
It's ok to be outnumbered – sub-stoichiometric modulation of homomeric protein complexes
Yoana N.
Dimitrova
a,
Jemy A.
Gutierrez
b and
Kim
Huard
 *a
*a
aGenentech, 1 DNA Way, South San Francisco, CA 94080, USA. E-mail: huardhak@gene.com
bPfizer, 1 Portland Street, Cambridge, MA 02139, USA
First published on 27th October 2022
Abstract
An arsenal of molecular tools with increasingly diversified mechanisms of action is being developed by the scientific community to enable biological interrogation and pharmaceutical modulation of targets and pathways of ever increasing complexity. While most small molecules interact with the target of interest in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 relationship, a noteworthy number of recent examples were reported to bind in a sub-stoichiometric manner to a homomeric protein complex. This approach requires molecular understanding of the physiologically relevant protein assemblies and in-depth characterization of the compound's mechanism of action. The recent literature examples summarized here were selected to illustrate methods used to identify and characterize molecules with such mechanisms. The concept of one small molecule targeting a homomeric protein assembly is not new but the subject deserves renewed inspection in light of emerging technologies and increasingly diverse target biology, to ensure relevant in vitro systems are used and valuable compounds with potentially novel sub-stoichiometric mechanisms of action aren't overlooked.
:1 relationship, a noteworthy number of recent examples were reported to bind in a sub-stoichiometric manner to a homomeric protein complex. This approach requires molecular understanding of the physiologically relevant protein assemblies and in-depth characterization of the compound's mechanism of action. The recent literature examples summarized here were selected to illustrate methods used to identify and characterize molecules with such mechanisms. The concept of one small molecule targeting a homomeric protein assembly is not new but the subject deserves renewed inspection in light of emerging technologies and increasingly diverse target biology, to ensure relevant in vitro systems are used and valuable compounds with potentially novel sub-stoichiometric mechanisms of action aren't overlooked.
Introduction
In early stages of drug discovery programs, a strategy is put in place to identify chemical matter that can likely be developed into leads, and potentially into a clinical candidate. The definition of a promising starting point is relative to the desired profile of the drug candidate, which in turn is defined by the targeted disease indication, acceptable route of administration, and biological target or pathway. The nature of biological targets and pathways of interest to the drug discovery community is more diverse than ever, and an ever-expanding arsenal of modalities, including small molecules, monoclonal antibodies and antisense oligonucleotides, is being developed to address the associated needs and challenges.1,2 While we acknowledge an increasing overlap in applications that use different modalities, small molecules remain the preferred approach in scenarios tackling intracellular targets for example, or when oral administration or brain penetration is desired. Our understanding of the outer limits of chemical space in which orally absorbed and cell permeable compounds may be discovered has improved in recent years. With many reported examples, it is now well established that cellular permeability and oral absorption can be achieved by molecules weighting up to ∼1000 Da, which is how small molecules are defined in this work.3–8In vitro approaches to identify small molecules in drug discovery efforts span a range of complexity from simple target-based assays to more convoluted phenotypic models, and have been recently reviewed.9 Target-based screenings are typically set up to only identify hits with specific mechanisms of action (MoAs). While more complex cellular systems can capture various MoAs, identifying the exact nature of the mechanism and testing the tractability of the approach can prove challenging. Beyond direct modulation of receptors, enzymes, ion channels and transporters, examples of MoAs that have recently been reported for small molecules include disruption or stabilization of protein complexes,9 protein degradation,10–12 splicing modulation13–17 and translational stalling.18–20 Unless these MoAs are deliberately targeted during screening, highly valuable hits may be missed from the use of an overly simplified screening system. The merits and challenges of various screening approaches have been reviewed and debated,21 and ultimately, screening methods should be considered and selected based on their sensitivity, robustness and the appropriate balance between practicality (i.e., simplicity, throughput) and biological relevance. This balance can be program-specific and often depends on the level of understanding and knowledge about the target and the disease biology. It is worth noting that in some cases such as membrane proteins, high quality target-based assays are sometimes not readily available for technical reasons, and a more complex but reliable cellular system may be preferred even when a specific MoA is desired.
Even when considering newly characterized MoAs, most small molecules are believed to bind to their target in a 1:1 stoichiometry (or 1:1:1 when forming a ternary complex such as in the case of degraders and molecular glues). However, a growing number of examples report detailed characterization of small molecules binding to a homomeric complex of the targeted protein, leading to the desired biological response without achieving 1:1 stoichiometry between the small molecule and each protein in the homomeric complex. In this context, the small molecules are defined as sub-stoichiometric modulators of their respective targets. In these cases, the activity of the multimeric protein assembly is regulated by mechanisms that can be described by the biochemical principles of allostery, cooperativity and oligomerization.22,23 Building on early work studying biological modulation of aspartate transcarbamoylase,24–26 threonine deaminase27,28 and hemoglobin,29 these biochemical principles are now considered fundamental. However, small molecules acting in a sub-stoichiometric manner by affecting the quaternary structure of a homomeric complex can only be identified from a screen where the relevant complex is formed, again highlighting the importance of understanding the biology of the target at a molecular level in the selection of appropriate screening methods and protein constructs.30 Furthermore, small molecules that act cooperatively on a multimeric complex may have an initial in vitro profile similar to compounds interacting with the target in a non-specific manner such as aggregators,31 and be mistakenly classified as such during hit triage. As discussed by experts in the field,32 it is worth emphasizing the importance of orthogonal assays and a thorough characterization of small molecules' MoA before investing significant chemistry efforts or reporting potentially misassigned in vitro activity in the public domain.33
There are aspects to working with homomeric protein complexes, defined here as a protein complex assembled by multiple copies of a single type of polypeptide chain, that may enhance their druggability as pharmacological targets. Small molecules with sub-stoichiometric effects have the potential to achieve cellular modulation of proteins that are typically considered undruggable due to high cellular concentrations. Protein assemblies may also form functionally relevant binding sites that could expand options for targeting with small molecules beyond the ones found on the protomer, and these allosteric sites could provide strategic advantages in the quest for improved potency and selectivity. With these potential advantages in mind, and to make sure valuable compounds aren't overlooked during hit triage, we herein highlight recently published examples of small molecules acting in a sub-stoichiometric manner on a multimeric complex of the targeted protein. While it isn't our goal to provide a comprehensive review, we selected examples from recent literature that illustrate various approaches used to identify hits and to characterize their MoA. We also highlight aspects of hit identification that are particular to this MoA and offer our perspective on their impact on drug discovery programs in a more general sense. Even though small molecule degraders are typically thought to act catalytically, they are reported to interact with their target in a stoichiometric manner, have been recently reviewed, and will not be discussed here.34 Emerging hypotheses suggesting that biological condensates could lead to non-stoichiometric molecular interactions are also beyond the scope of this article.35 The concept of one small molecule targeting a homomeric protein assembly is not new, with one of the earliest examples from the 1990s being the clinically approved HIV protease inhibitor ritonavir.36,37 While pursuing chemical matter with this MoA may in certain cases be more complex than some other approaches, we see value in bringing attention to some of its implications and subtleties to help ensure small molecules acting in a sub-stoichiometric manner continue to contribute to the expanding molecular tool set available to study new and diverse biological targets.
Selected examples of small molecules reported to act sub-stoichiometrically on multimeric complexes of the protein target
Selected examples targeting a complex with defined stoichiometry
The following examples were selected from recent literature to exemplify a number of biochemical concepts as well as methods used to screen and determine the MoA of small molecules. This collection illustrates that small molecules acting in a sub-stoichiometric manner can target various gene families and are relevant in multiple disease areas.TNF-α monomers associate into a functional homotrimer with a 3-fold axis of symmetry forming a bell-shaped structure, with receptor binding sites located within the grooves formed between each of two monomers.54 Receptors TNFR1 and TNFR2 form homodimers in the absence of ligand but quickly assemble into trimers upon TNF-α binding.55 In 2019, O'Connell et al. reported a series of small molecule TNF-α inhibitors that bind at the center of the homotrimer and stabilize an asymmetric form of the protein resulting in non-productive receptor binding and impaired downstream signalling.51 The hit finding efforts started with a fragment screen using a surface plasmon resonance (SPR) binding assay and identified a hit, UCB-6876, which showed slow binding kinetics (KD = 22 μM; kon = 923 M−1 s−1; koff = 0.02 s−1), unusual for its small size. The crystal structure showed fragment binding with 1:3 stoichiometry, where one compound is trapped at the center of three TNF-α monomers forming an asymmetric trimer. Double electron–electron resonance (DEER) analysis supported the idea that the fragment could stabilize an asymmetric trimer conformation, and that the slow binding kinetics may be explained by the low abundance of this transient state as well as the deep buried location of the small molecule binding site.56 Using a mass spectrometry (MS)-based monomer subunit exchange assay they showed that the compound could stabilize the asymmetric trimer by preventing subunit exchange between human and mouse TNF-α, a process that normally occurs at low physiological concentrations.51 Compound treatment disrupted normal receptor binding as shown in their analytical size-exclusion chromatography (SEC) study where treatment with the more potent analog UCB-5307 caused a shift in the receptor population that bound the TNF-α trimer from the active three subunits to the inactive two, thereby acting as an allosteric inhibitor of TNF-receptor interaction. These findings are supported by more recent structural biology work from the same group including the use of a monoclonal antibody tool selective for the compound-stabilised asymmetric TNF-α trimer.57,58 Using UCB-9260, an analog optimized for cellular and in vivo studies, (KD = 13 nM; kon = 3175 M−1 s−1; koff = 4.39 × 10−5 s−1), they demonstrated TNF-α inhibition in cells by reducing levels of receptor-interacting protein 1 (RIP1) and phosphorylated NF-κB in Jurkat cells which are downstream proteins in the signalling cascade, as well as by inhibiting TNF-α induced cytotoxicity in mouse L929 cells. Oral dosing of UCB-9260 in mice resulted in dose-dependent decrease in neutrophil recruitment as measured by the number of CD45+GR1+ cells using flow cytometry, and in a mouse model of rheumatoid arthritis symptoms, a significant lower clinical score was obtained following oral administration of UCB-9260.51Fig. 1 shows the structure and proposed MoA for initial hit UCB-6876 and optimized analogs covered in this review, along with crystal structure of the TNF-α homotrimer before and after binding the most advanced lead UCB-9260.
 | ||
| Fig. 1 A: Chemical structure of UCB-6876, UCB-5307 and UCB-9260. B: Schematic representation of proposed MoA for the small molecules interacting with TNF-α. The small molecule is represented as a red diamond and the red arrows illustrate compound-induced shifts in equilibrium based on the proposed MoA. C: Crystal structures of the symmetric and UCB-9260-bound asymmetric TNF-α homotrimer in blue (PDB ID: 1TNF and 6OP0 respectively). The compound is shown in orange sticks. | ||
Similar lead-finding efforts for TNF-α have been reported by other groups before and since, with a similar MoA that is allosteric inhibition by stabilization of an inactive asymmetric form of the trimer.50,52,53 This demonstrates the value of understanding the nature of slow binding kinetics that appear in unexpected places as a possible signal of allostery at play, and the many benefits of structure-enabled drug design.
The binding site was elucidated for one of these small molecules, compound 1 (Fig. 2A), which was reported to interact with the IL-17A dimer and inhibit complex formation with the IL-17RA receptor.77,78 SPR, thermal shift assays and nuclear magnetic resonance (NMR) studies confirmed binding of compound 1 to the IL-17A homodimer (KD = 0.660 μM) and suggested that the binding event resulted in significant conformational shift of the protein complex.79 Of note, IL-17A was immobilized as a dimer for SPR studies, reflecting the species targeted by the small molecule interaction. Extensive co-crystallization and soaking efforts towards obtaining a crystal structure of compound 1 bound to the IL-17A dimer were unsuccessful. This aligns with the challenges reported by the same group in a separate publication, where crystallization efforts with a high affinity peptide were unsuccessful and required the development of a fragment antigen-binding (Fab) to enable the peptide/IL-17A dimer co-crystallization.80 Fortunately, using the same Fab along with the reported high affinity peptide as co-crystallization chaperones provided a high-resolution structure revealing that compound 1 binds at the interface of the IL-17A homodimer in an induced binding site (Fig. 2C). A conformational change in the protein was observed when compared to the apo structure, effectively perturbing the cytokine–receptor interaction (Fig. 2B).79 This binding site was determined to be highly druggable as assessed by computational methods.
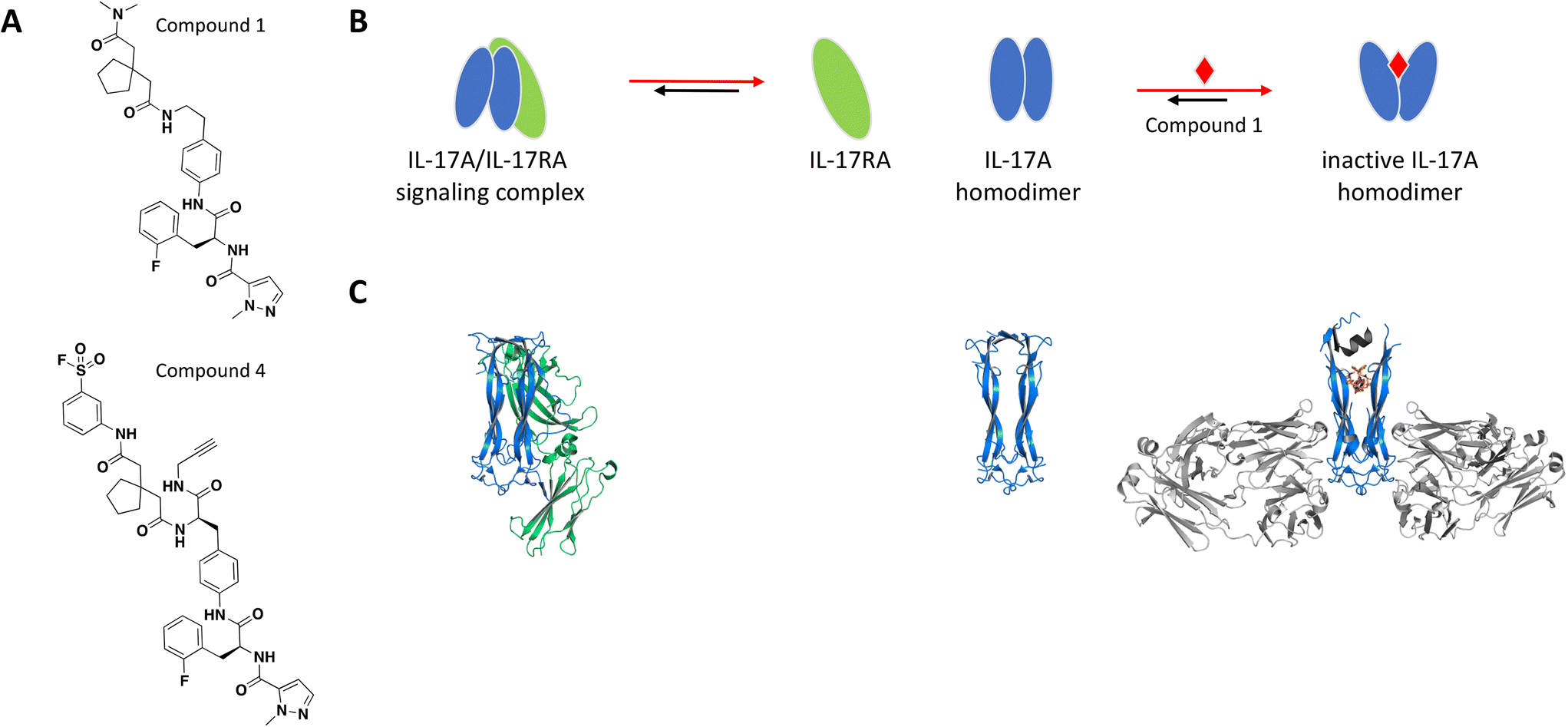 | ||
| Fig. 2 A: Chemical structures of compounds 1 and 4. B: Schematic representation of proposed MoA for compound 1 interacting with the IL-17A dimer. The small molecule is represented as a red diamond and the red arrows illustrate compound-induced shifts in equilibrium based on the proposed MoA. C: Crystal structures of the IL-17A dimer (blue) bound to IL-17RA (green), alone or bound to compound 1 (PDB ID: 4HSA, 4HR9 and 5HI3, respectively). Coloured in grey are the stabilizing IL-17A Fabs and peptide. The compound is shown in orange sticks. | ||
To confirm the binding mode of compound 1 in solution, analogs containing a sulfonyl fluoride group were prepared. Following incubation of IL-17A with the electrophilic analog compound 4 (Fig. 2A), electrospray/time-of-flight MS showed a major mass consistent with the IL-17A dimer being covalently modified with one equivalent of compound 4, confirming the 1:2 stoichiometry. Furthermore, peptide mapping of the conjugate was consistent with the crystal structure and modeling. Of note, the amino acid sequence in the regions of IL-17A forming the binding site are not strictly conserved in IL-17 cytokines which leads to specificity of this series of compounds for IL-17A.
Ultimately, this chemical series was optimized into macrocycles that inhibited the interaction between IL-17A and IL-17RA by SPR and fluorescence resonance energy transfer (FRET) assay with high affinity, and demonstrated activity in a keratinocyte cellular assay.79
A single glutamine-to-valine point mutation on the β-chain of adult Hb (HbA) is the cause of sickle cell disease (SCD), one of the most common genetic disorders.85 Under low oxygen conditions, the mutation promotes Hb polymerization and sickling of the red blood cells (RBC). An attractive approach to therapy is to identify an allosteric inhibitor that stabilizes Hb in its oxygenated state, as polymerization occurs only with deoxygenated Hb, under hypoxic conditions.86
The intracellular concentration of Hb in the body is estimated at 5 mM, posing a significant challenge in reaching an effective drug concentration in the RBC while preventing off-target toxicity. The two recent examples taken to the clinic are allosteric inhibitors with sub-stoichiometric mechanism of action: GBT440 (voxelotor), a reversible-covalent binder with recent FDA approval and PF-07059013, the first non-covalent inhibitor to reach clinical trials.86,87
Following earlier work that demonstrated aldehydes can stabilize deoxygenated Hb, Safo et al. identified naturally occurring 5-hydroxymethyl-2-furfural (5-HMF) as an allosteric effector of Hb (Fig. 3A).84,88–90 The co-crystal structure of Hb with 5-HMF illustrates the formation of a symmetrical Schiff base adduct between the N-terminal Val1 nitrogen atoms from each of the Hb α subunits, resulting in a stoichiometry of two molecules per Hb tetramer.84 Importantly, 5-HMF increases the affinity of Hb for oxygen and inhibits the sickling of homozygous RBCs expressing the disease-causing mutant Hb.84
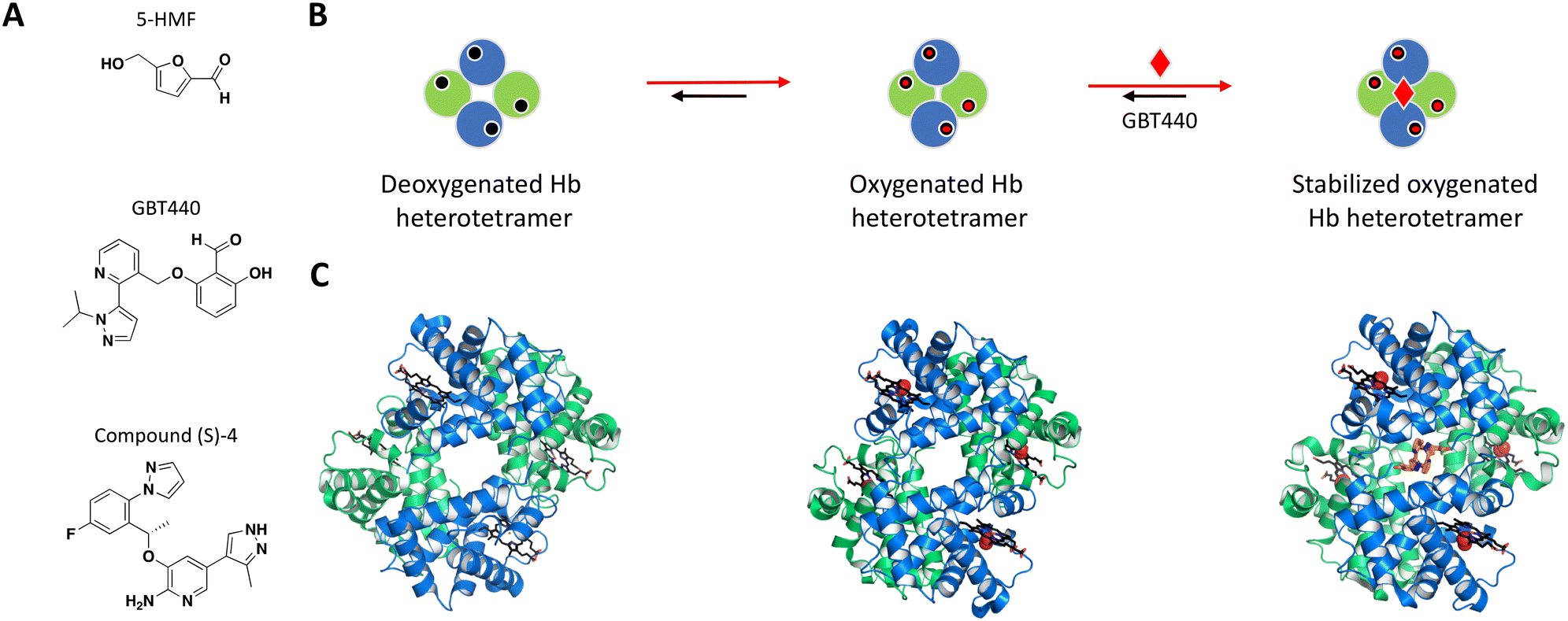 | ||
| Fig. 3 A: Chemical structures of 5-HMF, GBT440 and (S)-compound 4. B: Schematic representation of proposed MoA for GBT440 interacting with Hb. The small molecule is represented as a red diamond and the red arrows illustrate compound-induced shifts in equilibrium based on the proposed MoA. C: Crystal structures of the deoxygenated, oxygenated and GBT440-bound Hb (PDB ID: 2HHB, 1HHO and 5E83, respectively). Hb protomer shown in green and blue cartoon, oxygen as red spheres and the compound in orange sticks. | ||
Identification of GBT440 (voxelotor) began with a focused SAR analysis of 5-HMF-derived synthetic analogs using two parallel approaches: i) a hemoximetry assay calculating the allosteric modulation and the ability to stabilize purified Hb in the oxygenated state, and ii) X-ray co-crystal structures with lead compounds.86
The most potent compounds that confirmed activity in the polymerization assay were identified after an important switch of in vitro assays from using purified Hb to sickle disease blood in a whole blood assay. GBT440 had the most promising SAR and favorable pharmacokinetic properties, working as a covalent, allosteric effector of Hb.86,91 Interestingly, while occupying the same binding pocket as 5-HMF, the co-crystal structure shows that GBT440 forms hydrogen bonds with both of the Hb α subunits (Ser131 of the first and Val1 of the second α-chain), leading to a shift in stoichiometry from two molecules (5-HMF) to one molecule (GBT440) per Hb tetramer (Fig. 3B).84,86 Inhibiting a target with an estimated intracellular concentration of 5 mM is difficult to achieve, but GBT440 showed RBC preferential disposition with a RBC/plasma ratio of ∼150 and sub-stoichiometric binding to Hb tetramer of 1:1. GBT440 was shown to reach therapeutic concentrations in RBCs without toxic compromise and was approved by the FDA.
The second example is the reversible allosteric inhibitor developed by Pfizer. The hit finding efforts began with a virtual screening using the structure of Hb with 5-HMF, against the well-known α-chain binding pocket of aldehydes.84,87 The resulting compounds were selected based on their docking score, chemical diversity, and a small subset of hits were confirmed for binding with Hb by MS.87 Following hit triage by affinity selection mass spectrometry (ASMS), the wealth of knowledge from previous NMR studies of Hb in its different equilibrium states was used in one-dimensional 1H-NMR experiments to map binding sites, measure binding affinity and rank compounds for assay characterization and co-crystallization. The racemic compound 4 (rac-4) was one with the highest solubility that induced a significant chemical shift perturbation upon binding to Hb. Electrospray ionization native mass spectrometry (ESI-MS) showed binding stoichiometry of two rac-4 molecules to one Hb tetramer, and isothermal titration calorimetry (ITC) further demonstrated the first ligand being more entropically favorable than the second, suggesting a ditopic binding mode. Following HPLC purification of the rac-4 enantiomers, the rest of the experiments were performed with the pure (S)-4 enantiomer (Fig. 3A) with confirmed binding affinity by SPR (KD = 3.56 μM). High resolution crystal structure of (S)-4 with Hb confirmed the 1:2 stoichiometry, showing each (S)-4 molecule interacting at the interface of the α subunits, occupying the same binding pocket as 5-HMF and the covalent inhibitor GBT440.86,87 Assays such as delay release of oxygen from Hb, binding affinity (SPR) and blood binding were used during compound optimization, resulting in clinical candidate PF-07059013 that showed a 37.8 ± 9.0% decrease in sickling, compared to vehicle controls in an in vivo SCD mouse model.87
The crystal structure of the PD-1/PD-L1 complex reveals extensive contacts of PD-1 with PD-L1 through their Ig variable domains in a similar interface as antibodies and T cell receptors (Fig. 4C).92,93 Monoclonal antibodies targeting and disrupting the PD-1/PD-L1 interaction have become successful immunotherapy for the treatment of many forms of cancer.94–100 Mimicking this MoA with a small molecule would be difficult due to large contact surface area and high affinity interaction between PD-1 and PD-L. A few recent examples demonstrate a novel mechanism of small molecule-induced PD-L1 dimerization resulting in inhibition of the PD-1/PD-L1 complex formation.
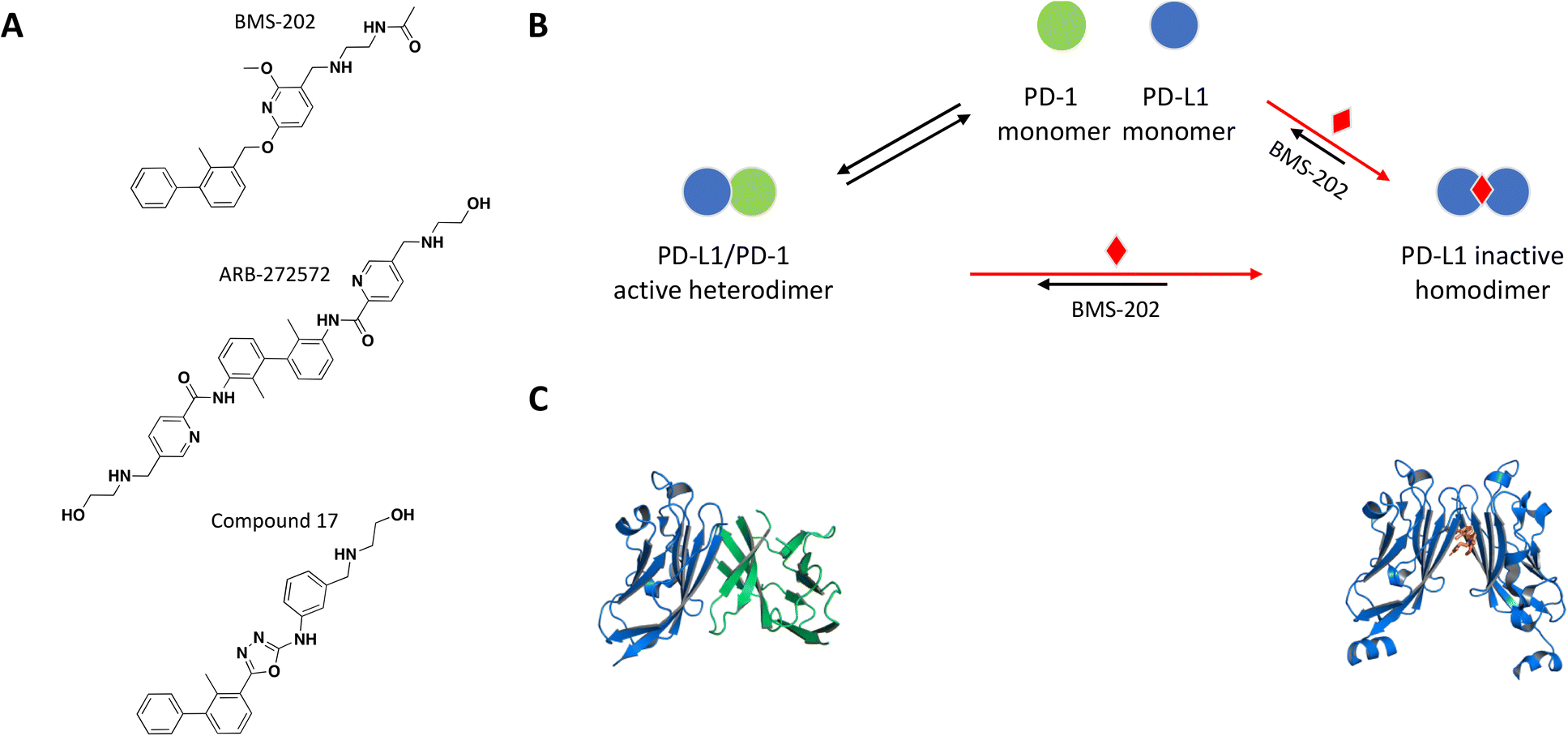 | ||
| Fig. 4 A: Chemical structures of BMS-202, ARB-272572, and compound 17. B: Schematic representation of proposed MoA for BMS-202 interacting with PD-L1. The small molecule is represented as a red diamond and the red arrows illustrate compound-induced shifts in equilibrium based on the proposed MoA. C: Crystal structures of the human PD-1/PD-L1 heterodimer and BMS-202-bound PD-L1 homodimer (PDB ID: 4ZQK and 5J89, respectively). Shown in cartoon PD-L1 (blue), PD-1 (green) and in sticks BMS-202 compound (orange). | ||
The first non-peptidic small molecule inhibitors of the PD-1/PD-L1 interaction were reported in a patent by Bristol Myers Squibb and identified by a homogeneous time-resolved fluorescence (HTRF) binding assay.101
As a follow up of the patent, Zak et al. used NMR 2D 1H–15N HMQC experiments and NMR based antagonist induced dissociation assay (AIDA), to demonstrate that four of the disclosed compounds bind specifically to PD-L1 only and BMS-202 (Fig. 4A) prevents the formation of a PD-L1/PD-1 complex.102,103 Using differential scanning fluorimetry (DSF), the relative affinity and selectivity of BMS-202 was demonstrated with a significant Tm shift of 13 °C for PD-L1 and not PD-L2. The co-crystal structure of PD-L1 with BMS-202 revealed a 2:1 stoichiometry, with a single small molecule at the interface of a PD-L1 homodimer.102 The intermolecular interactions within the dimer involve the PD-1 binding site of PD-L1, precluding interactions with PD-1 and providing a rationale of the MoA (Fig. 4B and C). The inhibitor-induced dimerization was also confirmed in solution by SEC and 1H resonance linewidth NMR experiments.102,103 The optimized small molecule inhibitors from this chemical series attenuated the inhibitory effect of soluble and cell surface associated PD-L1 (CHO) in a nuclear factor of activated T cells (NFAT) reporter assay developed with Jurkat cells constitutively expressing PD-1.104 While characterizing their own chemical series of PD-L1 inhibitors, scientists from Gilead made the interesting observation that the potency of their lead molecule GS-4224 increases with higher PD-L1 expression.105 This is presumably due to the molecule's dimerization MoA and is relevant considering that PD-L1 expression is upregulated in some cancers.106
Following the example of a successful PD-L1 small molecule inhibitor, Wang et al. performed a virtual screen using the co-crystal structure of PD-L1 dimer with BMS-202 as a model (PDB ID 5J89 and Fig. 4C), focusing their efforts on identifying compounds with the same MoA.107 The best hit from the screen inhibited the interaction of PD-1 with PD-L1 with an IC50 of 64 μM based on TR-FRET assay, and molecular docking suggested the same binding mode in the hydrophobic cleft of the PD-L1 dimer as with BMS-202. Extensive optimization resulted in the identification of compound 17 with an improved IC50 of 27.8 nM. The co-crystal structure of PD-L1 with compound 17 and SEC experiments confirmed compound induced dimerization of PD-L1 in a stoichiometry of 1:2.107 In flow cytometry experiments, cells pre-treated with compound 17 showed weakened PD-1 binding, compound 17-induced internalization and lysosome-dependent degradation of PD-L1. Compound 17 achieved a significant dose-dependent activation of antitumor immunity of peripheral blood mononuclear cells (PBMCs) in a T cell tumor co-culture assay, as well as antitumor activity in vivo.107
A new chemical series with a similar mechanism of inhibition was recently identified also by using an HTRF assay in the hit finding stage.108 Electrophoresis analysis of CHO cells expressing PD-L1 showed that treatment with ARB-272572 led to PD-L1 dimerization on cell membranes and subsequent cytosolic internalization as measured by flow cytometry. Aligned with PD-L1 internalization observed in vitro, small molecule treatment of in vivo tumor models promoted loss of cell surface PD-L1, and the decrease in PD-L1 expression in tumor cells directly correlated with the degree of tumor growth inhibition.108 The most advanced small molecule inhibitor of PD-L1 is currently in a phase 2 clinical trial (INCB086550). It was based on a rational chemical approach and has the same MoA as the other small molecule inhibiters discussed here.109
By competing for binding sites within αKG-dependent dioxygenases such as histone demethylases, prolyl hydroxylases and ten-eleven translocation (TET) 5-methylcytosine hydroxylases, 2-HG inhibits normal enzyme turnover resulting in genome-wide histone and DNA hypermethylation.111 This leads to large-scale epigenetic changes that contribute to impaired cell differentiation and tumorigenesis.110 Several small molecule drug campaigns targeting mutant IDH2 have made it to clinical trials for AML and solid tumors, including glioma. In 2017 Agios (in partnership with Celgene) received FDA approval for their first-in-class oral compound AG-221 (enasidenib) for late-stage AML harboring an IDH2 mutation,112,113 and their brain-penetrant dual mutant IDH1/IDH2 inhibitor AG-881 (vorasidenib) is currently in phase 1 for low-grade glioma.113,114
AGI-6780 was the first successful candidate that came out of a small molecule screen looking for inhibitors selective for the IDH2-R140Q mutant which occurs in 9% of AML cases (Fig. 5A).110,115 Wang et al. used an enzyme assay to monitor conversion of αKG to 2-HG by first preincubating compounds for 16 hours in the presence of saturating NADPH before initiating reaction by addition of αKG.115 The remaining cofactor was consumed in a diaphorase-catalyzed reaction which converts resazurin to resorufin with a proportional increase in fluorescence signal. AGI-6780 inhibited 2-HG production with nanomolar potency in both biochemical and cell-based assays, and Lineweaver–Burk plots revealed a binding modality that was non-competitive against αKG and uncompetitive with respect to NADPH. Shorter compound preincubation resulted in a significant reduction in potency (>7-fold) compared to 1 hour, indicative of time-dependent inhibition. Kinetic studies revealed slow, tight binding inhibition with long residence time (kon = 5.8 × 104 M−1 min−1; koff = 8.3 × 10−3 min−1), and evaluation of a targeted panel showed good selectivity against IDH1 (cytosolic IDH) and other related dehydrogenases.115 X-ray crystal structure of the R140Q mutant bound to NADP+, Ca2+, and AGI-6780 revealed a 1:2 stoichiometry with one inhibitor molecule bound to one IDH2 homodimer at the interface created by four α-helices from each of the protomers (Fig. 5C).115 This allosteric site sits at the dimer interface in a deep pocket covered by dynamic loops that move to allow ligand occupancy. Inhibitor binding locks the protein in an open conformation that separates the large cofactor binding domain from the small catalytic domain leading to reduced catalytic activity (Fig. 5B). In separate in vitro and ex vivo experiments, AGI-6780 induced cell differentiation in TF-1 erythroleukemia cells as well as in primary human AML cells harboring the IDH2-R14Q mutation, respectively, demonstrating the clinical relevance of targeting mutant IDH2 for AML and other cancers.115
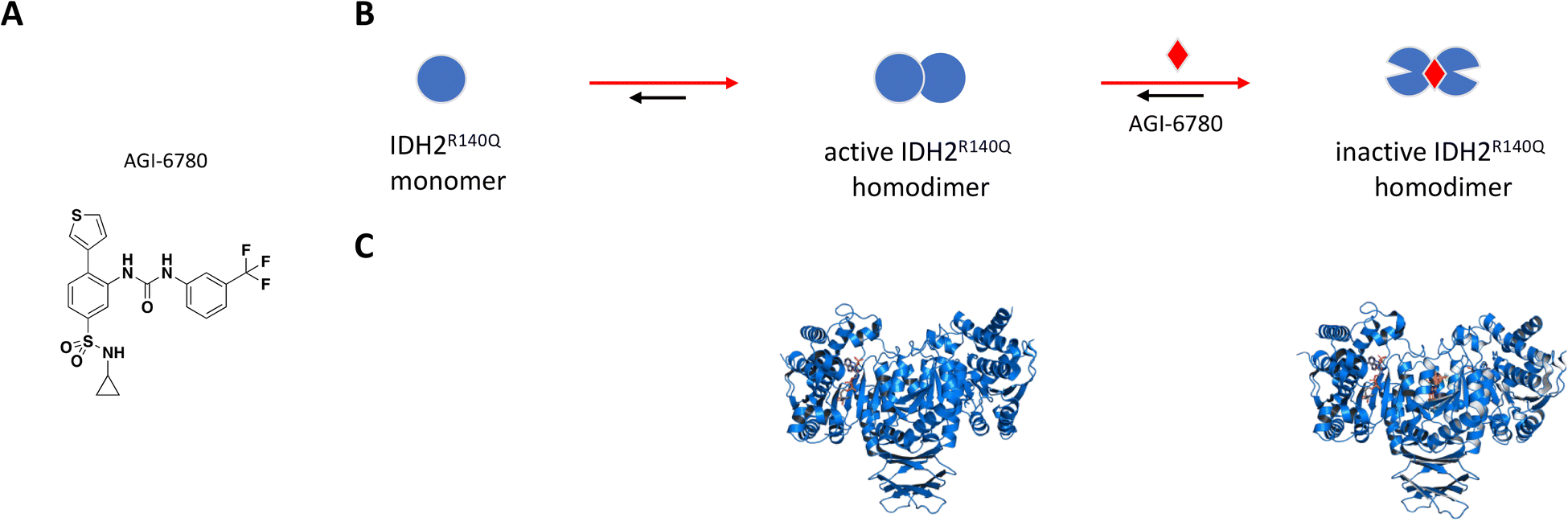 | ||
| Fig. 5 A: Chemical structure of AGI-6780. B: Schematic representation of proposed MoA for AGI-6780 interacting with IDH2. The small molecule is represented as a red diamond and the red arrows illustrate compound-induced shifts in equilibrium based on the proposed MoA. C: Crystal structures of IDH2 homodimer (blue), alone and bound to AGI-6780 (PDB ID: 5SVO and 5I96, respectively). The compound is shown in orange sticks. | ||
The long preincubation time built into the screening conditions aided by structure-based design allowed identification and optimization of allosteric hits that exhibit slow, tight binding inhibition despite the atypical 1:2 stoichiometry. This strategy emphasized the great potential of targeting the dimer interface in mutant IDH2 as well as in IDH1 for cancer therapeutics,116 and led to the successful development of AG-221 (enasidenib) and AG-881 (vorasidenib) which are now being tested in the clinic.117,118
Selected examples targeting protein oligomers with undefined stoichiometry
The concept of targeting disease relevant large oligomers with small molecules is not new and well-studied examples include modulators of viral capsid119–123 and inhibitors of tubulin polymerization.124–129 In this context, small molecules targeting large oligomers may be able to shift the oligomeric forms and impact cytotoxic properties or activity by interacting in a sub-stoichiometric manner.130–132 The following examples reinforce the importance of characterizing and understanding the biological target of interest at a molecular level in order to capture and characterize small molecules with relevant MoAs.While most reported small molecules require high doses to inhibit membrane-bound hIAPP fibrillation, Anle138b was reported to impact formation of amyloid fibrils,159 and structurally related small molecule Anle145c (Fig. 6A) protected INS-1E cells from hIAPP oligomerization-induced cell death at a concentration equivalent to 1/10 of the concentration of monomeric hIAPP.158 Thioflavin T (ThT), a fluorescent dye known to bind to fibrils but not monomeric hIAPP, was used to demonstrate complete inhibition of fibril formation in solution by Anle145c at a 1:5 molar ratio. Lag time and kinetics for fibril formation were unaffected suggesting the small molecule interacts with higher order species. Inhibition of fibril formation by Anle145c, as opposed to displacement of ThT binding on fibrils, was confirmed by negative stain transmission electron microscopy (TEM), which also showed preferential formation of smaller complexes. A number of biophysical approaches were used to characterize the various species formed. Dynamic light scattering (DLS) and atomic force microscopy (AFM), confirmed that incubation with the small molecule leads to the formation of oligomeric complexes of hIAPP that are significantly smaller than fibrils. Conformational behaviour was studied with circular dichroism (CD) spectroscopy, demonstrating that Anle138b treatment prevents formation of β-sheet like fibrils. Finally, these smaller oligomers were found to have decreased cellular toxicity when compared to fibrils. The preferential formation of these non-toxic oligomeric species was observed when Anle145c was incubated with monomeric hIAPP, pre-formed fibrils or at various time points during fibril growth, supporting a thermodynamic driving force (Fig. 6B).158
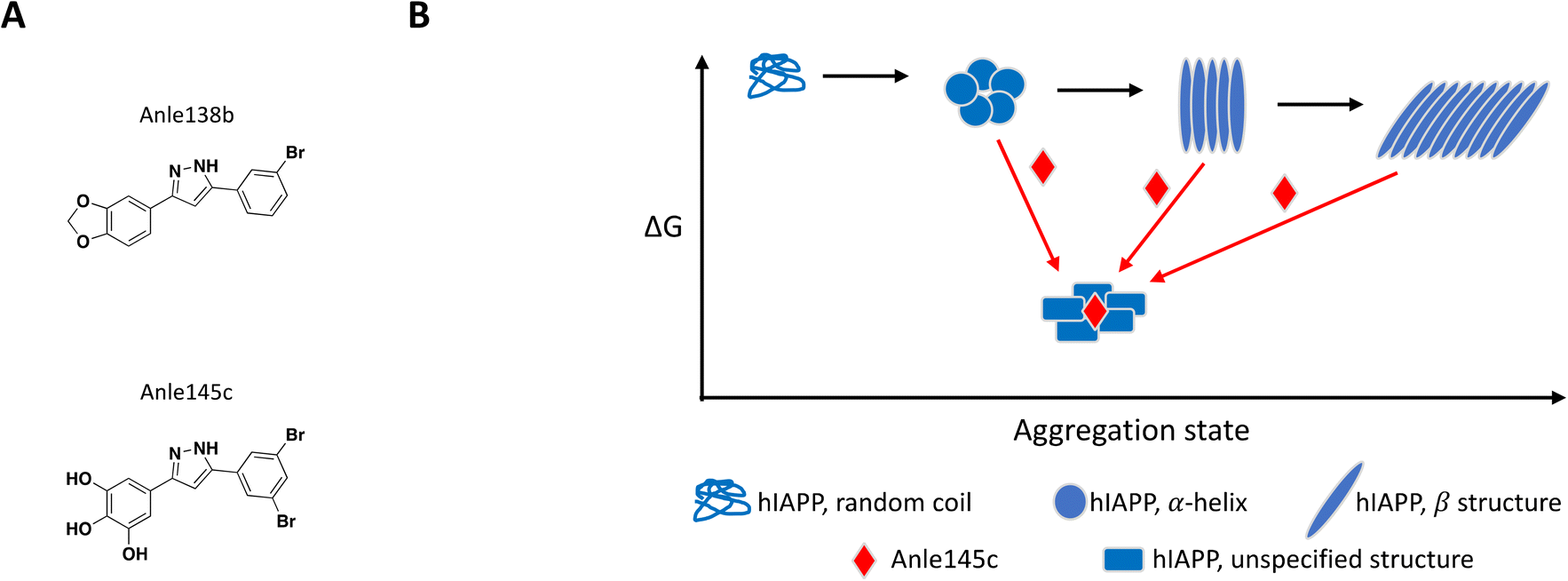 | ||
| Fig. 6 A: Chemical structure of Anle138b and Anle145c. B: Schematic representation of proposed MoA for Anle145c interacting with hIAPP (reproduced from ref. 158) the small molecule is represented as a red diamond and the red arrows illustrate compound-induced shifts in equilibrium based on the proposed MoA. | ||
It is important to note that the catechol functional group can be easily oxidized to a quinone motif, which in turn can covalently react with thiols and amines from amino acid side chains. This MoA has been demonstrated in vitro for a number of catechol-containing small molecules inhibiting amyloid fibrillation of various proteins, including hIAPP.160–166 The chemical series described here was also reported to inhibit fibrillation of other proteins,159 which suggests a lack of specific interactions and/or reactivity with hIAPP. For those reasons, further characterization of Anle145c's MoA will be critical to determine whether this MoA can be optimized to target hIAPP in a selective and safe manner.
As mentioned previously, the hIAPP peptide is mostly unstructured in solution but adopts a helical conformation after interaction with the cell membrane, which has been proposed to act as a template for fibril formation.140–142 In order to target hIAPP in its helical state, Maity et al. included large unilamellar vesicles (LUVs), a membrane model relevant to cell plasma membrane, in their experimental setup to evaluate compound effect on fibril formation.167 DM 1 is a small molecule peptidomimetic which was engineered to complement the charges exposed upon formation of the hIAPP α-helix and disrupt potentially propagating pi interactions (Fig. 7A).
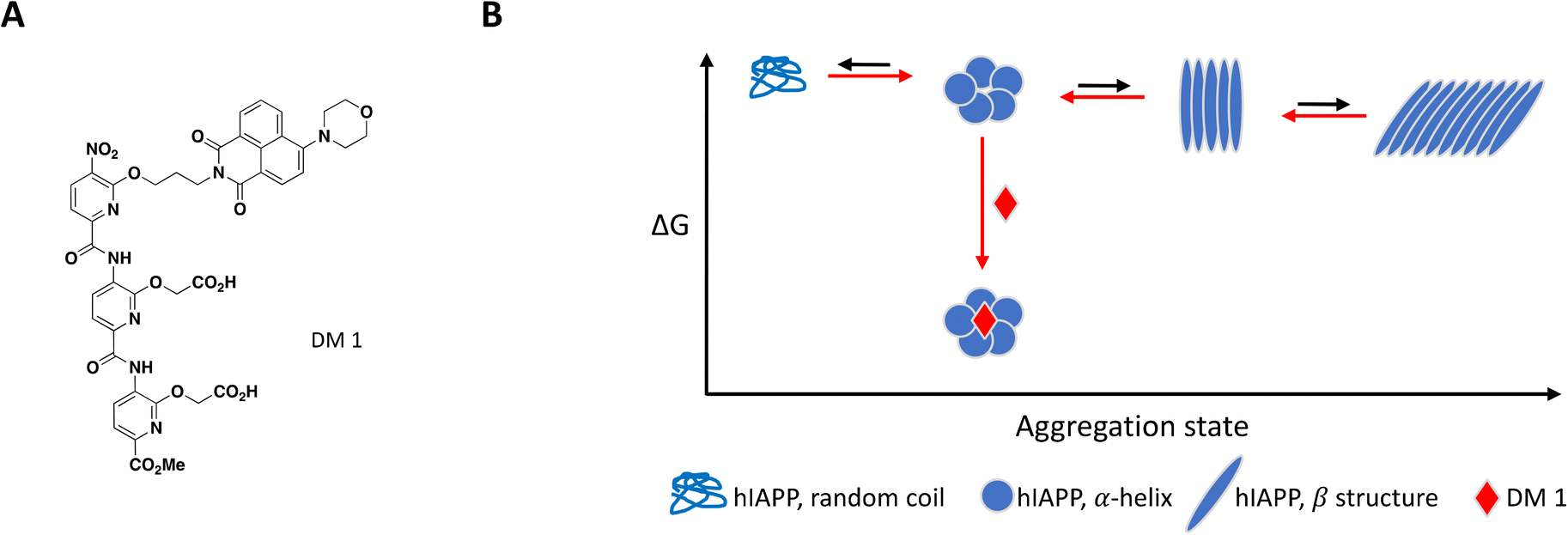 | ||
| Fig. 7 A: Chemical structure of DM 1. B: Schematic representation of proposed MoA for DM 1 interacting with hIAPP. The small molecule is represented as a red diamond and the red arrows illustrate compound-induced shifts in equilibrium based on the proposed MoA. | ||
At 0.2 equivalents, DM 1 completely inhibited fibril formation under these lipid-induced conditions, as demonstrated by ThT fluorescence readout and TEM. CD spectroscopy showed that hIAPP adopts a α-helical conformation in the presence of lipids which converts to β-sheet over time, a conformational change characteristic of amyloid formation. Addition of DM 1 stabilized the α-helix, preventing its conversion to β-sheet under these conditions. Similar to Anle145c, DM 1 was able to disaggregate preformed fibrils at a sub-stoichiometric concentration by preferentially forming smaller oligomeric species that are less toxic and rescue cells from hIAPP oligomerization-induced cell death (Fig. 7B).
As exemplified here with hIAPP, the ability to reproduce fibrillation processes in cell-free systems from a single recombinant protein led the scientific community to investigate diseases with fibril-centric approaches. However, several lines of evidence show that pathological aggregates have highly diverse morphologies and biochemical compositions. Aspects of the pathological aggregates' complexity that are missing in fibril formation models, such as additional biochemical components, post-translational modifications or polymorphic heterogeneity, may contribute to their clinical relevance as proposed by Lashuel.168
The catalytically active protease is a barrel-shaped complex with two components, a catalytic P subunit, ClpP, and a regulatory ATPase subunit, such as chaperone ClpX, responsible for the selection and unfolding of substrates. The main function of the ClpPX complex is to maintain cellular homeostasis during cell division, and in response to stress through degradation of damaged or misfolded proteins.175,176
Structurally, the proteolytic machinery of ClpP consists of two homo-heptameric rings stacked in a cylindrical shape to form an active tetradecamer (Fig. 8B) with 14 active sites aligned within its chamber, each of them comprised of the canonical Ser-His-Asp triad. While ClpP is able to degrade peptides, processing of large proteins requires binding of a hexameric ATPase chaperone such as ClpX.177 Inactivating mutations in S. aureus ClpP (SaClpP) were characterized and found to preferentially form a catalytically inactive heptamer rather than active tetradecameric complex.178
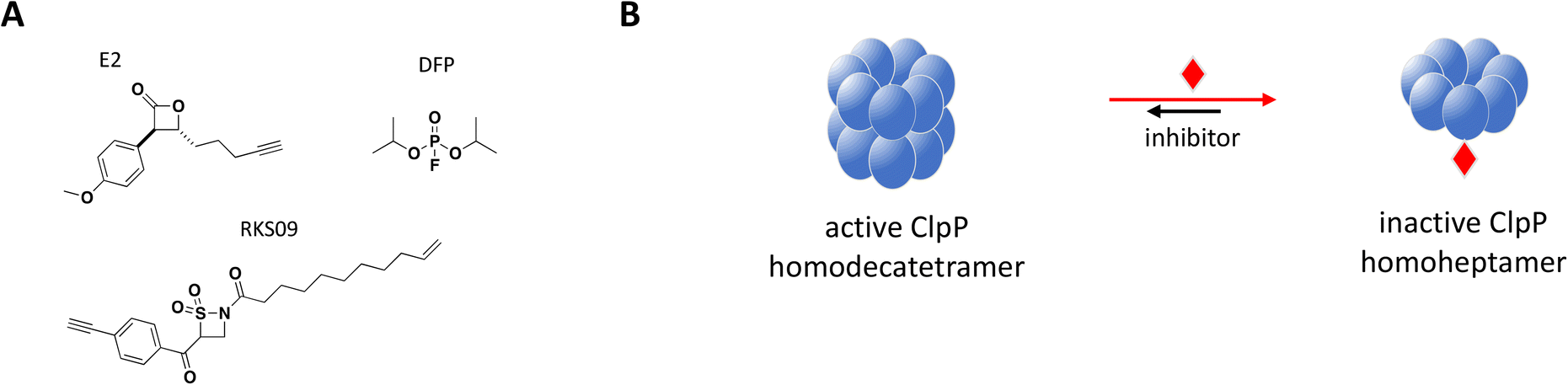 | ||
| Fig. 8 A: Chemical structures of E2, DFP and RKS09. B: Schematic representation of proposed MoA for inhibitors E2, DFP and RKS09 interacting with ClpP. The small molecule is represented as a red diamond and the red arrows illustrate compound-induced shifts in equilibrium based on the proposed MoA. | ||
Previously identified ClpP protease inhibitors are active site or allosteric binders that directly inhibit the catalytic activity, but are not known to influence the oligomeric state of the complex.179,180 Based on this new inhibitory mechanism identified with mutated SaClpP, screening efforts with a focused covalent library were undertaken and small molecule inhibitors that preferentially modify the active site and shift the oligomerization state of SaClpP towards an inactive heptamer were identified.181 Validated hits were carefully analyzed for this specific mode of inhibition: protein oligomerization was measured by SEC, protease activity by a fluorogenic substrate assay and the covalent protein modification determined by intact protein MS.
Complete inhibition of SaClpP in a sub-stoichiometric manner was demonstrated with three compounds, diisopropyl fluorophosphate (DFP), β-sultam RKS09, and β-lactone E2, showing active site modification of only 57%, 63% and 35%, respectively, by MS and dissociation of the active tetradecamer into inactive heptamer by SEC (Fig. 8).181 The shift in ClpP oligomerization induced by DFP, RKS09, and E2 was also confirmed by static light scattering (SLS). The results support that complete inhibition of the ClpP protease activity can be achieved with sub-stoichiometric small molecule inhibitors. Of note, complete inhibition of ClpP was achieved by varying degree of active site covalent modification by the three inhibitors (Fig. 8B). The sub-stoichiometric mode of inhibition was also demonstrated in Listeria monocytogenes and Escherichia coli.181
This last example does not fall under typical hit-finding efforts but is more of a cautionary tale that highlights the possible consequences of sub-stoichiometric binding to a highly cooperative protein complex system.
To date, there have been no successful drug development efforts targeting DegP but numerous mechanistic studies into protein dynamics that accompany enzyme activation paint a complex picture that could aide future campaigns, for this and other highly cooperative target classes. These mechanistic studies used synthetic chromogenic peptides as substrates,197,198 demonstrating sigmoidal instead of hyperbolic substrate titration curves with Hill slopes of 2 or higher, indicative of substrate-induced allostery and positive cooperativity.199 Time course traces showed an initial lag phase followed by faster and steadier rate of product formation, consistent with a slow interconversion between “resting” and “activated” states. In a biochemical assay using peptide inhibitor DPMFKLV-B(OH)2, Merdanovic et al. showed that DegP activity with SPMFKGV-pNA as substrate resembled a bell-shaped curve with inhibition preceded by up to 3-fold increase in activity at sub-stoichiometric concentrations of the inhibitor.198 ITC revealed that inhibitor occupancy was a 2-step binding event consistent with the cooperative nature of DegP, starting with weak binding of a resting state (KD = 6 μM) followed by much tighter interaction of an activated state (KD = 33 nM) at higher molar ratios of inhibitor. SEC and chemical cross-linking confirmed the existence of hexamer and dodecamer species under different inhibitor ratios, with sub-stoichiometric activation occurring in the hexameric fractions. While the “closed hexamer” represents the canonical resting state of DegP in the absence of ligands, an “open hexamer” conformation was also observed by crystallography wherein the triangle-shaped 45 Å cavity which houses the proteolytic site typically shielded from solvent opens up allowing ligand binding and initial catalysis.195 Recent solution-based biophysical characterization of DegP in the apo form also showed that the resting state is not restricted to hexamers but to an ensemble of sizes including larger complexes which are structurally distinct from their activated counterparts.199
Overall, the sub-stoichiometric binding of inhibitor at the PDZ domain provided sufficient occupancy to allosterically activate the transition from low to higher order oligomeric states and increase binding affinity and catalytic activity (Fig. 9B). This work highlights the importance of understanding the behavior of highly cooperative enzyme targets and the need to build screening strategies that would capture unique and potentially paradoxical pharmacology. It is also critical to understand target distribution and abundance in tissues as well as drug bioavailability in the clinic, to avoid undesirable off-target effects arising from inadvertent activation.
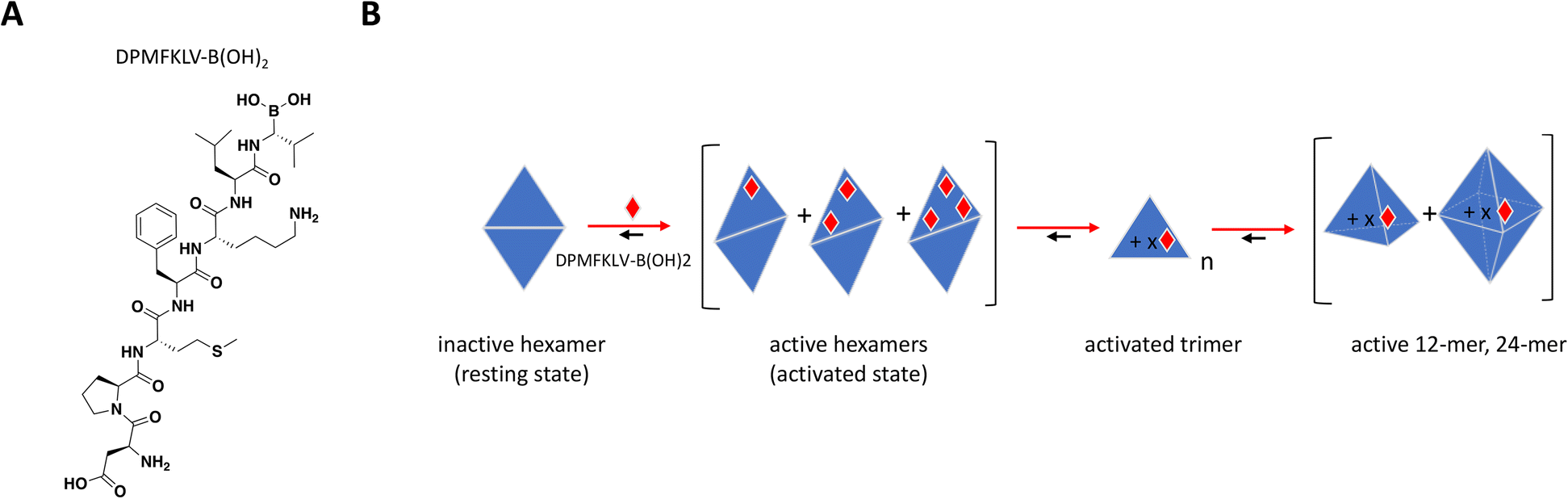 | ||
| Fig. 9 A: Chemical structure of DPMFKLV-B(OH)2. B: Schematic representation of proposed MoA for DPMFKLV-B(OH)2 interacting with DegP. The peptide is represented as a red diamond and the red arrows illustrate compound-induced shifts in equilibrium based on the proposed MoA. The notations n and x represent multiples of trimer, and compound, respectively. Active trimer, 12- and 24-mer could theoretically have multiple bound ligands. | ||
Conclusions and perspectives
The diversity and complexity of targets and pathways investigated by the drug discovery community is ever expanding and the selected examples here capture a variety of multimeric protein assemblies from different drug target classes, all of them now shown to be modulated by sub-stoichiometric small molecule inhibitors. While these drug discovery efforts are recent, the targets themselves have been studied for years: inhibition of TNF-α, IL-17A and PD-L1 was previously thought feasible only with biologics, Hb was deemed unattainable because of its high in vivo concentration, hIAPP presents pathological aggregates of high complexity, and the novel MoA for IDH2 and ClpP builds on in vivo mutagenesis data. Highlighted for each of these examples is the toolbox of biophysical and biochemical techniques, and their critical role in providing molecular, near-atomic resolution insights of the target and the molecular mechanisms driving function. Table 1 summarizes the examples covered in this review as well as the strategies and methods used to characterize their sub-stoichiometric MoAs. In addition to reviewing these techniques, in this section we highlight various aspects of hit finding and lead discovery activities that we believe are important to consider to ensure valuable compounds with a sub-stoichiometric MoA are not overlooked.| Target (protein class) | Reported cpd:protein stoichiometry |
Lead finding approach (screening method) | Characterization methods | Information generated | Compound (ref.) |
|---|---|---|---|---|---|
| Affinity selection mass spectrometry (ASMS), atomic force microscopy (AFM), circular dichroism (CD), double electron–electron resonance (DEER), differential scanning fluorimetry (DSF), dynamic light scattering (DLS), fluorescence resonance energy transfer (FRET), fragment antigen-binding (Fab), high performance liquid chromatography (HPLC), high throughput screen (HTS), homogeneous time resolved fluorescence (HTRF), isothermal titration calorimetry (ITC), mass spectrometry (MS), microscale thermophoresis (MST), molecular dynamics (MD), nuclear magnetic resonance (NMR), size exclusion chromatography (SEC), static light scattering (SLS), surface plasmon resonance (SPR), thermal shift assay (TSA), time-resolved fluorescence resonance energy transfer (TR-FRET), transmission electron microscopy (TEM), variational implicit solvent model (VISM). | |||||
| TNF-α (cytokine) | 1:3 |
Fragment-based drug discovery (SPR) | SPR | Binding affinity, kinetics | UCB-6876, UCB-5307, UCB-9260 (ref. 51 and 56) |
| Co-crystallization | Changes to trimer symmetry, compound stoichiometry, binding mode | ||||
| MS | Trimer stabilization | ||||
| Analytical SEC | Stoichiometry of receptor-ligand interaction, inhibition of normal receptor binding | ||||
| DEER and MD simulation | Conformational sampling, transition between trimer states | ||||
| IL-17A (cytokine) | 1:2 |
Not reported | FRET assay | Interaction between IL-17A dimer and IL-17RA | Compound 1 (ref. 77–79) |
| SPR (immobilized dimer), TSA, NMR | Binding affinity, binding kinetics, large conformational change | ||||
| Co-crystallization (required Fab and peptides targeting a different region) | Stoichiometry, binding site | ||||
| Covalent modification | Confirm stoichiometry and binding site in solution | ||||
| Keratinocyte cellular assay | IL-17A-dependent IL-8 production | ||||
| VISM and MD simulation | Computational identification and druggability assessment of binding site | ||||
| Hb (globin) | 2:4 |
Focused screen of naturally occurring furanic compounds | Oxygen equilibrium studies | Compound-induced shift of oxygen equilibrium in cells | 5-HMF |
| 1:4 |
HPLC | Confirm direct Hb-compound interaction | GBT-440 (ref. 84, 86 and 91) | ||
| Co-crystallization | Stoichiometry, binding mode | ||||
| Antisickling assay | Determine antisickling effect of tested compounds | ||||
| 2:4 |
Virtual screening (ASMS) | Molecular docking | Hit finding docking model with desired MoA | Compound 4 (ref. 87) | |
| ASMS, 1H NMR | Hit finding, qualitative compound binding assessment | ||||
| ESI-MS, ITC, SPR | Binding affinity and stoichiometry | ||||
| Co-crystallization | Stoichiometry, binding mode | ||||
| Oxygen dissociation assay | Levels of oxygenated Hb | ||||
| Polymerization assay | Hb polymerization | ||||
| PD-L1 (immunoglobulin) | 1:2 |
HTS (HTRF) | HTRF assay | Inhibition of PD-1/PD-L1 complex formation | BMS-202 & ARB-272572 (ref. 101–104 and 108) |
| NMR, DSF | Binding confirmation | ||||
| SEC, NMR, native PAGE gel electrophoresis | Confirm PD-L1 dimerization | ||||
| Co-crystallization | Compound stoichiometry, binding mode | ||||
| Flow cytometry | Cytosolic internalization of PD-L1 dimers | ||||
| Effector assay | Measuring T cell inhibition | ||||
| Virtual screening (molecular docking of ChEMBL database) | Molecular docking | Hit finding docking model with desired MoA | Compound 17 (ref. 107) | ||
| TR-FRET | Confirm inhibition of PD-1/PD-L1 | ||||
| Co-crystallization | Compound stoichiometry, binding mode | ||||
| SEC | Confirm PD-L1 dimerization | ||||
| MST | Binding affinity | ||||
| Flow cytometry, T cell tumor co-culture assay | Cell based binding assay, functional assay | ||||
| IDH2 (enzyme) | 1:2 |
HTS (biochemical assay) | Functional NADPH assay, fluorescence readout | Inhibitor potency, binding mode, kinetics (time-dependent inhibition) | AGI-6780 (ref. 115) |
| Co-crystallization | Compound stoichiometry, binding mode | ||||
| IAPP (secreted hormone) | ND | HTS (thioflavin T binding) or peptidomimetic engineered to complement α-helix of IAPP | Thioflavin T binding with fluorescent readout | Quantify fibril formation | Anle145c, DM 1 (ref. 158 and 167) |
| TEM, DLS, AFM | Evaluate the size of oligomers formed | ||||
| CD | Study transition to β-sheet conformation | ||||
| Cell rescue from hIAPP oligomerization-induced cell death | Relative toxicity of smaller aggregates vs. fibrils | ||||
| ClpP (enzyme, chaperone) | Sub-stoichiometric | Focused covalent library (SEC, fluorogenic substrate assay, MS) | SEC, SLS | Protein oligomerization | E2, RKS09, DFP181 |
| Fluorogenic substrate assay | Peptidase activity | ||||
| Intact protein MS | Protein covalent modification, binding site confirmation | ||||
| DegP (enzyme, chaperone) | 1:2 |
Established peptide inhibitor | Chromogenic substrate assay | Sigmoidal concentration response curve, Hill slope, cooperativity | DPMFKLV-B(OH)2 (ref. 196) |
| 1:3 |
|||||
| 1:6 |
|||||
| ITC | Binding events corresponding to conformational states, dissociation constants | ||||
| SEC, chemical crosslinking | Changes in oligomeric state | ||||
Protein characterization can influence the selection of in vitro systems used for hit identification, selection and optimization
Understanding a protein's biological function at a molecular level provides opportunities for pharmacological modulation with defined MoAs, and enables the selection of appropriate assays and conditions for screening. This is illustrated by the use of LUVs to mimic cell membranes in the identification of the fibril inhibitor DM 1, in order to specifically target the helical form of hIAPP. Similarly, when biologically relevant states of the target protein are well elucidated screening hits can be more easily triaged for a defined MoA. Inactivating mutations in the ClpP protein provided clues as to the functional consequences of shifting from catalytically active tetradecameric complex to inactive heptameric state, allowing the successful use of SEC in the hit triage strategy. Furthermore, as exemplified by IDH2 and Hb, understanding the disease-relevant biochemical context, in these cases disease-causing mutations, was critical in developing the most appropriate in vitro systems for screening such as in the choice of assay conditions and protein reagents. Indeed, the starting point for any successful screening campaign such as those highlighted in this review is the careful consideration of the most appropriate constructs for reagent generation, and that is particularly important when targeting protein oligomers or complexes. For example, in vitro assays used for screening frequently employ truncated versions of the protein and in-depth understanding of the target's regulation may influence construct selection. In the context of a protein complex, it is important that proteins used for in vitro assays include all domains involved in the formation of potential allosteric binding sites, and that the oligomerization state is not perturbed. While certain aspects of protein characterization are especially important to identifying sub-stoichiometric small molecule modulators, this is generally true and valuable for all targets and MoAs.The choice of screening conditions could expose or conceal sub-stoichiometric hits
In the context of setting up screening assay conditions, there are a variety of ways that sub-stoichiometric mechanisms of action could be missed in the course of a typical hit finding campaign. For instance, the choice of conditions for compound incubation may not capture the dynamics of protein binding and/or conformational changes necessary in the functional readout. In the case of IDH2, screening in the absence of a 16-hour compound preincubation would have precluded observation of time-dependent inhibition and would have yielded relatively weak IC50 values which may not have been investigated further.The order of reagent addition may also influence the subset of captured hits, as in the case of the HTRF screening for PD-1/PD-L1 inhibitor. Human PD-L1, small molecules and antibodies were first added to the assay plate, followed by recombinant PD-1. Considering the small molecule PD-L1 binding site and the PD-L1/PD-1 heterodimer interface are the same, this setup would allow for non-competitive binding and homodimerization of PD-L1, instead of competition with precomplexed PD-L1/PD-1. MoAs that require presentation of specific oligomeric states or dynamic changes to these states may not have been identified using assays or techniques that immobilize the target or use the wrong oligomeric form. The mechanistic studies for PD-L1 hits were performed using non-immobilized protein and solution-based techniques such as NMR, DSF, and SEC, as immobilizing the PD-1/PD-L1 heterodimer would have negatively impacted the proper characterization of the compound's MoA. The experimental setup for crystallography and the use of co-crystallization vs. soaking of apo-crystals should also be carefully considered, since the latter may not capture ligand induced changes in protein oligomerization or conformation. For example, the conformational change on TNF-α trimer induced upon ligand binding cracked the crystals and co-crystallization proved to be a successful approach, as in the case for PD-L1.
Sub-stoichiometric MoAs could emerge from typical screening campaigns and resemble features of conventional hits
It is important to note that not all of the examples highlighted in this review set out to specifically develop sub-stoichiometric MoAs in the course of their lead finding efforts, at least not by design. Some started out as typical programs using conventional screening strategies in order to find the best hits for their targets, and only with thorough biophysical and mechanistic characterization did the novel MoAs emerge. In the case of TNF-α, the primary goal was to identify small molecule inhibitors in a space dominated by biologics, and the fragment-based approach aimed at finding weak binders as starting points for chemistry optimization was like any other. Yet the decision to pursue a fragment hit that showed unusual binding kinetics made all the difference in the outcome. In much the same way as some of these examples may have started out with more conventional hit finding goals, profiling these hits could have yielded results that resemble stoichiometric binding or something else entirely, especially when interpreting data from limited characterization work. Slow binding kinetics described in a number of examples (e.g. TNF-α, IL-17A, IDH2) had been more widely reported for stoichiometric binders due to convention and the relative novelty of sub-stoichiometric interactions. High Hill slopes from concentration response curves of cooperative binders could be mistaken as a sign of aggregation. In the case of DegP the choice of compound concentration used to measure activity showed either activation or inhibition depending on the ratio to the target protein. All of these points emphasize the importance of conducting proper diligence when elucidating a compound's MoA and providing as much biophysical and mechanistic evidence and proper data interpretation before drawing conclusions or discarding potential leads.Complementary biophysical methods are essential in characterizing sub-stoichiometric binders
The range of biophysical techniques used in characterizing protein–ligand interactions for the above examples are not specialized for identifying sub-stoichiometric binders, but instead represent some of the most frequently used methods in research and drug discovery (Table 1). As discussed in detail by Renaud et al., the methods are highly complementary and provide limited information on their own, but when combined, insights from each method allow to fully characterize the target's MoA.200 This typically requires deep expertise and tight collaboration within diverse, multi-disciplinary teams.Until recently, high-resolution models for structure-based drug discovery were obtained primarily with well-established biophysical techniques such as X-ray crystallography. Major computational and technological advances have revolutionized both protein structure prediction and experimentally derived single particle cryo-electron microscopy (cryo-EM) structures. One important artificial intelligence (AI) advances for the life sciences is the first neural network-based structure prediction approach AlphaFold2 (AF2).201,202 Given the amino acid sequence of a protein target, AF2 can generate a structural model with unprecedented high accuracy and a confidence score based on a local, per-residue measurements as well as a global fold assessment, with a specific focus on domain packing. Comparison of AF2 models to experimentally derived structures that were not included in the training data set, show a medium backbone RMSD (Cα root-mean-square deviation at 95% residue coverage) of 1.46 Å, and an all-atom RMSD indicative of side-chain accuracy of 2 Å and 1 Å in 52% and 17% of cases, respectively.201,202 Interesting to note for this perspective is that AF2 accuracy increases especially for predictions of homomers compared to proteins with few intra-chain or homotypic contacts, such as bridging domains within the context of large complexes.201 And while AlphaFold has revolutionized protein structure prediction, provided insights for new binding pockets and mechanisms, models for molecular replacement of X-ray data and interpretation of cryo-EM density maps,201 the accuracy currently achieved at the side chain level in the most reliable structures is still considered a limiting factor for structure based drug discovery.203,204 While AlphaFold continues to optimize its algorithms, the drug discovery community has taken full advantage of the advances in single particle cryo-EM, providing a powerful method to the researcher's toolbox and many high resolution structures.205–208 Cryo-EM advances are also beginning to shed light on the high complexity samples of pathological aggregates, providing first high-resolution structures of hIAPP fibrils discussed here, tau and α-synuclein.209–212 One advantage of this structural technique is the ability to capture the conformational flexibility of a molecular target by providing multiple 3D reconstructions from a single cryo-EM dataset.213–215 This approach could potentially be used to reveal new dynamic mechanisms for modulation of proteins and protein complexes, otherwise difficult to study by static or low-resolution biophysical techniques such as X-ray crystallography and NMR, respectively. The success of each structural technique is highly dependent on the biochemical properties of the target and the complementary information obtained from each of them is key to characterizing sub-stoichiometric modulators.
Advances in computational approaches can help identify allosteric sites and characterize novel sub-stoichiometric MoAs
Computational chemistry is a staple in drug discovery programs which, when combined with structural biology, can provide molecular insights into a target's functionality and how it may be modulated. Again, while not specific to sub-stoichiometric MoAs, computational methods are highly valuable tools that can be applied towards the discovery and characterization of these mechanisms and some examples are highlighted here. Druggability assessment has become more powerful with the aid of molecular dynamics (MD) simulations such as the approach taken for IL-17A.79 Using a previously published structure, the group specifically probed the dimer surface using the variational implicit solvent model (VISM) algorithm216 in search of putative binding pockets, and identified one that is suitable to accommodate a drug-like ligand. Using MD simulations they assessed the flexibility of the IL-17A dimer with or without their lead compound docked in the central pocket and found the cavity to be highly druggable and a favorable starting point for small molecule design. In the case of TNF-α, the team used computational methods to better elucidate their lead's mechanism of action.51 Using enhanced MD simulation of the unbound protein, they studied the conformational transition between the symmetric trimer and the asymmetric open conformation representing the inhibitor-bound state. The simulations yielded a free-energy surface with two clear energy basins including one for the asymmetric apo-TNF-α suggesting its relative stability despite the absence of bound compound. This finding provided further evidence that the UCB compounds work by stabilizing a low-abundance naturally sampled transient TNF-α conformation that is incompetent of downstream signalling. This result aligns with previous biophysical work by the same group using DEER spectroscopy.56 Lastly, although not exemplified in this review, the recent success in identification of allosteric inhibitors of TRAP1 using computational design is worth mentioning. As a member of the heat shock protein 90 (HSP90) family whose function involves among other things the proper folding, trafficking, and regulation of client proteins, TRAP1 serves as an important bioenergetic regulator in the mitochondria.217–219 In order to achieve a high level of selectivity against other members of the protein family, pharmacological modulation of TRAP1 by small molecules targeting an allosteric site, rather than the ATP binding site, is desired. Binding of ATP to the TRAP1 dimer causes buckling of one of the protomers, inducing asymmetry and ATPase activity in the second protomer.220 Sanchez-Martin et al. designed sub-stoichiometric allosteric inhibitors using computational methods that exploit the dimer asymmetry in order to identify cryptic druggable pockets and then developed a pharmacophore model that would capture the interactions necessary for binding.221 In this manner they were able to identify inhibitors selective against HSP90 that can modulate downstream pathways in cancer cells and in zebrafish. Further reading can be found here.222,223Binding sites located at the protomer interface constitute differentiated options for small molecules to safely modulate a protein complex
As illustrated with the selected examples, a number of small molecules targeting homomeric complexes in a sub-stoichiometric manner do so by binding at a site located at the interface between two or more protomers. This type of binding sites can provide strategic differentiation and deserves further discussion. In some instances, such as TNF-α and IL-17A, no druggable pocket is observed on the protomer, however the oligomers form a binding site where a small molecule can influence protein conformation and function. Abrusán et al.'s recent analysis of the PDB augmented with computational simulations explored relationships between protein complex formation and evolution, and ligand binding. They concluded that homomeric complexes with binding sites formed by more than one protomer are much more likely to be allosterically modulated than other complexes.224 This conclusion is supported by the observed functional impact of small molecules binding at the interface of protomers such as TNF-α, IDH2 and IL-17A inhibitors. It is generally believed that allosteric binding sites do not face the same evolutionary pressure as orthosteric sites, resulting in higher diversity.225 One can potentially take advantage of this diversity to achieve greater selectivity and this argument was made for the GPCR226 and kinase227 families. However, Abrusán's analysis of the PDB concluded that binding sites formed by more than one protomer in a homomeric complex are more similar across homologs than binding sites formed by a single protein or other complexes, suggesting that selectively targeting a certain protein over its homologs may be more challenging to achieve through this type of binding site.228 That being said, both IL-17A and IDH2 inhibitors were able to achieve selectivity against close homologs and, based on the growing number of small molecules targeting such binding sites that entered clinical evaluation, achieving a safe and efficacious level of selectivity seems feasible, at least pre-clinically. Overall, binding sites formed by more than one protomer of a homomeric complex may offer properties that are well suited for a small molecule approach, are likely to have allosteric effect on function and provide selectivity.Small molecules with different MoAs may cause divergent phenotypes
An additional aspect to consider is that small molecules with different MoAs may impact differently other functions of the targeted protein. For instance, ClpP inhibitors shifting oligomeric populations and IDH2 inhibitors causing dimer conformational changes can potentially affect scaffolding functions, subcellular localization or stability of the protein complex, in addition to the intended inhibition of the enzymatic activity. This is illustrated with the PD-L1 targeting small molecules which inhibit the PD-L1/PD-1 interaction through dimerization and internalization of PD-L1. This MoA not only inhibits the PD-L1/PD-1 interaction, it also leads to increased potency of the compounds under disease relevant high expression of PD-L1 and their duration of action depends on the renewal of protein at the plasma membrane. Small molecules with distinct MoAs may thus lead to different phenotypes when compared to competitive inhibitors, which warrant the careful evaluation of downstream events.Concluding remarks
As described by the biochemical principles of allostery, cooperativity and oligomerization, the function of proteins forming homomeric complexes may be regulated in numerous ways, and benefits can come from an upfront investment in their careful characterization. The examples selected here illustrate how small molecules can regulate such protein complexes in sub-stoichiometric manners. This approach was employed to successfully target proteins with high cellular concentration, where reaching a 1:1 stoichiometry with a small molecule would be impractical and likely unsafe. Other examples show that by binding at a site formed at the interface of protomers, small molecules can effectively inhibit proteins that are known to be difficult to modulate due to the lack of suitable binding sites.
The examples were selected to demonstrate that sub-stoichiometric inhibition with small molecules can take various forms and is relevant to many target classes and disease areas. Using these examples, we aimed to provide a summary of the various methods used to identify hits and characterize their MoA, noting that these techniques are common in research laboratories. Some of these examples illustrate how a deep understanding of the target's regulation can Influence experimental setups in order to capture this MoA specifically, while others show the value of thoroughly characterizing a small molecule's MoA from an untargeted screen.
By bringing attention to recent examples of small molecules acting in a sub-stoichiometric manner as well as some technical aspects critical to their identification, we invite the scientific community to consider this MoA when homomeric protein complexes arise. While potentially increasing the level of complexity required in screening assay format and hit characterization, targeting a homomeric protein complex with sub-stoichiometric small molecules may also offer certain advantages. Hence, in this context it's ok to be outnumbered.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank Maureen Beresini, James Kiefer, Christian Gampe, Thomas Magee, Jessica Ward, Timothy Heffron, Charlene Bashore and Jeremy Murray for their thoughts on the topic and manuscript.Notes and references
- E. Valeur, F. Narjes, C. Ottmann and A. T. Plowright, Emerging modes-of-action in drug discovery, MedChemComm, 2019, 10, 1550–1568 RSC.
- M.-J. Blanco and K. M. Gardinier, New Chemical Modalities and Strategic Thinking in Early Drug Discovery, ACS Med. Chem. Lett., 2020, 11, 228–231 CrossRef CAS PubMed.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, Molecular Properties That Influence the Oral Bioavailability of Drug Candidates, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
- V. Poongavanam, B. C. Doak and J. Kihlberg, Opportunities and guidelines for discovery of orally absorbed drugs in beyond rule of 5 space, Curr. Opin. Chem. Biol., 2018, 44, 23–29 CrossRef CAS PubMed.
- B. C. Doak, B. Over, F. Giordanetto and J. Kihlberg, Oral Druggable Space beyond the Rule of 5: Insights from Drugs and Clinical Candidates, Chem. Biol., 2014, 21, 1115–1142 CrossRef CAS PubMed.
- D. A. DeGoey, H.-J. Chen, P. B. Cox and M. D. Wendt, Beyond the Rule of 5: Lessons Learned from AbbVie's Drugs and Compound Collection, J. Med. Chem., 2018, 61, 2636–2651 CrossRef CAS PubMed.
- P. Matsson, B. C. Doak, B. Over and J. Kihlberg, Cell permeability beyond the rule of 5, Adv. Drug Delivery Rev., 2016, 101, 42–61 CrossRef CAS PubMed.
- G. Ermondi, M. Vallaro, G. Goetz, M. Shalaeva and G. Caron, Updating the portfolio of physicochemical descriptors related to permeability in the beyond the rule of 5 chemical space, Eur. J. Pharm. Sci., 2020, 146, 105274 CrossRef CAS PubMed.
- S. A. Busby, S. Carbonneau, J. Concannon, C. E. Dumelin, Y. Lee and S. Numao, et al., Advancements in Assay Technologies and Strategies to Enable Drug Discovery, ACS Chem. Biol., 2020, 15, 2636–2648 CrossRef CAS PubMed.
- K. Raina and C. M. Crews, Targeted protein knockdown using small molecule degraders, Curr. Opin. Chem. Biol., 2017, 39, 46–53 CrossRef CAS PubMed.
- J. M. Kastl, G. Davies, E. Godsman and G. A. Holdgate, Small-Molecule Degraders beyond PROTACs—Challenges and Opportunities, SLAS Discovery, 2021, 26, 524–533 CrossRef CAS PubMed.
- M. Békés, D. R. Langley and C. M. Crews, PROTAC targeted protein degraders: the past is prologue, Nat. Rev. Drug Discovery, 2022, 21, 1–20 CrossRef PubMed.
- J. Palacino, S. E. Swalley, C. Song, A. K. Cheung, L. Shu and X. Zhang, et al., SMN2 splice modulators enhance U1–pre-mRNA association and rescue SMA mice, Nat. Chem. Biol., 2015, 11, 511–517 CrossRef CAS PubMed.
- A. K. Cheung, B. Hurley, R. Kerrigan, L. Shu, D. N. Chin and Y. Shen, et al., Discovery of Small Molecule Splicing Modulators of Survival Motor Neuron-2 (SMN2) for the Treatment of Spinal Muscular Atrophy (SMA), J. Med. Chem., 2018, 61, 11021–11036 CrossRef CAS PubMed.
- H. Ratni, M. Ebeling, J. Baird, S. Bendels, J. Bylund and K. S. Chen, et al., Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 (SMN2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA), J. Med. Chem., 2018, 61, 6501–6517 CrossRef CAS PubMed.
- J. Wang, P. G. Schultz and K. A. Johnson, Mechanistic studies of a small-molecule modulator of SMN2 splicing, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 201800260 Search PubMed.
- T. A. Chappie, M. Abdelmessih, C. W. Ambroise, M. Boehm, M. Cai and M. Green, et al., Discovery of Small-Molecule CD33 Pre-mRNA Splicing Modulators, ACS Med. Chem. Lett., 2022, 13, 55–62 CrossRef CAS PubMed.
- D. N. Petersen, J. Hawkins, W. Ruangsiriluk, K. A. Stevens, B. A. Maguire and T. N. O'Connell, et al., A Small-Molecule Anti-secretagogue of PCSK9 Targets the 80S Ribosome to Inhibit PCSK9 Protein Translation, Cell Chem. Biol., 2016, 23, 1362–1371 CrossRef CAS PubMed.
- A. T. Londregan, L. Wei, J. Xiao, N. G. Lintner, D. Petersen and R. G. Dullea, et al., Small Molecule Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Inhibitors: Hit to Lead Optimization of Systemic Agents, J. Med. Chem., 2018, 61, 5704–5718 CrossRef CAS PubMed.
- A. T. Londregan, G. Aspnes, C. Limberakis, P. M. Loria, K. F. McClure and D. N. Petersen, et al., Discovery of N-(piperidin-3-yl)-N-(pyridin-2-yl)piperidine/piperazine-1-carboxamides as small molecule inhibitors of PCSK9, Bioorg. Med. Chem. Lett., 2018, 28, 3685–3688 CrossRef CAS PubMed.
- D. C. Swinney, Phenotypic vs. Target-Based Drug Discovery for First-in-Class Medicines, Clin. Pharmacol. Ther., 2013, 93, 299–301 CrossRef CAS PubMed.
- H. Prinz, Hill coefficients, dose–response curves and allosteric mechanisms, J. Chem. Biol., 2010, 3, 37–44 CrossRef PubMed.
- J. Monod, J. Wyman and J.-P. Changeux, On the nature of allosteric transitions: A plausible model, J. Mol. Biol., 1965, 12, 88–118 CrossRef CAS PubMed.
- J. Gerhart, From feedback inhibition to allostery: the enduring example of aspartate transcarbamoylase, FEBS J., 2014, 281, 612–620 CrossRef CAS PubMed.
- N. Kresge, R. D. Simoni and R. L. Hill, The Enzymology of Feedback Inhibition by Arthur B, Pardee, J. Biol. Chem., 2005, 280, e38–e40 CrossRef CAS.
- A. B. Pardee and G. P.-V. Reddy, Beginnings of feedback inhibition, allostery, and multi-protein complexes, Gene, 2003, 321, 17–23 CrossRef CAS PubMed.
- C. P. Dunne and W. A. Wood, L-Threonine Dehydrase as a Model of Allosteric Control Involving Ligand-lnduced Oligomerization, Curr. Top. Cell. Regul., 1975, 9, 65–101 CAS.
- Y. Shizuta and O. Hayaishi, Regulation of Biodegradative Threonine Deaminase, Curr. Top. Cell. Regul., 1976, 11, 99–146 CAS.
- T. Yonetani and M. Laberge, Protein dynamics explain the allosteric behaviors of hemoglobin, Biochim. Biophys. Acta, Proteins Proteomics, 2008, 1784, 1146–1158 CrossRef CAS PubMed.
- L. Thabault, M. Liberelle and R. Frédérick, Targeting protein self-association in drug design, Drug Discovery Today, 2021, 26, 1148–1163 CrossRef CAS PubMed.
- B. Y. Feng, A. Simeonov, A. Jadhav, K. Babaoglu, J. Inglese and B. K. Shoichet, et al., A High-Throughput Screen for Aggregation-Based Inhibition in a Large Compound Library, J. Med. Chem., 2007, 50, 2385–2390 CrossRef CAS PubMed.
- M. Klumpp, Non-stoichiometric inhibition in integrated lead finding – a literature review, Expert Opin. Drug Discovery, 2015, 11, 149–162 CrossRef PubMed.
- C. H. Arrowsmith, J. E. Audia, C. Austin, J. Baell, J. Bennett and J. Blagg, et al., The promise and peril of chemical probes, Nat. Chem. Biol., 2015, 11, 536–541 CrossRef CAS PubMed.
- S. A. Busby, S. Carbonneau, J. Concannon, C. E. Dumelin, Y. Lee and S. Numao, et al., Advancements in Assay Technologies and Strategies to Enable Drug Discovery, ACS Chem. Biol., 2020, 15, 2636–2648 CrossRef CAS PubMed.
- P. Bhat, D. Honson and M. Guttman, Nuclear compartmentalization as a mechanism of quantitative control of gene expression, Nat. Rev. Mol. Cell Biol., 2021, 1–18 CAS.
- D. J. Kempf, L. Sham Hing, K. C. Marsh, C. A. Flentge, D. Betebenner and B. E. Green, et al., Discovery of Ritonavir, a Potent Inhibitor of HIV Protease with High Oral Bioavailability and Clinical Efficacy, J. Med. Chem., 1998, 41, 602–617 CrossRef CAS PubMed.
- N. A. Roberts, J. A. Martin, D. Kinchington, A. V. Broadhurst, J. C. Craig and I. B. Duncan, et al., Rational Design of Peptide-Based HIV Proteinase Inhibitor, Science, 1990, 248, 358–361 CrossRef CAS PubMed.
- E. A. Carswell, L. J. Old, R. L. Kassel, S. Green, N. Fiore and B. Williamson, An endotoxin-induced serum factor that causes necrosis of tumors, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 3666–3670 CrossRef CAS PubMed.
- H. W. Ziegler-Heitbrock, A. Möller, R. P. Linke, J. G. Haas, E. P. Rieber and G. Riethmüller, Tumor necrosis factor as effector molecule in monocyte mediated cytotoxicity, Cancer Res., 1986, 46, 5947–5952 CAS.
- L. M. Sedger and M. F. McDermott, TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants – past, present and future, Cytokine Growth Factor Rev., 2014, 25, 453–472 CrossRef CAS PubMed.
- R. A. Black, C. T. Rauch, C. J. Kozlosky, J. J. Peschon, J. L. Slack and M. F. Wolfson, et al., A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells, Nature, 1997, 385, 729–733 CrossRef CAS PubMed.
- L. A. Tartaglia, R. F. Weber, I. S. Figari, C. Reynolds, M. A. Palladino and D. V. Goeddel, The two different receptors for tumor necrosis factor mediate distinct cellular responses, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 9292–9296 CrossRef CAS PubMed.
- M. S. Hayden and S. Ghosh, Regulation of NF-κB by TNF family cytokines, Semin. Immunol., 2014, 26, 253–266 CrossRef CAS PubMed.
- T. Bouwmeester, A. Bauch, H. Ruffner, P.-O. Angrand, G. Bergamini and K. Croughton, et al., A physical and functional map of the human TNF-α/NF-κB signal transduction pathway, Nat. Cell Biol., 2004, 6, 97–105 CrossRef CAS PubMed.
- R. Janssen, A. van Wengen, E. Verhard, T. de Boer, T. Zomerdijk and T. H. M. Ottenhoff, et al., Divergent Role for TNF-α in IFN-γ-Induced Killing of Toxoplasma gondii and Salmonella typhimurium Contributes to Selective Susceptibility of Patients with Partial IFN-γ Receptor 1 Deficiency, J. Immunol., 2002, 169, 3900–3907 CrossRef CAS PubMed.
- L. Alexopoulou, K. Kranidioti, S. Xanthoulea, M. Denis, A. Kotanidou and E. Douni, et al., Transmembrane TNF protects mutant mice against intracellular bacterial infections, chronic inflammation and autoimmunity, Eur. J. Immunol., 2006, 36, 2768–2780 CrossRef CAS PubMed.
- L. S. Guedes-Barbosa, I. P. da Costa, V. Fernandes, L. M. H. da Mota, I. de Menezes and M. A. Scheinberg, Leishmaniasis during anti-tumor necrosis factor therapy: Report of 4 cases and review of the literature (additional 28 cases), Semin. Arthritis Rheum., 2013, 43, 152–157 CrossRef PubMed.
- M. A. V. Willrich, D. L. Murray and M. R. Snyder, Tumor necrosis factor inhibitors: clinical utility in autoimmune diseases, Transl. Res., 2015, 165, 270–282 CrossRef CAS PubMed.
- J. R. Kalden and H. Schulze-Koops, Immunogenicity and loss of response to TNF inhibitors: implications for rheumatoid arthritis treatment, Nat. Rev. Rheumatol., 2017, 13, 707–718 CrossRef CAS PubMed.
- M. M. He, A. S. Smith, J. D. Oslob, W. M. Flanagan, A. C. Braisted and A. Whitty, et al., Small-Molecule Inhibition of TNF-α, Science, 2005, 310, 1022–1025 CrossRef CAS PubMed.
- J. O'Connell, J. Porter, B. Kroeplien, T. Norman, S. Rapecki and R. Davis, et al., Small molecules that inhibit TNF signalling by stabilising an asymmetric form of the trimer, Nat. Commun., 2019, 10, 5795 CrossRef PubMed.
- J. D. Dietrich, K. L. Longenecker, N. S. Wilson, C. Goess, S. C. Panchal and S. L. Swann, et al., Development of Orally Efficacious Allosteric Inhibitors of TNFα via Fragment-Based Drug Design, J. Med. Chem., 2021, 64, 417–429 CrossRef CAS PubMed.
- W. Sun, Y. Wu, M. Zheng, Y. Yang, Y. Liu and C. Wu, et al., Discovery of an Orally Active Small-Molecule Tumor Necrosis Factor-α Inhibitor, J. Med. Chem., 2020, 63, 8146–8156 CrossRef CAS PubMed.
- M. J. Eck and S. R. Sprang, The Structure of Tumor Necrosis Factor-alpha at 2.6 Å Resolution: Implications for Receptor Binding, J. Biol. Chem., 1989, 29, 17595–17605 CrossRef.
- J. H. Naismith, T. Q. Devinell, B. J. Brandhuberll and S. R. Sprang, Crystallographic Evidence for Dimerization of Unliganded Tumor Necrosis Factor Receptor, J. Biol. Chem., 1995, 22, 13303–13307 CrossRef PubMed.
- B. Carrington, W. K. Myers, P. Horanyi, M. Calmiano and A. D. G. Lawson, Natural Conformational Sampling of Human TNFα Visualized by Double Electron-Electron Resonance, Biophys. J., 2017, 113, 371–380 CrossRef CAS PubMed.
- D. J. Lightwood, R. J. Munro, J. Porter, D. McMillan, B. Carrington and A. Turner, et al., A conformation-selective monoclonal antibody against a small molecule-stabilised signalling-deficient form of TNF, Nat. Commun., 2021, 12, 583 CrossRef CAS PubMed.
- D. McMillan, C. Martinez-Fleites, J. Porter, D. Fox, R. Davis and P. Mori, et al., Structural insights into the disruption of TNF-TNFR1 signalling by small molecules stabilising a distorted TNF, Nat. Commun., 2021, 12, 582 CrossRef CAS PubMed.
- M. J. McGeachy, D. J. Cua and S. L. Gaffen, The IL-17 Family of Cytokines in Health and Disease, Immunity, 2019, 50, 892–906 CrossRef CAS PubMed.
- V. Sud, A. Abboud, S. Tohme, Y. Vodovotz, R. L. Simmons and A. Tsung, IL-17A – A regulator in acute inflammation: Insights from in vitro, in vivo and in silico studies, Cytokine, 2021, 139, 154344 CrossRef CAS PubMed.
- S. Liu, X. Song, B. A. Chrunyk, S. Shanker, L. R. Hoth and E. S. Marr, et al., Crystal structures of interleukin 17A and its complex with IL-17 receptor A, Nat. Commun., 2013, 4, 1888 CrossRef PubMed.
- W. Wang, M. R. Groves and A. Dömling, Artificial macrocycles as IL-17A/IL- 17RA antagonists, MedChemComm, 2017, 9, 22–26 RSC.
- S. L. Alexander, T. F. Briggs, F. G. Favaloro Jr, S. P. Hale, B. A. Seigal and K. C. Shortsleeves, et al., Compounds modulating IL-17, WO Pat., 2014US066726, 2014 Search PubMed.
- M. Taylor, N. K. Terrett, W. H. Connors, K. C. Shortsleeves, B. A. Seigal and C. Snedeker, et al., Preparation of macrocyclic compounds as IL-17 modulators, Patent WO2013-US24386, 2013.
- G. N. Brace, R. E. Chappell, H. J. C. Deboves, A. M. Foley, G. Foulkes and E. P. Jones, et al., Spirocyclic indolines as IL-17 modulators, Patent WO2018-EP65558, 2018.
- P. T. Chovatia, A. M. Foley, G. W. Haslett, M. C. Hutchings, J. A. Johnson and F. C. Lecomte, et al., Preparation of difluorocyclohexyl derivatives as IL-17 modulators as treatment and prevention of inflammatory and autoimmune disorders, Patent WO2021-EP58940, 2021.
- P. T. Chovatia, G. W. Haslett, F. C. Lecomte, J. Madden, N. J. T. Monck and T. J. Norman, et al., Preparation of difluorocyclohexyl derivatives as IL-17 modulators as treatment and prevention of inflammatory and autoimmune disorders, WO2021-EP58937, 2021.
- R. L. Connelly, J. R. Frost, E. O. Gallimore, G. W. Haslett, H. T. Horsley and J. R. Quincey, et al., Preparation of difluorocyclohexyl derivatives as IL-17 modulators for the treatment and prevention of diseases, Patent WO2021-EP54519, 2021.
- G. N. Brace, R. E. Chappell, P. T. Chovatia, E. O. Gallimore, H. T. Horsley and J. A. Johnson, et al., Preparation of difluorocyclohexyl derivatives as IL-17 modulators for the treatment and prevention of diseases, Patent WO2021-EP54523, 2021.
- K. N. Dack, X. Liang, M. Larsen, M. Andrews, A. S. Jessiman and M. N. Burhardt, et al., Amino-acid anilides as small molecule modulators of IL-17, Patent WO2019-EP86239, 2020.
- D. A. Coates, K. Frimpong, W. G. Holloway, S. B. Jones, A. M. Levinson and C. Willis, et al., Imidazo[1,2-b]pyridazines as IL-17a inhibitors and their preparation, WO2020-US12115, 2020.
- M. Andrews, K. N. Dack and M. Larsen, Amidospirocyclic compounds as small molecule modulators of IL-17 and their preparation, Patent WO2021-EP66225, 2021.
- M. Andrews, K. N. Dack and M. N. Burhardt, Benzimidazolylalkanamide derivatives as small molecule modulators of IL-17 and their preparation, Patent WO2021-EP66226, 2021.
- M. Andrews, M. D. Soerensen, M. Larsen, N. Devaux, V. B. R. Silva and Q. Perron, et al., N-Heteroaryl and N-aryl amidoamides as small molecule modulators of IL-17 and their preparation, WO2021-EP65690, 2021.
- P. R. Fatheree, M. S. Linsell, J. R. Jacobsen, W. V. D. Linden, T. J. Church and C. Aquino, et al., Amidoamide ligands as IL-17 modulators and their preparation, Patent US2020-16783268, 2020.
- P. R. Fatheree, M. S. Linsell, J. R. Jacobsen, W. A. V. D. Linden, T. J. Church and C. Aquino, et al., Preparation of carboxamides as IL-17A modulators and uses thereof, Patent WO2020-US50924, 2021.
- M. Taylor, N. K. Terrett, W. H. Connors, C. Snedeker, K. C. Shortsleeves and B. A. Seigal, et al., Macrocyclic compounds for modulating IL-17, Patent US20150005319, 2015.
- S. L. Alexander, T. F. Briggs, G. F. Favaloro Jr, S. P. Hale and B. A. Seigal, et al., Compounds for modulating IL-17, WO Pat., 066726A2, 2014 Search PubMed.
- S. Liu, L. A. Dakin, L. Xing, J. M. Withka, P. V. Sahasrabudhe and W. Li, et al., Binding site elucidation and structure guided design of macrocyclic IL-17A antagonists, Sci. Rep., 2016, 6, 30859 CrossRef CAS PubMed.
- S. Liu, J. Desharnais, P. V. Sahasrabudhe, P. Jin, W. Li and B. D. Oates, et al., Inhibiting complex IL-17A and IL-17RA interactions with a linear peptide, Sci. Rep., 2016, 6, 26071 CrossRef CAS PubMed.
- H. F. Bunn, Pathogenesis and Treatment of Sickle Cell Disease, N. Engl. J. Med., 1997, 337, 762–769 CrossRef CAS PubMed.
- J. Baldwin and C. Chothia, Haemoglobin: the structural changes related to ligand binding and its allosteric mechanism, J. Mol. Biol., 1979, 129, 175–220 CrossRef CAS PubMed.
- W. Bolton and M. F. Perutz, Three Dimensional Fourier Synthesis of Horse Deoxyhaemoglobin at 2.8 Å Resolution, Nature, 1970, 228, 551–552 CrossRef CAS PubMed.
- M. K. Safo, O. Abdulmalik, R. Danso-Danquah, J. C. Burnett, S. Nokuri and G. S. Joshi, et al., Structural Basis for the Potent Antisickling Effect of a Novel Class of Five-Membered Heterocyclic Aldehydic Compounds, J. Med. Chem., 2004, 47, 4665–4676 CrossRef CAS PubMed.
- D. C. Rees, T. N. Williams and M. T. Gladwin, Sickle-cell disease, Lancet, 2010, 376, 2018–2031 CrossRef CAS PubMed.
- B. Metcalf, C. Chuang, K. Dufu, M. P. Patel, A. Silva-Garcia and C. Johnson, et al., Discovery of GBT440, an Orally Bioavailable R-State Stabilizer of Sickle Cell Hemoglobin, ACS Med. Chem. Lett., 2017, 8, 321–326 CrossRef CAS PubMed.
- A. Gopalsamy, A. E. Aulabaugh, A. Barakat, K. C. Beaumont, S. Cabral and D. P. Canterbury, et al., PF-07059013: A Noncovalent Modulator of Hemoglobin for Treatment of Sickle Cell Disease, J. Med. Chem., 2021, 64, 326–342 CrossRef CAS PubMed.
- D. J. Abraham, A. S. Mehanna, F. C. Wireko, J. Whitney, R. P. Thomas and E. P. Orringer, Vanillin, a Potential Agent for the Treatment of Sickle Cell Anemia, Blood, 1991, 77, 1334–1341 CrossRef CAS PubMed.
- C. R. Beddell, P. J. Goodford, G. Kneen, R. D. White, S. Wilkinson and R. Wootton, Substituted benzaldehydes designed to increase the oxygen affinity of human haemoglobin and inhibit the sickling of sickle erythrocytes, Br. J. Pharmacol., 1984, 82, 397–407 CrossRef CAS PubMed.
- P. Fitzharris, A. McLean, R. Sparks, B. Weatherley, R. White and R. Wootton, The effects in volunteers of BW12C, a compound designed to left-shift the blood-oxygen saturation curve, Br. J. Clin. Pharmacol., 1985, 19, 471–481 CrossRef CAS PubMed.
- D. Oksenberg, K. Dufu, M. P. Patel, C. Chuang, Z. Li and Q. Xu, et al., GBT440 increases haemoglobin oxygen affinity, reduces sickling and prolongs RBC half-life in a murine model of sickle cell disease, Br. J. Haematol., 2016, 175, 141–153 CrossRef CAS PubMed.
- D. Y. Lin, Y. Tanaka, M. Iwasaki, A. G. Gittis, H.-P. Su and B. Mikami, et al., The PD-1/PD-L1 complex resembles the antigen-binding Fv domains of antibodies and T cell receptors, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3011–3016 CrossRef CAS PubMed.
- K. M. Zak, R. Kitel, S. Przetocka, P. Golik, K. Guzik and B. Musielak, et al., Structure of the Complex of Human Programmed Death 1, PD-1, and Its Ligand PD-L1, Structure, 2015, 23, 2341–2348 CrossRef CAS PubMed.
- G. Scapin, X. Yang, W. W. Prosise, M. McCoy, P. Reichert and J. M. Johnston, et al., Structure of full-length human anti-PD1 therapeutic IgG4 antibody pembrolizumab, Nat. Struct. Mol. Biol., 2015, 22, 953–958 CrossRef CAS PubMed.
- M. K. Callahan, M. A. Postow and J. D. Wolchok, Targeting T Cell Co-receptors for Cancer Therapy, Immunity, 2016, 44, 1069–1078 CrossRef CAS PubMed.
- Y. Jiang, M. Chen, H. Nie and Y. Yuan, PD-1 and PD-L1 in cancer immunotherapy: clinical implications and future considerations, Hum. Vaccines Immunother., 2019, 15, 1111–1122 CrossRef PubMed.
- K. Liu, S. Tan, Y. Chai, D. Chen, H. Song and C. W.-H. Zhang, et al., Structural basis of anti-PD-L1 monoclonal antibody avelumab for tumor therapy, Cell Res., 2017, 27, 151–153 CrossRef PubMed.
- Y. Y. Syed, Durvalumab: First Global Approval, Drugs, 2017, 77, 1369–1376 CrossRef CAS PubMed.
- S. Antonia, S. B. Goldberg, A. Balmanoukian, J. E. Chaft, R. E. Sanborn and A. Gupta, et al., Safety and antitumour activity of durvalumab plus tremelimumab in non-small cell lung cancer: a multicentre, phase 1b study, Lancet Oncol., 2016, 17, 299–308 CrossRef CAS PubMed.
- X. Lin, X. Lu, G. Luo and H. Xiang, Progress in PD-1/PD-L1 pathway inhibitors: From biomacromolecules to small molecules, Eur. J. Med. Chem., 2019, 186, 111876 CrossRef PubMed.
- L. S. Chupak and X. Zheng, Compounds useful as immunomodulators, Patent WO2015034820 A1, 2015.
- K. M. Zak, P. Grudnik, K. Guzik, B. J. Zieba, B. Musielak and A. Dömling, et al., Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1), Oncotarget, 2016, 7, 30323–30335 CrossRef PubMed.
- K. Guzik, K. M. Zak, P. Grudnik, K. Magiera, B. Musielak and R. Törner, et al., Small-Molecule Inhibitors of the Programmed Cell Death-1/Programmed Death-Ligand 1 (PD-1/PD-L1) Interaction via Transiently Induced Protein States and Dimerization of PD-L1, J. Med. Chem., 2017, 60, 5857–5867 CrossRef CAS PubMed.
- L. Skalniak, K. M. Zak, K. Guzik, K. Magiera, B. Musielak and M. Pachota, et al., Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells, Oncotarget, 2017, 8, 72167–72181 CrossRef PubMed.
- M. Graupe, 264th Am Chem Soc (ACS) Natl Meet, Abstract number 3729357, 2022.
- X. Wang, F. Teng, L. Kong and J. Yu, PD-L1 expression in human cancers and its association with clinical outcomes, OncoTargets Ther., 2016, 9, 5023–5039 CrossRef CAS PubMed.
- T. Wang, S. Cai, Y. Cheng, W. Zhang, M. Wang and H. Sun, et al., Discovery of Small-Molecule Inhibitors of the PD-1/PD-L1 Axis That Promote PD-L1 Internalization and Degradation, J. Med. Chem., 2022, 65, 3879–3893 CrossRef CAS PubMed.
- J.-J. Park, E. P. Thi, V. H. Carpio, Y. Bi, A. G. Cole and B. D. Dorsey, et al., Checkpoint inhibition through small molecule-induced internalization of programmed death-ligand 1, Nat. Commun., 2021, 12, 1222 CrossRef CAS PubMed.
- H. K. Koblish, L. Wu, L.-C. S. Wang, P. C. C. Liu, R. Wynn and J. Rios-Doria, et al., Characterization of INCB086550, a potent and novel small-molecule PD-L1 inhibitor alternative to mAbs, Cancer Discovery, 2022, 12, 1482–1499 CrossRef PubMed.
- P. S. Ward, J. Patel, D. R. Wise, O. Abdel-Wahab, B. D. Bennett and H. A. Coller, et al., The Common Feature of Leukemia-Associated IDH1 and IDH2 Mutations Is a Neomorphic Enzyme Activity Converting α-Ketoglutarate to 2-Hydroxyglutarate, Cancer Cell, 2010, 17, 225–234 CrossRef CAS PubMed.
- W. Xu, H. Yang, Y. Liu, Y. Yang, P. Wang and S.-H. Kim, et al., Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases, Cancer Cell, 2011, 19, 17–30 CrossRef CAS PubMed.
- Celgene, A Phase 3, Multicenter, Open-label, Randomized Study Comparing the Efficacy and Safety of AG-221 (CC-90007) Versus Conventional Care Regimens in Older Subjects With Late Stage Acute Myeloid Leukemia Harboring an Isocitrate Dehydrogenase 2 Mutation, 2022, https://clinicaltrials.gov/ct2/show/NCT02577406, (accessed 16 Oct 2015).
- Institut de Recherches Internationales Servier, A Phase 1, Multicenter, Randomized, Controlled, Open-Label, Perioperative Study of AG-120 and AG-881 in Subjects With Recurrent, Non-Enhancing, IDH1 Mutant, Low Grade Glioma, 2022, https://ClinicalTrials.gov/show/NCT03343197, (accessed 17 Nov 2017).
- Institut de Recherches Internationales Servier, A Phase 1, Multicenter, Open-Label, Dose-Escalation and Expansion, Safety, Pharmacokinetic, Pharmacodynamic, and Clinical Activity Study of Orally Administered AG-881 in Patients With Advanced Solid Tumors, Including Gliomas, With an IDH1 and/or IDH2 Mutation, 2022, https://ClinicalTrials.gov/show/NCT02481154, (accessed 25 Jun 2015).
- F. Wang, J. Travins, B. DeLaBarre, V. Penard-Lacronique, S. Schalm and E. Hansen, et al., Targeted Inhibition of Mutant IDH2 in Leukemia Cells Induces Cellular Differentiation, Science, 2013, 340, 622–626 CrossRef CAS PubMed.
- D. Golub, N. Iyengar, S. Dogra, T. Wong, D. Bready and K. Tang, et al., Mutant Isocitrate Dehydrogenase Inhibitors as Targeted Cancer Therapeutics, Front. Oncol., 2019, 9, 417 CrossRef PubMed.
- K. Yen, J. Travins, F. Wang, M. D. David, E. Artin and K. Straley, et al., AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations, Cancer Discovery, 2017, 7, 478–493 CrossRef CAS PubMed.
- Z. Konteatis, E. Artin, B. Nicolay, K. Straley, A. K. Padyana and L. Jin, et al., Vorasidenib (AG-881): A First-in-Class, Brain-Penetrant Dual Inhibitor of Mutant IDH1 and 2 for Treatment of Glioma, ACS Med. Chem. Lett., 2020, 11, 101–107 CrossRef CAS PubMed.
- Y. Wang, Z. Wang, J. Liu, Y. Wang, R. Wu and R. Sheng, et al., Discovery of novel HBV capsid assembly modulators by structure-based virtual screening and bioassays, Bioorg. Med. Chem., 2021, 36, 116096 CrossRef CAS PubMed.
- S. D. Kuduk, B. Stoops, R. Alexander, A. M. Lam, C. Espiritu and R. Vogel, et al., Identification of a new class of HBV capsid assembly modulator, Bioorg. Med. Chem. Lett., 2021, 39, 127848 CrossRef CAS PubMed.
- Y. H. Lee, H.-M. Cha, J. Y. Hwang, S. Y. Park, A. G. Vishakantegowda and A. Imran, et al., Sulfamoylbenzamide-based Capsid Assembly Modulators for Selective Inhibition of Hepatitis B Viral Replication, ACS Med. Chem. Lett., 2021, 12, 242–248 CrossRef CAS PubMed.
- Y. Ren, Y. Ma, S. Cherukupalli, J. E. Tavis, L. Menéndez-Arias and X. Liu, et al., Discovery and Optimization of Benzenesulfonamide-based HBV Capsid Modulators via Contemporary Medicinal Chemistry Strategies, Eur. J. Med. Chem., 2020, 206, 112714 CrossRef CAS PubMed.
- C. Ko, R. Bester, X. Zhou, Z. Xu, C. Blossey and J. Sacherl, et al., A New Role for Capsid Assembly Modulators To Target Mature Hepatitis B Virus Capsids and Prevent Virus Infection, Antimicrob. Agents Chemother., 2019, 64, e01440-19 CrossRef PubMed.
- S. Banerjee, F. Mahmud, S. Deng, L. Ma, M.-K. Yun and S. O. Fakayode, et al., X-ray Crystallography-Guided Design, Antitumor Efficacy, and QSAR Analysis of Metabolically Stable Cyclopenta-Pyrimidinyl Dihydroquinoxalinone as a Potent Tubulin Polymerization Inhibitor, J. Med. Chem., 2021, 64, 13072–13095 CrossRef CAS PubMed.
- T. Zhu, S.-H. Wang, D. Li, S.-Y. Wang, X. Liu and J. Song, et al., Progress of tubulin polymerization activity detection methods, Bioorg. Med. Chem. Lett., 2021, 37, 127698 CrossRef CAS PubMed.
- Y. Ren, Y. Wang, G. Li, Z. Zhang, L. Ma and B. Cheng, et al., Discovery of Novel Benzimidazole and Indazole Analogues as Tubulin Polymerization Inhibitors with Potent Anticancer Activities, J. Med. Chem., 2021, 64, 4498–4515 CrossRef CAS PubMed.
- H. Chen, S. Deng, N. Albadari, M.-K. Yun, S. Zhang and Y. Li, et al., Design, Synthesis, and Biological Evaluation of Stable Colchicine-Binding Site Tubulin Inhibitors 6-Aryl-2-benzoyl-pyridines as Potential Anticancer Agents, J. Med. Chem., 2021, 64, 12049–12074 CrossRef CAS PubMed.
- E. Sisco and K. L. Barnes, Design, Synthesis, and Biological Evaluation of Novel 1,3-Oxazole Sulfonamides as Tubulin Polymerization Inhibitors, ACS Med. Chem. Lett., 2021, 12, 1030–1037 CrossRef CAS PubMed.
- T. Mühlethaler, D. Gioia, A. E. Prota, M. E. Sharpe, A. Cavalli and M. O. Steinmetz, Comprehensive Analysis of Binding Sites in Tubulin, Angew. Chem., Int. Ed., 2021, 60, 13331–13342 CrossRef PubMed.
- J. Pujols, S. Peña-Díaz, D. F. Lázaro, F. Peccati, F. Pinheiro and D. González, et al., Small molecule inhibits α-synuclein aggregation, disrupts amyloid fibrils, and prevents degeneration of dopaminergic neurons, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 201804198 CrossRef PubMed.
- D. L. Price, M. A. Koike, A. Khan, W. Wrasidlo, E. Rockenstein and E. Masliah, et al., The small molecule alpha-synuclein misfolding inhibitor, NPT200-11, produces multiple benefits in an animal model of Parkinson's disease, Sci. Rep., 2018, 8, 16165 CrossRef PubMed.
- S. Peña-Díaz, J. Pujols, M. Conde-Giménez, A. Čarija, E. Dalfo and J. García, et al., ZPD-2, a Small Compound That Inhibits α-Synuclein Amyloid Aggregation and Its Seeded Polymerization, Front. Mol. Neurosci., 2019, 12, 306 CrossRef PubMed.
- A. E. Butler, J. Janson, S. Bonner-Weir, R. Ritzel, R. A. Rizza and P. C. Butler, Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes, Diabetes, 2003, 52, 102–110 CrossRef CAS PubMed.
- J. J. Meier, R. Kayed, C.-Y. Lin, T. Gurlo, L. Haataja and S. Jayasinghe, et al., Inhibition of human IAPP fibril formation does not prevent β-cell death: evidence for distinct actions of oligomers and fibrils of human IAPP, Am. J. Physiol., 2006, 291, E1317–E1324 CAS.
- R. A. Ritzel, J. J. Meier, C.-Y. Lin, J. D. Veldhuis and P. C. Butler, Human Islet Amyloid Polypeptide Oligomers Disrupt Cell Coupling, Induce Apoptosis, and Impair Insulin Secretion in Isolated Human Islets, Diabetes, 2007, 56, 65–71 CrossRef CAS PubMed.
- L. Haataja, T. Gurlo, C. J. Huang and P. C. Butler, Islet Amyloid in Type 2 Diabetes, and the Toxic Oligomer Hypothesis, Endocr. Rev., 2008, 29, 303–316 CrossRef CAS PubMed.
- J. Cho, J. Kim, J. Shin, J. Shin and K. Yoon, β-cell mass in people with type 2 diabetes, J. Diabetes Invest., 2011, 2, 6–17 CrossRef CAS PubMed.
- C. A. Jurgens, M. N. Toukatly, C. L. Fligner, J. Udayasankar, S. L. Subramanian and S. Zraika, et al., β-Cell Loss and β-Cell Apoptosis in Human Type 2 Diabetes Are Related to Islet Amyloid Deposition, Am. J. Pathol., 2011, 178, 2632–2640 CrossRef CAS PubMed.
- J. J. Meier and R. C. Bonadonna, Role of Reduced β-Cell Mass Versus Impaired β-Cell Function in the Pathogenesis of Type 2 Diabetes, Diabetes Care, 2013, 36, S113–S119 CrossRef CAS PubMed.
- J. D. Knight and A. D. Miranker, Phospholipid Catalysis of Diabetic Amyloid Assembly, J. Mol. Biol., 2004, 341, 1175–1187 CrossRef CAS PubMed.
- J. A. Williamson, J. P. Loria and A. D. Miranker, Helix Stabilization Precedes Aqueous and Bilayer-Catalyzed Fiber Formation in Islet Amyloid Polypeptide, J. Mol. Biol., 2009, 393, 383–396 CrossRef CAS PubMed.
- J. A. Hebda and A. D. Miranker, The Interplay of Catalysis and Toxicity by Amyloid Intermediates on Lipid Bilayers: Insights from Type II Diabetes, Annu. Rev. Biophys., 2009, 38, 125–152 CrossRef CAS PubMed.
- A. K. Bishoyi, P. H. Roham, K. Rachineni, S. Save, M. A. Hazari and S. Sharma, et al., Human islet amyloid polypeptide (hIAPP) - a curse in type II diabetes mellitus: insights from structure and toxicity studies, Biol. Chem., 2021, 402, 133–153 CAS.
- L.-M. Yan, M. Tatarek-Nossol, A. Velkova, A. Kazantzis and A. Kapurniotu, Design of a mimic of nonamyloidogenic and bioactive human islet amyloid polypeptide (IAPP) as nanomolar affinity inhibitor of IAPP cytotoxic fibrillogenesis, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2046–2051 CrossRef CAS PubMed.
- A. Spanopoulou, L. Heidrich, H. Chen, C. Frost, D. Hrle and E. Malideli, et al., Designed Macrocyclic Peptides as Nanomolar Amyloid Inhibitors Based on Minimal Recognition Elements, Angew. Chem., Int. Ed., 2018, 57, 14503–14508 CrossRef CAS PubMed.
- V. Armiento, K. Hille, D. Naltsas, J. S. Lin, A. E. Barron and A. Kapurniotu, The Human Host-Defense Peptide Cathelicidin LL-37 is a Nanomolar Inhibitor of Amyloid Self-Assembly of Islet Amyloid Polypeptide (IAPP), Angew. Chem., Int. Ed., 2020, 59, 12837–12841 CrossRef CAS PubMed.
- F. Meng, A. Abedini, A. Plesner, C. B. Verchere and D. P. Raleigh, The Flavanol (−)-Epigallocatechin 3-Gallate Inhibits Amyloid Formation by Islet Amyloid Polypeptide, Disaggregates Amyloid Fibrils, and Protects Cultured Cells against IAPP-Induced Toxicity, Biochemistry, 2010, 49, 8127–8133 CrossRef CAS PubMed.
- A. Pithadia, J. R. Brender, C. A. Fierke and A. Ramamoorthy, Inhibition of IAPP Aggregation and Toxicity by Natural Products and Derivatives, J. Diabetes Res., 2016, 2016, 2046327 Search PubMed.
- D. E. Ehrnhoefer, J. Bieschke, A. Boeddrich, M. Herbst, L. Masino and R. Lurz, et al., EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers, Nat. Struct. Mol. Biol., 2008, 15, 558–566 CrossRef CAS PubMed.
- A. R. A. Ladiwala, J. C. Lin, S. S. Bale, A. M. Marcelino-Cruz, M. Bhattacharya and J. S. Dordick, et al., Resveratrol Selectively Remodels Soluble Oligomers and Fibrils of Amyloid Aβ into Off-pathway Conformers, J. Biol. Chem., 2010, 285, 24228–24237 CrossRef CAS PubMed.
- P. Nedumpully-Govindan, A. Kakinen, E. H. Pilkington, T. P. Davis, P. C. Ke and F. Ding, Stabilizing Off-pathway Oligomers by Polyphenol Nanoassemblies for IAPP Aggregation Inhibition, Sci. Rep., 2016, 6, 19463 CrossRef CAS PubMed.
- P. C. Ke, E. H. Pilkington, Y. Sun, I. Javed, A. Kakinen and G. Peng, et al., Mitigation of Amyloidosis with Nanomaterials, Adv. Mater., 2020, 32, 1901690 CrossRef CAS PubMed.
- S. Sinha, D. H. J. Lopes, Z. Du, E. S. Pang, A. Shanmugam and A. Lomakin, et al., Lysine-Specific Molecular Tweezers Are Broad-Spectrum Inhibitors of Assembly and Toxicity of Amyloid Proteins, J. Am. Chem. Soc., 2011, 133, 16958–16969 CrossRef CAS PubMed.
- D. H. J. Lopes, A. Attar, G. Nair, E. Y. Hayden, Z. Du and K. McDaniel, et al., Molecular Tweezers Inhibit Islet Amyloid Polypeptide Assembly and Toxicity by a New Mechanism, ACS Chem. Biol., 2015, 10, 1555–1569 CrossRef CAS PubMed.
- I. Saraogi, J. A. Hebda, J. Becerril, L. A. Estroff, A. D. Miranker and A. D. Hamilton, Synthetic α-Helix Mimetics as Agonists and Antagonists of Islet Amyloid Polypeptide Aggregation, Angew. Chem., Int. Ed., 2010, 49, 736–739 CrossRef CAS PubMed.
- S. Kumar, D. E. Schlamadinger, M. A. Brown, J. M. Dunn, B. Mercado and J. A. Hebda, et al., Islet Amyloid-Induced Cell Death and Bilayer Integrity Loss Share a Molecular Origin Targetable with Oligopyridylamide-Based α-Helical Mimetics, Chem. Biol., 2015, 22, 369–378 CrossRef CAS PubMed.
- S. Kumar, M. Birol, D. E. Schlamadinger, S. P. Wojcik, E. Rhoades and A. D. Miranker, Foldamer-mediated manipulation of a pre-amyloid toxin, Nat. Commun., 2016, 7, 11412 CrossRef CAS PubMed.
- M. S. Saravanan, S. Ryazanov, A. Leonov, J. Nicolai, P. Praest and A. Giese, et al., The small molecule inhibitor anle145c thermodynamically traps human islet amyloid peptide in the form of non-cytotoxic oligomers, Sci. Rep., 2019, 9, 19023 CrossRef CAS PubMed.
- J. Wagner, S. Ryazanov, A. Leonov, J. Levin, S. Shi and F. Schmidt, et al., Anle138b: a novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson's disease, Acta Neuropathol., 2013, 125, 795–813 CrossRef CAS PubMed.
- P. Velander, L. Wu, F. Henderson, S. Zhang, D. R. Bevan and B. Xu, Natural product-based amyloid inhibitors, Biochem. Pharmacol., 2017, 139, 40–55 CrossRef CAS PubMed.
- M. Sato, K. Murakami, M. Uno, Y. Nakagawa, S. Katayama and K. Akagi, et al., Site-specific Inhibitory Mechanism for Amyloid β42 Aggregation by Catechol-type Flavonoids Targeting the Lys Residues, J. Biol. Chem., 2013, 288, 23212–23224 CrossRef CAS PubMed.
- L. Wu, P. Velander, A. M. Brown, Y. Wang, D. Liu and D. R. Bevan, et al., Rosmarinic Acid Potently Detoxifies Amylin Amyloid and Ameliorates Diabetic Pathology in a Transgenic Rat Model of Type 2 Diabetes, ACS Pharmacol. Transl. Sci., 2021, 4, 1322–1337 CrossRef CAS PubMed.
- P. Velander, L. Wu, W. K. Ray, R. F. Helm and B. Xu, Amylin Amyloid Inhibition by Flavonoid Baicalein: Key Roles of Its Vicinal Dihydroxyl Groups of the Catechol Moiety, Biochemistry, 2016, 55, 4255–4258 CrossRef CAS PubMed.
- S. Feng, X.-H. Song and C.-M. Zeng, Inhibition of amyloid fibrillation of lysozyme by phenolic compounds involves quinoprotein formation, FEBS Lett., 2012, 586, 3951–3955 CrossRef CAS PubMed.
- T.-T. An, S. Feng and C.-M. Zeng, Oxidized epigallocatechin gallate inhibited lysozyme fibrillation more strongly than the native form, Redox Biol., 2016, 11, 315–321 CrossRef PubMed.
- V. T. Huong, T. Shimanouchi, N. Shimauchi, H. Yagi, H. Umakoshi and Y. Goto, et al., Catechol derivatives inhibit the fibril formation of amyloid-β peptides, J. Biosci. Bioeng., 2010, 109, 629–634 CrossRef CAS PubMed.
- D. Maity, S. Kumar, R. AlHussein, L. Gremer, M. Howarth and L. Karpauskaite, et al., Sub-stoichiometric inhibition of IAPP aggregation: a peptidomimetic approach to anti-amyloid agents, RSC Chem. Biol., 2020, 1, 225–232 RSC.
- H. A. Lashuel, Rethinking protein aggregation and drug discovery in neurodegenerative diseases: Why we need to embrace complexity?, Curr. Opin. Chem. Biol., 2021, 64, 67–75 CrossRef CAS PubMed.
- D. Frees, S. N. A. Qazi, P. J. Hill and H. Ingmer, Alternative roles of ClpX and ClpP in Staphylococcus aureus stress tolerance and virulence, Mol. Microbiol., 2003, 48, 1565–1578 CrossRef CAS PubMed.
- H. Brötz-Oesterhelt, D. Beyer, H.-P. Kroll, R. Endermann, C. Ladel and W. Schroeder, et al., Dysregulation of bacterial proteolytic machinery by a new class of antibiotics, Nat. Med., 2005, 11, 1082–1087 CrossRef PubMed.
- T. Böttcher and S. A. Sieber, β-Lactones as Specific Inhibitors of ClpP Attenuate the Production of Extracellular Virulence Factors of Staphylococcus aureus, J. Am. Chem. Soc., 2008, 130, 14400–14401 CrossRef PubMed.
- K. Nouri, Y. Feng and A. D. Schimmer, Mitochondrial ClpP serine protease-biological function and emerging target for cancer therapy, Cell Death Dis., 2020, 11, 841 CrossRef CAS PubMed.
- A. Cole, Z. Wang, E. Coyaud, V. Voisin, M. Gronda and Y. Jitkova, et al., Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia, Cancer Cell, 2015, 27, 864–876 CrossRef CAS PubMed.
- S. Bhaskaran, G. Pharaoh, R. Ranjit, A. Murphy, S. Matsuzaki and B. C. Nair, et al., Loss of mitochondrial protease ClpP protects mice from diet-induced obesity and insulin resistance, EMBO Rep., 2018, 19, e45009 CrossRef PubMed.
- D. Frees, U. Gerth and H. Ingmer, Clp chaperones and proteases are central in stress survival, virulence and antibiotic resistance of Staphylococcus aureus, Int. J. Med. Microbiol., 2013, 304, 142–149 CrossRef PubMed.
- R. T. Sauer, D. N. Bolon, B. M. Burton, R. E. Burton, J. M. Flynn and R. A. Grant, et al., Sculpting the Proteome with AAA+ Proteases and Disassembly Machines, Cell, 2004, 119, 9–18 CrossRef CAS PubMed.
- T. A. Baker and R. T. Sauer, ClpXP, an ATP-powered unfolding and protein-degradation machine, Biochim. Biophys. Acta, Mol. Cell Res., 2012, 1823, 15–28 CrossRef CAS PubMed.
- M. Gersch, A. List, M. Groll and S. A. Sieber, Insights into Structural Network Responsible for Oligomerization and Activity of Bacterial Virulence Regulator Caseinolytic Protease P (ClpP) Protein, J. Biol. Chem., 2012, 287, 9484–9494 CrossRef CAS PubMed.
- M. Drag and G. S. Salvesen, Emerging principles in protease-based drug discovery, Nat. Rev. Drug Discovery, 2010, 9, 690–701 CrossRef CAS PubMed.
- Y. Ju, Q. An, Y. Zhang, K. Sun, L. Bai and Y. Luo, Recent advances in Clp protease modulation to address virulence, resistance and persistence of MRSA infection, Drug Discovery Today, 2021, 26, 2190–2197 CrossRef CAS PubMed.
- M. Gersch, R. Kolb, F. Alte, M. Groll and S. A. Sieber, Disruption of oligomerization and dehydroalanine formation as mechanisms for ClpP protease inhibition, J. Am. Chem. Soc., 2013, 136, 1360–1366 CrossRef PubMed.
- B. Lipinska, S. Sharma and C. Georgopoulos, Sequence analysis and regulation of the htrA gene of Escherichia coli: a sigma 32-independent mechanism of heat-inducible transcription, Nucleic Acids Res., 1988, 16, 10053–10067 CrossRef CAS PubMed.
- C. Spiess, A. Beil and M. Ehrmann, A Temperature-Dependent Switch from Chaperone to Protease in a Widely Conserved Heat Shock Protein, Cell, 1999, 97, 339–347 CrossRef CAS PubMed.
- J. Skórko-Glonek, D. Zurawa, E. Kuczwara, M. Wozniak, Z. Wypych and B. Lipinska, The Escherichia coli heat shock protease HtrA participates in defense against oxidative stress, Mol. Gen. Genet., 1999, 262, 342–350 CrossRef PubMed.
- M. R. Leandro, L. de Souza Vespoli, L. F. Andrade, F. S. Soares, A. L. Boechat and V. R. Pimentel, et al., DegP protease is essential for tolerance to salt stress in the plant growth-promoting bacterium Gluconacetobacter diazotrophicus PAL5, Microbiol. Res., 2020, 243, 126654 CrossRef PubMed.
- T. Krojer, J. Sawa, R. Huber and T. Clausen, HtrA proteases have a conserved activation mechanism that can be triggered by distinct molecular cues, Nat. Struct. Mol. Biol., 2010, 17, 844–852 CrossRef CAS PubMed.
- J. Skórko-Glonek, D. Zurawa, F. Tanfani, A. Scirè, A. Wawrzynów and J. Narkiewicz, et al., The N-terminal region of HtrA heat shock protease from Escherichia coli is essential for stabilization of HtrA primary structure and maintaining of its oligomeric structure, Biochim. Biophys. Acta, 2003, 1649, 171–182 CrossRef PubMed.
- A. J. Bäumler, J. G. Kusters, I. Stojiljkovic and F. Heffron, Salmonella typhimurium loci involved in survival within macrophages, Infect. Immun., 1994, 62, 1623–1630 CrossRef PubMed.
- K. Johnson, I. Charles, G. Dougan, D. Pickard, P. O'Gaora and G. Costa, et al., The role of a stress-response protein in Salmonella typhimurium virulence, Mol. Microbiol., 1991, 5, 401–407 CrossRef CAS PubMed.
- N. Dorrell, S.-R. Li, P. H. Everest, G. Dougan and B. W. Wren, Construction and characterisation of a Yersinia enterocolitica O:8 ompR mutant, FEMS Microbiol. Lett., 1998, 165, 145–151 CrossRef CAS PubMed.
- G. E. Purdy, M. Hong and S. M. Payne, Shigella flexneri DegP Facilitates IcsA Surface Expression and Is Required for Efficient Intercellular Spread, Infect. Immun., 2002, 70, 6355–6364 CrossRef CAS PubMed.
- S. I. Resto-Ruiz, D. Sweger, R. H. Widen, N. Valkov and B. E. Anderson, Transcriptional Activation of the htrA (High-Temperature Requirement A) Gene from Bartonella henselae, Infect. Immun., 2000, 68, 5970–5978 CrossRef CAS PubMed.
- T. Krojer, M. Garrido-Franco, R. Huber, M. Ehrmann and T. Clausen, Crystal structure of DegP (HtrA) reveals a new protease-chaperone machine, Nature, 2002, 416, 455–459 CrossRef CAS PubMed.
- T. Krojer, M. Garrido-Franco, R. Huber, M. Ehrmann and T. Clausen, Correction: Corrigendum: Crystal structure of DegP (HtrA) reveals a new protease-chaperone machine, Nature, 2002, 417, 102–102 CrossRef CAS.
- T. Krojer, J. Sawa, E. Schäfer, H. R. Saibil, M. Ehrmann and T. Clausen, Structural basis for the regulated protease and chaperone function of DegP, Nature, 2008, 453, 885–890 CrossRef CAS PubMed.
- P. Hauske, M. Meltzer, C. Ottmann, T. Krojer, T. Clausen and M. Ehrmann, et al., Selectivity profiling of DegP substrates and inhibitors, Bioorg. Med. Chem., 2009, 17, 2920–2924 CrossRef CAS PubMed.
- M. Merdanovic, S. G. Burston, A. L. Schmitz, S. Köcher, S. Knapp and T. Clausen, et al., Activation by substoichiometric inhibition, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 1414–1418 CrossRef CAS PubMed.
- M. Merdanovic, N. Mamant, M. Meltzer, S. Poepsel, A. Auckenthaler and R. Melgaard, et al., Determinants of structural and functional plasticity of a widely conserved protease chaperone complex, Nat. Struct. Mol. Biol., 2010, 17, 837–843 CrossRef CAS PubMed.
- R. W. Harkness, Y. Toyama, Z. A. Ripstein, H. Zhao, A. I. M. Sever and Q. Luan, et al., Competing stress-dependent oligomerization pathways regulate self-assembly of the periplasmic protease-chaperone DegP, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2109732118 CrossRef CAS PubMed.
- J.-P. Renaud, C. Chung, U. H. Danielson, U. Egner, M. Hennig and R. E. Hubbard, et al., Biophysics in drug discovery: impact, challenges and opportunities, Nat. Rev. Drug Discovery, 2016, 15, 679–698 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov and O. Ronneberger, et al., Highly accurate protein structure prediction with AlphaFold, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- K. Tunyasuvunakool, J. Adler, Z. Wu, T. Green, M. Zielinski and A. Žídek, et al., Highly accurate protein structure prediction for the human proteome, Nature, 2021, 596, 590–596 CrossRef CAS PubMed.
- A. Mullard, What does AlphaFold mean for drug discovery?, Nat. Rev. Drug Discovery, 2021, 20, 725–727 CrossRef CAS PubMed.
- A. B. Tong, J. D. Burch, D. McKay, C. Bustamante, M. A. Crackower and H. Wu, Could AlphaFold revolutionize chemical therapeutics?, Nat. Struct. Mol. Biol., 2021, 28, 771–772 CrossRef CAS PubMed.
- J.-P. Renaud, A. Chari, C. Ciferri, W. Liu, H.-W. Rémigy and H. Stark, et al., Cryo-EM in drug discovery: achievements, limitations and prospects, Nat. Rev. Drug Discovery, 2018, 17, 471–492 CrossRef CAS PubMed.
- M. J. Robertson, J. G. Meyerowitz and G. Skiniotis, Drug discovery in the era of cryo-electron microscopy, Trends Biochem. Sci., 2021, 47, 124–135 CrossRef PubMed.
- J. H. V. Drie and L. Tong, Cryo-EM as a powerful tool for drug discovery, Bioorg. Med. Chem. Lett., 2020, 30, 127524 CrossRef PubMed.
- J. A. Terrett, H. Chen, D. G. Shore, E. Villemure, R. Larouche-Gauthier and M. Déry, et al., Tetrahydrofuran-Based Transient Receptor Potential Ankyrin 1 (TRPA1) Antagonists: Ligand-Based Discovery, Activity in a Rodent Asthma Model, and Mechanism-of-Action via Cryogenic Electron Microscopy, J. Med. Chem., 2021, 64, 3843–3869 CrossRef CAS PubMed.
- A. W. P. Fitzpatrick, B. Falcon, S. He, A. G. Murzin, G. Murshudov and H. J. Garringer, et al., Cryo-EM structures of tau filaments from Alzheimer's disease, Nature, 2017, 547, 185–190 CrossRef CAS PubMed.
- R. Guerrero-Ferreira, N. M. Taylor, D. Mona, P. Ringler, M. E. Lauer and R. Riek, et al., Cryo-EM structure of alpha-synuclein fibrils, eLife, 2018, 7, e36402 CrossRef PubMed.
- B. Li, P. Ge, K. A. Murray, P. Sheth, M. Zhang and G. Nair, et al., Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel, Nat. Commun., 2018, 9, 3609 CrossRef PubMed.
- S. Lövestam, F. A. Koh, B. van Knippenberg, A. Kotecha, A. G. Murzin and M. Goedert, et al., Assembly of recombinant tau into filaments identical to those of Alzheimer's disease and chronic traumatic encephalopathy, eLife, 2022, 11, e76494 CrossRef PubMed.
- B. J. Greber, D. B. Toso, J. Fang and E. Nogales, The complete structure of the human TFIIH core complex, eLife, 2019, 8, e44771 CrossRef PubMed.
- A. Ayoub, S. H. Park, Y.-T. Lee, U.-S. Cho and Y. Dou, Regulation of MLL1 Methyltransferase Activity in Two Distinct Nucleosome Binding Modes, Biochemistry, 2022, 61, 1–9 CrossRef CAS PubMed.
- J. Chen, Q. Wang, B. Malone, E. Llewellyn, Y. Pechersky and K. Maruthi, et al., Ensemble cryo-EM reveals conformational states of the nsp13 helicase in the SARS-CoV-2 helicase replication–transcription complex, Nat. Struct. Mol. Biol., 2022, 29, 250–260 CrossRef CAS PubMed.
- Z. Guo, B. Li, L.-T. Cheng, S. Zhou, J. A. McCammon and J. Che, Identification of Protein–Ligand Binding Sites by the Level-Set Variational Implicit-Solvent Approach, J. Chem. Theory Comput., 2015, 11, 753–765 CrossRef CAS PubMed.
- M. Sciacovelli, G. Guzzo, V. Morello, C. Frezza, L. Zheng and N. Nannini, et al. The Mitochondrial Chaperone TRAP1 Promotes Neoplastic Growth by Inhibiting Succinate Dehydrogenase, Cell Metab., 2013, 17, 988–999 CrossRef CAS PubMed.
- S. Yoshida, S. Tsutsumi, G. Muhlebach, C. Sourbier, M.-J. Lee and S. Lee, et al., Molecular chaperone TRAP1 regulates a metabolic switch between mitochondrial respiration and aerobic glycolysis, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E1604–E1612 CAS.
- M. Taipale, D. F. Jarosz and S. Lindquist, HSP90 at the hub of protein homeostasis: emerging mechanistic insights, Nat. Rev. Mol. Cell Biol., 2010, 11, 515–528 CrossRef CAS PubMed.
- E. Moroni, D. A. Agard and G. Colombo, The Structural Asymmetry of Mitochondrial Hsp90 (Trap1) Determines Fine Tuning of Functional Dynamics, J. Chem. Theory Comput., 2018, 14, 1033–1044 CrossRef CAS PubMed.
- C. Sanchez-Martin, E. Moroni, M. Ferraro, C. Laquatra, G. Cannino and I. Masgras, et al., Rational Design of Allosteric and Selective Inhibitors of the Molecular Chaperone TRAP1, Cell Rep., 2020, 31, 107531 CrossRef CAS PubMed.
- M. Ferraro, E. Moroni, E. Ippoliti, S. Rinaldi, C. Sanchez-Martin and A. Rasola, et al., Machine Learning of Allosteric Effects: The Analysis of Ligand-Induced Dynamics to Predict Functional Effects in TRAP1, J. Phys. Chem. B, 2021, 125, 101–114 CrossRef CAS PubMed.
- S. A. Serapian, C. Sanchez-Martín, E. Moroni, A. Rasola and G. Colombo, Targeting the mitochondrial chaperone TRAP1: strategies and therapeutic perspectives, Trends Pharmacol. Sci., 2021, 42, 566–576 CrossRef CAS PubMed.
- G. Abrusán and J. A. Marsh, Ligand-Binding-Site Structure Shapes Allosteric Signal Transduction and the Evolution of Allostery in Protein Complexes, Mol. Biol. Evol., 2019, 36, 1711–1727 CrossRef PubMed.
- A. S. Bhat, R. D. Schaeffer, L. Kinch, K. E. Medvedev and N. V. Grishin, Recent advances suggest increased influence of selective pressure in allostery, Curr. Opin. Struct. Biol., 2020, 62, 183–188 CrossRef CAS PubMed.
- A. Christopoulos, L. T. May, V. A. Avlani and P. M. Sexton, G-protein-coupled receptor allosterism: the promise and the problem(s), Biochem. Soc. Trans., 2004, 32, 873–877 CrossRef CAS PubMed.
- Y. Pan and M. M. Mader, Principles of Kinase Allosteric Inhibition and Pocket Validation, J. Med. Chem., 2022, 65, 5288–5299 CrossRef CAS PubMed.
- G. Abrusán and J. A. Marsh, Ligand Binding Site Structure Influences the Evolution of Protein Complex Function and Topology, Cell Rep., 2018, 22, 3265–3276 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2023 |