
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCrafting mono- and novel bis-methylated pyrroloquinoxaline derivatives from a shared precursor and its application in the total synthesis of marinoquinoline A†
Margarita Damai ,
Norman Guzzardi,
Viliyana Lewis,
Zenobia X. Rao,
Daniel Sykes and
Bhaven Patel*
,
Norman Guzzardi,
Viliyana Lewis,
Zenobia X. Rao,
Daniel Sykes and
Bhaven Patel*
School of Human Sciences, London Metropolitan University, 166-220 Holloway Road, London, N7 8DB, UK. E-mail: b.patel1@londonmet.ac.uk
First published on 10th October 2023
Abstract
The synthesis of mono- and novel bis-methylated pyrrolo[1,2-a]quinoxalines through the addition of unstable methyl radicals to aryl isocyanides is described contingent upon the reaction conditions employed. The strategy has been effectively employed in the total synthesis of the natural product marinoquinoline A.
Introduction
Nitrogen-containing heterocycles constitute a fundamental class of chemical compounds, playing a pivotal role in natural products, biologically active structures and compounds of significance in medicinal chemistry. The synthesis of pyrrolo[1,2-a]quinoxaline ring systems has been a subject of substantial research over the past century.1,2 These compounds exhibit significant biological activity, with notable emphasis on the introduction of substitutions at the C-4 position of the pyrroloquinoxaline motif.3 Such modifications yield structures that include having anticancer potential,4 antimalarial properties,5 and antiproliferative effects.6 Moreover, these molecular structures have been identified as inhibitors of the human protein kinase CK2,7 activators of glucagon receptors,8 and agonists for 5-HT3 receptors,9 while also serving as building blocks for synthesising gamma aminobutyric acid (GABA) benzodiazepine receptor agonists and antagonists10 thus prompting significant interest within the pharmaceutical sector.11Traditional synthetic methods to access pyrroloquinoxalines substituted at the 4-position are toxic and dangerous.12 Among the prevailing methodologies, the most prominent routes entail the reduction of 1-(2-nitrophenyl)pyrroles to their corresponding amino derivatives.13 Treatment of the amino group with acid chlorides to generate the corresponding acetamides, followed by intramolecular cyclisation under Bischler–Napieralski conditions, thus furnishing the core of the 4-substituted pyrrolo[1,2-a]quinoxaline structure.14 Another route encompasses the condensation of amino derivatives with aldehydes, followed by oxidation of the ensuing 4,5-dihydro pyrroloquinoxaline intermediate.15 Several methods utilising the modified Pictet–Spengler reaction followed by oxidation have been reported for constructing 4-arylpyrrolo[1,2-a]quinoxalines.16 Recently, a catalyst free electrochemical coupling of 2-isocyanobiphenyls and amines has been reported.17 The limitation with all of these methods is that the substituent is only efficient with aryl groups.
The use of radical insertion reactions within isonitriles has arisen as an effective alternative method for the formation of nitrogen-based heterocycles, including phenanthridines, indoles, quinolines, quinoxalines and isoquinolines.18 Addition of alkyl groups have been recently reported to isocyanides to form 4-substituted pyrrolo[1,2-a]quinoxalines.19–25 The methyl group is one of the most widespread functional groups in small biologically active molecules. Over the past decade, the methyl group has gained much interest in the pharmaceutical field due to its ability to increase a drug's binding affinity and potency.26 This phenomenon is known as the “magic-methyl effect” and is well established in medicinal chemistry.27 Motivated by the recent advancements in the creation of nitrogen heterocyclic derivatives using biaryl isocyanides as radical acceptors we were interested in exploring whether methyl radicals could be used in a similar approach to create 4-methyl substituted pyrrolo[1,2-a]quinoxalines.
Results and discussion
1-(2-Isocyanophenyl)-1H-pyrrole 4a was chosen as a model substrate for the radical addition of a methyl group. 4a was prepared in a four-step procedure according to the literature in good yields.19,28 A nucleophilic aromatic substitution between pyrrole and 2-fluoronitrobenzene, followed by a reduction using Pd/C and hydrazine hydrate furnished amine 2a. Formylation, with ethyl formate and formic acid, followed by dehydration with phosphorus oxychloride in the presence of diisopropylamine resulted in the required cyclisation precursor 4a in excellent yield (Scheme 1).29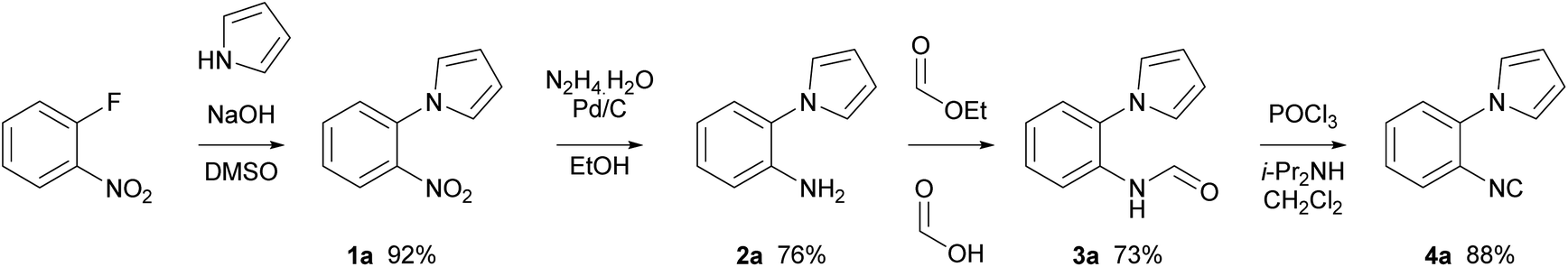 | ||
| Scheme 1 Synthesis of isocyanide 4a. | ||
With the cyclisation precursor in hand, reported methods for the generation of methyl radicals were screened. The application of the photoredox catalysis conditions described by Jamison, utilising visible light, led to the isolation of the desired pyrroloquinoxaline 5a in a yield of 30%, this was similar to that previously reported (Table 1, entry 1).21
|
|||
|---|---|---|---|
| Entry | Conditions | 5a (%) | 6a (%) |
| 1 | (1) PhI(OAc)2 (1 equiv.), MeCOOH (2 equiv.) | 30 | — |
| (2) [fac-Ir(ppy)3] (1 mol%), 26 W fluorescent bulb, DMF, rt | |||
| 2 | PhI(OAc)2 (2 equiv.) 2-nitropropane (1 equiv.), DBU (3 equiv.), CH3CN, rt | 45 | 10 |
| 3 | DTBP (3 equiv.), FeCl2 (20 mol%), Davephos (5 mol%), PhF, 130 °C | 48 | — |
| 4 | DCP (2 equiv.), KF (0.5 equiv.), t-BuOH, 120 °C | 67 | 4 |
| 5 | MeNHNH2·HCl (3 equiv.), K2CO3 (3 equiv.), eosin (5 mol%), 5 W blue LED, DMSO, air | 32 | 24 |
| 6 | MeSO2Na (4 equiv.), 24 W blue LED, EtOAc, rt, air | 33 | 26 |
| 7 | FeCl2 (0.2 equiv.), 30%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) H2O2 (3 equiv.), DMSO, rt H2O2 (3 equiv.), DMSO, rt |
— | 88 |
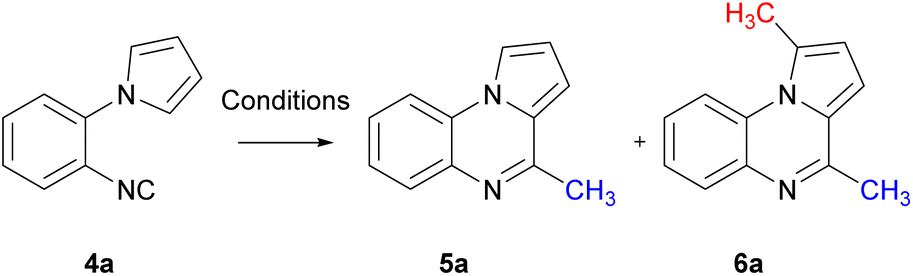
The use of phenyliodine diacetate in the presence of 2-nitropropane and DBU resulted in the desired pyrroloquinoxaline 5a in 45% yield. Notably, a minor amount of an unexpected bis-methylated pyrroloquinoxaline 6a was also obtained in a yield of 10% (entry 2);30 this is similar to the difluoromethyl radical addition reported previously in the 1 and 4 positions of a pyrroloquinoxaline.20 When the sequential methylation and intramolecular aromatisation conditions employed by Dai were applied to aryl isocyanide 4a, pyrroloquinoxaline 5a was isolated in an encouraging 48% yield with no bis-methyl adduct 6a observed (entry 3).31 Cyclisation with dicumyl peroxide (DCP) in the presence of KF in t-BuOH resulted in pyrroloquinoxaline 5a in 67% yield with 4% of 6a in the reaction mixture observed by 1H-NMR (entry 4).32 Upon exposure to blue LED light, the reaction of 4a with methylhydrazine hydrochloride in the presence of eosin B at room temperature, as well as blue LED-induced cyclisation in the absence of a photocatalyst with sodium methanesulfinate under aerobic conditions, yielded a mixture of pyrroloquinoxaline 5a and 6a (entries 5 and 6).33,34 Using Fenton reaction conditions reported by Zhang led exclusively to the formation of bis-methyl pyrroloquinoxaline 6a (entry 7).35
Encouraged by these results of selectively being able to favour mono-methyl radical addition over bis-methylation, these conditions were initially investigated. The use of DCP as a source of methyl radicals provided the greatest yield of pyrroloquinoxaline 5a, albeit with trace amounts of 6a. Conditions were further optimised for the methylation of 1-(2-isocyanophenyl)-1H-pyrrole 4a. Using the conditions reported by Xu et al. for varying time periods revealed that heating for 12 hours resulted in only pyrroloquinoxaline 5a (Table 2, entry 1).32 Prolonged heating led to increased pyrroloquinoxaline 5a yields accompanied by the formation of bis-methyl pyrroloquinoxaline 6a (entries 2–4). Although heating for 18 hours showed formation of bis-methyl pyrroloquinoxaline 6a, the desired product was formed in greatest yield. Altering the catalyst to CsF resulted in a decreased yield (entry 5) as observed previously.32
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (equiv.) | DCP equiv. | Time (h) | Temperature (°C) | 5a (%) | 6a (%) |
| a Slow addition of DCP over 4 h. | ||||||
| 1 | KF (0.5) | 2 | 12 | 120 | 52 | — |
| 2 | KF (0.5) | 2 | 18 | 120 | 74 | 3 |
| 3 | KF (0.5) | 2 | 24 | 120 | 67 | 4 |
| 4 | KF (0.5) | 2 | 48 | 120 | 65 | 12 |
| 5 | CsF (0.5) | 2 | 18 | 120 | 48 | 2 |
| 6 | KF (0.5) | 2 | 18 | 100 | 50 | — |
| 7 | KF (0.5) | 2 | 18 | 140 | 56 | 2 |
| 8 | KF (0.5) | 1.1 | 18 | 120 | 84 | — |
| 9 | KF (0.5) | 3 | 18 | 120 | 66 | 14 |
| 10a | KF (0.5) | 1.1 | 18 | 120 | 76 | — |
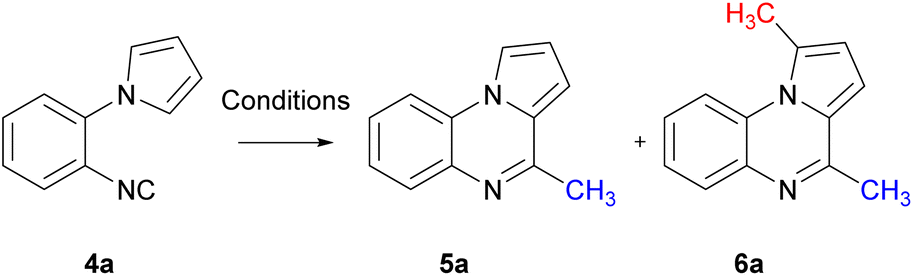
Given that the half-life of DCP is 10 hours at 117 °C and 1 hour at 137 °C,36 adjusting the temperature to 100 °C resulted in 50% of pyrroloquinoxaline 5a and recovered starting material 4a (entry 6). Raising the temperature to 140 °C led to a reduced mixture of the pyrroloquinoxalines 5a and 6a (entry 7). To curtail the bis-methylated product formation, the amount of DCP was explored. Using 1.1 equiv. of the initiator resulted in exclusive formation of 5a in 84% yield (entry 8). Conversely of excess DCP, 3 equiv., increased the formation of the bis-product 6a at the expense of 5a (entry 9). Finally, slow addition of the radical initiator, DCP, over 4 hours resulted in a slightly lower yield of 76% of 5a.
With optimised conditions for the methylation concomitant pyrroloquinoxaline formation, investigation of the cyclisation with analogous pyrrole and five-membered heterocycle substituted aryl isocyanides was conducted. The synthesis of arylamines involved three different approaches (Scheme 2). In the first method, arylamines were synthesised by nucleophilic aromatic substitution of nitrogen-containing ring systems with 2-fluoronitrobenzene. Subsequent reduction of the nitro intermediates (1b–e) using Pd/C and hydrazine hydrate yielded amines (2b–e) in yields ranging from 65–91%. Conversely, the Clauson–Kass reaction of electron-rich nitroanilines with 2,5-dimethoxytetrahydrofuran in acetic acid,8 followed by reduction using BiCl3–NaBH4 resulted in amines 2f and 2g. The final approach utilised the Suzuki–Miyaura reaction of aryl iodide and commercially available boronic acids to give the corresponding amines 2h and 2i in good yields.37
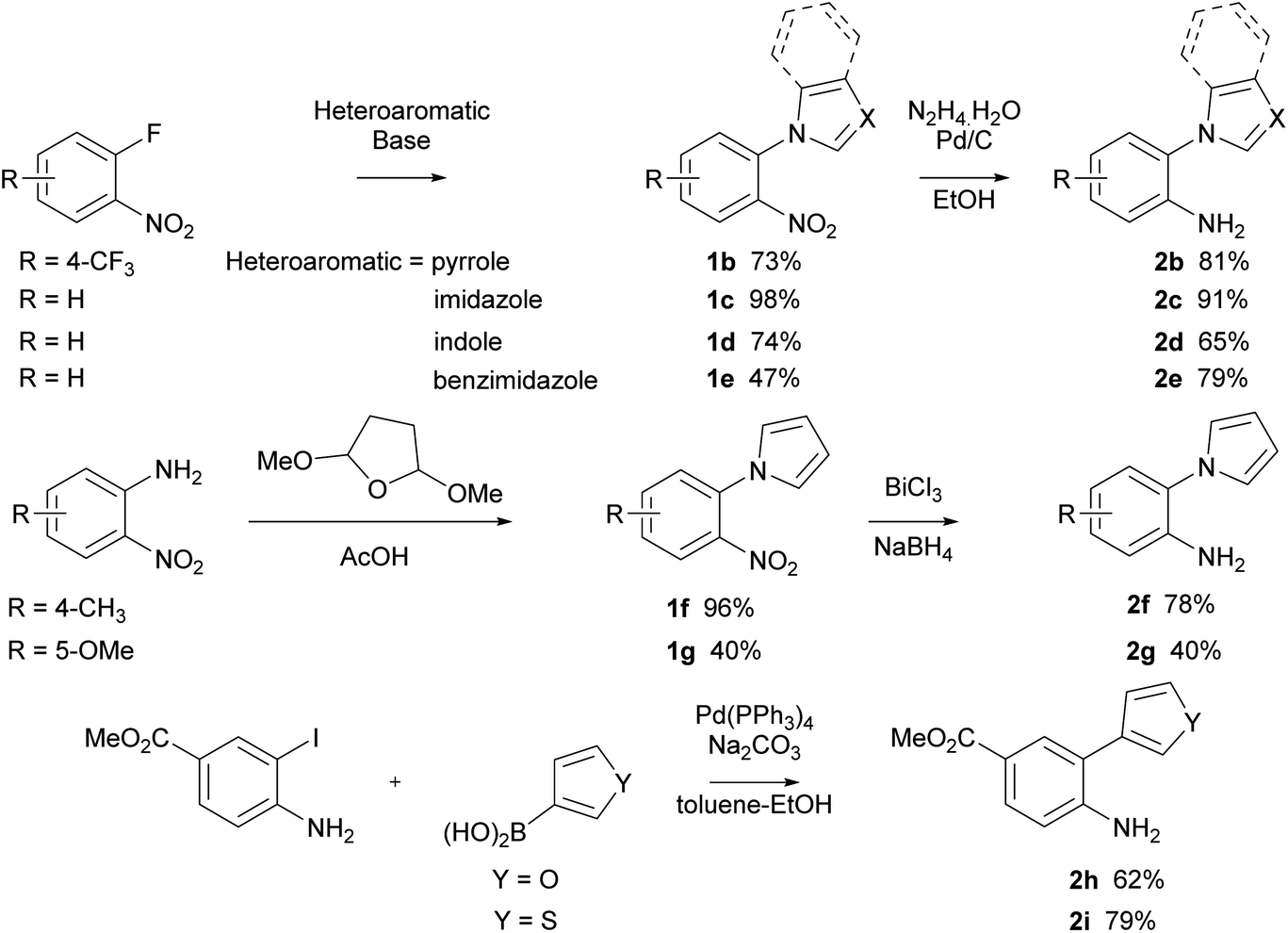 | ||
| Scheme 2 Synthesis of biaryl amines 2. | ||
Subsequently, all the arylamines underwent a two-step conversion to the respective isocyanides. Formylation with formic acetic anhydride followed by dehydration using phosphorus oxychloride and diisopropylamine furnished isocyanides 4 in good yields (Scheme 3).
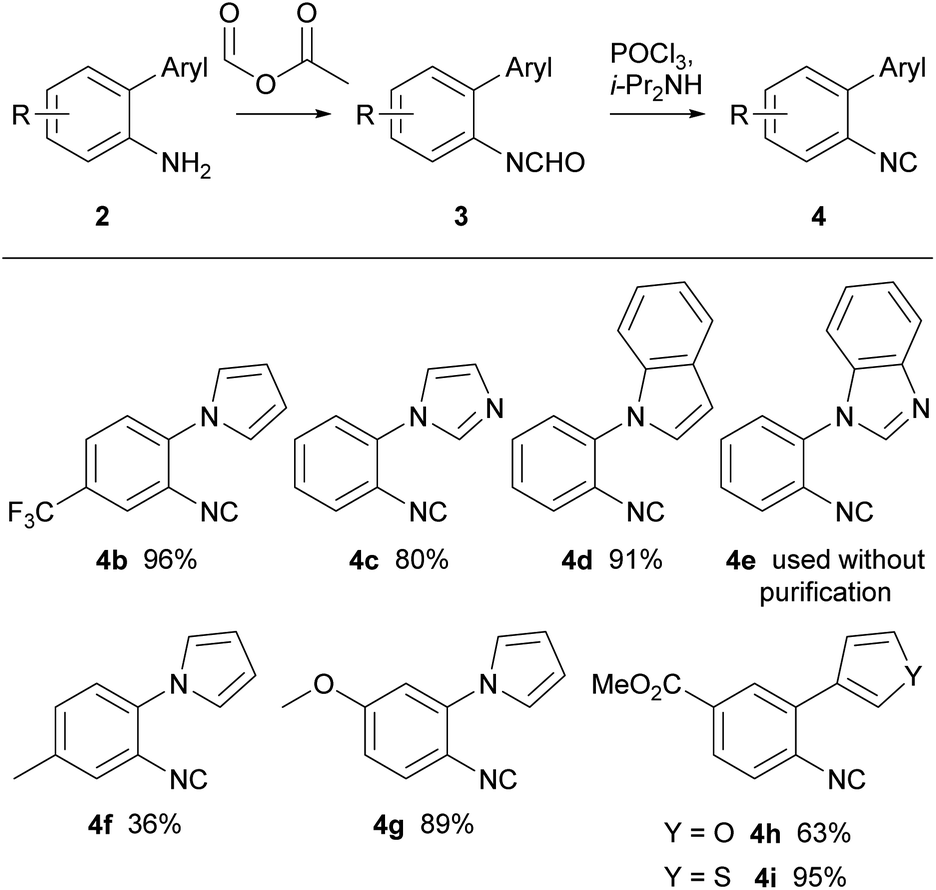 | ||
| Scheme 3 Synthesis of isocyanides 4. | ||
With the corresponding isocyanides 4 prepared, optimised mono-methylation/radical cyclisation conditions were employed using DCP and KF. The resulting 6-endo cyclisation gave a range of tri- and tetracyclic structures bearing a methyl group in the 2-position of the quinoline ring (Scheme 4).
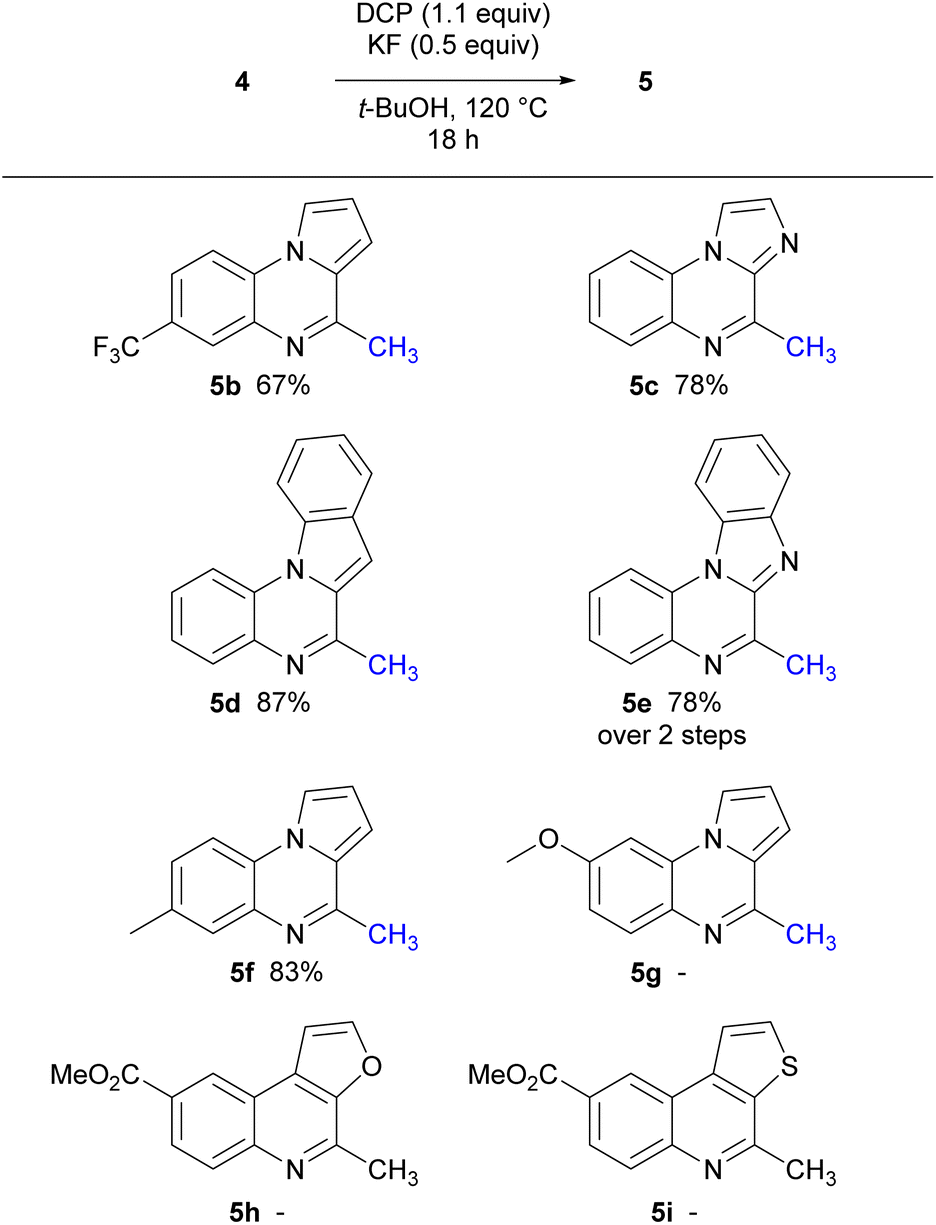 | ||
| Scheme 4 Cyclisation using DCP to give tricyclic and tetracyclic derivatives. | ||
Precursors, 4b, 4f, and 4g, were prepared to investigate the electronic effects of the cyclisation precursor and from the results attained it was evident cyclisation was favoured for most of the isocyanides apart from electron-rich isocyanide 4g. Following this preliminary screen, isocyanides 4c–e were reacted and successfully underwent 6-endo cyclisation to tricyclic and tetracyclic congeners. The slightly higher yields for 5d and 5e could be explained by the extended conjugation in comparison to the tricyclic counterparts. Furthermore, we explored oxygen and sulfur derivatives, 5h and 5i, although no discernible reactions were observed. The lack of reactivity is attributed to the presence of lone pairs of electrons on the heteroatom in thiophene and furan, which might impede the closure of the intermediate radical, thus explaining the absence of the expected ring-closing reactions.
As previously mentioned, bis-methylation was observed with isocyanide 4a when Fenton conditions were subjected to the reaction conditions (Table 1, entry 7). Employing Fenton reaction conditions, isocyanides 4a–c and 4f–h were treated, yielding bis-methyl pyrroloquinoxalines 6a, 6b and 6f in very good yields (Scheme 5). No mono-methylated products were observed in any case.
 | ||
| Scheme 5 Cyclisation using Fenton conditions for bis-methylation. | ||
Notably, decreasing the amount of DMSO used in the reaction below 10 equiv. resulted in lower yields. Interestingly, the imidazole isocyanide 4c exclusively produced 4-methylimidazo[1,2-a]quinoxaline 5c, even with prolonged reaction times and increased amount of radical initiator, possibly due to stabilisation of the resulting radical by the lone pair of electrons on the adjacent nitrogen atom. To date, only one example in the literature similar to electron rich methoxy isocyanide 4g that has undergone radical cyclisation reported,21 however the methoxy group is in the 7-position. In our hands, isocyanide 4g displayed a lack of reactivity, possibly attributed to the presence of the electron-rich methoxy group, which seems to render the isocyanide nucleophilic.38 This trend was similarly observed with the furan and thiophene derivatives.
The mechanism for the mono- and bis-methylation was investigated for the in situ generation of methyl radicals. Initially, an inhibition reaction was carried out in the presence of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) under standard reaction conditions. A complete suppression of methylation concomitant cyclisation was observed, and formation of a methyl-TEMPO adduct was observed by GC-MS. This indicates the methyl radical species is involved and initiates the reaction and is consistent with previous studies.39 Based on this data, a plausible mechanism for the reaction is proposed (Scheme 6). Initiation begins with the homolysis of DCP, yielding acetophenone and a methyl radical via β-cleavage.32 The methyl radical adds to the isocyanide to generate the α-imidoyl radical 7 which cyclises onto the aromatic ring to give radical 8. H-abstraction by the cumyloxy or methyl radical results in the mono-methylated product 5a. The bis-methylated pyrroloquinoxaline 6a was observed when excess DCP was used, however was more predominant with Fenton conditions.
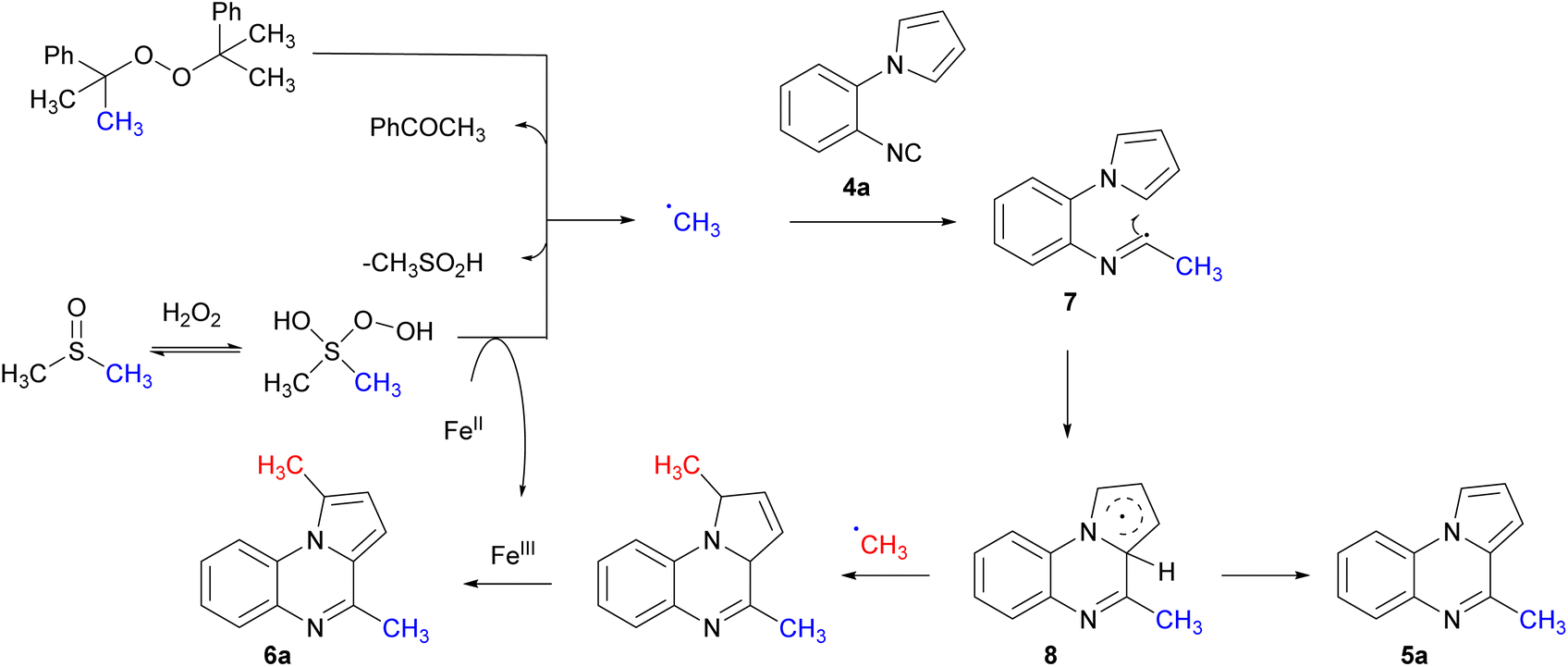 | ||
| Scheme 6 Proposed mechanism for methyl radical addition to form mono- or bis-methylated pyrroloquinoxaline. | ||
When mono-methylated pyrroloquinoxaline 5a was subjected to FeCl2, in H2O2 and DMSO only 17% of the bis-methylated pyrroloquinoxaline 6a was isolated. Wan et al. observed a radical addition to form a bis-alkylated pyrroloquinoxaline.20 This is proposed to occur via a direct C–H radical addition to the α-position of the pyrrole ring followed by a second radical addition to the isocyanide to form the α-imidoyl radical. This undergoes cyclisation followed by aromatisation to form the bis-alkylated pyrroloquinoxaline 6a. This study features radical addition to produce the mono-alkylated product with no bis-alkylation product. A second radical addition was not efficient from the pyrroloquinoxaline 5a. Thus, an alternative mechanism is proposed to form the bis-methylated pyrroloquinoxaline 6a. Initial addition of the methyl radical followed by cyclisation. The resulting π-radical 8 is stabilised by resonance and allows for another methyl radical to trap the radical and yield the bis-methylated pyrrolines; this undergoes aromatisation to the pyrrole in the presence of iron.40
Marinoquinoline A is a methylated 3H-pyrrolo[2,3-c]quinoline natural product isolated from the marine gliding bacteria Rapidithrix thailandica TISTR 1742 by Plubrukarn in 2008.41 and displays inhibition against acetylcholinesterase (IC50 = 4.9 μM against Torpedo californica AChE).42 To demonstrate the practical utility of the conditions developed herein, marinoquinoline A was successfully synthesised in five steps from commercially available starting materials (Scheme 7). Isocyanide 4j was prepared through a Suzuki–Miyaura coupling between 2-iodoaniline and 3-bromopyrrole-1-carboxylic acid tert-butyl ester in good yield, followed by formylation with formic acetic anhydride and dehydration with phosphorous oxychloride in the presence of diisopropylamine. The key step was the radical methylation of 4j using DCP to provide the 3H-pyrrolo[2,3-c]quinoline core structure 5j in excellent yield. Consequent deprotection of the N-Boc group using TFA in CH2Cl2 resulted in marinoquinoline A in excellent yield.
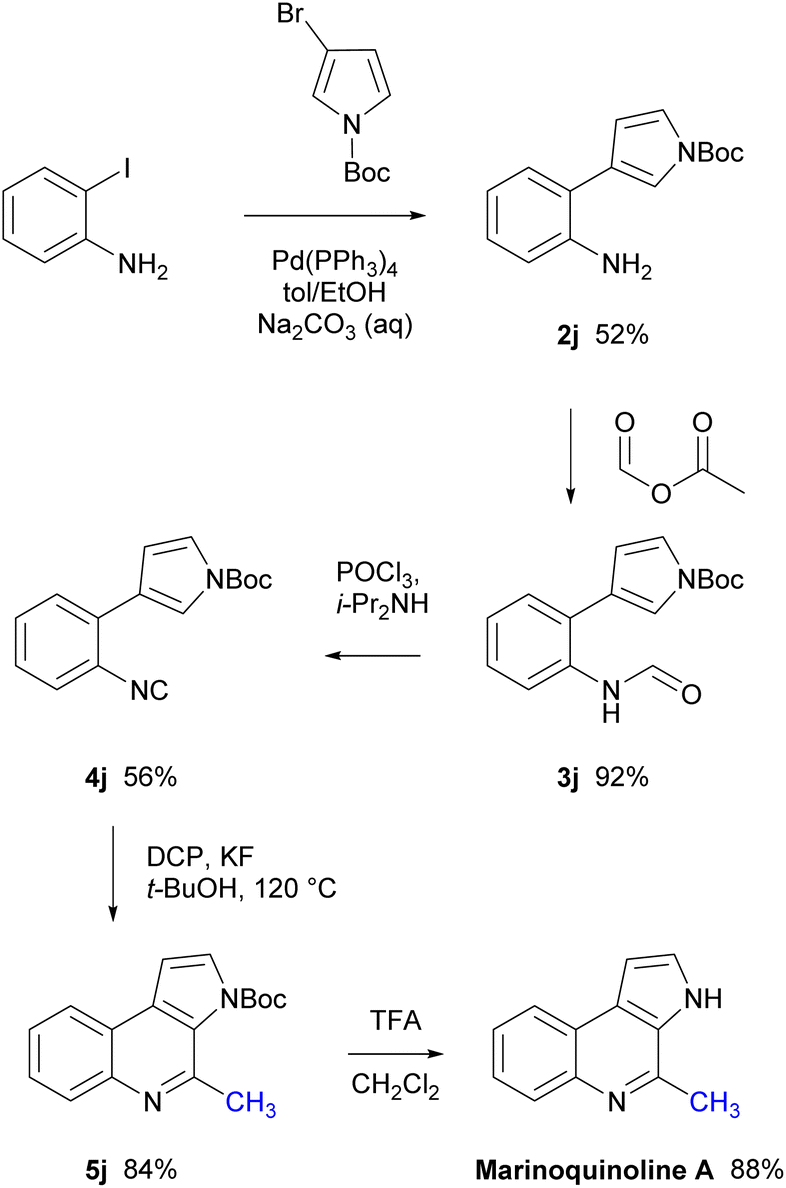 | ||
| Scheme 7 Total synthesis of marinoquinoline A. | ||
Conclusions
To summarise, an effective approach has been tuned to yield selective mono- and bis-methylated pyrroloquinoxaline derivatives, contingent on the specific reaction conditions employed. Insights from mechanistic studies indicate that methylation at the 1-position occurs subsequent to the initial addition of the methyl radical to the α-imidoyl radical, rather than via direct addition to a double bond. The methodology has been applied to the synthesis of the natural product marinoquinoline A in very good yield overall and provides a route to novel methylated N heterocycles. Exploration of the reaction mechanism, biological activity, further applications of the methodology and functionalisation are currently underway.43Experimental
General procedure for synthesis of compound 5
A mixture of isocyanide (1 equiv.), KF (0.5 equiv.), DCP (1.1 equiv.) and t-butanol (0.08 M) was heated at 120 °C in a sealed tube for 18 h. The mixture was allowed cool and concentrated under reduced pressure. The residue was purified by column chromatography (99:1 Hex:EtOAc) give mono-methylated products.
General procedure for synthesis of compound 6
A mixture of isocyanide (1 equiv.), FeCl3 (0.2 equiv.), 30% w/w H2O2 (3 equiv.) and DMSO (0.083 M) was stirred at 25 °C under inert atmosphere for 6 h in a sealed tube. The mixture was extracted with ethyl acetate (3 × 5 mL) and water (3 × 5 mL), dried over MgSO4, and concentrated under reduced pressure. The residue was purified by column chromatography (99:1 Hex:EtOAc) give bis-methylated products.
Author contributions
MD: investigation, methodology, writing – original draft, writing – review & editing. NG: methodology, writing – review & editing. VT: methodology, writing – review & editing. ZR: methodology, writing – review & editing. DS: methodology, supervision, writing – review & editing. BP: conceptualization, funding acquisition, supervision, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank London Metropolitan University and the Royal Society of Chemistry Research Fund grant (R20-3293) for funding. We thank the EPSRC UK National Mass Spectrometry Facility at the Swansea University for high resolution mass spectrometry analyses.Notes and references
- A. A. Culinin, L. N. Islamova and G. M. Fazleeva, Chem. Heterocycl. Compd., 2019, 55, 384 Search PubMed.
- H. Aiping and M. Chen, Mini-Rev. Med. Chem., 2013, 13, 607 CrossRef.
- C. Escude, C. H. Nguyen, S. Kukreti, Y. Janin, J. S. Sun, E. Bisagni, T. Garestier and C. Helene, Proc. Natl. Acad. Sci. U.S.A., 1998, 95, 3591 CrossRef CAS PubMed; L. W. Deady, A. J. Kaye, G. J. Finlay, B. C. Baguley and W. A. Denny, J. Med. Chem., 1997, 40, 2040 CrossRef; A. Huang and C. Ma, Mini-Rev. Med. Chem., 2013, 13, 607 CrossRef; F. A. R. Rodrigues, I. D. S. Bomfim, B. C. Cavalcanti, C. D. O. Pessoa, J. L. Wardell, S. M. S. V. Wardell, A. C. Pinheiro, C. R. Kaiser, T. C. M. Nogueira, J. N. Low, L. R. Gomes and M. V. N. D. Souza, Bioorg. Med. Chem. Lett., 2014, 24, 934 CrossRef PubMed.
- B. Parrino, A. Carbone, C. Ciancimino, V. Spano, A. Montalbano, P. Barraja, G. Cirrincione, P. Diana, C. Sissi, M. Palumbo, O. Pinato, M. Pennati, G. Beretta, M. Folini, P. Matyus, B. Balogh and N. Zaffaroni, Eur. J. Med. Chem., 2015, 94, 149 CrossRef CAS PubMed; V. Desplat, A. Geneste, M. A. Begorre and J. Guillon, J. Enzyme Inhib. Med. Chem., 2008, 23, 648 CrossRef PubMed; S. Alleca, P. Corona, M. Lorigo, G. Paglietti, R. Loddo, V. Mascia, B. Busonera and P. La Colla, Farmaco, 2003, 58, 639 CrossRef PubMed; W. Lv, B. Budke, M. Pawlowski, P. P. Connell and A. P. Kozikowski, J. Med. Chem., 2016, 59, 4511 CrossRef PubMed; F. Aiello, G. Carullo, F. Giordano, E. Spina, A. Nigro, A. Garofalo, S. Tassini, G. Costantino, P. Vincetti, A. Bruno and M. Radi, ChemMedChem, 2017, 12, 1279 CrossRef; W. You, D. Rotili, T.-M. Li, C. Kambach, M. Meleshin, M. Schutkowski, K. F. Chua, A. Mai and C. Steegborn, Angew. Chem., Int. Ed., 2017, 56, 1007 CrossRef.
- J. Guillon, A. Cohen, N. M. Gueddouda, R. N. Das, S. Moreau, L. Ronga, S. Savrimoutou, L. Basmaciyan, A. Monnier, M. Monget, S. Rubio, T. Garnerin, N. Azas, J.-L. Mergny, C. Mullie and P. Sonnet, J. Enzyme Inhib. Med. Chem., 2017, 32, 547 CrossRef CAS PubMed; L. Ronga, M. Del Favero, A. Cohen, C. Soum, P. Le Pape, S. Savrimoutou, N. Pinaud, C. Mullie, S. Daulouede, P. Vincendeau, N. Farvacques, P. Agnamey, F. Pagniez, S. Hutter, N. Azas, P. Sonnet, J. Guillon and J. Eur, J. Med. Chem., 2014, 81, 378 Search PubMed; J. Guillon, E. Mouray, S. Moreau, C. Mullie, I. Forfar, V. Desplat, S. Belisle-Fabre, N. Pinaud, F. Ravanello, A. Le-Naour, J. M. Leger, G. Gosmann, C. Jarry, G. Deleris, P. Sonnet and P. Grellier, Eur. J. Med. Chem., 2011, 46, 2310 CrossRef PubMed.
- V. Desplat, S. Moreau, A. Gay, S. B. Fabre, D. Thiolat, S. Massip, G. Macky, F. Godde, D. Mossalayi, C. Jarry and J. Guillon, J. Enzyme Inhib. Med. Chem., 2010, 25, 204 CrossRef CAS PubMed; V. Desplat, S. Moreau, S. Belisle-Fabre, D. Thiolat, J. Uranga, R. Lucas, L. de Moor, S. Massip, C. Jarry, D. M. Mossalayi, P. Sonnet, G. Déléris and J. Guillon, J. Enzyme Inhib. Med. Chem., 2011, 26, 657 CrossRef PubMed.
- J. Guillon, M. L. Borgne, C. Rimbault, S. Moreau, S. Savrimoutou, N. Pinaud, S. Baratin, M. Marchivie, S. Roche, A. Bollacke, A. Pecci, L. Alvarez, V. Desplat and J. Jose, Eur. J. Med. Chem., 2013, 65, 205 CrossRef CAS PubMed.
- J. Guillon, P. Dallemagne, B. Pfeiffer, P. Renard, D. Manechez, A. Kervran and S. Rault, Eur. J. Med. Chem., 1998, 33, 293 CrossRef CAS.
- G. Campiani, E. Morelli, S. Gemma, V. Nacci, S. Butini, M. Hamon, E. Novellino, G. Greco, A. Cagnotto, M. Goegan, L. Cervo, F. D. Valle, C. Fracasso, S. Caccia and T. Mennini, J. Med. Chem., 1999, 42, 4362 CrossRef CAS.
- E. J. Jacobsen, L. S. Stelzer, K. L. Belonga, D. B. Carter, W. B. Im, V. H. Sethy, A. H. Tang, P. F. Von Voigtlander and J. D. Petke, J. Med. Chem., 1996, 39, 3820 CrossRef CAS PubMed; V. Colotta, L. Cecchi, D. Catarzi, G. Filacchioini, C. Martini, P. Tacchi and A. Lucacchini, Eur. J. Med. Chem., 1995, 30, 133 CrossRef.
- A. A. Kalinin and V. A. Mamedov, Chem. Heterocycl. Compd., 2011, 46, 1423 CrossRef CAS.
- L. Quan, W. Wang and M. Liu, Asian J. Org. Chem., 2023, e202300277 Search PubMed.
- C. Wang, Y. Li, R. Guo, J. Tian, C. Tao, B. Cheng, H. Wang, J. Zhang and H. Zhai, Asian J. Org. Chem., 2015, 4, 866 CrossRef CAS; C. Xie, L. Feng, W. Li, X. Ma, Y. Liu and C. Ma, Org. Biomol. Chem., 2016, 14, 8529 RSC.
- J. Guillon, I. Forfar, M. Mamani-Matsuda, V. Desplat, M. Saliege, D. Thiolat, S. Massip, A. Tabourier, J.-M. Léger and B. Dufaure, Bioorg. Med. Chem., 2007, 15, 194 CrossRef CAS PubMed; G. W. H. Cheeseman and B. Tuck, J. Chem. Soc., Chem., 1966, 852 Search PubMed.
- A. Kamal, K. S. Babu, J. Kovvuri, V. Manasa, A. Ravikumar and A. Alarifi, Tetrahedron Lett., 2015, 56, 7012 CrossRef CAS; A. Preetam and M. Nath, RSC Adv., 2015, 5, 21843 RSC; C. Wang, Y. Li, J. Zhao, B. Cheng, H. Wang and H. Zhai, Tetrahedron Lett., 2016, 57, 3908 CrossRef.
- A. K. Verma, R. R. Jha, V. Kasi Sankar, T. Aggarwal, R. Singh and R. Chandra, Eur. J. Org Chem., 2011, 2011, 6998 CrossRef CAS; H.-R. Huo, X.-Y. Tang and Y. F. Gong, Synthesis, 2018, 50, 2727 CrossRef; T. Krishna, T. R. Reddy, E. Laxminarayana and D. Kalita, ChemistrySelect, 2019, 4, 250 CrossRef; P. N. Allan, M. I. Ostrowska and B. Patel, Synlett, 2019, 30, 2148 CrossRef.
- B. K. Malviya, K. Singh, P. K. Jaiswal, M. Karnatak, V. P. Verma, S. S. Badsara and S. Sharma, New J. Chem., 2021, 45, 6367 RSC.
- B. Zhang and A. Studer, Chem. Soc. Rev., 2015, 44, 3505 RSC.
- B. Patel and S. T. Hilton, Synlett, 2015, 26, 79 CAS.
- W. Wan, X. Xu, Y. Chen, H. Jiang, Y. Wang, H. Deng and J. Hao, Eur. J. Org Chem., 2017, 2017, 3145 CrossRef CAS.
- Z. He, M. Bae, J. Wu and T. F. Jamison, Angew. Chem., Int., 2014, 53, 14451 CrossRef CAS.
- S. Rohe, T. McCallum, A. O. Morris and L. Barriault, J. Org. Chem., 2018, 83, 10015 CrossRef CAS.
- Y. Wang, J. Wang, G. X. Li, G. He and G. Chen, Org. Lett., 2017, 19, 1442 CrossRef CAS.
- W. B. Qin, W. Xiong, X. Li, J. Y. Chen, L. T. Lin, H. N. Wong and G. K. Liu, J. Org. Chem., 2020, 85, 10479 CrossRef CAS PubMed.
- H. B. Ye, X. Y. Zhou, L. Li, X. K. He and J. Xuan, Org. Lett., 2022, 24, 6018 CrossRef CAS.
- J. Gui, Q. Zhou, C. Pan, Y. Yabe, A. C. Burns, M. R. Collins, M. A. Ornelas, Y. Ishihara and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 4853 CrossRef CAS; D. A. DiRocco, K. Dykstra, S. Krska, P. Vachal, D. V. Conway and M. Tudge, Angew. Chem., Int. Ed., 2014, 53, 4802 CrossRef PubMed.
- H. Schönherr and T. Cernak, Angew. Chem., Int. Ed., 2013, 52, 12256 CrossRef CAS PubMed; D. Aynetdinova, M. C. Callens, H. B. Hicks, C. Y. Poh, B. D. Shennan, A. M. Boyd, Z. H. Lim, J. A. Leitch and D. J. Dixon, Chem. Soc. Rev., 2021, 50, 5517 RSC.
- B. Patel, G. Saviolaki, C. Ayats, M. A. E. Garcia, T. Kapadia and S. T. Hilton, RSC Adv., 2014, 4, 18930 RSC.
- R. Obrecht, R. Herrmann and I. Ugi, Synthesis, 1985, 4, 400 CrossRef.
- S. C. Lu, H. S. Li, Y. L. Gong, S. P. Zhang, J. G. Zhang and S. Xu, J. Org. Chem., 2018, 83, 15415 CrossRef CAS.
- Q. Dai, J. T. Yu, X. Feng, Y. Jiang, H. Yang and J. Cheng, Adv. Synth. Catal., 2014, 356, 3341 CrossRef CAS.
- Z. Xu, C. Yan and Z.-Q. Liu, Org. Lett., 2014, 16, 5670 CrossRef CAS PubMed.
- T. Xiao, L. Li, G. Lin, Q. Wang, P. Zhang, Z. W. Mao and L. Zhou, Green Chem., 2014, 16, 2418 RSC.
- P. Natarajan, New J. Chem., 2022, 46, 22862 RSC.
- R. Zhang, X. Shi, Q. Yan, Z. Li, Z. Wang, H. Yu, X. Wang, J. Qi and M. Jiang, RSC Adv., 2017, 7, 38830 RSC.
- E. Gitzhofer, B. Vileno, M. Bouquey and D. Chan-Seng, Polym. Chem., 2023, 14, 934 RSC.
- S. Martina, V. Enkelmann, G. Wegner and A. D. Schlüter, Synthesis, 1991, 1991, 613 CrossRef.
- L. Luo, H. Li, J. Liu, Y. Zhou, L. Dong, Y.-C. Xiao and F.-E. Chen, RSC Adv., 2020, 10, 29257 RSC.
- G. Ma, W. Wan, J. Li, Q. Hu, H. Jiang, S. Zhu, J. Wang and J. Hao, Chem. Commun., 2014, 50, 9749 RSC; M. Tobisu, K. Koh, T. Furukawa and N. Chatani, Angew. Chem., Int. Ed., 2012, 51, 11363 CrossRef CAS PubMed; Q. Wang, X. Dong, T. Xiao and L. Zhou, Org. Lett., 2013, 15, 4846 CrossRef PubMed.
- A. Bunrit, S. Sawadjoon, S. Tsupova, P. J. Sjöberg and J. S. Samec, J. Org. Chem., 2016, 81, 1450 CrossRef CAS PubMed.
- Y. Sangnoi, O. Sakulkeo, S. Yuenyongsawad, A. Kanjana-opas, K. Ingkaninan, A. Plubrukarn and K. Suwanborirux, Mar. Drugs, 2008, 6, 578 CrossRef CAS.
- S. V. Stoddard, M. T. Hamann and R. M. Wadkins, Mar. Drugs, 2014, 12, 2114 CrossRef.
- K. T. Le, P. V. Duong, Y. N. Dong, H. T. Nguyen, D. D. Nguyen, V. H. Luu, H. X. Le and T. T. Nguyen, Tetrahedron Lett., 2023, 127, 154669 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and copies of 1H and 13C for novel compounds and cyclised product. See DOI: https://doi.org/10.1039/d3ra05952a |
| This journal is © The Royal Society of Chemistry 2023 |