
Aqueous electrocatalytic N2 reduction for ambient NH3 synthesis: recent advances in catalyst development and performance improvement
Xiaojuan
Zhu
ab,
Shiyong
Mou
a,
Qiling
Peng
a,
Qian
Liu
 a,
Yonglan
Luo
b,
Guang
Chen
c,
Shuyan
Gao
d and
Xuping
Sun
*a
a,
Yonglan
Luo
b,
Guang
Chen
c,
Shuyan
Gao
d and
Xuping
Sun
*a
aInstitute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu 610054, Sichuan, China. E-mail: xpsun@uestc.edu.cn
bChemical Synthesis and Pollution Control Key Laboratory of Sichuan Province, College of Chemistry and Chemical Engineering, China West Normal University, Nanchong 637002, Sichuan, China
cThe Key Laboratory of Life-Organic Analysis, Key Laboratory of Pharmaceutical Intermediates and Analysis of Natural Medicine, School of Chemistry and Chemical Engineering, Qufu Normal University, Qufu 273165, Shandong, China
dSchool of Materials Science and Engineering, Henan Normal University, Xinxiang 453007, Henan, China
First published on 16th December 2019
Abstract
Electrochemical N2 reduction has emerged as an environmentally benign alternative to the Haber–Bosch process for sustainable NH3 synthesis under ambient reaction conditions, and considerable recent attention has focused on electrocatalytic NH3 synthesis from N2 and H2O in aqueous media. In this Minireview, we summarize the recent advances in the development of electrocatalysts for the N2 reduction reaction (NRR). Strategies to boost the NRR performances are also discussed. Perspectives for further research directions are provided finally.
 Xuping Sun | Xuping Sun received his PhD degree from the Changchun Institute of Applied Chemistry (CIAC), Chinese Academy of Sciences in 2006. During 2006–2009, he carried out postdoctoral research at Konstanz University, the University of Toronto, and Purdue University. In 2010, he started his independent research career as a full professor at the CIAC and then moved to Sichuan University in 2015. In 2018, he joined the University of Electronic Science and Technology of China where he founded the Research Center of Nanocatalysis & Sensing. He was recognized as a highly cited researcher (2018 & 2019) in areas of both chemistry and materials science by Clarivate Analytics. He has published over 440 papers with total citations over |
1. Introduction
NH3 is not only widely utilized as an essential activated nitrogen source to manufacture agricultural fertilizers, dyes, polymers, explosives, etc., but also provides a carbon-free chemical energy carrier solution for the transportation sector.1–3 As the dominant route for industrial-scale NH3 production using N2 and H2 as the feed gases, the century-old Haber–Bosch process suffers from harsh reaction conditions, complicated factory infrastructure, high energy consumption, and serious CO2 emissions.4 One alternative approach to solve the NH3 synthesis problem is to use electricity to drive the NH3 production reaction,5 and this electrically driven process is compatible with intermittent operation and enables utilization of renewable electricity without needing transmission capacity expansion. Electrochemical N2 reduction has emerged as an attractive method for artificial N2-to-NH3 conversion under ambient conditions; however it is severely challenged by N2 activation and needs efficient electrocatalysts to break the rather inert molecular structure of N2 with an extremely high bond energy of about 941 kJ mol−1.6–11 One big issue for the NRR in aqueous electrolytes is that the competitive hydrogen evolution reaction (HER) limits its current efficiency and leads to a low overall reaction rate (Fig. 1).12 Although they have better NH3 selectivity, molecular catalysts are fragile, which can be circumvented by using heterogeneous NRR catalysts. | ||
Fig. 1 Competition between the electrocatalytic NRR and HER processes on the catalyst. The NRR process involves the transfer of six electrons for one N2 molecule: N2 (g) + 6H2O (l) + 6e− ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) 2NH3 (aq.) + 6OH− (aq.) (Eo = 0.092 V vs. the reversible hydrogen electrode, RHE). In this process, N2 reacts with protons from the electrolyte on the surface of the catalyst to form NH3 using electrons from the electrode. Because of the involvement of only two electrons per H2 molecule, 2H+ (aq.) + 2e− H2 (g) (Eo = 0.00 V vs. RHE), the HER is kinetically preferred over the multi-step six-electron NRR process. 2NH3 (aq.) + 6OH− (aq.) (Eo = 0.092 V vs. the reversible hydrogen electrode, RHE). In this process, N2 reacts with protons from the electrolyte on the surface of the catalyst to form NH3 using electrons from the electrode. Because of the involvement of only two electrons per H2 molecule, 2H+ (aq.) + 2e− H2 (g) (Eo = 0.00 V vs. RHE), the HER is kinetically preferred over the multi-step six-electron NRR process. | ||
Two basic mechanisms (Scheme 1) are involved in the NRR over heterogeneous catalysts: the dissociative and associative mechanisms.13 In the dissociative mechanism, the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond is broken before its hydrogenation, leaving individual N-adatoms on the catalyst surface which are transformed into NH3 independently (Scheme 1a). The NRR based on the associative mechanism proceeds via an alternating (Scheme 1b) and a distal (Scheme 1c) pathway. For the alternating pathway, each of the two N atoms of adsorbed N2 is hydrogenated in turn until one N atom is converted into NH3 and the triple bond is broken. For the distal one, preferential hydrogenation of the N atom furthest away from the surface releases one equivalent of NH3, followed by the hydrogenation of the other N-adatom to release a second equivalent of NH3.
N triple bond is broken before its hydrogenation, leaving individual N-adatoms on the catalyst surface which are transformed into NH3 independently (Scheme 1a). The NRR based on the associative mechanism proceeds via an alternating (Scheme 1b) and a distal (Scheme 1c) pathway. For the alternating pathway, each of the two N atoms of adsorbed N2 is hydrogenated in turn until one N atom is converted into NH3 and the triple bond is broken. For the distal one, preferential hydrogenation of the N atom furthest away from the surface releases one equivalent of NH3, followed by the hydrogenation of the other N-adatom to release a second equivalent of NH3.
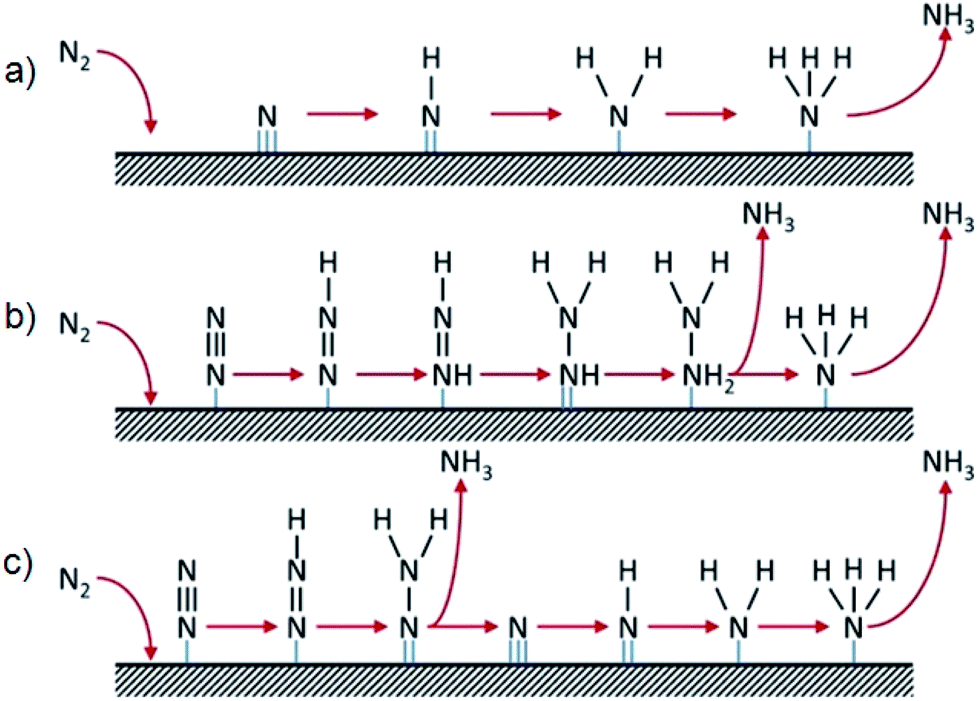 | ||
| Scheme 1 Generic NRR mechanisms on heterogeneous catalysts: (a) dissociative pathway; (b) associative alternating pathway; and (c) associative distal pathway. Reproduced from ref. 13 with permission from Elsevier, copyright 2018. | ||
In this Minireview, we summarize the recent advances in developing NRR electrocatalysts including noble-metal, non-noble-metal and non-metal catalysts. Following this, we discuss the strategies to boost the NRR performances including electronic structure tailoring, active site enrichment and HER suppression. Finally, future research directions are also proposed in the concluding remarks.
2. Advances in NRR electrocatalysts
2.1. Noble-metal catalyst
Based on previous density functional theory (DFT) calculations predicting a stronger binding of intermediates to the stepped facets than to the flat terraces on a N2-fixing metal catalyst,14 Yan and co-workers demonstrated a proof-of-concept that a Au nanorod with a tetrahexahedral structure is capable of catalyzing the NRR with an NH3 yield of 1.648 μg h−1 cm−2 and a faradaic efficiency (FE) of 4.02%.15 Wang et al. reported that Pd/C is superior in NRR activity to Au and Pt in phosphate buffer solution (PBS), attaining a FE of 8.2%.16 Theoretical calculations further reveal that the in situ formed α-PdH enables N2 activation through a thermodynamically more favorable Grotthuss-like hydride transfer pathway compared to direct surface hydrogenation or proton-coupled electron transfer steps. Ru is industrially used as an alternative to conventional Fe catalysts for more efficient NH3 synthesis at lower temperatures.17 Recent work also shows that Ru nanoparticles perform efficiently in ambient electrocatalytic N2 reduction to NH3 (5.5 mg h−1 m−2; 5.4%).18 Of note, Ag nanosheets also provides quite positive results (4.8%).192.2. Non-noble-metal catalysts
Compared with noble-metal materials, non-noble-metal ones have much higher earth abundance and hence hold greater promise for application as attractive NRR catalysts. Biological nitrogenases containing Mo, Fe and V are involved in natural N2 fixation,20,21 and a variety of NRR electrocatalysts made of these metals have been developed for artificial electrochemical NH3 synthesis under ambient conditions.Inspired by the fact that Mo and S elements play significant roles in nitrogenases, we performed theoretical calculations to study the electronic structures of MoS2 and mapped out the energy profile of the NRR on MoS2, which suggests that the positively charged Mo-edge is the key to polarizing and activating the N2 molecules (Fig. 2).22 To prove this, we hydrothermally grew a MoS2 nanosheet array on carbon cloth (MoS2/CC) to catalyze the NRR with a FE of 1.17%. Interestingly, other Mo compounds are also active for the NRR. For instance, an MoN catalyst can suppress electron transfer from Mo to the adsorbed N2, facilitating the release of produced NH3 and thus enhancing the NRR performance, and MoN nanosheets achieve an NH3 yield of 3.01 × 10−10 mo1 s−1 cm−2 with a FE of 1.15%.23 Superior NRR performances have been reported for Mo2C nanorods24 and Mo2C nanodots embedded in ultrathin carbon nanosheets25 with FEs of 8.13% and 7.8%, respectively. The exposed Mo atoms of the Mo2C(121) surface have large adsorption energies to dissociate N2 and the subsequent exothermic hydrogenation reactions are energetically preferable via the alternating pathway.25 Compared with the above Mo-based catalysts, its oxide counterpart can be more easily prepared on a large scale, and our study also verifies that MoO3 nanosheets are active for electrocatalytic N2-to-NH3 fixation (4.80 × 10−10 mol s−1 cm−2; 1.9%).26
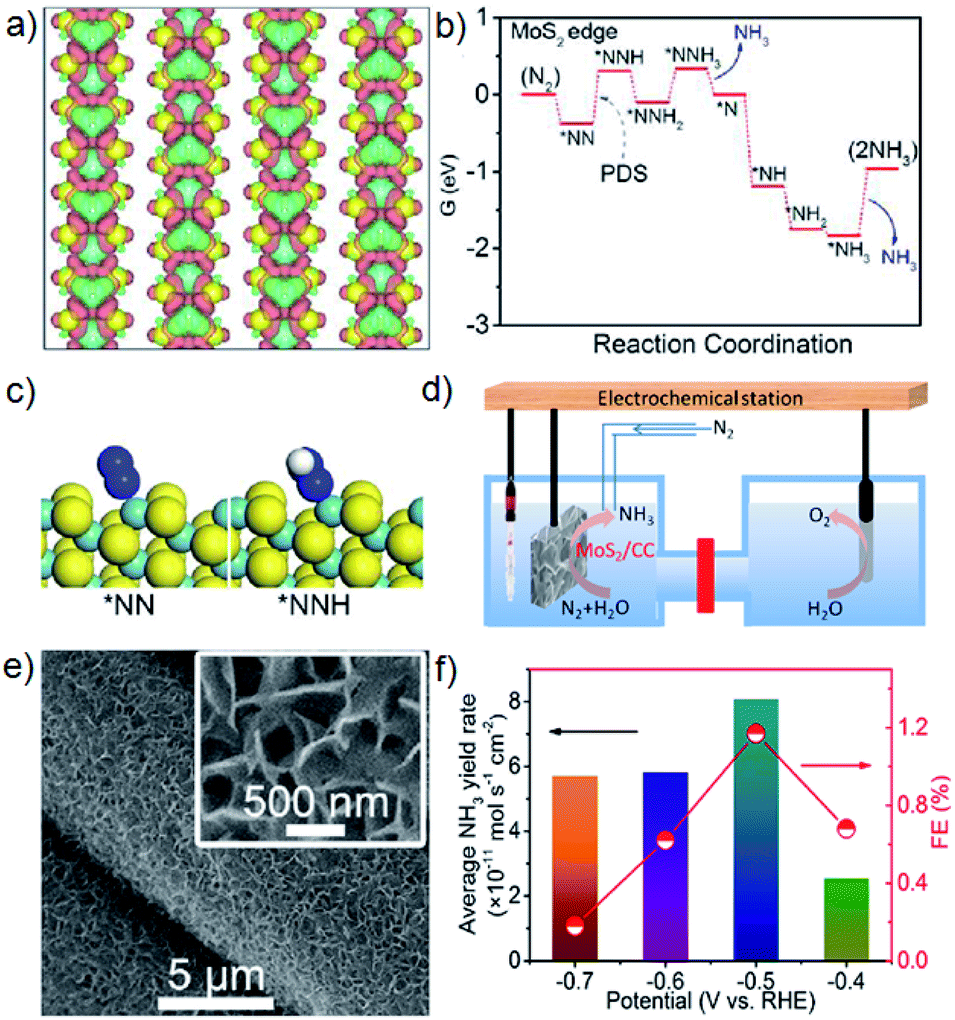 | ||
| Fig. 2 (a) Isosurface of deformation charge density as viewed from the top. Red and green represent charge accumulation and loss, respectively. The isosurface is 0.0025 a.u. (b) Free energy profiles for the NRR at the MoS2 edge site. The asterisk (*) denotes the adsorption site. (c) Structures of key intermediates of the potential-determining step (PDS). (d) A schematic diagram to illustrate the electrochemical setup for the NRR tests. (e) Scanning electron microscopy (SEM) images for MoS2/CC. (f) Average NH3 yields and FEs of MoS2/CC at different potentials in 0.1 M Na2SO4. Reproduced from ref. 22 with permission from Wiley-VCH, copyright 2018. | ||
As the cheapest and one of the most abundant transition metals, Fe-based NRR catalysts have also been widely explored. Licht et al. reported efficient N2 reduction using Fe2O3 in a molten hydroxide electrolyte cell at temperatures ≥200 °C.27 At room temperature and pressure, however, only a low FE of 0.15% was obtained for Fe2O3 nanoparticles supported on carbon nanotubes in a flow electrochemical cell operating in the gas phase.28 Since then, much effort has been made to explore Fe oxides with higher NRR activities in aqueous media.29–31 An Fe3S4 nanocatalyst was also shown by Zhao et al. to be active with a FE of 6.45% for NH3 formation.32 For the first time, we demonstrate that FeOOH nanorods achieve a FE of 6.7%, and DFT calculations evidence that the Fe-edge atom of the FeOOH(110) surface plays a key role in polarizing and activating the N2 molecules for both charge exchange and transfer.33 Quite surprisingly, a more recent study by us suggests that a P-rich FeP2 nanoparticle-reduced graphene oxide hybrid exhibits superhigh performances with a large NH3 yield of 35.26 μg h−1 mgcat.−1 and a high FE of 21.99%.34 DFT calculations reveal decreased HER activity, higher N2 adsorption energy, and a larger number of NRR active sites for FeP2 compared to FeP.
VN has previously been identified by DFT analysis as a promising candidate for the NRR under ambient conditions,35 but only recently has this prediction been experimentally verified.36–38 For transition metal nitrides, a Mars-van Krevelen mechanism is considered for NH3 generation: a surface N atom is reduced to form NH3 after which the resulting vacancy is replenished by a N2 molecule from the electrolyte. The N-vacancy must be stable at the surface, thus avoiding migration into the bulk of the catalyst. If not so, the reacted N on the surface is only replaced with more N atoms from the catalyst itself until all the N atoms of the metal nitrides are completely depleted, leaving only pure metal. V oxides are also able to catalyze the NRR.39,40 It should be noted that many other metals without biological implications, including Ti,41–43 Nb,44,45 Cr,46 Mn,47 Co,48 Cu,49 Bi,50,51 Sn,52 La,53etc., have also been identified to actively electrocatalyze the reduction.
2.3. Non-metal catalysts
From economic and environmental viewpoints, using non-metal catalysts could lower the cost and avoid the residues of metal ions. Nanocarbons, which are mainly composed of carbon and can even be made directly out of biomass, feature wide potential windows and structural diversity, which make them promising for electrochemical application as sustainable materials.54 Unfortunately, pristine nanocarbons have poor NRR activities and effective strategies must be implemented to improve their catalytic performances (see the following section for details).Interestingly, nanomaterials of elemental B55,56 and black P57 perform efficiently in electrochemical NH3 synthesis. Other non-metal materials including B4C,58 BP,59 BN,60,61 and polymeric carbon nitride (PCN)62 are highly active NRR catalysts. Fig. 3 presents the morphologies and the performance metrics are detailed in Table 1.
 | ||
| Fig. 3 Transmission electron microscopy (TEM) images of non-metal catalysts: (a) B nanosheet. Reproduced from ref. 56 with permission from The Royal Society of Chemistry, copyright 2019; (b) black P nanosheet. Reproduced from ref. 57 with permission from Wiley-VCH, copyright 2019; (c) B4C nanosheet. Reproduced from ref. 58 with permission from the Nature Publishing Group, copyright 2018; (d) BP nanoparticles. Reproduced from ref. 59 with permission from The Royal Society of Chemistry, copyright 2019; (f) PCN. Reproduced from ref. 62 with permission from Wiley-VCH, copyright 2018; (e) atomic force microscopy image and corresponding height profile for BN nanosheets. Reproduced from ref. 61 with permission from Tsinghua University Press and Springer-Verlag, copyright 2019. | ||
| Catalyst | Electrolyte | Potential (V) vs. RHE | NH3 yield | FE (%) | Ref. | ||
|---|---|---|---|---|---|---|---|
| Catalyst development | Noble-metal catalysts | Au nanorods | 0.10 M KOH | −0.2 | 1.648 μg h−1 cm−2 | 4.02 | Adv. Mater., 2017, 29, 1604799 |
| Pd/C | 0.01 M HCl | 4.5 μg h−1 mgPd−1 (−0.05 ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) V) V) |
8.2 (0.1 V) |
Nat. Commun., 2018, 9, 1795 | |||
| Ru nanoparticles | 0.01 M HCl | 5.5 μg h−1 cm−2 (−0.1 V) |
5.4 (0.01 V) |
ChemSusChem, 2018, 11, 3416−3422 | |||
| Ag nanosheets | 0.1 M HCl | −0.6 | 4.62 × 10−11 mol s−1 cm−2 | 4.8 | Chem. Commun., 2018, 54, 11427−11430 | ||
| Non-noble-metal catalysts | MoS2 nanosheet array | 0.1 M Na2SO4 | −0.5 | 8.08 × 10−11 mol s−1 cm−1 | 1.17 | Adv. Mater., 2018, 30, 1800191 | |
| MoN nanosheet array | 0.1 M HCl | −0.3 | 3.01 × 10−10 mo1 s−1 cm−2 | 1.15 | ACS Sustainable Chem. Eng., 2018, 6, 9550−9554 | ||
| Mo2C/C | 0.5 M Li2SO4 | −0.3 | 11.3 μg h−1 mgMo2C−1 | 7.8 |
Adv. Mater., 2018, 30, 1803694 | ||
| Mo2C nanorods | 0.1 M HCl | −0.3 | 91.5 μg h−1 mgcat.−1 | 8.31 | ACS Central Sci., 2019, 5, 116−121 | ||
| MoO3 nanosheets | 0.1 M HCl | 4.8 × 10−10 mol s−1 cm−2 (−0.5 V) | 1.9 (−0.3 V) | J. Mater. Chem. A, 2018, 6, 12974–12977 | |||
| Fe2O3-CNT | KHCO3 | −2.0 | 0.22 μg h−1 cm−2 | 0.15 | Angew. Chem., Int. Ed., 2017, 56, 2699–2703 | ||
| Non-noble-metal catalysts | Fe/Fe3O4 | 0.1 M PBS | −0.3 | 0.19 μg h−1 cm−2 | 8.29 | ACS Catal., 2018, 8, 9312–9319 | |
| Fe2O3 nanorods | 0.1 M Na2SO4 | −0.8 | 15.9 μg h−1 mg−1 | 0.94 | ChemCatChem, 2018, 10, 4530–4535 | ||
| Fe3O4 nanorods | 0.1 M Na2SO4 | −0.4 | 5.6 × 10−11 mol s−1 cm−2 | 2.6 | Nanoscale, 2018, 10, 14386–14389 | ||
| β-FeOOH nanorods | 0.5 M LiClO4 | 23.32 μg h−1 mgcat.−1 (−0.75 V) | 6.7 (−0.7 V) | Chem. Commun., 2018, 54, 11332−11335 | |||
| Fe3S4 nanosheets | 0.1 M HCl | −0.4 | 75.4 μg h−1 mgcat.−1 | 6.45 | Chem. Commun., 2018, 54, 13010–13013 | ||
| VN nanoparticles | 0.05 M H2SO4 | −0.1 | 3.3 × 10−10 mol s−1 cm−2 | 6.0 | J. Am. Chem. Soc., 2018, 140, 13387–13391 | ||
| VN nanosheet array | 0.1 M HCl | −0.5 | 8.4 × 10−11 mol s−1 cm−2 | 2.25 | ACS Sustainable Chem. Eng., 2018, 6, 9545–9549 | ||
| TiO2 nanosheet array | 0.1 M Na2SO4 | −0.7 | 9.16 × 10−11 mol s−1 cm−2 | 2.5 | ACS Appl. Mater. Interfaces, 2018, 10, 28251–28255 | ||
| Ti3C2Tx nanosheets | 0.1 M HCl | −0.4 | 20.4 μg h−1 mgcat.−1 | 9.3 | J. Mater. Chem. A, 2018, 6, 24031–24035 | ||
| Ti3C2Tx/FeOOH | 0.05 M H2SO4 | 0.53 μg·h−1·cm−2 (−0.5 V) | 5.78 (−0.2 V) | Joule, 2019, 3, 279–289 | |||
| NbO2 nanoparticles | 0.05 M H2SO4 | −0.65 | 11.6 μg h−1 mgcat.−1 | 32 | Small Methods, 2019, 3, 1800386 | ||
| Nb2O5 nanofibers | 0.1 M HCl | −0.55 | 43.6 μg h−1 mgcat.−1 | 9.26 | Nano Energy, 2018, 52, 264–270 | ||
| Hollow Cr2O3 microspheres | 0.1 M Na2SO4 | −0.9 | 25.3 μg h−1 mgcat.−1 | 6.78 | ACS Catal., 2018, 8, 8540–8544 | ||
| MnO particles | 0.1 M Na2SO4 | −0.39 | 1.11 × 10−10 mol s−1 cm−2 | 8.02 | Adv. Sci., 2019, 6, 1801182 | ||
| LaF3 nanoplates | 0.5 M LiClO4 | −0.45 | 55.9 μg h−1 mgcat.−1 | 16.0 | J. Mater. Chem. A, 2019, 7, 17761–17765 | ||
| Mosaic Bi nanosheets | 0.1 M Na2SO4 | −0.8 | 2.54 ± 0.16 μgNH3 cm−2 h−1 | 10.46 ± 1.45 | ACS Catal., 2019, 9, 2902–2908 | ||
| Bi nanosheet array | 0.1 M HCl | −0.5 | 6.89 × 10−11 mol s−1 cm−2 | 10.26 | Chem. Commun., 2019, 55, 5263–526 | ||
| CuO/rGO | 0.1 M Na2SO4 | −0.75 | 1.8 × 10−10 mol s−1 cm−2 | 3.9% | ChemCatChem, 2019, 11, 1441–1447 | ||
| Non-metal catalysts | B nanosheets | 0.1 M HCl | −0.14 | 31.37 μg h−1 mgcat.−1 | 4.84 | Chem. Commun., 2019, 55, 4246–4249 | |
| B nanosheets | 0.1 M Na2SO4 | −0.8 | 13.22 μg h−1 mgcat.−1 | 4.04 | ACS Catal., 2019, 9, 4609–4615 | ||
| Black P nanosheets | 0.01 M HCl | 31.37 μg h−1 mgcat.−1 (−0.7 V) | 5.07 (−0.6 V) | Angew. Chem., Int. Ed., 2019, 58, 2612−2616 | |||
| B4C nanosheets | 0.1 M HCl | −0.75 | 26.57 μg h−1 mgcat.−1 | 15.95 | Nat. Commun., 2018, 9, 3485 | ||
| BP nanoparticles | 0.1 M HCl | −0.6 | 26.42 μg h−1 mgcat.−1 | 12.7 | J. Mater. Chem. A, 2019, 7, 16117−16121 | ||
| BN nanosheets | 0.1 M HCl | −0.75 | 22.4 μg h−1 mgcat.−1 | 4.7 | Nano Res., 2019, 12, 919−924 | ||
| Mesoporous BN | 0.1 M Na2SO4 | −0.7 | 18.2 μg h−1 mgcat.−1 | 5.5 | Nanoscale, 2019, 11, 4231−4235 | ||
| PCN | 0.1 M HCl | −0.2 | 8.09 μg h−1 mgcat.−1 | 11.59 | Angew. Chem., Int. Ed., 2018, 57, 10246−10250 | ||
| Performance improvement | Tailoring electronic structures | Defective TiO2 | 0.1 M HCl | −0.15 | 1.24 × 10−10 mol s−1 cm−2 | 9.17 | Nanoscale, 2019, 11, 1555–1562 |
| MnOx nanowire array | 0.1 M Na2SO4 | −0.5 | 1.63 × 10−10 mol s−1 cm−2 | 11.40 | Chem. Commun., 2019, 55, 4627–4630 | ||
| R-WO3 nanosheets | 0.1 M HCl | −0.3 | 17.28 μg h−1 mgcat.−1 | 7.0 | Nanoscale, 2019, 11, 19274–19277 | ||
| r-CeO2 nanorods | 0.1 M Na2SO4 | 16.4 μg h−1 mgcat.−1 (−0.5 V) | 3.7 (−0.4 V) | ACS Sustainable Chem. Eng., 2019, 7, 2889–2893 | |||
| W2N3 nanosheets | 0.1 M KOH | −0.2 | 11.66 ± 0.98 μg h−1 mgcat.−1 | 11.67 ± 0.93 | Adv. Mater., 2019, 31, 1902709 | ||
| Defect-rich MoS2 nanoflowers | 0.1 M Na2SO4 | −0.4 | 29.28 μg h−1 mgcat.−1 | 8.34 | Adv. Energy Mater., 2018, 8, 1801357 | ||
| Defect-Bi nanoplates | 0.2 M Na2SO4 | −0.6 | 5.453 μg h−1 mgBi−1 | 11.6 | Angew. Chem., Int. Ed., 2019, 58, 9464–9469 | ||
| NPC | 0.05 M H2SO4 | −0.9 | 1.40 mmol g−1 h−1 | 1.42 | ACS Catal., 2018, 8, 1186–1191 | ||
| NCM-Au NPs | 0.1 M KOH | 0.36 g m−2 h−1 (−0.2 V) | 22 (−0.1 V) | Angew. Chem., Int. Ed., 2018, 57, 12360–12364 | |||
| B-Doped graphene | 0.05 M H2SO4 | −0.5 | 9.8 μg h−1 cm−2 | 10.8 | Joule, 2018, 2, 1610–1622 | ||
| O-Doped graphene | 0.1 M HCl | 21.3 μg h−1 mgcat.−1 (−0.55 V) | 12.6 (−0.45 V) | Chem. Commun., 2019, 55, 7502–7505 | |||
| S-Doped graphene | 0.1 M HCl | 27.3 μg h−1 mgcat.−1 (−0.6 V) | 11.5 (−0.5 V) | Chem. Commun., 2019, 55, 3371–3374 | |||
| Tailoring electronic structures | Defect-rich fluorographene | 0.1 M Na2SO4 | −0.7 | 9.3 μg h−1 mgcat.−1 | 4.2 | Chem. Commun., 2019, 55, 4266–4269 | |
| FeO(OH,F) nanorods | 0.5 M LiClO4 | −0.6 | 42.38 μg h−1 mgcat.−1 | 9.02 | Chem. Commun., 2019, 55, 3987–3990 | ||
| F-SnO2 nanosheets | 0.1 M Na2SO4 | −0.45 | 19.3 μg h−1 mgcat.−1 | 8.6 | Inorg. Chem., 2019, 58, 10424−10431 | ||
| Cu-CeO2−x nanorods | 0.1 M Na2SO4 | −0.45 | 5.3 × 10−10 mol s−1 cm−2 | 19.1 | Chem. Commun., 2019, 55, 2952–2955 | ||
| C-TixOy/C | 0.1 M LiClO4 | −0.4 | 14.8 μg h−1 mgcat.−1 | 17.8 | Angew. Chem., Int. Ed., 2019, 58, 13101–13106 | ||
| V-TiO2 nanorods | 0.5 M LiClO4 | 17.73 μg h−1 mgcat.−1 (−0.5 V) | 15.3 (−0.4 V) | Small Methods, 2019, 3, 1900356 | |||
| Zr-TiO2 nanotubes | 0.1 M KOH | −0.45 | 8.90 μg h−1 cm−2 | 17.3 | Nat. Commun., 2019, 10, 2877 | ||
| Fe-TiO2 nanoparticles | 0.5 M LiClO4 | −0.4 | 25.47 μg h−1 mgcat.−1 | 25.6 | Angew. Chem., Int. Ed., 2019, 131, 18620–18624 | ||
| O-CNT | 0.1 M LiClO4 | −0.4 | 32.33 μg h−1 mgcat.−1 | 12.50 | Chem. Commun., 2019, 55, 4997–5000 | ||
| TA-rGO | 0.5 M LiClO4 | −0.75 | 17.02 μg h−1 mgcat.−1 | 4.83 | ACS Sustainable Chem. Eng., 2019, 7, 14368−14372 | ||
| Pd-TA | 0.1 M Na2SO4 | −0.45 | 24.12 μg h−1 mgcat.−1 | 9.4 | J. Mater. Chem. A, 2019, 7, 21674−21677 | ||
| a-Au/CeOx-rGO | 0.1 M HCl | −0.2 | 8.31 μg h−1 mgcat.−1 | 10.10 | Adv. Mater., 2017, 29, 1700001 | ||
| Bi4V2O11/CeO2 | 0.1 M HCl | −0.2 | 23.21 μg h−1 mgcat.−1 | 10.16 | Angew. Chem., Int. Ed., 2018, 57, 6073–6073 | ||
| Au6/Ni | 0.05 M H2SO4 | −0.14 | 7.4 μg h−1 mgcat.−1 | 67.8 | J. Am. Chem. Soc., 2019, 141, 14976–14980 | ||
| Enriching active sites | TA-reduced Au/TiO2 | 0.1 M HCl | −0.2 | 21.4 μg h−1 mgcat.−1 | 8.11 | Adv. Mater., 2017, 29, 1606550 | |
| Au flowers | 0.1 M HCl | −0.2 | 25.57 μg h−1 mgcat.−1 | 6.05 | ChemSusChem, 2018, 11, 3480−3485 | ||
| AuHNCs | 0.5 M LiClO4 | 3.9 μg h−1 cm−2 (−0.5 V) | 30.2 (−0.4 V) | Nano Energy, 2018, 49, 316−323 | |||
| TiO2-rGO | 0.1 M Na2SO4 | −0.9 | 15.13 μg h−1 mgcat.−1 | 3.3 | J. Mater. Chem. A, 2018, 6, 17303–17306 | ||
| Mn3O4-rGO | 0.1 M Na2SO4 | −0.85 | 17.4 μg h−1 mgcat.−1 | 3.52 | Nano Res., 2019, 12, 1093–1098 | ||
| Cr2O3-rGO | 0.1 M HCl | 33.3 μg h−1 mgcat.−1 (−0.7 V) | 7.33 (−0.6 V) | Inorg. Chem., 2019, 58, 2257–2260 | |||
| SnO2/rGO | 0.1 M Na2SO4 | −0.5 | 25.6 μg h−1 mgcat.−1 | 7.1 | ACS Appl. Mater. Interfaces, 2019, 11, 31806–31815 | ||
| Pd0.2Cu0.8-rGO | 0.1 M KOH | −0.7 | 2.8 μg h−1 mgcat.−1 | 0.6 | Adv. Energy Mater., 2018, 8, 1800124 | ||
| S dots-rGO | 0.5 M LiClO4 | −0.85 | 28.56 μg h−1 mgcat.−1 | 7.07 | Chem. Commun., 2019, 55, 3152–3155 | ||
| PTCA-rGO | 0.1 M HCl | −0.5 | 24.7 μg h−1 mgcat.−1 | 6.9 | J. Mater. Chem. A, 2019, 7, 12446–12450 | ||
| MnO2–Ti3C2Tx | 0.1 M HCl | −0.55 | 34.12 μg h−1 mgcat.−1 | 11.39 |
J. Mater. Chem. A, 2019, 7, 18823–18827 |
||
| TiO2/Ti3C2Tx | 0.1 M HCl | 32.17 μg h−1 mgcat.−1 (−0.55 V) | 16.07 (−0.45 V) | Adv. Energy Mater., 2019, 9, 1803406 | |||
| BP/SnO2−x nanotubes | 0.1 M Na2SO4 | −0.4 | 48.87 μg h−1 mgcat.−1 | 14.6 | Angew. Chem., Int. Ed., 2019, 131, 1–7 | ||
| Au1/C3N4 | 0.05 M H2SO4 | −0.1 | 1.305 μg h−1 mgAu−1 | 11.1 | Sci. Bull., 2018, 63, 1246−1253 | ||
| AuSAs-NDPCs | 0.1 M HCl | −0.2 | 2.32 μg h−1 cm−2 | 12.3 | Small Methods, 2018, 2, 1800202 | ||
| Ru SAs/N–C | 0.05 M H2SO4 | −0.2 | 120.9 μg h−1 mgcat.−1 | 29.6 | Adv. Mater., 2018, 30, 1803498 | ||
| SA-Mo/NPC | 0.1 M KOH | −0.3 | 34.0 ± 3.6 μg h−1 mgcat.−1 | 14.6 ± 1.6 | Angew. Chem., Int. Ed., 2019, 58, 2321–2325 | ||
| Enriching active sites | FeSA-N–C | 0.1 M KOH | 0.0 | 7.48 μg h−1 mgcat.−1 | 56.55 | Nat. Commun., 2019, 10, 341 | |
| PEBCD | 0.5 M Li2SO4 | 2.01 μg h−1 cm−2 (−0.7 V) | 2.91 (−0.4 V) | J. Am. Chem. Soc., 2017, 139, 9771–9774 | |||
| MoS2/BCCF | 0.1 M Li2SO4 | −0.2 | 43.4 μg h−1 mgcat.−1 | 9.81 | Adv. Energy Mater., 2019, 9, 1803935 | ||
| BiNCs | Acidic 0.5 M K2SO4 (pH = 3.5) | −0.6 | 200 mmol g−1 h−1 | 66 | Nat. Catal., 2019, 2, 448−456 | ||
| Ag-Au@ZIF | 0.1 M HCl | −2.5 | 10 pmol cm−2 s−1 | 18 ± 4 | Sci. Adv., 2018, 4, eaar3208 | ||
| NPG@ZIF-8 | 0.1 M Na2SO4 | 28.7 ± 0.9 μg h−1 cm−2 (−0.8 V) | 44 (−0.6 V) | Angew. Chem., Int. Ed., 2019, 131, 15506−15510 |
3. Strategies for boosting the NRR performances
The catalytic performances of current catalysts are still far from meeting the needs of practical applications. From the point of view of thermodynamics, the NRR should proceed at negative potentials that are dominated by the competing HER, leading to a tough selectivity issue and thus unsatisfactory current efficiency. It is clear that strategies should be developed to enable more efficient N2 reduction electrocatalysis. In the following section, we will summarize the most recent progress in strategies to boost the NRR performances, which include: (1) tailoring the electronic structures to promote intrinsic NRR activity; (2) enriching active sites to increase the apparent NRR activity; (3) suppressing the HER to improve the selectivity.3.1. Tailoring the electronic structures of catalysts
Tailoring the electronic structure of a catalyst is the most common method to change the adsorption behaviour and tune the intrinsic activity of each active site.63 Defect engineering and heteroatom doping have been proven to be the two most effective methods to boost the intrinsic activity of NRR electrocatalysts. Surface functionalization and interface engineering provide us with two other good choices for this purpose.The Yu group reported experimentally and theoretically that electrochemical N2 reduction can be achieved by using PCN with N vacancies (VN) which chemisorbs and significantly increases the bond length of N2 resulting in strong N2 activation.62 Because of the lack of 15N2 isotopic experiments, one cannot confirm that the N of NH3 only comes from the N2 feed gas. A more recent study by the Qiao group shows that VN confined on 2D W2N3 nanosheets provide an electron-deficient environment to facilitate N2 adsorption and lower the thermodynamic limiting potential of the NRR.71 A nuclear magnetic resonance (NMR) test using 15N2 as the feed gas verifies that the NH3 was indeed generated via the NRR instead of decomposition of the catalyst or other contaminants.
The intrinsic high HER activity of MoS2 limits its current efficiency for NH3 formation.22,72 We have addressed this issue by designing S-rich defective MoS2 nanoflowers.73 This catalyst attains a much higher FE of 8.34% than its defect-free counterpart (2.18%). The DFT results suggest that the defects lead to the d-band center of Mo atoms moving close to the Fermi level (−0.26 eV → –0.14 eV) related to a stronger interaction between the catalyst surface and N2 molecule (−0.65 eV). Moreover, the defective MoS2 has a lower energy barrier of the PDS (0.60 eV) than the pristine one (0.65 eV).
For pure metal catalysts, the defect effect is rarely studied. A recent study by the Yan group demonstrates the modulation of the electronic properties of Bi via defect engineering (Fig. 4).74 In their study, Bi(110) nanoplates with a high fraction of isolated Bi vacancies were fabricated from Bi2O3 nanoplates using a low-temperature plasma bombardment approach. This defect-Bi achieves an NH3 yield of 5.453 μg h−1 mgcat.−1 and a FE of 11.68%. Theoretical calculations indicate that defect-Bi(110) has a much lower activation energy (0.56 eV) in comparison with perfect-Bi(110) (1.03 eV) for the rate-determining step.
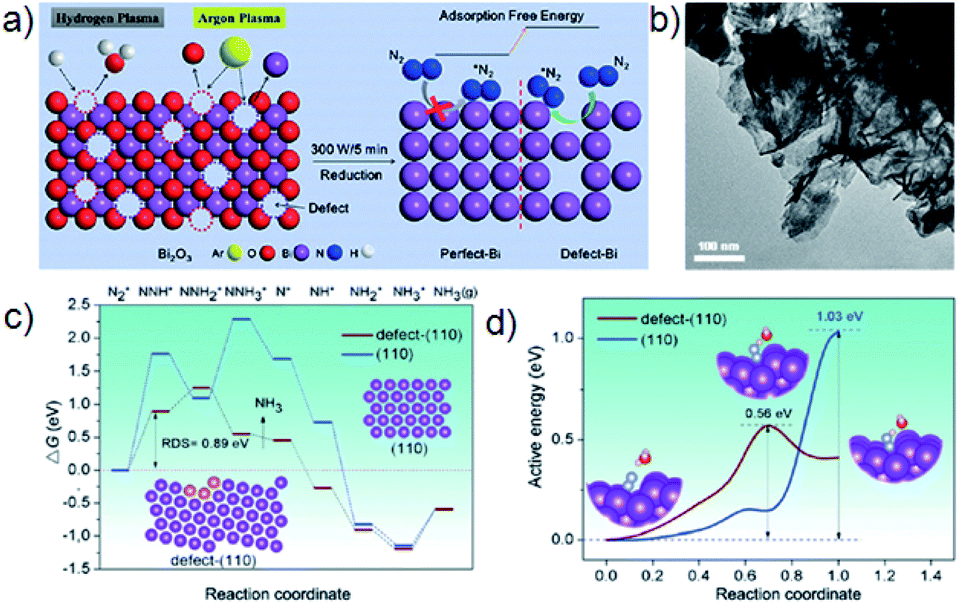 | ||
| Fig. 4 (a) Illustration of the synthesis of a defect-Bi nanoplate and its application for the NRR; (b) TEM image of defect-Bi nanoplates; (c) free–energy diagrams for the NRR on the perfect-Bi(110) and defect-Bi(110) facets via a reaction pathway; (d) activation energy of the rate-determining step. Reproduced from ref. 74 with permission from Wiley-VCH, copyright 2019. | ||
As a 2D nanocarbon with high conductivity and chemical stability, graphene with B doping attains an NH3 yield of 9.8 μg h−1 cm−2 and a FE of 10.8%.77 B incorporation causes redistribution of electron density and the as-formed electron-deficient B sites have higher N2-binding capability. The much larger electronegativity of O with respect to C endows it with a stronger ability to manipulate the electronic properties of carbon catalysts. O-doped graphene derived from sodium gluconate was also identified by us as a superior NRR catalyst with a larger NH3 yield of 21.3 μg h−1 mgcat.−1 and a higher FE of 12.6%.78 DFT data show that both C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and O–CO groups make a greater contribution to the NRR than the C–O group. Other heteroatoms like S79 and F80 also work effectively as dopants to boost the NRR performances of graphene.
O and O–CO groups make a greater contribution to the NRR than the C–O group. Other heteroatoms like S79 and F80 also work effectively as dopants to boost the NRR performances of graphene.
Like carbon materials, metal-based catalysts can also be doped with non-metal and metal heteroatoms to boost the NRR performances. F was utilized by us to dope FeOOH to decrease the reaction energy barrier, and the performance metrics are greatly boosted from 10.01 μg h−1 mgcat.−1 and 2.16% to 42.38 μg h−1 mgcat.−1 and 9.02%, respectively.81 The same idea was followed by Liu et al. for SnO2 mesoporous nanosheets,82 and it could also be applicable to other metal oxides. Cu doping in CeO2 was reported to form multiple VO for dramatically enhanced catalytic activities.83 For such a strategy, TiO2 is the most extensively studied metal oxide and several dopants like C,84,85 B,86 V,87 and Zr88 work effectively. The C-doped TiO2/C material derived from MIL-125(Ti) affords a high FE of 17.8%, which is attributed to the doping of C atoms into VO and the formation of Ti–C bonds, which enable energetically more favourable N2 activation.85 In Zheng's work, given that Zr4+ has a similar d-electron configuration and oxide structure but relatively larger ionic size, it was doped into TiO2 to induce the formation of adjacent bi-Ti3+ pairs as the most active centers for efficient N2 lying-down chemisorption and activation (8.90 μg h−1 mgcat.−1; 17.3%) (Fig. 5).88 Impressively, high-performance NRR catalysis can also be enabled over TiO2 using Fe as a dopant due to the synergistic effect of bi-Ti3+ and VO (25.47 μg h−1 mgcat.−1; 25.6%).89
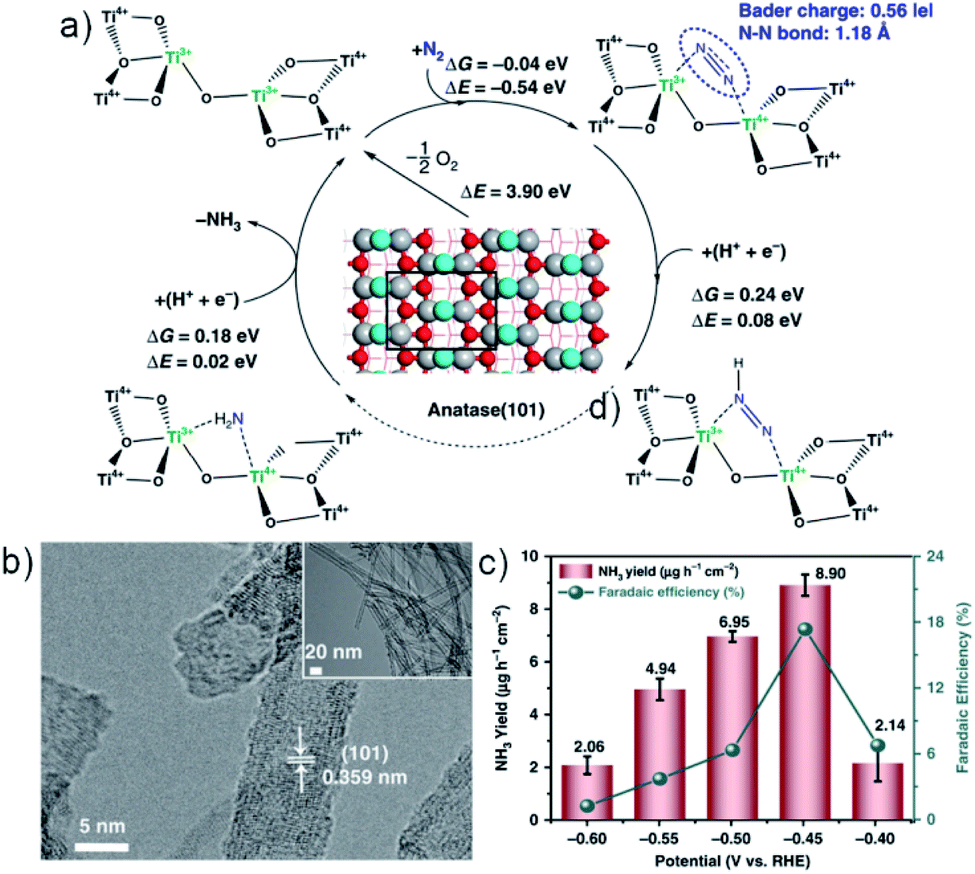 | ||
| Fig. 5 (a) DFT prediction of the NRR activity for adjacent bi-Ti3+ on anatase (101) with VO (light-blue spheres: lattice oxygen at the bridge sites where the surface VO are formed most easily; red spheres: other lattice oxygens on the surfaces; gray spheres: titanium cations; ΔG: free energy; ΔE: electronic energy. (b) TEM image for Zr–TiO2 nanotubes. (c) NH3 yields and FEs of Zr–TiO2 at each given potential. Reproduced from ref. 88 with permission from the Nature Publishing Group, copyright 2019. | ||
O) on their surface, and as a NRR catalyst, they exhibit superior performances (32.33 μg h−1 mgcat.−1; 12.50%).90 Our recent work further shows an enhancement in the NRR activity of reduced graphene oxide (rGO) after surface modification with oxygen-rich tannic acid (TA).91 The strong π–π stacking interactions between π-rich TA and rGO brings TA into very close proximity with rGO, which leads to intimate contact between the oxygen functional groups of TA and rGO favouring more effective manipulation of its electronic properties. The same strategy has also been used to improve the NRR performances of Pd catalysts (24.12 μg h−1 mgcat.−1; 9.49%).92 Compared to Pd, the much larger electronegativity of the O of TA could drive the formation of electron-deficient Pd with enhanced N2-adsorption ability,69 but the exact enhancement mechanism is not completely understood at the present time.
Because amorphous catalysts are in a metastable state with abundant unsaturated coordination sites, they exhibit higher catalytic activity than the crystalline ones. To this end, Shi et al. achieved the amorphization of Au nanoparticles anchored on rGO using CeOx as a trigger, and the resulting a-Au/CeOx-rGO hybrid shows superior NRR performances (8.31 μg h−1 mgcat.−1; 10.10%) to other control catalysts.93 The Yu group also reported highly active NRR electrocatalysis enabled by a CeO2/amorphous Bi4V2O11 hybrid (23.21 μg h−1 mgcat.−1; 10.16%).94 The amorphous phase has more abundant defective sites and lowers the energy barrier, and CeO2 triggers its amorphization during calcination. More recently, metal/metal interface engineering has also been applied to boost NRR catalysis, and the donor–acceptor couple of Ni and Au nanoparticles with a strong electronic connection attains a superhigh FE of 67.8%.95 In this study, Au nanoparticles were directly deposited on Ni nanoparticles via Galvanic replacement, and the electron-rich Au nanoparticles accepting electrons from Ni nanoparticles facilitate the adsorption and activation of N2 molecules.
3.2. Enriching the active sites of catalysts
Enriching the active sites to increase the apparent activity is regarded as the most straightforward way to boost the catalytic performances of electrocatalysts. To obtain a more active catalyst, one promising approach is nanostructuring of the catalyst to significantly increase the number of active sites. Previous studies point out that the size and shape of catalysts have a large influence on their NRR activities. The Yan group reported using Au sub-nanoclusters on TiO2 as a more efficient catalyst (21.4 μg h−1 cm−2; 8.11%) due to the fact that the small size of the sub-nanoclusters with high surface energy facilitates the formation of strong Au–O–Ti bonds and the resulting positively charged Au centers favour N2 adsorption.96 Au flowers were also reported to show higher performances than Au spheres, which is attributed to the exposure of more active sites by the dendritic structures of Au flowers.97 Using hollow gold nanocages as a NRR catalyst, Nazemi et al. attained a FE of 30.2% with an NH3 yield of 3.9 μg h−1 cm−2.98 Such a unique hollow nanostructure not only exposes abundant active sites, but also provides a confined reaction environment that increases the residence time of N2 molecules on its inner surface to enhance the conversion of N2 to NH3. In another study, we analyzed the performances of Cr2O3 catalysts, including hollow microspheres, solid microspheres, and nanoparticles with the following performance metrics: 25.3 μg h−1 mgcat.−1 & 6.78%, 11.4 μg h−1 mgcat.−1 & 2.94%, and 13.8 μg h−1 mgcat.−1 & 4.73%.46Another attractive approach is to disperse electrocatalysts on supports with a high surface area and conductivity. Using such a support can better disperse and reduce aggregation of the nanocatalysts on one hand and enhance the charge transport on the other hand. Thus far, graphene has been largely used to support CoO,48 CuO,49 Au/CeOx,93 TiO2,99 Mn3O4,100 Cr2O3,101 SnO2,102 Fe2O3,103 FeOOH,104 PdCu,105 PdP2,106 Ru2P,107 S dots,108 and perylene-3,4,9,10-tetracarboxylic acid nanorods.109 Graphene is electrocatalytically inert for the NRR, and its use as an inactive support would thus have an adverse effect on NH3 yield per unit mass. The high conductivity and large surface area of 2D Ti3C2Tx MXene,110 together with its intrinsic NRR activity,43,111 are promising for its use as an ideal support for NRR catalysts. We grew whisker-like MnO2 on Ti3C2Tx flakes and the MnO2–Ti3C2Tx hybrid attains a large NH3 yield of 34.12 μg h−1 mgcat.−1 and a high FE of 11.39%, superior to those of each component due to the synergistic catalytic effect.112 In another study, Ti3C2Tx MXene nanosheets act as both the support and Ti source for in situ solvothermal development of VO-rich TiO2 nanoparticles, and this hybrid efficiently catalyzes the NRR (32.17 μg h−1 mgcat.−1; 16.07%).113 More recently, black P quantum dots were stably confined on electrochemically active, electrically conductive black SnO2–x nanotubes via Sn–P coordination, and both components in the hybrid synergistically catalyze the NRR to afford amazing performance metrics (48.87 μg h−1 mgcat.−1; 14.6%).114
Single-atom catalysts (SACs) with maximum atom-utilization efficiency and unique properties enable reasonable use of metal resources and achieve atom economy, and they have recently emerged as a new Frontier in materials and catalysis sciences. Encouragingly, the establishment of reliable synthetic strategies to guarantee the stabilization of single metal atoms against migration and agglomeration has aroused huge recent interest in exploring their electrocatalytic applications.115 Several recent studies have described highly active N2 reduction electrocatalysis enabled by SACs. Wang et al. synthesized atomically dispersed Au catalysts on carbon nitride for the NRR with a FE of 11.1% and an NH3 yield of 1.305 μg h−1 mgAu−1, which is ∼22.5 times as high as that for supported Au nanoparticles.116 Au single sites stabilized on N-doped porous and highly oxidized carbon also enable efficient N2 electroreduction (2.32 μg h−1 cm−2; 12.3%).117 An NH3 yield of 120.9 μg h−1 mgcat.−1 was reported by Geng et al. over single Ru atoms anchored on N-doped carbon (Ru SAs/N–C).118 Han et al. prepared single Mo atoms anchored to N-doped porous carbon, and the high-density active sites and hierarchically porous carbon frameworks led to an NH3 yield of 34.0 ± 3.6 μg h−1 mgcat.−1 and a FE of 14.6 ± 1.6%.119 Another recent study by Wang et al. confirms that single-atom Fe on N-doped carbon achieves a much lower NH3 yield of 7.48 μg h−1 mgcat.−1 than Ru and Mo-based SACs.120 These studies would open up an exciting new avenue to explore using SACs for electrocatalytic N2 reduction, but care should be taken regarding these and other N-containing catalysts because they may decompose and leach N leading to inaccurate results.
3.3. Suppressing the HER
Suppressing the HER at the catalyst/electrolyte interface by manipulating the availability of H+ is an attractive route to improving NRR selectivity. DFT calculations predict that polyimide has intrinsic sluggish HER activity with a large activation barrier and Li+ association can further reduce the active sites to passivate the HER.121 Inspired by this, Chen et al. reported a Li+-incorporated poly(N-ethyl-benzene-1,2,4,5-tetracarboxylicdiimide) (PEBCD) to boost NRR selectivity, and the NRR and HER processes are effectively promoted and retarded, respectively, by associating Li+ ions with the O atoms in the PEBCD matrix.122 Although it achieves significant HER passivation, this catalyst attains a FE of only 2.85%, which could be due to its intrinsic low NRR activity. In another study by the Zhao group, in-operando created strong Li–S interactions endow S-rich MoS2 with superior NRR activity (43.4 μg h−1 mgcat.−1; 9.81%).123 Such interactions suppress the HER, facilitate N2 adsorption and increase NRR activity.Hao et al. recently reported a strategy to simultaneously promote NRR selectivity and activity using Bi nanocrystals and K+ cations (Fig. 6).124 The K+ cations not only stabilize the key nitrogen-reduction intermediates but also regulate proton transfer to increase the selectivity. A previous study suggests that supporting cations with high concentration can retard H+ migration from the bulk solution to the electrode surface leading to significant HER suppression.125 In Hao's work, optimized NRR performances (200 mmol g−1 h−1; 66%) were obtained in acidic 0.5 M K2SO4 electrolytes (pH = 3.5). Of note, such NRR promotion and HER suppression by K+ can also apply to Au and Pt catalysts.
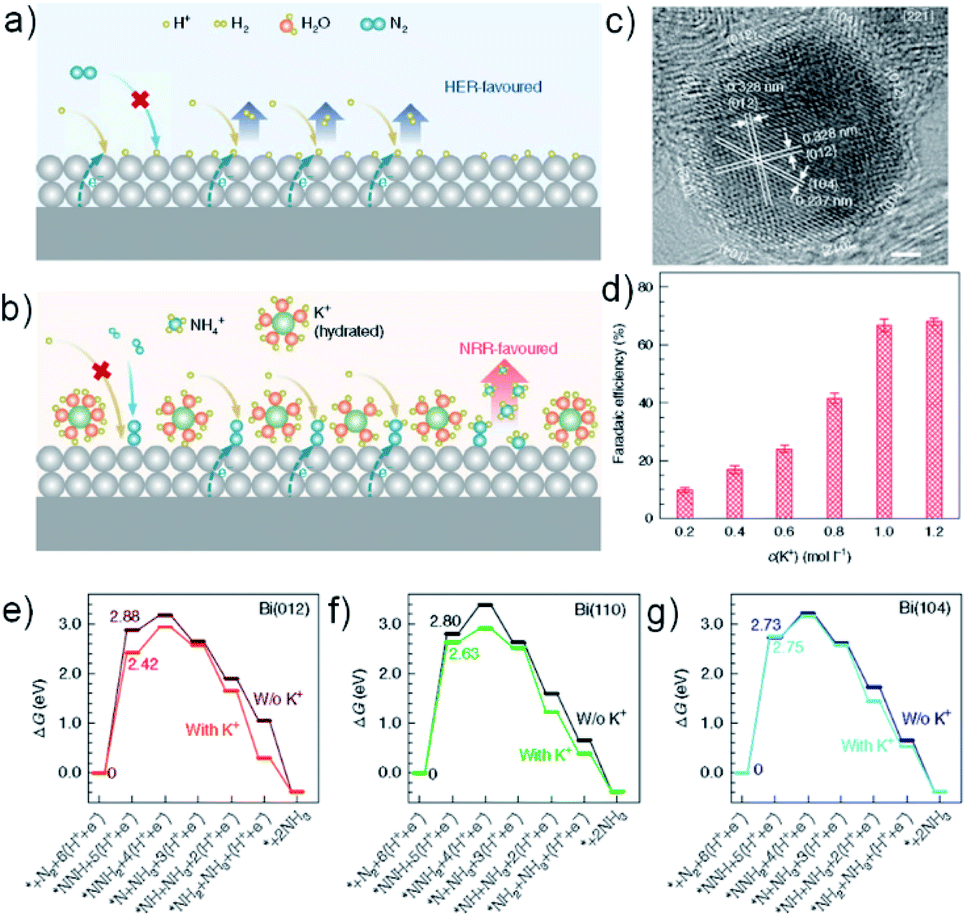 | ||
| Fig. 6 Mass transfer of H+ and N2 to the catalyst surface in electrolytes (a) without and (b) with K+. (a) In acidic solutions without K+, H+ can be transferred to the surface readily, and the HER will dominate. (b), K+ hinders H+ transfer to the catalyst surface. N2 will be adsorbed preferentially, and the NRR is promoted. (c) High-resolution TEM image of BiNCs, showing the exposed surfaces of {012}, {104} and {110}. (d) FE vs. c(K+). Gibbs free–energy diagrams for the NRR on (e) Bi(012), (f) Bi(110), and (g) Bi(104) facets in the presence or absence of K+ (pH 0 and U = 0 V vs. SHE). Reproduced from ref. 124 with permission from the Nature Publishing Group, copyright 2019. | ||
Coating a porous hydrophobic nanolayer over a NRR catalyst to repel water molecules could be another attractive alternative. This concept was first demonstrated in Ling's work.12 The authors deposited Ag nanocubes onto an Au electrode, followed by coating with a bifunctional ZIF-71 thin film as a sorption layer for confining N2 molecules to improve reactant–catalyst interactions and as a superhydrophobic barrier to inhibit water access. This structure affords a FE of 18 ± 4%, which is boosted by 10% compared to that of the uncoated catalyst. However, this system is neither cost-effective nor environmentally friendly because of the use of tetrahydrofuran and ethanol as the medium and proton source, respectively. An improved catalyst was recently designed by Yang et al.126 In their work, nanoporous gold (NPG) as the core was coated with a ZIF-8 shell which weakens hydrogen evolution and retards reactant diffusion, leading to a superb aqueous-based NPG@ZIF-8 catalyst (28.7 ± 0.9 μg h−1 cm−2; 44%). However, the authors have not commented on why the hydrophobic ZIF-8 can operate efficiently in an aqueous electrolyte.
The higher barrier for mass- and charge-transfer of neutral electrolytes leads to less favourable HER kinetics.127 Wang et al. tested a Pd/C catalyst in PBS with a FE of 8.2%, but this catalyst only affords a FE lower than 0.1% in both H2SO4 and NaOH.16 It is argued that PBS is a promising electrolyte for the electrochemical NRR due to its effective suppression of HER activity. Given the wide variety of experimental parameters tested in each study, this conclusion should be taken with caution because the large amount of research experience of our group suggests that the choice of electrolyte for optimized NRR performances also relies strongly on the catalyst itself.
4. Conclusions and outlook
The electrocatalysis community has made some recent progress in the design and development of catalytic systems to enable efficient electrochemical N2 reduction to NH3 under ambient conditions. An ideal catalyst should effectively adsorb and activate N2 molecules to overcome the slow kinetics. By manipulating the electronic structures of NRR catalysts, we can promote their intrinsic activities. Increasing the number of exposed active sites provides us with the most straightforward way to enhance the apparent catalytic activity. As the HER has lower overpotentials, the NRR in aqueous electrolytes always suffers from strong HER competition, leading to limited selectivity for NH3 formation. Despite some successful examples demonstrating the effective suppression of the HER to boost NRR selectivity, the versatility of this strategy has not been fully exploited. New strategies for catalyst design and HER suppression are still needed. Obviously, the selectivity issue will be better addressed by more general HER-retarding strategies like adding organic additives in the electrolytes or modifying the catalyst surface with water solvable H+-repelling molecules. We shoud point out that it is not appropriate to only emphasize current efficiency, and NH3 yield should also be treated seriously as an important metric for evaluating the overall performances of a NRR catalyst.Recently, the reliability of the NRR results has provoked discussions of experimental protocols,128 and strict standards have been set to obtain correct scientific reports.129 Control measurements with Ar under the exact same conditions and with N2 at open circuit potential are required to probe NH3 contamination within the cell, catalyst, and feed gas.130 Possible contaminants like NOx and other labile nitrogen compounds can be effectively removed through adsorption on a reduced Cu catalyst,129 and possible NH3 and NOx impurities can also be removed through a saturator filled with 0.05 M H2SO4 (aq.)130 Provided that all these protocols are carefully taken into consideration,129–131 both isotope-specific nuclear magnetic resonance and colorimetric measurements should give similar quantified results.129 As a well-established technique for ion analysis, ion chromatography (IC) also provides quantitative results which are consistent with those of colorimetry.47,66,80 Both colorimetry and IC measurements should also be considered as reliable alternatives to quantitatively gauge the NH3 product. The future development of electrochemical NH3 synthesis will heavily rely on establishing more efficient catalytic systems to achieve high selectivity and large current density for massive NH3 production, which however will still remain a big challenge for quite a long time.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21575137).References
- R. Schlögl, Angew. Chem., Int. Ed., 2003, 42, 2004–2008 CrossRef PubMed.
- S. Xu, D. C. Ashley, H.-Y. Kwon, G. R. Ware, C.-H. Chen, Y. Losovyj, X. Gao, E. Jakubikova and J. M. Smith, Chem. Sci., 2018, 9, 4950–4958 RSC.
- A. Klerke, C. Christensen, J. Nørskov and T. Vegge, J. Mater. Chem., 2008, 18, 2304–2310 RSC.
- I. Dybkjaer, Ammonia, Catalysis and Manufacture, ed. A. Nielsen, Springer, Heidelberg, 1995, pp. 199–308 Search PubMed.
- G. Marnellos and M. Stoukides, Science, 1998, 282, 98–100 CrossRef CAS PubMed.
- C. Guo, J. Ran, A. Vasileff and S. Qiao, Energy Environ. Sci., 2018, 11, 45–56 RSC.
- Q. Wang, Y. Lei, D. Wang and Y. Li, Energy Environ. Sci., 2019, 12, 1730–1750 RSC.
- R. Zhao, H. Xie, L. Chang, X. Zhang, X. Zhu, X. Tong, T. Wang, Y. Luo, P. Wei, Z. Wang and X. Sun, EnergyChem, 2019, 1, 100011 CrossRef.
- K. A. Brown, D. F. Harris, M. B. Wilker, A. Rasmussen, N. Khadka, H. Hamby, D. Keable, G. Dukovic, J. W. Peters, L. C. Seefeldt and P. W. King, Science, 2016, 352, 448–450 CrossRef CAS PubMed.
- M. A. Shipman and M. D. Symes, Catal. Today, 2017, 286, 57–68 CrossRef CAS.
- N. Cao and G. Zheng, Nano Res., 2018, 11, 2992–3008 CrossRef CAS.
- H. K. Lee, C. S. L. Koh, Y. H. Lee, C. Liu, I. Y. Phang, X. Han, C.-K. Tsung and X. Y. Ling, Sci. Adv., 2018, 4, eaar3208 CrossRef PubMed.
- M. A. Shipman and M. D. Symes, Catal. Today, 2017, 286, 57–68 CrossRef CAS.
- Á. Logadóttir and J. K. Nørskov, J. Catal., 2003, 220, 273–279 CrossRef.
- D. Bao, Q. Zhang, F. Meng, H. Zhong, M. Shi, Y. Zhang, J. Yan, Q. Jiang and X. Zhang, Adv. Mater., 2017, 29, 1604799 CrossRef PubMed.
- J. Wang, L. Yu, L. Hu, G. Chen, H. Xin and X. Feng, Nat. Commun., 2018, 9, 1795 CrossRef PubMed.
- H. Bielawa, O. Hinrichsen, A. Birkner and M. Muhler, Angew. Chem., Int. Ed., 2001, 40, 1061–1063 CrossRef CAS PubMed.
- D. Wang, L. M. Azofra, M. Harb, L. Cavallo, X. Zhang, B. H. R. Suryanto and D. R. MacFarlane, ChemSusChem, 2018, 11, 3416–3422 CrossRef CAS PubMed.
- H. Huang, L. Xia, X. Shi, A. M. Asiri and X. Sun, Chem. Commun., 2018, 54, 11427–11430 RSC.
- B. M. Hoffman, D. Lukoyanov, Z. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS PubMed.
- R. R. Eady, Chem. Rev., 1996, 96, 3013–3030 CrossRef CAS PubMed.
- L. Zhang, X. Ji, X. Ren, Y. Ma, X. Shi, Z. Tian, A. M. Asiri, L. Chen, B. Tang and X. Sun, Adv. Mater., 2018, 30, 1800191 CrossRef PubMed.
- L. Zhang, X. Ji, X. Ren, Y. Luo, X. Shi, A. M. Asiri, B. Zheng and X. Sun, ACS Sustainable Chem. Eng., 2018, 6, 9550–9554 CrossRef CAS.
- X. Ren, J. Zhao, Q. Wei, Y. Ma, H. Guo, Q. Liu, Y. Wang, G. Cui, A. M. Asiri, B. Li, B. Tang and X. Sun, ACS Cent. Sci., 2019, 5, 116–121 CrossRef CAS PubMed.
- H. Cheng, L. Ding, G. Chen, L. Zhang, J. Xue and H. Wang, Adv. Mater., 2018, 30, 1803694 CrossRef PubMed.
- J. Han, X. Ji, X. Ren, G. Cui, L. Li, F. Xie, H. Wang, B. Li and X. Sun, J. Mater. Chem. A, 2018, 6, 12974–12977 RSC.
- S. Licht, B. Cui, B. Wang, F. Li, J. Lau and S. Liu, Science, 2014, 345, 637–640 CrossRef CAS PubMed.
- S. Chen, S. Perathoner, C. Ampelli, C. Mebrahtu, D. Su and G. Centi, Angew. Chem., Int. Ed., 2017, 56, 2699–2703 CrossRef CAS PubMed.
- X. Xiang, Z. Wang, X. Shi, M. Fan and X. Sun, ChemCatChem, 2018, 10, 4530–4535 CrossRef CAS.
- Q. Liu, X. Zhang, B. Zhang, Y. Luo, G. Cui, F. Xie and X. Sun, Nanoscale, 2018, 10, 14386–14389 RSC.
- L. Hu, A. Khaniya, J. Wang, G. Chen, W. E. Kaden and X. Feng, ACS Catal., 2018, 8, 9312–9319 CrossRef CAS.
- X. Zhao, X. Lan, D. Yu, H. Fu, Z. Liu and T. Mu, Chem. Commun., 2018, 54, 13010–13013 RSC.
- X. Zhu, Z. Liu, Q. Liu, Y. Luo, X. Shi, A. M. Asiri, Y. Wu and X. Sun, Chem. Commun., 2018, 54, 11332–11335 RSC.
- X. Zhu, T. Wu, L. Ji, Q. Liu, Y. Luo, G. Cui, Y. Xiang, Y. Zhang, B. Zheng and X. Sun, Chem. Commun., 2020 10.1039/c9cc08352a.
- Y. Abghoui, A. L. Garden, J. G. Howalt, T. Vegge and E. Skúlason, ACS Catal., 2016, 6, 635–646 CrossRef CAS.
- R. Zhang, Y. Zhang, X. Ren, G. Cui, A. M. Asiri, B. Zheng and X. Sun, ACS Sustainable Chem. Eng., 2018, 6, 9545–9549 CrossRef CAS.
- X. Yang, J. Nash, J. Anibal, M. Dunwell, S. Kattel, E. Stavitski, K. Attenkofer, J. G. Chen, Y. Yan and B. Xu, J. Am. Chem. Soc., 2018, 140, 13387–13391 CrossRef CAS PubMed.
- X. Yang, S. Kattel, J. Nash, X. Chang, J. Lee, Y. Yan, J. Chen and B. Xu, Angew. Chem., Int. Ed., 2019, 131, 13906–13910 CrossRef.
- R. Zhang, H. Guo, L. Yang, Y. Wang, Z. Niu, H. Huang, H. Chen, L. Xia, T. Li, X. Shi, X. Sun, B. Li and Q. Liu, ChemElectroChem, 2019, 6, 1014–1018 CrossRef CAS.
- R. Zhang, J. Han, B. Zheng, X. Shi, A. M. Asiri and X. Sun, Inorg. Chem. Front., 2019, 6, 391–395 RSC.
- R. Zhang, X. Ren, X. Shi, F. Xie, B. Zheng, X. Guo and X. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 28251–28255 CrossRef CAS PubMed.
- G. Yu, H. Guo, W. Kong, T. Wang, Y. Luo, X. Shi, A. M. Asiri, T. Li and X. Sun, J. Mater. Chem. A, 2019, 7, 19657–19661 RSC.
- Y. Luo, G. Chen, L. Ding, X. Chen, L. Ding and H. Wang, Joule, 2019, 3, 279–289 CrossRef CAS.
- L. Huang, J. Wu, P. Han, A. M. Al-Enizi, T. M. Almutairi, L. Zhang and G. Zheng, Small Methods, 2019, 3, 1800386 CrossRef.
- J. Han, Z. Liu, Y. Ma, G. Cui, F. Xie, F. Wang, Y. Wu, S. Gao, Y. Xu and X. Sun, Nano Energy, 2018, 52, 264–270 CrossRef CAS.
- Y. Zhang, W. Qiu, Y. Ma, Y. Luo, Z. Tian, G. Cui, F. Xie, L. Chen, T. Li and X. Sun, ACS Catal., 2018, 8, 8540–8544 CrossRef CAS.
- Z. Wang, F. Gong, L. Zhang, R. Wang, L. Ji, Q. Liu, Y. Luo, H. Guo, Y. Li, P. Gao, X. Shi, B. Li, B. Tang and X. Sun, Adv. Sci., 2019, 6, 1801182 CrossRef PubMed.
- K. Chu, Y. Liu, Y. Li, H. Zhang and Y. Tian, J. Mater. Chem. A, 2019, 7, 4389–4394 RSC.
- C. Li, S. Mou, X. Zhu, F. Wang, Y. Wang, Y. Qiao, X. Shi, Y. Luo, B. Zheng, Q. Li and X. Sun, Chem. Commun., 2019, 55, 14474–14477 RSC.
- L. Li, C. Tang, B. Xia, H. Jin, Y. Zheng and S. Qiao, ACS Catal., 2019, 9, 2902–2908 CrossRef CAS.
- R. Zhang, L. Ji, W. Kong, H. Wang, R. Zhao, H. Chen, T. Li, B. Li, Y. Luo and X. Sun, Chem. Commun., 2019, 55, 5263–5266 RSC.
- L. Zhang, X. Ren, Y. Luo, X. Shi, A. M. Asiri, T. Li and X. Sun, Chem. Commun., 2018, 54, 12966–12969 RSC.
- P. Li, Z. Liu, T. Wu, Y. Zhang, L. Wang, L. Wang, L. Ji, Y. Zhang, Y. Luo, T. Wang, S. Liu, Y. Wu, M. Liu and X. Sun, J. Mater. Chem. A, 2019, 7, 17761–17765 RSC.
- J. Deng, M. Li and Y. Wang, Green Chem., 2016, 18, 4824–4854 RSC.
- X. Zhang, T. Wu, H. Wang, R. Zhao, H. Chen, T. Wang, P. Wei, Y. Luo, Y. Zhang and X. Sun, ACS Catal., 2019, 9, 4609–4615 CrossRef CAS.
- Q. Fan, C. Choi, C. Yan, Y. Liu, J. Qiu, S. Hong, Y. Jung and Z. Sun, Chem. Commun., 2019, 55, 4246–4249 RSC.
- L. Zhang, L. Ding, G. Chen, X. Yang and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 2612–2616 CrossRef CAS PubMed.
- W. Qiu, X. Xie, J. Qiu, W. Fang, R. Liang, X. Ren, X. Ji, G. Cui, A. M. Asiri, G. Cui, B. Tang and X. Sun, Nat. Commun., 2018, 9, 3485 CrossRef PubMed.
- X. Zhu, T. Wu, L. Ji, C. Li, T. Wang, S. Wen, S. Gao, X. Shi, Y. Luo, Q. Peng and X. Sun, J. Mater. Chem. A, 2019, 7, 16117–16121 RSC.
- J. Zhao, X. Ren, X. Li, D. Fan, X. Sun, H. Ma, Q. Wei and D. Wu, Nanoscale, 2019, 11, 4231–4235 RSC.
- Y. Zhang, H. Du, Y. Ma, L. Ji, H. Guo, Z. Tian, H. Chen, H. Huang, G. Cui, A. M. Asiri, F. Qu, L. Chen and X. Sun, Nano Res., 2019, 12, 919–924 CrossRef CAS.
- C. Lv, Y. Qian, C. Yan, Y. Ding, Y. Liu, G. Chen and G. Yu, Angew. Chem., Int. Ed., 2018, 57, 10246–10250 CrossRef CAS PubMed.
- Y. Yang, M. Luo, W. Zhang, Y. Sun, X. Chen and S. Guo, Chem, 2018, 4, 2054–2083 CAS.
- S. Dou, X. Wang and S. Wang, Small Methods, 2019, 3, 1800211 CrossRef.
- C. Li, T. Wang, Z. Zhao, W. Yang, J. Li, A. Li, Z. Yang, G. A. Ozin and J. Gong, Angew. Chem., Int. Ed., 2018, 57, 5278–5282 CrossRef CAS PubMed.
- L. Yang, T. Wu, R. Zhang, H. Zhou, L. Xia, X. Shi, H. Zheng, Y. Zhang and X. Sun, Nanoscale, 2019, 11, 1555–1562 RSC.
- L. Zhang, X. Xie, H. Wang, L. Ji, Y. Zhang, H. Chen, T. Li, Y. Luo, G. Cui and X. Sun, Chem. Commun., 2019, 55, 4627–4630 RSC.
- W. Kong, R. Zhang, X. Zhang, L. Ji, G. Yu, T. Wang, Y. Luo, X. Shi, Y. Xu and X. Sun, Nanoscale, 2019, 11, 19274–19277 RSC.
- X. Cui, C. Tang, X. Liu, C. Wang, W. Ma and Q. Zhang, Chem.–Eur. J., 2018, 24, 18494–18501 CrossRef CAS PubMed.
- B. Xu, L. Xia, F. Zhou, R. Zhao, H. Chen, T. Wang, Q. Zhou, Q. Liu, G. Cui, X. Xiong, F. Gong and X. Sun, ACS Sustainable Chem. Eng., 2019, 7, 2889–2893 CrossRef CAS.
- H. Jin, L. Li, X. Liu, C. Tang, W. Xu, S. Chen, L. Song, Y. Zheng and S. Qiao, Adv. Mater., 2019, 31, 1902709 CrossRef PubMed.
- J. D. Benck, T. R. Hellstern, J. Kibsgaard, P. Chakthranont and T. F. Jaramillo, ACS Catal., 2014, 4, 3957–3971 CrossRef CAS.
- X. Li, T. Li, Y. Ma, Q. Wei, W. Qiu, H. Guo, X. Shi, P. Zhang, A. M. Asiri, L. Chen, B. Tang and X. Sun, Adv. Energy Mater., 2018, 8, 1801357 CrossRef.
- Y. Wang, M. Shi, D. Bao, F. Meng, Q. Zhang, Y. Zhou, K. Liu, Y. Zhang, J. Wang, Z. Chen, D. Liu, Z. Jiang, M. Luo, L. Gu, Q. Zhang, X. Cao, Y. Yao, M. Shao, Y. Zhang, X. Zhang, J. Chen, J. Yan and Q. Jiang, Angew. Chem., Int. Ed., 2019, 58, 9464–9469 CrossRef CAS.
- Y. Liu, Y. Su, X. Quan, X. Fan, S. Chen, H. Yu, H. Zhao, Y. Zhang and J. Zhao, ACS Catal., 2018, 8, 1186–1191 CrossRef CAS.
- H. Wang, L. Wang, Q. Wang, S. Ye, W. Sun, Y. Shao, Z. Jiang, Q. Qiao, Y. Zhu, P. Song, D. Li, L. He, X. Zhang, J. Yuan, T. Wu and G. A. Ozin, Angew. Chem., Int. Ed., 2018, 57, 12360–12364 CrossRef CAS PubMed.
- X. Yu, P. Han, Z. Wei, L. Huang, Z. Gu, S. Peng, J. Ma and G. Zheng, Joule, 2018, 2, 1610–1622 CrossRef CAS.
- T. Wang, L. Xia, J. Yang, H. Wang, W. Fang, H. Chen, D. Tang, A. M. Asiri, Y. Luo, G. Cui and X. Sun, Chem. Commun., 2019, 55, 7502–7505 RSC.
- L. Xia, J. Yang, H. Wang, R. Zhao, H. Chen, W. Fang, A. M. Asiri, F. Xie, G. Cui and X. Sun, Chem. Commun., 2019, 55, 3371–3374 RSC.
- J. Zhao, J. Yang, L. Ji, H. Wang, H. Chen, Z. Niu, Q. Liu, Ti. Li, G. Cui and X. Sun, Chem. Commun., 2019, 55, 4266–4269 RSC.
- X. Zhu, Z. Liu, H. Wang, R. Zhao, H. Chen, T. Wang, F. Wang, Y. Luo, Y. Wu and X. Sun, Chem. Commun., 2019, 55, 3987–3990 RSC.
- Y. Liu, Y. Li, H. Zhang and K. Chu, Inorg. Chem., 2019, 58, 10424–10431 CrossRef CAS PubMed.
- S. Zhang, C. Zhao, Y. Liu, W. Li, J. Wang, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Chem. Commun., 2019, 55, 2952–2955 RSC.
- K. Jia, Y. Wang, Q. Pan, B. Zhong, Y. Luo, G. Cui, X. Guo and X. Sun, Nanoscale Adv., 2019, 1, 961–964 RSC.
- Q. Qin, Y. Zhao, M. Schmallegger, T. Heil, J. Schmidt, R. Walczak, G. Demner, H. Jiao and M. Oschatz, Angew. Chem., Int. Ed., 2019, 58, 13101–13106 CrossRef CAS PubMed.
- Y. Wang, K. Jia, Q. Pan, Y. Xu, Q. Liu, G. Cui, X. Guo and X. Sun, ACS Sustainable Chem. Eng., 2019, 7, 117–122 CrossRef CAS.
- T. Wu, W. Kong, Y. Zhang, Z. Xing, J. Zhao, T. Wang, X. Shi, Y. Luo and X. Sun, Small Methods, 2019, 3, 1900356 CrossRef CAS.
- N. Cao, Z. Chen, K. Zang, J. Xu, J. Zhong, J. Luo, X. Xu and G. Zheng, Nat. Commun., 2019, 10, 2877 CrossRef.
- T. Wu, X. Zhu, Z. Xing, S. Mou, C. Li, Y. Qiao, Q. Liu, Y. Luo, X. Shi, Y. Zhang and X. Sun, Angew. Chem., Int. Ed., 2019, 131, 18620–18624 CrossRef.
- J. Zhao, B. Wang, Q. Zhou, H. Wang, X. Li, H. Chen, Q. Wei, D. Wu, Y. Luo, J. You, F. Gong and X. Sun, Chem. Commun., 2019, 55, 4997–5000 RSC.
- Y. Song, T. Wang, J. Sun, Z. Wang, Y. Luo, L. Zhang, H. Ye and X. Sun, ACS Sustainable Chem. Eng., 2019, 7, 14368–14372 CrossRef CAS.
- G. Deng, T. Wang, A. A. Alshehri, K. A. Alzahrani, Y. Wang, H. Ye, Y. Luo and X. Sun, J. Mater. Chem. A, 2019, 7, 21674–21677 RSC.
- S. Li, D. Bao, M. Shi, B. Wulan, J. Yan and Q. Jiang, Adv. Mater., 2017, 29, 1700001 CrossRef.
- C. Lv, C. Yan, G. Chen, Y. Ding, J. Sun, Y. Zhou and G. Yu, Angew. Chem., Int. Ed., 2018, 57, 6073 CrossRef CAS PubMed.
- Z. Xue, S. Zhang, Y. Lin, H. Su, G. Zhai, J. Han, Q. Yu, X. Li, M. Antonietti and J. Chen, J. Am. Chem. Soc., 2019, 141, 14976–17980 CrossRef CAS PubMed.
- M. Shi, D. Bao, B. Wulan, Y. Li, Y. Zhang, J. Yan and Q. Jiang, Adv. Mater., 2017, 29, 1606550 CrossRef.
- Z. Wang, Y. Li, H. Yu, Y. Xu, H. Xue, X. Li, H. Wang and L. Wang, ChemSusChem, 2018, 11, 3480–3485 CrossRef CAS PubMed.
- M. Nazemi, S. R. Panikkanvalappil and M. A. El-Sayed, Nano Energy, 2018, 49, 316–323 CrossRef CAS.
- X. Zhang, Q. Liu, X. Shi, A. M. Asiri, Y. Luo, X. Sun and T. Li, J. Mater. Chem. A, 2018, 6, 17303–17306 RSC.
- H. Huang, F. Gong, Y. Wang, H. Wang, X. Wu, W. Lu, R. Zhao, H. Chen, X. Shi, A. M. Asiri, T. Li, Q. Liu and X. Sun, Nano Res., 2019, 12, 1093–1098 CrossRef CAS.
- L. Xia, B. Li, Y. Zhang, R. Zhang, L. Ji, H. Chen, G. Cui, H. Zheng, X. Sun, F. Xie and Q. Liu, Inorg. Chem., 2019, 58, 2257–2260 CrossRef CAS PubMed.
- K. Chu, Y. Liu, Y. Li, J. Wang and H. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 31806–31815 CrossRef CAS PubMed.
- J. Li, X. Zhu, T. Wang, Y. Luo and X. Sun, Inorg. Chem. Front., 2019, 6, 2682–2685 RSC.
- X. Zhu, J. Zhao, L. Ji, T. Wu, T. Wang, S. Gao, A. A. Alshehri, K. A. Alzahrani, Y. Luo, Y. Xiang, B. Zheng and X. Sun, Nano Res., 2019 DOI:10.1007/s12274-019-2600-8.
- M. Shi, D. Bao, S. Li, B. Wulan, J. Yan and Q. Jiang, Adv. Energy Mater., 2018, 8, 1800124 CrossRef.
- H. Xie, Q. Geng, X. Zhu, Y. Luo, L. Chang, X. Niu, X. Shi, A. M. Asiri, S. Gao, Z. Wang and X. Sun, J. Mater. Chem. A, 2019, 7, 24760–24764 RSC.
- R. Zhao, C. Liu, X. Zhang, X. Zhu, P. Wei, L. Ji, Y. Guo, S. Gao, Y. Luo, Z. Wang and X. Sun, J. Mater. Chem. A, 2019, 1, 100011 Search PubMed.
- H. Chen, X. Zhu, H. Huang, H. Wang, T. Wang, R. Zhao, H. Zheng, A. M. Asiri, Y. Luo and X. Sun, Chem. Commun., 2019, 55, 3152–3155 RSC.
- P. Li, J. Wang, H. Chen, X. Sun, J. You, S. Liu, Y. Zhang, M. Liu, X. Niu and Y. Luo, J. Mater. Chem. A, 2019, 7, 12446–12450 RSC.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS.
- J. Zhao, L. Zhang, X. Xie, X. Li, Y. Ma, Q. Liu, W. Fang, X. Shi, G. Cui and X. Sun, J. Mater. Chem. A, 2018, 6, 24031–24035 RSC.
- W. Kong, F. Gong, Q. Zhou, G. Yu, L. Ji, X. Sun, A. M. Asiri, T. Wang, Y. Luo and Y. Xu, J. Mater. Chem. A, 2019, 7, 18823–18827 RSC.
- Y. Fang, Z. Liu, J. Han, Z. Jin, Y. Han, F. Wang, Y. Niu, Y. Wu and Y. Xu, Adv. Energy Mater., 2019, 9, 1803406 CrossRef.
- Y. Liu, D. Li, J. Yu and B. Ding, Angew. Chem., Int. Ed., 2019, 58, 1–7 CrossRef.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- X. Wang, W. Wang, M. Qiao, G. Wu, W. Chen, T. Yuan, Q. Xue, M. Chen, Y. Zhang, X. Wang, J. Wang, J. Ge, X. Hong, Y. Li, Y. Wu and Y. Li, Sci. Bull., 2018, 63, 1246–1253 CrossRef CAS.
- Q. Qin, T. Heil, M. Antonietti and M. Oschatz, Small Methods, 2018, 2, 1800202 CrossRef.
- Z. Geng, Y. Liu, X. Kong, P. Li, K. Li, Z. Liu, J. Du, M. Shu, R. Si and J. Zeng, Adv. Mater., 2018, 30, 1803498 CrossRef.
- L. Han, X. Liu, J. Chen, R. Lin, H. Liu, F. Lü, S. Bak, Z. Liang, S. Zhao, E. Stavitski, J. Luo, R. R. Adzic and H. L. Xin, Angew. Chem., Int. Ed., 2019, 58, 2321–2325 CrossRef CAS.
- M. Wang, S. Liu, T. Qian, J. Liu, J. Zhou, H. Ji, J. Xiong, J. Zhong and C. Yan, Nat. Commun., 2019, 10, 341 CrossRef PubMed.
- Y. Wang, X. Cui, Y. Zhang, L. Zhang, X. Gong and G. Zheng, Adv. Mater., 2016, 28, 7626–7632 CrossRef CAS.
- G. Chen, X. Cao, S. Wu, X. Zeng, L. Ding, M. Zhu and H. Wang, J. Am. Chem. Soc., 2017, 139, 9771–9774 CrossRef CAS.
- Y. Liu, M. Han, Q. Xiong, S. Zhang, C. Zhao, W. Gong, G. Wang, H. Zhang and H. Zhao, Adv. Energy Mater., 2019, 9, 1803935 CrossRef.
- Y. Hao, Y. Guo, L. Chen, M. Shu, X. Wang, T. Bu, W. Gao, N. Zhang, X. Su, X. Feng, J. Zhou, B. Wang, C. Hu, A. Yin, R. Si, Y. Zhang and C. Yan, Nat. Catal., 2019, 2, 448–456 CrossRef CAS.
- Y. Mukouyama, R. Nakazato, T. Shiono, S. J. Nakanishi and H. Okamoto, J. Electroanal. Chem., 2014, 713, 39–46 CrossRef CAS.
- Y. Yang, S. Wang, H. Wen, T. Ye, J. Chen, C. Li and M. Du, Angew. Chem., Int. Ed., 2019, 131, 15506–15510 CrossRef.
- D. Strmcnik, P. P. Lopes, B. Genorio, V. R. Stamenkovic and N. M. Markovic, Nano Energy, 2016, 29, 29–36 CrossRef CAS.
- L. F. Greenlee, J. N. Renner and S. L. Foster, ACS Catal., 2018, 8, 7820–7827 CrossRef CAS.
- S. Z. Andersen, V. Čolić, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. McEnaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh, B. A. Rohr, M. J. Statt, S. J. Blair, S. Mezzavilla, J. Kibsgaard, P. C. K. Vesborg, M. Cargnello, S. F. Bent, T. F. Jaramillo, I. E. L. Stephens, J. K. Nørskovand and I. Chorkendorff, Nature, 2019, 570, 504–508 CrossRef CAS.
- C. Tang and S. Qiao, Joule, 2019, 3, 1573–1575 CrossRef.
- H. Du, T. R. Gengenbach, R. Hodgetts, D. R. MacFarlane and A. N. Simonov, ACS Sustainable Chem. Eng., 2019, 7, 6839–6850 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |