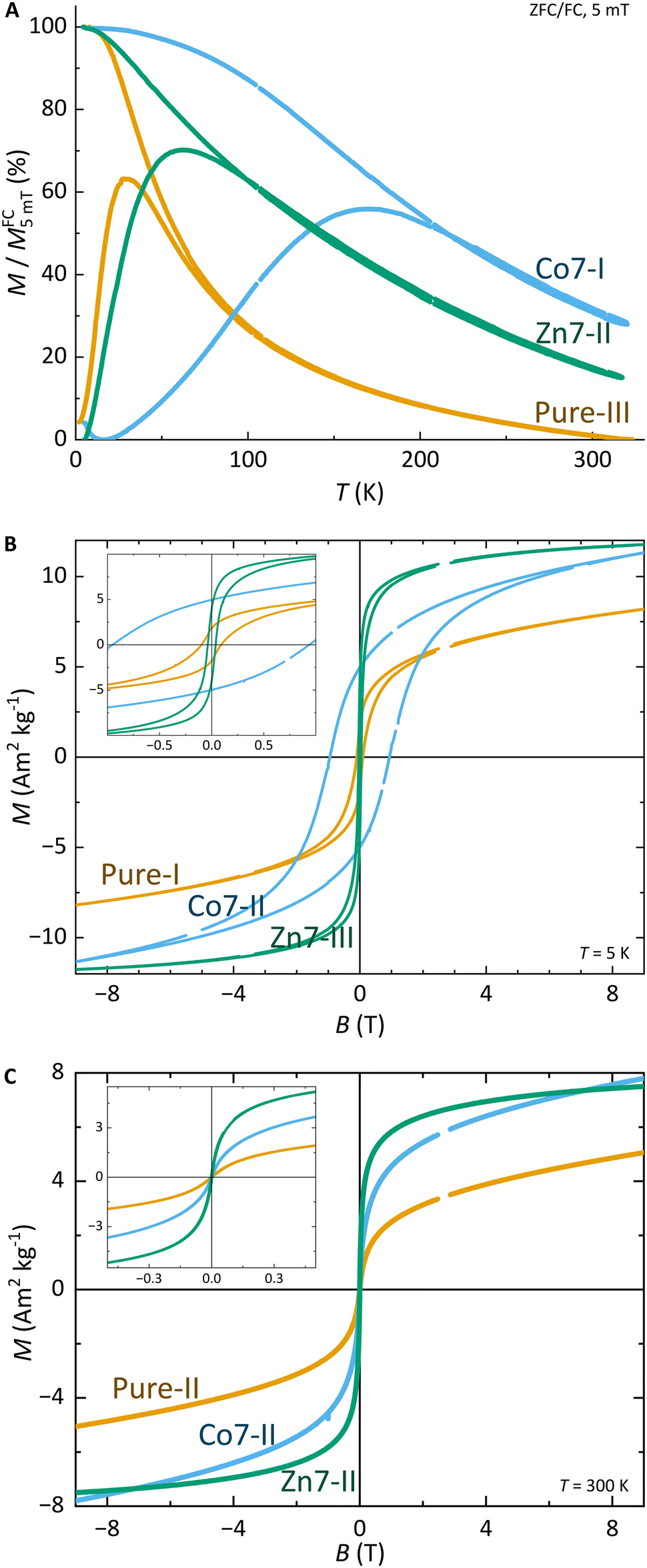 | ||
| Fig. 5 Magnetometry results of pure and Co7 and Zn7 doped samples. The run number behind the doping conditions reflects the individual sample. Statistics are given in the text. (A) ZFC/FC curves in B = 5 mT. (B) Hysteresis loops at T = 5 K and (C) superparamagnetic response at T = 300 K. Insets show magnification at the origin in identical units. Voids in the curves are a technical feature of VSM resetting the sample position by a touchdown to the end position switch giving no data while the field is constantly swept. | ||
Furthermore, we discuss the important features: (1) magnetisation and (2) coercive field dependence on doping. Fig. 5 presents hysteresis loops at (B) T = 5 K and (C) T = 300 K. All curves are normalised to the total mass of the dried sample powder including the organic shell and residual salts. For the whole particle, we obtained magnetisations of 7–13 Am2 kg−1 in B = 9 T and, as expected, the values decrease with increasing temperature. However, we cannot define the saturation magnetisation with regard to the magnetic cores only since the magnetic content is not exactly known. Instead, we used Langevin fitting as discussed below. None of the hysteresis loops reaches saturation, which points to additional paramagnetic Fe salts or antiferromagnetic contributions in the MFt MNPs. Taking the saturation magnetisation of 6 nm Fe3O4 MNPs of 45 (30) Am2 kg−1 at T = 5 K (300 K)57 as an upper limit, we obtained a ferrimagnetic fraction of at least 20–50 mass% in the samples. This is also in line with XRD measurements showing sharp peaks (large crystallites) of NaCl after drying (cf.Fig. 4), giving a significant contribution to the total mass.
The coercive fields μ0HC at T = 5 K are 99 ± 29 mT for pure MFt, 977 ± 64 mT for Co7 and 35 ± 4 mT for Zn7. These results follow earlier studies showing magnetic softening and higher magnetisation for Zn-doped ferrites and strong hardening at slightly reduced magnetisation for Co-doped ferrites.77,78 At 300 K, all MNPs show a superparamagnetic response as expected. MFts can thus be treated as single domain and weakly interacting particles following the Néel–Brown model.79
Finally, the magnetisation of the MNPs can be extracted from the Langevin fitting of superparamagnetic magnetisation at T = 300 K. Fig. S3† presents the results for pure MFt and Co7 samples. Here, we used the Langevin function and an additional linear slope accounting for add-on contributions of salts and organics and paramagnetic or antiferromagnetic contributions in the samples. For simplicity, we fit the data with a single parameter in the Langevin function omitting the distribution of the product of magnetisation and volume. The resulting volume magnetisation values are 2824 ± 704 μB per particle (200 ± 50 kA m−1) for pure MFt, 2973 ± 931 μB per particle (211 ± 66 kA m−1) for Co7 and 5620 ± 3611 μB per particle (398 ± 255 kA m−1) for Zn7. Pure MFt and Co7 yield similar values which are about 40% of bulk Fe3O4 magnetisation at ambient temperature. For Zn7, the larger value and larger variation of the fitting results of the three independent runs are ascribed to the larger number of agglomerates in Zn7 (cf.Fig. 2).
For further use of doped MFt in biomedical applications, magnetometry suggests that all the generated MNPs exhibit magnetisation values suitable for spatial manipulation in external fields. For applications in magnetic hyperthermia, however, especially the Co-doped MFts appear suitable due to their enhanced MAE, leading to magnetic blocking close to ambient temperature. This behaviour is consistent with the substitution of Co ions for Fe ions, since cobalt is well known to increase the magneto-crystalline anisotropy and correspondingly the blocking temperature.9,59,65
Magnetic hyperthermia
The heating capabilities of the MNPs were evaluated by recording their temperature kinetic curves and hyperthermic efficiency. To this end, the MFts in HEPES-1 buffer solution were exposed to alternating magnetic fields (AMFs) of 93.75 kHz and 104.5 kHz and B = 45 mT amplitude using a custom-made hyperthermia setup. Due to extensive cooling and mitigation of inductive losses in the electromagnet, this setup allows for accurate measurements of the MNPs’ dissipated power. The total particle concentration in the sample was 35 μM, corresponding to 55 mg mL−1 and 16% (w/w) of total metal ions. Fig. 6 and Fig. S4† show the average heating curves of three repeats for pure, Co7, or Zn7 MFts. From each measurement, the temperature kinetics of buffer measured under identical conditions were subtracted, yielding ΔT. Care was taken to measure each sample up to the steady-state conditions during the heating and cooling stages (10–15 min after switching the AMF on or off). At 93.75 kHz, the pure MFt sample exhibits a temperature increase above the buffer reference up to ΔT = 3.6 K during exposure to the AMF. The Zn7 sample exhibits a lower temperature increase of ΔT = 2.5 K during inductive heating. This lower response, however, is not significantly different from that of the pure MFt sample given the ±2 K absolute temperature accuracy of the IR-camera. Single heating curves were highly robust, since repetitions were near identical and hence did not contribute to the uncertainty (see Fig. 6A, where the coloured envelope indicates the error arising from repeated measurements). Intriguingly, Co7 samples led to an inductive heating of ΔT = 8 K after 300 s in the AMF. Such a significant temperature increase provides evidence that doped MFts can act as heat mediators. At 104.5 kHz AMF frequency, all three MFt samples exhibited slightly lower heating rates compared to those at 93.75 kHz (see Fig. S4†). While the pure MFt sample again heated up to ΔTmax = 3 K, the Zn7 sample showed ΔTmax = 1.2 K and the Co7 sample showed ΔTmax = 6.1 K. Previous tests using horse spleen ferritin as a reference led to no measurable heating,28 corroborating our finding that the synthesis of a particular magnetic core is critical to maximise the heating response.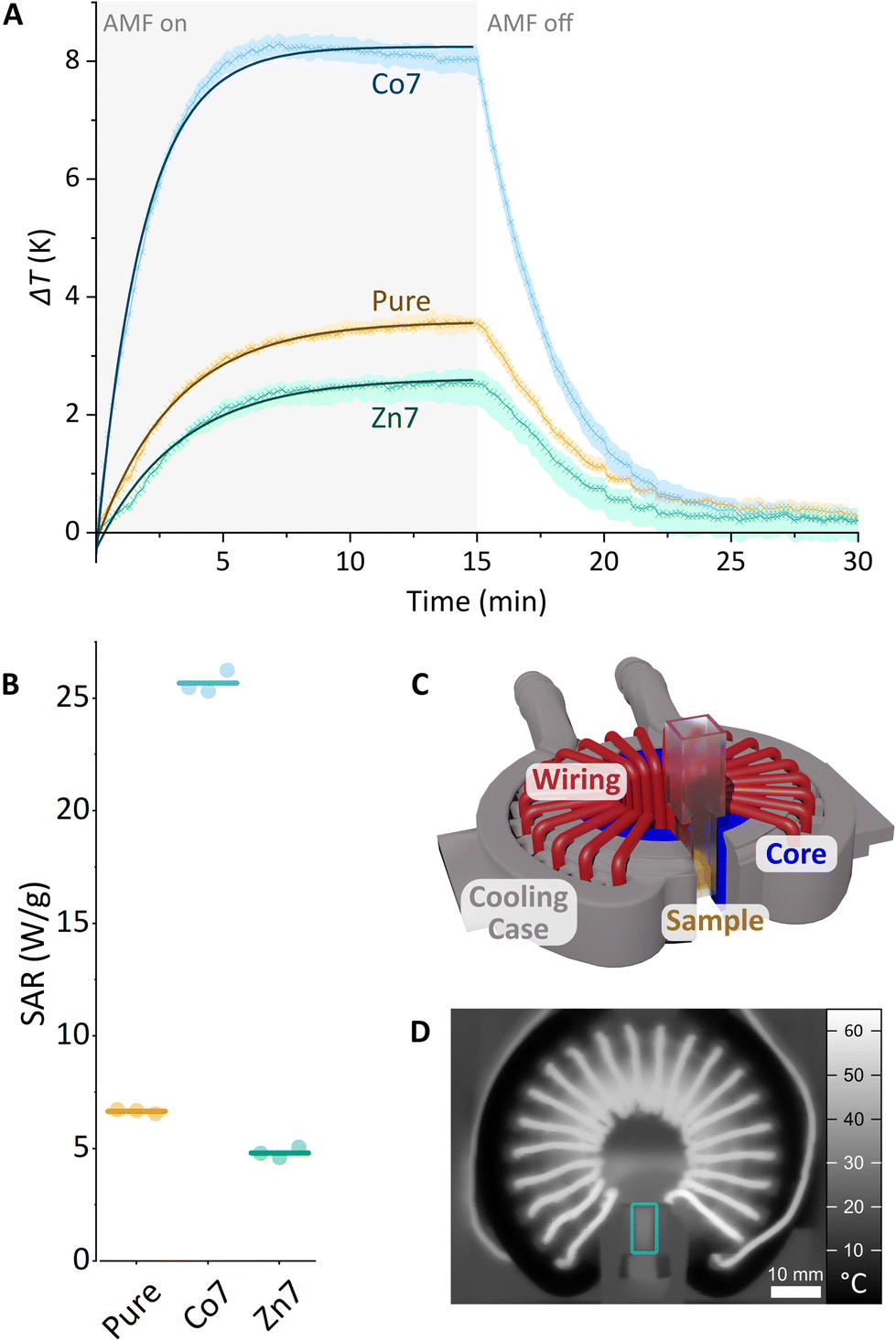 | ||
| Fig. 6 (A) Hyperthermia measurement. Temperature kinetics of pure, Co7, and Zn7 MFt samples (each 35 μM) during exposure to an alternating magnetic field (45 mT, 93.75 kHz). Each curve is an average of three runs. The absolute temperature change ΔT refers to the temperature increase above the temperature of pure buffer solution. All MFt and buffer solution samples were measured in an identical manner. (B) Specific absorption rate (W g−1) of each run (coloured dot) and average value (coloured line). (C) Custom-made hyperthermia setup. (D) Selected thermal image that was taken during the hyperthermia measurement with the sample (blue box) in the centre of the magnet gap. | ||
The trend observed in the calorimetric experiments can be explained with MFt's magnetic properties. In the linear response regime, the power losses are described by the specific absorption rate (SAR). It is proportional to the frequency, f, to the square of the external magnetic field amplitude, H0, and to the out-of-phase component of the magnetic susceptibility, χ′′(f):80
| SAR ∝ fμ0H02χ′′(f). |
Since all measurements were performed using identical settings, the slightly lower inductive heating in the case of Co7 and Zn7 at 104.5 kHz could arise from a lower AC susceptibility χ′′, which is directly proportional to the dissipated power.
Our data reveal an overall low inductive heating of MFts, which manifests itself in the SAR values per mass of total metal ions (see Fig. 6B). The SAR was evaluated from the initial slope of the fit to the heating curves amounting to 6.7 ± 0.1 W g−1 for pure MFt, 4.8 ± 0.2 W g−1 for pure Zn7, and 25.7 ± 0.5 W g−1 for Co7. The low inductive heating response and small SAR values are indeed expected for the MNPs of this diameter.54,81 SAR values are also low in comparison with the data reported for larger MNPs in the literature82–85 and our measurements using the same setup,66 typically amounting to ∼100 W g−1. However, the heating rate of the Co7 sample is the maximal value typically achieved for these types of magnetic cores measured under similar conditions60 and is promising to promote or even initiate heat sensitive cellular processes in the AMF. Moreover, the changes observed for different core doping and different frequency settings highlight that the inductive heating response is dependent on the experimental design for which here optimised AMF excitation settings and core compositions are found. Co7 MFts could hence only act as mild heat mediators when the nanoparticles are accumulated to high local concentrations (e.g., 35 μM), resulting in the surrounding buffer solution heated by 2–3 degrees per minute.
Finally, it should be noted that all measurements were performed using external field parameters which fall below the physiological tolerance threshold.86 Such settings are important when biological measurements are envisaged, where physiologically serviceable power losses are required.
MFt spatial manipulation
Since cell signalling processes were shown to rely on (i) the spatiotemporal regulation of molecular distribution14,15 or (ii) the application of mechanical forces,16,18 we here provide examples of how synthesised MFts are useful in this biological context. In particular, we show how MFts can be redistributed and accumulated in different intracellular compartments. On the one hand, this may be exploited for: (i) the coupling of MFts to signalling proteins and their active spatial redistribution to understand cell signalling processes and on the other hand, (ii) localised MFt accumulations may exert gentle forces to trigger mechanosensitive processes.To explore the suitability of MFts to respond to an applied magnetic field gradient, we first redistributed the MFts under idealised conditions, i.e., using droplets of MFts in a glycerol/water (25:75 V/V) mixture on PDMS-coated glass coverslips. A home-made magnetic tip consisting of spring steel wire connected to neodymium magnets was moved close to the droplet (see Fig. 7 and Video S1†). The magnetic tip generated magnetic gradients on the order of 103–104 T m−1. To determine such magnetic flux density gradients over small scales (∼μm), several assays have been developed. These include (i) The MNP attraction in solution using the Stokes equation17,31 or (ii) the optical magnetometry based on nitrogen vacancy colour centres in diamond to map the 3D magnetic field of micromagnets.87 Together with the magnetic moment reported in the previous section, MFts should hence encounter forces in the femto-Newton regime. This is too weak to produce clear ballistic MNP trajectories toward the magnetic tip, and instead the MNPs show a biased diffusion resulting from the superposition of Brownian motion with a magnetic drift. When the magnetic tip was positioned at a particular distance d from the surface of the droplet, MFts were attracted towards the tip and formed a sharp gradient from the edge towards the centre of the droplet up to the distance where the attractive magnetic potential was exceeded by the thermal Brownian motion (see Fig. 7B). At the steady state, the MNP distribution followed a Boltzmann distribution and was fitted with an approximate relationship to determine the applied force F:
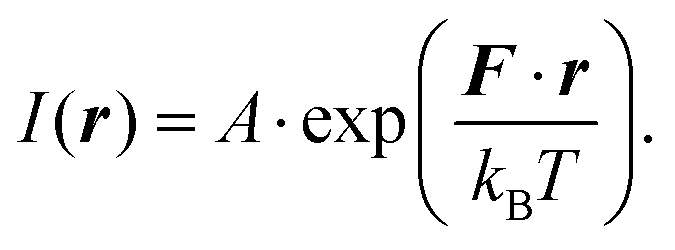
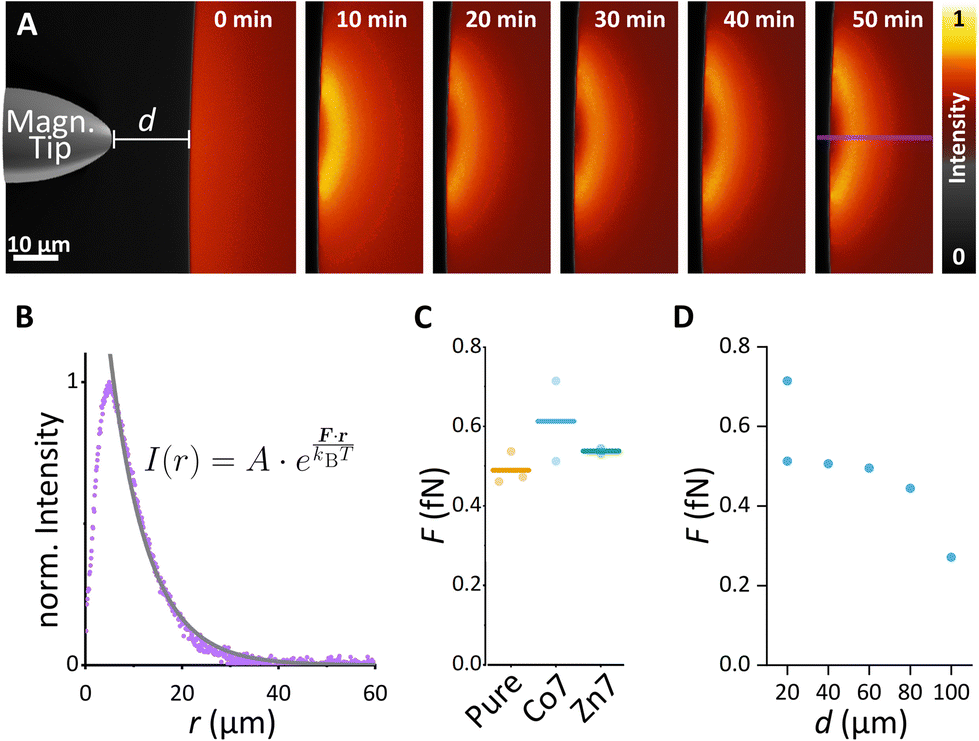 | ||
| Fig. 7 Droplet assay with differently doped magnetoferritin. (A) Exemplary assay with pure MFt. The magnetic tip was introduced at 0 min at a distance of 20 μm and left at the designated position for 50 min. (B) In the steady state, a line scan over 1 μm (purple line in A) was used to determine the force F affecting MFt using the displayed fit function. (C) Forces determined using droplet assay for all doping conditions. (D) Force–distance dependence for different distances d between the droplet and the magnetic tip using the Co7 MFt sample. | ||
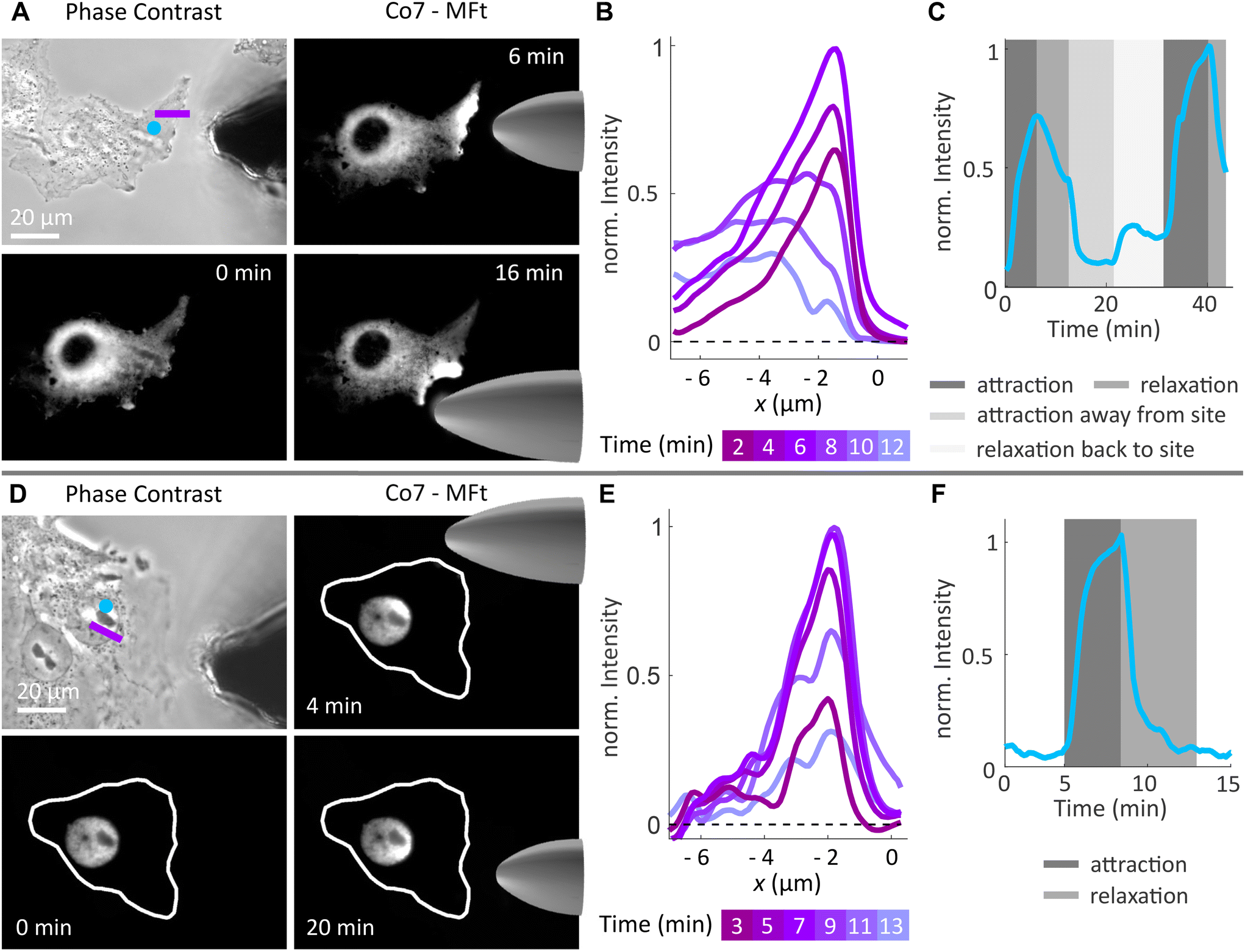 | ||
| Fig. 8 Spatial manipulation of magnetoferritin in cells. (A) Phase contrast image of Cos7 cells and the micromagnetic tip and fluorescence images of Co7-MFt's spatial redistribution in the cell cytoplasm placing the magnetic tip at different positions. (B) Fluorescence intensity profile along the pink line in (A) during attraction (2–6 min) and relaxation (8–12 min). (C) Average fluorescence intensity within the blue spot in (A) over time. The intensity increases and decreases according to the spatial manipulation mode. (D) Phase contrast image of Cos7 cells and the micromagnetic tip and fluorescence images of Co7-MFt's spatial redistribution in the cell nucleus placing the magnetic tip at different positions. (E) Fluorescence intensity profile along the pink line in (D) during attraction (3–10 min) and relaxation (11–13 min). (F) Average fluorescence intensity within the blue spot in (D) over time. The intensity profile exhibits the characteristic exponential increase and decrease during attraction and relaxation, respectively. In (B) and (E), the background intensity profile was subtracted from the data and spatial averaging was performed over a line with a width of 5 pixels. | ||
When MFts were manipulated in the cell nucleus (see Fig. 8D–F), particles were reversibly attracted inside the nucleus without observing any intensity change after the manipulation. Interestingly, the attraction was fully reversible and MFts remained inside the nucleus without any detectable escape to the cell cytoplasm.
We further tested the biocompatibility of MFts and found that cells that were incubated with MFt Co7 for 1 h did not show any increased mortality in comparison with buffer controls (see Fig. S5A†). Additionally, the cell death, which occurred only in microinjected cells, was similar between only the microinjected cells and cells after magnetic accumulation of MFts over the course of 6 h (see Fig. S5B†). Additional cell death, which was observed on longer time scales (>6 h), could arise from microinjection late effects, excessive magnetic MFt accumulation, and a change in sterile or imaging conditions (since the sample was not covered during imaging for microinjection and magnetic manipulation purposes), among others. While these long-time effects require further investigation, the Co2+-doped MFt, when brought into contact with the cells alone, suggested a high biocompatibility, in line with previous studies.9,31
Thus, provided the MFt transfer to the cellular environment is further improved, the spatial manipulation of MFts can serve as a method to actively redistribute MFt-coupled molecules for cell signalling studies. It may also be used to apply gentle mechanical stimuli to intracellular structures depending on the magnetic field gradient and the magnetic flux density applied.
Conclusion and outlook
We have demonstrated that ferritin-based magnetic nanoparticles (MFts) offer a versatile platform for spatial magnetic manipulation approaches, whereas an inductive heating of the nanoparticles is possible only for optimal alternating magnetic field settings and mineralised cores of high magnetic anisotropy (e.g., Co-doped cores). In particular, we synthesised a bioengineered ferritin-based magnetic nanoparticle (MNP) with mEGFP and PEG coupled to its surface. This enabled tracking of the particle and preventing any unspecific interaction of the particle within a synthetic or cellular environment. Three types of MFts were synthesised with an undoped, 7% Zn- or 7% Co-doped superparamagnetic core with high size homogeneity of the shell and core. Thus, MFts are addressable one particle at a time and allows the transmission of the same magnetic stimulus, an important prerequisite for many bio- and nanotechnological applications.7% cobalt doping yielded the highest effective magnetic anisotropy and significantly improved the hyperthermic heat performance. Here, inductive heating experiments at 93.75 kHz resulted in a temperature increase of 8 K above a buffer reference within a few minutes. This provided evidence that Co-doped MFts can act as mild heat mediators when optimised magnetic field settings and nanoparticle core properties are used. 7% zinc doping and the pure mineral core resulted in superparamagnetism down to ≤60 K and measurements at different frequencies only brought about a slight heating effect. Our rigorous measurements provide new insights into the recent discussion on whether (or under which conditions) MFts can act as heat mediators.
Next to probing the MFt applicability for magnetic hyperthermia, their suitability for spatial manipulation approaches in a biological context was tested. The performed experiments nicely showed the applicability of our doped MFts in solution and in a cellular context.
Here, all MFts performed very well and were comparable in terms of their spatial redistribution potential. The MFts were reversibly attracted in droplets or inside cells approaching a magnetic tip (i.e., a magnetic field gradient) and attraction forces were easily modulated by changing the MFt–magnetic tip distance. In agreement with theoretical calculations, the analysis of experimentally obtained MNP distributions revealed that nanoparticles exert mild forces in the fN regime. The MFts could hence be used to couple molecules to the MFt surface (i) to spatially redistribute the molecules to create local signalling hubs, (ii) to switch their (conformational) activity state, or (iii) to exploit the biological mechano-sensitivity in a mild force transduction approach. In conclusion, the Co-doped MFt is an extremely versatile and small functionalised targeting nanosystem, which offers the best magnetic properties for a spatial- or force-mediated manipulation of individual molecules along with some potential for nanotechnological hyperthermia applications.
Author contributions
Conceptualisation: AN, CM, and UW; methodology: AN, CM, DAK, IPN, NT, SS, PH, and UW; investigation: AN, CM, DAK, and UW; visualisation: AN, CM, DAK, and UW; supervision: CM, UW, and MF; writing – original draft: AN, CM, DAK, and UW.Data availability
The data supporting the manuscript “Semisynthetic ferritin-based nanoparticles with high magnetic anisotropy for spatial magnetic manipulation and inductive heating” (NR-ART-04-2024-001652) have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
CM acknowledges financial support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) through CRC1208 ‘Identity and Dynamics of Membrane Systems’ (A12, Project ID 267205415) and CRC1535 ‘Microbial Networks’ (A09, Project ID 458090666). CM and AN acknowledge financial support via the ‘Freigeist fellowship’ of Volkswagen Foundation. The authors acknowledge the DFG and the State of North Rhine–Westphalia for funding the cryo-TEM (INST 208/749-1 FUGG) hosted by the Centre of Advanced Imaging (CAi, Heinrich-Heine University). The authors thank the group of Prof. Claus Seidel (Physical Chemistry, Heinrich-Heine University) for providing lab space and laboratory equipment, the group of Prof. Lutz Schmitt (Biochemistry, Heinrich-Heine University) for providing the cell disruptor, and the group of Prof. Laura Hartmann (Macromolecular Chemistry, Heinrich-Heine University) for providing the Zetasizer.References
- Z. Yuan, B. Wang, Y. Teng, W. Ho, B. Hu, K. O. Boakye-Yiadom, X. Xu and X.-Q. Zhang, Nanoscale, 2022, 14, 6449–6464 RSC.
- I. Inoue, M. Chiba, K. Ito, Y. Okamatsu, Y. Suga, Y. Kitahara, Y. Nakahara, Y. Endo, K. Takahashi, U. Tagami and N. Okamoto, Nanoscale, 2021, 13, 1875–1883 RSC.
- X. Lin, J. Xie, G. Niu, F. Zhang, H. Gao, M. Yang, Q. Quan, M. A. Aronova, G. Zhang, S. Lee, R. Leapman and X. Chen, Nano Lett., 2011, 11, 814–819 CrossRef CAS PubMed.
- K. Fan, C. Cao, Y. Pan, D. Lu, D. Yang, J. Feng, L. Song, M. Liang and X. Yan, Nat. Nanotechnol., 2012, 7, 459–464 CrossRef CAS PubMed.
- K. Li, Z. P. Zhang, M. Luo, X. Yu, Y. Han, H. P. Wei, Z. Q. Cui and X. E. Zhang, Nanoscale, 2012, 4, 188–193 RSC.
- C. Massner, F. Sigmund, S. Pettinger, M. Seeger, C. Hartmann, N. P. Ivleva, R. Niessner, H. Fuchs, M. H. de Angelis, A. Stelzl, N. L. Koonakampully, H. Rolbieski, U. Wiedwald, M. Spasova, W. Wurst, V. Ntziachristos, M. Winklhofer and G. G. Westmeyer, Adv. Funct. Mater., 2018, 28, 1706793 CrossRef.
- H. J. Kang, Y. J. Kang, Y. M. Lee, H. H. Shin, S. J. Chung and S. Kang, Biomaterials, 2012, 33, 5423–5430 CrossRef CAS PubMed.
- Z. Wang, Y. Dai, Z. Wang, O. Jacobson, F. Zhang, B. C. Yung, P. Zhang, H. Gao, G. Niu, G. Liu and X. Chen, Nanoscale, 2018, 10, 1135–1144 RSC.
- E. Fantechi, C. Innocenti, M. Zanardelli, M. Fittipaldi, E. Falvo, M. Carbo, V. Shullani, L. Di Cesare Mannelli, C. Ghelardini, A. M. Ferretti, A. Ponti, C. Sangregorio and P. Ceci, ACS Nano, 2014, 8, 4705–4719 CrossRef CAS PubMed.
- R. Chen, G. Romero, M. G. Christiansen, A. Mohr and P. Anikeeva, Science, 2015, 347, 1477–1480 CrossRef CAS PubMed.
- K. Ohara, T. Moriwaki, K. Nakazawa, T. Sakamoto, K. Nii, M. Abe and Y. Ichiyanagi, AIP Adv., 2023, 13, 025238 CrossRef CAS.
- F. Etoc, D. Lisse, Y. Bellaiche, J. Piehler, M. Coppey and M. Dahan, Nat. Nanotechnol., 2013, 8, 193–198 CrossRef CAS PubMed.
- F. Etoc, C. Vicario, D. Lisse, J. M. Siaugue, J. Piehler, M. Coppey and M. Dahan, Nano Lett., 2015, 15, 3487–3494 CrossRef CAS PubMed.
- M. Aubry, W. A. Wang, Y. Guyodo, E. Delacou, J. M. Guigner, O. Espeli, A. Lebreton, F. Guyot and Z. Gueroui, ACS Synth. Biol., 2020, 9, 3030–3041 CrossRef CAS PubMed.
- Y. Shi, L. Huang, H. Dong, M. Yang, W. Ding, X. Zhou, T. Lu, Z. Liu, X. Zhou, M. Wang, B. Zeng, Y. Sun, S. Zhong, B. Wang, W. Wang, C. Yin, X. Wang and Q. Wu, Cell Res., 2024, 34, 193–213 CrossRef CAS PubMed.
- D. Seo, K. M. Southard, J. W. Kim, H. J. Lee, J. Farlow, J. U. Lee, D. B. Litt, T. Haas, A. P. Alivisatos, J. Cheon, Z. J. Gartner and Y. W. Jun, Cell, 2016, 165, 1507–1518 CrossRef CAS PubMed.
- D. Liße, C. Monzel, C. Vicario, J. Manzi, I. Maurin, M. Coppey, J. Piehler and M. Dahan, Adv. Mater., 2017, 29, 1–7 CrossRef PubMed.
- C. Monzel, C. Vicario, J. Piehler, M. Coppey and M. Dahan, Chem. Sci., 2017, 8, 7330–7338 RSC.
- A. S. Garanina, M. V. Efremova, A. E. Machulkin, E. V. Lyubin, N. S. Vorobyeva, O. A. Zhironkina, O. S. Strelkova, I. I. Kireev, I. B. Alieva, R. E. Uzbekov, V. N. Agafonov, I. V. Shchetinin, A. A. Fedyanin, A. S. Erofeev, P. V. Gorelkin, Y. E. Korchev, A. G. Savchenko and M. A. Abakumov, Magnetochemistry, 2022, 8, 185 CrossRef CAS.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS PubMed.
- Y. Zhu, Y. Zhu, T. Cao, X. Liu, X. Liu, Y. Yan, Y. Shi and J. C. Wang, Med. Rev., 2023, 3, 49–74 CrossRef PubMed.
- D. He and J. Marles-Wright, Nat. Biotechnol., 2015, 32, 651–657 CAS.
- M. Kumar, J. Markiewicz-Mizera, J. D. Janna Olmos, P. Wilk, P. Grudnik, A. P. Biela, M. Jemioła-Rzemińska, A. Górecki, S. Chakraborti and J. G. Heddle, Nanoscale, 2021, 13, 11932–11942 RSC.
- Y. Jin, J. He, K. Fan and X. Yan, Nanoscale, 2019, 11, 12449–12459 RSC.
- A. Mohanty, A. Parida, R. K. Raut and R. K. Behera, ACS Bio Med Chem Au, 2022, 2, 258–281 CrossRef CAS PubMed.
- A. N. Gabashvili, S. S. Vodopyanov, N. S. Chmelyuk, V. A. Sarkisova, K. A. Fedotov, M. V. Efremova and M. A. Abakumov, Int. J. Mol. Sci., 2021, 22, 12275 CrossRef CAS PubMed.
- M. Meister, eLife, 2016, 5, 1–14 CrossRef PubMed.
- H. C. Davis, S. Kang, J. H. Lee, T. H. Shin, H. Putterman, J. Cheon and M. G. Shapiro, Biophys. J., 2020, 118, 1502–1510 CrossRef CAS PubMed.
- Y. Zhang and B. P. Orner, Int. J. Mol. Sci., 2011, 12, 5406–5421 CrossRef CAS PubMed.
- P. M. Harrison and P. Arosio, Biochim. Biophys. Acta, Bioenerg., 1996, 1275, 161–203 CrossRef PubMed.
- I. P. Novoselova, A. Neusch, J. S. Brand, M. Otten, M. R. Safari, N. Bartels, M. Karg, M. Farle, U. Wiedwald and C. Monzel, Nanomaterials, 2021, 11, 1–20 CrossRef PubMed.
- E. Falvo, E. Tremante, R. Fraioli, C. Leonetti, C. Zamparelli, A. Boffi, V. Morea, P. Ceci and P. Giacomini, Nanoscale, 2013, 5, 12278–12285 RSC.
- D. Liße, C. P. Richter, C. Drees, O. Birkholz, C. You, E. Rampazzo and J. Piehler, Nano Lett., 2014, 14, 2189–2195 CrossRef PubMed.
- X. Li, L. Qiu, P. Zhu, X. Tao, T. Imanaka, J. Zhao, Y. Huang, Y. Tu and X. Cao, Small, 2012, 8, 2505–2514 CrossRef CAS PubMed.
- F. Raudzus, H. Schöneborn, S. Neumann, E. Secret, A. Michel, J. Fresnais, O. Brylski, C. Ménager, J. M. Siaugue and R. Heumann, Sci. Rep., 2020, 10, 22452 CrossRef CAS PubMed.
- M. Le Jeune, E. Secret, M. Trichet, A. Michel, D. Ravault, F. Illien, J.-M. Siaugue, S. Sagan, F. Burlina and C. Ménager, ACS Appl. Mater. Interfaces, 2022, 14, 15021–15034 CrossRef CAS PubMed.
- N. K. Lee, S. Cho and I. S. Kim, Exp. Mol. Med., 2022, 54, 1652–1657 CrossRef CAS PubMed.
- N. Aronovitz, M. Neeman and R. Zarivach, in Iron Oxides, ed. D. Faivre, John Wiley & Sons, Ltd, Hoboken, 2016, pp. 117–142 Search PubMed.
- E. C. Theil, Adv. Enzymol. Relat. Areas Mol. Biol., 1990, 63, 421–449 CAS.
- A. García-Prieto, J. Alonso, D. Muñoz, L. Marcano, A. Abad Díaz de Cerio, R. Fernández de Luis, I. Orue, O. Mathon, A. Muela and M. L. Fdez-Gubieda, Nanoscale, 2016, 8, 1088–1099 RSC.
- M. A. Kostiainen, P. Ceci, M. Fornara, P. Hiekkataipale, O. Kasyutich, R. J. M. Nolte, J. J. L. M. Cornelissen, R. D. Desautels and J. Van Lierop, ACS Nano, 2011, 5, 6394–6402 CrossRef CAS PubMed.
- M. A. Kostiainen, P. Hiekkataipale, A. Laiho, V. Lemieux, J. Seitsonen, J. Ruokolainen and P. Ceci, Nat. Nanotechnol., 2013, 8, 52–56 CrossRef CAS PubMed.
- S. Gider, D. D. Awschalom, T. Douglas, S. Mann and M. Chaparala, Science, 1995, 268, 77–80 CrossRef CAS PubMed.
- Q. A. Pankhurst, S. Betteridge, D. P. E. Dickson, T. Douglas, S. Mann and R. B. Frankel, Hyperfine Interact., 1994, 91, 847–851 CrossRef CAS.
- M. Allen, D. Willits, M. Young and T. Douglas, Inorg. Chem., 2003, 42, 6300–6305 CrossRef CAS PubMed.
- D. A. Resnick, K. Gilmore, Y. U. Idzerda, M. T. Klem, M. Allen, T. Douglas, E. Arenholz and M. Young, J. Appl. Phys., 2006, 99, 2006–2008 CrossRef.
- P. Mackle, J. M. Charnock, C. D. Garner, F. C. Meldrum and S. Mann, J. Am. Chem. Soc., 1993, 115, 8471–8472 CrossRef CAS.
- B. Warne, O. I. Kasyutich, E. L. Mayes, J. A. L. Wiggins and K. K. W. Wong, IEEE Trans. Magn., 2000, 36, 3009–3011 CrossRef CAS.
- T. Ueno, M. Suzuki, T. Goto, T. Matsumoto, K. Nagayama and Y. Watanabe, Angew. Chem., Int. Ed., 2004, 43, 2527–2530 CrossRef CAS PubMed.
- R. M. Kramer, C. Li, D. C. Carter, M. O. Stone and R. R. Naik, J. Am. Chem. Soc., 2004, 126, 13282–13286 CrossRef CAS PubMed.
- K. K. W. Wong and S. Mann, Adv. Mater., 1996, 8, 928–932 CrossRef CAS.
- I. Yamashita, J. Hayashi and M. Hara, Chem. Lett., 2004, 33, 1158–1159 CrossRef CAS.
- K. Iwahori, K. Yoshizawa, M. Muraoka and I. Yamashita, Inorg. Chem., 2005, 44, 6393–6400 CrossRef CAS PubMed.
- J. P. Fortin, C. Wilhelm, J. Servais, C. Ménager, J. C. Bacri and F. Gazeau, J. Am. Chem. Soc., 2007, 129, 2628–2635 CrossRef CAS PubMed.
- M. Gonzales-Weimuller, M. Zeisberger and K. M. Krishnan, J. Magn. Magn. Mater., 2009, 321, 1947–1950 CrossRef CAS PubMed.
- L. Lartigue, C. Innocenti, T. Kalaivani, A. Awwad, M. D. M. Sanchez Duque, Y. Guari, J. Larionova, C. Gueírin, J. L. G. Montero, V. Barragan-Montero, P. Arosio, A. Lascialfari, D. Gatteschi and C. Sangregorio, J. Am. Chem. Soc., 2011, 133, 10459–10472 CrossRef CAS PubMed.
- M. V. Efremova, Y. A. Nalench, E. Myrovali, A. S. Garanina, I. S. Grebennikov, P. K. Gifer, M. A. Abakumov, M. Spasova, M. Angelakeris, A. G. Savchenko, M. Farle, N. L. Klyachko, A. G. Majouga and U. Wiedwald, Beilstein J. Nanotechnol., 2018, 9, 2684–2699 CrossRef CAS PubMed.
- M. Comes Franchini, G. Baldi, D. Bonacchi, D. Gentili, G. Giudetti, A. Lascialfari, M. Corti, P. Marmorato, J. Ponti, E. Micotti, U. Guerrini, L. Sironi, P. Gelosa, C. Ravagli and A. Ricci, Small, 2010, 6, 366–370 CrossRef CAS PubMed.
- E. Skoropata, R. D. Desautels, E. Falvo, P. Ceci, O. Kasyutich, J. W. Freeland and J. Van Lierop, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 174424 CrossRef.
- R. Das, N. P. Kim, S. B. Attanayake, M.-H. Phan and H. Srikanth, Appl. Sci., 2021, 11, 930 CrossRef CAS.
- S. Singhal, T. Namgyal, S. Bansal and K. Chandra, J. Electromagn. Anal. Appl., 2010, 2, 376–381 CAS.
- D. S. Mathew and R. S. Juang, Chem. Eng. J., 2007, 129, 51–65 CrossRef CAS.
- M. G. Christiansen, N. Mirkhani, W. Hornslien and S. Schuerle, J. Appl. Phys., 2022, 132, 174304 CrossRef CAS.
- S. Chakrabarti, S. K. Mandal and S. Chaudhuri, Nanotechnology, 2005, 16, 506–511 CrossRef CAS.
- M. T. Klem, D. A. Resnick, K. Gilmore, M. Young, Y. U. Idzerda and T. Douglas, J. Am. Chem. Soc., 2007, 129, 197–201 CrossRef CAS PubMed.
- D. A. Kuckla, J.-S. Brand, B. Czech, A. Asharion, J. V. Jüttner, I. P. Novoselova, A. Neusch, P. Hagemann, M. Getzlaff and C. Monzel, J. Phys. D: Appl. Phys., 2023, 56, 505002 CrossRef.
- A. L. Thangawng, R. S. Ruoff, M. A. Swartz and M. R. Glucksberg, Biomed. Microdevices, 2007, 9, 587–595 CrossRef CAS PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- M. A. Hink, R. A. Griep, J. W. Borst, A. Van Hoek, M. H. M. Eppink, A. Schots and A. J. W. G. Visser, J. Biol. Chem., 2000, 275, 17556–17560 CrossRef CAS PubMed.
- F. Etoc, E. Balloul, C. Vicario, D. Normanno, D. Liße, A. Sittner, J. Piehler, M. Dahan and M. Coppey, Nat. Mater., 2018, 17, 740–746 CrossRef CAS PubMed.
- J. I. Langford and A. J. C. Wilson, J. Appl. Crystallogr., 1978, 11, 102–113 CrossRef CAS.
- K. Zeth, E. Hoiczyk and M. Okuda, Trends Biochem. Sci., 2016, 41, 190–203 CrossRef CAS PubMed.
- V. A. Drits, B. A. Sakharov, A. L. Salyn and A. Manceau, Clay Miner., 1993, 28, 185–207 CrossRef CAS.
- E. Jansen, A. Kyek, W. Schäfer and U. Schwertmann, Appl. Phys. A: Mater. Sci. Process., 2002, 74, 1004–1006 CrossRef.
- W. Kim, C. Y. Suh, S. W. Cho, K. M. Roh, H. Kwon, K. Song and I. J. Shon, Talanta, 2012, 94, 348–352 CrossRef CAS PubMed.
- Y. A. Nalench, I. V. Shchetinin, A. S. Skorikov, P. S. Mogilnikov, M. Farle, A. G. Savchenko, A. G. Majouga, M. A. Abakumov and U. Wiedwald, J. Mater. Chem. B, 2020, 8, 3886–3895 RSC.
- P. Saha, R. Rakshit, M. Alam and K. Mandal, Phys. Rev. Appl., 2019, 11, 024059 CrossRef.
- S. Liébana-Viñas, K. Simeonidis, U. Wiedwald, Z.-A. Li, Z. Ma, E. Myrovali, A. Makridis, D. Sakellari, G. Vourlias, M. Spasova, M. Farle and M. Angelakeris, RSC Adv., 2016, 6, 72918–72925 RSC.
- L. Néel, Ann. Geophys. (C. N. R. S.), 1949, 5, 99–136 Search PubMed.
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167–R181 CrossRef CAS.
- S. Purushotham and R. V. Ramanujan, J. Appl. Phys., 2010, 107, 114701 CrossRef.
- A. Lascialfari, M. Filibian, C. Sangregorio and P. Carretta, La Riv. del Nuovo Cim., 2013, 36, 211–271 CAS.
- P. Guardia, R. Di Corato, L. Lartigue, C. Wilhelm, A. Espinosa, M. Garcia-Hernandez, F. Gazeau, L. Manna and T. Pellegrino, ACS Nano, 2012, 6, 3080–3091 CrossRef CAS PubMed.
- J.-H. Lee, J. Jang, J. Choi, S. H. Moon, S. Noh, J. Kim, J.-G. Kim, I.-S. Kim, K. I. Park and J. Cheon, Nat. Nanotechnol., 2011, 6, 418–422 CrossRef CAS PubMed.
- H. Gavilán, K. Simeonidis, E. Myrovali, E. Mazarío, O. Chubykalo-Fesenko, R. Chantrell, L. Balcells, M. Angelakeris, M. P. Morales and D. Serantes, Nanoscale, 2021, 13, 15631–15646 RSC.
- R. Hergt and S. Dutz, J. Magn. Magn. Mater., 2007, 311, 187–192 CrossRef CAS.
- L. Toraille, K. Aïzel, É. Balloul, C. Vicario, C. Monzel, M. Coppey, E. Secret, J. M. Siaugue, J. Sampaio, S. Rohart, N. Vernier, L. Bonnemay, T. Debuisschert, L. Rondin, J. F. Roch and M. Dahan, Nano Lett., 2018, 18, 7635–7641 CrossRef CAS PubMed.
- T. Häberle, F. Haering, H. Pfeifer, L. Han, B. Achinuq, U. Wiedwald, U. Herr and B. Koslowski, New J. Phys., 2012, 14, 043044 CrossRef.
- A. H. B. De Vries, B. E. Krenn, R. Van Driel and J. S. Kanger, Biophys. J., 2005, 88, 2137–2144 CrossRef CAS PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01652a |
| ‡ Present address: Institute of Human Genetics, University Hospital Düsseldorf, Heinrich-Heine-University Düsseldorf, 40225 Düsseldorf, Germany. |
| § Present address: Cell Biology and Cancer, UMR 144 CNRS, Institute Curie, 75248 Paris cedex 05, France. |
| This journal is © The Royal Society of Chemistry 2024 |