
Layered metallic vanadium diboride as an active cocatalyst for efficient dye-sensitized photocatalytic hydrogen evolution†
Wenbo
Li
abc,
Shixiong
Min
 *abc,
Fang
Wang
abc and
Zhengguo
Zhang
abc
*abc,
Fang
Wang
abc and
Zhengguo
Zhang
abc
aSchool of Chemistry and Chemical Engineering, Key Laboratory of Electrochemical Energy Conversion Technology and Application, North Minzu University, Yinchuan, 750021, P. R. China. E-mail: sxmin@nun.edu.cn
bKey Laboratory of Chemical Engineering and Technology, State Ethnic Affairs Commission, North Minzu University, Yinchuan, 750021, P. R. China
cNingxia Key Laboratory of Solar Chemical Conversion Technology, North Minzu University, Yinchuan 750021, P. R. China
First published on 26th November 2019
Abstract
Here, we report that layered metallic vanadium diboride (VB2) can function as an active cocatalyst for efficiently catalyzing photocatalytic hydrogen evolution in a dye-sensitized system under visible light irradiation. In an Erythrosin B–triethanolamine (ErB–TEOA) molecular system, the highest turnover number (TON) of hydrogen evolution based on ErB reaches 40. In addition, VB2 outperforms V2O5, VS2, VN, and VC for photocatalytic hydrogen evolution.
Hydrogen (H2) is being pursued as the most promising energy carrier in the future because it has a high-energy density and its combustion product is only water. Photocatalytic water splitting has been regarded as one of the most ideal technologies for sustainable production of H2.1–4 To lower the kinetic barriers of the photocatalytic H2 evolution reaction (HER) on semiconductors or in dye-sensitized systems, the use of a cocatalyst that can reduce the overpotential and consequently increase the efficiency of the photocatalytic HER is typically essential.4 So far, noble metal-based materials are still proven to be the most efficient cocatalysts and widely used for the photocatalytic HER due to their low overpotential of the HER and excellent photochemical stability.4,5 However, the practical application of noble metals in a large-scale photocatalytic HER is greatly restricted because of their high cost and scarcity. Therefore, developing highly active and stable H2 evolution cocatalysts (HECs) based on earth-abundant elements is highly desirable and has recently been extensively pursued.
Recently, inspired by the progress in the electrocatalytic HER, a large number of electrocatalysts based on transition-metal-based compounds including sulfides,6 selenides,7 phosphides,8 carbides,9 and nitrides10 have been successfully employed as HECs for the photocatalytic HER. Among them, transition metal carbides (TMCs), nitrides (TMNs), and diborides (TMBs) are more promising as HECs as they possess high electronic conductivity and a high density of Pt-like active sites for the HER.11–13 On the other hand, recently various vanadium-based catalysts, such as VS2,14 VSe2,15 VN,16 VC,17 and VB2,13 have been identified as promising alternatives to noble-metal-based electrocatalysts in catalyzing the electrocatalytic HER with high performance, and this research wave has transitioned to photocatalysis. Specifically, it has been shown that VS2 can function as an efficient HEC to greatly promote the visible light photocatalytic HER activity of g-C3N4.18 In addition, in our previous studies, we have also shown that VC can act as a versatile, active, and robust cocatalyst when integrated with CdS and dye-sensitized systems under visible light irradiation.17,19 In comparison with other vanadium-based catalysts, VB2 not only has excellent chemical stability, but also inherently possesses graphene-like boron sheet motifs (borophene subunits) in its structure (Fig. 1a),19–21 endowing VB2 with high electronic conductivity and a large density of active sites, which make VB2 highly promising as a HEC for the photocatalytic HER. However, to the best of our knowledge, there is no study on employing VB2 as a HEC for promoting the photocatalytic HER.
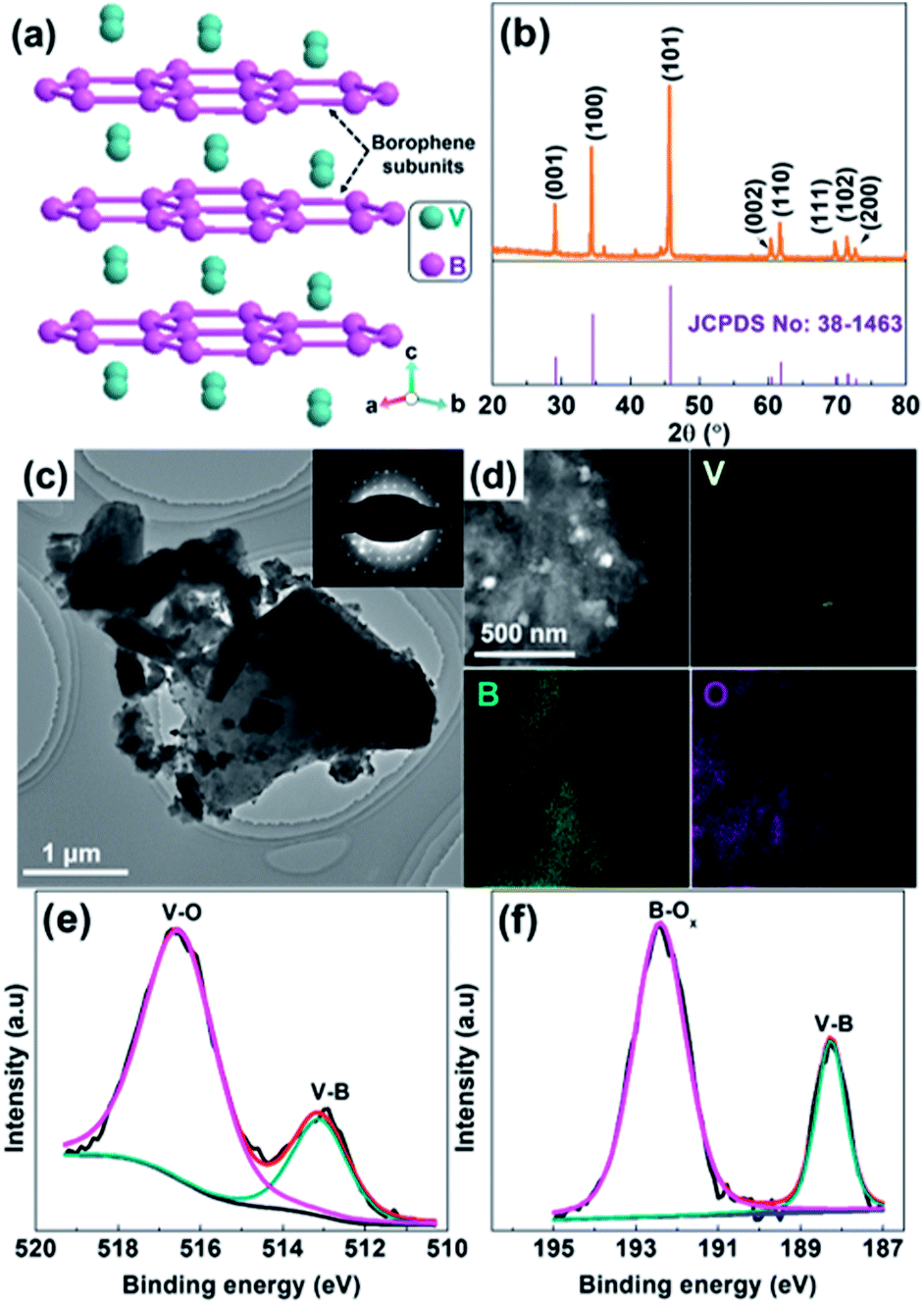 | ||
| Fig. 1 (a) The crystal structures of VB2. (b) XRD pattern of as-received VB2. (c) TEM image of VB2 and corresponding SAED pattern (inset). (d) HAADF-STEM image of VB2 and corresponding EDX elemental maps (V, B, and O). (e) V 2p and (f) B 1s XPS spectra of VB2. | ||
Herein, we report that layered metallic VB2 could function as a superior HEC for catalyzing the visible-light-driven photocatalytic HER in an organic dye-based sensitization system for the first time. In an Erythrosin B–triethanolamine (ErB–TEOA) system, VB2 can efficiently catalyze H2 evolution, affording a H2 evolution turnover number (TON) of 40 based on VB2. Moreover, VB2 exhibits much higher activity than V2O5, VS2, VN, and VC in catalyzing photocatalytic H2 evolution.
Fig. 1b shows the XRD pattern of as-received VB2 powder, where the main XRD peaks located at 29.19°, 34.53°, 45.82° 60.53°, 61.84°, 69.83°, 71.60° and 72.81° can be assigned to the (001), (100), (101), (002), (110), (111), (102) and (200) crystal planes of hexagonal VB2 (JCPDS 38-1463),19 respectively. No additional peaks related to impurities such as V and B oxides can be observed, suggesting the high purity of VB2. The transmission electron microscopy (TEM) image shown in Fig. 1c indicates that VB2 possesses a sheet-like structure with a lateral size up to 0.5–5 μm. The corresponding selected area electron diffraction (SAED) pattern (inset of Fig. 1c) shows well-resolved diffraction spots, further manifesting that VB2 has a single-crystal structure. From its high resolution TEM (HRTEM) image (Fig. S1, ESI†), the observed interplanar distance of the lattice fringes is 0.260 nm, which corresponds to the (100) planes of VB2 nanoparticles. Furthermore, the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image of VB2 and the corresponding energy-dispersive X-ray (EDX) elemental maps (Fig. 1d) further demonstrate that VB2 is mainly composed of V and B elements. The presence of O element is probably due to the surface oxidation of VB2 since no characteristic diffraction peaks of V and B oxides can be observed in its XRD pattern. The Brunauer–Emmett–Teller (BET) specific surface areas, average pore size, and total pore volume for VB2 were determined to be 9.3 m2 g−1, 5.24 nm, and 0.012 cm3 g−1, respectively (Fig. S2, ESI†). In addition, VB2 is a black powder and its UV-vis diffusion reflectance spectrum (UV-vis DRS) exhibits a strong light absorption from 300 to 800 nm but without an obvious absorption edge (Fig. S3, ESI†), which implies its metallic character.17 Survey X-ray photoelectron spectroscopy (XPS) reveals that VB2 consists of V, B and O elements (Fig. S4, ESI†). The V 2p envelope (Fig. 1e) can be fitted into two main peaks with different chemical environments. The V 2p peak at 513.2 eV is assigned to V–B, while the high binding energy peak at 516.6 eV is attributed to surface V–O species.19–21 Accordingly, the fitting of the B 1s XPS spectrum also shows the presence of both V–B (188.3 eV) and surface B–Ox species (192.4 eV) (Fig. 1f). The presence of V–O and B–Ox species is a result of the surface oxidation of VB2 when exposed to air and will passivate VB2 from further oxidation.
The catalytic activity of VB2 for photochemical H2 evolution was evaluated in a dye-sensitized system using xanthene dyes as the photosensitizer and triethanolamine (TEOA) as the electron donor under visible light. Control experiments indicated that no H2 was evolved in the absence of light or any of the other components. As shown in Fig. 2a, VB2 can efficiently catalyze photocatalytic H2 evolution with various xanthene dyes including Erythrosin B (ErB), Eosin Y (EY), Rose Bengal (RB), Fluorescein sodium (FS), and Rhodamine B (RhB), and ErB outperforms the others in photosensitizing VB2 for H2 evolution. The activity of dye/VB2 systems follows an order of ErB > EY ≈ FS > RB ≫ RhB. The best performance obtained with ErB as the photosensitizer can be attributable to the heavy-atom effect of I substituents in the xanthene ring, which can improve the quantum yield of long-lived triplet states compared to their light-atom-substituted counterparts.22 The pH value of the TEOA solution was also found to significantly influence the photocatalytic activity of the ErB/VB2/TEOA system, and the activity of H2 evolution reaches the highest at pH 11 (Fig. S5, ESI†). The decrease in activity at much higher pH values (>11) is likely due to the lower H+ concentration in solution and the fact that H2 evolution becomes less favorable with increasing pH values.23 In contrast, decreasing the pH value from 11 to 9 leads to much lower H2 evolution activity because the decomposition of protonated TEOA (TEOA+) becomes unfavorable, which will diminish the ability of TEOA as an electron donor.
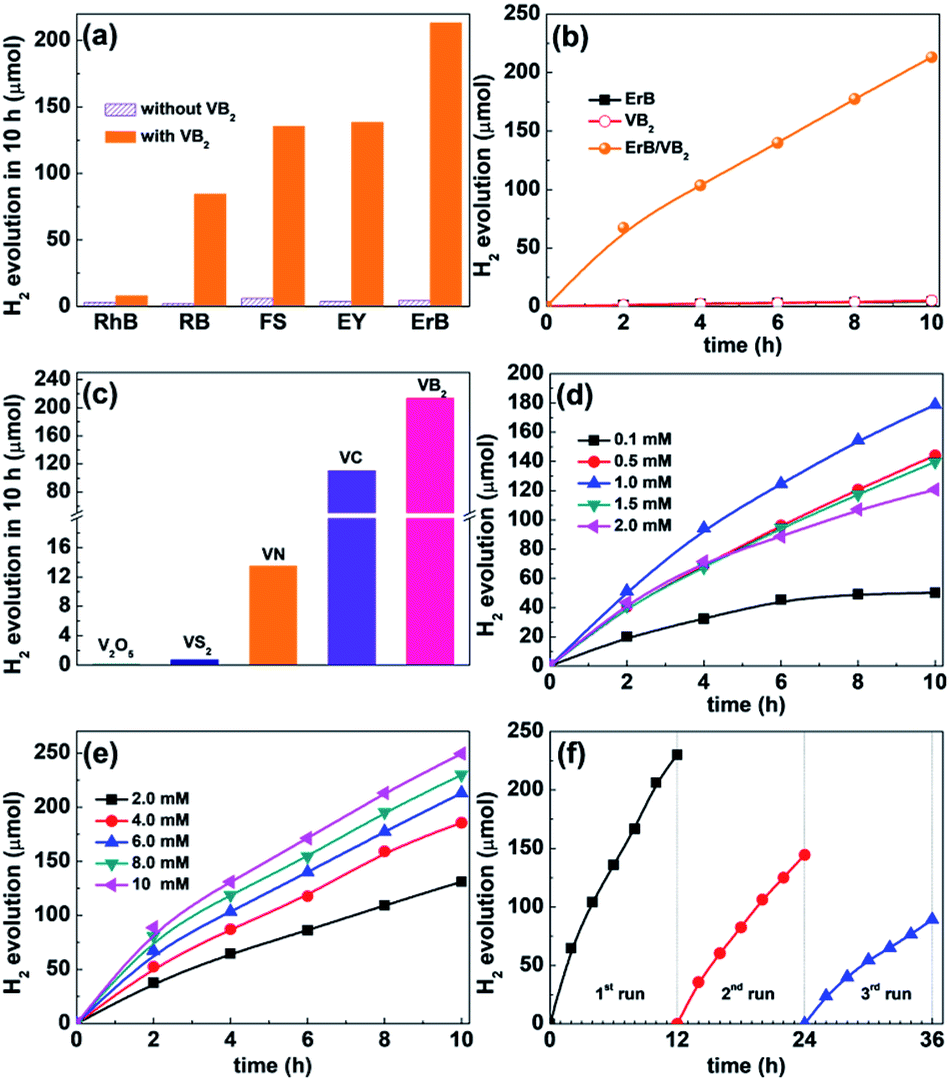 | ||
| Fig. 2 (a) H2 evolution from TEOA solution (10 vol%, 25 mL, pH 11) containing 6.0 mM VB2 as the catalyst and various xanthene dyes (1.0 mM) as the photosensitizers. (b) Time courses of H2 evolution from TEOA solution (10 vol%, 25 mL, pH 11) containing VB2 (6.0 mM) alone, ErB (1.0 mM) alone, and both ErB (1.0 mM) and VB2 (6.0 mM). (c) Comparison of the H2 evolution activity of V-base materials (6.0 mM) in the ErB (1.0 mM)–TEOA (10 vol%, 25 mL, pH 11) system under visible light irradiation. (d) H2 evolution from systems containing 4.0 mM VB2 and different concentrations of ErB. (e) H2 evolution from TEOA solution (10 vol%, 25 mL, pH 11) containing 1.0 mM ErB and different concentrations of VB2. (f) Stability of VB2 (6 mM) for catalyzing H2 evolution from TEOA solution (10 vol%, 25 mL, pH 11) containing ErB (1 mM). Light source: white LED lamp (380 nm ≤ λ ≤ 780 nm). | ||
Fig. 2b presents the time course of H2 evolution in the ErB/VB2 system under visible light irradiation, including those of VB2 and ErB systems for comparison. It is found that the ErB/VB2 system can rapidly produce H2 gas with a high initial H2 evolution rate of 34 μmol h−1. The total amount of H2 evolved from this system is 213.1 μmol in a 10 h reaction, and the calculated turnover numbers (TONs) of H2 evolution are about 17 and 7 based on ErB and VB2, respectively, confirming that the H2 evolution reaction in the ErB/VB2 system proceeds photocatalytically.17 In contrast, the ErB system in the absence of VB2 produces only a trace amount of H2 (4.3 μmol in 10 h) along with rapid degradation of ErB, while VB2 alone also shows negligible photocatalytic H2 evolution activity (5.1 μmol in 10 h), although it has strong absorption toward visible light. These results clearly indicate that VB2 could function as an efficient HEC in a dye-sensitized system for catalyzing H2 evolution under visible light, but its activity is not comparable to that of most recently reported noble-metal-free cocatalysts in both dye sensitized and semiconductor-based photocatalytic H2 evolution systems, probably due to its large size and thus limited active sites (Table S1†). In addition, when Ru(bpy)2Cl2 was used as a photosensitizer, VB2 also shows a substantial catalytic activity for H2 evolution in the presence of ascorbic acid as the sacrificial donor (Fig. S6, ESI†) under visible light. Furthermore, it was also found that VB2 can efficiently catalyze photocatalytic H2 evolution when integrated with inorganic semiconductors such as CdS and TiO2 (Fig. S7, ESI†) under visible and UV-visible light, respectively, again verifying the versatility of VB2 as an active HEC for solar photocatalytic H2 evolution.
Most recently, it has been reported that various V-based materials such as V2O5, VS2, VN, and VC could function as efficient (co)catalysts for electrocatalytic and photocatalytic H2 evolution.12,16,17,24 For comparison, the catalytic H2 evolution activity of these V-based cocatalysts (XRD patterns shown in Fig. S8, ESI†) was tested and compared with that of VB2 under the same reaction conditions. As shown in Fig. 2c, among the V-based cocatalysts tested, VB2 exhibits the highest efficiency for photocatalytic H2 evolution in the ErB–TEOA system. Typically, VB2 shows 2 and 16 times higher activity than metallic VC and VN, respectively, while V2O5 and VS2 are almost inactive for photocatalytic H2 evolution. In addition, the catalytic activity of VB2 was also compared with that of the most commonly studied transition metal diborides such as Mo and W borides and the obtained results are provided in Fig. S9, ESI.† Among the borides tested, VB2 outperforms Mo2B5 and WB in catalyzing H2 evolution in ErB–TEOA solution under visible light.
Furthermore, the effect of the concentrations of ErB and VB2 on the photocatalytic H2 evolution activity of the ErB/VB2 system was investigated. As shown in Fig. 2d, at a fixed VB2 concentration (4.0 mM) the amount of H2 evolved and the system lifetime increase with the concentration of ErB and reach a maximum at 1.0 mM ErB, corresponding to a TON of 14.3 based on ErB, which suggests that at this value of [ErB] the system becomes limited by the concentration of VB2.23 Further increasing the concentration of ErB will lead to a decrease in the activity and TON probably due to the severe light-shield effect caused by excessive ErB present in solution.22 Notably, at 0.1 mM ErB, the highest TON for H2 evolution is found to be as high as 40.1 based on ErB (Fig. S10, ESI†). On the other hand, as [VB2] is increased at fixed [ErB] (Fig. 2e), the initial rate of H2 evolution has a first-order dependence on [VB2] (Fig. S11, ESI†) and the amount of H2 produced monotonously increases with the concentration of VB2 in a 10 h reaction. At 2 mM VB2, the highest TON of H2 evolution achieved in the ErB/VC system is 5.2 after a 10 h reaction.
The catalytic stability of VB2 for photocatalytic H2 evolution in the ErB–TEOA system was then evaluated by performing three cycles of H2 evolution reaction under continuous light irradiation for 36 h. As shown in Fig. 2f, during the first 12 h reaction, although the ErB/VB2 system shows a high initial H2 evolution activity, the activity is gradually decreased with reaction time, which might be caused by the degradation of ErB and deactivation of VB2. In order to clarify these hypotheses, the UV-vis absorption spectra of ErB during the photocatalytic H2 evolution reaction were first monitored, as shown in Fig. S12, ESI.† As expected, the results show that ErB is not stable and degrades over time, which partially accounts for the observed decrease in H2 evolution. On the other hand, after the first 12 h reaction, the used VB2 was recovered by centrifugation, washed with water, and then redispersed in a reaction solution containing fresh ErB and TEOA for the subsequent H2 evolution cycles. It is found that the H2 evolution activity of the ErB/VB2 system could only be partially revived in the subsequent cyclic reaction even with fresh ErB and TEOA added. This result suggests that VB2 is not very stable during long-term photocatalytic H2 evolution. The used VB2 catalyst was characterized. As shown in Fig. S13, ESI,† although all the characteristic diffraction peaks ascribed to VB2 can be observed in the XRD pattern of used VB2, these peaks are slightly shifted to high 2θ values and their intensities become lower. These results indicate that the bulk phase structure of VB2 almost keeps unchanged, while the surfaces of VB2 might undergo oxidization to form amorphous V and/or B oxides. This hypothesis was confirmed by XPS analysis (Fig. S14, ESI†), revealing that the V–B species totally vanishes and only V–O could be observed in the XPS spectrum of V 2p. In addition, TEM and HAADF-STEM analyses (Fig. S15, ESI†) indicate that the surface of VB2 was covered by 30–40 nm thick amorphous oxides. To further exclude the possibility that the VOx or BOx species on the surfaces of VB2 may act as active sites, a series of air-annealed VB2 samples obtained by annealing VB2 at different temperatures (XRD patterns shown in Fig S16a, ESI†) with increased surface VOx or BOx species were also tested for the photocatalytic HER in the ErB–TEOA system (Fig S16b, ESI†). It is found that the activities of all the a-VB2 samples are lower than that of unannealed VB2 and decrease with increasing annealing temperature, clearly revealing that the surface VOx or BOx species cannot act as the active sites for the photocatalytic HER. These results clearly indicate that the decrease in catalytic stability is due to the surface oxidation of VB2 during the photocatalytic H2 evolution and catalyst recovery processes.
The photoinduced electron transfer and the photocatalytic H2 evolution mechanism in the ErB/VB2 system were explored by photoluminescence (PL) spectroscopy. As shown in Fig. 3a and b, it is found that the PL emission of ErB (excited at 480 nm) could be either reductively quenched by TEOA or oxidatively quenched by VB2. These results suggest that when both TEOA and VB2 cocatalysts are present in the system, reductive and oxidative quenching may simultaneously occur and compete with each other in the electron transfer processes for H2 evolution from the excited state of ErB.17,22,23 However, the oxidative quenching of excited ErB by VB2 would dominate because of the much larger kq (9.17 × 1011 M−1 s−1) for oxidative quenching compared to kq (8.57 × 109 M−1 s−1) for reductive quenching by TEOA. Therefore, it could be proposed that the photocatalytic H2 evolution in the ErB/VB2/TEOA system most likely proceeds via an oxidative quenching mechanism.17 Upon visible light irradiation, ErB absorbs visible light photons to form excited state ErB*, which was then oxidatively quenched by directly transferring electrons to the VB2 cocatalyst, where H+/H2O is reduced to form H2 gas. At the same time, the oxidative ErB cation radicals are regenerated to ground state ErB by TEOA as an electron donor, thereby completing the catalytic cycles. The cocatalyst role of VB2 is further confirmed by electrochemical measurements. As shown in the polarization curves (Fig. 3c), the observed cathodic current in the range of 0 to −0.6 V versus RHE can be ascribed to the H2 evolution. In comparting to pristine carbon paper (CP), VB2 loaded CP (VB2/CP) shows a much higher cathodic current. This result indicates that VB2 indeed is a highly active cocatalyst for catalyzing photocatalytic H2 evolution. Moreover, VB2/CP exhibits a much smaller semicircle diameter than CP (Fig. 3d), suggesting an obviously reduced charge transfer resistance due to the metallic character of VB2,13 which is favorable for accepting electrons from photoexcited ErB, in turn enhancing the charge separation efficiency and thus the photocatalytic H2 evolution performance of the ErB/VB2 system under visible light irradiation.
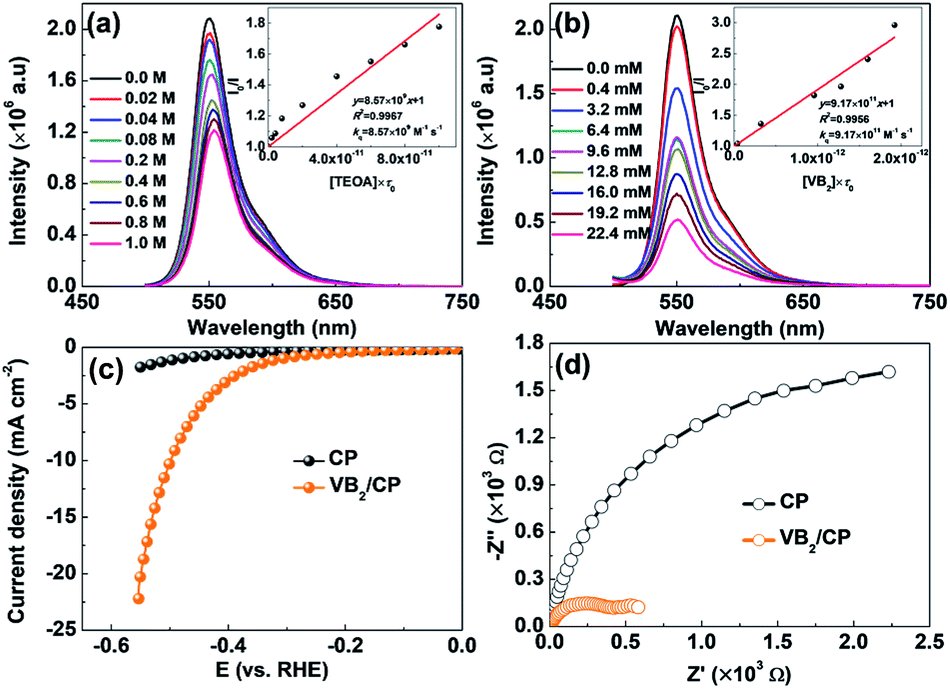 | ||
| Fig. 3 (a) PL mission quenching of ErB solution (10 μM) with TEOA and corresponding Stern–Volmer plot. (b) PL mission quenching of ErB solution (10 μM) with VB2 and corresponding Stern–Volmer plot. (c) Catalytic HER polarization curves of carbon paper (CP) and VB2 loaded carbon paper (VB2/CP) recorded in Na2SO4 (0.5 M) containing TEOA (10 vol%, pH 11) at a scan rate of 10 mV s−1. (d) EIS Nyquist plots of CP and VB2/CP recorded in Na2SO4 (0.5 M) containing TEOA (10 vol%, pH 11). Light source: white LED lamp (380 nm ≤ λ ≤ 780 nm). | ||
In summary, metallic VB2 has been identified as an efficient cocatalyst for catalyzing photocatalytic H2 evolution in a dye-sensitized system under visible light irradiation. VB2 was also found to catalyze the H2 evolution reaction more efficiently than various potential V-based cocatalysts such as V2O5, VS2, VN, and VC. This work not only confirms the feasibility of using metallic and earth-abundant VB2 as a H2 evolution cocatalyst in molecular systems, but also could be expected to stimulate research on employing and developing V-based materials in large-scale light energy conversion devices for practical applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (21763001 and 21463001), the Natural Science Foundation of Ningxia Province (2018AAC02011), the West Light Foundation of the Chinese Academy of Sciences (XAB2018AW13), the Foundation of Training Program for Young and Middle-aged Talents of State Ethnic Affairs Commission of China, and the Foundation of Key Laboratory of Electrochemical Energy Conversion Technology and Application, North Minzu University.Notes and references
- Z. Q. Cao, H. B. Hu, M. Z. Wu, C. J. Tu, D. W. Zhang and Z. Q. Wu, Sustainable Energy Fuels, 2019, 3, 2409 RSC.
- S. T. Wu, X. W. Shi and M. S. Zhu, Sustainable Energy Fuels, 2019, 3, 1461 RSC.
- X. Chen, S. Shen, L. Guo and S. Mao, Chem. Rev., 2010, 110, 6503 CrossRef CAS.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900 CrossRef CAS.
- S. X. Min, Y. Xue, F. Wang, Z. G. Zhang and H. T. Zhu, Chem. Commun., 2019, 55, 10631 RSC.
- X. Zong, H. Yan, G. Wu, G. Ma, F. Wen, L. Wang and C. Li, J. Am. Chem. Soc., 2008, 130, 7176 CrossRef CAS.
- M. L. Liu, B. B. Chen, R. S. Li, C. M. Li, H. Y. Zou and C. Z. Huang, ACS Sustainable Chem. Eng., 2017, 5, 4154 CrossRef CAS.
- A. Indra, A. Acharjya, P. W. Menezes, C. Merschjann, D. Hollmann, M. Schwarze, M. Aktas, A. Friedrich, S. Lochbrunner, A. Thomas and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 1653 CrossRef CAS.
- Q. S. Gao, W. B. Zhong, Z. P. Shi, L. C. Yang and Y. Tang, Adv. Mater., 2019, 31, 1802880 CrossRef.
- R. Godin, Y. O. Wang, M. A. Zwijnenburg, J. W. Tang and J. R. Durrant, J. Am. Chem. Soc., 2017, 5, 4154 Search PubMed.
- S. Meyer, A. V. Nikiforov, I. M. Petrushina, K. Kohler, E. Christensen, J. Jensen and N. Bjerrum, Int. J. Hydrogen Energy, 2015, 40, 2905 CrossRef CAS.
- Y. Zhong, X. H. Xia, F. Shi, J. Y. Zhan, J. P. Tu and H. J. Fan, Adv. Sci., 2016, 3, 1500286 CrossRef.
- F. F. Guo, Y. Y. Wu, X. Ai, H. Chen, G.-D. Li, W. Chen and X. X. Zou, Chem. Commun., 2019, 55, 8627 RSC.
- J. Yuan, J. Wu, W. J. Hardy, P. Loya, M. Lou, Y. Yang, S. Najma ei, M. Jiang, F. Qin, K. Keyshar, H. Ji, W. Gao, J. Bao, J. Kono, D. Natelson, P. M. Ajayan and J. Lou, Adv. Mater., 2015, 27, 5605 CrossRef CAS.
- M. Yan, X. Pan, P. Wang, F. Chen, L. He, G. Jiang, J. Wang, J. Z. Liu, X. Xu, X. Liao, J. Yang and L. Mai, Nano Lett., 2017, 17, 4109 CrossRef CAS.
- B. B. Bin, G. S. Tang, H. F. Liang, Z. B. Qi, D. F. Zhang, W. S. Hu, H. Shen and Z. C. Wang, Electrochem. Commun., 2018, 93, 166 CrossRef.
- L. Tian, S. X. Min, Y. G. Lei, S. S. Chen and F. Wang, Chem. Commun., 2019, 55, 6870 RSC.
- M. M. Shao, Y. F. Shao, S. J. Ding, J. W. Wang, J. C. Xu, Y. J. Qu, X. W. Zhong, X. M. Chen, W. Fai, N. Wang, B. M. Xu, X. Q. Shi, X. E. Wang and H. Pan, Appl. Catal., B, 2018, 237, 295 CrossRef CAS.
- L. Tian, S. X. Min and F. Wang, Appl. Catal., B, 2019, 259, 118029 CrossRef CAS.
- V. Mazánek, H. Nahdi, J. Luxa, Z. Sofer and M. Pumera, Nanoscale, 2018, 10, 11544 RSC.
- P. Wang, R. Kumar, E. M. Sankaran, X. T. Qi, X. Y. Zhang, D. Popov, A. L. Cornelius, B. S. Li, Y. S. Zhao and L. P. Wang, Inorg. Chem., 2018, 57, 1096 CrossRef CAS.
- J. H. Hou, Y. G. Lei, F. Wang, X. H. Ma, S. X. Min, Z. L. Jin and J. Xu, Int. J. Hydrogen Energy, 2017, 42, 11118 CrossRef CAS.
- Z. J. Han, W. R. McNamara, M.-S. Eum, P. L. Holland and R. Eisenberg, Angew. Chem., Int. Ed., 2012, 51, 1667 CrossRef CAS.
- J. Yuan, J. Wu, W. J. Hardy, P. Loya, M. Lou, Y. Yang, S. Najmaei, M. Jiang, F. Qin, K. Keyshar, H. Ji, W. Gao, J. Bao, J. Kono, D. Natelson, P. M. Ajayan and J. Lou, Adv. Mater., 2015, 27, 5605 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional data. See DOI: 10.1039/c9se00820a |
| This journal is © The Royal Society of Chemistry 2020 |