
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZinc ion interactions in a two-dimensional covalent organic framework based aqueous zinc ion battery†
Abdul
Khayum M
 ab,
Meena
Ghosh
ab,
Vidyanand
Vijayakumar
ab,
Arjun
Halder
ab,
Maryam
Nurhuda
c,
Sushil
Kumar
b,
Matthew
Addicoat
c,
Sreekumar
Kurungot
*ab and
Rahul
Banerjee
*d
ab,
Meena
Ghosh
ab,
Vidyanand
Vijayakumar
ab,
Arjun
Halder
ab,
Maryam
Nurhuda
c,
Sushil
Kumar
b,
Matthew
Addicoat
c,
Sreekumar
Kurungot
*ab and
Rahul
Banerjee
*d
aAcademy of Scientific and Innovative Research (AcSIR), Sector 19, Kamla Nehru Nagar, Ghaziabad, Uttar Pradesh-201002, India
bPhysical and Materials Chemistry Division, CSIR-National Chemical Laboratory, Dr. Homi Bhabha Road, Pune-411008, India. E-mail: k.sreekumar@ncl.res.in
cSchool of Science and Technology, Nottingham Trent University, Clifton Lane, NG11 8NS Nottingham, UK
dDepartment of Chemical Sciences, Indian Institute of Science Education and Research (IISER), Mohanpur Campus, Mohanpur, Kolkata, 741252 India. E-mail: r.banerjee@iiserkol.ac.in
First published on 6th August 2019
Abstract
The two-dimensional structural features of covalent organic frameworks (COFs) can promote the electrochemical storage of cations like H+, Li+, and Na+ through both faradaic and non-faradaic processes. However, the electrochemical storage of cations like Zn2+ ion is still unexplored although it bears a promising divalent charge. Herein, for the first time, we have utilized hydroquinone linked β-ketoenamine COF acting as a Zn2+ anchor in an aqueous rechargeable zinc ion battery. The charge-storage mechanism comprises of an efficient reversible interlayer interaction of Zn2+ ions with the functional moieties in the adjacent layers of COF (−182.0 kcal mol−1). Notably, due to the well-defined nanopores and structural organization, a constructed full cell, displays a discharge capacity as high as 276 mA h g−1 at a current rate of 125 mA g−1.
Introduction
Two-dimensional (2D) porous crystalline covalent organic frameworks (COFs) are integrated from pre-designed symmetric organic building units and self-assembled by π–π stacking.1 Due to the structural tunability, well-defined porosity, and high chemical stability, COFs have been widely explored as electrode materials for electrochemical energy storage.2 However, until now, the energy storage applications in COFs were mostly limited to the monovalent ions like H+, Li+, and Na+ that could only carry forward single electron reactions. Although, divalent aqueous zinc ion batteries are observed to be cost-effective and superior in terms of safety and recyclability than the monovalent Li+, Na+ metal-ion batteries,3 it is extremely challenging to design an organic polymeric electrode that can act as a Zn2+ acceptor. A few literature reports deal with small molecules as cathode materials for accepting the electrophilic Zn2+ ions through coordination bonds.4 However, limited chemical and thermal stabilities of these organic molecules and the undesirable leaching of the active electrode materials during the electrochemical cycling hampers their performance and creates a need for a polymeric electrode capable of hosting Zn2+ ions. Keeping this in perspective, herein, we have demonstrated hydroquinone based covalent organic framework5 (HqTp) acting as an efficient polymeric cathode material for an aqueous zinc ion battery. We anticipated that a COF must be enriched with several nucleophilic centers so that it can anchor to a significant number of divalent Zn2+ ions. Furthermore, the framework must be constructed from such building blocks, which can act as nucleophilic centers for efficient interaction with Zn2+ under electrochemical conditions.It is well documented that the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H functionalities in polymers are capable of coordinating Zn2+ ions in an electrochemical cell.4,6 Both CO⋯Zn and Zn⋯N–H interactions are reversible in nature and can “make and break” during the electrochemical redox process. Thus, an electron-rich backbone in the HqTp facilitates efficient coordination with Zn2+ ions. HqTp exhibits an excellent discharge capacity of 276 mA h g−1 at a current of 125 mA g−1 in an operating potential of 0.2 to 1.6 V vs. Zn/Zn2+ due to the efficient intermolecular O⋯Zn (∼2.0 Å) and N⋯Zn (∼2.0 Å) interactions.4a,7 To the best of our knowledge, this is the first report of a COF based aqueous zinc ion battery and puts forward a novel concept based on the zinc ion interaction with a porous crystalline and polymeric organic cathode.
O and N–H functionalities in polymers are capable of coordinating Zn2+ ions in an electrochemical cell.4,6 Both CO⋯Zn and Zn⋯N–H interactions are reversible in nature and can “make and break” during the electrochemical redox process. Thus, an electron-rich backbone in the HqTp facilitates efficient coordination with Zn2+ ions. HqTp exhibits an excellent discharge capacity of 276 mA h g−1 at a current of 125 mA g−1 in an operating potential of 0.2 to 1.6 V vs. Zn/Zn2+ due to the efficient intermolecular O⋯Zn (∼2.0 Å) and N⋯Zn (∼2.0 Å) interactions.4a,7 To the best of our knowledge, this is the first report of a COF based aqueous zinc ion battery and puts forward a novel concept based on the zinc ion interaction with a porous crystalline and polymeric organic cathode.
Results and discussion
The HqTp COF is synthesized by a solid-state mechano-mixing of 2,5-diaminohydroquinone dihydrochloride (Hq) and 1,3,5-triformylphloroglucinol (Tp) in the presence of p-toluenesulfonic acid (PTSA·H2O) as a catalyst (Fig. 1a; ESI, S1†). A black coloured product was obtained after removing the impurities upon wash with water, N,N-dimethylacetamide (DMAc) and acetone. The crystalline framework structure and an ordered integration of the building blocks throughout the network is confirmed by the PXRD characterization. The powder X-ray diffraction (PXRD) profile exhibits sharp peaks at the 2θ position of 4.6°, 8.0°, 9.4°, 12.4° and 27.3° (Fig. 1b). The sharp and intense peaks at 4.6 and 27.3° (2θ) correspond to the reflections from 100 and 001 planes, which indicates the existence of a periodic structure. Moreover, the experimental PXRD pattern matches with the simulated PXRD pattern with a slipped eclipsed orientation as modeled using SCC-DFTB (Fig. 1b; ESI S2 and S3†).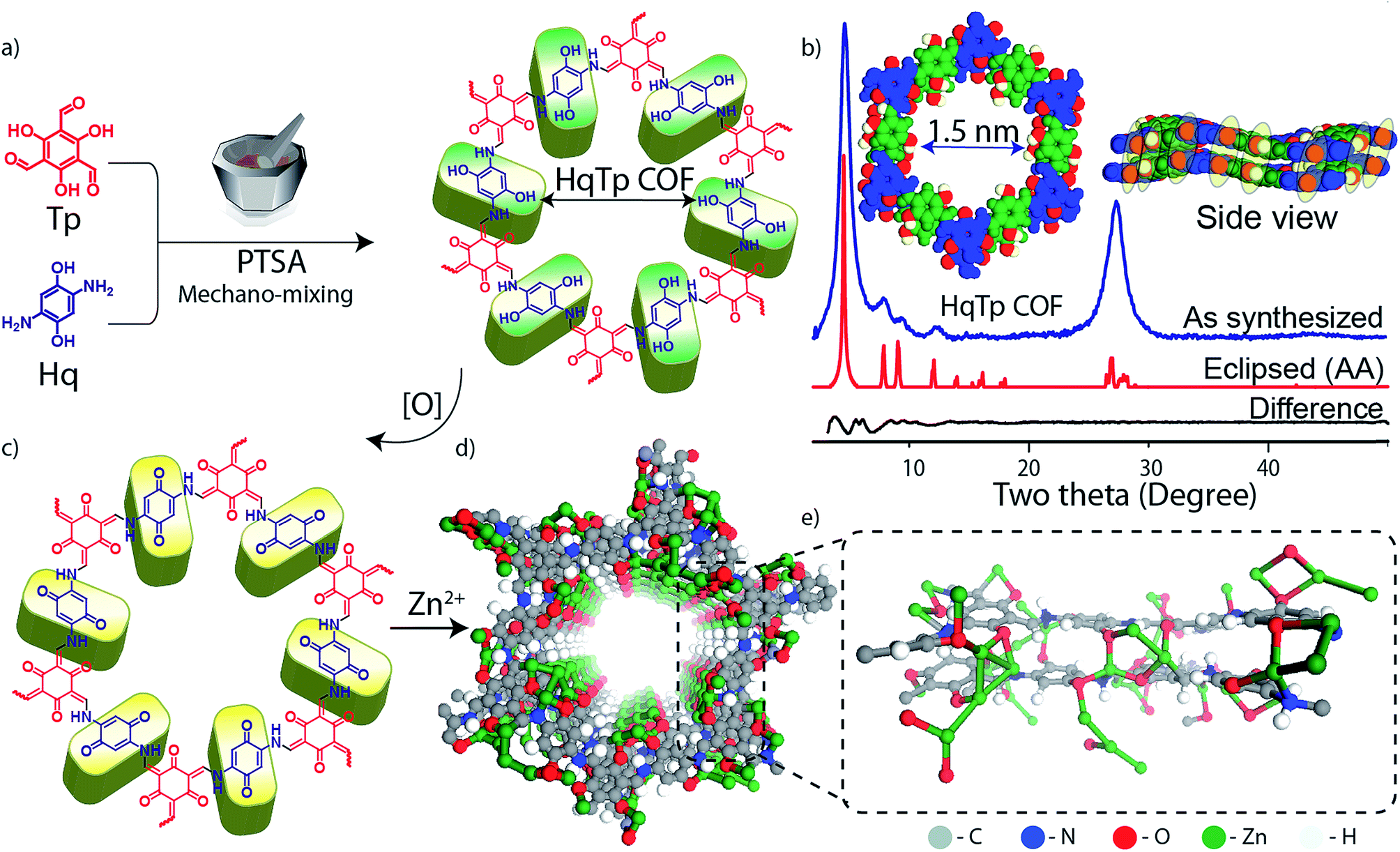 | ||
| Fig. 1 (a) Schematic representation of the synthesis of HqTp (Hq-2,5-diaminohydroquinone, Tp-1,3,5-triformylphloroglucinol). (b) Powder X-ray diffraction pattern with the AA eclipsed slipped SCC-DFT model. (c) The electrochemical oxidation of hydroquinone to quinone in HqTp (d and e). The DFTB model of interlayer interaction of Zn2+ cations with the adjacent layers of HqTp. | ||
The β-ketoenamine framework formation is confirmed by the new peaks in FT-IR spectra at 1583, 1551 and 1244 cm−1, which correspond to CO, CC and C–N bonds (all generated after enol–keto tautomerization; ESI; Fig. S6†). Additionally, the 13C solid-state CP-MAS NMR shows enamine and α-enamine carbon peak resonances at 142 and 105 ppm respectively and similarly CO carbon peak at 184 ppm (Fig. 2c; ESI, Fig. S7†). The thermogravimetric analysis (TGA) of HqTp displays thermal stability up to 380 °C (ESI, Fig. S9†). Brunauer–Emmett–Teller (BET) analysis, using N2 adsorption at 77 K shows a moderate surface area of 113 m2 g−1 (ESI, Fig. S10†). The non-local density functional theorem (NLDFT) provides a sharp pore size distribution around 1.5 nm (ESI, Fig. S11†).
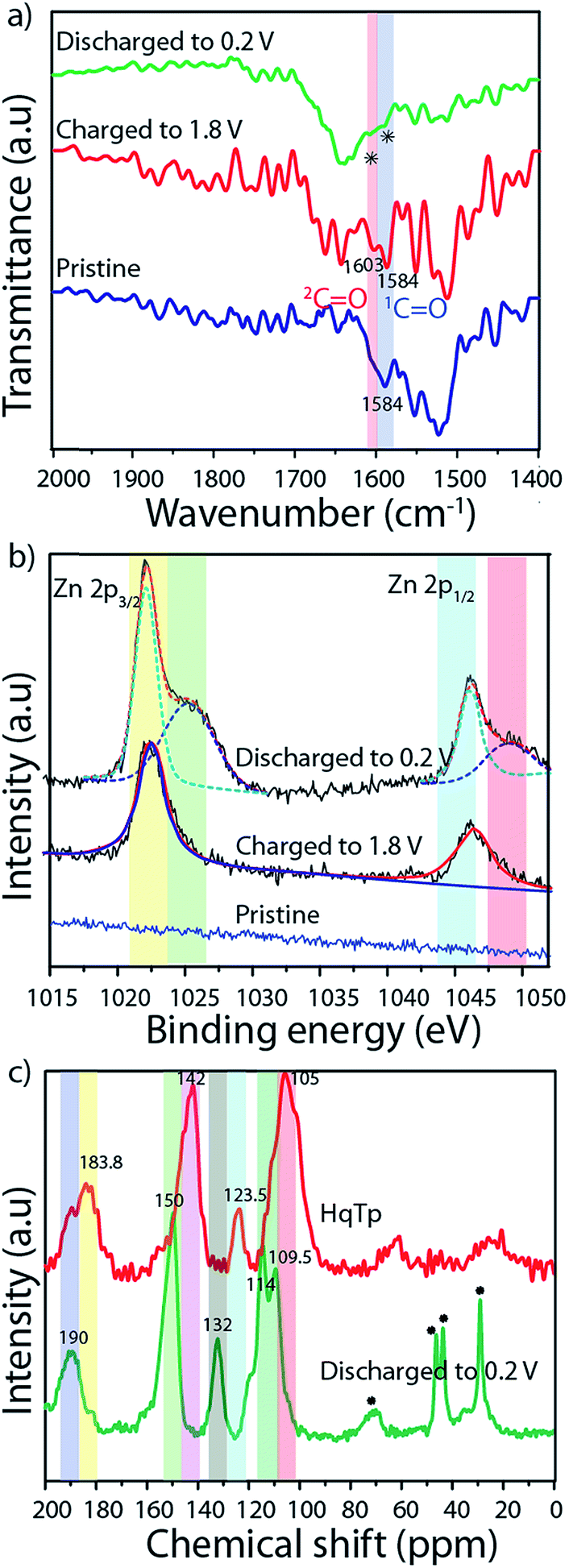 | ||
| Fig. 2 (a) FT-IR of pristine; charged and discharged cathodes. (b) XPS analysis of pristine; charged and discharged cathodes. (c) 13C CP-MAS NMR of HqTp and the discharged cathode (all the charging and discharge potential are represented vs. Zn/Zn2+ reference). | ||
The presence of CO moieties in HqTp COF are principally responsible for the storage of Zn2+ ions. These are the C3 symmetric CO groups resulting from the enol to keto tautomerism and the C2 symmetric CO group from electrochemically oxidized hydroquinone linker8 (Fig. 1a, c, and 3a; ESI, Fig. S8†). The hexagonal 2D lattice enriched with a large number of CO groups, stacked with an interlayer distance of 3.4 Å, can efficiently host a significant amount of Zn2+ in between the two-dimensional layers. Also, the crystalline honey-comb structure of HqTp provides a unique pore size of 1.5 nm, which, we believe, further boosts the lucid movement of Zn2+ ions through the entire organic cathode without any interruptions.
 | ||
| Fig. 3 (a) Diagrammatic representation of the aqueous Zn/HqTp unit cell. SEM image of the (b) HqTp; (c) the pristine HqTp organic cathode. (d) The TEM image of HqTp. (e) TEM elemental mapping images of carbon and zinc. (f) SEM elemental mapping of carbon (grey) and (g) zinc (green) of organic cathode discharged to 0.2 V vs. Zn/Zn2+. (h) The TEM image of HqTp organic cathode at the discharged state. It shows the presence of COF as well as CNF present in the electrode (all the charging and discharge potential are represented vs. Zn/Zn2+ reference). | ||
DFTB calculations were used to probe the nature of the interaction of Zn2+ with the COF backbone. They suggest the possibility of the prominent interlayer interactions of Zn2+ ions with nucleophilic centers of COF (Fig. 1e, ESI, Fig. S3 and S4†). The CO⋯Zn (2–2.3 Å) and Zn⋯N–H distances (∼2.0 Å) agree with the possibility of intermolecular interactions. The DFTB models of AA and slipped AA COF⋯Zn2+ interaction show various structural possibilities including a formation of a Zn2+ ion cluster inside the pores. Also, a large enhancement in the per-layer stabilization is observed for the discharged HqTp⋯Zn2+ adduct (−182.0 kcal mol−1) than the pristine COF (−40.0 kcal mol−1) which indicates favorable Zn2+ ion interaction. To understand the role of electrochemical active sites for Zn2+ anchoring in concerned COF, we have performed and compared the electrostatic potential mapping (ESP) of HqTp with benzene linked analog PaTp COF (ESI, Fig. S5 and S30†). ESP of the unit cell of HqTp displays a potential range from −0.33 au (red) to 0.061 au (blue). Herein, the negative ESP (red) reflects the affinity of the sites toward the Zn2+ uptake. Therefore, from ESP, it is clear that the quinone ‘CO’ and Tp ‘CO’ in HqTP exhibits more nucleophilicity compared to the ‘N–H’ sites. Whereas, the ESP of benzene linked PaTp COF showcases only Tp ‘CO’ groups as active sites for the uptake of Zn2+.
We have analyzed the working electrode (cathode) at three different electrochemical conditions to elucidate the probable mechanism of the Zn2+ ion interactions within the HqTp COF during the electrochemical charging and discharging process in a zinc ion unit cell; viz: (1) a pristine cathode (an electrode prior to any electrochemical perturbation/cycling); (2) a cathode at a fully discharged state (up to 0.2 V vs. Zn/Zn2+; where Zn2+ do interact with HqTp) and (3) fully charged up to 1.8 V vs. Zn/Zn2+ (at this stage, Zn2+ are withdrawn from the HqTp cathode). Then, we have characterized the electrodes, followed by washing with water.
FT-IR spectroscopy of the pristine organic cathode is found to be similar to the pristine HqTp (Fig. 2a; ESI, Fig. S6b†). The CO group from β-ketoenamine framework appeared at 1584 cm−1. After charging up to 1.8 V vs. Zn/Zn2+, the hydroquinones are electrochemically oxidized to quinones. This has been confirmed by the observation of a new stretching vibrational peak formed at 1603.0 cm−1. When the system is discharged to 0.2 V vs. Zn/Zn2+, a simultaneous diminishing of both CO groups is evident from the FT-IR spectra. Still, a diligent observation shows the presence of a very weak stretching peak at the CO region of the discharged cathode which indicates that the first cycle of discharging is not sufficient for the full utilization of the COs in the framework. Additionally, to figure out the efficiency of the COF based organic cathode, we have strategically performed 500 charge–discharge cycles for a cell. After complete discharging up to 0.2 V vs. Zn/Zn2+, the cathode was again subjected to FT-IR analysis (ESI, Fig. S6b†). The full disappearance of the CO stretching peaks suggests the prospect of the effective utilization of active groups in HqTp for the interaction with Zn2+.
We have performed 13C CP-MAS NMR spectroscopy of HqTp and discharged (0.2 V vs. Zn/Zn2+) HqTp cathode to investigate for any chemical modifications at the atomic level of the framework (Fig. 2c; ESI, Fig. S7†). It showcases a down field chemical shift of the CO peak from 184.0 ppm (for HqTp) to 190.0 ppm (for discharged HqTp). Similarly, the distinct 5–7 ppm downfield chemical shifts have been observed for every peak of the discharged HqTp cathode compared to pristine HqTp COF (ESI, Fig. S7 and S8†). It could be due to the appearance of the deshielding effect because of intercalated electropositive Zn2+ ions, which decrease the electron density in the framework.9
The X-ray photoelectron spectroscopy (XPS) provides information about the chemical state of the concerned elements in the pristine, charged, and discharged conditions of HqTp. In the XPS profile of the discharged HqTp electrode (0.2 V vs. Zn/Zn2+), the sharp peaks are visible at the binding energy of 1022.0 and 1045.0 eV, which correspond to Zn 2p3/2 and Zn 2p1/2. Meanwhile, the intensity of the XPS peaks of Zn2+ in the charged electrode diminished by half concerning the discharged peak due to the removal of Zn2+ ions from the cathode (Fig. 2b; ESI, Fig. S17–S19†).
Moreover, the ex situ analysis is extended to electronic imaging techniques with elemental mapping. The TEM images revealed the layered ribbon-like morphology of HqTp organic cathode with the lateral dimension of ∼200 nm length and ∼50 nm width (Fig. 3d and h; ESI, S12 and S13†). It also displayed the presence of carbon nanofiber (CNF) which has been used as an electrical conductivity amplifier in the cathode. The elemental mapping of carbon and zinc shows an efficient distribution of carbon (red color) both in COFs as well as CNF (Fig. 3e; ESI, Fig. S13†). However, the zinc distribution is solely present in COF (green color) and not in CNF. It indicates that the Zn2+ ions only interact with COF due to the specific functional moieties and, here, CNF is free from any interaction with Zn2+ ions. It also points out that the role of CNF is limited only for improving electrical conductivity.
Furthermore, to explore the morphological evolution of HqTp organic cathode; the discharged; charged and the pristine electrodes were subjected to the SEM analysis. Notably, in the pristine organic cathode, the COF samples are well distinguished as the previous sheet-like morphology of HqTp COF (Fig. 3b and c; ESI, Fig. S14 & 15†). Also, we have recorded the elemental mapping of C, N, O, and Zn in the discharged HqTp which suggests the uniform distribution of Zn2+ within the HqTp cathode (Fig. 3f and g; ESI, Fig. S15†). Moreover, the intactness of morphology even after 500 continuous charge–discharge cycles proves the good stability of the organic cathode without any leaching in harsh electrochemical conditions (ESI, Fig. S16†).
The reversible and efficient intermolecular interaction of HqTp⋯Zn2+ has further allowed us to fabricate an aqueous rechargeable zinc ion battery (Zn/HqTP unit cell) (Fig. 4a). The electrochemical impedance spectroscopy (EIS) analysis of the fabricated cell at OCV condition has been provided in Fig. 4b. From the plot, an equivalent series resistance (ESR) value of 0.8 Ω and a charge transfer resistance value of 91 Ω were obtained. Exploiting the over-potential advantages evolved because the usage of the zinc-salt, there are reports on aqueous zinc-ion batteries operable between the potential window of 0.2 to 2.2 V vs. Zn/Zn2+.4,10 Considering this, the cathode materials should be compatible within the stability window offered by the aqueous electrolyte.4,10 Moreover, it is highly desirable for the cathode material to exhibit its redox properties at high potentials (≥0.6 V) vs. Zn/Zn2+ to position it for practical applications. However, herein, we have chosen the potential range of 0.2 V to 1.8 V vs. Zn/Zn2+ to showcases the complete charge-storage features of HqTp COF.
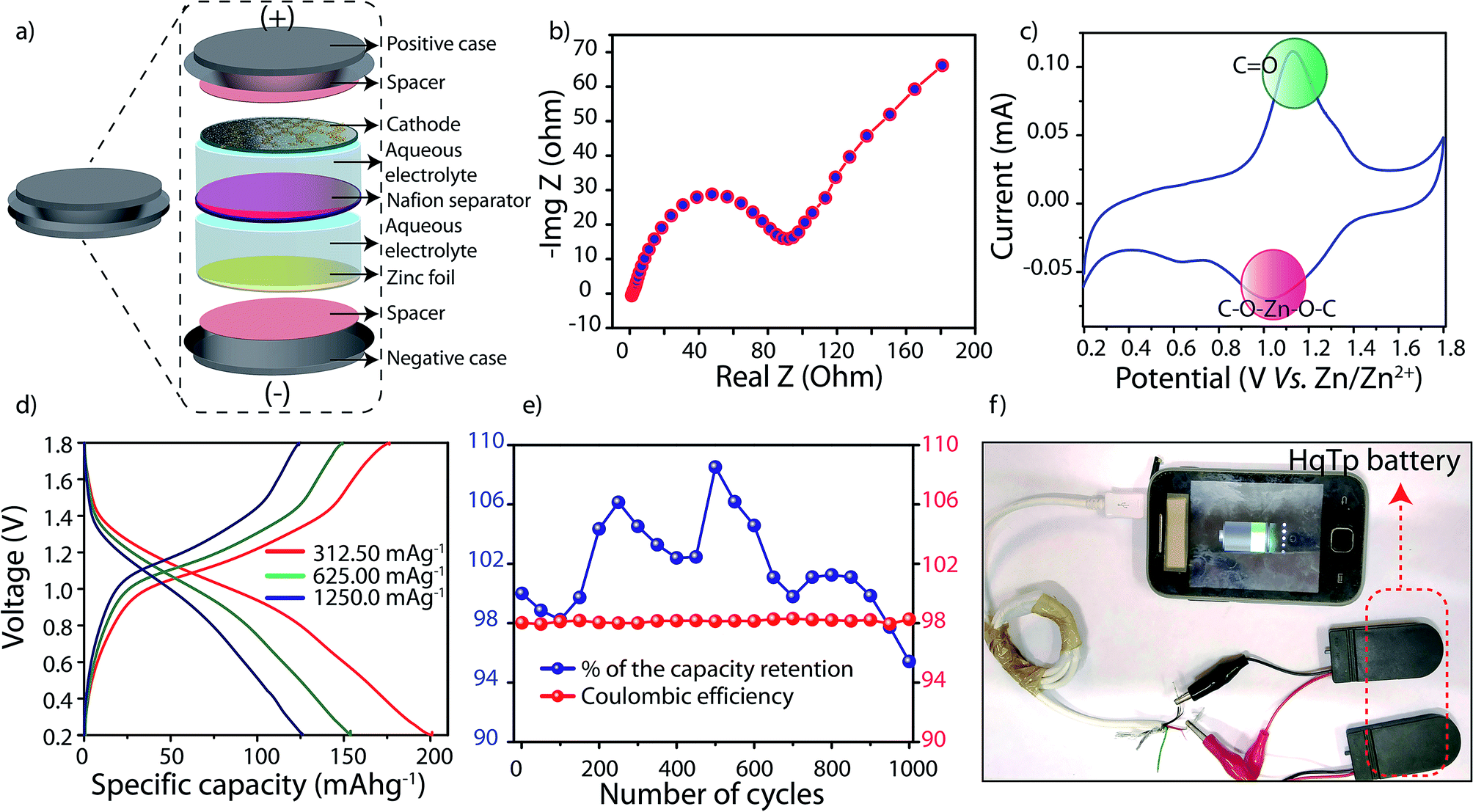 | ||
| Fig. 4 (a) Diagrammatic representation of the fabrication of lab scale aqueous Zn/HqTP unit cell. Electrochemical characterizations of Zn/HqTP cell. (b) The impedance analysis of Zn/HqTP cell. (c) CV profile of Zn/HqTP cell. (d) Charge–discharge profile of Zn/HqTP cell. (e) Long-life cyclic stability and coulombic efficiency plot at 3750 mA g−1. (f) Digital photograph of charging a smart-phone by Zn/HqTP cell. | ||
From the cyclic voltammetry (CV)-profile obtained at the scan rate of 0.1 mV sec−1, a distinct pair of redox peaks has been observed (Fig. 4c; ESI, Fig. S20†). The prominent sharp redox peaks at 1.12/1.0 V vs. Zn/Zn2+ correspond to the quinone oxidation and reduction (Onset potentials: 0.9/1.4 V vs. Zn/Zn2+). Also, an enhancement of the current gain has been observed at the higher CV scan rates of 0.5 and 1 mV sec−1 (Fig. S21†). To decipher the role of hydroquinone in the specific capacity of the COF, we have assembled a Zn/HqTP unit cell with a non-hydroquinone analog of HqTp, i.e., PaTp as an organic cathode (Zn/PaTp unit cell) (ESI, Fig. S30†). However, a significantly less current response was noted from the CV, i.e., only 18.0% compared to the Zn/HqTp cell at the same scan rate (ESI, Fig. S23†). Such observation, in turn, signifies the Zn2+ receptor capability of the hydroquinone functionality in the host framework.
The galvanostatic charge–discharge (GCD) profile of the Zn/HqTP cell displays its charge-storage properties in the adopted potential window without a voltage plateau. Herein, the (GCD) analysis shows that the HqTp cathode presents a significantly high discharge capacity of 276.0 mA h g−1 at the current rate of 125.0 mA g−1 (Fig. 4d; ESI, Fig. S22†). Notably, many cathode materials have been explored for zinc-ion batteries showing charge-storage properties despite the absence of sharp voltage plateaus.11 Similarly, the same trend is observed for COF based Li or Na ion batteries as well.2c,e The lack of a voltage plateau of HqTp could be due to the polymeric porous nature of the material and it further indicates a hybrid charge-storage behavior.11 Moreover, it is worth mentioning that HqTp exhibits good performance in terms of the obtained discharge capacity as well as cyclic stability as an organic polymer cathode in aqueous zinc ion batteries (Table S4†).3,12
Meanwhile, the PaTp COF exhibits only 121.0 mA h g−1 discharge capacity at the same current rate (125 mA g−1) (ESI, Fig. S24†). Considering the discharge capacity of 276.0 mA h g−1, we have found that 7.5 number of Zn2+ ions interact in the unit cell of HqTp (ESI, S-9†). Moreover, at a current rate of 3750.0 mA g−1 where the organic cathode displays the specific capacity of 85.0 mA h g−1, we have carried out a long-term cyclic stability experiment of the HqTp zinc ion battery. The cyclic stability exceeded beyond 1000 cycles with 95% retention of its initial capacity (Fig. 3d). Moreover, the coulombic efficiency of the cell is well maintained to 98% throughout the 1000 charge–discharge cycles (Fig. 4e). Although the symmetric shape of reversible peaks are absent in the CV profile, the coulombic efficiency over 1000 cycles indicate the high reversible charge storage characteristics of the HqTp COF. Besides, considering the average voltage of 0.87 V (Fig. S27†), a high energy density (240.0 W h kg−1) has been obtained at the power density of 109.0 W kg−1 as provided in the Ragone plot at the energy density of 75.0 W h kg−1, the power density increases to 3262.0 W kg−1 (ESI, Fig. S26†). We could assemble four 1.75 V HqTp-zinc ion cells in a series connection and directly used for charging a smart-phone device (Fig. 4f; ESI, Fig. S29†).
Conclusions
In summary, we have demonstrated hydroquinone stitched HqTp COF as a new class of organic cathode material for rechargeable aqueous zinc ion batteries. Herein, HqTp COF serves as a rich functional platform for binding Zn2+ ions. The efficient inter-layer interaction of these divalent Zn2+ ions with CO and N–H from the adjacent layers provides an excellent discharge capacity (276.0 mA h g−1 at 125.0 mA g−1). We believe, the utility of COF as an organic cathode in such devices may lead to the further invention of more powerful crystalline polymer organic cathodes in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. K. M, V. V, M. G., and A. H acknowledge UGC and CSIR for SRF. M. A. A. thanks the Materials Chemistry Consortium for computational resources on THOMAS (EP/P020194). S. K. acknowledges CSIR for funding through the project TLP003526. R. B. acknowledges IISER-Kolkata start-up research and DST- Swarnajayanti Fellowship grant for funding.Notes and references
- (a) A. P. Cote, A. I. Benin, N. W. Ockwig, A. J. Matzger, M. O'Keeffe and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed; (b) P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed; (c) S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC; (d) S. Wang, Q. Wang, P. Shao, Y. Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2017, 139, 4258–4261 CrossRef CAS PubMed; (e) J. W. Crowe, L. A. Baldwin and P. L. McGrier, J. Am. Chem. Soc., 2016, 138, 10120–10123 CrossRef CAS PubMed; (f) A. Sun, B. Aguila, J. Perman, N. Nguyen and S. Ma, J. Am. Chem. Soc., 2016, 138, 15790–15796 CrossRef PubMed; (g) X. Han, Q. Xia, J. Huang, Y. Liu, C. Tan and Y. Cui, J. Am. Chem. Soc., 2017, 139, 8693–8697 CrossRef CAS PubMed; (h) X.-H. Liu, C.-Z. Guan, S.-Y. Ding, W. Wang, H.-J. Yan, D. Wang and L.-J. Wan, J. Am. Chem. Soc., 2013, 135, 10470–10474 CrossRef CAS PubMed; (i) M. Dogru, A. Sonnauer, A. Gavryushin, P. Knochel and T. Bein, Chem. Commun., 2011, 47, 1707–1709 RSC; (j) L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC; (k) E. Q. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. H. Chen and D. L. Jiang, Science, 2017, 357, 673–676 CrossRef CAS PubMed.
- (a) J. Zhou and B. Wang, Chem. Soc. Rev., 2017, 46, 6927–6945 RSC; (b) C. R. DeBlase, K. E. Silberstein, T. T. Truong, H. D. Abruna and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS PubMed; (c) S. Wang, Q. Wang, P. Shao, Y. Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2017, 139, 4258–4261 CrossRef CAS PubMed; (d) M. -S. Kim, W.-J. Lee, S.-M. Paek and J. K. Park, ACS Appl. Mater. Interfaces, 2018, 10, 32102–32111 CrossRef CAS PubMed; (e) S. Gu, S. Wu, L. Cao, M. Li, N. Qin, J. Zhu, Z. Wang, Y. Li, Z. Li, J. Chen and Z. Lu, J. Am. Chem. Soc., 2019, 141(24), 9623–9628 CrossRef CAS PubMed.
- (a) G. Fang, J. Zhou, A. Pan and S. Liang, ACS Energy Lett., 2018, 3, 2480–2501 CrossRef CAS; (b) D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 CrossRef CAS; (c) A. Konarov, N. Voronina, J. H. Jo, Z. Bakenov, Y.-K. Sun and S.-T. Myung, ACS Energy Lett., 2018, 3, 2620–2640 CrossRef CAS; (d) M. Ghosh, V. Vijayakumar and S. Kurungot, Energy Technol., 2019, 1900442 CrossRef.
- (a) Q. Zhao, W. Huang, Z. Luo, L. Liu, Y. Lu, Y. Li, L. Li, J. Hu, H. Ma and J. Chen, Sci. Adv., 2018, 4, eaao1761 CrossRef PubMed; (b) Z. Guo, Y. Ma, X. Dong, J. Huang, Y. Wang and Y. Xia, Angew. Chem., Int. Ed., 2018, 57, 11737–11741 CrossRef CAS PubMed; (c) D. Kundu, P. Oberholzer, C. Glaros, A. Bouzid, E. Tervoort, A. Pasquarello and M. Niederberger, Chem. Mater., 2018, 30, 3874–3881 CrossRef CAS; (d) F. Wan, L. Zhang, X. Wang, S. Bi, Z. Niu and J. Chen, Adv. Funct. Mater., 2018, 1804975 CrossRef; (e) L. Ma, S. Chen, H. Li, Z. Ruan, Z. Tang, Z. Liu, Z. Wang, Y. Huang, Z. Pei, J. A. Zapien and C. Zhi, Energy Environ. Sci., 2018, 11, 2521–2530 RSC.
- (a) S. Kandambeth, A. Mallick, B. Lukose, M. V. Mane, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2012, 134, 19524 CrossRef CAS PubMed; (b) S. Chandra, D. R. Chowdhury, M. Addicoat, T. Heine, A. Paul and R. Banerjee, Chem. Mater., 2017, 29, 2074–2080 CrossRef CAS.
- (a) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed; (b) S. D. Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461–1479 CrossRef.
- (a) A. Haaland, Angew. Chem., Int. Ed., 1989, 28, 992–1007 CrossRef; (b) M. Yamakawa and R. Noyori, Organometallics, 1999, 18, 128–133 CrossRef CAS.
- (a) E. W. L. Chan and M. N. Yousaf, J. Am. Chem. Soc., 2006, 128, 15542–15546 CrossRef CAS PubMed; (b) C. Nicosia and J. Huskens, Mater. Horiz., 2014, 1, 32–45 RSC.
- (a) J. C. Hammel and J. A. S. Smith, J. Chem. Soc. A, 1969, 2883–2887 RSC; (b) C. A. Wilkie and D. T. Harworth, J. Inorg. Nucl. Chem., 1978, 40, 195–197 CrossRef CAS.
- (a) M. H. Lee, S. J. Kim, D. Chang, J. Kim, S. Moon, K. Oh, K.-Y. Park, W. M. Seong, H. Park, G. Kwon, B. Lee and K. Kang, Mater. Today, 2019 DOI:10.1016/j.mattod.2019.02.004; (b) L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- (a) H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Nat. Energy, 2016, 1, 16039 CrossRef CAS; (b) D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119–16126 CrossRef CAS; (c) S. Chen, L. Ma, K. Zhang, M. Kamruzzaman, C. Zhia and J. A. Zapien, J. Mater. Chem. A, 2019, 7, 7784–7790 RSC; (d) C. Xia, J. Guo, Y. Lei, H. Liang, C. Zhao and H. N. Alshareef, Adv. Mater., 2018, 30, 1705580 CrossRef PubMed; (e) Z. Luo, L. Liu, J. Ning, K. Lei, Y. Lu, F. Li and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 9443–9446 CrossRef CAS PubMed; (f) S. Wang, Q. Wang, P. Shao, Y. Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2017, 139(12), 4258–4261 CrossRef CAS PubMed.
- (a) H. Karami, M. F. Mousavi and M. Shamsipur, J. Power Sources, 2003, 117, 255–259 CrossRef CAS; (b) H.-Y. Shi, Y.-J. Ye, K. Liu, Y. Song and X. Sun, Angew. Chem., Int. Ed., 2018, 57, 16359–16363 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section. See DOI: 10.1039/c9sc03052b |
| This journal is © The Royal Society of Chemistry 2019 |