
Irreversible inhibition of BoNT/A protease: proximity-driven reactivity contingent upon a bifunctional approach†
Lewis D.
Turner‡
 a,
Alexander L.
Nielsen‡
ab,
Lucy
Lin
a,
Sabine
Pellett
c,
Takashi
Sugane
a,
Margaret E.
Olson
ad,
Eric A.
Johnson
c and
Kim D.
Janda
*a
a,
Alexander L.
Nielsen‡
ab,
Lucy
Lin
a,
Sabine
Pellett
c,
Takashi
Sugane
a,
Margaret E.
Olson
ad,
Eric A.
Johnson
c and
Kim D.
Janda
*a
aDepartments of Chemistry and Immunology, The Skaggs Institute for Chemical Biology, Worm Institute of Research and Medicine (WIRM), Scripps Research, 10550 N Torrey Pines Road, La Jolla, CA 92037, USA. E-mail: kdjanda@scripps.edu
bCenter for Biopharmaceuticals & Department of Drug Design and Pharmacology, Faculty of Health and Medical Sciences, University of Copenhagen, Universitetsparken 2, DK-2100, Copenhagen, Denmark
cDepartment of Bacteriology, University of Wisconsin, 1550 Linden Drive, Madison, WI 53706, USA
dCollege of Pharmacy, Roosevelt University, Schaumburg, IL 60173, USA
First published on 19th May 2021
Abstract
Botulinum neurotoxin A (BoNT/A) is categorized as a Tier 1 bioterrorism agent and persists within muscle neurons for months, causing paralysis. A readily available treatment that abrogates BoNT/A's toxicity and longevity is a necessity in the event of a widespread BoNT/A attack and for clinical treatment of botulism, yet remains an unmet need. Herein, we describe a comprehensive warhead screening campaign of bifunctional hydroxamate-based inhibitors for the irreversible inhibition of the BoNT/A light chain (LC). Using the 2,4-dichlorocinnamic hydroxamic acid (DCHA) metal-binding pharmacophore modified with a pendent warhead, a total of 37 compounds, possessing 13 distinct warhead types, were synthesized and evaluated for time-dependent inhibition against the BoNT/A LC. Iodoacetamides, maleimides, and an epoxide were found to exhibit time-dependent inhibition and their kGSH measured as a description of reactivity. The epoxide exhibited superior time-dependent inhibition over the iodoacetamides, despite reacting with glutathione (GSH) 51-fold slower. The proximity-driven covalent bond achieved with the epoxide inhibitor was contingent upon the vital hydroxamate–Zn2+ anchor in placing the warhead in an optimal position for reaction with Cys165. Monofunctional control compounds exemplified the necessity of the bifunctional approach, and Cys165 modification was confirmed through high-resolution mass spectrometry (HRMS) and ablation of time-dependent inhibitory activity against a C165A variant. Compounds were also evaluated against BoNT/A-intoxicated motor neuron cells, and their cell toxicity, serum stability, and selectivity against matrix metalloproteinases (MMPs) were characterized. The bifunctional approach allows the use of less intrinsically reactive electrophiles to intercept Cys165, thus expanding the toolbox of potential warheads for selective irreversible BoNT/A LC inhibition. We envision that this dual-targeted strategy is amenable to other metalloproteases that also possess non-catalytic cysteines proximal to the active-site metal center.
Introduction
Botulinum neurotoxins (BoNTs) are produced by a pathogenic Gram-positive, anaerobic bacterium known as Clostridium botulinum, and are amongst the most neurotoxic agents known.1 To date, seven immunologically distinct serotypes of BoNT (A–G) have been identified and confirmed to be potent neurotoxins, with serotypes A, B, E and F causing botulism in humans.2,3 In addition, several naturally occurring hybrid toxins (CD, DC, F5A/HA) have been described, as well as more distantly related putative BoNTs.4–6 BoNT/A has the highest incidence rate amongst its serotype counterparts, is the most toxic, and causes the most severe botulism,7 with an estimated intravenous LD50 of 1–2 ng kg−1 and ∼10 ng kg−1 by inhalation.8 Furthermore, it has a half-life of several months once internalized in neurons.9 The toxicity, persistence, ease of production, and dissemination of BoNT/A, coupled with no effective post-intoxication remedy, has warranted its designation by the Centers for Disease Control and Prevention (CDC) as a Tier 1 bioterrorism agent alongside smallpox, plague, and anthrax.10In contrast to its potential misuse in bioterrorism, BoNT/A is used for a wide array of therapeutic applications. Disorders that are characterized by aberrant muscular spasticity have been a focus of BoNT/A treatment, including migraines, post-stroke injury, spinal cord injury and more recently, cerebral palsy.11–14 BoNT/A is also used cosmetically for a variety of aesthetic procedures, with a staggering 7.7 million procedures carried out in 2019, up 878% from the year 2000.15 The huge increase in BoNT/A cosmetic usage over the past two decades comes with health concerns; consistent and long-term use of BoNT/A has been associated with adverse side-effects including necrotizing fasciitis, respiratory compromise, iatrogenic botulism, and even anaphylactic shock, leading to death.16
Both the toxicity and the therapeutic potential of BoNT/A arise from its ability to disrupt vesicle fusion at neuronal termini, thus preventing acetylcholine release and synaptic transmission, which results in flaccid paralysis and botulism.17 The BoNT/A holotoxin is a large protein complex of 150 kDa that consists of a heavy chain (HC, 100 kDa) and a Zn2+ metalloprotease light chain (LC, 50 kDa). The HC associates with the neuronal membrane through recognition of specific gangliosides and receptor proteins resulting in endocytosis of the toxin.18 Endosomal acidification results in conformation changes leading to membrane insertion of N-terminus of the HC, which subsequently leads to translocation of the LC to the cytosol.19 Once internalized, the LC is released from the HC via disulfide bond reduction, and is able to selectively bind synaptosomal-associated protein 25 (SNAP-25) and implement site-specific cleavage through Zn2+-mediated proteolysis. This disrupts the action of the soluble N-ethylmaleimide sensitive factor (NSF) attachment protein receptor (SNARE) complex, a crucial component of vesicular fusion to the synaptosomal membrane.20
The potential misuse of BoNT/A in a wide-scale bioterrorism event, coupled with adverse events stemming from its prolific therapeutic and cosmetic use, means that a readily available, non-toxic, and effective reversal agent is vital. Currently, the only approved treatment for BoNT/A-induced botulism is an equine-derived antitoxin, which targets and neutralizes the BoNT/A holotoxin before neuronal internalization has taken place. However, this treatment method has a limited therapeutic window (12–24 h post-intoxication) as the antibody-based antitoxin cannot enter neuronal compartments.21 This pitfall outlines the necessity to develop a treatment that can not only inhibit internalized BoNT/A but also address its longevity.
To this end, our group has discovered several examples of reversible small-molecule inhibitors of the BoNT/A LC, with the majority possessing metal-binding groups (MBGs) that disrupt SNAP-25 cleavage through coordination to the active site Zn2+.22,23 One of the most potent inhibitors to date is DCHA (Fig. 1A) which exhibits a Ki of 0.3 μM against the BoNT/A LC.24 Structure–activity relationships (SARs) suggest that subtle changes to the DCHA core (e.g. removal of the transcinnamic double bond, removal of Cl atoms from phenyl ring) result in a significant loss in activity.24 Despite displaying promising in vitro activity, and modest activity in vivo against BoNT/A-challenged mice, DCHA (and other reversible inhibitors) have not progressed beyond pre-clinical studies,25 due in part to the transient nature of reversible inhibition. Because of the longevity of the BoNT/A LC in neurons, a long-term consistent dosing regimen of a reversible inhibitor would need to be employed. Additionally, metalloenzyme inhibitors often exhibit cross-target inhibition and associated side-effects, meaning that dosing of such molecules over several months is not a viable strategy.26,27
 | ||
| Fig. 1 (A) Previously reported BoNT/A inhibitors based on the cinnamic hydroxamate scaffold. (B) X-ray co-crystal structures outlining the basis for a bifunctional approach, overlaying DCHA (green; PDB 2IMA)31,33 and MTSEA (yellow; PDB 4ELC)29 bound to Cys165 near the active site Zn2+ (purple sphere). (C) Overview of research explored in this project. | ||
Irreversible inhibition has the potential to abrogate BoNT/A's toxicity and longevity by means of permanent deactivation. Cys165, which lies approximately 5–8 Å away from the Zn2+ center, has become the focus of several targeted covalent inhibition strategies.28–31 Stura et al. reported the first X-ray crystal structure of the BoNT/A LC covalently modified on Cys165 with (2-aminoethyl)methanethiosulfonate (MTSEA), a small molecule containing the highly reactive methanethiosulfonate (MTS) warhead.29 Recently, we sought to integrate both reversible and irreversible aspects in a bifunctional fashion, whereby DCHA was modified with a pendent MTS moiety that had the potential to simultaneously chelate the Zn2+ metal and react with Cys165.30 An overlay (Fig. 1B) of both X-ray co-crystal structures of DCHA and MTSEA bound to the BoNT/A LC shows how the active site may simultaneously accommodate both a small covalent molecule and a reversible MBG inhibitor. Compound 1 (Fig. 1A) showed excellent enzyme inhibition with a kinact/Ki value of 418 M−1 s−1. However, this promising biochemical data was not reflected in a BoNT/A-implicated cell assay.30 The exaggerated disconnect between the enzymatic and cellular efficacy likely stemmed from the promiscuous nature of the MTS warhead.30,32 To address this issue, we describe an extensive screening campaign in search of intrinsically less reactive warheads that were capable of forming proximity-driven covalent bonds with Cys165 for the irreversible inhibition of the BoNT/A LC.
Results and discussion
Compound design and synthesis
Acrylamides are well known for their safety and selectivity profiles when forming selective, proximity-driven covalent bonds and therefore were selected for synthesis. An additional ten distinct warhead categories, possessing a variety of reactivities and sizes, were also synthesized (Fig. 2A). Beginning with 2,4-dichlorocinnamic acid, O-tetrahydropyran (THP)-protected linkers possessing warhead precursor functionality, e.g. alkene, amine, alcohol, were coupled to the carboxylic acid using simple amide coupling conditions. Key chemical transformations such as amide couplings or Mitsunobu reactions34 were then performed to install the electrophilic species, with subsequent THP deprotection to afford the final hydroxamate compounds (Fig. 2A). In the case of condition-sensitive warheads such as the epoxides and aldehydes, deprotection occurred before functionalization of the reactive warhead (Scheme S5†).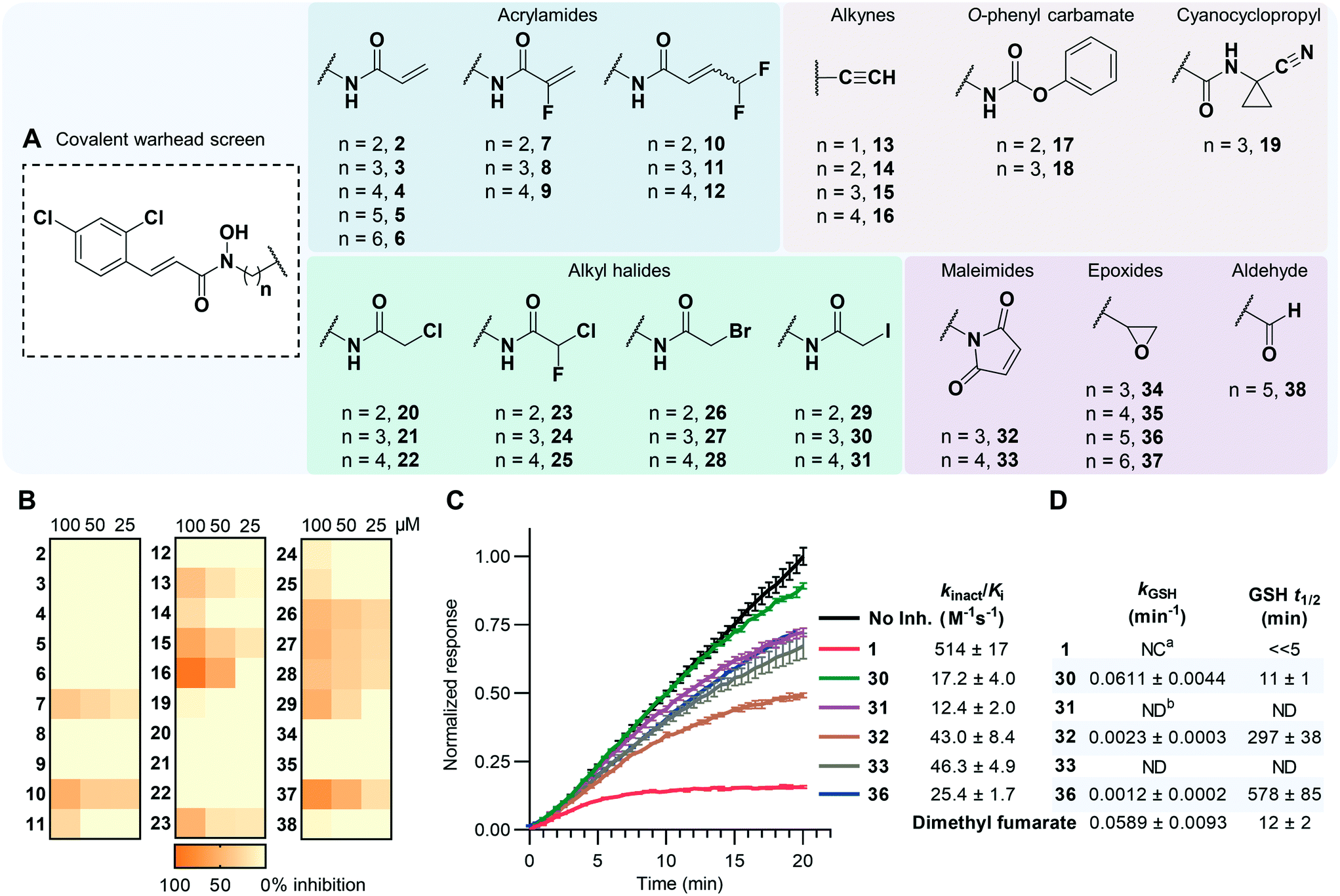 | ||
| Fig. 2 (A) Summary of compounds synthesized. See ESI-1,† Section S4.0 for full synthetic details. (B) Heatmap of BoNT/A LC inhibition exhibited by reversible compounds expressed as percent inhibition. Data for 17 and 18 omitted due to solubility issues. See Table S1† for full details. (C) Representative data from the continuous SNAPtide assay showing time-dependent inhibition of the BoNT/A LC from covalent compounds with their calculated kinact/Ki values outlined. Displayed data was obtained at 37 °C at a compound concentration of 32 μM, excluding 1, which was screened at 8 μM. Data was normalized to the maximal fluorescence of the no inhibitor control at 20 min. Error bars represent mean ± SD, n = 3. See Fig. S1† for additional data. (D) GSH reactivity data for covalent hits. Errors reported as SEM, n = 4. See Fig. S2† for additional data. a NC = not calculable, b ND = not determined. | ||
Optimization of warheads for irreversible inhibition
Compounds were evaluated for time-dependent inhibition using a continuous SNAPtide fluorescence resonance energy transfer (FRET) assay, which allowed for facile identification of covalent inhibitors by their characteristic non-linear inhibition profile of BoNT/A LC activity.30 Acrylamides 2–12, alkynes 13–16, O-phenyl carbamates 17–18, and cyanocyclopropyl 19 did not exhibit time-dependent inhibition and, with the exception of the alkynes, appeared to inhibit the BoNT/A LC poorly (Fig. 2B). It was hypothesized that these warheads lacked the necessary reactivity to form a covalent bond with Cys165. Previous reports have postulated that Cys165 exists as a thiolate anion under physiological coniditions.28,31 To investigate this, we performed covalent docking of compound 2 with the BoNT/A LC (Fig. S3A†) to predict if warheads larger than the MTS-warhead could be accommodated within the active site; the results clearly indicated that a covalent bond could be formed with Cys165. However, contrary to previous postulation, computational evaluation of protonation states using PROPKA35 predicted a pKa of 12.6 for Cys165 (Fig. S3B†). This suggests it possesses poor nucleophilic potential, which could explain the lack of time-dependent inhibition observed for compounds 2–19.Our predicted low reactivity of Cys165 suggested that warheads possessing higher reactivities were required to form the desired covalent bond. Thus, a series of haloacetamide (20–31) and maleimide (32–33) containing inhibitors were evaluated, as both groups are regularly used as cysteine capping reagents in bioconjugation.36,37 In fact, we observed that the less reactive chloro- and bromo-acetamides 20–28 did not show time-dependent inhibition within the continuous assay, whereas iodo-acetamides 30 and 31 exhibited moderate kinact/Ki values of 17.2 and 12.4 M−1 s−1, respectively (Fig. 2C). Although compound 29 also contained the iodoacetamide group, its shorter linker length may not have allowed simultaneous Zn2+ coordination and Cys165 engagement, thus preventing the covalent reaction. We have observed in the past that covalent bond formation with bifunctional inhibitors not only requires sufficient reactivity but also proximity of the warhead to Cys165.30 Maleimides 32 and 33 also exhibited time-dependent inhibition with slightly improved kinact/Ki values of 43.0 and 46.3 M−1 s−1, respectively. Intriguingly, the less reactive epoxide 36 displayed covalent inhibition with a kinact/Ki value of 25.4 M−1 s−1. The lack of time-dependent inhibition observed for compounds 35 and 37 emphasized the importance of linker length in the epoxide series, with 36 possessing optimum positioning for a proximity-driven covalent bond to form with Cys165. The subtle SAR for the epoxide series was not reflected in the iodoacetamides (30 and 31) and maleimides (32 and 33), with both compounds for each warhead type exhibiting equipotent time-dependent inhibition. This suggests that the requirements for successful covalent bond formation between Cys165 and 30–33 were biased towards the inherent reactivity of the warheads as opposed to optimal alignment of the reactive vectors.
To directly measure reactivity differences between each warhead type, we quantified the GSH reactivity of compounds 1, 30, 32 and 36 (Fig. 2D).38 We also included US Food and Drug Administration (FDA)-approved covalent inhibitor dimethyl fumarate as a positive control, as it has been shown to readily react with GSH.39 Notably, compound 1 reacted near-instantaneously with GSH, meaning the rate of depletion could not be confidently quantified. Compound 30 was also shown to be highly reactive (t1/2 = 11 min) with comparable reactivity with dimethyl fumarate, while both 32 and 36 were less reactive towards GSH with t1/2 ∼ 5 h and 10 h, respectively. Moreover, epoxide-containing compound 36 reacted with GSH ∼51-fold slower than iodoacetamide 30 but exhibited a superior kinact/Ki value. This emphasizes the importance of Cys165 proximity on the success of these bifunctional inhibitors; clearly the advantage conferred by having higher intrinsic reactivity can be matched by improving other interactions. Furthermore, it was observed that inhibitors 16 and 37, both possessing less bulky linkers, exhibited greater reversible inhibition against the BoNT/A LC over compounds that possessed bulkier amide-based linkers (Fig. 2B). We posit that the restricted conformations of the amide bond result in unfavorable trajectory of the reactive centers with respect to Cys165. More flexible alkyl linkers may allow for improved binding interactions and optimal placement of the warhead within the small cleft that presides between Cys165 and the catalytic Zn2+ center. This observation reflects a core advantage of the bifunctional strategy; less reactive warheads can be utilized for covalent modification if they can be effectively directed.
Confirmation of covalent modification of Cys165
Due to substrate depletion, the linear range of the continuous SNAPtide assay is limited to approximately 20 min at 37 °C. To investigate irreversible inhibition of the BoNT/A LC over a longer incubation time, we employed a conventional method of monitoring covalent kinetics, whereby enzyme and inhibitor are preincubated and aliquots are quenched at given time points and enzyme activity monitored. Thus, compounds 30, 32 and 36 were evaluated for covalent inhibition over the course of 5 h (Fig. 3A). | ||
| Fig. 3 (A) Preincubation assay showing covalent inhibition over time. (B) Dialysis studies of covalent inhibitors. Inhibitors screened using same conditions as for preincubation assay. Error bars represent ± SD, n = 3. See Fig. S4† for full data set. | ||
When incubated at a concentration of 50 μM with 500 nM BoNT/A LC, both 30 and 32 exhibited time-dependent inhibition, reaching a maximum of ∼95% inhibition after ∼3 h. Epoxide 36 exhibited considerably slower time-dependent inhibition, reflecting its lower reactivity, reaching a maximum of ∼70% inhibition with 100 μM compound after ∼8 h (Fig. S4D†). To confirm irreversible inhibition, preincubated enzyme/compound mixtures were exhaustively dialyzed (Fig. 3B), removing excess unreacted inhibitor from the enzyme, and the enzyme activity re-evaluated. BoNT/A LC activity remained unchanged for 30, 32 and 36, indicating an irreversible binding mechanism. DCHA, which was included as a non-covalent control, showed recovery of BoNT/A LC activity after dialysis, confirming its reversible binding mechanism. In addition to kinetic analysis of covalent modification, HRMS was used to detect formation of covalent adducts of all bifunctional inhibitors on the BoNT/A LC (see ESI-1,† Section S6.2 for raw deconvoluted traces). Compounds exhibiting time-dependent behavior within the continuous assay showed adduct masses consistent with the addition of one labelling event within one hour of incubation with the BoNT/A LC (Fig. S5†). The slower-acting epoxide 36 required longer incubation to exhibit significant time-dependent labelling, which was consistent with our kinetic evaluation.
Metal chelating anchor directs the warhead toward Cys165
To validate the hypothesized mechanism of inhibition, five control compounds were synthesized (Fig. 4A). Compounds 39–41 lack the key hydroxamate anchor and would establish the importance of the MBG in directing the covalent warhead toward Cys165. Compounds 42 and 43 were designed as non-covalent versions of compounds 30 and 32 respectively, whereby the electrophilic potential of the warhead was replaced with inert functionality. | ||
| Fig. 4 (A) Control compounds 39–43. (B) Kinetic profiles of control compounds in the continuous assay. Compounds were run at 32 μM, data was normalized to the maximal fluorescence of the no inhibitor control at 20 min. Error bars represent ± SEM, n = 3. See Fig. S6† for expansion of B. | ||
Both compounds 39 and 40 exhibited slight time-dependent inhibition but were drastically reduced when compared to the hydroxamate parent compounds 30 and 32 (Fig. 2C). Moreover, compound 41 showed no time-dependent inhibition. This indicated that the bidentate metal-binding interaction between the hydroxamate and the active-site Zn2+ is fundamental in directing the covalent appendage towards Cys165. As expected, non-covalent controls 42 and 43 displayed no activity consistent with covalent inhibition. In addition, both compounds showed poor competitive inhibition, bolstering the earlier hypothesis that sterically bulky linkers and warheads may not allow for optimal binding. To explore the role of Cys165 in covalent bond formation, we expressed a variant of the BoNT/A LC in which Cys165 was replaced with an alanine residue. Compounds 30, 32 and 36 were evaluated against the C165A variant in the preincubation assay (Fig. S7†), all of which exhibited no time-dependent inhibition, outlining the importance of Cys165 in covalent bond formation. Notably, we found that the C165A variant was less active and less stable than the WT, which is in agreement with previous reports.29,31 Although it may not be directly involved in the catalytic machinery, Cys165 likely plays an important structural role for LC activity.
Epoxide 36 possesses superior pharmacological properties
The promising biochemical data for covalent molecules 30, 32, and 36 led us to evaluate them against BoNT/A in a cell assay that employs the use of human induced pluripotent stem cell (hiPSC)-derived neurons.40 BoNT/A intoxicated neurons were incubated with various concentrations of compound and SNAP-25 cleavage was analyzed by Western blot analysis (Fig. 5A). Solubility of compounds was determined to be ≤200 μM when assessed by UV/vis spectroscopy (Fig. S8†). Compounds 30 and 32 completely abrogated BoNT/A activity at the highest tested concentration (100 μM), showing dose dependent behavior. The high kGSH values of 30 and 32 (Fig. 2D) suggest that reaction with endogenous GSH may be exhausting the inhibitors, highlighting the need for a less reactive warhead. Furthermore, moderate cell rounding, which is indicative of cytotoxicity, was observed at higher concentrations of 30 and 32. Unexpectedly, compound 36 showed no protection of SNAP-25 cleavage across all tested concentrations, even with longer incubation times. As such, we prepared lysate from primary rat spinal cord (RSC) cells, whereby cell lysis was carried out prior to toxin addition and compound evaluation. In this in vitro model, compound 36 completely protected SNAP-25 cleavage at 100 μM, though it did not protect at lower tested concentrations. This outlines that the inhibitory behavior of compound 36 against native BoNT/A LC is somehow attenuated in the cellular environment. We suggest that poor cellular permeability could account for the lack of potency within the hiPSC assay.41 | ||
| Fig. 5 (A) Western blot evaluation of 30 and 32 against post-exposure BoNT/A in hiPCS-derived neurons. BoNT/A cleavage of SNAP-25 was measured by quantifying the lower molecular weight band, which corresponds to the SNAP-25 breakdown product, n = 3. (B) Western blot evaluation of 36 in hiPCS-derived neurons cells and in RSC cell lysate, n = 3. Arrow depicts image crop of lower compound concentrations. See ESI-1,† Appendix S6.1 for full immunoblots. (C) Residual activity of various MMPs in the presence of covalent hits. Compounds tested at 25 μM. See Table S2† for full data. (D) Cell viability of HEK293 cells post 24 h compound incubation, n = 3. See Fig. S9A† for viability of additional compounds. (E) Stability assay of compounds in neat human serum at 37 °C, n = 3. See Fig. S9B† for longer incubation times. Error bars represent ± SD. | ||
One particular class of metalloenzyme, matrix metalloproteinases (MMPs), are involved in governing the structure of the extracellular matrix and are frequently targeted by hydroxamates.42–44 To test for cross-metalloenzyme interference, compounds 30, 32 and 36 were screened against a panel of MMPs to establish their selectivity for the BoNT/A LC (Fig. 5C). In general, compounds 30 and 36 did not show inhibition against MMPs at the tested concentration. The selectivity imparted by the pendent covalent warhead likely overrides the inherent affinity of the hydroxamate for Zn2+, as there is no corresponding cleft or reactive cysteine residue in the active site of these MMPs. Interestingly, at the tested concentration, compound 32 inhibited the majority of MMPs by over 50%. A previous report described the use of a photoaffinity probe for the irreversible inhibition of MMP-12, whereby a lysine residue near to the active site was covalently modified.45 Maleimides are known to react with various nucleophilic amino acids,46 including lysine, and suggests a potential role of 32 in covalent modification of MMPs. The use of maleimides for the modification of Cys165 in the BoNT/A LC poses potential selectivity issues and further outlines the need for less reactive, more selective warheads.
To further explore any off-target potential and toxicity associated with compounds 30, 32 and 36, we evaluated their effect on cell viability in HEK293 cells (Fig. 5D). Control compounds 39, 40, 42 and 43 were also included (Fig. S9A†). Compound 30 and iodoacetamide control 39 were toxic across all tested concentrations after 24 h, suggesting off-target alkylation was the likely cause for the observed toxicity in the cellular assay. Both compound 32 and maleimide control 40 showed mild toxicity, with IC50 values of 28 ± 3 and 6 ± 2 μM, respectively. Control compounds 42 and 43, which lack the electrophilic warhead, did not exhibit appreciable toxicity indicating that any toxicity imparted by compounds likely stemmed from the warhead and its associated reactivity. This was supported by the observation that compound 36 was the least toxic, retaining ∼75% cell viability at 100 μM.
As a further consideration, we evaluated the stability of compounds 30, 32 and 36 in human serum (Fig. 5E). Compounds 30 and 32 showed rapid degradation, with t1/2 < 30 min. Compound 36 exhibited excellent serum stability, with t1/2 = 11 h (Fig. S9B†). These results reflected the trend observed for GSH reactivity and toxicity; more inherently reactive warheads (i.e. MTS, iodoacetamide and maleimide) possess higher potential for off-target effects than the less reactive epoxide warhead of 36.
Significance
Classical approaches to covalent inhibition of cysteine proteases utilize the innate high nucleophilicity of the cysteine residue, which often has a low pKa value (4–7) and predominates as the thiolate anion.47–49 Electrophilic compounds, known as suicide inactivators, directly modify the active site cysteine and irreversibly interrupt the endogenous mechanism of the enzyme. Contrary to cysteine proteases, metalloproteases employ the use of a metal center to catalyze the cleavage of peptide bonds by association and activation of an active-site water co-factor – no nucleophilic catalytic residues are present in the active site. Direct interruption of the catalytic machinery involved in metalloproteases has proven difficult, with a limited number of examples reported to reversibly and irreversibly covalently modify gelatinases and carboxypeptidases, respectively.50–53 Strategies utilizing allosteric modification sites to irreversibly inhibit metalloproteases, namely targeted covalent inhibition, have seen success,45,54,55 though the number of such reports remains low relative to the field of small-molecule metalloprotease inhibition. The potential of targeted covalent inhibition is illustrated by its prolific use in kinase inhibitor design, which has boasted several successful drug discovery efforts leading to FDA-approved covalent kinase inhibitors.56,57 These inhibitors have undergone extensive optimization of enzyme/inhibitor interactions such that the electrophilic moieties (i.e. acrylamides) needed for successful covalent reaction possess very little intrinsic reactivity, resulting in exquisite selectivity for their targets. Our work has demonstrated that this approach can be used to leverage the design of selective irreversible inhibitors of the BoNT/A LC. By allowing the use of intrinsically less reactive electrophiles, our bifunctional covalent inhibitor approach has expanded the toolbox of Cys-reactive warheads for the BoNT/A LC, allowing the consideration of warheads that possess less off-target inhibition and improved pharmacological potential.The obtained kinact/Ki values contrasted with the large difference in GSH reactivity between iodoacetamide- and epoxide-containing compounds illustrate the complex interplay between intrinsic reactivity, binding affinity, and covalent reaction that contributes to the success of these covalent inhibitors. In compound 36, the hydroxamate–Zn2+ interaction acts as a fundamental anchor that is crucial in improving both the effective molarity and proximity of the epoxide to Cys165. Thus, this lowers the entropy of activation and increases the rate of reaction resulting in a selective covalent bond. To further exemplify the importance of the metal-chelating anchor, previous work reported by Garland et al. looked at the use of cysteine-reactive warheads for the covalent modification of Cys165 in the BoNT/A LC.31 Simple scaffolds containing a range of warheads that were devoid of a MBG were shown to have minimal irreversible behavior against the BoNT/A LC. Most importantly, epoxyketone compounds, which possess intrinsically higher reactivity than epoxides alone, did not show modification of Cys165. Only highly reactive selenide-based warheads were capable of modifying Cys165.31 Analysis of the apo active site X-ray crystal structure (PDB 4EJ5)29 shows that Cys165 is sequestered deep within a narrow cleft, which in combination with the high predicted pKa of Cys165, would explain the need for highly electrophilic groups. This outlines that the requirements for successful modification of Cys165 using less reactive warheads is contingent upon BoNT/A LC-specific binding functionality, i.e. a metal-binding group, in directing the warhead toward Cys165.
We envision that this strategy can be utilized amongst other metalloproteases that possess non-catalytic cysteines proximal to the active site metal center. For example, one important class of Zn2+-dependent metalloproteases are the histone deacetylases (HDACs), which are primarily implicated in the epigenetic regulation of the chromatin landscape.58 Their prominent role in regulation of gene expression means they are highly sought-after targets for a multitude of diseases, including cancer, inflammatory disease, and metabolic disorders.59 The conserved proximal cysteine ∼5–7 Å away from the Zn2+ center has been targeted for covalent modification – in 2015, Daniel et al. reported a prodrug of HDAC inhibitor suberoylanilide hydroxamic acid (SAHA) that released SAHA upon modification of the proximal cysteine. Notably, neither species remained covalently tethered to one another, and the thiol-reactive quinine demonstrated rapid reactivity with GSH and non-specific labelling of other cysteines.55 We suggest that the bifunctional strategy could serve to functionalize SAHA and other metal-chelating inhibitor scaffolds, thus allowing the use of less intrinsically reactive warheads to develop irreversible HDAC inhibitors with more favorable pharmacological properties.
Conclusion
In summary, a comprehensive exploratory search for intrinsically less reactive and more pharmacologically relevant warheads for the covalent modification of Cys165 and subsequent irreversible inhibition of the BoNT/A LC protease was carried out. Covalent inhibition of the BoNT/A LC was challenging given the predicted poor nucleophilicity of Cys165 near the Zn2+-containing active site. Thus, mild electrophiles (i.e. acrylamides and chloroacetamides) furnished no covalent reactivity, making the BoNT/A LC seemingly more difficult to inhibit than other targets reported in literature.60 Despite this, covalent labelling was achieved using iodoacetamide-, maleimide- and epoxide-containing inhibitors 30–33 and 36. As outlined with the epoxide series, flexible alkyl linkers offered optimal positioning of the warhead to Cys165, resulting in a directed, proximity-driven covalent bond. This was exemplified through the superior covalent inhibition of epoxide 36 over iodoacetamides 30 and 31, despite their huge differences in warhead reactivity. Linker length and flexibility was crucial to successful covalent modification, however only simple alkyl linkers were employed. Heteroatom incorporation within the linker could be leveraged to improve reversible binding interactions through additional H-bonding contacts with residues that reside within the narrow cleft between Cys165 and the Zn2+ metal center. Covalent mechanism of inhibition was confirmed through exhaustive dialysis studies with confirmation of covalent adducts using HRMS. The importance of the bifunctional approach was investigated through synthesis of key control compounds 39–43 with successful covalent modification contingent upon the crucial metal chelating anchor. Modification of Cys165 was confirmed through HRMS and kinetic evaluation of compounds against the C165A variant. A trend in GSH reactivity, serum stability, and toxicity was observed, with compound 36 possessing superior pharmacological properties. The drop in potency between enzymatic and cellular assays may be attributed to poor cell permeability rather than off-target reactivity, as exemplified by the RSC lysate assay. We postulate that this bifunctional strategy could be easily translated to other metalloproteases that possess non-catalytic reactive residues near the active site, allowing medicinal chemists to increase the fine margin between potency and toxicity of irreversible covalent inhibitors.Abbreviations
| BoNT/A | Botulinum neurotoxin A |
| CDC | Center for Disease Control and Prevention |
| DCHA | 2,4-Dichlorocinnamic hydroxamic acid |
| ESI | Electronic supplementary information |
| FDA | Food and Drug Administration |
| FRET | Fluorescence resonance energy transfer |
| GSH | Glutathione |
| HC | Heavy chain |
| HDAC | Histone deacetylase |
| hiPSC | Human induced pluripotent stem cell |
| HRMS | High-resolution mass spectrometry |
| LC | Light chain |
| MBGs | Metal-binding groups |
| MMPs | Matrix metalloproteinases |
| MTS | Methanethiosulfonate |
| MTSEA | (2-Aminoethyl)methanethiosulfonate |
| NC | Not calculable |
| ND | Not determined |
| NSF | N-Ethylmaleimide sensitive factor |
| RSC | Rat spinal cord |
| SAHA | Suberoylanilide hydroxamic acid |
| SAR | Structure–activity relationship |
| SNARE | NSF attachment protein factor |
| SNAP-25 | Synaptosomal-associated protein-25 |
| THP | Tetrahydropyran |
Author contributions
L. D. T., M. E. O., and K. D. J. conceptualized the study; L. D. T., A. L. N., and T. S. performed chemical synthesis. L. D. T., A. L. N., and L. L. performed biochemical characterization and analysis of raw data; A. L. N. carried out computational modelling; L. D. T. performed mutagenesis and expression of the variant; S. P. and A. L. N. performed cell-based assays; L. D. T. wrote the original draft of the manuscript with input from A. L. N. and L. L., and all authors reviewed and edited the final manuscript; E. A. J. and K. D. J. acquired funding and supervised the study.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Dr. Jiaxing Wang for provision of chemical building blocks, Bill Webb and Elizabeth Billings for technical protein HRMS support, Dr. Joseph Barbieri for kindly supplying the BoNT/A LC, Dr. Lisa Eubanks for mutagenesis guidance, William H. Tepp for production of BoNT/A holotoxin, and Dr. Jason Chen for useful discussions regarding synthesis. This work was supported by the National Institutes of Health grants R01 AI153298 and R21 AI137709 (K. D. J.) and F32 DA044692 (M. E. O.); the Fulbright Scholar Program (A. L. N.); the Natural Sciences and Engineering Research Council of Canada PGSD3-502274 (L. L.) and the Skaggs Institute for Chemical Biology (L. L. and K. D. J.). This is Scripps Research manuscript #30077.Notes and references
- M. W. Peck, Adv. Microb. Physiol., 2009, 55, 183–265 CrossRef CAS , 320.
- R. L. Shapiro, C. Hatheway and D. L. Swerdlow, Ann. Intern. Med., 1998, 129, 221–228 CrossRef CAS.
- M. D. Collins and A. K. East, J. Appl. Microbiol., 1998, 84, 5–17 CrossRef CAS.
- E. M. Hansbauer, M. Skiba, T. Endermann, J. Weisemann, D. Stern, M. B. Dorner, F. Finkenwirth, J. Wolf, W. Luginbühl, U. Messelhäußer, L. Bellanger, C. Woudstra, A. Rummel, P. Fach and B. G. Dorner, Analyst, 2016, 141, 5281–5297 RSC.
- S. E. Maslanka, C. Lúquez, J. K. Dykes, W. H. Tepp, C. L. Pier, S. Pellett, B. H. Raphael, S. R. Kalb, J. R. Barr, A. Rao and E. A. Johnson, J. Infect. Dis., 2016, 213, 379–385 CrossRef CAS.
- J. Brunt, A. T. Carter, S. C. Stringer and M. W. Peck, FEBS Lett., 2018, 592, 310–317 CrossRef CAS.
- K. A. Jackson, B. E. Mahon, J. Copeland and R. P. Fagan, Botulinum J., 2015, 3, 6–17 CrossRef.
- S. S. Arnon, R. Schechter, T. V. Inglesby, D. A. Henderson, J. G. Bartlett, M. S. Ascher, E. Eitzen, A. D. Fine, J. Hauer, M. Layton, S. Lillibridge, M. T. Osterholm, T. O'Toole, G. Parker, T. M. Perl, P. K. Russell, D. L. Swerdlow, K. Tonat and the Working Group on Civilian Biodefense, JAMA, J. Am. Med. Assoc., 2001, 285, 1059–1070 CrossRef CAS PubMed.
- P. G. Foran, N. Mohammed, G. O. Lisk, S. Nagwaney, G. W. Lawrence, E. Johnson, L. Smith, K. R. Aoki and J. O. Dolly, J. Biol. Chem., 2003, 278, 1363–1371 CrossRef CAS PubMed.
- M. Oliveira, G. Mason-Buck, D. Ballard, W. Branicki and A. Amorim, Forensic Sci. Int., 2020, 314, 110366 CrossRef.
- R. B. Lipton, N. L. Rosen, J. Ailani, R. E. DeGryse, P. J. Gillard and S. F. Varon, Cephalalgia, 2016, 36, 899–908 CrossRef.
- S. Ozcakir and K. Sivrioglu, Clin. Med. Res., 2007, 5, 132–138 CrossRef CAS.
- X. Yan, J. Lan, Y. Liu and J. Miao, Med. Sci. Monit., 2018, 24, 8160–8171 CrossRef CAS.
- S. M. Farag, M. O. Mohammed, T. A. El-Sobky, N. A. ElKadery and A. K. ElZohiery, JBJS Rev., 2020, 8, e0119 CrossRef.
- American Society of Plastic Surgeons, Plastic Surgery Statistics Report, https://www.plasticsurgery.org/documents/News/Statistics/2019/plastic-surgery-statistics-full-report-2019.pdf, (accessed 10/05/2020, 2020) Search PubMed.
- E. Yiannakopoulou, Pharmacology, 2015, 95, 65–69 CrossRef CAS.
- R. Pellizzari, O. Rossetto, G. Schiavo and C. Montecucco, Philos. Trans. R. Soc., B, 1999, 354, 259–268 CrossRef CAS.
- A. Rummel, T. Karnath, T. Henke, H. Bigalke and T. Binz, J. Biol. Chem., 2004, 279, 30865–30870 CrossRef CAS PubMed.
- S. Sun, S. Suresh, H. Liu, W. H. Tepp, E. A. Johnson, J. M. Edwardson and E. R. Chapman, Cell Host Microbe, 2011, 10, 237–247 CrossRef CAS PubMed.
- V. V. Vaidyanathan, K. Yoshino, M. Jahnz, C. Dörries, S. Bade, S. Nauenburg, H. Niemann and T. Binz, J. Neurochem., 1999, 72, 327–337 CrossRef CAS PubMed.
- C. O. Tacket, W. X. Shandera, J. M. Mann, N. T. Hargrett and P. A. Blake, Am. J. Med., 1984, 76, 794–798 CrossRef CAS.
- L. Lin, M. E. Olson, L. M. Eubanks and K. D. Janda, Acc. Chem. Res., 2019, 52, 2322–2331 CrossRef CAS PubMed.
- L. Lin, L. D. Turner, P. Šilhár, S. Pellett, E. A. Johnson and K. D. Janda, RSC Med. Chem., 2021, 12, 137–143 RSC.
- G. E. Boldt, J. P. Kennedy and K. D. Janda, Org. Lett., 2006, 8, 1729–1732 CrossRef CAS PubMed.
- L. M. Eubanks, M. S. Hixon, W. Jin, S. Hong, C. M. Clancy, W. H. Tepp, M. R. Baldwin, C. J. Malizio, M. C. Goodnough, J. T. Barbieri, E. A. Johnson, D. L. Boger, T. J. Dickerson and K. D. Janda, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2602 CrossRef.
- J. A. Day and S. M. Cohen, J. Med. Chem., 2013, 56, 7997–8007 CrossRef.
- A. Y. Chen, R. N. Adamek, B. L. Dick, C. V. Credille, C. N. Morrison and S. M. Cohen, Chem. Rev., 2019, 119, 1323–1455 CrossRef CAS.
- B. Li, S. C. Cardinale, M. M. Butler, R. Pai, J. E. Nuss, N. P. Peet, S. Bavari and T. L. Bowlin, Bioorg. Med. Chem., 2011, 19, 7338–7348 CrossRef.
- E. A. Stura, L. Le Roux, K. Guitot, S. Garcia, S. Bregant, F. Beau, L. Vera, G. Collet, D. Ptchelkine, H. Bakirci and V. Dive, J. Biol. Chem., 2012, 287, 33607–33614 CrossRef PubMed.
- L. Lin, M. E. Olson, T. Sugane, L. D. Turner, M. A. Tararina, A. L. Nielsen, E. K. Kurbanov, S. Pellett, E. A. Johnson, S. M. Cohen, K. N. Allen and K. D. Janda, J. Med. Chem., 2020, 63, 11100–11120 CrossRef CAS PubMed.
- M. Garland, B. M. Babin, S.-I. Miyashita, S. Loscher, Y. Shen, M. Dong and M. Bogyo, ACS Chem. Biol., 2019, 14, 76–87 CrossRef CAS PubMed.
- M. Lo Conte, J. Lin, M. A. Wilson and K. S. Carroll, ACS Chem. Biol., 2015, 10, 1825–1830 CrossRef CAS.
- N. R. Silvaggi, G. E. Boldt, M. S. Hixon, J. P. Kennedy, S. Tzipori, K. D. Janda and K. N. Allen, Chem. Biol., 2007, 14, 533–542 CrossRef CAS PubMed.
- M. A. Walker, Tetrahedron Lett., 1994, 35, 665–668 CrossRef.
- C. R. Søndergaard, M. H. M. Olsson, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 2284–2295 CrossRef PubMed.
- Y. Kim, S. O. Ho, N. R. Gassman, Y. Korlann, E. V. Landorf, F. R. Collart and S. Weiss, Bioconjugate Chem., 2008, 19, 786–791 CrossRef.
- C. Smythe, J. Biol. Chem., 1936, 114, 601–612 CrossRef.
- A. Böhme, D. Thaens, A. Paschke and G. Schüürmann, Chem. Res. Toxicol., 2009, 22, 742–750 Search PubMed.
- T. J. Schmidt, M. Ak and U. Mrowietz, Bioorg. Med. Chem., 2007, 15, 333–342 CrossRef CAS.
- S. Pellett, W. H. Tepp, C. M. Clancy, G. E. Borodic and E. A. Johnson, FEBS Lett., 2007, 581, 4803–4808 CrossRef CAS.
- P. Silhár, L. M. Eubanks, H. Seki, S. Pellett, S. Javor, W. H. Tepp, E. A. Johnson and K. D. Janda, J. Med. Chem., 2013, 56, 7870–7879 CrossRef PubMed.
- G. Murphy and H. Nagase, Mol. Aspects Med., 2008, 29, 290–308 CrossRef CAS.
- J. A. Jacobsen, J. L. Major Jourden, M. T. Miller and S. M. Cohen, Biochim. Biophys. Acta, 2010, 1803, 72–94 CrossRef.
- J. M. Cathcart and J. Cao, Front. Biosci., 2015, 20, 1164–1178 CrossRef PubMed.
- A. S. Dabert-Gay, B. Czarny, E. Lajeunesse, R. Thai, H. Nagase and V. Dive, Bioconjugate Chem., 2009, 20, 367–375 CrossRef PubMed.
- N. E. Sharpless and M. Flavin, Biochemistry, 1966, 5, 2963–2971 CrossRef PubMed.
- F. Hofer, J. Kraml, U. Kahler, A. S. Kamenik and K. R. Liedl, J. Chem. Inf. Model., 2020, 60, 3030–3042 CrossRef.
- T. K. Harris and G. J. Turner, IUBMB Life, 2002, 53, 85–98 CrossRef PubMed.
- J. C. Powers, J. L. Asgian, Ö. D. Ekici and K. E. James, Chem. Rev., 2002, 102, 4639–4750 CrossRef CAS PubMed.
- S. Brown, M. M. Bernardo, Z.-H. Li, L. P. Kotra, Y. Tanaka, R. Fridman and S. Mobashery, J. Am. Chem. Soc., 2000, 122, 6799–6800 CrossRef.
- M. M. Bernardo, S. Brown, Z. H. Li, R. Fridman and S. Mobashery, J. Biol. Chem., 2002, 277, 11201–11207 CrossRef PubMed.
- C. Forbes, Q. Shi, J. F. Fisher, M. Lee, D. Hesek, L. I. Llarrull, M. Toth, M. Gossing, R. Fridman and S. Mobashery, Chem. Biol. Drug Des., 2009, 74, 527–534 CrossRef CAS.
- S. A. Testero, C. Granados, D. Fernández, P. Gallego, G. Covaleda, D. Reverter, J. Vendrell, F. X. Avilés, I. Pallarès and S. Mobashery, ACS Med. Chem. Lett., 2017, 8, 1122–1127 CrossRef.
- N. Sin, L. Meng, M. Q. W. Wang, J. J. Wen, W. G. Bornmann and C. M. Crews, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 6099–6103 CrossRef PubMed.
- K. B. Daniel, E. D. Sullivan, Y. Chen, J. C. Chan, P. A. Jennings, C. A. Fierke and S. M. Cohen, J. Med. Chem., 2015, 58, 4812–4821 CrossRef CAS PubMed.
- D. Kazandjian, G. M. Blumenthal, W. Yuan, K. He, P. Keegan and R. Pazdur, Clin. Cancer Res., 2016, 22, 1307–1312 CrossRef CAS.
- H. Singh, A. J. Walker, L. Amiri-Kordestani, J. Cheng, S. Tang, P. Balcazar, K. Barnett-Ringgold, T. R. Palmby, X. Cao, N. Zheng, Q. Liu, J. Yu, W. F. Pierce, S. R. Daniels, R. Sridhara, A. Ibrahim, P. G. Kluetz, G. M. Blumenthal, J. A. Beaver and R. Pazdur, Clin. Cancer Res., 2018, 24, 3486–3491 CrossRef CAS PubMed.
- G. Milazzo, D. Mercatelli, G. Di Muzio, L. Triboli, P. De Rosa, G. Perini and F. M. Giorgi, Genes, 2020, 11, 556 CrossRef CAS.
- D. F. Tough, P. P. Tak, A. Tarakhovsky and R. K. Prinjha, Nat. Rev. Drug Discovery, 2016, 15, 835–853 CrossRef CAS.
- E. Resnick, A. Bradley, J. Gan, A. Douangamath, T. Krojer, R. Sethi, P. P. Geurink, A. Aimon, G. Amitai, D. Bellini, J. Bennett, M. Fairhead, O. Fedorov, R. Gabizon, J. Gan, J. Guo, A. Plotnikov, N. Reznik, G. F. Ruda, L. Díaz-Sáez, V. M. Straub, T. Szommer, S. Velupillai, D. Zaidman, Y. Zhang, A. R. Coker, C. G. Dowson, H. M. Barr, C. Wang, K. V. M. Huber, P. E. Brennan, H. Ovaa, F. von Delft and N. London, J. Am. Chem. Soc., 2019, 141, 8951–8968 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: ESI-1: ESI figures and tables, biochemical materials and methods, chemical schemes and experimental, full immunoblots, raw deconvoluted HRMS spectra, and compound HPLC chromatograms. ESI-2: NMR spectra of reported compounds. See DOI: 10.1039/d1md00089f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |