
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBalancing computational chemistry's potential with its environmental impact
Oliver
Schilter
 *abc,
Philippe
Schwaller
bc and
Teodoro
Laino
ab
*abc,
Philippe
Schwaller
bc and
Teodoro
Laino
ab
aIBM Research Europe, Säumerstrasse 4, 8803 Rüschlikon, Switzerland. E-mail: oli@zurich.ibm.com
bNational Center for Competence in Research-Catalysis (NCCR-Catalysis), Switzerland
cLIAC, EPFL Lausanne, Rte Cantonale, 1015 Lausanne, Switzerland
First published on 8th July 2024
Abstract
Computational chemistry techniques offer tremendous potential for accelerating the discovery of sustainable chemical processes and reactions. However, the environmental impacts of the substantial computing power required for these digital methods are often overlooked. This review provides a comprehensive analysis of the carbon footprint associated with molecular simulations, machine learning, optimization algorithms, and the required data center and research activities within the field of digital chemistry. Successful applications of these methods tackling climate-related issues like CO2 conversion and storage are highlighted, contrasted with assessments of their environmental burden. Strategies to minimize the carbon emissions from computational efforts are evaluated, including sustainable data center practices, efficient coding, reaction optimization, and sustainable research culture. Additionally, we surveyed tools and methodologies for tracking and reporting environmental impacts. Overall, guidelines and best practices are distilled for balancing the green potential of computational chemistry with responsible management of its environmental costs. Assessing and mitigating the field's carbon footprint is crucial for ensuring digital chemical discoveries truly contribute to sustainability goals.
1 Introduction
The need for a more sustainable future is undeniable, and a necessary shift towards carbon-neutral processes within the chemical industry is imperative. This transition can only be achieved through an increased emphasis on research and development, accelerating the discovery of more efficient processes, alternative chemical routes, green solvent replacements, new synthesis pathways, and more efficient catalysts that will lead to a reduction of the environmental footprint. One key tool that has gained popularity in recent years to achieve these discoveries is the field of digital chemistry, composed of molecular dynamics (MD), density functional theory (DFT), machine learning (ML), Bayesian optimization (BO), design of experiments (DOE) and artificial intelligence (AI). These computational techniques allow for a deeper understanding of the underlying chemistry and enable modeling real-world scenarios. Crucially, they facilitate the minimization of physical experiments, thereby streamlining the discovery process and reducing resource consumption. As chemical experiments often involve using hazardous substances, specialized laboratory equipment, and significant time investment from researchers, the ability to reduce the reliance on physical experimentation offers substantial advantages.The success of such methods has already extensively showcased their potential,1–3 also directly tackling CO2 conversion4–8 and storage. However, even when directly addressing climate change-related issues, scientists often overlook the environmental consequences of their direct actions; their research efforts have an associated environmental cost. From the energy consumed to run computations, to the resources needed for the production of hardware, to the land use for building data centers, storing data, to traveling to conferences,9 there are significant environmental impacts to consider. This study examines the current state of digital chemistry and explores how its environmental implications are currently being addressed. We showcase the success of such methods in tackling sustainability issues within the field of chemistry, highlighting their positive research impact. Furthermore, we aim to provide an overarching assessment and distill recommendations for more sustainable computing practices, sensitizing digital chemists to the importance of minimizing the environmental footprint of their computational and research efforts.
2 Simulations and their impact
The insight that simulations can provide into the mechanistic understanding of chemical processes is invaluable. For instance, density functional theory (DFT) calculations have been employed in numerous studies4–8 involving the conversion of CO2 to methanol, which stands as one of the most crucial pathways for defossilization of various chemical value chains.10–12 Furthermore, investigations into the production of green hydrogen, required for this methanol production, have been carried out via DFT13,14 and molecular dynamics.15,16 There is no shortage of publications utilizing these computational tools for a variety of other climate-relevant processes, such as CO2 absorption with metal–organic frameworks,17,18 recycling of chlorine for polyvinyl chloride production,19,20 biofuel production,21,22 and material discovery.23 Additionally, Jain et al.24 provided a comprehensive review of the application of DFT for energy materials like batteries, photovoltaics, and capacitors.One common trend we noticed analyzing these studies is, that even if they often focus directly or indirectly on climate change relevant issues, the climate impact of their calculations is not considered. The required information for reproducing results is frequently provided, but the resources used for the calculations are seldom mentioned. In the few notable cases, they are usually composed of the CPU model and hours, which could be used by environmental impact tools (see section 7) to estimate the direct impact of their calculations. Knowing the CPU model allows estimating the processor's efficiency, which when multiplied by the CPU hours, can provide a rough approximation of the energy required for running experiments. To minimize both factors, computational time and utilizing more efficient hardware, several advancements were made. From changing methods for calculating free energies in molecular dynamics reducing the number of CPU hours an order of magnitude,25 to utilizing GPUs and other hardware accelerators for the required computations.26–30 In the latter, Lin and Gavini26 demonstrated that switching to the GPU for matrix–matrix multiplications for computing the ground-state DFT calculations on large-scale systems can result in an 8-fold speedup compared to CPU. Jiang et al.30 utilized GPUs in a similar fashion, adapting the scaling of calculations to multiple GPUs accelerating the DFT calculations even further. As reported in related fields, the decision to use hardware acceleration often results in faster simulations. However, this does not necessarily lead to a lower carbon footprint, since the additional hardware requires energy for computation as well. As a single GPU, even providing more single floating point operation per watt, if not utilized efficiently can consume up 700 W of energy (max thermal design power).31,32 A wider study by Grealey et al.33 demonstrated that using a GPU for phylogeographic modeling of the Ebola virus while providing a 6× speedup, also increased the carbon footprint by 84%.
Despite the widespread use of computational tools like DFT and molecular dynamics in studying chemical and climate-relevant processes, the environmental impact of these calculations is not considered, highlighting the need for more awareness and reporting of the resources utilized in computational studies.
3 Machine learning and AI and their impact
In addition to traditional computational methods like DFT and MD, another rapidly growing area within digital chemistry is the field of machine learning (ML) or, more broadly, artificial intelligence (AI). This surge in popularity can be attributed to the advent of foundation models such as AlphaFold,34 AlphaGo,35 GPT,36 Gemini,37 and Claude.38 However, the integration of ML and AI into the field of chemistry has been an ongoing process for quite some time. Examples of early adoption include the use of quantitative structure–activity relationship (QSAR) models,39,40 molecular design algorithms,41–48 retrosynthesis planning49–52 and property prediction.53Machine learning was successfully employed for exploring sustainability-focused reactions and design processes, such as the design of electrocatalysts for the CO2 reduction,54–56 the design of metal–organic frameworks to capture CO2,2,57–59 helping to find greener solvent alternatives60,61 and the synthesis methanol from CO2.62
One area where machine learning shows the potential to lower the computational power required is training ML algorithms to approximate more computationally expensive calculations requiring DFT. While initial DFT calculations are often necessary to build a training dataset, and the training process itself requires computational resources, the inference from ML models, once trained, is generally computationally cheaper and faster compared to performing full DFT calculations. Wengert et al.53 demonstrated the potential of these approximation methods by training a machine learning model to predict organic crystal structures. Their approach combined machine learning models trained on high-quality quantum mechanical (QM) data for short-range interactions with computationally cheaper physics-based density functional tight binding methods for long-range interactions. The authors showed that for predicting the structures of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 molecules, factoring in the training of the machine learning algorithm and the generation of training QM data, their approach resulted in a 375-fold reduction in computational cost (from 30000000 CPU hours to 80000 CPU hours) compared to performing direct calculations at the target level theory. This translates to an approximate reduction of 40 metric tons of CO2 equivalents if ML is used as a surrogate model (see section 8.2 for the detailed calculations).
000 molecules, factoring in the training of the machine learning algorithm and the generation of training QM data, their approach resulted in a 375-fold reduction in computational cost (from 30000000 CPU hours to 80000 CPU hours) compared to performing direct calculations at the target level theory. This translates to an approximate reduction of 40 metric tons of CO2 equivalents if ML is used as a surrogate model (see section 8.2 for the detailed calculations).
Similarly, the Open Catalyst Project63 utilized 200 million CPU hours to compute the adsorption energy for 1.2 M adsorbate molecules such as CO2 and catalyst surfaces via DFT. For this data, benchmarks are organized annually to develop machine learning models that can approximate the costly DFT calculations, and it is worth noting that the authors claim the DFT calculations were performed using 100% renewable energy.
Again, a similar trend was noted in the DFT-related works: the main part of these publications does not mention the carbon emissions caused by the calculations.
The trend to larger models fueled technological advancements, such as the utilization of GPU for the training of neural networks64,65 and the specific architectures that tend to increase performance with model size such as transformers66,67 tend to also come with an increase of resource consumption during for training and inference. A study by Korolev and Mitrofanov68 showed this trend by retraining the state-of-the-art model architectures between 2016 and 2022 on the predicting the CO2 working capacity of metal–organic frameworks while measuring their greenhouse gas emission required for training them with the GPU model most commonly used at the time. They showcased that decreasing the mean absolute error of the prediction task by 28% comes at the cost of a 15000% increase in greenhouse gas emission, even when adjusted by the more efficient hardware available at the time.
This phenomenon bears a striking resemblance to the Jevons Paradox,69–71 which states that technological progress aimed at increasing the efficiency of resource use can paradoxically lead to an overall increase in resource consumption. In the context of neural network training, the pursuit of more accurate and capable models has resulted in architectures that are exponentially more computationally intensive, thereby offsetting the efficiency gains from hardware improvements.
As the most recent trend is towards large language models (LLMs), the number of parameters that need to be trained and, consequently, the energy consumption grows rapidly. A study by Strubell et al.72 estimated the CO2 eq. of training a LLM for a language translation task can cause carbon emission in the magnitude of 284′019 kg and cost around 150k USD in cloud computing. They also highlighted the quickly growing computational, therefore, environmental cost of running even small-scale hyperparameter grid search. Hence, there is a need for more efficient hyperparameter screening approaches (see section 4 how optimization techniques can help).
Another aspect of the environmental footprint was investigated by Luccioni et al.,73 where they examined the environmental impact of BLOOM, a 176B parameter large language model (LLM).74 They conducted a life cycle assessment for the broader scope of LLMs, from manufacturing the GPU server hardware to the energy required for model training and infrastructure such as networking, also factoring in model deployment to enable other scientists to utilize their trained model. Some key findings of their study are that only 54.5% of the energy was used for training, while the rest was consumed by idle operations (32%) and infrastructure (13.5%). The embodied emissions (hardware production emissions) were responsible for 11.2 tonnes of CO2 equivalent, while the dynamic consumption (model training) caused 24.69 tonnes of CO2 equivalent emissions. Infrastructure and idle operations accounted for 14.6 tonnes of CO2 equivalent, totaling a staggering 50.5 tonnes of CO2 equivalent for the full final training of BLOOM. It should be noted that the final training run of the model was responsible for only 37.24% of the project emissions, as other model variants were trained alongside.
Further impact LLM beyond climate emission should be noted, such as the postulated socioeconomic implications, but are outside of this review scope.75,76
4 Optimizers and their impact
The pursuit of accelerating the discovery process and reducing the number of experiments required for new discoveries with digital tools has been an ongoing endeavor for a long time. The overarching idea is that certain strategies can be employed to explore a vast space of possible experiments more methodically and identify patterns and correlations in previously conducted experiments to optimally determine the next set of reaction parameters that should be experimentally validated. Often in experimental chemistry, for example, if a new reaction mechanism is discovered, it is optimized in a one factor at a time (OFAT) fashion, e.g., first screening a variety of solvents, then with the highest-yielding solvent, screening the equivalents of reactants, followed by temperature variations.77,78 This approach strongly assumes variable independence, which is often not guaranteed. We will first discuss the potential of more methodical approaches to reduce the number of needed experiments before exploring the computational cost of one such method.Design of experiments (DoE), a statistical technique with origins dating back to the 1920s,79 focuses on maximizing information gain from a limited number of experiments by carefully designing experimental points according to specific criteria, such as orthogonality or space-filling properties. The multidimensional design space is explored simultaneously, screening different combinations of solvents, temperatures, and reactants. This approach allows for more effective interpolation of parameter interactions compared to one-factor-at-a-time (OFAT) experimentation80 and quantifies the effects of various factors on the response. DoE can reduce the number of experiments required to find more optimal reaction conditions, such as identifying more effective conditions for CO2 to CO conversion,81 electrochemical CO2 reduction,82 solvent-based CO2 capture systems,83 and CO2 methanation.84
One popular alternative to DOE is Bayesian optimization (BO), which is a sequential model-based technique that aims to find the global optimum of a black-box function by selecting the next reaction parameters to be evaluated based on the previous observations and a probabilistic model, so-called surrogate model. The new parameters are chosen to strike a balance between exploitation, using the already run experiments to select promising points, and exploration to improve the model's accuracy in unknown regions. An acquisition function guides the balance between these two strategies. Typical surrogate models used for BO are Gaussian processes,85–87 Bayesian neural networks87–89 and Random forest.87,90
The potential of BO has been explored in climate-focused studies. Zhang et al.91 employed BO to optimize the partial pressures of CO2 and H2O, as well as the reaction time, aiming to maximize the reaction rate of the photocatalytic reduction of CO2. Their approach reached optimal conditions faster compared to DOE and kinetic modeling. Similarly, Iwama et al.92 utilized BO to optimize the conversion of CO2 to CO via the reverse water–gas shift reaction. BO also helped to optimize the reaction conditions for biocatalytic C–C bond formation reactions,93 synthesis of methanol from syngas,94 and the CO2 to methanol conversion using heterogeneous catalysis.95 Several other studies show the reduction of the number of run experiments to achieve more optimal reaction conditions in also digital chemistry experiments, e.g. Ward and Pini96 used BO and ML to design pressure-vacuum swing adsorption processes for CO2 capture 14 faster compared to the classical optimization algorithm. Furthermore, BO is a commonly used method to optimize other digital chemistry approaches such as most commonly used to tune the hyperparameters of machine learning algorithms97–100 and DFT methods.101 Additionally, specific frameworks have been developed to facilitate the use of BO in the chemistry context. Gryffin102 optimizer was introduced to improve the selection of categorical variables, while Phoenics103 was developed to handle limited experimental objective evaluations. Furthermore, libraries86,104 and web applications105 have been created to facilitate the adoption of BO by laboratory chemists who may not have coding expertise, providing user-friendly interfaces to leverage computational tools.
The overall benefit of such optimizers lies in reducing required experiments, as demonstrated by Shields et al.106 through a benchmarking study comparing expert chemists against BO. The aim of study was to find reaction conditions leading to the highest yielding direct arylation of imidazoles. The full grid of 1728 reactions based 12 ligands, four bases, four solvents, three temperatures, and three concentrations, was run on a robotic platform and the objective of this project was then to let a chemist pick five reaction conditions at a time, returning the recorded yields of the picked reactions, allowing them to gain insight and pick the next conditions. Initially, humans tend to outperform BO's random parameter selection in the first few experiments. However, after 15 experiments (three batches of five experiments each), the optimizer surpasses human performance and finds the global optimal conditions (100% yield) within the first 50 experiments. Our analysis of their published data revealed that to achieve a desired yield of 95% (see Fig. 1), the average human performed around 60 experiments (12 batches of five experiments), whereas BO required only 25 experiments (5 batches). This reduction in the number of necessary experiments, equivalent to running 2.4 times fewer experiments, corresponds to a reduction in emissions, assuming an equal climate impact for each chemical involved. The energy consumption of running this particular BO campaign roughly corresponds to an emission of 0.0294 g CO2 eq. This is negligible compared to the emission from running chemical experiments since the equivalent of CO2 produced corresponds to using 0.8 mg of methanol (see section 8.1 for details).
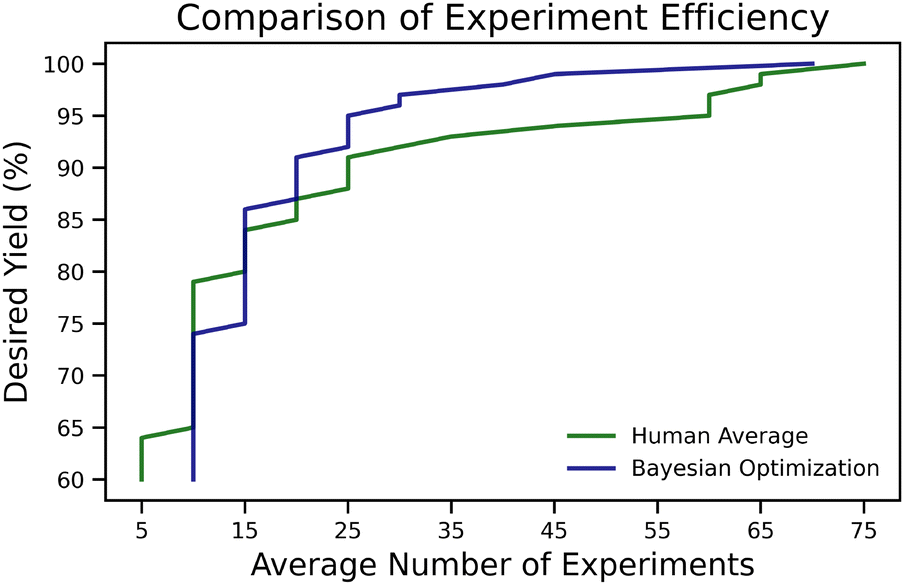 | ||
| Fig. 1 The average number of experiments needed to achieve a desired yield for the direct arylation of imidazoles.106 It can be seen that if a higher yield is desired the BO optimizer on average outperforms an expert chemist. | ||
5 Data centers and their impact
While the computational techniques discussed so far offer significant benefits for sustainability-focused research, it is crucial to consider the environmental impact of the underlying infrastructure that enables these digital chemistry endeavors researchers often face constraints in selecting the location for their computational workloads and data storage. Institutional resources, such as high-performance computing clusters and data centers, are typically provided by their university or employers, limiting their ability to choose more environmentally friendly options. Moreover, data sovereignty regulations may prohibit certain data from leaving the country's boundaries,107 further restricting the choice of data center locations. Nevertheless, when leveraging cloud computing services, researchers can exercise greater control over their environmental impact by carefully evaluating the geographic locations of cloud providers’ data centers and opting for regions with a higher proportion of renewable energy sources powering their operations.108–111 A study by Lacoste et al.112 showed that the CO2 emitted for producing 1 kW h of electrical energy can vary between 20 g to 736 g CO2 eq. changing just by changing the data center location from Iowa in the USA to Quebec Canada. This showcases that even in a single cloud provider region (both regions called North America) the variance can be significant. Similarly,109 found that changing the data center location from Australia to Switzerland can reduce emissions caused by power consumption by over 70 times, simply by choosing a location where a higher percentage of energy is produced from renewable sources (see Fig. 2).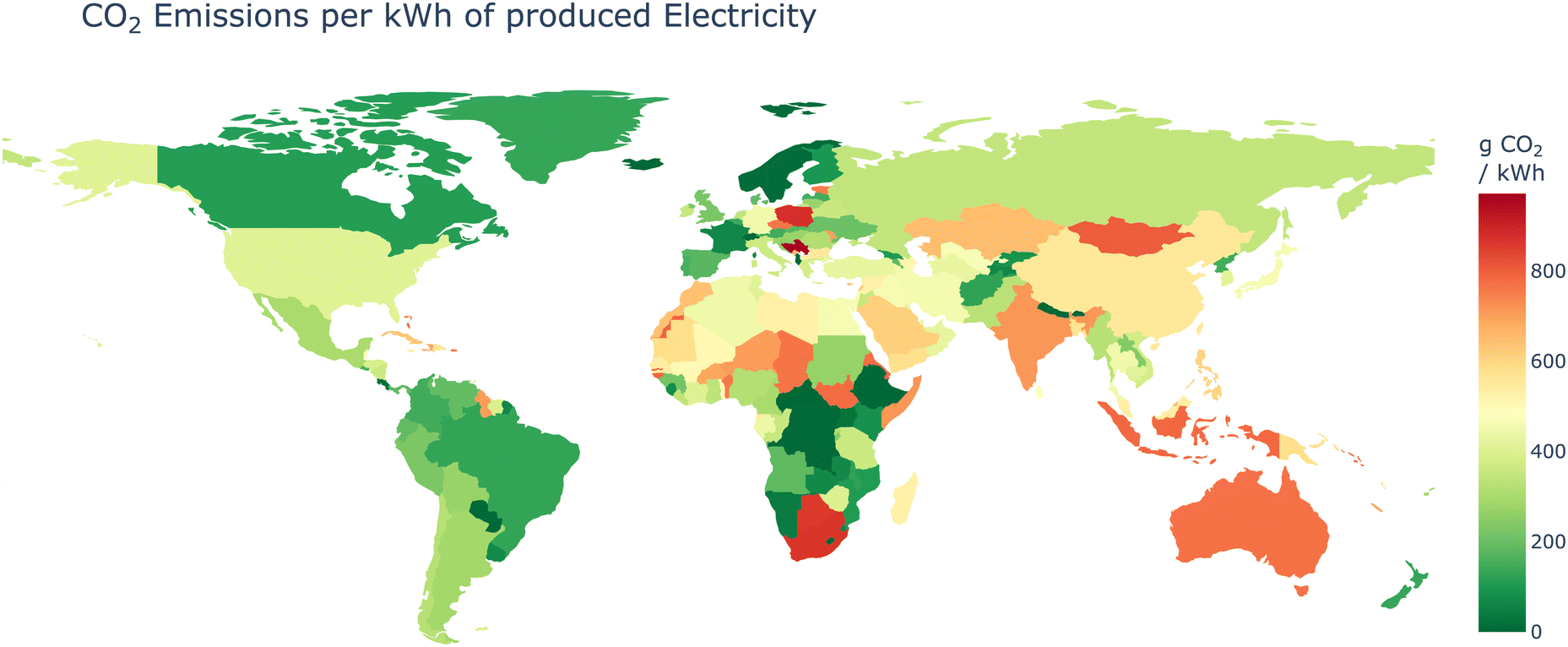 | ||
| Fig. 2 Average CO2 emission per produced kW h of electricity based on the international electricity factors of 2023.121 The energy source of a data center is a crucial factor in determining the environmental burden of computing. | ||
One key drawback of renewable energy is the unpredictability of its supply. However, this presents an opportunity to dynamically switch workloads from one region of the world to another based on the time and availability of the resource.111,113–117 Xu and Buyya111 simulated that switching between data center locations in California, Virginia, and Dublin could reduce carbon emissions by 43% while ensuring average response time for services. As simulations are often less time-sensitive, there is significant potential to implement similar approaches and take advantage of renewable resources by dynamically choosing locations where a surplus of renewable energy is produced and making them carbon-aware.
The location also influences broader aspects of environmental impact such as water footprint118–120 and land footprint.120 The water footprint of computing is mainly contributed by the power production and cooling used by the data center, as a study by Ristic et al.118 found.
6 Researchers and their impact
Beyond the direct environmental consequences of digital chemistry techniques and infrastructure, it is imperative to consider the broader impacts associated with the practices and behaviors of researchers themselves. One notable contributor to the environmental footprint is the travel required for attending conferences and sharing research findings. Conferences play a crucial role in gaining eminence and contributing to the advancement of a field. Presenting findings at conferences is crucial for gaining eminence and contributing to a field's success. However, the locations of these events often necessitate long-distance travel, typically achieved through air transportation, which can have a significant carbon footprint. A study by Klöwer et al.9 revealed that the average attendance at the American Geophysical Union conference resulted in 3 tonnes of CO2 emissions, primarily attributed to the conference's location on the US west coast. Since popular chemistry conferences such as the ACS Fall& Spring meetings have similar locations and attendance numbers, we can postulate that their travel emissions are comparable. The study found that if 36% of attendees who required intercontinental flights opted for virtual participation instead, the carbon footprint of the conference could be reduced by a significant 77%. The report highlighted several key recommendations for mitigating the environmental impact of conferences, including selecting venues that are easily accessible, transitioning to a biennial conference model, and actively encouraging researchers to attend conferences predominantly through virtual means. Implementing these strategies would directly reduce the carbon footprint associated with conference participation. Similar findings Arsenault et al.122 concluded that the average professor at Université de Montréal is responsible for emitting 10.7 tonnes of CO2 mainly caused by research-related travel. The study from Achten et al.123 agrees with these findings and deduces that traveling is responsible for 35% of the environmental footprint of the average Ph.D. student, followed by infrastructure and commuting as the two next big factors.We see one other key related to a researcher's behavior is the practice of sharing usable code. A study from Samuel and Mietchen124 found that in the research field of bioinformatics only around 7.6% of the Jupyter notebooks, a common file format to share Python code, from peer-reviewed publications were executable without error and of these, an even smaller percentage (5.5%) had the postulated reproducible output.
Lastly, we should be aware that storing data, that is no longer needed has an associated environmental footprint. Al Kez et al.120 found that only the unused data storage, so-called dark data, contributes to 5.26 million tons of CO2 eq., 41.65 gigaliters of water consumption, and 59.45 square kilometers of land usage.
7 Tools for impact assessment
Given the various environmental impacts discussed in the previous sections, it is imperative to have tools and methodologies for monitoring and assessing the environmental impact of our research efforts to identify areas for future enhancement. Various tools have been devised to track the carbon footprint generated at multiple levels, such as data center/cloud provider, operating system level, energy consumption during code execution, and retrospective analysis. If computing is run in the cloud, most cloud providers extensively offer estimations about the environmental footprint. Google,125 Amazon AWS,126 Microsoft Azure,127 and IBM Cloud128 provide carbon emission estimations that go beyond simple energy consumption, also considering factors such as the environmental impact of hardware production, data center operations, and even business travel-related climate impact of their employees. If on-premise computing is used, operating system-level energy consumption tracking can be utilized, such as PowerAPI129 or FreeIPMI.130 These tools, as they require to be run on bare metal not virtual machines or containers, showcase the need for more software-level tracking. For some programming languages,131–133 packages exist that allow tracking of resource and carbon emissions directly during execution. For Python, packages like TraCarbon131 and CodeCarbon132 are available. The latter tracks resource consumption for each code execution and includes a dashboard for interactive visualization of the recorded data and the corresponding environmental impact. For websites, there are also several tools available to investigate their efficiency134 as well as carbon emission caused by the data transfer, estimated visitor number, and energy source.135,136 If emission tracking was not done during code execution or hardware level, there is still the possibility to do a carbon emission estimation retro activity with the help of emission calculators. There are several online tools available112,137,138 for these calculations, requiring the CPU/GPU model used, the compute time, and the cloud region respectively, energy efficiency of the data center used to run the calculation (see Fig. 3). With these tools, we can also asses part of the emissions of the calculations mentioned in publications. Unfortunately, so far, most of these tools are limited to simple carbon emission based on energy consumption and don't factor in other aspects like a more complete life cycle assessment also of hardware production.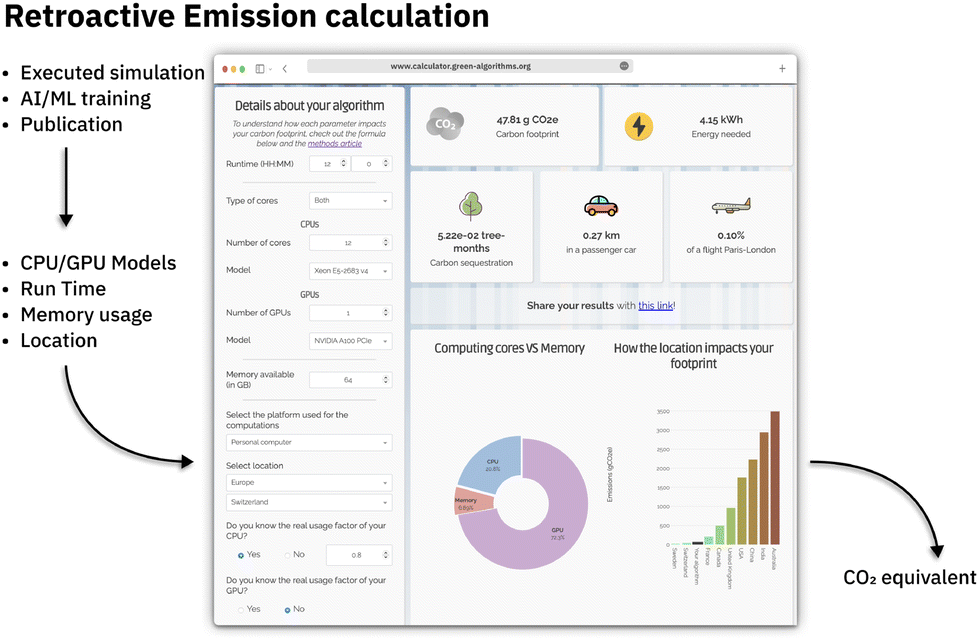 | ||
| Fig. 3 Tool such as green algorithm calculator (https://calculator.green-algorithms.org138) can be used to retroactively calculate the CO2 equivalent form already run computations or publications providing the number of CPU/GPU hours and model. | ||
We postulate that carbon tracking tools, which monitor emissions from digital chemistry endeavors, can help change researchers’ behavior similarly to how carbon footprint tracking apps influence consumers’ behavior. A study by Hoffmann et al.139 indicates that feedback from carbon tracking apps can decrease carbon emissions by 23% by continuously educating consumers about their carbon footprint. We hope similar effects can be achieved by tracking emissions of code executions, raising awareness, and ultimately lowering the emission caused by the necessary computations.
8 Methods
8.1 Emission tracking of BO experiments
To estimate the electricity consumption of the BO campaign, we used the package CodeCarbon132 to track the emission used for running the full Jupyter Notebook provided by Shields et al.106 in the GitHub repository experiments/edbo_demo_and_simulations.ipynb, we decided to run the full notebook including the plotting and hyperparameter search for the acquisition function and other experiments to have the worst-case scenario of somebody wanting to recreate the full pipeline. We only modified the notebook by including a first cell starting the CodeCarbon tracker and stopping the tracker in the last cell, the rest of the code is unchanged. The code was run on a MacBook Pro 2021, 32 Gb, and with an M1 Max chip running on grid power in Switzerland assuming an emission of 0.00278 per CO2 kg per kW h.121 The overall usage of electricity to execute the code was 0.0106 kW h which results in an average 0.0294 g CO2 eq. This is negligible compared to emission from running chemical experiments since the equivalent of CO2 produced corresponds to roughly 0.8 mg methanol (if 1 g of produced methanol from natural gas corresponds to 33 g of CO2 eq. emission140).8.2 Emission calculation of ML as surrogate models
We used the GreenAlgorithm138 website to retroactively calculate emissions based on the information provided by Wengert et al.53 They reported using an Intel® Xeon® CPU E5-2697 v3 @ 2.60 GHz, and we assumed their location was Germany based on the first author's affiliation. We estimated a total system memory of 64 GB, which when divided by the number of cores (14), results in approximately 4.6 GB per CPU core – a value used for the calculation. Assuming their reported 30000000 CPU hours would require a total of 294.40 MW h of energy and cause 43.2 metric tons of CO2 equivalent emissions, comparable to 61.8 flights from New York to Melbourne. In contrast, their full ML pipeline required 80000 CPU hours, causing 265.88 kg of CO2 equivalent emissions from 785.08 kW h of energy consumption. This represents a reduction of approximately 40 metric tons of CO2 equivalent emissions compared to running full simulations instead of their ML approximations.
9 Future research opportunities
As the application of AI in the field of digital chemistry emerges, there are opportunities for future studies regarding the environmental impact. For example, recently, the shift from adapting currently existing foundation models to a specific task, such as retraining a foundation model for material discovery,141 to a new set of tasks can be much more computationally efficient compared to training the model from scratch, which could be quantitatively investigated. Similarly, few-shot prompting142 could be explored as a cost-efficient alternative to retraining or training AI models. Furthermore, the use of external services, such as consuming large language models (LLMs) through the use of application programming interfaces (APIs) as an interface to control other digital tools to solve complex tasks, should have its environmental burden investigated.143–145 Additionally, environmental impact assessments should expand from primarily focusing on energy consumption to a comprehensive life cycle assessment as a standard practice. Interestingly, AI could automate these life cycle assessments for both physical and digital chemical experiments. Such an application of AI would ease the often complicated calculation and could amplify the number of conducted life cycle analysis.10 Conclusions
To summarize, the computational approaches employed in digital chemistry, although resource-intensive, offer a unique opportunity to reduce the overall environmental impact of chemical research. The judicious application of optimization techniques, machine learning surrogate models, and density functional theory calculations can drastically curtail the need for physical experiments, which often carry a more substantial environmental burden. However, these computational efforts must be conducted in an environmentally responsible manner. Researchers can adopt several best practices to achieve this synergy between computational power and sustainability:• If possible, implement carbon tracking into your codebase when doing digital chemistry to enable the estimation of emissions (e.g. with CodeCarbon132). We encourage you to publish the estimated emissions to allow future researchers to have a benchmark for comparison. Be specific and mention the hardware model, energy source, compute time, and software version employed for calculations in publications.
• Share source code, trained machine learning models, and raw data following the FAIR (findability, accessibility, interoperability, and reuse of digital assets) principle. Reusability lowers duplication of efforts, e.g., repetition of computations.
• Using machine learning as surrogate models can greatly reduce the environmental impact of more computationally costly simulations. It is important to keep an eye on the chosen architecture and the resulting computational complexity.
• Optimization techniques can significantly reduce the number of physical experiments and are generally worth the associated computational cost.
• Reduce unnecessary exhaustive screening of hyperparameters of simulations and machine learning, as they can notably contribute to the overall carbon emissions of a project.
• If possible, choose time and data center locations where the energy used for computations is largely generated by renewable sources. Making your code carbon-aware allows you to dynamically shift workloads to achieve this.
• Be aware that storing data has an associated emissions cost. Delete no longer needed data and services.
• As travel-associated emissions are one of the largest contributors to a researcher's greenhouse gas emissions, choose to participate in conferences virtually whenever possible.
Data availability
This study was carried out using publicly available data from https://www.carbonfootprint.com/international_electricity_factors.html. The code used in the method section “emission calculation of ML as surrogate models” can be found at https://www.green-algorithms.org with https://doi.org/10.1002/advs.202100707. The code used in the method section “emission tracking of BO experiments” can be found can be found at https://github.com/b-shields/edbo with https://doi.org/10.1038/s41586-021-03213-y as well as we utilized CodeCarbon for the carbon tracking available at https://github.com/mlco2/codecarbon with https://doi.org/10.5281/zenodo.11171501.Author contributions
Oliver Schilter: methodology, investigation, conceptualization, visualization, data curation, writing – original draft, writing – review & editing. Teodoro Laino: methodology, writing – original draft, writing – review & editing, project administration, supervision. Philippe Schwaller: methodology, writing – original draft, writing – review & editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This publication was created as part of NCCR Catalysis (grant number 180544), a National Centre of Competence in Research funded by the Swiss National Science Foundation.References
- R. Hardian, Z. Liang, X. Zhang and G. Szekely, Green Chem., 2020, 22, 7521–7528 RSC.
- H. C. Gulbalkan, G. O. Aksu, G. Ercakir and S. Keskin, Ind. Eng. Chem. Res., 2024, 63, 37–48 CrossRef CAS PubMed.
- D. P. Tabor, L. M. Roch, S. K. Saikin, C. Kreisbeck, D. Sheberla, J. H. Montoya, S. Dwaraknath, M. Aykol, C. Ortiz and H. Tribukait, et al. , Nat. Rev. Mater., 2018, 3, 5–20 CrossRef CAS.
- T. Pinheiro Araújo, J. Morales-Vidal, T. Zou, R. García-Muelas, P. O. Willi, K. M. Engel, O. V. Safonova, D. Faust Akl, F. Krumeich, R. N. Grass, C. Mondelli, N. López and J. Pérez-Ramírez, Adv. Energy Mater., 2022, 12, 2103707 CrossRef.
- T. Pinheiro Araújo, J. Morales-Vidal, T. Zou, M. Agrachev, S. Verstraeten, P. O. Willi, R. N. Grass, G. Jeschke, S. Mitchell, N. López and J. Pérez-Ramírez, Adv. Energy Mater., 2023, 13, 2204122 CrossRef.
- X. Yang, ACS Catal., 2014, 4, 1129–1133 CrossRef CAS.
- J. Ye, C.-J. Liu, D. Mei and Q. Ge, J. Catal., 2014, 317, 44–53 CrossRef CAS.
- J. Ye, C. Liu, D. Mei and Q. Ge, ACS Catal., 2013, 3, 1296–1306 CrossRef CAS.
- M. Klöwer, D. Hopkins, M. Allen and J. Higham, Nature, 2020, 583, 356–359 CrossRef PubMed.
- A. González-Garay, M. S. Frei, A. Al-Qahtani, C. Mondelli, G. Guillén-Gosálbez and J. Pérez-Ramírez, Energy Environ. Sci., 2019, 12, 3425–3436 RSC.
- J. Sehested, J. Catal., 2019, 371, 368–375 CrossRef CAS.
- K. Narine, J. Mahabir, N. Koylass, N. Samaroo, S. Singh-Gryzbon, A. Baboolal, M. Guo and K. Ward, J. CO2 Util., 2021, 44, 101399 CrossRef CAS.
- J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- J. Rossmeisl, A. Logadottir and J. K. Nørskov, Chem. Phys., 2005, 319, 178–184 CrossRef CAS.
- N. Burton, R. Padilla, A. Rose and H. Habibullah, Renewable Sustainable Energy Rev., 2021, 135, 110255 CrossRef CAS.
- F. Hofbauer and I. Frank, Chem. – Eur. J., 2012, 18, 277–282 CrossRef CAS PubMed.
- L. Grajciar, A. D. Wiersum, P. L. Llewellyn, J.-S. Chang and P. Nachtigall, J. Phys. Chem. C, 2011, 115, 17925–17933 CrossRef CAS.
- G. Alonso, D. Bahamon, F. Keshavarz, X. Giménez, P. Gamallo and R. Sayós, J. Phys. Chem. C, 2018, 122, 3945–3957 CrossRef CAS.
- N. López, J. Gómez-Segura, R. P. Marín and J. Perez-Ramirez, J. Catal., 2008, 255, 29–39 CrossRef.
- J. Pérez-Ramírez, C. Mondelli, T. Schmidt, O. F.-K. Schlüter, A. Wolf, L. Mleczko and T. Dreier, Energy Environ. Sci., 2011, 4, 4786–4799 RSC.
- K. Kumar, V. Khatri, S. Upadhyayula and H. K. Kashyap, Appl. Catal., A, 2021, 610, 117951 CrossRef CAS.
- S. Gueddida, S. Lebègue and M. Badawi, J. Phys. Chem. C, 2020, 124, 20262–20269 CrossRef CAS.
- G. Hautier, Comput. Mater. Sci., 2019, 163, 108–116 CrossRef CAS.
- A. Jain, Y. Shin and K. A. Persson, Nat. Rev. Mater., 2016, 1, 1–13 Search PubMed.
- L. Yang, A. Ahmed and S. I. Sandler, J. Comput. Chem., 2013, 34, 284–293 CrossRef CAS PubMed.
- C.-C. Lin and V. Gavini, Comput. Phys. Commun., 2023, 282, 108516 CrossRef CAS.
- N. Vaughn and University of Michigan, PhD thesis, 2020.
- S. Seritan, C. Bannwarth, B. S. Fales, E. G. Hohenstein, C. M. Isborn, S. I. Kokkila-Schumacher, X. Li, F. Liu, N. Luehr and J. W. Snyder Jr, et al. , Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11, e1494 CAS.
- A. Mathiasen, H. Helal, P. Balanca, K. Klaeser, J. Dean, C. Luschi, D. Beaini, A. W. Fitzgibbon and D. Masters, 1st Workshop on the Synergy of Scientific and Machine Learning Modeling@ ICML2023, 2023.
- Q. Jiang, L. Wan, S. Jiao, W. Hu, J. Chen and H. An, 2020 IEEE 22nd International Conference on High Performance Computing and Communications; IEEE 18th International Conference on Smart City; IEEE 6th International Conference on Data Science and Systems (HPCC/SmartCity/DSS), 2020, pp. 197–205.
- Y. Sun, N. B. Agostini, S. Dong and D. Kaeli, arXiv, 2019, preprint, arXiv:1911.11313, DOI:10.48550/arXiv.1911.11313.
- Nvidia, NVIDIA H100 Tensor-Core-GPU, https://www.nvidia.com/de-de/data-center/h100/, accessed on 25th March 2024.
- J. Grealey, L. Lannelongue, W.-Y. Saw, J. Marten, G. Méric, S. Ruiz-Carmona and M. Inouye, Mol. Biol. Evol., 2022, 39, msac034 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek and A. Potapenko, et al. , Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- D. Silver, A. Huang, C. J. Maddison, A. Guez, L. Sifre, G. Van Den Driessche, J. Schrittwieser, I. Antonoglou, V. Panneershelvam and M. Lanctot, et al. , Nature, 2016, 529, 484–489 CrossRef CAS PubMed.
- OpenAI, ChatGPT, https://www.https://chat.openai.com, note = accessed on 25th March 2024.
- G. Team, R. Anil, S. Borgeaud, Y. Wu, J.-B. Alayrac, J. Yu, R. Soricut, J. Schalkwyk, A. M. Dai and A. Hauth, et al., arXiv, 2023, preprint, arXiv:2312.11805, DOI:10.48550/arXiv.2312.11805.
- Aanthropic, Claude 3, https://claude.ai/, note = accessed on 25th March 2024.
- C. Hansch and T. Fujita, J. Am. Chem. Soc., 1964, 86, 1616–1626 CrossRef CAS.
- S. M. Free and J. W. Wilson, J. Med. Chem., 1964, 7, 395–399 CrossRef CAS PubMed.
- N. Brown, P. Ertl, R. Lewis, T. Luksch, D. Reker and N. Schneider, J. Comput.-Aided Mol. Des., 2020, 34, 709–715 CrossRef CAS PubMed.
- O. Schilter, A. Vaucher, P. Schwaller and T. Laino, Digital Discovery, 2023, 2, 728–735 RSC.
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 268–276 CrossRef PubMed.
- M. Manica, J. Born, J. Cadow, D. Christofidellis, A. Dave, D. Clarke, Y. G. N. Teukam, G. Giannone, S. C. Hoffman and M. Buchan, et al. , npj Comput. Mater., 2023, 9, 69 CrossRef.
- M. H. Segler, T. Kogej, C. Tyrchan and M. P. Waller, ACS Cent. Sci., 2018, 4, 120–131 CrossRef CAS PubMed.
- M. Olivecrona, T. Blaschke, O. Engkvist and H. Chen, J. Cheminf., 2017, 9, 1–14 Search PubMed.
- P. Ertl, R. Lewis, E. Martin and V. Polyakov, arXiv, 2017, preprint, arXiv:1712.07449, DOI:10.48550/arXiv.1712.07449.
- B. Sanchez-Lengeling, C. Outeiral, G. L. Guimaraes and A. Aspuru-Guzik, ChemRxiv, 2017, preprint, DOI:10.26434/chemrxiv.5309668.v3.
- P. Schwaller, R. Petraglia, V. Zullo, V. H. Nair, R. A. Haeuselmann, R. Pisoni, C. Bekas, A. Iuliano and T. Laino, Chem. Sci., 2020, 11, 3316–3325 RSC.
- C. W. Coley, D. A. Thomas III, J. A. Lummiss, J. N. Jaworski, C. P. Breen, V. Schultz, T. Hart, J. S. Fishman, L. Rogers and H. Gao, et al. , Science, 2019, 365, eaax1566 CrossRef CAS PubMed.
- S. Genheden, A. Thakkar, V. Chadimová, J.-L. Reymond, O. Engkvist and E. Bjerrum, J. Cheminf., 2020, 12, 70 Search PubMed.
- V. S. Gil, A. M. Bran, M. Franke, R. Schlama, J. S. Luterbacher and P. Schwaller, arXiv, 2023, preprint, arXiv:2312.09004, DOI:10.48550/arXiv.2312.09004.
- S. Wengert, G. Csányi, K. Reuter and J. T. Margraf, Chem. Sci., 2021, 12, 4536–4546 RSC.
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C.-T. Dinh, P. De Luna, Z. Yu, A. S. Rasouli and P. Brodersen, et al. , Nature, 2020, 581, 178–183 CrossRef CAS PubMed.
- N. Zhang, B. Yang, K. Liu, H. Li, G. Chen, X. Qiu, W. Li, J. Hu, J. Fu and Y. Jiang, et al. , Small Methods, 2021, 5, 2100987 CrossRef CAS PubMed.
- Z. Sun, H. Yin, K. Liu, S. Cheng, G. K. Li, S. Kawi, H. Zhao, G. Jia and Z. Yin, SmartMat, 2022, 3, 68–83 CrossRef CAS.
- R. Anderson, J. Rodgers, E. Argueta, A. Biong and D. A. Gómez-Gualdrón, Chem. Mater., 2018, 30, 6325–6337 CrossRef CAS.
- A. Chen, X. Zhang, L. Chen, S. Yao and Z. Zhou, J. Phys. Chem. C, 2020, 124, 22471–22478 CrossRef CAS.
- H. Dureckova, M. Krykunov, M. Z. Aghaji and T. K. Woo, J. Phys. Chem. C, 2019, 123, 4133–4139 CrossRef CAS.
- D. Meng and Z. Liu, J. Mol. Liq., 2023, 392, 123286 CrossRef CAS.
- H. Gao, T. J. Struble, C. W. Coley, Y. Wang, W. H. Green and K. F. Jensen, ACS Cent. Sci., 2018, 4, 1465–1476 CrossRef CAS PubMed.
- M. Suvarna, T. P. Araujo and J. Pérez-Ramírez, Appl. Catal., B, 2022, 315, 121530 CrossRef CAS.
- R. Tran, J. Lan, M. Shuaibi, B. M. Wood, S. Goyal, A. Das, J. Heras-Domingo, A. Kolluru, A. Rizvi and N. Shoghi, et al. , ACS Catal., 2023, 13, 3066–3084 CrossRef CAS.
- R. Raina, A. Madhavan and A. Y. Ng, Proceedings of the 26th annual international conference on machine learning, 2009, pp. 873–880.
- J. Schmidhuber, Neural Networks, 2015, 61, 85–117 CrossRef PubMed.
- A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, L. Jones, A. N. Gomez, L. U. Kaiser and I. Polosukhin, Advances in Neural Information Processing Systems, 2017.
- A. Radford, K. Narasimhan, T. Salimans and I. Sutskever, Improving language understanding by generative pre-training, https://openai.com/index/language-unsupervised/, 2018.
- V. Korolev and A. Mitrofanov, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-zctn1-v3.
- B. Alcott, M. Giampietro, K. Mayumi and J. Polimeni, The Jevons paradox and the myth of resource efficiency improvements, Routledge, 2012, pp. 150–187 Search PubMed.
- E. I. Shumskaia, Industry 4.0: Fighting climate change in the economy of the future, Springer, 2022, pp. 359–365 Search PubMed.
- H. D. Saunders, Energy J., 1992, 13, 131–148 CrossRef.
- E. Strubell, A. Ganesh and A. McCallum, arXiv, 2019, preprint, arXiv:1906.02243, DOI:10.48550/arXiv.1906.02243.
- A. S. Luccioni, S. Viguier and A.-L. Ligozat, J. Mach. Learn. Res., 2023, 24, 1–15 Search PubMed.
- T. Le Scao, A. Fan, C. Akiki, E. Pavlick, S. Ilić, D. Hesslow, R. Castagné, A. S. Luccioni, F. Yvon, M. Gallé, et al., arXiv, 2023, DOI:10.48550/arXiv.2211.05100.
- E. M. Bender, T. Gebru, A. McMillan-Major and S. Shmitchell, Proceedings of the 2021 ACM conference on fairness, accountability, and transparency, 2021, pp. 610–623.
- M. C. Rillig, M. Ågerstrand, M. Bi, K. A. Gould and U. Sauerland, Environ. Sci. Technol., 2023, 57, 3464–3466 CrossRef CAS PubMed.
- B. Abtahi and H. Tavakol, Appl. Organomet. Chem., 2020, 34, e5895 CrossRef CAS.
- C. J. Taylor, A. Pomberger, K. C. Felton, R. Grainger, M. Barecka, T. W. Chamberlain, R. A. Bourne, C. N. Johnson and A. A. Lapkin, Chem. Rev., 2023, 123, 3089–3126 CrossRef CAS PubMed.
- J. F. Box, Am. Stat., 1980, 34, 1–7 Search PubMed.
- C. J. Taylor, A. Baker, M. R. Chapman, W. R. Reynolds, K. E. Jolley, G. Clemens, G. E. Smith, A. J. Blacker, T. W. Chamberlain and S. D. Christie, et al. , J. Flow Chem., 2021, 11, 75–86 CrossRef CAS.
- D. Mei, Y.-L. He, S. Liu, J. Yan and X. Tu, Plasma Processes Polym., 2016, 13, 544–556 CrossRef CAS.
- M. Dunwell, W. Luc, Y. Yan, F. Jiao and B. Xu, ACS Catal., 2018, 8, 8121–8129 CrossRef CAS.
- J. C. Morgan, A. S. Chinen, C. Anderson-Cook, C. Tong, J. Carroll, C. Saha, B. Omell, D. Bhattacharyya, M. Matuszewski and K. S. Bhat, et al. , Appl. Energy, 2020, 262, 114533 CrossRef CAS.
- C.-E. Yeo, M. Seo, D. Kim, C. Jeong, H.-S. Shin and S. Kim, Energies, 2021, 14, 8414 CrossRef CAS.
- P. I. Frazier, arXiv, 2018, preprint, arXiv:1807.02811, DOI:10.48550/arXiv.1807.02811.
- R.-R. Griffiths, L. Klarner, H. Moss, A. Ravuri, S. Truong, Y. Du, S. Stanton, G. Tom, B. Rankovic, A. Jamasb, et al., Advances in Neural Information Processing Systems, 2024, 36.
- J. Guo, B. Ranković and P. Schwaller, Chimia, 2023, 77, 31–38 CrossRef CAS PubMed.
- J. Snoek, O. Rippel, K. Swersky, R. Kiros, N. Satish, N. Sundaram, M. Patwary, M. Prabhat and R. Adams, International conference on machine learning, 2015, pp. 2171–2180.
- J. T. Springenberg, A. Klein, S. Falkner and F. Hutter, Advances in Neural Information Processing Systems, 2016, 29.
- J. Guo, X. Zan, L. Wang, L. Lei, C. Ou and S. Bai, Eng. Fract. Mech., 2023, 293, 109714 CrossRef.
- Y. Zhang, X. Yang, C. Zhang, Z. Zhang, A. Su and Y.-B. She, Processes, 2023, 11, 2614 CrossRef CAS.
- R. Iwama, K. Takizawa, K. Shinmei, E. Baba, N. Yagihashi and H. Kaneko, ACS Omega, 2022, 7, 10709–10717 CrossRef CAS PubMed.
- R. Tachibana, K. Zhang, Z. Zou, S. Burgener and T. R. Ward, ACS Sustainable Chem. Eng., 2023, 11, 12336–12344 CrossRef CAS PubMed.
- A. Kumar, K. K. Pant, S. Upadhyayula and H. Kodamana, ACS Omega, 2022, 8, 410–421 CrossRef PubMed.
- A. Ramirez, E. Lam, D. P. Gutierrez, Y. Hou, H. Tribukait, L. M. Roch, C. Copéret and P. Laveille, Chem. Catal., 2024, 4, 100888 CrossRef CAS.
- A. Ward and R. Pini, Ind. Eng. Chem. Res., 2022, 61, 13650–13668 CrossRef CAS.
- J. Snoek, H. Larochelle and R. P. Adams, Advances in neural information processing systems, 2012, 25.
- J. Wu, X.-Y. Chen, H. Zhang, L.-D. Xiong, H. Lei and S.-H. Deng, J. Electron. Sci. Technol., 2019, 17, 26–40 Search PubMed.
- A. H. Victoria and G. Maragatham, Evol. Syst., 2021, 12, 217–223 CrossRef.
- A. Klein, S. Falkner, S. Bartels, P. Hennig and F. Hutter, Artificial intelligence and statistics, 2017, pp. 528–536 Search PubMed.
- R. A. Vargas-Hernández, J. Phys. Chem. A, 2020, 124, 4053–4061 CrossRef PubMed.
- F. Häse, M. Aldeghi, R. J. Hickman, L. M. Roch and A. Aspuru-Guzik, Appl. Phys. Rev., 2021, 8, 31406 Search PubMed.
- F. Hase, L. M. Roch, C. Kreisbeck and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 1134–1145 CrossRef CAS PubMed.
- Y. Wang, T.-Y. Chen and D. G. Vlachos, J. Chem. Inf. Model., 2021, 61, 5312–5319 CrossRef CAS PubMed.
- J. A. G. Torres, S. H. Lau, P. Anchuri, J. M. Stevens, J. E. Tabora, J. Li, A. Borovika, R. P. Adams and A. G. Doyle, J. Am. Chem. Soc., 2022, 144, 19999–20007 CrossRef CAS PubMed.
- B. J. Shields, J. Stevens, J. Li, M. Parasram, F. Damani, J. I. M. Alvarado, J. M. Janey, R. P. Adams and A. G. Doyle, Nature, 2021, 590, 89–96 CrossRef CAS PubMed.
- C. R. Baudoin , 2018 32nd International Conference on Advanced Information Networking and Applications Workshops (WAINA), 2018, pp. 430–435.
- B. Whitehead, D. Andrews, A. Shah and G. Maidment, Build. Environ., 2014, 82, 151–159 CrossRef.
- L. Lannelongue, J. Grealey, A. Bateman and M. Inouye, PLoS Comput. Biol., 2021, 17, 1009324 CrossRef PubMed.
- A. Shehabi, S. Smith, D. Sartor, R. Brown, M. Herrlin, J. Koomey, E. Masanet, N. Horner, I. Azevedo and W. Lintner, United states data center energy usage report, 2016.
- M. Xu and R. Buyya, J. Parallel Distrib. Comput., 2020, 135, 191–202 CrossRef.
- A. Lacoste, A. Luccioni, V. Schmidt and T. Dandres, arXiv, 2019, preprint, arXiv:1910.09700, DOI:10.48550/arXiv.1910.09700.
- Z. Zhou, F. Liu, Y. Xu, R. Zou, H. Xu, J. C. Lui and H. Jin, 2013 IEEE 21st International Symposium on Modelling, Analysis and Simulation of Computer and Telecommunication Systems, 2013, pp. 232–241.
- G. Neglia, M. Sereno and G. Bianchi, ACM SIGMETRICS Performance Evaluation Review, 2016, 44, 64–69.
- A. Radovanović, R. Koningstein, I. Schneider, B. Chen, A. Duarte, B. Roy, D. Xiao, M. Haridasan, P. Hung and N. Care, et al. , IEEE Trans. Power Syst., 2022, 38, 1270–1280 Search PubMed.
- W.-T. Lin, G. Chen and H. Li, IEEE Trans. Cloud Comput., 2023, 11, 1111–1121 Search PubMed.
- P. Wiesner, M. Steinke, H. Nickel, Y. Kitana and O. Kao, Software, 2023, 53, 2362–2376 Search PubMed.
- B. Ristic, K. Madani and Z. Makuch, Sustainability, 2015, 7, 11260–11284 CrossRef.
- M. A. B. Siddik, A. Shehabi and L. Marston, Environ. Res. Lett., 2021, 16, 064017 CrossRef.
- D. Al Kez, A. M. Foley, D. Laverty, D. F. Del Rio and B. Sovacool, J. Cleaner Prod., 2022, 371, 133633 CrossRef.
- Carbon Footprint Ltd, International Electricity Factors, https://www.carbonfootprint.com/international_electricity_factors.html, accessed on 14th March 2024.
- J. Arsenault, J. Talbot, L. Boustani, R. Gonzalès and K. Manaugh, Environ. Res. Lett., 2019, 14, 095001 CrossRef.
- W. M. Achten, J. Almeida and B. Muys, Ecol. Indic., 2013, 34, 352–355 CrossRef CAS.
- S. Samuel and D. Mietchen, GigaScience, 2024, 13, giad113 CrossRef PubMed.
- Google Cloud, Carbon Footprint reporting methodology, https://cloud.google.com/carbon-footprint/docs/methodology, accessed on 14th March 2024.
- Amazon, Carbon Footprint Tool, https://aws.amazon.com/blogs/aws/new-customer-carbon-footprint-tool/, accessed on 14th March 2024.
- Microsoft, Emissions Impact Dashboard, https://www.microsoft.com/en-us/sustainability/emissions-impact-dashboard, accessed on 14th March 2024.
- IBM Cloud, IBM Cloud Carbon Calculator, https://cloud.ibm.com/media/docs/downloads/billing-usage/carbon-calc-method-v2.pdf, accessed on 14th March 2024.
- A. Bourdon, A. Noureddine, R. Rouvoy and L. Seinturier, ERCIM News, 2013, 92, 43–44.
- FreeIPMI, FreeIPMI, https://www.gnu.org/software/freeipmi/, accessed on 15th March 2024.
- TraCarbon, tracarbon, https://github.com/fvaleye/tracarbon, accessed on 18th March 2024.
- CodeCarbon, Code Carbon, https://github.com/mlco2/codecarbon, accessed on 18th March 2024.
- A. Noureddine , 18th International Conference on Intelligent Environments (IE2022), Biarritz, France, 2022.
- ec0lint, ec0lint, https://github.com/ec0lint/ec0lint, accessed on 18th March 2024.
- websitecarbon, Website Carbon calculator, https://www.websitecarbon.com, accessed on 18th March 2024.
- Ecograder, Ecograder, https://ecograder.com, accessed on 18th March 2024.
- P. Henderson, J. Hu, J. Romoff, E. Brunskill, D. Jurafsky and J. Pineau, J. Mach. Learn. Res., 2020, 21, 10039–10081 Search PubMed.
- L. Lannelongue, J. Grealey and M. Inouye, Adv. Sci., 2021, 8, 2100707 CrossRef PubMed.
- S. Hoffmann, W. Lasarov, H. Reimers and M. Trabandt, J. Cleaner Prod., 2024, 434, 139981 CrossRef CAS.
- Methanol Institute, Carbon footprint of methanol, https://www.methanol.org/wp-content/uploads/2022/01/CARBON-FOOTPRINT-OF-METHANOL-PAPER_1-31-22.pdf, accessed on 25th March 2024.
- I. Batatia, P. Benner, Y. Chiang, A. M. Elena, D. P. Kovács, J. Riebesell, X. R. Advincula, M. Asta, W. J. Baldwin, N. Bernstein, et al., arXiv, 2023, preprint, arXiv:2401.00096, DOI:10.48550/arXiv.2401.00096.
- T. Brown, B. Mann, N. Ryder, M. Subbiah, J. D. Kaplan, P. Dhariwal, A. Neelakantan, P. Shyam, G. Sastry, A. Askell, et al., Advances in neural information processing systems, 2020, 33, 1877–1901.
- A. M. Bran, S. Cox, O. Schilter, C. Baldassari, A. D. White and P. Schwaller, Nat. Mach. Intell., 2024, 1–11 Search PubMed.
- T. Erdmann, S. Zecevic, S. Swaminathan, B. Ransom, K. Lionti, D. Zubarev, S. Kunde, S. Houde, J. Hedrick, N. Park, et al., American Chemical Society (ACS) Spring Meeting, 2024.
- D. A. Boiko, R. MacKnight, B. Kline and G. Gomes, Nature, 2023, 624, 570–578 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |