
Intercalation and flexibility chemistries of soft layered materials
Yuya
Oaki

Department of Applied Chemistry, Faculty of Science and Technology, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama 223-8522, Japan. E-mail: oakiyuya@applc.keio.ac.jp
First published on 29th September 2020
Abstract
Layered materials, alternate stackings of two or more components, are found in a wide range of scales. Chemists can design and synthesize layered structures containing functional units. The soft-type layered materials exhibit characteristic dynamic functions originating from two-dimensional (2D) anisotropy and structure flexibility. This feature article focuses on “intercalation” and “flexibility” as two new perspectives for designing soft layered materials. Intercalation of guests is a characteristic approach for design of layered structures. Flexibility is an important factor to control the dynamic functions of the layered structures. As a model case, the intercalation-induced tunable stimuli-responsive color-change properties of layered polydiacetylene (PDA) are introduced to study the impact of the intercalation and flexibility on the dynamic functions. Recently, layered materials have drastically expanded the research area from conventional rigid inorganic compounds to new self-assembled nanostructures consisting of organic components, such as polymers, metal–organic frameworks, and covalent-organic frameworks. These new layered architectures have potentials for exhibiting dynamic functions originating from the structure flexibility beyond the static properties originating from classical intercalation and host–guest chemistries. Therefore, intercalation and flexibility chemistries of soft layered materials are regarded as new perspectives for design of advanced dynamic functional materials.
 Yuya Oaki | Yuya Oaki is an Associate Professor of Department of Applied Chemistry, Keio University, Japan. He received his PhD in 2006 from Keio University under the supervision of Prof. Hiroaki Imai and then joined the group of Prof. Takashi Kato at The University of Tokyo as a postdoctoral fellow. His research interest is layered materials, 2D materials, and conjugated polymers for their energy and sensing applications. He also studies materials informatics to accelerate these works. He was awarded The Young Scientists’ Prize of The Commendation for Science and Technology by the Minister of Education, Culture, Sports, Science and Technology (2017). |
1. Introduction
Two-dimensional (2D) materials are one of the recent hot topics in chemistry, physics, and materials science.1–12 The ultrathin nanostructures, such as monolayers and few-layers, exhibit the unique properties originating from the characteristic architectures.13–21 In recent years, 2D materials based on organic compounds and organic–inorganic composites, such as network polymers, metal–organic frameworks (MOFs), and covalent organic frameworks (COFs), have attracted much interest as new families of 2D materials.22–31 Therefore, new perspectives are required to understand the syntheses, structures, and properties of diverse 2D materials. For example, Ariga et al., summarized recent advances about organic ultrathin nanostructures and their potential applications as a new perspective of ‘soft’ 2D nanoarchitectonics.10 However, recent developments of “stacked and layered structures” have not been fully studied in contrast to those of “ultrathin 2D materials.” This feature article focuses on new intercalation and flexible chemistries of ‘soft layered materials’ with dynamic structures and functions. Fig. 1 summarizes the structural advantages of the flexible soft layered materials. Soft layered materials can be defined as layered structures exhibiting molecular motion. The flexibility plays important roles for emergence of the dynamic properties and functions based on the molecular motion. In addition, the 2D anisotropy affords to provide the specific morphologies, such as anisotropic shapes and homogeneous coating on substrates. Molecular design and intercalation are important approaches to control the structures and functions of soft layered materials. Langmuir–Blodgett (LB) films and layer-by-layer assemblies as soft layered materials with dynamic functions are well established and summarized in previous reviews.32,33 This feature article focuses on the other types of soft layered materials.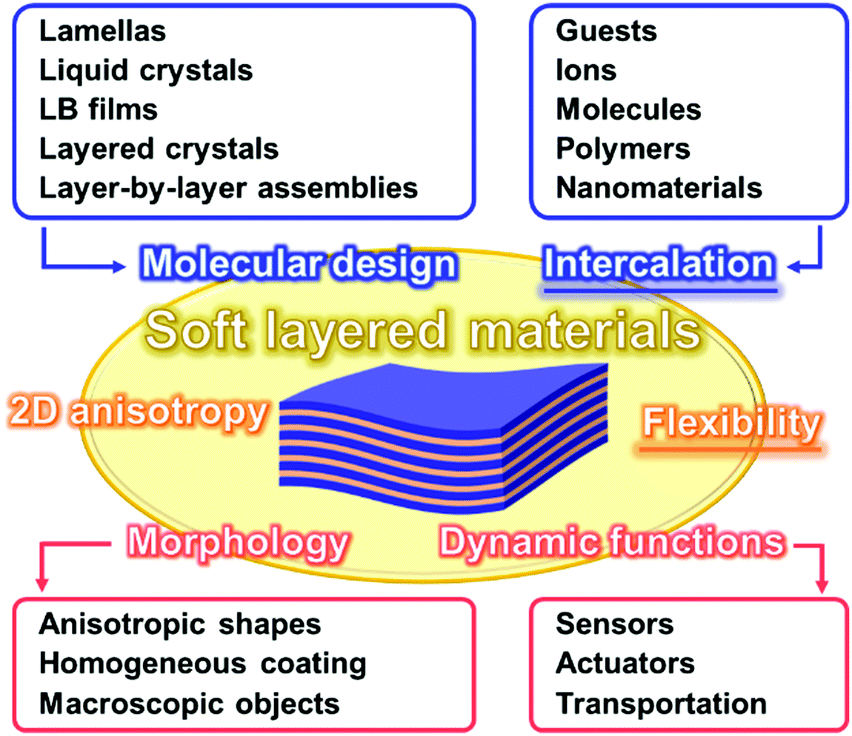 | ||
| Fig. 1 Conceptual illustration of soft layered materials with their characteristic design strategies including molecular design, structures including 2D anisotropy and flexibility, and properties including morphology and dynamic function. | ||
Classical inorganic layered compounds accommodate guests in the interlayer space. The static host–guest chemistry based on intercalation has been well studied for these layered compounds.34,35 The functional guests in the nanoscale space exhibit the characteristic properties and functions different from the bulk states.36–40 However, soft layered materials including intercalation and flexibility chemistries are not reviewed in the previous literatures. Therefore, this feature article focuses on soft layered materials toward design of advanced dynamic functional materials. The intercalated guests and flexibility play important roles for the dynamic properties and applications, such as sensors, actuators, and transportations. Section 2 briefly summarizes the structure advantages of the soft layered materials. Section 3 introduces the nice works about design of functional soft layered materials. Section 4 shows our recent works about functional soft layered materials, such as layered polydiacetylene (PDA) with tuned stimuli-responsive color-change properties by intercalation. Finally, Section 5 mentions the summary and future directions. The present article inspires a wide variety of researchers to develop new molecules and functional materials.
2. Soft and rigid natures of the layered structures
Section 2 focuses on the softness and rigidness of the layered materials. Soft layered materials are mainly found on organic materials, such as lamellar crystal structures, liquid crystals, and layer-by-layer assemblies (Fig. 1).41–45 Although the host–guest chemistry of organic layered materials were studied in a couple of previous works,46–48 the control of the flexibility and emergence of the dynamic functions were not demonstrated. Herein, an intrinsic question is what is structure softness and rigidness in layered materials. Although the soft or rigid is a common sense in general chemistry and materials science, those are now not quantitative factors but qualitative ones based on the experience and intuition by chemists. Here the typical layered compounds are classified into the soft and/or rigid types according to the interactions between the layers (Fig. 2). A number of layered compounds, such as graphite, black phosphorus, and transition-metal chalcogenides, consist of van der Waals interaction. On the other hand, layered compounds, such as transition-metal oxides, clays, and layered-double hydroxides, have the interlayer interaction based on electrostatic interaction. According to general chemistry, the interlayer interaction is assumed to be stronger for layered compounds based on electrostatic interaction and weaker for those based on van der Waals interaction (Fig. 2). Since these interactions are compositive, the soft and rigid natures are tuned depending on each layered material.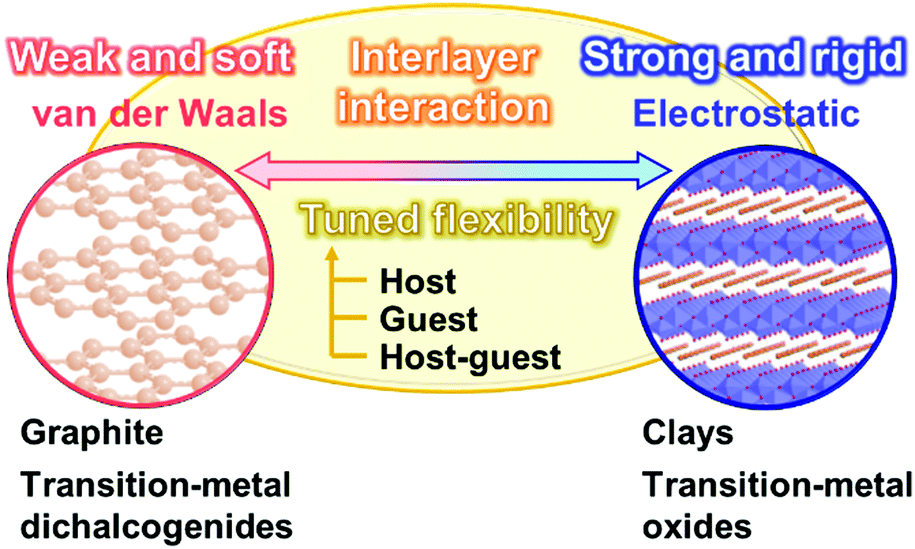 | ||
| Fig. 2 Flexibility of the layered structures based on the interlayer interactions. Typical layered compounds are classified into the soft and rigid types originating from van der Waals and electrostatic interactions between the layers, respectively. | ||
Our group has prepared the layered composites of transition-metal oxide and interlayer organic guest for tuning the flexibility. The exfoliation into the nanosheets is actually promoted by softening the layered structures with intercalation of the organic guests.49–56 The results imply that the flexibility of the layered structures is tuned by the guests. Therefore, the flexibility can be changed and tuned by the structures and types of the hosts, guests, and host–guest combinations. The structure flexibility can be an important factor for dynamic functions of soft layered materials.
3. Impact of flexibility for emergence of dynamic functions from soft layered materials
Section 3 shows three representative studies of soft layered materials with dynamic functions in previous works. These studies nicely applied the flexibility and intercalation for emergence of the dynamic functions as the devices.3.1. Liquid crystals exhibiting transportation properties
Liquid crystal is a typical soft material with dynamic functions. Smectic phase has layered structures consisting of lamellar molecular arrangement. Kato and co-workers have studied the self-organized soft layered structures for anisotropic transportation of ions and electrons (Fig. 3).57–64 Self-organization of the designed rod-like molecules consisting of ionic and nonionic parts forms the layered structures (Fig. 3a). The rigid nonionic part contributes to form the stable host layers. The interlayer flexible ionic part acts as functional units for intercalation and transportation of guest ions.57–63 The flexible interlayer domain exhibited the anisotropic 2D conduction of the intercalated ions, such as lithium ions and ionic liquids (Fig. 3b). In addition, the anisotropic electron conduction was demonstrated by introduction of the π-conjugated moieties in the host nonionic domain.64 These soft layered materials are applied to quasi solid-state electrolyte for lithium-ion battery and solar cells (Fig. 3c).62,65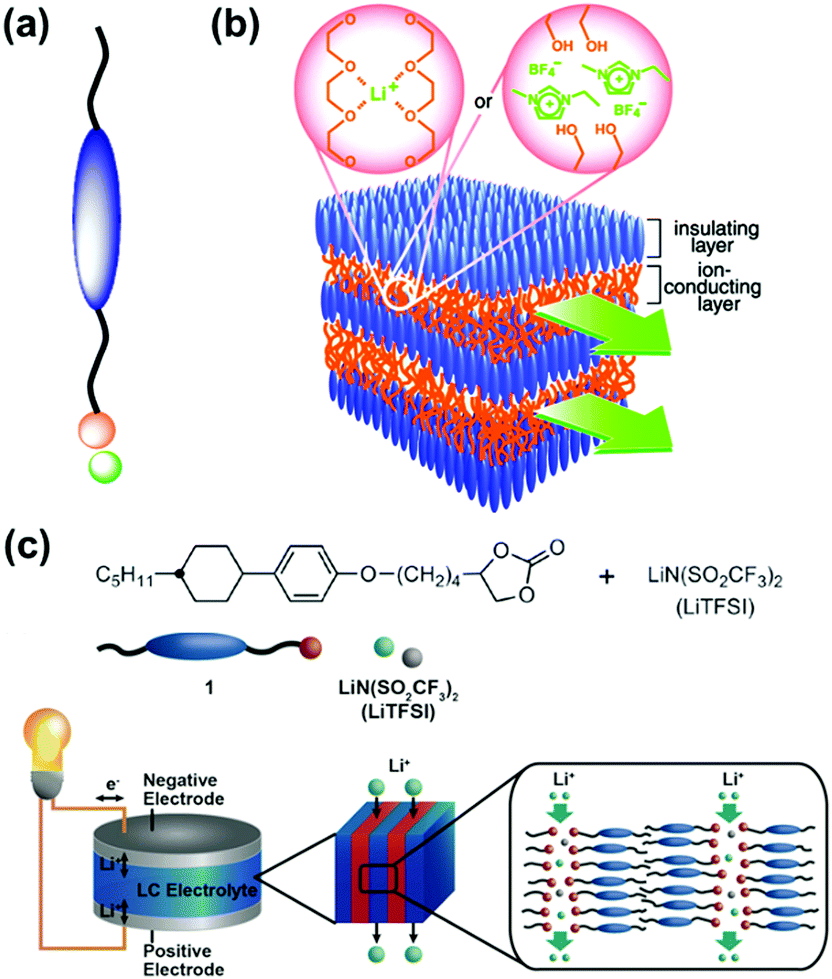 | ||
| Fig. 3 Soft layered materials based on liquid crystals exhibiting transportation properties.57,62 (a) Rod-like molecule with ionic and nonionic parts. (b) Schematic illustration of 2D nanosegregated structures containing poly(ethylene oxide) or ionic-liquid domain for anisotropic ion transportation. Reprinted with permission from the American Association of the Advancement of Science.57 (c) Molecular structures, their self-organized structures, and their application to the electrolyte for quasi solid-state lithium-ion battery.62 Reprinted with permission from Wiley-VCH.62 | ||
These works indicate general design strategies of functional soft layered materials. The designed molecules with ionic and nonionic moieties can provide the segregated layered structures. Although the overall layered structures are flexible, both the soft and rigid domains are coexistent in the layered structures. The more rigid part contributes to generate and preserve the stable layered structures. The more soft interlayer domain plays an important role for the dynamic motion of the molecules exhibiting dynamic functions, such as transportation. Moreover, these liquid-crystalline soft layered materials enable control of the macroscopic orientation and morphology on substrates. These works clearly show the design strategies how to design soft layered materials with anisotropic dynamic function based on the intercalation and flexibility.
3.2. Designer layered organopolysiloxane with photomechanical properties
Organopolysiloxane with layered structures is a good model case to study the importance of the flexibility. Shimojima and co-workers have shown the importance of the flexibility in the layered structures based on organosiloxanes.66–68 A number of the organoalkoxysilanes containing azobenzene moiety were designed and synthesized for generation of the lamellar structures (Fig. 4a).66–68 The structure flexibility can induce reversible cis–trans transitions of the azobenzene moiety and macroscopic motion of the layered material itself. Brinker et al., previously reported the layered structures of organoalkoxysilane 1 containing azobenzene and intermolecular hydrogen-bonding units (Fig. 4a).69 The photochromism of the azobenzene moiety was not observed for the self-organized layered structure derived from the organosilane 1. The strong intermolecular interaction via the hydrogen bonding causes the lack of the flexibility. Shimojima et al., showed the microscopic reversible changes in the basal spacing of the lamellar structures, originating from the cis–trans isomerization, derived from the designed organosilanes 2 and 3.66 However, the macroscopic motion was not observed. These results imply that the cis–trans isomerization can be induced by the weaker intermolecular interaction between the organosilane units. Although layered inorganic compounds with intercalation of the guests containing azobenzene units showed reversible changes of interlayer spacing upon irradiation of UV light,70,71 the macroscopic bending and unbending motions were not observed for these layered composites. Shimojima et al., reported the newly designed organosilanes 4 and 5 having both the –CH2–O–CH2– and –CH2–S–CH2– moieties in the alkyl spacer chain between azobenzene and silane moieties (Fig. 4a and b).68 The longer alkyl chain with –CH2–S–CH2– linker inhibits the close packing and lowers the crystallinity. Therefore, both the reversible changes in the microscopic basal spacing and macroscopic motion were observed on the free-standing film of the soft layered materials derived from the mixture of the organosilanes 4 and 5 (Fig. 4c and d).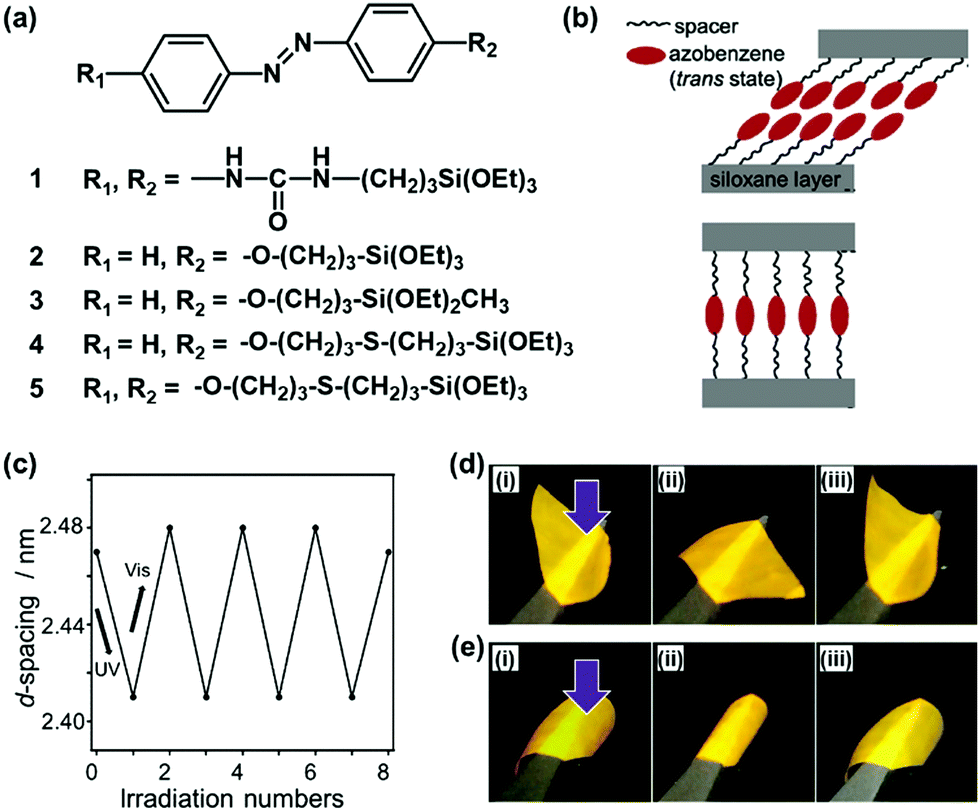 | ||
| Fig. 4 Soft layered materials based on organopolysiloxane with photomechanical properties.66–69 (a) Molecular structures of organosiloxanes 1–5. (b) Schematic illustration of the layered structures derived from the organosiloxanes 4 (upper) and 5 (lower) after hydrolyzation and spin-coating on substrate. (c) Microscopic reversible variation of the interlayer distance, namely d-spacing on the X-ray diffraction pattern, with irradiation of UV and visible lights on the thin-film derived from organosiloxane 4. (d) and (e) Macroscopic bending and unbending behaviors of a free-standing film derived from the mixture of the organosiloxanes 4 and 5 with UV-vis irradiation from the upper (d) and reverse (e) sides. Reprinted with permission from ref. 66. Copyright 2020 American Chemical Society. | ||
These works indicate that the appropriate flexibility is significant for emergence of both the microscopic and macroscopic dynamic motions from the soft layered materials. Although the typical inorganic layered materials are rigid, the flexibility is changed to the more soft by the designed organic spacer in the layers. In addition, the 2D anisotropic structures with tuned flexibility provide the macroscopic free-standing film. This softening strategy is applied to design the other organic–inorganic hybrid layered materials with dynamic functions.
3.3. Layered inorganic materials with flexibility in vertical direction for sensing applications
As discussed in Section 2, inorganic layered compounds consisting of electrostatic interlayer interaction are regarded as the rigid type (Fig. 2). In general, these layered compounds can intercalate guest ions. The intercalation initiates osmotic swelling with introduction of the solvent. The osmotic swelling eventually induces exfoliation into the nanosheets in liquid phase. Lotsch and co-workers applied the swelling behavior as the flexibility in vertical stacking direction to develop photonic sensors for humidity and vapor (Fig. 5a and b).72–75 The exfoliated nanosheets, such as phosphatoantimonic acid and tantalum phosphate, formed the thin films as the host layer several-hundred nanometer in thickness with re-stacking by spin coating (Fig. 5a).73 Then, the nanoparticles were deposited as the guest layer. The alternate stackings of the host nanosheet layers and guest nanoparticles formed the thin films exhibiting the structural colors on substrates (Fig. 5c). The restacked nanosheets as the host layer showed swelling with introduction of water and guest ions (Fig. 5b). This microscopic expansion with swelling yields to macroscopic changes in the thickness of the host layers. Therefore, the visible structural color was changed by the swelling with intercalation of the target molecules, such as water (Fig. 5b and d). The structural color was gradually changed depending on the humidity (Fig. 5d). The sensing devices enabled the colorimetric quantification and real-time imaging of humidity.73–75 In addition, the types of the organic amines and their diffusion behavior were successfully visualized by the structural colors.76,77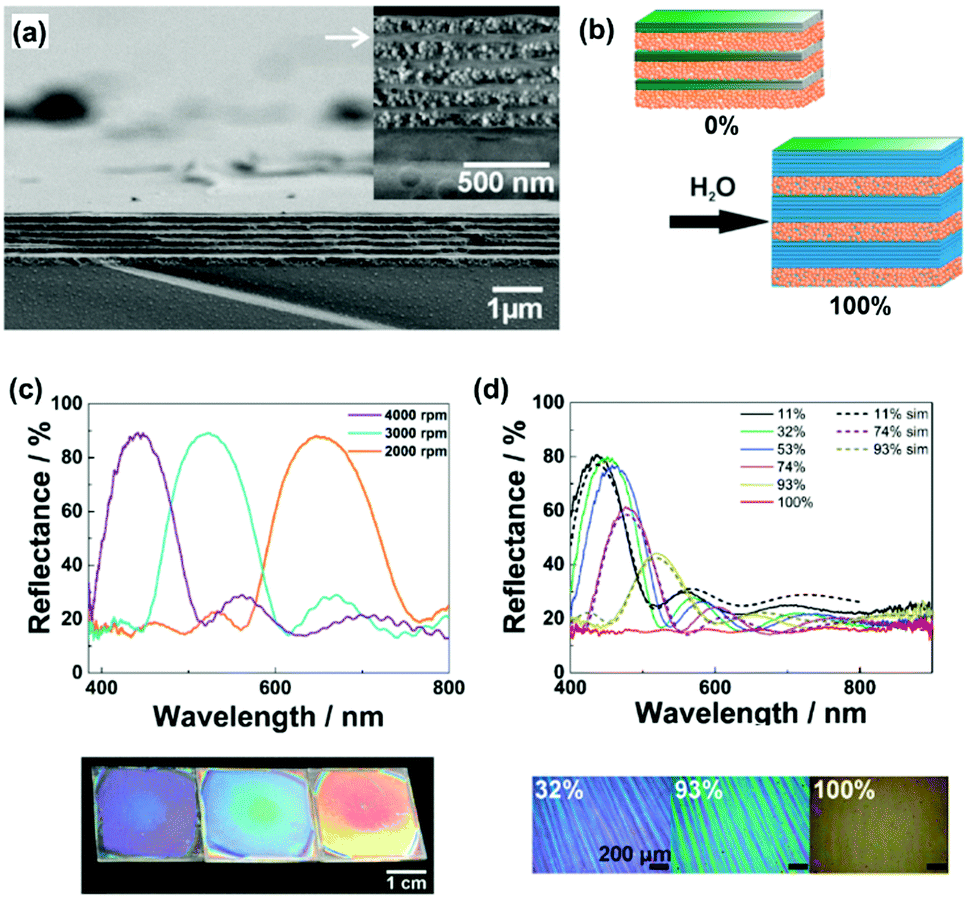 | ||
| Fig. 5 Soft layered materials with flexibility in vertical direction for sensing applications based on the structural color.73 (a) Cross-sectional scanning electron microscopy images of the layered architectures consisting of the exfoliated phosphatoantimonic-acid nanosheets and silica particles fabricated by spin coating at 4000 rpm. (b) Schematic illustration of the humidity sensing mechanism. (c) Optical spectra and photographs of the stackings fabricated by spin-coating at 2000, 3000, and 4000 rpm (from left to right in the photographs), respectively. (d) Experimental (solid spectra) and simulated (dashed spectra) reflectance spectra of the stacking at different humidity. (e) Microscope images at humidity 32%, 93%, and 100%, respectively. Reprinted with permission from Wiley-VCH.73 | ||
These works show that the dynamic motion is induced by the vertical flexibility originating from the swelling behavior with intercalation of the target molecules. The microscopic dynamic motion is transferred to the macroscopic one leading to changes in the structural colors. In addition, these nanosheets-based vertically soft layered materials enable formation of the large-area thin-film devices. The vertical flexibility is also an important strategy to design the dynamic functions using soft layered materials.
4. Soft-layered materials for tunable stimuli-responsive color-change properties
Section 3 reviewed three previous works indicating the impact of the flexibility in the soft layered materials on the dynamic properties. Section 4 shows tunable structure flexibility and dynamic properties of the soft layered materials in our recent works.78–89 The layered conjugated polymers with tunable flexibility enable design of a variety of stimuli-responsive colorimetric sensing materials. The flexibility of the soft layered materials is controlled by intercalation of the guest ions and molecules.4.1. Layered conjugated polymers
Conjugated macromolecules have a variety of properties, such as electronic, photochemical, and electrochemical properties.90–95 These macromolecules show visible and fluorescent colors depending on the effective conjugation length. If the effective conjugation length is changed by an application of external stimuli, the color changes can be applied to detection and sensing. In addition, the tunable stimuli responsivity is required for applications to sensing materials. Our group has focused on conjugated polymers with the layered structure for development of tunable stimuli-responsive color-change materials (Fig. 6).78–89 The structure flexibility of the layered conjugated polymers is designed and controlled by the intercalation of a variety of guests. This section focuses on the flexibility control and its impact.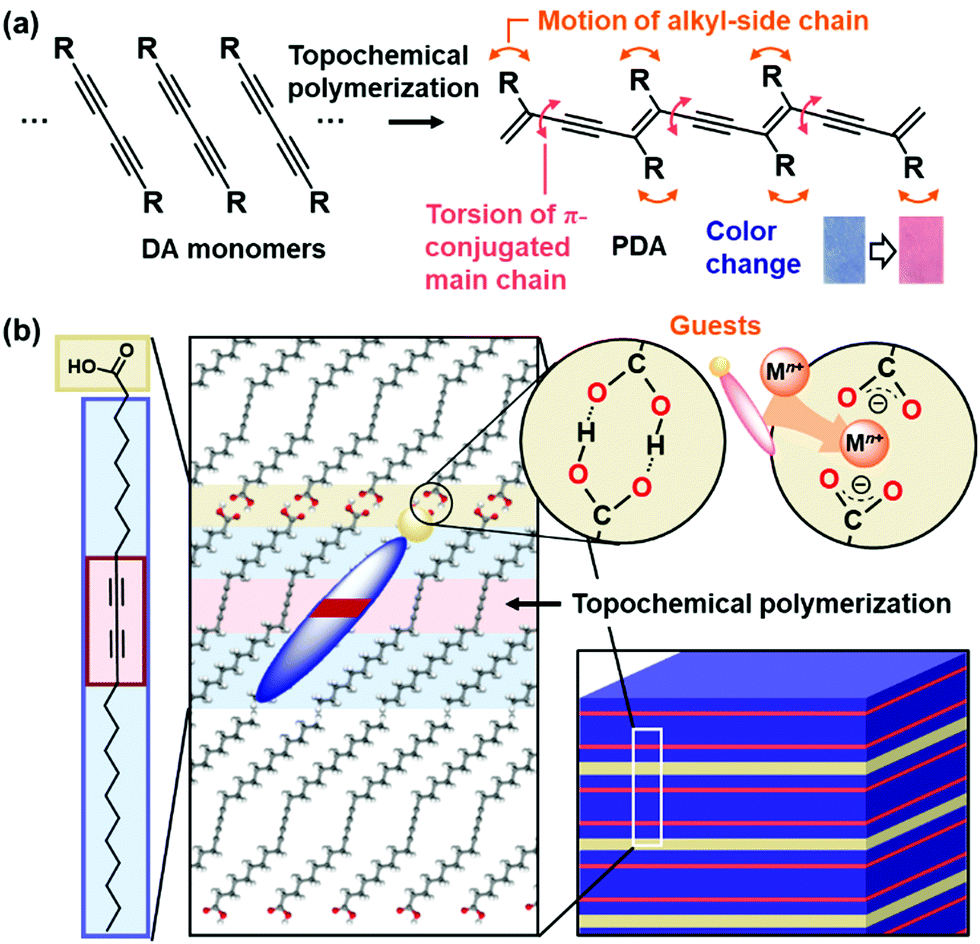 | ||
| Fig. 6 Schematic illustration of the layered PDA. (a) DA monomer, its topochemical polymerization, and molecular motions with the application of external stimuli. (b) Layered crystal structure of PCDA, an amphiphilic DA monomer, intercalation of cations in the interlayer space consisting of the carboxy groups, and topochemical polymerization. | ||
Polydiacetylene (PDA) with ene–yne conjugated main chain is synthesized by polymerization of diacetylene (DA) monomers.96–105 When DA moieties are organized in the condensed state with the distance shorter than 0.5 nm, the topochemical polymerization proceeds with irradiation of UV light and X-ray (Fig. 6a).106 PDA derivatives have color-change properties with application of external stimuli, such as thermal and chemical stresses. As external stimuli induce distortion of the PDA main chain through motion of the side chain, the color change is observed by shortening the effective conjugation length. The color changes have been applied to detection and sensing external stimuli, such as thermal, optical, mechanical, and chemical stresses.96–105 A number of the organized states, such as vesicles, were formed by DA monomers.107–109 In previous works, the designed DA monomers were synthesized for tuning the responsivity and sensing the desired targets.110–123 Kim et al., designed and synthesized a variety of DA monomers to achieve control of the color-change properties and their versatile applications.98,99,101,103,110–112,116,120,121,123 Among a variety of monomers, amphiphilic DA monomer forms the lamellar-like layered structures (Fig. 6b). The alkyl chain length and functional group actually have impacts on the stimuli-responsive color-change properties. Our group has focused on the layered crystal structure of commercially available 10,12-pentacosadiynoic acid (PCDA), an amphiphilic DA monomer, to demonstrate control of stimuli-responsive color-change properties by the intercalation. The flexibility of the layered structures was tuned by the types of the intercalated guests.
4.2. Intercalation chemistry of layered PDA
Two methods, such as intercalation and self-organization methods, were developed for intercalation of guests in the interlayer space consisting of the dimerized carboxy groups (Fig. 7).78–89 The layered composites consisting of the host DA monomers and guests are polymerized with irradiation of UV light. The intercalation and subsequent topochemical polymerization are the important sequence for syntheses of the layered composites based on PDA. If the guests are introduced in the interlayer space of the polymerized DA, the color change from blue to red occurs during the intercalation process. The resultant red-color PDA is not fully used for the sensing. In fact, since the intercalation of the guests itself acts as an external stimulus for PDA, the previous works used this color-change scheme for detection of the target molecules.124,125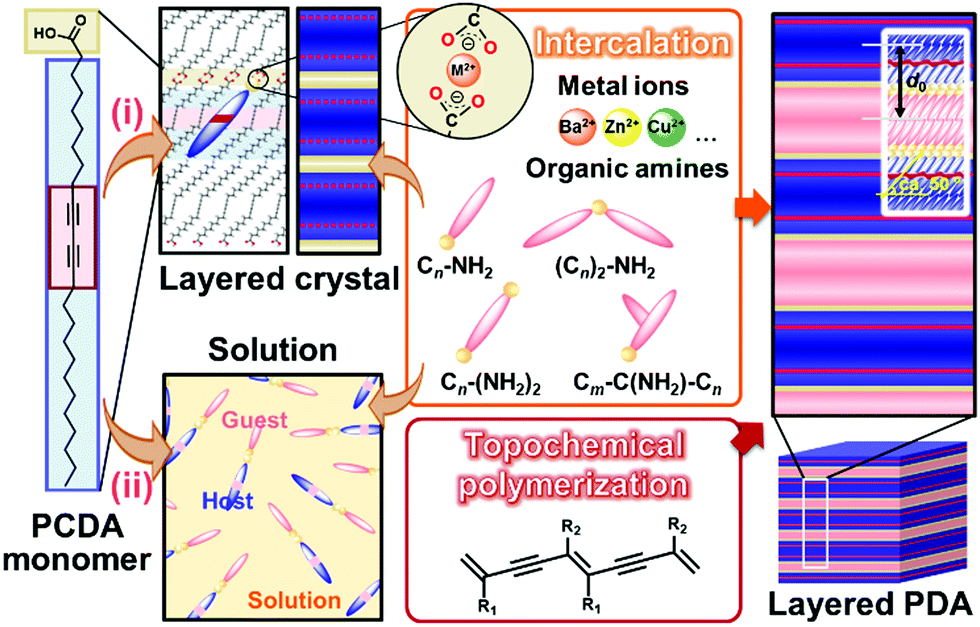 | ||
| Fig. 7 Schematic illustration of the intercalation route (i) and self-organization route (ii) for preparation of the layered composites based on PDA with topochemical polymerization. Reprinted with permission from Wiley-VCH and Elsevier.79,82,85 | ||
The layered PCDA monomer crystal accommodates cationic guests, such as metal cations and organic amines, in the interlayer space consisting of the intermolecular dimerized carboxy groups through the intercalation process (the route (i) in Fig. 7).78–84 The powdered sample and thin coating of the layered monomer crystal were immersed in an aqueous solution containing guests and tetrahydrofuran (THF). The guest ions and molecules were spontaneously introduced in the interlayer space. The guest molecules are introduced through exchange of proton to metal and ammonium cations with softening the layered monomer crystal by THF. However, the types of the intercalated guests were limited for this intercalation route. For example, the bulky amines, such as alkyl amines having the branches, were not intercalated in the interlayer space because of the weaker intermolecular interaction and lower packing density.
The self-organization approach was developed as the more general approach for preparation of the layered composites (the route (ii) in Fig. 7).85,88,89 The precursor solution containing PCDA monomer and organic guests were prepared with organic solvents. The precipitates were obtained before or after evaporation of the solvent. When substrates were immersed in the precursor solution, the thin and homogeneous coating was obtained by dipping. In the self-organization method, the complex between host PCDA monomer and guest amines was formed via the electrostatic interaction in the solution phase.85 Then, the host–guest complexes were organized in the layered structures. Whereas a limited type of the guest molecules is introduced in the interlayer space by the intercalation approach for several days, the self-organization route facilitates formation of the layered composites with a variety of guest amines within a day.
The original PCDA had the interlayer distance (d0) around 4.6 nm corresponding to the bilayer structure. The d0 increased with intercalation of the guests depending on the types (Fig. 8a).79 The original PCDA had the dimerized carboxy group in the interlayer space. The layered composites had the carboxylate ones after introduction of the guest ions and molecules (Fig. 8b). The facts mean that the guests are intercalated in the interlayer space of the PCDA host layer via electrostatic interaction. The normal alkyl amines are accommodated in the interlayer space with the interdigitated state (the right panel in Fig. 7). In this manner, the layered composites of monomer PCDA and guests are prepared by the intercalation and self-organization methods. The resultant layered composites are topochemicaly polymerized with irradiation of UV light.
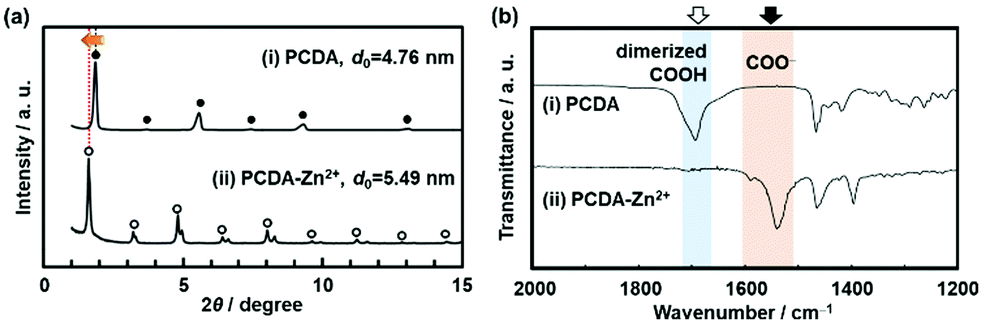 | ||
| Fig. 8 XRD patterns (a) and FT-IR spectra (b) of the layered PCDA (i) and PCDA-Zn2+ (ii) samples. Reprinted with permission from Wiley-VCH.79 | ||
4.3. Stimuli-responsive color-change properties of the layered PDA
The layered PDA shows blue color in the initial state at room temperature (Fig. 9a). The color change was observed by an application of external stimuli, such as heat, force, light, and chemical. The color-change properties, such as color, responsivity, and reversibility, varied by the types of the guests. Our group has used thermo-responsive color-change properties for the quantitative characterization of the responsiveness. PDA derived from PCDA without interlayer guests showed the color transition from blue to red around 65 °C (Fig. 9a).79,80 The reversibility was not observed after cooling. In Fig. 9, the sample was heated to the target temperature (i), then cooled at 25 °C (ii), heated to the next target temperature (iii), and cooled at 25 °C (iv). The heating and subsequent cooling operations were repeated according to the protocol displayed in the top of Fig. 9. When metal ions, such as zinc (Zn2+) and barium (Ba2+) ions, were introduced in the interlayer space, the blue color was gradually changed to red via purple in response to the temperature (Fig. 9b and c).79,80 The red color recovered to the original blue one with cooling. These color-change properties are regarded as the temperature-dependent and gradual type with reversibility. The characteristic color-change properties were observed even when the guest Zn2+ ions were partially introduced, namely occupancy around 30%, in the interlayer space.81 The other types of the metal ions, such as copper (Cu2+) and nickel (Ni2+) ions, induced the irreversible color transition from blue to red at the threshold temperature (Fig. 9d and e).79,80 A couple of the metal ions showed the different color other than red, such as yellow for Ni2+ and green for Cu2+ ion (Fig. 9d and e). The absorption originating from the intercalated metal ions themselves have also an effect on the color. | ||
| Fig. 9 Photographs of PDA and PDA-M2+ with changes of the temperature by the protocol including heating (the upper panels) and subsequent cooling (the lower panels) as shown in the top part. (a) PDA, (b) PDA-Zn2+, (c) PDA-Ba2+, (d) PDA-Cu2+, and (e) PDA-Ni2+. The temperature of the samples in the panels (a)–(e) is referred to the same position in the top part. Reprinted with permission from Wiley-VCH.79 | ||
When the monoamines with the normal alkyl chains (CnH2n+1NH2, Cn-NH2, n = 1–18) were intercalated in the layered PDA, the irreversible color transition was observed (Fig. 10).82,85 The color-change temperature showed the odd–even effects of the alkyl-chain length.85 The temperature-dependent gradual color changes with reversibility were achieved by the introduction of the alkyl diamines (CnH2n(NH2)2, Cn-(NH2)2, n = 4, 6, 8, 12) and p-xylylendiamine (Ph-(CH2NH2)2) (Fig. 11).83 As the alkyl chain length increased, the temperature-dependency and reversibility gradually appeared as shown in the yellow frames in Fig. 11.83
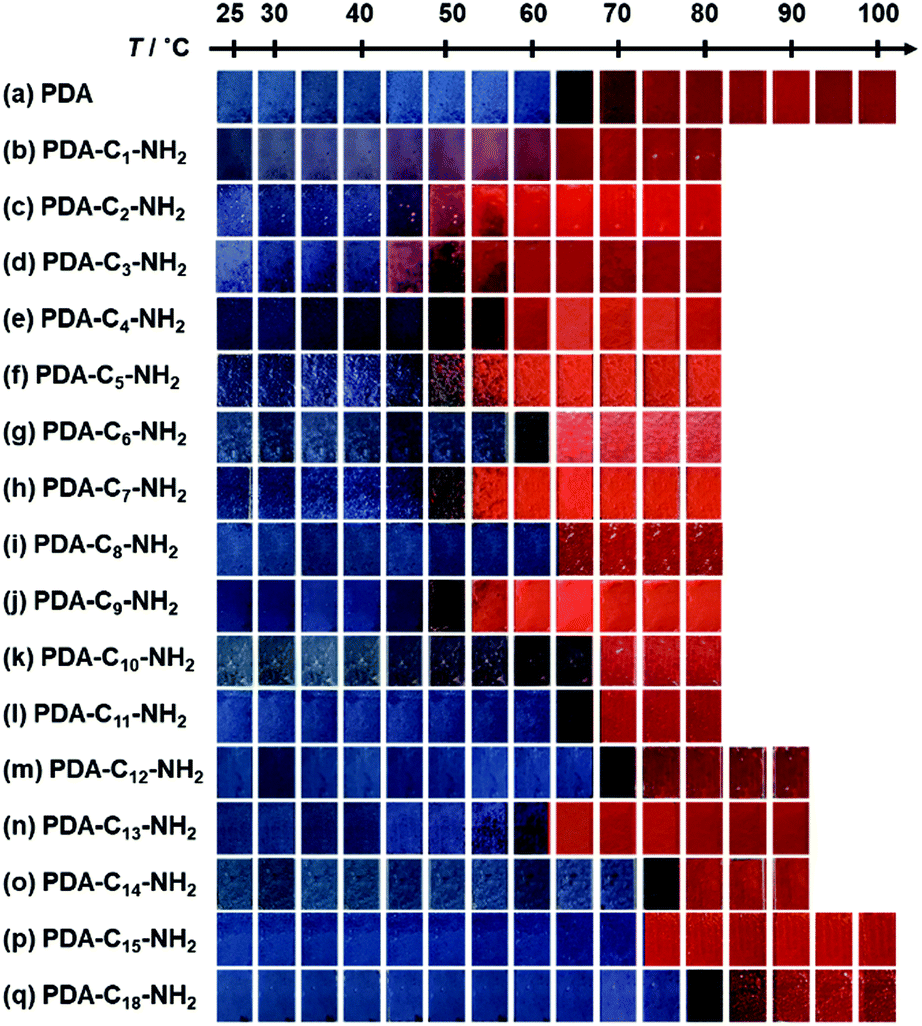 | ||
| Fig. 10 Photographs of the alkyl-amine-intercalated PDA samples with heating. Reprinted with permission from Wiley-VCH.85 | ||
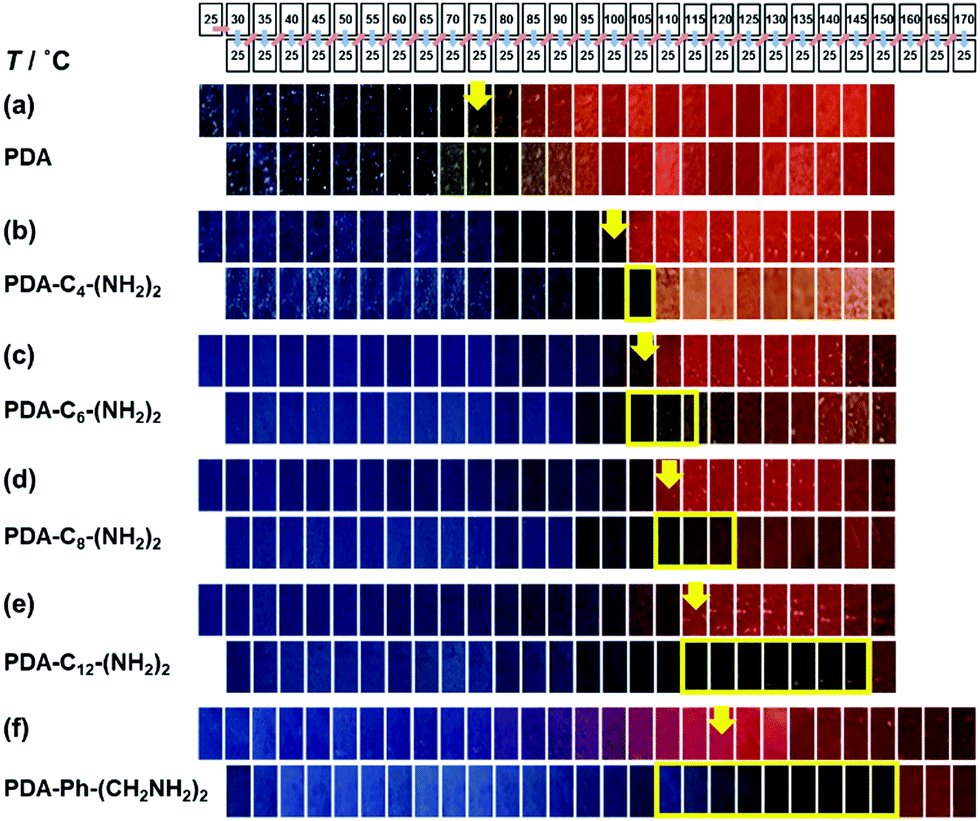 | ||
| Fig. 11 Photographs of the alkly-diamine-intercalated PDA samples with changes of the temperature by the protocol including heating (the upper panels) and subsequent cooling (the lower panels) as shown in the top part. The yellow arrows indicate the color-change temperature to red recognized by the naked eye. The yellow frames in the lower panels show the reversible color change to the original blue. Reprinted with permission from Springer Nature.83 | ||
The temperature-responsive color-changing properties were characterized using the red-color intensity (x) of the photographs by the image analysis based on an international standard (Fig. 12a).82 The x values as a red-color intensity were estimated from the images at each temperature. The color-transition temperature (Ttrs) was defined as the temperature to achieve 0.5Δx (Fig. 12a), where Δx was the differences in the x values in the original and final states. Fig. 12b summarizes the Ttrs of the layered PDA with intercalation of a variety of the guests. Ttrs of the layered PDA with intercalation of dialkyl amines ((Cn)2-NH: H2n+1CnNHCnH2n+1, n = 4, 6, 8, 10, 12) and branched amines (Cm-C(NH2)-Cn: H2m+1CmCH(NH2)CnH2n+1, m = 1, n = 5, and m = 3, n = 3) was lower than that of the PDA-Cn-(NH2)2 with the same carbon number. Ttrs varies in the range of 40–150 °C depending on the types of the interlayer guests (Fig. 12b). These results imply that the color-change properties are controlled by the flexibility of the layered structures originating from the intercalated guests.
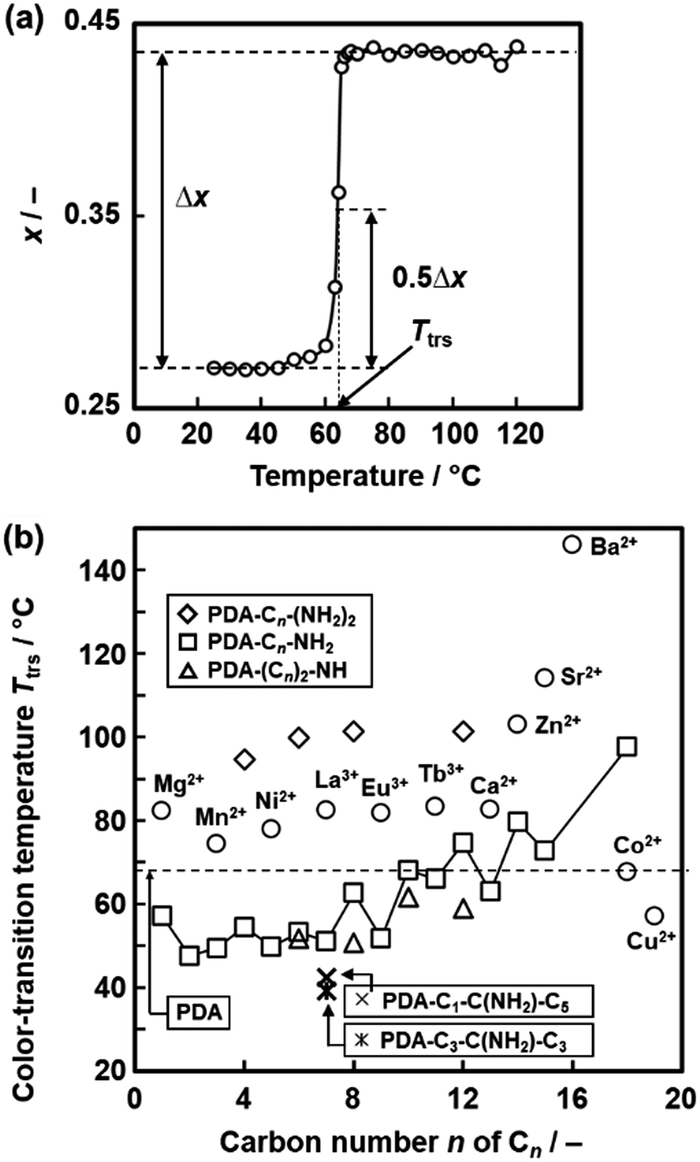 | ||
| Fig. 12 Quantitative characterization of the color-transition temperature (Ttrs). (a) Relationship between the temperature and x value of the PDA-C8-NH2.82 The x and Δx represent the intensity of the red color and the overall color change, respectively. The Ttrs is defined as the temperature to achieve 0.5Δx. Reprinted with permission from Elsevier.82 (b) Summary of the Ttrs values of the layered PDA with intercalation of a variety of the guests.79,82,83,85 Reprinted with permission from Wiley-VCH.85 | ||
4.4. Impact of soft and rigid natures on the stimuli-responsive color-change properties
The more rigid layered structures induce the temperature-dependent reversible color-changing with the higher Ttrs. On the other hand, the more soft layered structures show the irreversible color transition at lower Ttrs.78,82–85 The packing states of the layered structures determine the soft and rigid natures. The host–host, host–guest, and guest–guest interactions have effects on the structure flexibility. The densely packed states of the hosts and guests provide the more rigid layered composites because of the host–guest electrostatic interaction and stronger host–host and guest–guest van der Waals interaction. Therefore, the amine and diamine with the longer alkyl chain form the more rigid layered structure (Fig. 12b). In addition, the densely packed states are achieved by the metal ions. The metal-ion-coordinated DA monomers with the lower tilted angle perpendicular to the layer provide the more densely packed organization state. In contrast, the amines with shorter and branched alkyl chains form the loosely packed states. In these cases, the more soft layered structures are formed via weaker van der Waals interaction. Recently, our group has reported that the flexible polymer guests provide the more soft layered structure with lower Ttrs.88 In this manner, the soft and rigid natures of the layered composites are controlled by the types of the intercalated guests.The color-change processes of the soft and rigid layered structures are explained as follows (Fig. 13).84 PDA with blue color has a stable original conformation. An application of external stimuli, such as heating, induces molecular motion of the PDA side chain and remaining monomer and oligomer themselves at TIs and TIr for the soft and rigid types, respectively (TIs < TIr, Stage I in Fig. 13a and b). Then, the PDA main chain changes to the torsional conformation with shortening the effective conjugation length at TIIs and TIIr (TIIs < TIIr, Stage II in Fig. 13b and c). The layered structures are finally deformed by heating at TIIIs and TIIIr (TIIIs < TIIIr, Stage III in Fig. 13c and d). When the torsional conformation is stabilized by the guests in the more rigid layered structures, the conformation changes gradually proceed with heating at higher temperature (Fig. 13b and c). The distorted conformation can recover to the most stable original one with cooling (Fig. 13b). Therefore, the gradual color-changing behavior with reversibility and higher Ttrs is observed on the more rigid type. In contrast, the more soft types exhibit the molecular motions and distorted conformation with heating at lower temperature. However, the recovery to the original conformation is not achieved by the cooling because of the lack of the stability and rigidity of the original layered structures. In this manner, the temperature-responsive color-change properties are tuned by the soft and rigid natures of the layered composites, namely the types of the guests. Moreover, the present strategy is applied to control the responsivity to the other external stimuli, such as mechanical stress.85,88
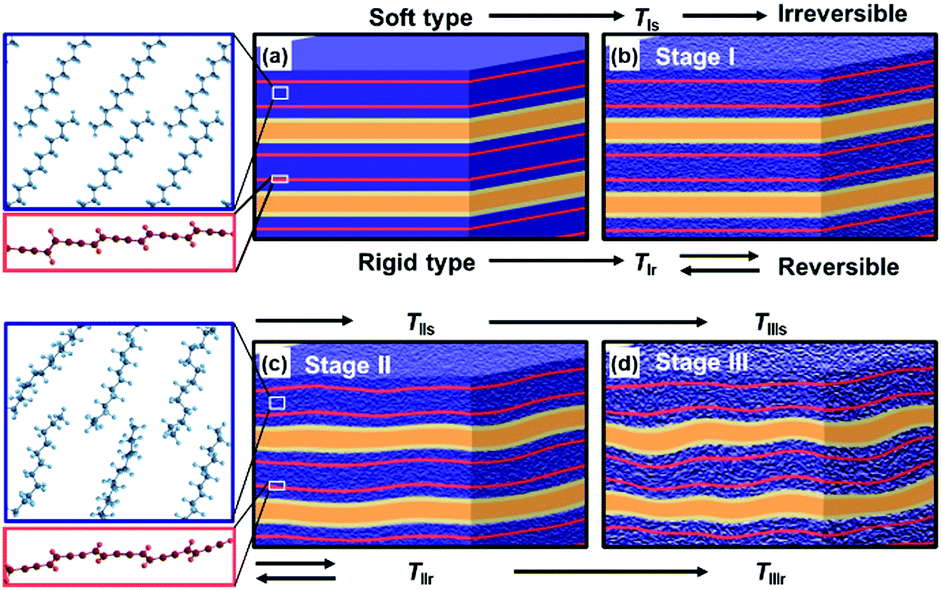 | ||
| Fig. 13 Schematic illustration of the structure changes of the more soft and rigid layered PDA by heating. (a) Initial state of the layered structure. (b) Layered structure including the disordered alkyl domains (density change in the blue part) with thermal motion. (c) Torsion of the PDA main chain (the distortion in the red band) caused by thermal motion of alkyl side chain. (d) Deformation of the layered structure with lowering the crystallinity. The left magnified pannels represent the local structure changes in molecular scale. Reprinted with permission from Wiley-VCH.84 | ||
4.5. Applications of the soft layered materials to visualization and colorimetric quantification of external stimuli
The soft layered PDA are applied to visualization and colorimetric quantification of external stimuli, such as heat, force, and light. As mentioned in the Section 4.3, the layered composites based on PDA have two types of the color-change properties, i.e., reversible temperature-dependent gradual and irreversible transition types (Fig. 9). The stimuli-responsive color-change properties are tuned by the guest ions and molecules. Moreover, the layered composites based on PDA are homogeneously coated on substrates and substances through intercalation and self-organization methods (Fig. 14).80,85 Therefore, the soft layered composites based on PDA are applied to visualization and quantification of a variety of external stimuli. | ||
| Fig. 14 Scanning electron microscopy (SEM) images of the PDA-coated papers. (a) Secondary-electron image of the PDA-Zn2+-coated (lower-left part indicated by blue arrow) and -uncoated (upper-right part indicated by white arrow) parts. The inset shows the photograph of the filter paper. Reprinted with permission from ref. 78. Copyright 2020 American Chemical Society. (b) Back-scattered electron image of the uncoated (left) and PDA-C6-NH2 coated (right) parts. Reprinted with permission from Wiley-VCH.85 | ||
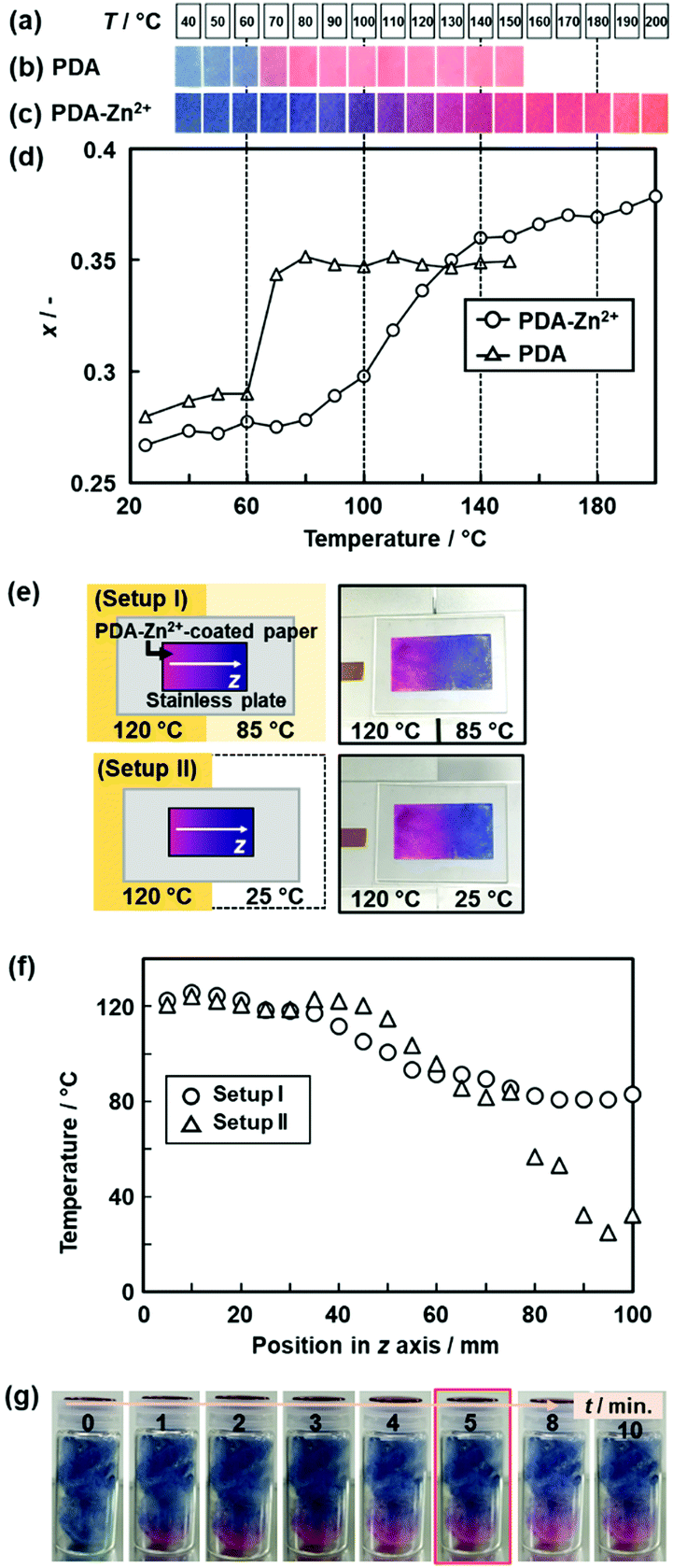 | ||
| Fig. 15 Real-time 2D and 3D temperature-distribution imaging by the PDA-Zn2+-coated devices. (a) Sample temperature. (b) and (c) Photographs of the PDA- and the PDA-Zn2+-coated filter paper with changes of temperature. (d) Relationship between the temperature and x value of the PDA (triangles) and PDA-Zn2+ (circles) estimated from the image analysis. (e) Experimental setup and photographs of the two different temperature distributions on the 2D surface, namely the setups (i) and (ii). (f) Relationship between the position in z axis and temperature in the setups (i) (circles) and (ii) (triangles). The x values of the PDA-Zn2+-coated filter paper were converted to temperature by using the relationship in Fig. 15d. (g) Time-course color change of the PDA-Zn2+-coated cotton in a glass bottle after the setting on the temperature-controlled stage at 150 °C. Reprinted with permission from ref. 78. Copyright 2020 American Chemical Society. | ||
A resin containing the layered PDA exhibited the color change with molecular motion of the glass transition.87 When styrene (St) monomer containing PCDA monomer was polymerized, polystyrene (PSt) and PCDA formed the segregated structures in micrometer scale through simultaneous polymerization and crystallization processes.95,126 The composite of PSt and PDA was obtained by the subsequent topochemical polymerization of the PCDA monomer. As molecular motion of PSt domain proceeded with heating, PDA showed the color change from blue to red near glass transition of PSt.
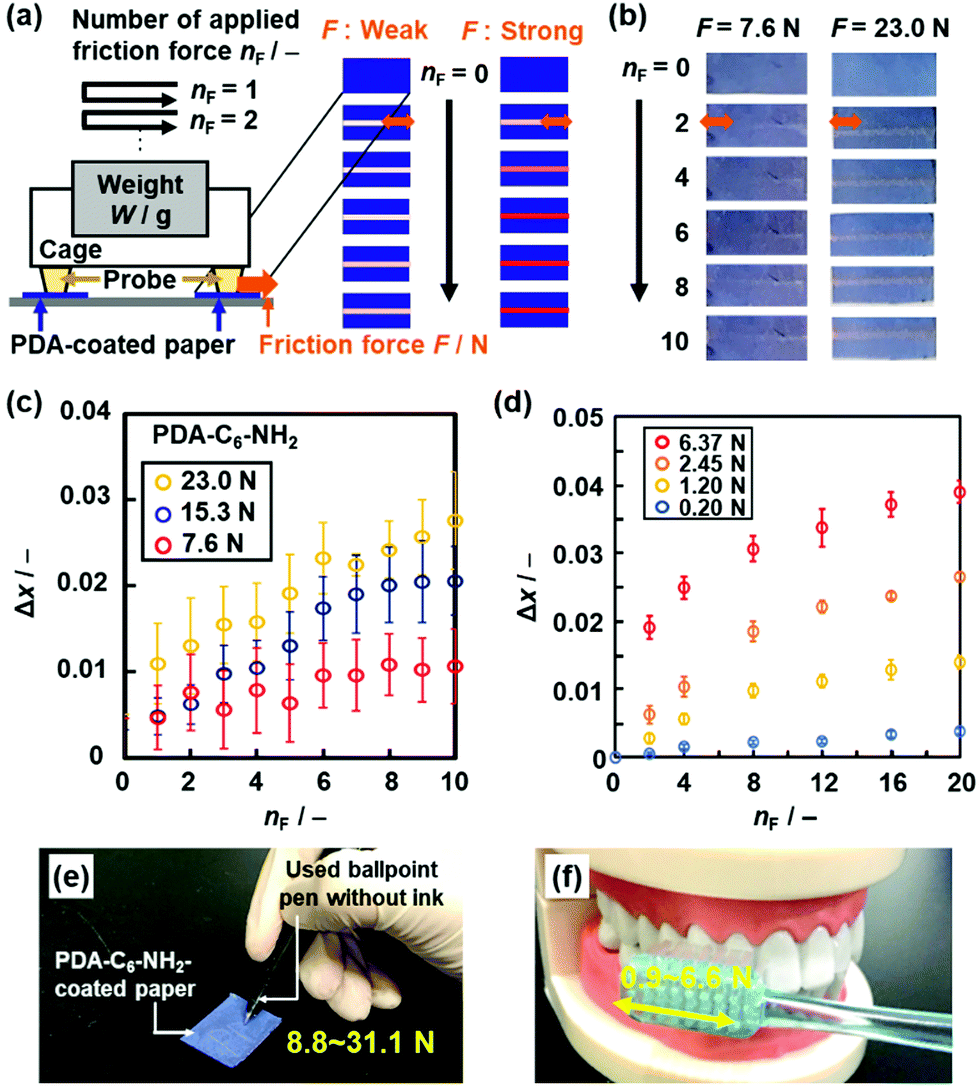 | ||
| Fig. 16 Visualization and quantification of strong and weak friction force using the PDA-C6-NH2- and PDA-PEI-coated papers, respectively. (a) Schematic illustration of the experimental setup for quantitative measurement of the applied friction force (F). (b) Photographs of the PDA-C6-NH2-coated paper with the application of F = 7.6 N (left panels) and 23.0 N (right panels) at nF = 0, 2, 4, 6, 8, 10. (c) Relationship between nF and Δx for the PDA-C6-NH2-coated paper with the application of F = 7.6, 15.3, and 23.0 N. (d) Relationship between nF and Δx for PDA-PEI-coated paper with the application of F = 0.20, 1.20, 2.45, and 6.37 N. (e) and (f) Colorimetric quantification of the writing pressure (e) and toothbrushing force (f) using PDA-C6-NH2- and PDA-PEI-coated papers, respectively. Reprinted with permission from Wiley-VCH.85 Reprinted from ref. 86 published by The Royal Society of Chemistry. | ||
The stimuli responsivity is controlled by the types of the guests. The weaker and stronger friction forces are visualized and quantified by the more soft and rigid layered composites, respectively. The layered composites of PDA and normal alkyl amines (C6-NH2) were used for visualization and quantification of friction force in the range 5–30 N (Fig. 16c).85 The guest molecules were changed to polyethyleneimine (PEI) to obtain the more soft layered composites.88 The layered composites of PDA and PEI showed the color changes by application of friction force less than 5 N (Fig. 16d).88 The paper-based devices of PDA-C6-NH2 and PDA-PEI realized the colorimetric quantification of writing pressure and toothbrushing forces as unknown strong and weak friction forces, respectively (Fig. 16e and f),85,88 respectively. In these cases, the applied friction force directly induces motion of the alkyl-side chains and subsequent distortion of the conjugated main chain.
We studied coating of polypyrrole (PPy) with photothermal conversion properties to sensing of near-infrared light (NIR) (Fig. 17).86 A number of previous works showed the photo-thermal conversion properties of the conjugated macromolecules and nanocarbons.146–149 Fujii et al., used the photo-thermal conversion properties from NIR light to heat for motion of liquid marble consisting of core liquid droplet and shell solid powder.150,151 The thin PPy coating was performed by our vapor-phase method on the PDA-coated substrates.152 When NIR was irradiated to the substrate, the generated thermal stress induced the color change of the PDA (Fig. 17a and b). The PPy layer changes NIR to thermal stress. Moreover, the red-color intensity was increased with an increase in the irradiated power of NIR (Fig. 17c and d).
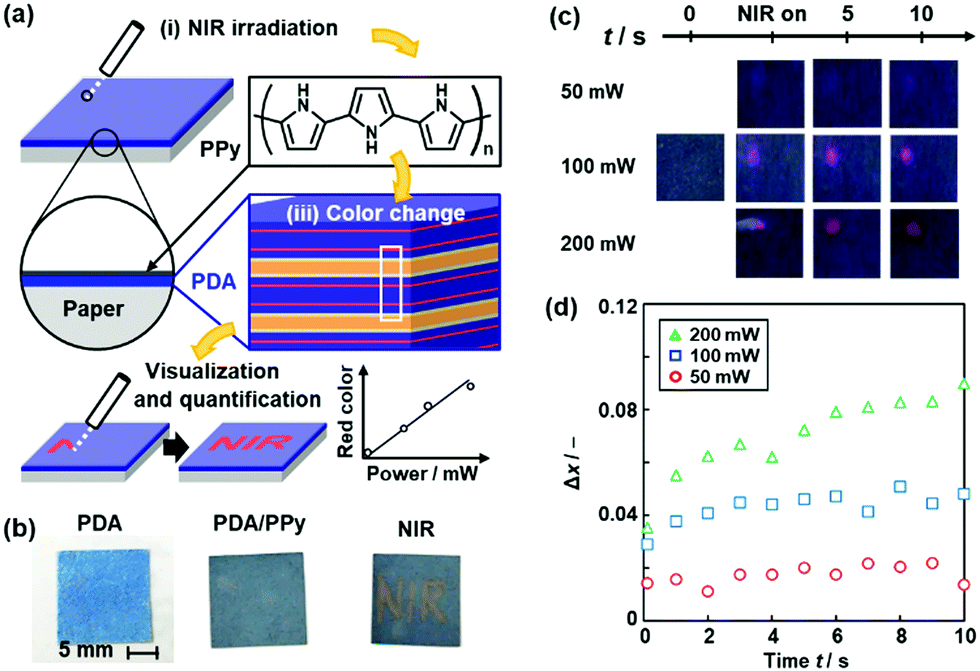 | ||
| Fig. 17 Visualization and quantification of NIR using PPy/PDA-coated paper. (a) Structure of the device consisting of layered PDA with PPy coating and its NIR visualization mechanisms with the irradiation (i), photothermal generation (ii), and color change (iii). (b) Photographs of the PDA-coated paper (left), after the PPy coating for 15 min (center), and after writing the characters with irradiation using a NIR laser pointer (808 nm) (right). (c) Photographs before (t = 0) and after the irradiation (NIR on t < 0.5 s and t = 5, 10 s) of NIR (50, 100, 200 mW). (d) Time-dependent changes of the Δx value with irradiation of 50 (red circle), 100 (blue square), 200 mW (green triangle) NIR. Reprinted from ref. 84 published by The Royal Society of Chemistry. | ||
The PDA-containing hydrogel was prepared for visualization and quantification of microwave.89 The layered PCDA monomer crystals containing vinylbenzylamine (VBA) were prepared for grafting of poly-N-isopropylacrylamide (PNIPAAm) from the vinyl group of the interlayer VBA (Fig. 18a). Then, the stretchable PNIPAAm hydrogel containing PDA was obtained by the topochemical polymerization of PCDA (Fig. 18b). When microwave heating was performed on the PNIPAAm hydrogel containing PDA, the color was changed from blue to red (Fig. 18c). The temperature distribution was visualized by the color. The irradiation time and power of microwave were visualized and quantified by the color (Fig. 18c and d). Since the volume change of the PNIPAAm gel is gradually induced by the heating with irradiation of microwave, the molecular motion of PDA proceeds with that of the PNIPAAm gel network. These model cases indicate that integration of another stimuli-responsive material to layered PDA has potentials for sensing a variety of external stimuli.
 | ||
| Fig. 18 Visualization and quantification of microwave using PNIPAAm gel containing PDA-VBA. (a) PNIPAAm/PDA-VBA thermoresponsive color-change gel synthesized by preparation of the layered composite of PCDA and VBA, grafting and polymerization of NIPAAm to the interlayer VBA, and topochemical polymerization of PCDA. (b) Stretching behavior of the PNIPAAm/PDA-VBA gel. (c) Photographs with irradiation of microwave (210 W) for 5, 10, 20, and 33 s. (d) Relationship between the irradiation power (P/W) and time to complete the color change (tf). Reprinted with permission from ref. 87. Copyright 2020 American Chemical Society. | ||
5. Summary and outlook
This feature article focuses on intercalation and flexibility chemistry of soft layered materials exhibiting dynamic functions. Inorganic layered compounds and their intercalation chemistry were well studied in previous works. 2D materials derived from layered structures is one of the most promising nanostructures in recent materials science. Compared with these well-established chemistry and materials, soft layered materials are an emerging research area for design and development of dynamic functional materials. Section 2 introduced a potential new perspective for the soft and rigid natures of the layered materials. A variety of well-known layered materials can be classified by the softness and rigidness of the structures. Section 3 showed a couple of the representative studies about syntheses, structures, and applications of soft layered materials inspiring the design strategies. The flexibility and interaction are efficiently used for emergence of the dynamic functions. Section 4 focused on the tunable stimuli-responsive color-change properties of layered PDA and their applications in our recent works. The soft and rigid natures of the layered PDA are controlled by the intercalated guests. These facts indicate that the controlled flexibility is an important factor to tune the dynamic properties. Intercalation is an important and characteristic approach to control the flexibility and dynamic functions of soft layered materials, as well as designing the structure units and their organized states (Fig. 19). In addition, the 2D anisotropy contributes to control the macroscopic morphologies favorable to prepare the devices. Therefore, soft layered materials based on intercalation and flexibility chemistries have potentials for development of advanced dynamic functional materials.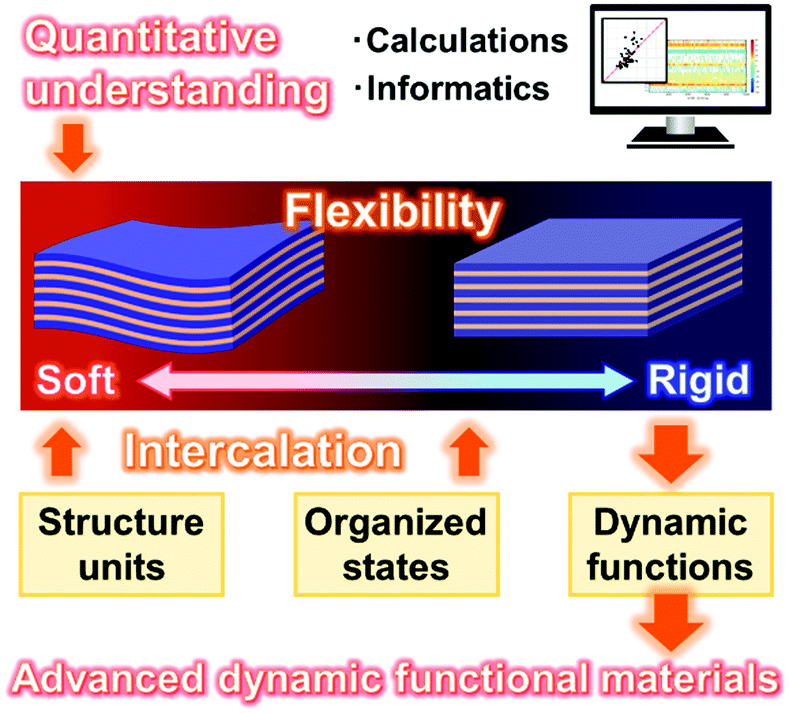 | ||
| Fig. 19 Overview of the summary and future directions for designing of the advanced dynamic functional materials and quantitative understanding of the structure flexibility. | ||
One of the important remaining challenges is how to quantify soft and rigid natures of layered materials (Fig. 19). Structural softness and rigidness of molecules and materials are usually discussed in general chemistry and materials science. However, the flexibility is still a qualitive factor based on experience and intuition of professional researchers. The quantification is required for further understanding of organized materials including layered materials. In this respect, layered materials are good model case to explore the quantitative factors regarding the rigidness and softness because of the structure definitude (Fig. 19). The calculational study is required for exploration of the quantitative factors determining the soft and rigid natures. Moreover, our group suggests that application of data-scientific approach including machine learning is helpful for extraction of the descriptors from dataset to explain the target properties and phenomena.153–155 Machine learning will assist extraction of the descriptors to explain the flexibility and dynamic function of soft layered materials. Quantitative understanding the true natures of soft layered materials will contribute to design the dynamic properties. Moreover, the factors are applied to understanding the structure softness and rigidness of a wide variety of molecules and organized materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
I appreciate all the collaborators as listed in the literature cited in the reference section. This work is partially supported by Grant-in-Aid for Scientific Research on Innovative Areas of “Fusion Materials: Creative Development of Materials and Exploration of Their Function through Molecular Control” (No. 2206) from the Ministry of Education, Culture, Sports, Science and Technology, by Grant-in-Aid for Young Scientist (A, No. 22685022) from Japan Society of the Promotion of Science, by JST PRESTO (JPMJPR16N2), by Izumi Science and Technology Foundation, Asahi Glass Foundation, and Takahashi Industrial and Economic Research Foundation.Notes and references
- K. S. Novoselov, Angew. Chem., Int. Ed., 2011, 50, 6986 CrossRef CAS.
- H. Okamoto, Y. Sugiyama and H. Nakano, Chem. – Eur. J., 2011, 17, 9864 CrossRef CAS.
- V. Nicolosi, M. Chhowalla, M. G. Kanatzidis, M. S. Strano and J. N. Coleman, Science, 2013, 340, 1226419 CrossRef.
- H. P. Cong, J. F. Chen and S. H. Yu, Chem. Soc. Rev., 2014, 43, 7295 RSC.
- M. Renzhi and T. Sasaki, Acc. Chem. Res., 2015, 48, 136 CrossRef.
- J. E. ten Elshof, H. Yuan and P. G. Rodriguez, Adv. Energy Mater., 2016, 6, 1600355 CrossRef.
- A. A. Tedstone, D. J. Lewis and P. O’Brien, Chem. Mater., 2016, 28, 1965 CrossRef CAS.
- M. Servalli and A. D. Schlüter, Annu. Rev. Mater. Res., 2017, 47, 361 CrossRef CAS.
- C. Tan, X. Cao, X. J. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han, G. H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225 CrossRef CAS.
- K. Ariga, S. Watanabe, T. Mori and J. Takeya, NPG Asia Mater., 2018, 10, 90 CrossRef.
- Q. Zhang, J. Zhang, S. Wan, W. Wang and L. Fu, Adv. Funct. Mater., 2018, 28, 1802500 CrossRef.
- C. N. R. Rao and K. Paramoda, Bull. Chem. Soc. Jpn., 2019, 92, 441 CrossRef CAS.
- M. Osada and T. Sasaki, Adv. Mater., 2012, 24, 210 CrossRef CAS.
- B. Mendoza-Sánchez and Y. Gogotsi, Adv. Mater., 2016, 28, 6104 CrossRef.
- B. Luo, G. Liu and L. Wang, Nanoscale, 2016, 8, 6904 RSC.
- A. H. Khan, S. Ghosh, B. Pradhan, A. Dalui, L. K. Shrestha, S. Acharya and K. Ariga, Bull. Chem. Soc. Jpn., 2017, 90, 627 CrossRef.
- B. T. Hogan, E. Kovalska, M. F. Craciun and B. Baqidycheva, J. Mater. Chem. C, 2017, 5, 11185 RSC.
- Q. Zhang, J. Zhang, S. Wan, W. Wang and L. Fu, Adv. Funct. Mater., 2018, 28, 1802500 CrossRef.
- X. Liu and M. C. Hersam, Adv. Mater., 2018, 30, 1801586 CrossRef.
- Y. K. Ryu, R. Frisenda and A. Castelianos-Gomez, Chem. Commun., 2019, 55, 11498 RSC.
- S. Ippolito, A. Clesielski and P. Samori, Chem. Commun., 2019, 55, 8900 RSC.
- J. Sakamoto, J. v. Heijst, O. Lukin and A. D. Schlüter, Angew. Chem., Int. Ed., 2009, 48, 1030 CrossRef CAS.
- I. Berlanga, M. L. Ruiz-González, J. González-Calbet, J. L. G. Fierro, R. M. Ballesté and F. Zamora, Small, 2011, 7, 1207 CrossRef CAS.
- D. N. Bunck and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 14952 CrossRef CAS.
- R. Bhola, P. Payamyar, D. J. Murray, B. Kumar, A. J. Teator, M. U. Schmidt, S. M. Hammer, A. Saha, J. Sakamoto, A. D. Schlüter and B. T. King, J. Am. Chem. Soc., 2013, 135, 14134 CrossRef CAS.
- P. Kissel, R. Ernil, W. B. Schweizer, M. D. Rossell, B. T. King, T. Bauer, S. Götzinger, A. D. Schlüter and J. Sakamoto, Nat. Chem., 2012, 4, 287 CrossRef CAS.
- J. W. Colson and W. R. Dichtel, Nat. Chem., 2013, 5, 453 CrossRef CAS.
- X. Zhuang, Y. Mai, D. Wu, F. Zhang and X. Feng, Adv. Mater., 2015, 27, 403 CrossRef CAS.
- R. Sakamoto, N. Fukui, H. Maeda, R. Matsuoka, R. Toyoda and H. Nishihara, Adv. Mater., 2019, 31, 1804211 CrossRef CAS.
- S. Yano, J. Suzuki, K. Sato, H. Imai and Y. Oaki, Commun. Chem., 2019, 2, 97 CrossRef.
- J. Suzuki, A. Ishizone, K. Sato, H. Imai, Y. J. Tseng, C. H. Peng and Y. Oaki, Chem. Sci., 2020, 11, 7003 RSC.
- K. Ariga, Langmuir, 2020, 36, 7158 CrossRef CAS.
- J. J. Richardson, J. Cui, M. Björnmalm, J. A. Braunger, H. Ejima and F. Caruso, Chem. Rev., 2016, 116, 14828 CrossRef CAS.
- Intercalation chemistry, ed. M. S. Whittingham and A. J. Jacobson, Academic Press, New York, 1982 Search PubMed.
- T. E. Mallouk and J. Gavin, Acc. Chem. Res., 1998, 31, 209 CrossRef CAS.
- G. A. Ozin, Adv. Mater., 1992, 4, 612 CrossRef CAS.
- M. Ogawa and K. Kuroda, Chem. Rev., 1995, 95, 399 CrossRef CAS.
- R. Schollhörn, Chem. Mater., 1996, 8, 1747 CrossRef.
- E. Ruiz-Hitzky, M. Darder and P. Aranda, J. Mater. Chem., 2005, 15, 3650 RSC.
- M. Ogawa, K. Saito and M. Sohmiya, Dalton Trans., 2014, 43, 10340 RSC.
- T. Kato, N. Mizoshita and K. Kishimoto, Angew. Chem., Int. Ed., 2006, 45, 38 CrossRef CAS.
- T. Kato, M. Yoshio, T. Ichikawa, B. Soberats, H. Ohno and M. Funahashi, Nat. Rev. Mater., 2017, 2, 17001 CrossRef.
- T. Kato, J. Uchida, T. Ichikawa and T. Sakamoto, Angew. Chem., Int. Ed., 2018, 57, 4355 CrossRef CAS.
- K. Ariga, Y. Yamauchi, G. Rydzek, Q. Ji, Y. Yonamine, K. C. W. Wu and J. P. Hill, Chem. Lett., 2014, 43, 36 CrossRef CAS.
- K. Ariga, M. Ishii and T. Mori, Chem. – Eur. J., 2020, 26, 6461 CrossRef CAS.
- A. W. Coleman, S. G. Bott, S. D. Morley, C. M. Means, K. D. Robinson, H. Zhang and J. L. Atwood, Angew. Chem., Int. Ed. Engl., 1988, 27, 1361 CrossRef.
- A. Matsumoto, T. Odani, T. Sada, M. Miyata and K. Tashiro, Nature, 2000, 405, 328 CrossRef CAS.
- A. Matsumoto and T. Odani, Macromol. Rapid Commun., 2001, 22, 1195 CrossRef CAS.
- M. Honda, Y. Oaki and H. Imai, Chem. Mater., 2014, 26, 3579 CrossRef CAS.
- M. Honda, Y. Oaki and H. Imai, Chem. Commun., 2015, 51, 10046 RSC.
- H. Matsui, Y. Oaki and H. Imai, Chem. Commun., 2016, 52, 9466 RSC.
- Y. Ishijima, M. Okaniwa, Y. Oaki and H. Imai, Chem. Sci., 2017, 8, 647 RSC.
- G. Nakada, H. Imai and Y. Oaki, Chem. Commun., 2018, 54, 244 RSC.
- G. Nakada, Y. Igarashi, H. Imai and Y. Oaki, Adv. Theory Simul., 2019, 2, 1800180 CrossRef CAS.
- Y. Yamamoto, H. Imai and Y. Oaki, Bull. Chem. Soc. Jpn., 2019, 92, 779 CrossRef CAS.
- K. Noda, Y. Igarashi, H. Imai and Y. Oaki, Adv. Theory Simul., 2020, 3, 200084 Search PubMed.
- T. Kato, Science, 2002, 295, 2414 CrossRef CAS.
- M. Yoshio, T. Mukai, K. Kanie, M. Yoshizawa, H. Ohno and T. Kato, Adv. Mater., 2002, 14, 351 CrossRef CAS.
- K. Hoshino, M. Yoshio, T. Mukai, K. Kishimoto, H. Ohno and T. Kato, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3486 CrossRef CAS.
- M. Yoshio and T. Kato, Mol. Cryst. Liq. Cryst., 2004, 413, 2235 CAS.
- K. Kishimoto, M. Yoshio, T. Mukai, M. Yoshizawa, H. Ohno and T. Kato, J. Am. Chem. Soc., 2003, 125, 3196 CrossRef CAS.
- J. Sakuda, E. Hosono, M. Yoshio, T. Ichikawa, T. Matsumoto, H. Ohno, H. Zhou and T. Kato, Adv. Funct. Mater., 2015, 25, 1206 CrossRef CAS.
- D. Högberg, B. Soberats, R. Yatagai, S. Uchida, M. Yoshio, L. Kloo, H. Segawa and T. Kato, Chem. Mater., 2016, 28, 6493 CrossRef.
- S. Yazaki, M. Funahashi, J. Kagimoto, H. Ohno and T. Kato, J. Am. Chem. Soc., 2010, 132, 7702 CrossRef CAS.
- A. Kuwabara, M. Enomoto, E. Hosono, K. Hamaguchi, T. Onuma, S. Kajiyama and T. Kato, Chem. Sci., 2020 10.1039/D0SC01646B.
- S. Guo, A. Sugawara-Narutaki, T. Okubo and A. Shimojima, J. Mater. Chem. C, 2013, 1, 6989 RSC.
- S. Guo, W. Chjaikittisilp, T. Okubo and A. Shimojima, RSC Adv., 2014, 4, 25391 Search PubMed.
- S. Guo, K. Matsukawa, T. Miyata, T. Okubo, K. Kuroda and A. Shimojima, J. Am. Chem. Soc., 2015, 137, 15434 CrossRef CAS.
- N. Liu, K. Yu, B. Smarsly, D. R. Dauphy, Y. B. Jian and C. J. Brinker, J. Am. Chem. Soc., 2002, 124, 14540 CrossRef CAS.
- M. Ogawa, T. Ishii, N. Miyamoto and K. Kuroda, Adv. Mater., 2001, 13, 1107 CrossRef CAS.
- Y. Nabetani, H. Takamura, Y. Hayasak, T. Shimada, S. Takagi, H. Tachibana, D. Masui, Z. Tong and H. Inoue, J. Am. Chem. Soc., 2011, 133, 17130 CrossRef CAS.
- P. Ganter and B. V. Lotsch, Mol. Syst. Des. Eng., 2019, 4, 566 RSC.
- K. Szendrei, P. Ganter, O. Sànchez-Sobrado, R. Eger, A. Kuhn and B. V. Lotsch, Adv. Mater., 2015, 27, 6341 CrossRef CAS.
- P. Ganter, K. Szendrei and B. V. Lotsch, Adv. Mater., 2016, 28, 7436 CrossRef CAS.
- K. Szendrei-Temesi, O. Sanchez-Sobrado, S. B. Betzler, K. M. Durner, T. Holzmann and B. V. Lotsch, Adv. Funct. Mater., 2018, 28, 1705740 CrossRef.
- K. Szendrei-Temesi, A. Jiménez-Solano and B. V. Lotsch, Adv. Mater., 2018, 30, 1803730 CrossRef.
- P. Ganter, L. M. Schoop, M. Däntl and B. V. Lotsch, Chem. Mater., 2018, 30, 2557 CrossRef CAS.
- M. Okaniwa, Y. Oaki, S. Kaneko, K. Ishida, H. Maki and H. Imai, Chem. Mater., 2015, 27, 2627 CrossRef CAS.
- M. Okaniwa, Y. Oaki and H. Imai, Adv. Funct. Mater., 2016, 26, 2463 CrossRef.
- M. Takeuchi, H. Imai and Y. Oaki, ACS Appl. Mater. Interfaces, 2017, 9, 16546 CrossRef CAS.
- M. Takeuchi, H. Imai and Y. Oaki, J. Mater. Chem. C, 2017, 5, 8250 RSC.
- Y. Ishijima, H. Imai and Y. Oaki, Chem, 2017, 3, 509 CAS.
- Y. Oaki, Y. Ishijima and H. Imai, Polym. J., 2018, 50, 319 CrossRef CAS.
- M. Takeuchi, K. Gnanasekaran, H. Friedrich, H. Imai, N. A. J. M. Sommerdijk and Y. Oaki, Adv. Funct. Mater., 2018, 28, 1804906 CrossRef.
- H. Terada, H. Imai and Y. Oaki, Adv. Mater., 2018, 30, 1801121 CrossRef.
- M. Takeuchi, H. Kawashima, H. Imai, S. Fujii and Y. Oaki, J. Mater. Chem. C, 2019, 7, 4089 RSC.
- S. Ishioka, K. Watanabe, H. Imai, Y. J. Tseng, C. H. Peng and Y. Oaki, Chem. Commun., 2019, 55, 11725 RSC.
- K. Watanabe, H. Imai and Y. Oaki, J. Mater. Chem. C, 2020, 8, 1265 RSC.
- M. Nakamitsu, H. Imai and Y. Oaki, ACS Sens., 2020, 5, 133 CrossRef CAS.
- J. Roncali, Macromol. Rapid Commun., 2007, 28, 1761 CrossRef CAS.
- S. Kirchmeyer and K. Reuter, J. Mater. Chem., 2005, 15, 2077 RSC.
- G. Snook, P. Kao and A. Best, J. Power Sources, 2011, 196, 112 CrossRef.
- A. Rudge, I. Raistrick, S. Gottesfeld and J. Ferraris, Electrochim. Acta, 1994, 39, 273 CrossRef CAS.
- K. Wang, H. Wu, Y. Meng and Z. Wei, Small, 2014, 10, 14 CrossRef CAS.
- Y. Oaki and K. Sato, J. Mater. Chem. A, 2018, 6, 23197–23219 RSC.
- T. Ogawa, Prog. Polym. Sci., 1995, 20, 943 CrossRef CAS.
- R. W. Carpick, D. Y. Sasaki, M. S. Marcus, M. A. Eriksson and A. R. Burns, J. Phys.: Condens. Matter, 2004, 16, R679 CrossRef CAS.
- D. J. Ahn and J. M. Kim, Acc. Chem. Res., 2008, 41, 805 CrossRef CAS.
- D. J. Ahn, S. Lee and J. M. Kim, Adv. Funct. Mater., 2009, 19, 1483 CrossRef CAS.
- X. Sun, T. Chen, S. Huang, L. Li and H. Peng, Chem. Soc. Rev., 2010, 39, 4244 RSC.
- O. Yarimaga, J. Jaworski, B. Yoon and J. M. Kim, Chem. Commun., 2012, 48, 2469 RSC.
- R. Jelinek and M. Ritenberg, RSC Adv., 2013, 3, 21192 RSC.
- D. H. Park, B. J. Park and J. M. Kim, Acc. Chem. Res., 2016, 49, 1211 CrossRef CAS.
- J. Huo, Q. Deng, T. Fan, G. He, X. Hu, X. Hong, H. Chen, S. Luo, Z. Wang and D. Chen, Polym. Chem., 2017, 8, 7438 RSC.
- X. Qian and B. Städler, Chem. Mater., 2019, 31, 1196 CrossRef CAS.
- B. Tieke, G. Lieser and G. Wegner, J. Polym. Sci. Polym. Chem. Ed., 1979, 17, 1631 CrossRef CAS.
- S. Okada, S. Peng, W. Spevak and D. Charych, Acc. Chem. Res., 1998, 31, 229 CrossRef CAS.
- X. Chen, G. Zhou, X. Peng and J. Yoon, Chem. Soc. Rev., 2012, 41, 4610 RSC.
- M. Wetson, A. D. Tjandra and R. Chandrawati, Polym. Chem., 2020, 11, 166 RSC.
- J. M. Kim, Y. B. Lee, D. H. Yang, J. S. Lee, G. S. Lee and D. J. Ahn, J. Am. Chem. Soc., 2005, 127, 17580 CrossRef CAS.
- D. J. Ahn, E. H. Chae, G. S. Lee, H. Y. Shim, T. E. Chang, K. D. Ahn and J. M. Kim, J. Am. Chem. Soc., 2003, 125, 8976 CrossRef CAS.
- S. Lee and J. M. Kim, Macromolecules, 2007, 40, 9201 CrossRef CAS.
- S. Dei, M. Matsumoto and A. Matsumoto, Macromolecules, 2008, 41, 2467 CrossRef CAS.
- S. Ampornpun, S. Montha, G. Tumcharern, V. Vchirawongkwin, M. Sukwattanasinitt and S. Wacharasindhu, Macromolecules, 2012, 45, 9038 CrossRef CAS.
- C. Tanioku, K. Matsukawa and A. Matsumoto, ACS Appl. Mater. Interfaces, 2013, 5, 940 CrossRef CAS.
- I. S. Park, H. J. Park and J. M. Kim, ACS Appl. Mater. Interfaces, 2013, 5, 8805 CrossRef CAS.
- S. Lee, J. Lee, M. Lee, Y. K. Cho, J. Baek, J. Kim, S. Park, M. H. Kim, R. Chang and J. Yoon, Adv. Funct. Mater., 2014, 24, 3699 CrossRef CAS.
- K. P. Kooter, H. Jiang, S. Kolusheva, T. P. Vinod, M. Ritenberg, L. Zeiri, R. Volinsky, D. Malferrari, P. Galletti, E. Tagliavini and R. Jelinek, ACS Appl. Mater. Interfaces, 2014, 6, 8613 CrossRef.
- S. Dolai, S. K. Bhunia, S. B. Beglaryan, S. Kolusheva, L. Zein and R. Jelinek, ACS Appl. Mater. Interfaces, 2017, 9, 2891 CrossRef CAS.
- I. S. Park, H. J. Park, W. Jeong, J. Nam, Y. Kang, K. Shin, H. Chung and J. M. Kim, Macromolecules, 2016, 49, 1270 CrossRef CAS.
- W. Jeong, M. I. Khazi, D. G. Lee and J. M. Kim, Macromolecules, 2018, 51, 10312 CrossRef CAS.
- B. Hu, S. Sun, B. Wu and P. Wu, Small, 2019, 15, 1804975 CrossRef.
- G. Shin, M. I. Khazi, U. Kundapur, B. Kim, Y. Kim, C. W. Lee and J. M. Kim, ACS Macro Lett., 2019, 8, 610 CrossRef CAS.
- T. Shimogaki and A. Matsumoto, Macromolecules, 2011, 44, 3323 CrossRef CAS.
- B. Yoon, J. Jaworski and J. M. Kim, Supramol. Chem., 2013, 25, 54 CrossRef CAS.
- K. Sato, Y. Oaki and H. Imai, NPG Asia Mater., 2017, 9, e377 CrossRef CAS.
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755 CrossRef CAS.
- Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605 CrossRef CAS.
- D. R. T. Roberts and S. J. Holder, J. Mater. Chem., 2011, 21, 8256 RSC.
- Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073 CrossRef CAS.
- S. Stauss and I. Honma, Bull. Chem. Soc. Jpn., 2018, 91, 492 CrossRef CAS.
- R. Zhang, Q. Wang and X. Zhaeng, J. Mater. Chem. C, 2018, 6, 3182 RSC.
- S. Hayashi, Polym. J., 2019, 51, 813 CrossRef CAS.
- B. R. Crenshaw and C. Weder, Chem. Mater., 2003, 15, 4717 CrossRef CAS.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. V. Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martínez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68 CrossRef CAS.
- K. Sakakibara, L. A. Joyce, T. Mori, T. Fujisawa, S. H. Shabbir, J. P. Hill, E. V. Anslyn and K. Ariga, Angew. Chem., Int. Ed., 2012, 51, 9643 CrossRef CAS.
- H. Yuan, K. Wang, K. Yang, B. Liu and B. Zou, J. Phys. Chem. Lett., 2014, 5, 2968 CrossRef CAS.
- Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2008, 47, 5175 CrossRef CAS.
- H. Ito, M. Muromoto, S. Ishizaki, N. Kitamura, H. Sato and T. Seki, Nat. Commun., 2013, 4, 2009 CrossRef.
- K. Nagura, S. Saito, H. Yusa, H. Yamawaki, H. Fujihisa, H. Sato, Y. Shimoikeda and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 10322 CrossRef CAS.
- R. Yoshii, K. Suenaga, K. Tanaka and Y. Chujo, Chem. – Eur. J., 2015, 21, 7231 CrossRef CAS.
- L. Y. Hsu, S. Maity, Y. Matsunaga, Y. F. Hsu, Y. H. Liu, S. M. Peng, T. Shinmyozu and J. S. Yang, Chem. Sci., 2018, 9, 8990 RSC.
- S. Chae, J. P. Lee and J. M. Kim, Adv. Funct. Mater., 2016, 26, 1769 CrossRef CAS.
- J. P. Lee, H. Hwang, S. Chae and J. M. Kim, Chem. Commun., 2019, 55, 9395 RSC.
- L. Polacchi, A. Brosseau, R. Métivier and C. Allain, Chem. Commun., 2019, 55, 14566 RSC.
- F. Li, M. A. Winnik, A. Matvienko and A. Mandelis, J. Mater. Chem., 2007, 17, 4309 RSC.
- K. M. Au, M. Chen, S. P. Armes and N. Zheng, Chem. Commun., 2013, 49, 10525 RSC.
- H. S. Ju, P. Verwlist, A. Sharma, J. Shin, J. L. Sessler and J. S. Kim, Chem. Soc. Rev., 2018, 47, 2280 RSC.
- O. A. Savchuk, J. J. Carvajal, J. Massons, M. Aguiló and F. Díaz, Carbon, 2016, 103, 134 CrossRef CAS.
- M. Paven, H. Mayama, T. Sekido, H. J. Butt, Y. Nakamura and S. Fujii, Adv. Funct. Mater., 2016, 26, 3199 CrossRef CAS.
- H. Kawashima, M. Paven, H. Mayama, H. J. Butt, Y. Nakamura and S. Fujii, ACS Appl. Mater. Interfaces, 2017, 9, 33351 CrossRef CAS.
- R. Muramatsu, Y. Oaki, K. Kuwabara, K. Hayashi and H. Imai, Chem. Commun., 2014, 50, 11840 RSC.
- G. Nakada, Y. Igarashi, H. Imai and Y. Oaki, Adv. Theory Simul., 2019, 2, 1800180 CrossRef CAS.
- H. Numazawa, Y. Igarashi, K. Sato, H. Imai and Y. Oaki, Adv. Theory Simul., 2019, 2, 1900130 CrossRef CAS.
- K. Noda, Y. Igarashi, H. Imai and Y. Oaki, Adv. Theory Simul., 2020, 3, 2000084 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |