
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Guidelines for performing lignin-first biorefining
Mahdi M.
Abu-Omar
 a,
Katalin
Barta
b,
Gregg T.
Beckham
*cd,
Jeremy S.
Luterbacher
*e,
John
Ralph
*f,
Roberto
Rinaldi
g,
Yuriy
Román-Leshkov
*h,
Joseph S. M.
Samec
*i,
Bert F.
Sels
j and
Feng
Wang
k
a,
Katalin
Barta
b,
Gregg T.
Beckham
*cd,
Jeremy S.
Luterbacher
*e,
John
Ralph
*f,
Roberto
Rinaldi
g,
Yuriy
Román-Leshkov
*h,
Joseph S. M.
Samec
*i,
Bert F.
Sels
j and
Feng
Wang
k
aDepartment of Chemical Engineering and Department of Chemistry & Biochemistry, University of California, Santa Barbara, California 93106, USA
bStratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands
cRenewable Resources and Enabling Sciences Center, National Renewable Energy Laboratory, Golden, CO 80401, USA. E-mail: gregg.beckham@nrel.gov
dCenter for Bioenergy Innovation, Oak Ridge, TN 37830, USA
eLaboratory of Sustainable and Catalytic Processing, Institute of Chemical Sciences and Engineering, École Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland. E-mail: jeremy.luterbacher@epfl.ch
fU.S. Department of Energy Great Lakes Bioenergy Research Center, University of Wisconsin-Madison, Madison, WI 53726, USA. E-mail: jralph@wisc.edu
gDepartment of Chemical Engineering, Imperial College London, South Kensington Campus, London SW7 2AZ, UK
hDepartment of Chemical Engineering, MIT, Cambridge, MA 02139, USA. E-mail: yroman@mit.edu
iDepartment of Organic Chemistry, Stockholm University, SE-106 91 Stockholm, Sweden. E-mail: joseph.samec@su.se
jCenter for Sustainable Catalysis and Engineering, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium
kState Key Laboratory of Catalysis, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023 Liaoning, China
First published on 15th October 2020
Abstract
The valorisation of the plant biopolymer lignin is now recognised as essential to enabling the economic viability of the lignocellulosic biorefining industry. In this context, the “lignin-first” biorefining approach, in which lignin valorisation is considered in the design phase, has demonstrated the fullest utilisation of lignocellulose. We define lignin-first methods as active stabilisation approaches that solubilise lignin from native lignocellulosic biomass while avoiding condensation reactions that lead to more recalcitrant lignin polymers. This active stabilisation can be accomplished by solvolysis and catalytic conversion of reactive intermediates to stable products or by protection-group chemistry of lignin oligomers or reactive monomers. Across the growing body of literature in this field, there are disparate approaches to report and analyse the results from lignin-first approaches, thus making quantitative comparisons between studies challenging. To that end, we present herein a set of guidelines for analysing critical data from lignin-first approaches, including feedstock analysis and process parameters, with the ambition of uniting the lignin-first research community around a common set of reportable metrics. These guidelines comprise standards and best practices or minimum requirements for feedstock analysis, stressing reporting of the fractionation efficiency, product yields, solvent mass balances, catalyst efficiency, and the requirements for additional reagents such as reducing, oxidising, or capping agents. Our goal is to establish best practices for the research community at large primarily to enable direct comparisons between studies from different laboratories. The use of these guidelines will be helpful for the newcomers to this field and pivotal for further progress in this exciting research area.
Broader contextConversion of polysaccharides from lignocellulose to biofuels and chemicals has long been the primary objective of the biomass conversion community. However, for the success of a global bio-based circular economy that employs lignocellulosic biomass as a feedstock, it is imperative to also derive value from the lignin component, beyond low-value heat and power. Despite a century of lignin research, this goal remains elusive, and yet is increasingly important. The international research community is now vigorously pursuing lignin valorisation approaches in response to the need for a more sustainable carbon economy. The use of active stabilisation methods for lignin fractionation and valorisation, generally dubbed “lignin-first” biorefining, is receiving significant attention. Here we provide our perspective on how to unify the lignin-first community around a common set of research practices to accelerate progress in this field. |
1. Introduction
It is now clear that humankind must rapidly transition from the use of fossil resources, given concerns over anthropogenic climate change. Accordingly, the development of alternative and sustainable feedstocks for energy, chemicals, and materials has become one of this century's most important societal challenges.1,2 To that end, plant-based feedstocks are one of the most well-studied and promising green carbon sources available in sufficient quantities to contribute to a more sustainable global carbon economy without negatively affecting the climate.3Large amounts of lignocellulose are processed today for pulp and paper production and, in some parts of the world, for heat and power. In recent decades, substantial governmental and industrial investments have created a strong driving force to commercialise the production of cellulosic biofuels and bioproducts, which has mobilised a large global research community to focus on this problem. Most of the leading lignocellulosic biofuel production paradigms that have employed selective approaches in the last two decades focused on the conversion of plant polysaccharides to biofuels, such as ethanol, gasoline, diesel, and jet fuel, and have slated the high energy density aromatic biopolymer, lignin, for on-site heat and power production. This approach is mirrored by pulp and paper mills that also combust lignin for power and to recover inorganic pulping chemicals.4,5 Lignin combustion has long been thought to be the most viable approach for dealing with this biopolymer, given the challenges associated with isolating lignin from polysaccharides without making it more recalcitrant, and the difficulty in overcoming the inherent heterogeneity of lignin – the two main challenges facing lignin valorisation research efforts spanning the last century.
The canonical lignin building block is generally a phenylpropanoid group that usually exhibits one of three aromatic structures: syringyl (S), guaiacyl (G), and hydroxyphenyl (H) units. Each of these units derives from one of the monolignols: sinapyl alcohol, coniferyl alcohol, and p-coumaryl alcohol. Several types of C–O and C–C linkages are formed between these units during lignin biosynthesis to build the native polymer. In its native state in the plant, lignin is now regarded as being less branched than previously surmised,6–9 and more tractable. Additional building blocks have been shown in the past decade to be incorporated into lignin, including flavonoids, hydroxystilbenes, and others.10 Several lignin model structures are shown in Fig. 1 to illustrate some of the key features.
 | ||
| Fig. 1 Model lignin structures containing 11 units for: (a) a softwood, (b) a hardwood, and (c) a grass. These structures in no way represent the proportion of the various functionalities that are found analytically, but rather show the major and more interesting lignin units with ‘legal’ inter-unit bonding. Notable features include: (a) a dibenzodioxocin unit D, free-phenolic, from 5-5-coupling of oligomer units followed by 4-O-β-coupling with a single coniferyl alcohol monomer (thus not really creating a “Y-type” branchpoint), a spirodienone F from β-1 coupling, and a 4-O-5 (biphenyl ether) unit E also from the coupling of oligomer units but, again, not producing a real branchpoint because it is found only in its free-phenolic form shown; (b) the same range of units as for the softwood, but involving S-units where appropriate, and also showing, acylating a γ-OH, one acetate and one p-hydroxybenzoate, the latter being a feature in primarily poplar/aspen, willow, and palms; (c) a similar range of units again, except with the β-β-coupled unit arising from initial dimerisation of sinapyl p-coumarate to produce a tetrahydrofuran C′ rather than the resinol C that results from dimerisation of (un-acylated) monolignols, showing the natural lignin acylation by acetate and p-coumarate, features in all grasses; and inserts: (d) one of many structures involving ferulate units FA derived from lignification with monolignol ferulate conjugates that is a feature of all grasses and some hardwoods; (e) one of many units derived from the cross-linking of lignin and arabinoxylan via ferulates acylating the latter, a feature of all grasses (and commelinid monocots); (f) the tricin T, a flavone, that acts as a chain-starter in ‘all’ grasses. The figure was modified from Ralph et al.8 | ||
As lignin represents the largest source of sustainable aromatics on the planet, it is increasingly being recognised as foolhardy to ignore its potential value.11,12 As a result, many researchers today are pursuing more holistic strategies for biomass utilisation that place substantial value on both lignin and polysaccharides, in many cases using selective fractionation technologies. Much of the motivation for this work is underpinned by technoeconomic analyses (TEA) and life-cycle assessments (LCA) that indicate that lignin valorisation can improve the overall biorefinery economics and sustainability footprint.13–16 A fundamental challenge in selectively fractionating biomass into its constituents is the propensity of carbohydrates, lignin, or both to degrade to undesirable products during processing. More efficient methodologies to fractionate and conserve cellulose, hemicelluloses, and lignin are therefore desirable.
For selective lignin removal from intact lignocellulosic biomass, many fractionation strategies have been developed over decades of research.17–19 By far the most common are organosolv processes, generally defined as processes that employ an organic solvent, most commonly with an acid co-catalyst and water.20,21 These pretreatment strategies often liberate substantial amounts of lignin from biomass. Given the typical requirement for acidity, ether and ester bonds may be cleaved and, through reasonably well-understood mechanisms, condensation reactions occur to an extent dependent on the fractionation conditions (pH, residence time, solvent). As a result, non-native lignin-derived polymers (a.k.a. technical lignins) are formed.9,22–31 The rationale for this condensation is that the solvolysis conditions, catalysed by acid, in addition to being able to cleave weak C–O linkages prevalent in the lignin, protonate the α-OH in various lignin structures, leading to the ready formation of benzylic carbocations (benzyl cations, Scheme 1). These reactive intermediates readily participate in electrophilic aromatic substitution reactions on the electron-rich aryl groups of lignin to form recalcitrant C–C bonds that are not found in the native lignin; an equivalent chemical explanation is that the electron-rich aromatic rings readily participate in nucleophilic attack on the carbocation centres.24 The insidious aspect of this reaction sequence is that, although phenolic benzyl cations are more stable than their etherified counterparts, the latter still easily form, so condensation can therefore occur within the chain, not just on endgroups. Similarly, although the nucleophilicity of a free-phenolic unit is higher, and will therefore more rapidly attack such carbonium ions, etherified units also readily undergo the condensation reaction. The propensity to spontaneously undergo these reactions in acid explains how condensed lignins result and why conditions that are sufficiently harsh to extract the lignin generally produce particularly intractable polymers. These types of organosolv processes, for which there is a huge number of variations, typically yield low-quality lignin streams that are, because of these condensed units, not suitable for subsequent depolymerisation processes. Typical thermochemical pretreatment methods, such as hot water, steam explosion, and acid pretreatment, that focus almost solely on maximising monomeric carbohydrates, lead to many of the same condensed lignin structures (in part because of the native acids released from esters in the cell wall during such pretreatments), rendering lignin into a more recalcitrant polymer than its starting native form in the plant cell wall. High-severity alkaline conditions will similarly lead to undesirable lignin condensation.9,32–36
 | ||
| Scheme 1 Mechanism for the presumed major condensation reaction occurring under acidic aqueous conditions. A lignin β-ether unit 1 readily forms resonance-stabilized benzylium (benzyl carbocation, or benzyl carbonium) ions 3 following protonation of the α-OH to produce intermediate 2. Nucleophilic attack of a general lignin G-unit on this carbonium ion or, as alternatively viewed, electrophilic aromatic substitution by the carbonium ion on the G-unit, produces an intermediate 4 that rearomatizes by losing a proton to produce 5, a new ‘condensed unit’ with a particularly recalcitrant non-native 6-α-bond in so-called condensed lignins. G-units are favoured over S-units for attack by carbonium ions because of their more accessible (less sterically encumbered) 6-positions. Lignins that have undergone such condensation are difficult to degrade to monomers. Note: the numbering here is specific to this figure. | ||
Lignin-first processing is the broadly accepted umbrella term for solvent-based methods in which lignin preservation, together with that of the polysaccharides, is considered upfront, moving away from the current practice of having to deal with an intractable lignin product at the end of a biorefining process. Here we define ‘lignin-first’ as an active stabilisation approach that liberates lignin from the plant cell wall and prevents condensation reactions through either catalysis or protection-group chemistry. Importantly, lignin-first biorefining is not a synonym for lignin valorisation, but rather an integral approach that derives value from both lignin and polysaccharides, towards an atom-efficient and more sustainable utilisation of lignocellulosic biomass. Most commonly, lignin-first processes involve three steps: (i) the lignin is removed from whole biomass using an organic solvent through solvolysis or acid-catalysed reactions (similarly to organosolv pretreatment); (ii) the resulting intermediates are stabilised, with the intention of preventing condensation of reactive species generated by lignin depolymerisation, and (iii) further depolymerisation occurs if not fully depolymerised at the stabilisation stage.9,37–39
To date, there have been several approaches reported for lignin-first refining, the chemical steps of which are illustrated in Fig. 2. The most common methodology comprises solvent-based lignin extraction from biomass in the presence of a transition metal under hydrogen atmosphere or with the aid of a hydrogen-donor solvent or another reducing agent.40–44 This methodology, which has been termed ‘Catalytic Upstream Biorefinery’ (CUB) or ‘Early-stage Catalytic Conversion of Lignin’ (ECCL) for the process using 2-propanol as an H-donor,9,40,45–50 is generally now termed Reductive Catalytic Fractionation regardless of the H-source (RCF, Fig. 2a). It should be noted that RCF carried out under H2 pressure was first practiced/developed in the late 1930s and 1940s as a methodology to study lignin,51–57 and as a means for pulping and high-yield production of lignin-based chemicals, but this was not commercialized at the time.58–61 Variants of the RCF approach include systems in which sugars in the biomass operate as reducing agents,62 and recently flow-through operations or catalyst baskets have been applied such that biomass solvolysis and hydrogenation/hydrogenolysis reactions on lignin intermediates have been separated in time and space.63–67 The primary roles of the metal catalyst are reductive stabilisation of reactive intermediates from lignin that result from the solvolysis process and depolymerisation of the solubilised lignin oligomers. Solid(biomass)/solid(catalyst) contact is therefore not essential, as demonstrated in reactor set-ups that physically separate the solid biomass and catalyst.63,65,66 The solvent mixture and the potential presence of an additional acid or base catalyst can influence the yield of monophenolic compounds and (hemi)cellulose retention (Fig. 2).58,68 The solvent determines not only the delignification degree (i.e., solvolysis and extraction of lignin) but also the retention of (hemi-)cellulose as a pulp, as well as the selectivity and distribution of monophenolics.38 Usage of water, or protic solvents with a high proportion of water, may produce high delignification but will also hydrolyse and solubilise carbohydrates, which can be hydrogenated by the catalyst or react with the solvent.40,46,47,69
 | ||
| Fig. 2 Three lignin-first strategies reported to date using solvolysis and catalytic stabilization of reactive intermediates to stable products or protection-group chemistry and subsequent depolymerisation. (a) Reductive catalytic fractionation (RCF) relies on the use of a metal catalyst and H2 (or a hydrogen donor) in a polar, protic solvent to selectively extract lignin from the cell wall, which is depolymerised and further stabilised via reduction chemistry. This results in the direct production of monophenolics as well as the reduction of any carbohydrates that are solubilized to hydrogenated sugars and sometimes polyols. (b) Diol-assisted fractionation (DAF) relies on the use of an appropriate solvent (typically non-protic), acid catalyst (triflic acid, metal triflates, or sulphuric acid) and diol (typically ethylene glycol).79 During this process lignin is depolymerized by acidolysis of the β-O-4 moiety (C2 and C3 pathways) followed by the trapping of the unstable C2-aldehyde species in the form of their more stable (typically cyclic) acetals.76 (c) Aldehyde-assisted fractionation (AAF) similarly involves fractionation using a non-protic solvent and an acid (typically HCl or H2SO4) but in the presence of an aldehyde, which reacts with the diol on the β-O-4 structure to form an acetal. The acetal prevents condensation reactions during fractionation to yield stabilized lignin oligomers that can then be depolymerized to lignin oligomers at yields comparable to RCF.83 | ||
Other active stabilisation approaches during acidic fractionation in the lignin-first sphere comprise the rational use of protection-group chemistries.70,71 Acid-catalysed depolymerization of lignin via acidolysis of the β-O-4 linkage proceeds through two pathways,72,73 one of which results in the formation of the so called Hibbert's ketones,74,75 and the other delivers a C2 aldehyde (as mixture of H, G or S, depending on wood), which is unstable under the reaction conditions. Trapping this C2 aldehyde by using diols, typically ethylene glycol, to form more stable cyclic acetals results in suppressing recondensation phenomena and very high selectivity to the corresponding C2-acetals.76 This has been shown on a variety of organosolv lignins,77,78 but also in a ‘metal-free’ lignin first process, dubbed here diol-assisted fractionation (DAF), where C2-acetals are directly obtained from lignocellulose under carefully selected reaction conditions.79 Newly generated aldehydes produced in a process may be protected as acetals by using ethylene glycol (Fig. 2b).76,79
Alternatively capping the benzylic alcohol, for example by exploiting the natural 1,3-diol in lignin sidechains to produce an acetal with formaldehyde or other simple aldehydes, stops benzylic cation production, dubbed aldehyde-assisted fractionation (AAF) (Fig. 2c).24,80 The latter strategy, which involves capping with stoichiometric reagents, enables further downstream depolymerisation and transformations of chemically stabilised lignin. Hydrogenolysis reactions for example, can be used to yield narrow product distributions.24,59,80,81
Efforts towards developing the lignin-first concept for fractionation of lignocellulosic biomass have significantly accelerated in the past several years and new studies are continuously being published in this area by the global biomass conversion community. A challenge in this emerging field is that there are no commonly accepted standards for the choice of feedstocks, product analysis, or evaluation of process performance.82 This is a severe limitation as: (1) quantitative comparison of methodologies and results between laboratories is challenging, (2) reproduction of other research groups’ procedures becomes difficult or even unfeasible, and (3) standardised lignocellulosic materials are not available. We therefore posit that to advance this research field from fundamental studies to a de-risked technology that industry could harness to fractionate and valorise lignocellulosic biomass, best practices should be established and implemented across the lignin-first research field. In this perspective, we thus present a set of recommended guidelines to establish common practices in this promising research direction for lignin valorisation. This perspective is organised into sections describing feedstock preparation and analysis, reactor configurations for performing lignin-first processing, measuring the efficiency of the catalyst performance, and determining product yields and mass balances. We conclude with future perspectives and propose several next steps to further advance the growing lignin-first biorefining field.
2. Feedstock preparation and characterisation
In simple chemical reactions, mass or mole balances are straightforward to establish both for products and reactants, which makes the calculation of typical reaction parameters, such as conversion, yield, and selectivity, straightforward to accomplish. With biomass conversion, however, these parameters are more difficult to establish due to the solid, heterogeneous nature of the substrate. Nevertheless, to accurately quantify yields, it is crucial to characterise the biomass thoroughly before conducting further experimental work. The following subsections propose guidelines to report the feedstock characteristics accurately.We note that the US Department of Energy's National Renewable Energy Laboratory (NREL) has published Laboratory Analytical Procedures (LAPs) for the relevant techniques described in Sections 2.1 and 2.2.84–91 Overall, we suggest that these methods be followed as a consistent means to prepare and characterize biomass feedstocks. The NREL website also contains Microsoft Excel-based spreadsheets to use these LAPs. While ASTM International has published similar procedures, NREL's LAPs are periodically updated to reflect new advances in the field of biomass analytics.
2.1 Sourcing and preparing the feedstock
The raw material understandably has a profound effect on the outcome of any transformation. In the case of lignocellulosic biomass, this is even more important as the biomass will differ depending on factors that, for woody biomass, include: species, age of the wood, sapwood or heartwood, bark content, regional factors, and time of harvesting. For agricultural residues, it is necessary to again know if the biomass consists of stems and/or leaves, contains seeds or cobs, and the physiological state of the harvested plants. For all biomass feedstocks, the source must be specified. When possible, referencing the plantation establishment and maintenance should be included: namely, watering practices, fertiliser, and herbicide use, as well as growth stimulation. Harvesting, initial processing, handling, and storage conditions of biomass should be described including the dimensions of the sample and what anatomical fractions of the plant have been used, and whether it is whole (above-ground) biomass.Before storing biomass in the laboratory for extended periods of time, the feedstock is typically dried, according to the NREL/TP-510-42620.86 This method employs air drying, a convection oven, or lyophilisation. Fortunately, cell wall material is reasonably stable, but care should be taken to dry the material, to <10% moisture content, before fungal infection can occur. Biomass is subsequently knife-milled (Wiley-mill or other) to pass through a 2 mm screen. This biomass is then milled further to pass through a 20 mesh (1 mm) screen affording ∼300–600 μm particles. This sizing usually obeys the rules of the minimal suspension criteria during its processing in stirred tank reactors typically used in the laboratory (>50 mL).
The next step is to determine moisture content on a mass basis, via NREL/TP-510-42621 at 105 °C.88 Oven-drying to a constant weight, in triplicate, is the recommended method and can be combined with NREL/TP-510-4262287 to determine ash content and limit sample consumption. Each sample requires ∼0.50 g (accurately weighed) of dried biomass. Even after the initial moisture content of the samples is determined, this measurement must be taken before each of the following procedures to accurately correct to a dry-weight basis and account for any changes to the moisture content due to humidity, storage, or location changes.
Extractives may be removed in ethanol/water by simple sonication treatment, Soxhlet extraction, or by using Dionex™ accelerated solvent extraction (ASE) via NREL/TP-510-42619.84,92–94 The use of flow-through systems in which the solvent composition can be varied systematically is advantageous for the selective removal of extractives.95 The typical procedure involves Soxhlet extraction with water and then ethanol.84,92,93 Also popular (but not in the aforementioned NREL LAP) is a method in which the ground biomass is suspended in 80% v/v EtOH/H2O and sonicated.94 The amount of water and ethanol extractives is determined by mass loss in the recovered biomass sample. In poplar wood, for example, these extractives account for 7–8% of the biomass by weight, ranging from roughly 2–10% for various woods.96 It is not necessary to analyse extractives in detail, although metabolite profiling may be useful, especially for transgenic materials or if extractives themselves are targets for further valorisation.97–100 For seed or other fatty-acid-rich samples, a hexane or chloroform extraction is also necessary. Protein-rich samples benefit from initial water extraction as proteins may aggregate, denature, and precipitate, so becoming difficult to subsequently remove if initially treated with organic solvents.101 We note that protein content can be estimated as well via NREL/TP-510-42625.85
After determination of all other measurable components, lignin, cellulose, and hemicellulose content in the biomass feedstock sample (vide infra), the remaining mass is assumed to be ash inorganics, which can be estimated via NREL/TP-510-42622.87,89 Elemental determination of ash content is not necessary unless there is indication that it might affect the reaction chemistry; above 10% ash content in the extracted sample may cause problems with hydrolysis. For example, palm wood and wastes may contain significant levels of iron102 that may interfere with catalysis and certainly makes NMR analysis challenging.103 In such an instance, atomic emission or absorption spectroscopy can be used for determining the element of interest.104,105
|
Minimum reporting requirements for feedstock sourcing and preparation:
• Origin and species of the feedstock. • Biomass particle size used for reaction studies. • Moisture, extractives, and ash content, and the methodology used to determine them. Preferred reporting recommendations for feedstock sourcing and preparation: • Part of the feedstock used, growth location, and harvest parameters. • Procedures for drying, sizing, and storing (and duration). |
2.2 Compositional analysis
Reliable standard methods are available for identifying and quantifying the carbohydrate constituents of biomass, described in detail in NREL/TP-510-42618.89 Even when the focus is placed on lignin deconstruction, quantifying the carbohydrate fractions is essential for the calculation of overall mass balances and yields to specific components. Furthermore, preserving and/or valorising the carbohydrate fraction is vital to make any lignin-first biorefinery economical. In these techniques, biomass is hydrolysed in the presence of a mineral acid in two stages. The first step involves the dissolution of polysaccharides in 72% w/w sulfuric acid at 30 °C, which is analogous to the first step of a Klason lignin determination.106 Therefore, it is convenient to perform both analyses simultaneously. The dissolved polysaccharides are diluted to form a dilute acid mixture (4%) and hydrolysed to monosaccharides at a higher temperature. At this stage, standards are treated under the same conditions to account for sugar degradation. The final yields are then corrected for degradation by comparison with the standards. Once the acid-insoluble fraction is filtered out, quantification follows HPLC separation of the monomeric sugars in the resulting liquor. Depending on the source of biomass, different monomers should be used as standards besides D-glucose. For example, D-xylose, D-galactose, D-mannose, and L-arabinose are used as standards to quantify hemicellulosic sugars in the case of most hardwoods and grasses. Alternatively, GC can be used. In this case, the sugars are first reduced and then acetylated to their alditol acetates.107 Here, the monosaccharide standards also need to undergo this pretreatment. For both HPLC and GC data processing, quantification can be performed by analysing the resulting sugar peaks using a calibration curve built using a dilution series with external standards. NREL/TP-510-42618 contains detailed quality control and error analysis methods.89The quantified and corrected values can then be used to calculate the various polysaccharide fractions. The measured glucose is largely from biomass’ cellulose fraction, but a fraction also derives from hemicelluloses; xylose, galactose, arabinose, and mannose are assumed to have been produced from hemicelluloses. Researchers should also consider free sugars and starch, if applicable. When calculating the original mass of these simple sugars in the native biomass, the water molecule added during hydrolysis has to be taken into account such that, rather than the monosaccharides themselves (glucose and xylose, for example) their forms in the polymer (glucan, i.e. (C6H10O5)n; and xylan, (C5H8O4)n) are used.
The percent by mass content of cellulose and hemicelluloses in the original biomass sample is calculated as follows:
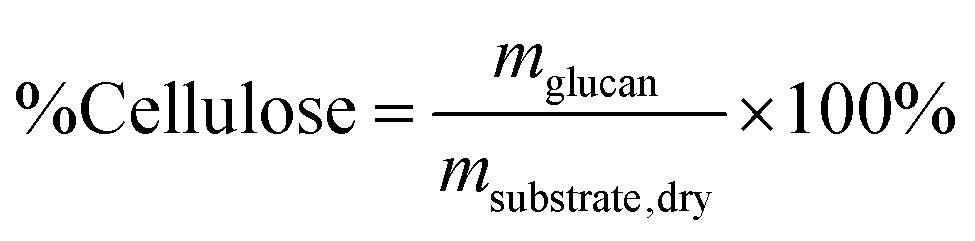 | (1) |
 | (2) |
|
Minimum reporting requirements for structural carbohydrate quantification:
• Fraction of major structural carbohydrates (cellulose and hemicelluloses) of the feedstock biomass based on at least three replicates with the resulting standard deviation. |
2.3 Lignin characterisation
It is noteworthy that the Klason lignin method is considerably more problematic for materials with bark and leaves, and for grasses and legumes that may contain proteins, suberins, and other complex components not found in extracted wood.101 The source of the non-lignin components in ‘Klason lignin’ has only occasionally been delineated; such is the case for cereal grains and other plant-based foods in which the limitations of “non-specific lignin determination methods”111 was recognised.111,112 A quote from an abstract notes the extent of the problem: “Estimation of the contribution of non-lignin compounds to the Klason lignin contents reduced the noncorrected Klason lignin contents of the insoluble fibres from 28.7% (kale), 22.8% (pear), 14.8% (wheat), and 9.9% (corn) to maximum lignin contents of 6.5% (kale), 16.4% (pear), 4.9% (wheat), and 2.3% (corn).”112 A seedcoat material containing an intriguing C-lignin (derived from the novel monomer, caffeyl alcohol) was originally reported to be 90% lignin from Klason lignin determination,113 but was later revised down to only 10% lignin in a subsequent study,28,114 which used methods that are unfortunately not viable for widespread corrections of Klason values. For now, Bunzel's methods appear to be the best for elucidating interfering components derived from fats, waxes, cutin, and suberin.112 Other methods exist for lignin determination, but all have their own limitations.101 This long-term unsatisfactory situation makes it difficult to make strong recommendations here except to note that Klason lignin remains the gold standard for woody biomass,89 and may also be reasonable for forage grasses and legumes.101,115
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :G:H ratio.
Measurement of the ratio of S, G, and H units within lignin is important because of the ramifications for depolymerisation and the value of the products. Having two, one, or zero methoxyls ortho to the phenolic group (that may be etherified in the polymer) affects more than just the electronic characteristics of the S, G, and H aromatics. Guaiacyl units are typically more condensed (i.e., have more C–C links with other lignin units) than S units because the availability of the aromatic C5 position for radical coupling leads to their being involved in 5-β-, 5-O-4-, and 5-5-coupled units that have no S counterparts;116 obviously S units may still be involved in 4-O-5-coupled units (with the G-unit linked at C5). S units are therefore more heavily involved in the β-ether units that are the weaker bonds that are cleaved in essentially all of the depolymerisation methods, including those used for analytics. S units are, however, also more heavily involved in (condensed) β-β units, because of the long lifetime and favourable dimerisation of the sinapyl alcohol radical during lignification.116 Incidentally, 4-O-5 and 5-5 units (Fig. 1) are difficult to quantify, and may not contribute to S:G:H measurements, depending on the method. They are important because they represent connections formed during lignification from two growing polymer chains.8,116 However, these were always thought to produce real ‘Y-type’ branchpoints in the polymer chain but, to date, evidence only for free-phenolic units can be obtained, meaning that the lignin polymer chains may be less branched than previously thought.6–8,117
:G:H ratio.
Measurement of the ratio of S, G, and H units within lignin is important because of the ramifications for depolymerisation and the value of the products. Having two, one, or zero methoxyls ortho to the phenolic group (that may be etherified in the polymer) affects more than just the electronic characteristics of the S, G, and H aromatics. Guaiacyl units are typically more condensed (i.e., have more C–C links with other lignin units) than S units because the availability of the aromatic C5 position for radical coupling leads to their being involved in 5-β-, 5-O-4-, and 5-5-coupled units that have no S counterparts;116 obviously S units may still be involved in 4-O-5-coupled units (with the G-unit linked at C5). S units are therefore more heavily involved in the β-ether units that are the weaker bonds that are cleaved in essentially all of the depolymerisation methods, including those used for analytics. S units are, however, also more heavily involved in (condensed) β-β units, because of the long lifetime and favourable dimerisation of the sinapyl alcohol radical during lignification.116 Incidentally, 4-O-5 and 5-5 units (Fig. 1) are difficult to quantify, and may not contribute to S:G:H measurements, depending on the method. They are important because they represent connections formed during lignification from two growing polymer chains.8,116 However, these were always thought to produce real ‘Y-type’ branchpoints in the polymer chain but, to date, evidence only for free-phenolic units can be obtained, meaning that the lignin polymer chains may be less branched than previously thought.6–8,117
Other factors can influence bond speciation in lignin, which is an active area of research.118,119 We comment only briefly on H units because they are minor and may often be neglected, except in targeted transgenics, and in softwood compression-wood zones in which the level may reach some 30%.120 In principle, H-units have even more options for radical coupling than G and S units because of the additional open C3 position on the ring but, although H units may be involved in more extensive 5/3 bonding, the longstanding assumption that H-lignins are substantially more condensed is not borne out experimentally – β-ether units still predominate even in H-rich lignins or H-only synthetic lignins.121 Unfortunately, due to conflation from other units that are not polymer units derived from p-coumaryl alcohol,122–124 H units are often reported at a far higher level than they actually are in lignin (vide infra). Care in analyses must therefore be taken to exclude compounds arising from components in lignin that are not derived from the prototypical monolignols. Examples include the vinylphenol and vinylguaiacol that efficiently arise from abundant p-coumarate and ferulate esters in grasses as analysed by analytical pyrolysis followed by mass spectrometric detection such as GC/MS or Molecular Beam Mass Spectrometry (MBMS).125–127 The common refrain that softwoods are G-lignins (with low H levels), hardwoods are S/G-lignins (with very low H levels), but grasses are H/G/S lignins largely results from this mischaracterisation; grass lignins rarely contain more than 5% H units.
We illustrate below how to obtain (more) reliable values but acknowledge that none of the methods listed below is capable of determining the actual distribution in the polymer. Although the NMR methods can in principle, limitations that are discussed below persist in practice. All of the degradative methods release only a fraction of the polymeric units for quantification. We will not discuss secondary spectroscopic methods such as FT-IR, Raman, or NIR because they are all compromised by their reliance on the other methods here for their calibration or have insufficient ‘peak purity’ to allow single- or multiple-peak direct quantification at this point.
The peak contours can be volume-integrated to provide comparative S:G:H estimates based on peak ratios. As far as anyone can discern (as there is no method to provide independent and accurate data), even when HSQC spectra are acquired under qualitative conditions, the ratios are reliable. Recent advances such as the more energy-efficient adiabatic-pulse variants of HSQC-type experiments are recommended as standard experiments on newer high-field instruments, particularly those equipped with cryogenically cooled probes. They offer the advantages of a wide inversion bandwidth meaning minimized 13C-pulse offset effects, lower power, and a wider decoupling range using various decoupling schemes, as reviewed in the context of lignin spectra.128 They are also less sensitive to spin–spin coupling effects, for which there are other solutions.132–134 To obtain S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :G:H ratios, it is strongly recommended to volume-integrate only the well-dispersed S2/6, G2, and H2/6 (which is unfortunately compromised by coincidence with phenylalanine) because these all have similar coupling environments – all are coupled only to the 6-proton with a small JH–H of 1.6–2.0 Hz, all have a similar 1JC–H coupling constant, and all are in the same small region of the spectrum.128
:G:H ratios, it is strongly recommended to volume-integrate only the well-dispersed S2/6, G2, and H2/6 (which is unfortunately compromised by coincidence with phenylalanine) because these all have similar coupling environments – all are coupled only to the 6-proton with a small JH–H of 1.6–2.0 Hz, all have a similar 1JC–H coupling constant, and all are in the same small region of the spectrum.128
Extrapolating the reliability of qualitative HSQC for S:G:H quantitation to other units or structures of interest is not, however, sound. Regular HSQC data are not quantitative. For example, it is well known that mobile end-units, with their longer relaxation times, become over-represented vs. units buried within the lignin backbone because the fast relaxation of the latter units during the actual NMR pulse sequence (and therefore after excitation but before the actual signal detection) is significant.128,131 Mobile, pendent units such as p-coumarates (in grasses) and p-hydroxybenzoates (in poplar, aspen, palms) adorning some lignins (but that should not be quantified as actual lignin units) are therefore seriously overestimated by such HSQC methods. As this longer relaxation giving higher signals is contrary to expectations for researchers familiar with 1D NMR, it is worth a simple illustration of why this is so. In such sophisticated NMR experiments, the length of the pulse sequence applied following the initial excitation of the proton signals, prior to the signal acquisition, is a non-trivial fraction of the relaxation time. If a long-relaxing end group's resonance has decayed to 90% of its original level before the start of the acquisition, but an internal unit's rapidly-relaxing nuclei have decayed to 10%, the former will be over-represented relative to the latter by a factor of 9.
Although current HSQC NMR methods must be used cautiously to quantitatively analyse whole cell wall samples, isolated lignins are more soluble and have sufficiently improved relaxation properties to allow utilisation of a quantitative method such as HSQC0.135 In this experiment, three 2D spectra with different starting points are acquired, the peak volumes are measured, and these are projected back to a theoretical time-zero (before relaxation has occurred). It works well in some cases, as illustrated recently,136 but we stress that quantification remains non-trivial. The recommendation for original biomass, therefore, is to use the described qualitative HSQC methods130,131 on whole-cell-wall (whole biomass) or lignin samples for reasonable S:G:H estimates. If accurate values are needed, the lignin must be isolated for the use of HSQC0 methods.135,136 The most straightforward way is to produce a so-called enzyme lignin (EL) by digesting away most of the polysaccharides while retaining essentially the entire lignin component.123,137 If keeping all structures in their native form is not considered crucial, a more rapid and convenient method that provides a large fraction of the lignin in rather clean form [but with spirodienones (β-1-linked units) hydrolysed to their open form, for example] is a mild acidolysis method.138,139 Solution-state NMR is indispensable for validating and profiling the incorporation of non-canonical monomers, including those from beyond the monolignol biosynthetic pathway (such as tricin and hydroxystilbenes) into the lignin polymer.10,126,128,140–142 Modern solid-state NMR (being currently used more for understanding polysaccharide interactions),143,144 and Dynamic Nuclear Polarisation-enhanced methods,145 are producing exciting new insights and are worth monitoring in the quantification space.
• Nitrobenzene oxidation produces the highest monomer yields as certain C–C-bonded structures may cleave to produce a monomer.151 Micro-methods have been developed to improve both throughput and safety,152 as nitrobenzene explosions are well-known. The product hydroxybenzaldehydes and hydroxybenzoic acids are all available as standard compounds so they are easily quantified. Researchers need to be aware that certain lignins (poplar, aspen, willow, and palms) contain p-hydroxybenzoate units on their lignins, and grasses (and all commelinid monocots) similarly contain p-coumarates that will produce the same monomers. Conflating all these molecular species into a single H-level number elevates the apparent H-levels in a way that does not reflect the true lignin composition; methods (vide infra) that do not produce the same products as lignin should be used in such cases. Grass cell walls also contain ferulates, mostly on arabinoxylan polysaccharides, and their oxidation to the same monomers may also artificially inflate G-levels.
• RCF conditions have been used to explore lignin structure since the 1930s,51,153 and may yet become a preferred method;24,42,57,60,62,118,154–159 comparisons have already been made with thioacidolysis.118,160 The reactions are simple, need little optimising, and produce a modest array of simple products largely retaining their H, G, and S signatures, products for which standards may be available (for authentication and as quantification standards). Catalyst choice (e.g., Ru vs. Pd) allows selectivity for primarily arylpropanes vs. arylpropanols.24,28,59,61,80,81 The only complication is that 4-O-5-coupled units may partially cleave to contribute (in only a small way) to the monomers, but also produce novel ‘rearrangement monomers’ and dimers diagnostic of their origin.161 Care should be taken when using RCF for analytical approaches to avoid demethoxylation reactions, which would skew the resulting S:G:H ratio. We stress that RCF is not yet a fully established analytical method and note that opportunities exist to develop this approach further.
• Thioacidolysis is perhaps today's premier diagnostic method for characterising lignin, releasing p-hydroxyphenyl-trithioethyl-propyl monomers solely by cleaving β-ethers.150,162,163 Again, small-scale methods are available.164,165 Unfortunately, the standards are not available and need to be synthesised.166 The desulphurisation method that produces the arylpropanes is not generally used for monomers analysis, but is used for dimers characterisation.147,150,162,167 In addition to the foul smell of the reagents, thioacidolysis has one shortcoming – many units in lignins are acylated (primarily by acetate, p-hydroxybenzoate, p-coumarate, or ferulate) but these are neither fully retained nor fully cleaved during the procedure, resulting in some distortion of values.168,169
• DFRC was invented to possibly circumvent the noxious odour from thioacidolysis by using a different mechanism, reductive cleavage, to specifically cleave β-ethers.149,170 In fact, thioacidolysis generally produces higher monomer yields, but DFRC is particularly useful for certain determinations. Like thioacidolysis, it leaves a diagnostic signature that a β-ether has been cleaved, specifically the double bond in the product monolignol acetates. It is somewhat attractive that the products are the monomers (although as their peracetates) from which the lignin was originally biosynthesised in the plant. Its huge advantage or disadvantage, depending on the level of information required, is that it absolutely does not cleave esters in the acylated monolignols noted above.169,171 That means that any unit that is acylated, except by acetate, will not yield its H, G, or S monomer, but will instead be released as a monolignol conjugate. Quantification therefore needs to either be measured with the normal and acylated monomers summed, or the esters should be first cleaved in a separate step (below). The DFRC procedure for cleaving ethers but leaving esters intact has become a powerful tool for studying such conjugates in lignin.149
In principle, it is straightforward to assure that acylated lignins do not pose a problem for nitrobenzene oxidation, thioacidolysis, DFRC, or RCF. Adding a saponification (and extraction) step prior to the procedure, removes these esters. It is not completely straightforward, however, and reliable protocols do not appear to have been developed to date.
|
Minimum reporting requirements for lignin characterisation:
• Klason lignin content based on at least three replicates along with the resulting standard deviation. • Based on carbohydrate and lignin analysis, the overall mass balance of the substrate (lignin, cellulose hemicelluloses, ash, extractives, protein, etc.). Preferred reporting recommendations for lignin characterisation: • S • 2D NMR volume-integrals for S:G:H from whole-cell-wall or isolated lignin samples. |
3. Reactor design for lignin-first biorefining
For any chemical transformation, selecting the appropriate type of reactor is crucial for the overall process design. Of all process equipment, reactor design requires consideration of rate constants, reaction enthalpies, heat and mass transfer coefficients, and phase equilibria data. In addition, other factors, such as process economics, scale-up, and safety of operation, influence this choice.172The two primary reactor configurations reported to date in lignin-first biorefining are batch and flow-through (Fig. 3). Although both reactor designs can be used to extract experimental data, it is imperative to understand the benefits and limitations of each configuration when delineating experimental goals. In this section, we describe innate advantages and common pitfalls for each reactor type and present guidelines for reactor selection, operation, and reporting. We provide recommendations and best practices for data collection, including heuristics for identifying heat and mass transfer limitations. We note that lignin-first processes often require high temperature and pressure; thus, operators must receive proper training and adhere to strict safety protocols and standards in the construction and utilization of chemical reactors.
 | ||
| Fig. 3 Reactor configurations reported to date for lignin-first biorefining. (a) Batch reactors wherein whole biomass, catalyst, solvents, and other species (e.g., H2) are mixed and reacted. (b) The use of a catalyst basket in a batch reactor system can physically separate the catalyst and biomass particles, thus simplifying post-reaction biomass and catalyst analyses.66 (c) Flow-through systems are also useful to physically separate the biomass and catalyst during lignin-first processing and to study solvolysis-limiting or hydrogenolysis-limiting reaction conditions. Therein, a heated solvent flows over a switchable biomass bed, and the liberated, lignin-rich liquor passes over a catalyst for depolymerization and stabilisation.63–65,118 Image credit: Gregory Facas, NREL. | ||
3.1 Batch reactors
A stirred autoclave vessel, commonly known as a batch reactor, has been the typical reactor used to investigate lignin-first fractionation processes in the condensed phase (Fig. 3a). In this type of reactor, the reagents, solvents, and catalysts are sealed and heated for a predetermined amount of time and can feature intermittent sampling to track reaction progress. Batch reactors are simple to operate and are used commercially to produce low-volume, high-value chemicals. To date, most biomass processing in the pulping and bioethanol industries is performed in batch or semi-batch mode. Understanding both the inherent practical advantages and the limitations of batch reactors is important to avoid data inconsistencies. The following suggested guidelines can be used to improve reporting and reproducibility across laboratories.Reactions carried out in batch systems are transient in nature, complicating the collection of reaction rate data, as reactant and product concentrations change as a function of time. Instantaneous rates that are not only specific to a particular set of reaction conditions (e.g., partial pressure, temperature, and initial concentrations), but also to the extent of reaction, are not readily available. Initial rate data can be obtained, for example, by calculating the slope of regressed datapoints collected in the near-linear, low-conversion regime (typically <10%) in a conversion as a function of time plot. Data fitting may be another way to obtain rate data, but this requires a reliable kinetic and/or mass transfer model that might not be trivial to develop.119 Reporting individual yields might generate misleading data given that product yields, particularly at high conversion extents, can be almost identical at two different timepoints as reactions rates slow down with reactant depletion or as reactions approach equilibrium. These drawbacks are exacerbated when trying to collect rate data for more complex reaction networks (e.g., A → B → C), which is always the case for lignin-first processes.
Ideally, the reaction should start being timed (t = 0) when the reactants are put into contact with the catalyst at the reaction temperature. However, in practice, this is often not possible as reagents and catalysts are mixed together and heated to the reaction temperature with the initial time chosen as the point at which heating starts; this heating period can take minutes to tens of minutes, depending on the laboratory setup. Using the starting time when the reactor is at reaction temperature is similarly problematic as reactions already occur earlier in the process during the heat-up period. Adding the biomass later after proper heating seems like a solution, but this is not commonly practical at laboratory scales. Construction materials, vessel volume, use of liners, heating and cooling method, stirring regime, and total mass loaded into the reactor all drastically alter the heating and cooling profiles, thereby introducing large errors particularly at the crucial early reaction times needed to calculate initial rate data. Similarly, although adding the catalyst at reaction temperature may be an option in specialised reactor configurations, depolymerisation and condensation can occur even in the absence of catalyst as a result of solvolysis. Regardless of the batch reactor setup, the heating and cooling profiles for the vessel should be reported.
In typical batch lignin-first processing, the solid biomass is mixed with a solid heterogeneous catalyst, which makes post-reaction biomass and catalyst characterisation a substantial challenge. One solution to overcome this problem includes the use of catalyst baskets in an autoclave (Fig. 3b).66 Using catalyst baskets and pre-sized catalyst pellets in these reactor types enables practical separation of catalysts and pulp after reaction. Such systems can also be used to investigate the stability of the metal catalysts by renewing the solvent and biomass loaded into the reactor with the same basket. Sizing the catalyst particles is important to prevent catalyst escape into the bulk liquid with the biomass. However, the size cannot to be too large or diffusion limitations will occur, hampering catalytic depolymerisation and the required rapid intermediate stabilisation.66
Non-uniform stirring caused by vessel geometry or the use of baffles, and/or the type of stirring mechanism (magnetic, mechanical), can cause the formation of dead spots with poor mixing that potentially introduce heat and mass transfer artefacts (vide infra). Although in some cases, liquid-phase experiments can be carried out under static conditions, in most biomass fractionation processes (particularly when dealing with three-phase systems), mixing is an important factor for obtaining reproducible data. Depending on the size of the biomass particles, slow stirring could cause particles to sink to the bottom of the reactor, thus changing reactivity profiles. The same occurs when using baffle systems that are beneficial to increase gas/liquid mass transfer. Similarly, using high biomass-to-solvent ratios, that are required for the economics of lignin-first processing, may result in highly viscous slurries that are difficult to mix without powerful overhead stirrers. For example, although Sels and co-workers were able to use very high biomass to solvent ratios (e.g., 60 gbiomass in 240 mL of solvent) in batch RCF experiments using an autoclave with overhead motorised stirring,43 similar conditions would have resulted in less effective mixing in vessels relying on stir bars powered by magnetic stir-plates. For this reason, small reactors, below 50 mL, with similar loadings generally give irreproducible results. For experiments involving three-phase systems (e.g., those involving solid biomass, solid catalyst, solvent, and reactant gas), the use of gas-entrained impellers is ideal to maximise gas–liquid interfacial area. However, caution must be exercised when operating at supercritical conditions, at which solid particles can enter and clog the impeller due to the loss of a significant density difference between the gas and liquid phases needed for gas entrainment.
Many autoclaves are equipped with a sampling port in which a small volume of the slurry (containing the solvent, liquid products, solid biomass, and the catalyst) is isolated from the rest of the reactor for collection. However, if the reaction volume loaded into the reactor is low compared to the volume of the sampling tube, then frequent sampling will change the reaction profile producing non-uniformities in the slurry and changes to the gas–liquid proportions in the vessel; preferably not more than 5–10% volume should be removed from the reactor (in total). As an alternative, several identical reactors can be run in parallel, stopping each one at a different time interval to construct a reaction time profile.
|
Minimum reporting requirements for batch reactor use in lignin-first processes:
• Reagents, catalysts, and solvents and their nominal quantities. • Vessel geometry, material of construction, reactor liner, stirring method (if used), and stirring rate. • Heating, cooling, and pressure profiles. • Quantity and frequency of sampling. • Definition of the reaction time. Preferred reporting recommendations for batch reactor use in lignin-first processes: • For kinetic studies, the initial rate data or full reaction time profiles rather than single/final yields. Best practices: • Total sampling volumes should not exceed 5–10% of the reaction volume. • Reaction vessels above 50 mL are recommended, especially for high solid loadings (above 10 wt%). • The use of overhead motorised stirring is recommended especially for high solid loadings (above 10 wt%). • Use Hastelloy reactors or an inert reactor liner to avoid leaching of reactive metal species from the reactor walls. |
3.2 Flow-through reactors
Disadvantages with batch reactors include: (1) slow heating, (2) inability to optimise and also study solvolysis and hydrogen-transfer reactions separately, (3) tedious separations of catalyst from the pulp, and (4) mechanical disruption of the pulp. A strategy to separate the solvolytic extraction of the lignin from the metal-catalysed reactions is to separate these two processes in space and time by employing a flow-through reactor (Fig. 3c).63–65,118 In such a system, the biomass is loaded into a percolation chamber and the metal catalyst is loaded into a downstream reactor. Solvent, which can be pre-heated to the desired reaction temperature, is then pumped through the percolation chamber to extract lignin (and some polysaccharides) and transfer them to the reactor containing the transition metal catalyst bed. In between the percolation chamber and the reactor, a T-coupling with an outlet may be fitted to allow real-time analysis of both solvolysis and hydrogen transfer reactions. A back-pressure regulator stabilises the pressure in the system.In contrast to batch reactors, flow-through reactors with switchable beds convert substrates in a semi-continuous manner (Fig. 3c) Using this configuration, steady-state can be approximated by switching biomass beds such that the concentration of extracted lignin reaching the catalyst bed remains relatively constant within a range that does not significantly alter reaction rates. In its simplest form, a packed-bed flow reactor consists of a tube packed with a catalyst bed held in place by a frit, mesh screen, or plugs of inert material (e.g., glass beads, quartz wool). Under ideal conditions, all substrate elements flow at the same velocity, parallel to the reactor axis, without back-mixing; plug flow conditions can be measured and reported as the dimensionless Reynolds number, Re. This scenario allows the assumption that all material present at any given reactor cross-section has had an identical residence time. Depending on the application, these reactors can be operated either in isothermal (i.e., constant temperature throughout the reactor) or adiabatic (i.e., varying temperature across the reactor length) modes to process feedstock over a wide range of throughput volumes. The advantages of flow systems were recently demonstrated for RCF of poplar using flow-through setups with separated biomass and catalyst beds to obtain intrinsic kinetic data and identify mass-transfer limiting conditions for the entire process.64,118 Decoupling the conditions for solvolysis from those of reductive stabilization in flow enabled the interrogation and optimization of crucial aspects of the RCF process that are difficult to access with batch reactors. For instance, operating at the limiting conditions for solvolysis (i.e., when lignin fragment detachment is slow relative to the time scale of the total number of turnovers for reductive bond cleavage at the catalyst surface) allowed the influence of the solvent composition on lignin solubilization to be isolated, while operating at limiting conditions for reductive stabilization (i.e., when lignin solvolysis is fast relative to the time scale of reductive bond cleavage of lignin fragments) allowed catalyst activity and stability to be studied. The following suggested guidelines for flow-through lignin-first reactors can be used to improve reporting and reproducibility across laboratories.
Although turbulent flow regimes are preferred to laminar flows for improved mixing and heat transfer normal to the flow direction, achieving high Reynolds numbers (Re) could require excessively high flowrates. Under laminar flow regimes, the flowrate is proportional to the pressure drop, which is a function of bed height, linear flowrate, and dynamic viscosity of the fluid. The pressure drop can increase with bed deformation caused by high flowrates, pellet dissolution, or structural changes in the bed, eventually leading to over-pressurisation and even complete loss of flow through the reactor.
Catalyst beds act as deep-bed filters, capturing and precipitating colloidal material that can cause bed fouling and clogging. Researchers must pay close attention to flowrate/pressure drop effects and always install the appropriate safety measures (guard beds, rupture disks, pressure monitors, as well as emergency over-pressurisation alarms and shutdown valves) to prevent accidental releases in the case of partial or full blockage.
Non-uniformity in the bed packing is a major culprit for flow non-idealities and should be evaluated when working with solid biomass substrates. For example, channelling (in which fluid (solvent or gas) passes through one part of the reactor bed more rapidly than other parts) or hold-up (in which a fraction of the substrate resides in stagnant areas with reduced flow) can drastically change reaction profiles. For catalytic beds in microreactors, it is recommended to pelletise the powdered catalyst and sieve it to a size large enough to prevent large pressure drops or add in inert particles of a larger diameter, in both cases to avoid external mass-transfer limitations (vide infra). A common heuristic is to allow space for ≥100 particle diameters in the axial direction and maintaining at least a 4:1 height-to-width aspect ratio of the bed to ensure uniform contact of all substrates with the solids.173 This is generally accomplished using inert fillers (e.g., quartz chips or silicon carbide particles) having a similar particle size to the sieved catalyst to reach this ratio for cases in which a lower amount of catalyst is required. Similarly, for the biomass beds, the use of inert filler particles may be necessary to avoid bed collapse, especially in cases where high extraction extents occur.174
Deviations from ideal plug flow due to back-mixing will cause product streams to have a distribution of residence times. For extremes of back-mixing profiles, the plug flow reactor approaches the behaviour of a continuously stirred reactor (CSTR), potentially limiting expected conversion values – the mathematical description of this phenomenon is beyond the scope of this work and can be found in standard reaction engineering textbooks.173,175,176 When using fixed biomass beds in flow-through reactors, as in the flow-through RCF studies described above, it is crucial to check the integrity of the bed during solubilisation. Indeed, although some solvents (e.g., methanol, ethanol, and dioxane) might selectively extract lignin from the cell wall, the use of water, acid, or base additives may also remove the carbohydrate fraction and cause severe structural changes to the biomass bed, which can cause bed collapse and plugging. Lastly, we note that changes in residence times in a flow-through reactor may result in different degrees of undesirable reaction products. For example, when performing flow-through RCF in reactors with separated biomass and catalyst beds, the flow profiles and residence time distribution between the beds may cause higher condensation rates compared to batch reactors unless a solvent system is used that temporarily stabilises the intermediates.177
In a sealed autoclave, the pressure is established by the vapor-liquid equilibrium at the reaction temperature of the liquid and gas substrates loaded into the reactor, whereas in a flow-through system the pressure is regulated by the user (e.g., by using a back-pressure regulator). If the appropriate pressure is not selected when desiring to work exclusively in the liquid phase at the reaction temperature, the feed may exist in a mixed liquid–vapor or entirely in the vapor phase when flowing through the catalyst bed, generating irreproducible data. For this reason, it is recommended to carry out the appropriate vapor–liquid equilibrium calculations prior to the design of the experimental stage using, for example, tabulated data or a software package such as ASPEN Plus.
3.3 Reactor selection for lignin-first studies
Reactor selection and experimental design must be performed in concert to obtain the desired information accurately, with the minimum number of measurements and the least expense. This is particularly important for lignin-first processes, such as RCF, that comprise two independent consecutive steps, each featuring intrinsic parameters that can dominate the apparent behaviour of the overall system. In this section, RCF will be used to illustrate the importance of reactor selection to meet experimental goals.Depending on reaction conditions, the overall performance of typical lignin-first processes can be limited either by the solvolysis or the stabilisation step. In RCF, solvolysis-limited conditions occur when lignin detachment from the cell wall is slow relative to the timescale of catalytic turnover, whereas the stabilisation-limited conditions are observed when lignin solvolysis is fast compared to the timescale of reduction.64 A solvolysis-limiting condition is required when investigating the dynamics of lignin detachment from the cell wall, including, for example, comparisons of product distributions across biomass types, extraction solvents, and additives (e.g., inorganic acids or co-solvents). AAF is almost always operated in solvolysis-limiting conditions, ensuring rapid stabilisation and condensation prevention, whereas for RCF, the limiting conditions depend on the goals of the study. Batch reactors are ideally suited for solvolysis-limited studies because different variables, including substrates, solvents, and temperatures can be screened with ease across a wide range of lignin extraction extents. Using low biomass-to-catalyst ratios is common practice to ensure operation in the solvolysis-limiting regime. For this reason, however, it is also common to observe invariant monomer yields and fully saturated monomer sidechains regardless of catalyst type used. Note that lignin solvolysis from the cell wall can be limited by mass transfer depending on, among other factors, biomass type and particle size.119 Mass transfer effects must be identified and reported, as it may cause data irreproducibility across experiments and laboratories.
Operation in a reduction-limited regime is necessary to study catalyst activity and stability, as well as to perform comparisons across catalyst types. This is always the case for AAF because stabilisation is temporally and spatially decoupled from downstream catalytic depolymerisation. Typically, intrinsic kinetic data (e.g., activation barriers) are used to assess catalyst performance, including reaction rates and product selectivity, whereas catalyst stability is evaluated from deactivation rate profiles coupled with catalyst characterisation. Extracting these data with batch reactors is cumbersome due to difficulties in measuring initial rates accurately and separating the catalyst from the biomass particles. Flow reactors, on the other hand, are ideally suited to this task. Having precise control over residence time allows collection of steady-state data under differential conditions, which is necessary to calculate initial rates and to identify primary vs. secondary products in complex reaction networks. Similarly, catalyst stability studies are readily accessible by measuring deactivation profiles at steady-state intermediate conversion levels and performing characterization studies post-reaction (or even under operando conditions) on the catalyst bed that is physically separated from the biomass bed. Section 3.4 outlines several strategies to check for the presence of transport artefacts in experimental data.
3.4 Transport effects
The rate of diffusion of reactive substrates across phases can alter the observed rate of chemical reactions. Similarly, the rate of heat transport into (for endothermic reactions) or away from (for exothermic reactions) a catalyst can influence the temperature-dependent rate constants for the system. To properly evaluate or compare lignin-first methods operated either in solvolysis- or reduction-limited regimes (vide supra), intrinsic kinetic data that are free from heat and mass transport artefacts must be measured.119 If the observed rate is modified by these artefacts, then new conditions must be selected to either lower the reaction rates or increase the rate of heat/mass transfer. Rigorous calculations of reaction-diffusion profiles to extract intrinsic parameters, (e.g., Thiele moduli and effectiveness factors) require numerical techniques and high-fidelity models, as recently shown by Thornburg and co-workers for lignin-first RCF.119 In this section, we show simplified methods to assess the influence of mass transfer limitations in an approximate manner.Gas–liquid diffusion is important when consuming gaseous reagents in a liquid solvent. For example, the transport of hydrogen gas across the interface between the headspace and the agitated solvent in a batch reactor performing RCF could limit the apparent rate if the catalyst is starved of hydrogen. It is recommended to always perform a “maximum rate of gas transfer” analysis on the reactor that will be used for the lignin fractionation studies under identical conditions of liquid volume, gas pressure, temperature, and agitation to those used in the actual fractionation experiment. This measurement is particularly important when working with systems featuring very fast apparent rates (typically those exceeding 1 turnover per site per second). Several methods exist to evaluate the maximum rate of gas transfer. For hydrogenation reactions using molecular H2, Meille et al.178 and Chaudhari et al.179 outlined experiments using either physical adsorption or reaction of a substrate with well-established kinetics (e.g., styrene or α-methylstyrene) to evaluate the gas-to-liquid mass transfer coefficients. A comparison of the experimentally determined maximum rate of transfer coefficients to those observed under catalytic conditions will indicate if the catalyst will be gas-starved. Note that if the system is gas–liquid mass transfer limited, increasing the stirring rate alone may not be able to overcome this limitation.
The transport of liquid substrates and dissolved gases to the external surface of the solid catalyst pellet can also disrupt intrinsic reaction rates. As the substrates approach the catalyst pellet, they must cross a stagnant boundary layer in which the flux across this layer will be a function of the concentration difference between the bulk phase and the external catalyst surface and the mass transfer coefficient. This phenomenon is often modelled by assuming a linear concentration profile across these two regions by assuming that no reactions take place at the boundary layer. A simple mathematical analysis combined with engineering correlations (not shown here) generates the following relationship between mass transfer coefficient and important process variables when flowing a fluid around a spherical catalyst particle:173
 | (3) |
If kc is known, then a simple criterion to evaluate if external mass transfer does not affect the rate is as follows:
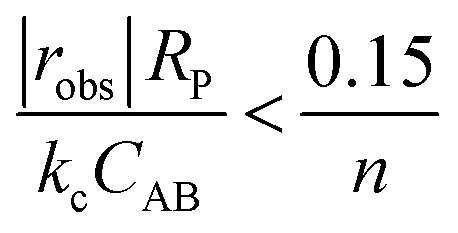 | (4) |
|
Minimum reporting requirements for flow-through reactor use in lignin-first processes:
• Reagents, catalysts, and solvents and their quantities. • Detailed diagram of the system (including bed aspect ratio, volume of reaction system, thermocouple location, and flow configurations). • Flowrates of liquids and gases. • System pressure and temperature. • Catalyst pellet size. • Type of solid diluent (e.g., silicon carbide). Preferred reporting recommendations for flow-through reactor use in lignin-first processes: • Description of reactor packing profile. • Description of product collection and quenching system. • Description of start-up phase, biomass and catalyst pretreatment. Best practices for flow-through reactor use in lignin-first processes: • Pressure in the flow system should be monitored actively during operation. • Pressure drop measurements across the catalyst bed reactor must be performed to identify appropriate fluid flow and mixing regimes. • Installation of pressure relief valves to avoid over-pressure from plugging (especially during bed compaction phase) is strongly recommended. • The use of check-valves to mitigate back-flow issues during plugging is recommended. • Back-pressure regulators to ensure that pressure is controlled and maintained are recommended. • Appropriate catalyst bed packing procedures should be followed to avoid data discrepancies (e.g., at least 10 particles in the radial and 50 particles in the axial direction to prevent stochastic effects). • The use of a flow profile that approaches an ideal plug flow, which requires turbulent flow. |
Once substrates reach the surface of the catalyst, they need to diffuse through the internal pore structure of the catalyst to reach the active sites. Depending on the size of the pores and the intrinsic rate of reaction, the observed rate might be internal-transport-limited if the substrates are consumed before reaching all the active sites within the particle. The non-linear analysis coupling the simultaneous diffusion and reaction events required to evaluate the extent of catalyst pellet utilisation (i.e., the effectiveness factor) is complex. Fortunately, Weisz and Prater developed a simple criterion based on measurable quantities to determine whether the observed rate is not influenced by internal mass transfer artefacts.180
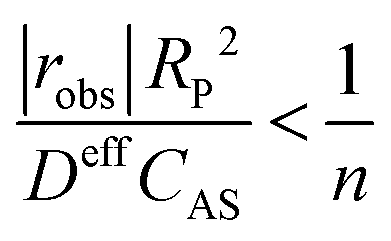 | (5) |
For highly exothermic or endothermic reactions, the temperature within a catalyst pellet might be very different from that measured by the thermocouple in the bulk phase if heat cannot be transported quickly enough within the boundary layer around the pellet (external limitation) or within the pellet (internal limitation). Fortunately, an external criterion developed by Mears (eqn (6)) and an internal criterion developed by Anderson (eqn (7)) can be used to evaluate whether heat transfer effects influence the observed reaction rate as follows:181,182
 | (6) |
 | (7) |
Diagnosing transport effects with eqn (3)–(7) above requires prior knowledge of several critical parameters, such as effective diffusivity and thermal conductivity, that may not be straightforward to obtain for a given system. For this reason, a simple, yet powerful, experiment to verify the presence of both heat and mass transfer limitations is known as the Madon-Boudart test183 (also known as the Koros–Nowak criterion). This test involves measuring rates on catalysts in which the concentration of active material is changed deliberately, comparing both concentrations at two different temperatures. In the absence of heat and mass transfer effects, the reaction rate is proportional to the concentration of active material (i.e., the TOF should be invariant) and this proportionality should hold at different temperatures. A common way of varying the concentration of the active material is simply by diluting the bed with an inert material while keeping the total bed volume constant, making sure that the inert packing has the same diffusional characteristics and comparable particle size to that of the active catalyst. This way, in the absence of transport effects, reducing the active material concentration by half, should result in a 50% reduction in rates. Importantly, this test does not require measuring the number of active sites in the catalyst. Examples applying the Madon–Boudart test for liquid-phase and three-phase systems can be found in the literature.184,185
|
Minimum reporting requirements for assessing transport effects for lignin-first processes:
• Assessment of mass and heat transfer limitations via either of the following criteria: – The Weisz–Prater, Mears, and Anderson criteria which should be applied for both external and internal heat and mass transport. – The Madon–Boudart test at two different temperatures. • Verification that the gas loading or gas flows are not limiting unless so intended. Best practices for assessing transport effects for lignin-first processes: • Rule out gas–liquid mass transfer limitations by evaluating the maximum rate of gas transfer to the liquid. |
4. Catalyst performance in lignin-first fractionation of biomass
The lignin-first process comprises two catalytic operations: (1) the first is solvolysis, which may be catalysed (usually by an acid), leading to fractionation of the biomass via liberation of lignin and varying amounts of polysaccharides. This initial step can occur in the presence or absence of stabilising reagents. However, in their absence, the resulting intermediates will eventually undergo condensation. Subsequently, (2) catalytic reactions occur that stabilise reactive intermediates and/or cleavage of ether bonds. These processes can occur simultaneously as is the case for RCF, or subsequently when stabilisation/capping agents are used. Each of these processes needs to be adequately characterised.4.1 Catalysed solvolysis
The solvolysis step is crucial in lignin-first methods and can be promoted by a catalyst. Therefore, reporting solvent mixture, reaction concentration, temperature, and amount of (generally acid) catalyst or stabilising reagent is necessary. In addition, both solvents and stabilisation reagents can degrade and or be further transformed during subsequent or simultaneous hydrogenolysis. Therefore, although not commonly reported, we recommend that the field moves toward reporting recovery of solvent mixtures, including the catalyst and stabilisation reagents.4.2 Catalytic depolymerisation and intermediate product stabilization
Lignin-first processes are usually conducted on feedstocks with inorganics and other impurities that can be detrimental to transition metal catalysts due to poisoning reactions. These impurities are usually present in significant quantities when the catalyst is in the presence of whole biomass, as is the case with RCF, and are somewhat reduced when in the presence of isolated lignin such as those extracted in processes such as AAF. The high temperature solvent at high pressure, present during hydrogenation/hydrogenolysis can also facilitate catalyst deactivation including via leaching, sintering, and strong adsorption of heavy degradation products.65 Stirring is required for batch lignin-first processes, whether solid biomass or soluble lignin is used, to limit external mass transfer limitations. Under these conditions, the mechanical force might cause catalyst attrition and result in a loss in performance. Therefore, the thermal, catalytic, and mechanical stability of the catalyst needs to be considered. Ultimately, catalyst regeneration will become an important consideration as the lignin-first field moves toward integration and scale-up, as these processes lead to both reversible and irreversible deactivation.When no stabilisation agents are used, the solubilised lignin is unstable at high temperatures and must be rapidly brought into contact with the catalyst. In batch RCF processes, this implies having the catalyst in close proximity with solid biomass, after which separation and recycling of the solid catalyst from the solid residue is a challenge. Although using granular catalyst in a basket66 can solve the downstream separation challenge, the mechanical stirring can lead to attrition and mass loss in the form of fine particles. These fine powders are mixed with the solid residue or lost in the filtration. Alternatively, some catalysts (such as RANEY® Ni) can be magnetically separated from solid polysaccharide-rich residues.40,46 The chemical and thermal stability of a catalyst determines its lifetime in industrial reactors and thus its recyclability is an important factor in determining process feasibility. The stability and recyclability of the heterogeneous catalyst should therefore be considered for such batch reactions. However, when recyclability studies are undertaken, care should be taken to demonstrate recyclability with subsequent batch runs below full conversion, otherwise a catalyst might appear artificially to be stable due to its deactivation not being sufficient to drop the reaction below maximum conversion/yield. For reactions in flow-through conditions, runs at less than maximal conversion/yield should be compared to ensure that catalyst deactivation can be measured. To ensure that recyclability is assessed in a statistically significant manner, we recommend at least three runs, specifying any regeneration operation, to assess the stability of catalyst both in batch and flow-through operation. Additionally, pre- and post-reaction catalyst characterisations (e.g., with microscopy, X-ray photoelectron spectroscopy, inductively-coupled plasma/mass spectrometry, chemisorption, and physisorption) should be conducted. Notably, microscopy and chemisorption together can be used to understand changes in metal particle size, whereas physisorption can be used to assess lignin deposition effects on catalyst porosity.
When evaluating catalyst systems for lignin-first processes, proper control experiments should be performed. For example, when using heterogeneous catalysts, and especially when developing new catalyst systems, reactions that employ only the catalyst support should be reported. In all cases, reactions with no catalyst should also be performed as a negative control.
In typical kinetic reactions involving a single known reaction with a heterogeneous catalyst, TOF (i.e., the moles of converted substrate/number of active centres/time) is used to describe the catalyst efficiency and can be measured repeatedly to determine its stability. However, determining heterogeneous catalyst active centres for real lignin conversion is quite difficult due to the complex substrates and different reactions that are involved. Different active centres may be involved and affect the overall process simultaneously. For real lignin, a simple measurement of the catalyst productivity (i.e., moles of desired products/weight of whole catalyst/time) and its evolution over time can be used to compare catalysts. Including the mass of the support in this calculation can be justified given that it may play a role in the catalytic cycle. TOF can in turn be used to describe the catalytic efficiency especially for well-defined catalysts (e.g., homogeneous or single-site catalysts) and model compounds of lignins (e.g., model lignin dimers), for which the substrate and active centre are easier to identify.
|
Minimum reporting requirements for assessing catalyst performance in lignin-first processes:
• Catalyst loading, biomass:catalyst ratio, and use and loading of any capping agents in the solvolysis step. • Depolymerisation catalyst properties including composition and its preparation method. • When using a new catalyst, control experiments performed with no catalyst and with only the support (for a heterogeneous catalyst). Preferred reporting recommendations for assessing catalyst performance in lignin-first processes: • Stability and recovery of the catalysts, any stabilising agents, and solvents involved in the solvolysis reactions. • Depolymerisation catalyst productivity = moles of monophenolics/(total mass of catalyst × time). • With well-defined catalysts and reactants (e.g., lignin models), TOF can be reported in addition to productivity. • The structure (i.e., appearance under microscopy, and pore size distribution) and composition (i.e., the metal content) of any heterogeneous catalysts used in the process must be characterised before and after reaction. |
5. Mass balances and product yields
One of the most acute challenges in lignin-first research is the comparison of data across different research articles. However, such comparisons are key because they facilitate the direct evaluation of the research field's progress by providing a common basis for expressing mass and carbon balances, and product yields. Calculating these yields also facilitates an assessment of the extent to which a lignin-first biorefinery uses the renewable carbon in lignocellulosic biomass to its fullest.In this section, we distinguish yields that describe the recovery of different component fractions and the yields of individual products. In some processes, the production of component fractions occurs simultaneously with the deconstruction of these fractions to individual products (i.e., specific molecular products). In these cases, yields and balances can be calculated for fractions and individual products for the same product mixture. One such example includes lignin-derived products (e.g., specific arylpropanols or arylpropanes) and some polysaccharide-derived products (e.g., xylose, glucose) resulting from RCF of whole biomass, in which mass balance, component fraction yields, and individual molecular yields can be estimated from analysis of the liquid-phase RCF products. In other processes (Fig. 2c), fractions will first be isolated but not fully deconstructed. After this initial fractionation step, only initial mass balance and fraction yields can be calculated rather than individual product yields. This is the case for AAF, in which stabilised lignin oligomers are first separated from other biomass fractions. Following this step, an initial yield of isolated lignin can be calculated. The stabilised lignin is subsequently depolymerised in a second process, the analysis of which allows the determination of individual product yields. If subsequent upgrading/refining processes occur, molecular product yields can also be established for these transformations.
In the two subsequent sections, we detail overall and fraction yields and balances and discuss individual product yields that result from depolymerisation and any further transformation.
5.1 Overall component balances and fractionation yields
It is important to determine the mass and carbon fluxes throughout process stages. Beginning with lignocellulosic materials, RCF and the initial fractionation stage of other lignin-first processes usually generate two to three main streams: polysaccharide-rich pulp, a possible carbohydrate-derived soluble stream, and a lignin-rich stream. The pulp typically contains cellulose, with variable quantities of hemicelluloses and some residual lignin. In several RCF procedures, this pulp will be physically mixed with the catalyst and difficult to separate. If separation is not possible, an analysis of this entire mixed solid fraction (residual biomass solids and catalyst) can be performed using a standard biomass analysis procedure (see Section 2.2) to measure the carbohydrates. The resulting glucan can be assumed to correspond to the cellulose, and other sugars can be assumed to correspond to the remaining hemicellulose fraction in this pulp. The total solids to which is subtracted the catalyst loading and this cellulose and hemicellulose fraction can give an estimate of the residual lignin. However, this residual lignin content estimate should be considered imprecise because it can be influenced by ash content and any mass loss from the catalyst.The liquor typically contains solubilised lignin possibly together with carbohydrate derivatives.47,69 This stream can generally be further purified to isolate the lignin and carbohydrate-derived streams. The lignin stream generally takes the form of a lignin oil in the case of RCF, whereas it can be precipitated as a solid in the case of AAF.40,83 We recommend at minimum calculating an overall mass balance, and recommend performing an overall carbon balance following any initial conversion stage from native biomass using eqn (8) and (9), respectively:
 | (8) |
 | (9) |
These equations can be adapted to calculate the mass yields of component fractions compared to their respective fractions in the original feedstock. As the carbohydrate-rich pulp and sugars or their derivatives solubilised in the liquor can originate from either cellulose or hemicelluloses, we only recommend estimating the yield of isolated lignin.
 | (10) |
When using a fractionation with stoichiometric capping agents such as AAF, the formation of an additional functionality can increase the mass of the recovered lignin beyond its original mass.83,186 For AAF, the mass of isolated lignin can be corrected by estimating the fraction of acetal-functionalised groups using the HSQC NMR spectra of the isolated lignin. Specifically, this acetal-functionalised fraction is estimated by taking volume-integral ratios of the peaks corresponding to the appropriate lignin units in the HSQC spectra and then subtracting the calculated mass of the additional functionality from that of the isolated lignin.186
Care should also be taken when using biomass with high extractives content because it can interfere with lignin yield estimation. Inadvertently including some extractives in the isolated lignin mass is common and will overestimate the delignification yield. Using pre-extracted biomass (vide supra) is a useful control experiment to check for any yield over-estimation.
Given that lignin and carbohydrate substrates may undergo catalytic deoxygenation – often resulting in the production of water – a carbon balance should accompany a mass balance. Specifically, even if no mass losses occur during lignin isolation and solids workup, water release may represent a significant fraction of the missing mass in the lignin-first mass balance. Additional reactions can also occur, including hydrodeoxygenation, cracking of the propyl sidechain of the lignin C9-units, demethoxylation, or reactions with C5 and C6 sugars, further increasing the complexity of the product distribution. Moreover, these catalytic processes can also lead to the formation of gaseous products, such as CO2 and CH4, that are difficult to distinguish from those formed by the catalytic decomposition of alcohol solvents often employed in the RCF processes or AAF hydrogenolysis. We note more generally that solvent conversion to less-volatile products and solvent-substrate cross reactivity can also occur, generating species that are difficult to distinguish from those of substrate conversion, but should be considered in overall mass and carbon yields.
Following the catalytic process, work-up losses can also constitute a substantial portion of carbon and mass losses for lignin upgrading processes. For example, successful isolation of the lignin from the liquor will invariably remove water and other volatile components. Rotary evaporation of solvents (under reduced pressure) can also remove some light components of the crude lignin stream.
|
Minimum reporting requirements for mass balances and fractionation yields for lignin-first processes:
• Overall mass balance for biomass components. • Mass yield of the isolated lignin stream. Preferred reporting requirements for overall mass and carbon balances for lignin-first processes: • Total mass balance, including all solvents, catalysts, and capping agents. • Carbon balance for biomass components. Best practices for estimating overall mass balances and fractionation yields: • Account for addition of any functionality by a stoichiometric capping agent when calculating the mass of isolated lignin or oil. • Conduct control experiments with and without extraction to account for extractives in mass balances, especially in lignin. |
5.2 Lignin depolymerisation and transformation
In this section, we discuss the reporting of yields to individual, identifiable products derived from lignin after its depolymerisation. Lignin depolymerisation to monophenolic compounds in RCF occurs simultaneously with fractionation, whereas other methods isolate a lignin fraction to be subsequently depolymerised. For depolymerisation and subsequent catalytic transformations of the resulting monophenolics, mole- and carbon-balances are more straightforward to calculate, as the elemental composition of reactants and products can be determined either via elemental analysis or, for simple mixtures, using chromatographic techniques to quantify individual components.101,187–189 Here, we strongly recommend that yields of individual products be reported whenever possible, rather than reporting more loosely identified fractions (e.g., ethanol-soluble fractions, gel-permeation chromatography (GPC) isolated fractions, or functional groups measured using chemical reagents, etc.).As previously discussed, a pragmatic basis for calculating the yield of lignin-derived monophenolics is the total content of original lignin estimated using the Klason method.89,187 However, as this method is based on the degradation of the lignocellulosic materials under severely acidic conditions that substantially alter the structure of the native lignin, estimating the initial share of carbon in native lignin is difficult. Although this can limit the understanding of the molecular mechanisms underlying the deconstruction of native lignin, the expression of yields (vide infra) of lignin products relative to the total Klason lignin content of the lignocellulosic substrate is useful. In this context, another way to express product yields is relative to 100 kg of lignocellulosic substrate (on a dry basis).
In both cases, it is essential to base the yields on the lignin content or total weight of the original biomass. This is especially important in cases in which fractionation and depolymerisation are run separately. When lignin is progressively separated from biomass, the first fractions of lignin that are removed from biomass typically give higher monophenolic yields than those removed subsequently.24,186 If only a small fraction of the lignin was extracted from the original biomass sample, this phenomenon leads to artificially high yields when expressed on an isolated lignin basis.
The next step in calculating lignin yields is to analyse lignin-derived products. The vast majority of lignin monophenolics (or monomer derivatives) that require identification and quantification have structures for which standards can be purchased or synthesised. Furthermore, monophenolics are usually volatile or can be rendered volatile by derivatisation, such as by trimethylsilylation.24,43,190 Therefore, we recommend initial identification of lignin-derived monophenolics by comparison of their retention times in gas chromatography (GC) or liquid chromatography (LC) with those of authentic standards and by comparison of the mass spectra to those of authentic standards (Fig. 4). To authenticate new compounds, high-resolution MS data and NMR spectra are required. Mass spectra for a large number of lignin monomers have also been published in the literature, which can also be used as a basis for identification.191 When possible, the similarity index of the EI-MS identification and the calculated vs. observed high-resolution mass (of the parent ion) should be reported.
 | ||
| Fig. 4 Representative monophenolic identification by MS comparison combined with retention time similarity in GC-for propyl syringol found in lignin-derived liquor. The top two panels represent the comparison of MS spectra obtained by GC-MS of an authenticated standard and that of the lignin monophenolic within the liquor resulting from hydrogenolysis of AAF lignin. The bottom two panels compare the retention time in GC-FID of the authenticated standard and that of the monophenolic within that same liquor resulting from AAF lignin hydrogenolysis. Other peaks in the bottom GC-FID trace include lignin monophenolics and the internal standard. Adapted from Shuai et al..24 | ||
Quantification is often straightforward with GC using a flame-ionisation detector (FID). In this technique, peak areas of identified monomers are converted to concentrations using either calibrations built with external or internal standards (e.g., 2-isopropylphenol or decane). Where standards are available, we recommend using authentic standards with their respective calibration curves. However, as shown several times for lignin monomers,24,43,186 the ratio of the peak area of the monomer to that of the internal standard can be used to adequately estimate their molar ratio times a correction factor based on the ratio of their effective carbon number (ECN).192 (Caution should be exercised when using the ECN method if the response factors (RFs) of the internal standard to the target compound are >2). Nevertheless, RFs are significantly affected by the concentration of the internal standard and the target compounds. RFs therefore need to be determined/optimised based on the real concentration of the products. The ECN can also vary slightly depending on the solvent used,5 so checking the accuracy with standards that are close in structure to the molecules that require quantification is advised. RFs should be measured for the principal monophenolics and reported. For LC, similar calibrations can be used with UV, diode-array detection (DAD), or MS; external standard calibration curves are generated using pure authentic compounds (which can further aid confirmation of structural assignments), and ideally an internal standard is used. The calibration curves need to be updated frequently as the UV lamp and MS conditions change over time. Calibration standards therefore need to be carefully maintained. Quantification by MS for both LC and GC is fraught with difficulties and, unlike GC-FID RFs, MS response factors are neither universal (across machines) nor stable over time. Use of an internal standard added to the reaction mixture is viable, as long as the internal standard has similar ionization efficiency in MS (and consequently has a relative RF of <2 again). For MS-based quantification, the gold standard approach is therefore to use stable-isotope-labelled versions of the exact same compounds injected (in known amounts) into the sample.171,193,194 As these labelled compounds must often be synthesised in-house, it is hard to make a generic recommendation.
Once the monophenolics are quantified, mass yields can be calculated directly using the mass of the original lignin or biomass (eqn (11)). Typically, RCF or AAF with subsequent hydrogenolysis can produce monophenolics at total individual yields exceeding 5–10 kg per 100 kg of lignocellulosic substrate.
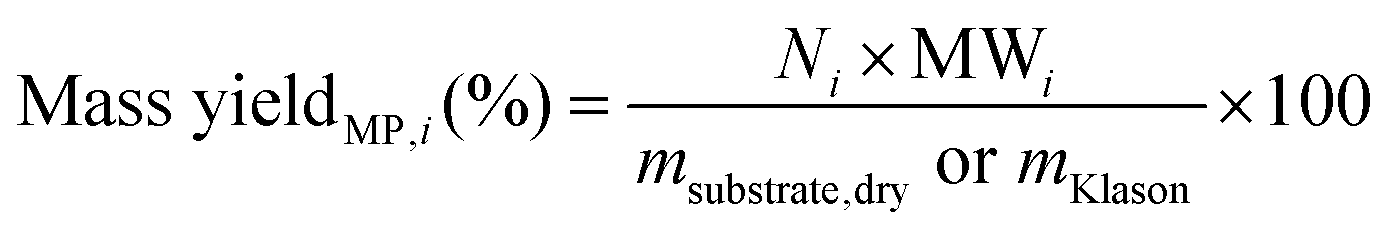 | (11) |
It may also be useful to calculate yields that consider the chemical modifications during depolymerisation (i.e., that account for the transformation and its associated change in mass without affecting the yield). Lignin-first depolymerisation processes will yield monophenolics that differ from their original form in the native lignin structure. Thus, yield calculations can be adapted to account for this transformation and avoid artificially decreasing or increasing the yield. Specifically, this is done by relating the targeted monophenolic to its original structure, which can be assumed to be within a β-ether unit or an ester and using its original molecular weight within native lignin (MW0). The original molecular weights for various common monophenolic products of lignin depolymerisation are shown in Fig. 5. Eqn (12) can then be used to calculate this yield (YieldMP,i).
 | (12) |
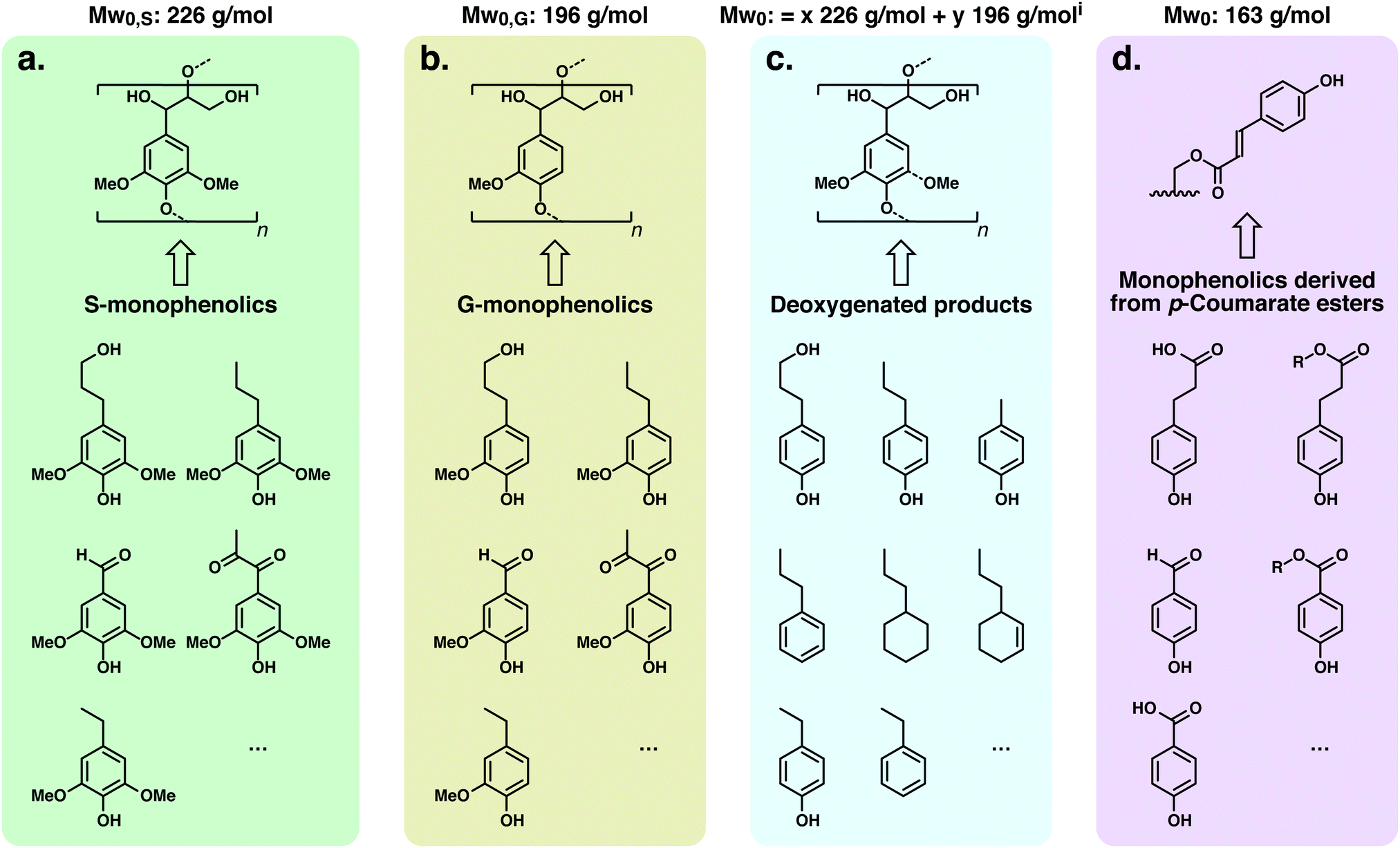 | ||
| Fig. 5 Approaches to estimating the presumed original molecular weight of units within the native lignin (MW0) for (a) syringyl, (b) guaiacyl, or (c) ill-defined monophenolics. i, where x and y represent the fractions of syringyl and guaiacyl functionalities measured in the original lignin. Fractions of syringyl or guaiacyl functionalities can be measured by various methods including NMR, thioacidolysis, DFRC, nitrobenzene oxidation, or RCF (Section 2.2). Various phenolic esters are associated with lignin (or, in fact, the cell wall polysaccharides) including p-hydroxybenzoate in poplar/aspen, willow, and palms, and p-coumarate and ferulate in grasses (or commelinid monocots in general) and will produce monophenolics or derivatives from degradative lignin-first methods. (d) p-Coumarate as an example (MW0 = 163); the products all derive from p-coumarate esters, the level of which is best determined by saponification (and quantified as the p-coumaric acid (MW = 164) released, by GC-FID, GC-MS, or LC-MS), or perhaps, as the dihydro analog (MW = 166) by RCF; estimates from 2D NMR, at least without careful use of quantitative methods, will always be high because p-coumarates are (free-phenolic) end-groups that relax much more slowly than internal polymer units. A similar approach can be taken for monophenolics derived from ferulate (MW0 = 193) and p-hydroxybenzoate (MW0 = 137) units, if products can be accurately traced back to their corresponding esters on the native lignin. Note: not all possible products from lignin-first methods are shown, as indicated by the ellipsis in each box. | ||
These total adjusted yields of monophenolic compounds have a strong correlation to β-O-4 bond content. Several models have attempted to calculate a theoretical monophenolic yield based on the cleavage of ether linkages in lignin. Models that assume a random distribution of C–C and ether linkages within a linear lignin polymer and neglect end-group effects, predict theoretical yields that are equal to the square of the linkage fraction within native lignin that are ethers.37,44,68,195 These estimates are often surprisingly close to the yields that result from RCF or stabilised lignin on a Klason lignin basis, which are in the low 20% for softwoods, up to ∼50% of Klason lignin for hardwoods, and as high as almost 80 or even 90% for specially engineered trees containing linear lignins high in ether linkages, such as ∼100% S-lignin poplar,24,80 or 100% C-lignin seedcoats.23,28 Thus, it is also recommended to report yields of monophenolic compounds with respect to the β-O-4 ether content determined by NMR, thioacidolysis, or nitrobenzene oxidation (see Section 2.2) and, where applicable, yields of esters from saponification or RCF.196–200
To expand the analysis to heavy species, GPC and more elaborate methods can be used. Dimer and oligomer identification and quantification is challenging without model studies to identify their derivation and authentic standards for quantification. GPC can provide an apparent distribution of dimers and oligomers, which is useful as a guide for comparative analyses. However, the data should be analysed with caution. When applied to product mixtures obtained from lignin, direct information regarding the content of species cannot be retrieved from a UV-vis detector as the detector response is not universal. The use of polystyrene standards for GPC calibration is useful for comparisons but may not be accurately translated to lignin, which can be directly addressed by use of MALS.201 Moreover, in samples obtained from reductive treatment of lignin streams that contain aliphatic hydrocarbons, compounds may be invisible to the UV-vis detector. Despite these limitations, GPC coupled with UV-Vis spectroscopy provides useful information on the apparent distribution of Mw and chemical structural uniformity of the eluting species, even if it provides little insight into accurate yields of dimers and higher fractions, or their structures.
Finally, we also recommend performing 1D and 2D NMR, including HSQC, of the product mixture after depolymerization to determine what inter-unit linkage motifs remain in the lignin-derived stream. Model compound experiments are often required to determine what units (with their characteristic inter-unit linkages) have reacted during the depolymerization process, and to provide a reference for assigning resonances in NMR. Examples of NMR-identified and assigned products from RCF are becoming more common.24,28,67,118,136
|
Minimum reporting requirements for yields of lignin-derived molecules:
• Monophenolic yields based on the quantification of individually identified products. • Precise description of the method for formally identifying said molecules (e.g., NMR spectrum, comparison of GC or HPLC retention time to an authenticated standard, comparison of MS spectra with an authenticated spectrum). Preferred reporting requirements for yields of lignin-derived molecules: • Comparison of yields on a Klason lignin basis with the β-ether content (and ester content, where applicable) of the original biomass, and the associated theoretical maximum yield. • If targeting individual molecule yields, reporting yields based on the original molecular weight (MW0), based on eqn (12). • GPC spectra to characterise lignin Mw distribution, but not for quantification purposes or to calculate yields. • 2D NMR of product mixtures to gain insight into the composition of product mixtures; caution should be used when deriving quantitative information. • Response factor calculations for quantified compounds. Best practices for estimating yields of lignin-derived molecules: • Yields based on the weight of the original biomass or original lignin fraction within that biomass, rather than on the basis of an isolated fraction. |
5.3 Pulp yields and composition
The lignin-first processes also generate a polysaccharide-rich fraction for further valorisation as a pulp or via catalytic, biological, or thermal conversion processes. When performing lignin-first biorefining in batch mode without catalyst baskets, the catalyst and the polysaccharide-rich pulp will be mixed (Fig. 3). Purifying cellulose from the catalyst requires extensive washing, but this separation is needed to determine the degree of delignification and the quality of the polysaccharides. Alternatively, a catalyst basket, magnetic catalyst, or flow-through setup can be used to avoid tedious separations of catalyst and pulp after the reaction (Section 3).We recommend reporting the weight of the pulp and its composition, which can be determined with the same methods as for whole biomass (Section 2.2). Additionally, we also recommend reporting the digestibility by commercial cellulase cocktails (an NREL LAP is available for this protocol as well at low solids loadings).91 Determining the digestibility of the pulp with enzymes is useful for several reasons. First, it can be used as a secondary confirmation of the pulp composition. Second, it can demonstrate the potential of the pulp for use in a cellulosic biorefinery, which can be useful for performing initial techno-economic analyses.16,202 Third, it can indirectly verify the occurrence of reactions between fractionation solvents or capping agents and cellulose, to which enzymes are sometimes sensitive.83,203
5.4 Product yields from liberated carbohydrates
Lignin-first experiments are usually performed in a solvent mixture containing water and sometimes acid, which typically hydrolyses some or most of the polysaccharide fraction, resulting in simple sugars, their dehydration and hydrogenation products,69 or carbohydrate oligomers in the liquor. Sugars can also be present in modified forms due to reaction with the solvent (e.g., methyl xyloside when performing RCF in MeOH),43 or with a capping agent (e.g., acetal-functionalised sugars).24,204 If simple sugars need to be isolated, the lignin-derived oil resulting from RCF can be decanted and/or extracted, whereas stabilised lignin can generally be precipitated from an aqueous phase containing sugar-derived products. Sugars can be quantified as described in Section 2.2. However, for modified sugars, including acetal-functionalised sugars, the corresponding external standards need to be used to build a calibration curve. Such standards will not necessarily be available commercially and so may have to be synthesised and purified. Once these sugars or their derivatives are quantified, yields can be calculated using the following equation: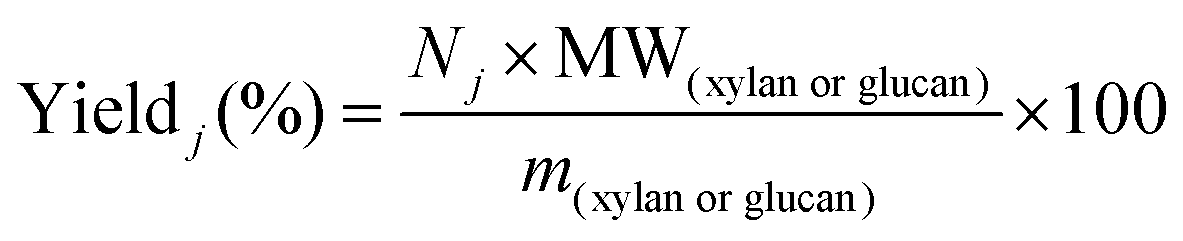 | (13) |
orglucan) is the total mass of xylan or glucan in the original biomass; and MW(xylanorglucan) is the molecular weight of this product in the original polysaccharide. For instance, xylan could be converted into xylose, methyl xyloside, or furfural, but all originate from a single xylan unit.
|
Minimum reporting requirements for yields of pulp, sugar, or sugar-derived molecules
• The mass of recovered purified pulp. • The carbohydrate and Klason lignin composition of the pulp. • Yields of major monosaccharides or their derivatives. Preferred reporting requirements for yields of pulp, sugar or sugar-derived molecules • Enzymatic digestibility of the pulp and the yield of the resulting sugars. • Enzymes (or enzyme cocktail) used, and enzyme loading per mass of polysaccharides. |
6. Outlook on the analysis and development of lignin-first biorefining
The lignin-first biorefining field has expanded dramatically in the past decade,9,23,38,47,50,70,79,159,205–209 spurred by the realisation that reductive catalytic processes, although long practiced on whole biomass, are active stabilisation methods that enable production of narrow product slates from lignin to a degree simply unmatched in other lignin deconstruction processes. The advent of protection-group chemistries as alternative lignin-first approaches, such as AAF and DAF, to hydrogenolysis has provided new options to achieve similar process outcomes. Alternative stabilisation chemistries and approaches beyond these three have begun to emerge, including oxidative and photocatalytic routes,207,210 and undoubtedly many more approaches will be reported in the coming years. The applicability of the guidelines presented here, although considered here in the context of the now well-accepted methods of RCF, AAF, and DAF, should be broadly applicable to other lignin-first biorefining approaches. In this last section, we briefly discuss the outlook for unifying the lignin-first field around these guidelines.The chemical composition of whole biomass feedstocks, as described in detail in Section 2, is typically measured via summative chemical methods that are now considered standard in the biomass conversion field. Many of these wet chemistry approaches have a long history of development over many decades (and indeed, Klason lignin analysis is over a century old), and are documented in open-access LAPs from NREL.84–91 These LAPs contain information that researchers should be able to carefully follow to quantify polysaccharide, lignin, protein, ash, extractives, and moisture content in biomass. All equipment and materials in the standard NREL compositional analysis LAP89 are readily available in most modern chemistry laboratories. This in turn should ideally present a low barrier to entry for laboratories new to the broader biomass conversion field, and certainly lignin-first biorefining, to accomplish these analyses. We stress that for all substrates treated in a lignin-first context, it is imperative to use these standard compositional analysis methodologies to ensure direct comparison of studies over time, across feedstocks, and, as stressed throughout this perspective, across different laboratories.
Eventual accessibility to standardised feedstocks for lignin-first biorefining would also be of significant benefit for the research community, as it would provide a benchmark substrate for all lignin-first researchers to compare new innovations against existing processes. The availability of a benchmark substrate would find utility, whether a researcher is focused on solvolysis, stabilisation chemistry, the catalytic process or catalyst materials development, downstream processing of either the lignin oil or pulp, or both, or overall process development. NIST supplies Biomass Reference Materials that were originally created in the 90s. Moreover, some large government-funded research organisations have established well-characterised feedstock repositories, such as in the US Department of Energy Bioenergy Technologies Office (via Idaho National Laboratory) or in the US Department of Energy's Bioenergy Research Centers. However, these substrates are typically not readily available to researchers around the world, which inherently limits the ability to have a truly “universal” feedstock for benchmarking. Going forward, this will become an important need for the growing lignin-first biorefining community – we anticipate pursuing efforts to this end.
Beyond analysing overall biomass composition, quantitative analytics for lignin chemistry have and continue to be a longstanding challenge.101,211 The RCF version of lignin-first biorefining approach was actually first proposed as an analytical tool to essentially study lignin structure, and recent results42,62,118,157–160 have shown that thioacidolysis and RCF provide similar C–O bond content measurements from the same feedstocks. Given the potential for an easier analytical approach, we propose that the lignin community might shift away from thioacidolysis and DFRC as routine approaches, and use simpler, less labour-intensive RCF-based chemical methods for the same measurements. Additional work remains to be done to definitively show that RCF-based chemical methods can substitute for well-accepted approaches, and this work is currently ongoing.
The lignin oil resulting from lignin-first chemistry typically contains monophenolics associated with the cleavage of ether and, where relevant, ester bonds. Beyond monomers, there are dimers and oligomers associated with lignin-first biorefining that derive from C–C-bonded lignin units that are not cleaved in essentially any RCF approaches tested to date, and that may not participate in the protection-group chemistries, as well as unreacted β-ether units. Several reports have used GC-MS to identify dimers,43,57,212–219 but a re-evaluation is required to thoroughly identify and quantify dimers and oligomers. As an example, products from 4-O-5-linked units using proper lignin models have recently revealed some interesting pathways and a unique dimer.161 Full analysis of dimers and oligomers represents a major analytical undertaking, as has been accomplished for thioacidolysis recently,147 but will be crucial to both understanding the impact of lignin-first processes on lignin more comprehensively and for designing downstream separations and valorisation strategies more holistically for the resulting lignin-rich streams.
Similarly, in most lignin-first contexts, some polysaccharides and extractives are liberated along with the lignin. The fate of the carbohydrate-derived compounds has been little explored to date, but here again, more inclusive analyses remain to be done on these compounds as well. For both lignin and off-target products, detailed experimental approaches combined with emerging computational tools,220–222 will be a key component of a more holistic characterisation of the resulting products from lignin-first biorefining.
Increased standardisation will improve comparisons and process performance across laboratories conducting research around the world. In tandem, improved analytics will allow us to gain better insights into aspects that are not yet understood in lignin-first processing including the fate of more minor products such as certain carbohydrates or extractives. The fate of minor products, small losses of solvents, purification and separation steps, recycling of catalysts, solvents and capping agents, and energy demand are usually ignored at the laboratory scale. However, these aspects are likely to become increasingly important as lignin-first processes move towards industrial implementation. Improved analytics and experimental standardisation, as described here, will allow more effective and quantitative comparisons of studies across the literature, which will in turn enable generation of rigorous process inputs for economic and sustainability modelling, towards robust and realistic scale-up.172 To this end, industrial implementation of lignin-first processes would represent the ultimate success for this field.
Conflicts of interest
The authors declare the following competing financial interest(s): MMAO is founder and part owner of Spero Renewables, LLC, a technology company making plant-based alternatives to petrochemicals. JSL is part owner of Bloom Biorenewables Ltd, which is exploring commercial opportunities for AAF lignin. JSMS is part owner of RenFuel AB and RenFuel Materials AB, that are exploring opportunities for products obtained by lignin-first approaches.Acknowledgements
All authors thank the members of their research groups for engaging discussions and exciting research efforts towards enabling the lignin-first biorefining concepts. The concept for this paper originated from discussions at the 2018 Lignin Gordon Research Conference. Specific thanks go to Brenna Black, David Brandner, Gregory Facas, Renee Happs, Rui Katahira, Jacob Kenny, Jacob Kruger, Ana Morais, Kelsey Ramirez, Michelle Reed, Justin Sluiter, Edward Wolfrum (NREL), Yanding Li (UW, Madison), Maxim V. Galkin (UG), Elias Cooreman, Yuhe Liao, Korneel Van Aelst, Joost Van Aelst, Sander Van den Bosch, Gil Van den Bossche, Thijs Vangeel (KUL), Ning Li (DICP/SU), Michael Stone, and Amber Phillips (MIT) for input on the manuscript and helpful discussions. We thank Gregory Facas (NREL) for making Fig. 3. MMAO thanks the US Department of Energy and the Mellichamp Sustainability Initiative for supporting our research in biomass conversion. KB thanks the European Research Council, ERC Starting Grant 2015 (CatASus) 638076. This work is part of the research programme Talent Scheme (Vidi) with project number 723.015.005, which is partly financed by the Netherlands Organization for Scientific Research (NWO). This work was authored in part by the National Renewable Energy Laboratory, operated by the Alliance for Sustainable Energy, LLC, for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308. Funding to GTB and YRL was provided by the U.S. Department of Energy Office of Energy Efficiency and Renewable Energy Bioenergy Technologies Office. GTB and YRL also acknowledge funding from the Center for Bioenergy Innovation, a U.S. Department of Energy Research Center supported by the Office of Biological and Environmental Research in the DOE Office of Science. The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes. JSL thanks the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (Starting grant: CATACOAT, No. 758653), the Swiss Competence Center for Bioenergy Research (SCCER-BIOSWEET), the Swiss National Foundation, and École Polytechnique Fédérale de Lausanne for financial support. JR acknowledges funding by the DOE Great Lakes Bioenergy Research Center (DOE BER Office of Science DE-SC0018409). RR acknowledges the financial support provided by the ERC Consolidator Grant (LIGNINFIRST, project number 725762). YRL thanks the National Science Foundation, CBET Award No. 1454299 for support. JSMS thanks the Swedish Energy Agency, STINT and Stockholm university for financial support. BFS acknowledges KULeuven, the Flemish and Belgian government for financial support through SB-FWO support (BIOFACT), SBO support (BIOWOOD) and Catalysti (NIBCON, PADDLE, ARBOREF, MAIA), and the EU via the HORIZON 2020 program (BBI SMARTBOX). FW thanks the National Science Foundation of China (21721004, 21690080, 21991090).References
- S. Pacala and R. Socolow, Stabilization wedges: Solving the climate problem for the next 50 years with current technologies, Science, 2004, 305, 968–972 CrossRef CAS.
- C. O. Tuck, E. Perez, I. T. Horvath, R. A. Sheldon and M. Poliakoff, Valorization of biomass: Deriving more value from waste, Science, 2012, 337, 695–699 CrossRef CAS.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, The path forward for biofuels and biomaterials, Science, 2006, 311, 484–489 CrossRef CAS.
- N. Mosier, C. Wyman, B. Dale, R. Elander, Y. Y. Lee, M. Holtzapple and M. Ladisch, Features of promising technologies for pretreatment of lignocellulosic biomass, Bioresour. Technol., 2005, 96, 673–686 CrossRef CAS.
- C. E. Wyman, B. E. Dale, R. T. Elander, M. Holtzapple, M. R. Ladisch and Y. Y. Lee, Coordinated development of leading biomass pretreatment technologies, Bioresour. Technol., 2005, 96, 1959–1966 CrossRef CAS.
- C. Crestini, F. Melone, M. Sette and R. Saladino, Milled wood lignin: A linear oligomer, Biomacromolecules, 2011, 12, 3928–3935 CrossRef CAS.
- F. Yue, F. Lu, S. Ralph and J. Ralph, Identification of 4–O–5-units in softwood lignins via definitive lignin models and NMR, Biomacromolecules, 2016, 17, 1909–1920 CrossRef CAS.
- J. Ralph, C. Lapierre and W. Boerjan, Lignin structure and its engineering, Curr. Opin. Biotechnol, 2019, 56, 240–249 CrossRef CAS.
- R. Rinaldi, R. Jastrzebshi, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Paving the way for lignin valorisation: Recent advances in bioengineering, biorefining and catalysis, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS.
- J. C. del Río, J. Rencoret, A. Gutiérrez, T. Elder, H. Kim and J. Ralph, Lignin monomers from beyond the canonical monolignol biosynthetic pathway: Another brick in the wall, ACS Sustainable Chem. Eng., 2020, 8, 4997–5012 CrossRef.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, The catalytic valorization of lignin for the production of renewable chemicals, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS.
- C. Z. Li, X. C. Zhao, A. Q. Wang, G. W. Huber and T. Zhang, Catalytic transformation of lignin for the production of chemicals and fuels, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS.
- A. Corona, M. J. Biddy, D. R. Vardon, M. Birkved, M. Z. Hauschild and G. T. Beckham, Life cycle assessment of adipic acid production from lignin, Green Chem., 2018, 20, 3857–3866 RSC.
- R. Davis, L. Tao, E. C. D. Tan, M. J. Biddy, G. T. Beckham, C. Scarlata, J. Jacobson, K. Cafferty, J. Ross, J. Lukas, D. Knorr and P. Schoen, Process design and economics for the conversion of lignocellulosic biomass to hydrocarbons: Dilute-acid and enzymatic deconstruction of biomass to sugars and biological conversion of sugars to hydrocarbons, Report NREL/TP-5100-60223, National Renewable Energy Lab. (NREL), Golden, CO, USA, 2013. https://www.nrel.gov/docs/fy14osti/60223.pdf.
- R. E. Davis, N. J. Grundl, L. Tao, M. J. Biddy, E. C. Tan, G. T. Beckham, D. Humbird, D. N. Thompson and M. S. Roni, Process design and economics for the conversion of lignocellulosic biomass to hydrocarbon fuels and coproducts: 2018 Biochemical design case update; Biochemical deconstruction and conversion of biomass to fuels and products via integrated biorefinery pathways, Report NREL/TP-5100-71949, National Renewable Energy Lab. (NREL), Golden, CO, USA, 2018. https://www.nrel.gov/docs/fy19osti/71949.pdf.
- Y. H. Liao, S. F. Koelewijn, G. Van den Bossche, J. Van Aelst, S. Van den Bosch, T. Renders, K. Navare, T. Nicolai, K. Van Aelst, M. Maesen, H. Matsushima, J. M. Thevelein, K. Van Acker, B. Lagrain, D. Verboekend and B. F. Sels, A sustainable wood biorefinery for low-carbon footprint chemicals production, Science, 2020, 367, 1385–1390 CrossRef CAS.
- X. Zhao, K. Cheng and D. Liu, Organosolv pretreatment of lignocellulosic biomass for enzymatic hydrolysis, Appl. Microbiol. Biotechnol., 2009, 82, 815–827 CrossRef CAS.
- P. J. Deuss, K. Barta and J. G. de Vries, Homogeneous catalysis for the conversion of biomass and biomass-derived platform chemicals, Catal.: Sci. Technol., 2014, 4, 1174–1196 RSC.
- Y. Liao, B. O. de Beeck, K. Thielemans, T. Ennaert, J. Snelders, M. Dusselier, C. M. Courtin and B. F. Sels, The role of pretreatment in the catalytic valorization of cellulose, Mol. Catal., 2020, 487, 110883 CrossRef CAS.
- T. N. Kleinert, Organosolvent pulping with aqueous alcohol, Tappi, 1974, 57, 99–102 CAS.
- A. Johansson, O. Aaltonen and P. Ylinen, Organosolv pulping – methods and pulp properties, Biomass, 1987, 13, 45–65 CrossRef CAS.
- M. R. Sturgeon, S. Kim, K. Lawrence, R. S. Paton, S. C. Chmely, M. Nimlos, T. D. Foust and G. T. Beckham, A mechanistic investigation of acid-catalyzed cleavage of aryl-ether linkages: implications for lignin depolymerization in acidic environments, ACS Sustainable Chem. Eng., 2013, 2, 472–485 CrossRef.
- W. Schutyser, T. Renders, S. Van den Bosch, S.-F. Koelewijn, G. T. Beckham and B. F. Sels, Chemicals from lignin: an interplay of lignocellulose fractionation, depolymerisation, and upgrading, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Héroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Formaldehyde stabilization facilitates lignin monomer production during biomass depolymerization, Science, 2016, 354, 329–333 CrossRef CAS.
- K. Shimada, S. Hosoya and T. Ikeda, Condensation reactions of softwood and hardwood lignin model compounds under organic acid cooking conditions, J. Wood Chem. Technol., 1997, 17, 57–72 CrossRef CAS.
- T. Shioya, T. Akiyama, T. Yokoyama and Y. Matsumoto, Formation rate of benzyl cation intermediate from p-hydroxyphenyl, guaiacyl, or syringyl nucleus in acidolysis of lignin, J. Wood Chem. Technol., 2017, 37, 75–86 CrossRef CAS.
- W. Lan and J. S. Luterbacher, Preventing lignin condensation to facilitate aromatic monomer production, Chimia, 2019, 73, 591–598 CrossRef CAS.
- Y. Li, L. Shuai, H. Kim, A. H. Motagamwala, J. K. Mobley, F. Yue, Y. Tobimatsu, D. Havkin-Frenkel, F. Chen, R. A. Dixon, J. S. Luterbacher, J. A. Dumesic and J. Ralph, An “ideal lignin” facilitates full biomass utilization, Sci. Adv., 2018, 4, eaau2968 CrossRef.
- S. Yasuda and K. Ota, Chemical structures of sulfuric acid lignin. Part X. Reaction of syringylglycerol-β-syringyl ether and condensation of syringyl nucleus with guaiacyl lignin model compounds in sulfuric acid, Holzforschung, 1987, 41, 59–65 CrossRef CAS.
- S. Yasuda, N. Terashima and T. Ito, Chemical structures of sulfuric acid lignin. II. Chemical structures of condensation products from arylglycerol-β-aryl ether type structures, Mokuzai Gakkaishi, 1981, 27, 216–222 CAS.
- R. M. Ede and G. Brunow, Formic acid/peroxyformic acid pulping. III. Condensation reactions of β-aryl ether model compounds in formic acid, Holzforschung, 1989, 43, 317–322 CrossRef CAS.
- J. Marton, Reactions in alkaline pulping, in Lignins, Occurrence, Formation, Structure and Reactions, ed. K. V. Sarkanen and C. H. Ludwig, Wiley-Interscience, New York, 1971, ch. 16, vol. 1, pp. 639–689 Search PubMed.
- T. J. Fullerton, The condensation reactions of lignin model compounds in alkaline pulping liquors, J. Wood Chem. Technol., 1987, 7, 441–462 CrossRef CAS.
- J. Gierer, I. Norén and S. Waennstroem, Formation of condensation products on treatment of non-phenolic lignin units of the β-aryl ether type with alkali: model studies on a novel mode of alkaline lignin condensation, Holzforschung, 1987, 41, 79–82 CrossRef CAS.
- V. L. Chiang and M. Funaoka, The dissolution and condensation reactions in guaiacyl and syringyl units in residual lignin during Kraft delignification of sweetgum, Holzforschung, 1990, 44, 140–155 Search PubMed.
- D. R. Dimmel and G. Gellerstedt, Chemistry of alkaline pulping, in Lignins and lignans: Advances in chemistry, ed. C. Heitner, D. R. Dimmel and J. A. Schmidt, CRC Press (Taylor & Francis Group), Boca Raton, FL, 2010, ch. 10, pp. 349–391 DOI:10.1201/EBK1574444865.
- M. V. Galkin and J. S. M. Samec, Lignin valorization through catalytic lignocellulose fractionation: A fundamental platform for the future biorefinery, ChemSusChem, 2016, 9, 1544–1558 CrossRef CAS.
- T. Renders, S. Van den Bosch, S.-F. Koelewijn, W. Schutyser and B. F. Sels, Lignin-first biomass fractionation: The advent of active stabilisation strategies, Energy Environ. Sci., 2017, 10, 1551–1557 RSC.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Bright side of lignin depolymerization: Toward new platform chemicals, Chem. Rev., 2018, 118, 614–678 CrossRef CAS.
- P. Ferrini and R. Rinaldi, Catalytic biorefining of plant biomass to non-pyrolytic lignin bio-oil and carbohydrates through hydrogen transfer reactions, Angew. Chem., Int. Ed., 2014, 53, 8634–8639 CrossRef CAS.
- T. Parsell, S. Yohe, J. Degenstein, T. Jarrell, I. Klein, E. Gencer, B. Hewetson, M. Hurt, J. Im Kim, H. Choudhari, B. Saha, R. Meilan, N. Mosier, F. Ribeiro, W. N. Delgass, C. Chapple, H. I. Kenttamaa, R. Agrawal and M. M. Abu-Omar, A synergistic biorefinery based on catalytic conversion of lignin prior to cellulose starting from lignocellulosic biomass, Green Chem., 2015, 17, 1492–1499 RSC.
- Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yu and J. Xu, Lignin depolymerization (LDP) in alcohol over nickel-based catalysts via a fragmentation–hydrogenolysis process, Energy Environ. Sci., 2013, 6, 994–1007 RSC.
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S.-F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Reductive lignocellulose fractionation into soluble lignin-derived phenolic monomers and dimers and processable carbohydrate pulps, Energy Environ. Sci., 2015, 8, 1748–1763 RSC.
- N. Yan, C. Zhao, P. J. Dyson, C. Wang, L. T. Liu and Y. Kou, Selective degradation of wood lignin over Noble-metal catalysts in a two-step process, ChemSusChem, 2008, 1, 626–629 CrossRef CAS.
- P. Ferrini, C. Chesi, N. Parkin and R. Rinaldi, Effect of methanol in controlling defunctionalization of the propyl side chain of phenolics from catalytic upstream biorefining, Faraday Discuss., 2017, 202, 403–413 RSC.
- P. Ferrini, C. A. Rezende and R. Rinaldi, Catalytic upstream biorefining through hydrogen transfer reactions: Understanding the process from the pulp perspective, ChemSusChem, 2016, 9, 3171–3180 CrossRef CAS.
- I. Graça, R. T. Woodward, M. Kennema and R. Rinaldi, Formation and fate of carboxylic acids in the lignin-first biorefining of lignocellulose via H-transfer catalyzed by Raney Ni, ACS Sustainable Chem. Eng., 2018, 6, 13408–13419 CrossRef.
- M. Kennema, I. B. D. de Castro, F. Meemken and R. Rinaldi, Liquid-phase H-transfer from 2-propanol to phenol on Raney Ni: Surface processes and inhibition, ACS Catal., 2017, 7, 2437–2445 CrossRef CAS.
- G. H. Wang, Z. Cao, D. Gu, N. Pfander, A. C. Swertz, B. Spliethoff, H. J. Bongard, C. Weidenthaler, W. Schmidt, R. Rinaldi and F. Schuth, Nitrogen-doped ordered mesoporous carbon supported bimetallic PtCo nanoparticles for upgrading of biophenolics, Angew. Chem., Int. Ed., 2016, 55, 8850–8855 CrossRef CAS.
- R. Rinaldi, R. Woodward, P. Ferrini and H. Rivera, Lignin-first biorefining of lignocellulose: the impact of process severity on the uniformity of lignin oil composition, J. Braz. Chem. Soc., 2018, 30, 479–491 Search PubMed.
- H. P. Godard, J. L. McCarthy and H. Hibbert, Hydrogenation of wood, J. Am. Chem. Soc., 1940, 62, 988 CrossRef CAS.
- H. P. Godard, J. L. McCarthy and H. Hibbert, Studies on lignin and related compounds. LXII. High pressure hydrogenation of wood using copper chromite catalyst (Part 1), J. Am. Chem. Soc., 1941, 63, 3061–3066 CrossRef CAS.
- J. R. Bower, J. L. McCarthy and H. Hibbert, Studies on lignin and related compounds. LXIII. Hydrogenation of wood (Part 2), J. Am. Chem. Soc., 1941, 63, 3066–3068 CrossRef CAS.
- J. R. Bower, L. M. Cooke and H. Hibbert, Studies on lignin and related compounds. LXXI. The course of formation of native lignin in spruce buds, J. Am. Chem. Soc., 1943, 65, 1195–1198 CrossRef CAS.
- C. P. Brewer, L. M. Cooke and H. Hibbert, Studies on lignin and related compounds. 84. The high pressure hydrogenation of maple wood: Hydrol lignin, J. Am. Chem. Soc., 1948, 70, 57–59 CrossRef CAS.
- J. M. Pepper and H. Hibbert, Studies on lignin and related compounds. LXXXVII. High pressure hydrogenation of maple wood, J. Am. Chem. Soc., 1948, 70, 67–71 CrossRef CAS.
- A. Sakakibara, Hydrogenolysis, in Methods in Lignin Chemistry, ed. C. W. Dence and S. Y. Lin, Springer-Verlag, Berlin-Heidelberg, 1992, pp. 350–368 DOI:10.1007/978-3-642-74065-7.
- I. Sobolev, H. G. Arlt and C. Schuerch, Alkaline hydrogenation pulping, Ind. Eng. Chem., 1957, 49, 1399–1400 CrossRef CAS.
- J. M. Pepper and Y. W. Lee, Lignin and related compounds. I. A comparative study of catalysts for lignin hydrogenolysis, Can. J. Chem., 1969, 47, 723–727 CrossRef CAS.
- J. M. Pepper and M. D. Rahman, Lignin and related compounds. XI. Selective degradation of aspen poplar lignin by catalytic hydrogenolysis, Cellul. Chem. Technol., 1987, 21, 233–239 CAS.
- J. M. Pepper and P. Supathna, Lignin and related compounds. VI. Study of variables affecting hydrogenolysis of spruce wood lignin using a rhodium-on-charcoal catalyst, Can. J. Chem., 1978, 56, 899–902 CrossRef CAS.
- M. V. Galkin and J. S. M. Samec, Selective route to 2-propenyl aryls directly from wood by a tandem organosolv and palladium-catalysed transfer hydrogenolysis, ChemSusChem, 2014, 7, 2154–2158 CrossRef CAS.
- I. Kumaniaev, E. Subbotina, J. Sävmarker, M. Larhed, M. V. Galkin and J. S. Samec, Lignin depolymerization to monophenolic compounds in a flow-through system, Green Chem., 2017, 19, 5767–5771 RSC.
- E. M. Anderson, M. L. Stone, M. J. Hülsey, G. T. Beckham and Y. Román-Leshkov, Kinetic studies of lignin solvolysis and reduction by reductive catalytic fractionation decoupled in flow-through reactors, ACS Sustainable Chem. Eng., 2018, 6, 7951–7959 CrossRef CAS.
- E. M. Anderson, M. L. Stone, R. Katahira, M. Reed, G. T. Beckham and Y. Román-Leshkov, Flowthrough reductive catalytic fractionation of biomass, Joule, 2017, 1, 613–622 CrossRef CAS.
- S. Van den Bosch, T. Renders, S. Kennis, S.-F. Koelewijn, G. Van den Bossche, T. Vangeel, A. Deneyer, D. Depuydt, C. M. Courtin and J. M. Thevelein, Integrating lignin valorization and bio-ethanol production: on the role of Ni-Al2O3 catalyst pellets during lignin-first fractionation, Green Chem., 2017, 19, 3313–3326 RSC.
- Y. Li, B. Demir, L. M. Vázquez Ramos, M. Chen, J. A. Dumesic and J. Ralph, Kinetic and mechanistic insights into hydrogenolysis of lignin to monomers in a continuous flow reactor, Green Chem., 2019, 21, 3561–3572 RSC.
- T. Renders, W. Schutyser, S. Van den Bosch, S.-F. Koelewijn, T. Vangeel, C. M. Courtin and B. F. Sels, Influence of acidic (H3PO4) and alkaline (NaOH) additives on the catalytic reductive fractionation of lignocellulose, ACS Catal., 2016, 6, 2055–2066 CrossRef CAS.
- T. Renders, E. Cooreman, S. Van den Bosch, W. Schutyser, S.-F. Koelewijn, T. Vangeel, A. Deneyer, G. Van den Bossche, C. Courtin and B. Sels, Catalytic lignocellulose biorefining in n-butanol/water: a one-pot approach toward phenolics, polyols, and cellulose, Green Chem., 2018, 20, 4607–4619 RSC.
- Y. M. Questell-Santiago, M. V. Galkin, K. Barta and J. S. Luterbacher, Stabilization strategies in biomass depolymerization using chemical functionalization, Nat. Rev. Chem., 2020, 4, 311–330 CrossRef CAS.
- X. Luo, Y. Li, N. K. Gupta, B. Sels, J. Ralph and L. Shuai, Protection strategies enable selective conversion of biomass, Angew. Chem., Int. Ed., 2020, 59, 11704–11716 CrossRef CAS.
- T. Yokoyama, Revisiting the mechanism of β-O-4 bond cleavage during acidolysis of lignin. Part 6: A review, J. Wood Chem. Technol., 2014, 35, 27–42 CrossRef CAS.
- K. Lundquist, Acid degradation of lignin. II. Separation and identification of low-molecular weight phenols, Acta Chem. Scand., 1970, 24, 889–907 CrossRef CAS.
- W. H. Steeves and H. Hibbert, Studies on lignin and related compounds. XLII. The isolation of a bisulfite soluble “extracted lignin”, J. Am. Chem. Soc., 1939, 61, 2194–2195 CrossRef CAS.
- M. Kulka and H. Hibbert, Studies on lignin and related compounds. LXVII. Isolation and identification of 1-(4-hydroxy-3,5-dimethoxyphenyl)-2-propanone and 1-(4-hydroxy-3-methoxyphenyl)-2-propanone from maple wood ethanolysis products – Metabolic changes in lower and higher plants, J. Am. Chem. Soc., 1943, 65, 1180–1185 CrossRef CAS.
- P. J. Deuss, M. Scott, F. Tran, N. J. Westwood, J. G. de Vries and K. Barta, Aromatic monomers by in situ conversion of reactive intermediates in the acid-catalyzed depolymerization of lignin, J. Am. Chem. Soc., 2015, 137, 7456–7467 CrossRef CAS.
- P. J. Deuss, C. S. Lancefield, A. Narani, J. G. de Vries, N. J. Westwood and K. Barta, Phenolic acetals from lignins of varying compositions via iron(iii) triflate catalysed depolymerisation, Green Chem., 2017, 19, 2774–2782 RSC.
- P. J. Deuss, C. W. Lahive, C. S. Lancefield, N. J. Westwood, P. C. J. Kamer, K. Barta and J. G. de Vries, Metal triflates for the production of aromatics from lignin, ChemSusChem, 2016, 9, 2974–2981 CrossRef CAS.
- A. De Santi, M. V. Galkin, C. W. Lahive, P. J. Deuss and K. Barta, Lignin-first fractionation of softwood lignocellulose using a mild dimethyl carbonate and ethylene glycol organosolv process, ChemSusChem, 2020, 13(17), 4468–4477 CrossRef CAS.
- W. Lan, M. Talebi Amiri, C. M. Hunston and J. S. Luterbacher, Protection group effects during α,γ-diol lignin stabilization promote high-selectivity monomer production, Angew. Chem., Int. Ed., 2018, 57, 1356–1360 CrossRef CAS.
- S. Van den Bosch, W. Schutyser, S. F. Koelewijn, T. Renders, C. M. Courtin and B. F. Sels, Tuning the lignin oil OH-content with Ru and Pd catalysts during lignin hydrogenolysis on birch wood, Chem. Commun., 2015, 51, 13158–13161 RSC.
- M. V. Galkin, D. Di Francesco, U. Edlund and J. S. M. Samec, Sustainable sources need reliable standards, Faraday Discuss., 2017, 202, 281–301 RSC.
- M. Talebi Amiri, G. R. Dick, Y. M. Questell-Santiago and J. S. Luterbacher, Fractionation of lignocellulosic biomass to produce uncondensed aldehyde-stabilized lignin, Nat. Protoc., 2019, 14, 921–954 CrossRef CAS.
- A. Sluiter, R. Ruiz, C. Scarlata, J. Sluiter and D. Templeton, Determination of extractives in biomass, Report NREL/TP-510-42619, National Renewable Energy Laboratory, Golden, CO, USA, 2008. https://www.nrel.gov/docs/gen/fy08/42619.pdf.
- B. Hames, C. Scarlata and A. Sluiter, Determination of protein content in biomass, Report NREL/TP-510-42625, National Renewable Energy Laboratory, Golden, CO, USA, 2008. https://www.nrel.gov/docs/gen/fy08/42625.pdf.
- B. Hames, R. Ruiz, C. Scarlata, A. Sluiter, J. Sluiter and D. Templeton, Preparation of samples for compositional analysis, Report NREL/TP-510-42620, National Renewable Energy Laboratory, Golden, CO, USA, 2008. https://www.nrel.gov/docs/gen/fy08/42620.pdf.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Determination of ash in biomass, National Renewable Energy Laboratory, Golden, 2008. http://www.nrel.gov/docs/gen/fy08/42622.pdf.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Determination of total solids in biomass and total dissolved solids in liquid process samples, National Renewable Energy Laboratory, Golden, 2008. http://www.nrel.gov/docs/gen/fy08/42621.pdf.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Determination of structural carbohydrates and lignin in biomass, Report NREL/TP-510-42618, National Renewable Energy Laboratory, Golden, CO, USA, 2012. https://www.nrel.gov/docs/gen/fy13/42618.pdf.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter and D. Templeton, Determination of sugars, byproducts, and degradation products in liquid fraction process samples, Report NREL/TP-510-42623, National Renewable Energy Laboratory (NREL), 2008. http://www.nrel.gov/docs/gen/fy08/42623.pdf.
- M. G. Resch, J. O. Baker and S. R. Decker, Low solids enzymatic saccharification of lignocellulosic biomass, Report NREL/TP-5100-63351, National Renewable Energy Laboratory, Golden, CO, USA, 2015. https://www.nrel.gov/docs/fy15osti/63351.pdf.
- J. B. Sluiter, R. O. Ruiz, C. J. Scarlata, A. D. Sluiter and D. W. Templeton, Compositional analysis of lignocellulosic feedstocks. 1. Review and description of methods, J. Agric. Food Chem., 2010, 58, 9043–9053 CrossRef CAS.
- D. W. Templeton, C. J. Scarlata, J. B. Sluiter and E. J. Wolfrum, Compositional analysis of lignocellulosic feedstocks. 2. Method uncertainties, J. Agric. Food Chem., 2010, 58, 9054–9062 CrossRef CAS.
- O. Theander, Chemical analysis of lignocellulose materials, Anim. Feed Sci. Technol., 1991, 32, 35–44 CrossRef CAS.
- M. L. Stone, E. M. Anderson, K. M. Meek, M. Reed, R. Katahira, F. Chen, R. A. Dixon, G. T. Beckham and Y. Román-Leshkov, Reductive catalytic fractionation of C-lignin, ACS Sustainable Chem. Eng., 2018, 6, 11211–11218 CrossRef CAS.
- R. C. Pettersen, The chemical composition of wood, in The Chemistry of Solid Wood, Advances in Chemistry, American Chemical Society, Wahsington, DC, USA, 1984, ch. 2, vol. 207, pp. 57–126 Search PubMed.
- K. Morreel, Y. Saeys, O. Dima, F. Lu, Y. Van de Peer, R. Vanholme, J. Ralph, B. Vanholme and W. Boerjan, Systematic structural characterization of metabolites in Arabidopsis via candidate substrate-product pair networks, Plant Cell, 2014, 26, 929–945 CrossRef CAS.
- R. Dauwe, K. Morreel, G. Goeminne, B. Gielen, A. Rohde, J. Van Beeumen, J. Ralph, A.-M. Boudet, J. Kopka, S. Rochange, C. Halpin, E. Messens and W. Boerjan, Molecular phenotyping of lignin-modified tobacco reveals associated changes in cell wall metabolism, primary metabolism, stress metabolism and photorespiration, Plant J., 2007, 52, 263–285 CrossRef CAS.
- S. F. Chen, R. A. Mowery, C. J. Scarlata and C. K. Chambliss, Compositional analysis of water-soluble materials in corn stover, J. Agric. Food Chem., 2007, 55, 5912–5918 CrossRef CAS.
- S. F. Chen, R. A. Mowery, R. S. Sevcik, C. J. Scarlata and C. K. Chambliss, Compositional analysis of water-soluble materials in switchgrass, J. Agric. Food Chem., 2010, 58, 3251–3258 CrossRef CAS.
- R. Hatfield and R. S. Fukushima, Can lignin be accurately measured?, Crop Sci., 2005, 45, 832–839 CrossRef CAS.
- S. Wanasuria, H. Steyobudi, I. B. Mayun and B. Suprihatno, Iron deficiency of oil plam in Sumatra, Better Crops Int., 1999, 13, 33–35 Search PubMed.
- F. Lu, S. D. Karlen, M. Regner, H. Kim, S. A. Ralph, R.-C. Sun, K.-I. Kuroda, M. A. Augustin, R. Mawson, H. Sabarez, T. Singh, G. Jimenez-Monteon, S. Hill, P. J. Harris, W. Boerjan, S. D. Mansfield and J. Ralph, Naturally p-hydroxybenzoylated lignins in palms, BioEnergy Res., 2015, 8, 934–952 CrossRef CAS.
- W. de Jong, Biomass composition, properties, and characterization, in Biomass as a Sustainable Energy Source for the Future: Fundamentals of Conversion Processes, ed. W. de Jong and J. R. Van Ommen, Wiley, 2015, ch. 2, pp. 36–68 DOI:10.1002/9781118916643.ch2.
- D. W. Templeton, E. J. Wolfrum, J. H. Yen and K. E. Sharpless, Compositional analysis of biomass reference materials: Results from an interlaboratory study, BioEnergy Res., 2016, 9, 303–314 CrossRef.
- G. A. Adams, Lignin determination, in Methods in Carbohydrate Chemistry, ed. R. L. Whistler, Academic Press, New York, 1965, vol. 5, pp. 185–187 Search PubMed.
- A. B. Blakeney, P. J. Harris, R. J. Henry and B. A. Stone, A simple and rapid preparation of alditol acetates for monosaccharide analysis, Carbohydr. Res., 1983, 113, 291–299 CrossRef CAS.
- O. Theander and E. Westerlund, Quantitative analysis of cell wall components, in Forage Cell Wall Structure and Digestibility, ed. H. G. Jung, D. R. Buxton, R. D. Hatfield and J. Ralph, Am. Soc. Agronomy, Madison, 1993, pp. 83–104 Search PubMed.
- J. M. De Ruiter and J. C. Burns, Cell wall carbohydrates of flaccidgrass plant parts. I. Neutral sugar composition of fermented residues, Crop Sci., 1987, 27, 1057–1063 CrossRef CAS.
- R. D. Hatfield, Carbohydrate composition of alfalfa cell walls isolated from stem sections differing in maturity, J. Agric. Food Chem., 1990, 40, 424–430 CrossRef.
- M. Bunzel and J. Ralph, NMR characterization of lignins isolated from fruit and vegetable insoluble dietary fiber, J. Agric. Food Chem., 2006, 54, 8352–8361 CrossRef CAS.
- M. Bunzel, A. Schussler and G. T. Saha, Chemical characterization of Klason lignin preparations from plant-based foods, J. Agric. Food Chem., 2011, 59, 12506–12513 CrossRef CAS.
- F. Chen, Y. Tobimatsu, D. Havkin-Frenkel, R. A. Dixon and J. Ralph, A polymer of caffeyl alcohol in plant seeds, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1772–1777 CrossRef CAS.
- S. Z. Wang, S. H. Su, L. P. Xiao, B. Wang, R. C. Sun and G. Y. Song, Catechyl lignin extracted from Castor seed coats using deep eutectic solvents: Characterization and depolymerization, ACS Sustainable Chem. Eng., 2020, 8, 7031–7038 CrossRef CAS.
- R. D. Hatfield, H. G. Jung, J. Ralph, D. R. Buxton and P. J. Weimer, A comparison of the insoluble residues produced by the Klason lignin and acid detergent lignin procedures, J. Sci. Food Agric., 1994, 65, 51–58 CrossRef CAS.
- J. Ralph, K. Lundquist, G. Brunow, F. Lu, H. Kim, P. F. Schatz, J. M. Marita, R. D. Hatfield, S. A. Ralph, J. H. Christensen and W. Boerjan, Lignins: natural polymers from oxidative coupling of 4-hydroxyphenylpropanoids, Phytochem. Rev., 2004, 3, 29–60 CrossRef CAS.
- Y. Li, T. Akiyama, T. Yokoyama and Y. Matsumoto, NMR assignment for diaryl ether structures (4−O−5 structures) in pine wood lignin, Biomacromolecules, 2016, 17, 1921–1929 CrossRef CAS.
- E. M. Anderson, M. L. Stone, R. Katahira, M. Reed, W. Muchero, K. J. Ramirez, G. T. Beckham and Y. Roman-Leshkov, Differences in S/G ratio in natural poplar variants do not predict catalytic depolymerization monomer yields, Nat. Commun., 2019, 10, 2033 CrossRef.
- N. E. Thornburg, M. B. Pecha, D. G. Brandner, M. L. Reed, J. V. Vermaas, W. E. Michener, R. Katahira, T. B. Vinzant, T. D. Foust, B. S. Donohoe, Y. Roman-Leshkov, P. N. Ciesielski and G. T. Beckham, Mesoscale reaction–diffusion phenomena governing lignin-first biomass fractionation, ChemSusChem, 2020, 13(17), 4495–4509 CrossRef CAS.
- T. E. Timmel, Compression wood in gymnosperms, Springer, Heidelberg, 1986 Search PubMed.
- J. Ralph, T. Akiyama, H. D. Coleman and S. D. Mansfield, Effects on lignin structure of coumarate 3-hydroxylase downregulation in Poplar, BioEnergy Res., 2012, 5, 1009–1019 CrossRef CAS.
- J. C. del Río, P. Prinsen, J. Rencoret, L. Nieto, J. Jiménez-Barbero, J. Ralph, Á. T. Martínez and A. Gutiérrez, Structural characterization of the lignin in the cortex and pith of elephant grass (Pennisetum purpureum) stems, J. Agric. Food Chem., 2012, 60, 3619–3634 CrossRef.
- H. Kim, D. Padmakshan, Y. Li, J. Rencoret, R. D. Hatfield and J. Ralph, Characterization and elimination of undesirable protein residues in plant cell wall materials for enhancing lignin analysis by solution-state NMR, Biomacromolecules, 2017, 18, 4184–4195 CrossRef CAS.
- W. Boerjan, J. Ralph and M. Baucher, Lignin biosynthesis, Annu. Rev. Plant Biol., 2003, 54, 519–546 CrossRef CAS.
- J. C. del Río, A. G. Lino, J. L. Colodette, C. F. Lima, A. Gutiérrez, A. T. Martínez, F. Lu, J. Ralph and J. Rencoret, Differences in the chemical structure of the lignins from sugarcane bagasse and straw, Biomass Bioenergy, 2015, 81, 322–328 CrossRef.
- J. C. del Río, J. Rencoret, P. Prinsen, Á. T. Martínez, J. Ralph and A. Gutiérrez, Structural characterization of wheat straw lignin as revealed by analytical pyrolysis, 2D-NMR, and reductive cleavage methods, J. Agric. Food Chem., 2012, 60, 5922–5935 CrossRef.
- S. R. Decker, A. E. Harman-Ware, R. M. Happs, E. J. Wolfrum, G. A. Tuskan, D. Kainer, G. B. Oguntimein, M. Rodriguez, D. Weighill, P. Jones and D. Jacobson, High throughput screening technologies in biomass characterization, Front. Energy Res., 2018, 6, 120 CrossRef.
- J. Ralph and L. L. Landucci, NMR of lignins, in Lignin and Lignans; Advances in Chemistry, ed. C. Heitner, D. R. Dimmel and J. A. Schmidt, CRC Press (Taylor & Francis Group), Boca Raton, FL, 2010, ch. 5, pp. 137–234 DOI:10.1201/EBK1574444865.
- F. Lu and J. Ralph, Non-degradative dissolution and acetylation of ball-milled plant cell walls; high-resolution solution-state NMR, Plant J., 2003, 35, 535–544 CrossRef CAS.
- H. Kim and J. Ralph, Solution-state 2D NMR of ball-milled plant cell wall gels in DMSO-d6/pyridine-d5, Org. Biomol. Chem., 2010, 8, 576–591 RSC.
- S. D. Mansfield, H. Kim, F. Lu and J. Ralph, Whole plant cell wall characterization using solution-state 2D-NMR, Nat. Protoc., 2012, 7, 1579–1589 CrossRef CAS.
- E. Kupče and R. Freeman, Compensation for spin-spin coupling effects during adiabatic pulses, J. Magn. Reson., 1997, 127, 36–48 CrossRef.
- E. Kupče and R. Freeman, Compensated adiabatic inversion pulses: Broadband INEPT and HSQC, J. Magn. Reson., 2007, 187, 258–265 CrossRef.
- S. Heikkinen, M. M. Toikka, P. T. Karhunen and I. A. Kilpeläinen, Quantitative 2D HSQC (Q-HSQC) via suppression of J-dependence of polarization transfer in NMR spectroscopy: Application to wood lignin, J. Am. Chem. Soc., 2003, 125, 4362–4367 CrossRef CAS.
- K. Hu, W. M. Westler and J. L. Markley, Simultaneous quantification and identification of individual chemicals in metabolite mixtures by two-dimensional extrapolated time-zero 1H–13C HSQC (HSQC0), J. Am. Chem. Soc., 2011, 133, 1662–1665 CrossRef CAS.
- M. Talebi Amiri, S. Bertella, Y. M. Questell-Santiago and J. S. Luterbacher, Establishing lignin structure-upgradeability relationships using quantitative 1H–13C heteronuclear single quantum coherence nuclear magnetic resonance (HSQC-NMR) spectroscopy, Chem. Sci., 2019, 10, 8135–8142 RSC.
- S. D. Karlen, C. Zhang, M. L. Peck, R. A. Smith, D. Padmakshan, K. E. Helmich, H. C. A. Free, S. Lee, B. G. Smith, F. Lu, J. C. Sedbrook, R. Sibout, J. H. Grabber, T. M. Runge, K. S. Mysore, P. J. Harris, L. E. Bartley and J. Ralph, Monolignol ferulate conjugates are naturally incorporated into plant lignins, Sci. Adv., 2016, 2, e1600393 CrossRef.
- T. Hibino, D. Shibata, T. Ito, D. Tsuchiya, T. Higuchi, B. Pollet and C. Lapierre, Chemical properties of lignin from Aralia cordata, Phytochemistry, 1994, 37, 445–448 CrossRef CAS.
- M. Rafiee, M. Alherech, S. D. Karlen and S. Stahl, Electrochemical aminoxyl-mediated oxidation of primary alcohols in lignin to carboxylic acids: Polymer modification and depolymerization, J. Am. Chem. Soc., 2019, 141, 15266–15276 CrossRef CAS.
- Y. Tobimatsu, T. Takano, T. Umezawa and J. Ralph, Solution-state multidimensional NMR of lignins: Approaches and applications, in Lignin: Biosynthesis, Functions, and Economic Significance, ed. F. Lu and F. Yue, Nova Science Publisher, Inc., Hauppauge, NY, USA, 2019, ch. 4, pp. 79–110 Search PubMed.
- W. Lan, F. Lu, M. Regner, Y. Zhu, J. Rencoret, S. A. Ralph, U. I. Zakai, K. Morreel, W. Boerjan and J. Ralph, Tricin, a flavonoid monomer in monocot lignification, Plant Physiology, 2015, 167, 1284–1295 CrossRef CAS.
- J. C. del Río, J. Rencoret, A. Gutiérrez, H. Kim and J. Ralph, Hydroxystilbenes are monomers in palm fruit endocarp lignins, Plant Physiol., 2017, 174, 2072–2082 CrossRef.
- T. J. Simmons, J. C. Mortimer, O. D. Bernardinelli, A. C. Poppler, S. P. Brown, E. R. deAzevedo, R. Dupree and P. Dupree, Folding of xylan onto cellulose fibrils in plant cell walls revealed by solid-state NMR, Nat. Commun., 2016, 7, 13902 CrossRef CAS.
- M. Dick-Perez, Y. A. Zhang, J. Hayes, A. Salazar, O. A. Zabotina and M. Hong, Structure and interactions of plant cell-wall polysaccharides by two- and three-dimensional magic-angle-spinning solid-state NMR, Biochemistry, 2011, 50, 989–1000 CrossRef CAS.
- F. A. Perras, H. Luo, X. Zhang, N. S. Mosier, M. Pruski and M. M. Abu-Omar, Atomic-level structure characterization of biomass pre- and post-lignin treatment by Dynamic Nuclear Polarization-enhanced solid-state NMR, J. Phys. Chem. A, 2017, 121, 623–630 CrossRef CAS.
- C. Lapierre, B. Pollet and B. Monties, Heterogeneous distribution of diarylpropane structures in spruce lignins, Phytochemistry, 1991, 30, 659–662 CrossRef CAS.
- F. Yue, F. Lu, M. Regner, R. Sun and J. Ralph, Lignin-derived thioacidolysis dimers: Reevaluation, new products, authentication, and quantification, ChemSusChem, 2017, 10, 830–835 CrossRef CAS.
- F. Lu and J. Ralph, The DFRC method for lignin analysis. Part 1. A new method for β-aryl ether cleavage: lignin model studies, J. Agric. Food Chem., 1997, 45, 4655–4660 CrossRef CAS.
- F. Lu and J. Ralph, The DFRC (Derivatization Followed by Reductive Cleavage) method and its applications for lignin characterization, in Lignin: Structural Analysis, Applications in Biomaterials, and Ecological Significance, ed. F. Lu, Nova Science Publishers, Inc, Hauppauge, New York, USA, 2014, ch. 2, pp. 27–65 Search PubMed.
- C. Rolando, B. Monties and C. Lapierre, Thioacidolysis, in Methods in Lignin Chemistry, ed. C. W. Dence and S. Y. Lin, Springer-Verlag, Berlin-Heidelberg, 1992, pp. 334–349 Search PubMed.
- C.-L. Chen, Nitrobenzene and cupric oxide oxidations, in Methods in Lignin Chemistry, ed. S. Y. Lin and C. W. Dence, Spring-Verlag, Berlin, 1992, ch. 6.2, pp. 301–321 DOI:10.1007/978-3-642-74065-7.
- M. Yamamura, T. Hattori, S. Suzuki, D. Shibata and T. Umezawa, Microscale alkaline nitrobenzene oxidation method for high-throughput determination of lignin aromatic components, Plant Biotechnol., 2010, 27, 305–310 CrossRef CAS.
- E. E. Harris, J. D’Ianni and H. Adkins, Reaction of hardwood lignin with hydrogen, J. Am. Chem. Soc., 1938, 60, 1467–1470 CrossRef CAS.
- B. F. Hrutfiord, Reduction and hydrogenolysis, in Lignins, Occurrence, Formation, Structure and Reactions, ed. K. V. Sarkanen and C. H. Ludwig, Wiley-Interscience, New York, 1971, pp. 487–509 Search PubMed.
- K. Sudo and A. Sakakibara, Hydrogenolysis of protolignin. XI, Mokuzai Gakkaishi, 1974, 20, 396–401 CAS.
- P. Hoffmann and W. Schweers, Hydrogenation of lignin. VI. Application of a semimicro-method to investigations of the system of hydrogenolysis, Holzforschung, 1975, 29, 18–24 CrossRef CAS.
- K. M. Torr, D. J. van de Pas, E. Cazeils and I. D. Suckling, Mild hydrogenolysis of in-situ and isolated Pinus radiata lignins, Bioresour. Technol., 2011, 102, 7608–7611 CrossRef CAS.
- E. M. Anderson, R. Katahira, M. Reed, M. G. Resch, E. M. Karp, G. T. Beckham and Y. Roman-Leshkov, Reductive catalytic fractionation of corn stover lignin, ACS Sustainable Chem. Eng., 2016, 4, 6940–6950 CrossRef CAS.
- T. Renders, G. Van den Bossche, T. Vangeel, K. Van Aelst and B. Sels, Reductive catalytic fractionation: state of the art of the lignin-first biorefinery, Curr. Opin. Biotechnol, 2019, 56, 193–201 CrossRef CAS.
- D. J. van de Pas, B. Nanayakkara, I. D. Suckling and K. M. Torr, Comparison of hydrogenolysis with thioacidolysis for lignin structural analysis, Holzforschung, 2014, 68, 151–155 CAS.
- Y. Li, S. D. Karlen, B. Demir, H. Kim, J. S. Luterbacher, J. A. Dumesic, S. S. Stahl and J. Ralph, Mechanistic study of diaryl ether bond cleavage during palladium-catalyzed lignin hydrogenolysis, ChemSusChem, 2020, 13, 4487–4494 CrossRef CAS.
- C. Lapierre, Application of new methods for the investigation of lignin structure, in Forage Cell Wall Structure and Digestibility, ed. H. G. Jung, D. R. Buxton, R. D. Hatfield and J. Ralph, American Society of Agronomy, Crop Science Society of America, Soil Science Society of America, Madison, WI, 1993, pp. 133-166 Search PubMed.
- C. Lapierre, B. Monties and C. Rolando, Thioacidolysis of poplar lignins: identification of monomeric syringyl products and characterization of guaiacyl-syringyl lignin fractions, Holzforschung, 1986, 40, 113–118 CrossRef CAS.
- A. R. Robinson and S. D. Mansfield, Rapid analysis of poplar lignin monomer composition by a streamlined thioacidolysis procedure and near-infrared reflectance-based prediction modeling, Plant J., 2009, 58, 706–714 CrossRef CAS.
- A. E. Harman-Ware, C. Foster, R. M. Happs, C. Doeppke, K. Meunier, J. Gehan, F. Yue, F. Lu and M. F. Davis, A thioacidolysis method tailored for higher-throughput quantitative analysis of lignin monomers, Biotechnol. J., 2016, 11, 1–6 CrossRef.
- F. Yue, F. Lu, R.-C. Sun and J. Ralph, Syntheses of lignin-derived thioacidolysis monomers and their uses as quantitation standards, J. Agric. Food Chem., 2012, 60, 922–928 CrossRef CAS.
- F. X. Yue, W. Lan, S. N. Hu, K. L. Chen and F. C. Lu, Structural modifications of sugarcane bagasse lignins during wet-storage and soda-oxygen pulping, ACS Sustainable Chem. Eng., 2016, 4, 5311–5318 CrossRef CAS.
- J. H. Grabber, S. Quideau and J. Ralph, p-Coumaroylated syringyl units in maize lignin; implications for β-ether cleavage by thioacidolysis, Phytochemistry, 1996, 43, 1189–1194 CrossRef CAS.
- F. Lu and J. Ralph, Detection and determination of p-coumaroylated units in lignins, J. Agric. Food Chem., 1999, 47, 1988–1992 CrossRef CAS.
- F. Lu and J. Ralph, Derivatization followed by reductive cleavage (DFRC method), a new method for lignin analysis: Protocol for analysis of DFRC monomers, J. Agric. Food Chem., 1997, 45, 2590–2592 CrossRef CAS.
- M. Regner, A. Bartuce, D. Padmakshan, J. Ralph and S. D. Karlen, Reductive cleavage method for quantitation of monolignols and low-abundance monolignol conjugates, ChemSusChem, 2018, 11, 1600–1605 CrossRef CAS.
- E. Cooreman, T. Vangeel, K. Van Aelst, J. Van Aelst, J. Lauwaert, J. W. Thybaut, S. Van den Bosch and B. F. Sels, Perspective on overcoming scale-up hurdles for the reductive catalytic fractionation of lignocellulose biomass, Ind. Eng. Chem. Res., 2020, 59(39), 17035–17045 CrossRef CAS.
- M. E. Davis and R. J. Davis, Fundamentals of chemical reaction engineering, McGraw-Hill, Boston, MA, 2003 Search PubMed.
- J. S. Luterbacher, J. M. Rand, D. M. Alonso, J. Han, J. T. Youngquist, C. T. Maravelias, B. F. Pfleger and J. A. Dumesic, Nonenzymatic sugar production from biomass using biomass-derived γ-valerolactone, Science, 2014, 343, 277–280 CrossRef CAS.
- L. D. Schmidt, The engineering of chemical reactions, Oxford University Press, Oxford, UK, 2nd edn, 2004 Search PubMed.
- H. F. Scott, Elements of chemical reaction engineering, Prentice Hall, Boston, MA, USA, 2016 Search PubMed.
- H. Luo and M. M. Abu-Omar, Lignin extraction and catalytic upgrading from genetically modified poplar, Green Chem., 2018, 20, 745–753 RSC.
- V. Meille, N. Pestre, P. Fongarland and C. de Bellefon, Gas/liquid mass transfer in small laboratory batch reactors: comparison of methods, Ind. Eng. Chem. Res., 2004, 43, 924–927 CrossRef CAS.
- R. V. Chaudhari, R. V. Gholap, G. Emig and H. Hofmann, Gas-liquid mass transfer in “dead-end” autoclave reactors, Can. J. Chem. Eng., 1987, 65, 744–751 CrossRef CAS.
- P. B. Weisz and C. D. Prater, Interpretation of measurements in experimental catalysis, Advances in Catalysis, Elsevier, 1954, vol. 6, pp. 143–196 Search PubMed.
- D. E. Mears, Tests for transport limitations in experimental catalytic reactors, Ind. Eng. Chem. Process Des. Dev., 1971, 10, 541–547 CrossRef CAS.
- J. B. Anderson, A criterion for isothermal behaviour of a catalyst pellet, Chem. Eng. Sci., 1963, 18, 147–148 CAS.
- R. J. Madon and M. Boudart, Experimental criterion for the absence of artifacts in the measurement of rates of heterogeneous catalytic reactions, Ind. Eng. Chem. Fundam., 1982, 21, 438–447 CrossRef CAS.
- J. W. Shabaker, R. R. Davda, G. W. Huber, R. D. Cortright and J. A. Dumesic, Aqueous-phase reforming of methanol and ethylene glycol over alumina-supported platinum catalysts, J. Catal., 2003, 215, 344–352 CrossRef CAS.
- E. P. Maris, W. C. Ketchie, V. Oleshko and R. J. Davis, Metal particle growth during glucose hydrogenation over Ru/SiO2 evaluated by X-ray absorption spectroscopy and electron microscopy, J. Phys. Chem. B, 2006, 110, 7869–7876 CrossRef CAS.
- W. Lan, J. Behaghel de Bueren and J. S. Luterbacher, Highly selective oxidation and depolymerization of α,γ-diol protected lignin, Angew. Chem., Int. Ed., 2019, 58, 2649–2654 CrossRef CAS.
- C. W. Dence, The determination of lignin, in Methods in Lignin Chemistry, ed. S. Y. Lin and C. W. Dence, Springer-Verlag, Heidelberg, 1992, pp. 33–61 Search PubMed.
- W. G. Campbell and I. R. C. McDonald, 608. The chemistry of the wood cell wall. Part II. The isolation of beech and spruce acid-soluble and modified lignins, J. Chem. Soc., 1952, 3180–3183, 10.1039/JR9520003180.
- J. R. Fedenko, J. E. Erickson, K. R. Woodard, L. E. Sollenberger, J. M. B. Vendramini, R. A. Gilbert, Z. R. Helsel and G. F. Peter, Biomass production and composition of perennial grasses grown for bioenergy in a subtropical climate across Florida, USA, BioEnergy Res., 2013, 6, 1082–1093 CrossRef CAS.
- S. Ahuja, Derivatization in gas-chromatography, J. Pharm. Sci., 1976, 65, 163–182 CrossRef CAS.
- J. Ralph and R. D. Hatfield, Pyrolysis-GC-MS characterization of forage materials, J. Agric. Food Chem., 1991, 39, 1426–1437 CrossRef CAS.
- J. T. Scanlon and D. E. Willis, Calculation of flame ionization detector relative response factors using the effective carbon number concept, J. Chromatogr. Sci., 1985, 23, 333–340 CAS.
- J. Schäfer, F. Urbat, K. Rund and M. Bunzel, A stable-isotope dilution GC-MS approach for the analysis of DFRC (derivatization followed by reductive cleavage) monomers from low-lignin plant materials, J. Agric. Food Chem., 2015, 63, 2668–2673 CrossRef.
- W. Lan, J. Rencoret, F. Lu, S. D. Karlen, B. G. Smith, P. J. Harris, J. C. del Río and J. Ralph, Tricin-lignins: Occurrence and quantitation of tricin in relation to phylogeny, Plant J., 2016, 88, 1046–1057 CrossRef CAS.
- T. Phongpreecha, N. C. Hool, R. J. Stoklosa, A. S. Klett, C. E. Foster, A. Bhalla, D. Holmes, M. C. Thies and D. B. Hodge, Predicting lignin depolymerization yields from quantifiable properties using fractionated biorefinery lignins, Green Chem., 2017, 19, 5131–5143 RSC.
- W. S. Borneman, R. D. Hartley, W. H. Morrison, D. E. Akin and L. G. Ljungdahl, Feruloyl and p-coumaroyl esterase from anaerobic fungi in relation to plant cell wall degradation, Appl. Microbiol. Biotechnol., 1990, 33, 345–351 CrossRef CAS.
- J. H. Grabber, J. Ralph and R. D. Hatfield, Model studies of ferulate-coniferyl alcohol cross-product formation in primary maize walls: implications for lignification in grasses, J. Agric. Food Chem., 2002, 50, 6008–6016 CrossRef CAS.
- J. Ralph, M. Bunzel, J. M. Marita, R. D. Hatfield, F. Lu, H. Kim, P. F. Schatz, J. H. Grabber and H. Steinhart, Peroxidase-dependent cross-linking reactions of p-hydroxycinnamates in plant cell walls, Phytochem. Rev., 2004, 3, 79–96 CrossRef CAS.
- E. M. Karp, C. T. Nimlos, S. Deutch, D. Salvachua, R. M. Cywar and G. T. Beckham, Quantification of acidic compounds in complex biomass-derived streams, Green Chem., 2016, 18, 4750–4760 RSC.
- S. D. Karlen, P. Fasahati, M. Mazaheri, J. Serate, R. A. Smith, S. Sirobhushanam, M. Chen, V. Tymokhin, C. L. Cass, S. Liu, D. Padmakshan, D. Xie, Y. Zhang, M. Mcgee, J. D. Russell, J. J. Coon, H. Kaeppler, N. de Leon, C. T. Maravelias, T. M. Runge, S. M. Kaeppler, J. C. Sedbrook and J. Ralph, Assessing the viability of recovering hydroxycinnamic acids from lignocellulosic biorefinery alkaline pretreatment waste streams, ChemSusChem, 2020, 13, 2012–2024 CrossRef CAS.
- G. Zinovyev, I. Sulaeva, S. Podzimek, D. Rossner, I. Kilpelainen, I. Sumerskii, T. Rosenau and A. Potthast, Getting Closer to Absolute Molar Masses of Technical Lignins, ChemSusChem, 2018, 11, 3259–3268 CrossRef CAS.
- M. Tschulkow, T. Compernolle, S. Van den Bosch, J. Van Aelst, I. Storms, M. Van Dael, G. Van den Bossche, B. Sels and S. Van Passel, Integrated techno-economic assessment of a biorefinery process: The high-end valorization of the lignocellulosic fraction in wood streams, J. Cleaner Prod., 2020, 266, 122022 CrossRef CAS.
- L. Shuai, Y. M. Questell-Santiago and J. S. Luterbacher, A mild biomass pretreatment using γ-valerolactone for concentrated sugar production, Green Chem., 2016, 18, 937–943 RSC.
- D. C. Johnson, M. D. Nicholson and F. C. Haigh, Dimethyl sulfoxide/paraformaldehyde: A nondegrading solvent for cellulose, Report IPC Number 5, Institute of Paper Chemistry, Appleton, Wisconsin, USA, Appleton, Wisconsin, USA, 1975. https://smartech.gatech.edu/bitstream/handle/1853/2900/tps-005.pdf.
- H. B. Yang, X. M. Zhang, H. Luo, B. Y. Liu, T. M. Shiga, X. Li, J. I. Kim, P. Rubinelli, J. C. Overton, V. Subramanyam, B. R. Cooper, H. P. Mo, M. M. Abu-Omar, C. Chapple, B. S. Donohoe, L. Makowski, N. S. Mosier, M. C. McCann, N. C. Carpita and R. Meilan, Overcoming cellulose recalcitrance in woody biomass for the lignin-first biorefinery, Biotechnol. Biofuels, 2019, 12, 171 CrossRef.
- G. F. Leal, S. Lima, I. Graca, H. Carrer, D. H. Barrett, E. Teixeira-Neto, A. A. S. Curvelo, C. B. Rodella and R. Rinaldi, Design of nickel supported on water-tolerant Nb2O5 catalysts for the hydrotreating of lignin streams obtained from lignin-first biorefining, iScience, 2019, 15, 467–488 CrossRef CAS.
- X. Wu, X. Fan, S. Xie, J. Lin, J. Cheng, Q. Zhang, L. Chen and Y. Wang, Solar energy-driven lignin-first approach to full utilization of lignocellulosic biomass under mild conditions, Nat. Catal., 2018, 1, 772–780 CrossRef CAS.
- Z. Cao, M. Dierks, M. T. Clough, I. B. Daltro de Castro and R. Rinaldi, A convergent approach for a deep converting lignin-first biorefinery rendering high-energy-density drop-in fuels, Joule, 2018, 2, 1118–1133 CrossRef CAS.
- W. Schutyser, S. Van den Bosch, T. Renders, T. De Boe, S. F. Koelewijn, A. Dewaele, T. Ennaert, O. Verkinderen, B. Goderis, C. M. Courtin and B. F. Sels, Influence of bio-based solvents on the catalytic reductive fractionation of birch wood, Green Chem., 2015, 17, 5035–5045 RSC.
- Y. T. Zhu, Y. H. Liao, W. Lv, J. Liu, X. B. Song, L. G. Chen, C. G. Wang, B. F. Sels and L. L. Ma, Complementing vanillin and cellulose production by oxidation of lignocellulose with stirring control, ACS Sustainable Chem. Eng., 2020, 8, 2361–2374 CrossRef CAS.
- J. S. Lupoi, S. Singh, R. Parthasarathi, B. A. Simmons and R. J. Henry, Recent innovations in analytical methods for the qualitative and quantitative assessment of lignin, Renewable Sustainable Energy Rev., 2015, 49, 871–906 CrossRef CAS.
- M. Matsukura and A. Sakakibara, Hydrogenolysis of protolignin. V. Isolation of some dimeric compounds with carbon to carbon linkage, Mokuzai Gakkaishi, 1973, 19, 131–135 CAS.
- M. Matsukura and A. Sakakibara, Hydrogenolysis of protolignin. VI. Isolation of a new trimeric compound with carbon linkage, Mokuzai Gakkaishi, 1973, 19, 137 CAS.
- M. Matsukura and A. Sakakibara, Hydrogenolysis of protolignin. VIII. Isolation of a dimer with CβCβ' linkage and a biphenyl, Mokuzai Gakkaishi, 1973, 19, 171–176 CAS.
- K. I. Sudo and A. Sakakibara, Hydrogenolysis of protolignin. VII. Isolation of D,L-syringaresinol, biphenyl, and diarylpropane from hardwood lignin, Mokuzai Gakkaishi, 1973, 19, 165–169 CAS.
- S. Yasuda and A. Sakakibara, Hydrogenolysis of protolignin in compression wood. IV. Isolation of a diphenyl ether and three dimeric compounds with carbon to carbon linkage, Mokuzai Gakkaishi, 1977, 23, 383–387 CAS.
- S. Yasuda and A. Sakakibara, Hydrogenolysis of protolignin in compression wood. III. Isolation of four dimeric compounds with carbon to carbon linkage, Mokuzai Gakkaishi, 1977, 23, 114–119 CAS.
- A. Sakakibara, A structural model of softwood lignin, Wood Sci. Technol., 1980, 14, 89–100 CrossRef CAS.
- B. H. Hwang, A. Sakakibara and K. Miki, Hydrogenolysis of protolignin. XVII. Isolation of three dimeric compounds with γ-O-4, β-1, and β-O-4 linkages from hardwood lignin, Holzforschung, 1981, 35, 229–232 CrossRef CAS.
- T. Z. H. Gani, M. J. Orella, E. M. Anderson, M. L. Stone, F. R. Brushett, G. T. Beckham and Y. Román-Leshkov, Computational evidence for kinetically controlled radical coupling during lignification, ACS Sustainable Chem. Eng., 2019, 7, 13270–13277 CrossRef CAS.
- M. J. Orella, T. Z. H. Gani, J. V. Vermaas, M. L. Stone, E. M. Anderson, G. T. Beckham, F. R. Brushett and Y. Roman-Leshkov, Lignin-KMC: A toolkit for simulating lignin biosynthesis, ACS Sustainable Chem. Eng., 2019, 7, 18313–18322 CrossRef CAS.
- J. V. Vermaas, L. D. Dellon, L. J. Broadbelt, G. T. Beckham and M. F. Crowley, Automated transformation of lignin topologies into atomic structures with LigninBuilder, ACS Sustainable Chem. Eng., 2018, 7, 3443–3453 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |