
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of relative humidity on the gas transport properties of zeolite A/PTMSP mixed matrix membranes†
Ana Fernández-Barquín a,
Riccardo Reab,
Davide Venturib,
Marco Giacinti-Baschettib,
Maria Grazia De Angelisb,
Clara Casado-Coterillo*a and
Ángel Irabiena
a,
Riccardo Reab,
Davide Venturib,
Marco Giacinti-Baschettib,
Maria Grazia De Angelisb,
Clara Casado-Coterillo*a and
Ángel Irabiena
aDepartment of Chemical and Biomolecular Engineering, ETSIIT. Universidad de Cantabria, Avda Los Castros S/N, 39005 Santander, Spain. E-mail: casadoc@unican.es
bDipartimento di Ingegneria Civile Chimica Ambientale e dei Materiali, Alma Mater Studiorum-Universita di Bologna, Via Terracini 28, Bologna, Italy
First published on 17th January 2018
Abstract
Increasing the knowledge of the influence of water vapor in new mixed matrix membranes (MMMs) could favor the integration of novel membrane materials in the recovery of CO2 from wet industrial streams. In this work, the water vapor effect on the N2, CH4 and CO2 permeability through MMMs comprised of 20 wt% hydrophilic zeolite 4A in hydrophobic PTMSP polymer were investigated in the relative humidity range 0–75%. While in the pure PTMSP membranes, the permeability of all gases decreases with water vapor activity, with almost unchanged CO2/N2 and CO2/CH4 selectivities, in zeolite A/PTMSP MMMs, the CO2 permeability increases with increasing water content in the system up to 50% R.H., resulting in an increase in CO2/N2 and CO2/CH4 selectivities with respect to pure PTMSP. Gas sorption was studied so that the effect the residual humidity in the zeolite 4A has on the sorption of the different gases helped explaining the permeability observations. The sorption and humid permeation behavior were evaluated by a simple model equation based on the NELF theory, taking into account the multicomponent gas sorption and diffusion in the presence of humidity, as well as the counteracting effects of the hydrophobic PTMSP and hydrophilic zeolite A in a very accurate way.
1. Introduction
Carbon dioxide emissions from fossil fuel combustion are a major contributor to climate change and global warming. One way to reduce CO2 emissions to the atmosphere is carbon capture, utilization and storage (CCUS). Nowadays, post-combustion CO2 capture is the strategy that can be implemented with the lowest technical risk and thus has the greatest near-term potential in the CO2 capture.1–3Among novel concepts for post-combustion capture, membrane separation is emerging as a promising technology to solve the drawbacks of conventional chemical absorption.4,5 Compared to the traditional technologies, membrane gas separation shows better prospects for industrial application due to its module compactness and modularity, operation simplicity and reliability, cost efficiency, energy saving, and environmental impact.6,7
Polymeric membranes show the highest maturity among the existing membrane materials for CO2 separation, although relatively few polymeric membranes have been commercialized for gas separation, compared to the number of polymeric materials commercially available.8 This is attributed to various reasons such as the lack of performance on productivity and separation selectivity, as polymeric membranes normally present a trade-off between selectivity and permeability,9 as well as module fabrication and lack of information regarding the performance in the presence of impurities in the CO2 streams, such as water vapour.10
Mixed matrix membranes (MMMs) constitute one of the ways of enhancing the performance of polymeric membranes. MMM combine the molecular sieving effect of inorganic fillers with the good processability of polymers achieving new materials with synergistic functional properties.11 Material selection for both polymeric matrices and sieve phases is a key aspect in the synthesis of MMMs. As the performance of the MMM is limited by the polymer performance, a highly performing polymer should be used as continuous matrix.12,13 In this work, poly(1-trimethylsilyl-1-propyne) (PTMSP), a polymer with one of the highest gas permeabilities, was chosen. PTMSP is a glassy polymer with extremely high fractional free volume (0.29),14,15 whose glassy structure leads to a low chain mobility and a glass transition temperature higher than 523 K, making it a good material for high temperature gas separations.16,17 However, its large gas permeability is coupled with low ideal selectivity9 and a tendency to undergo physical ageing,18 which limits the applicability of PTMSP as pure membrane material. We have overcome these drawbacks by adding small-pore zeolites with low Si/Al ratio to the hydrophobic PTMSP polymer, and both components showed good compatibility so that no defects were found in the interphase between the polymer and the filler.19
The effect of minor compounds frequently present in industrial separations can affect significantly the polymer membrane behaviour.20,21 In particular, the influence of water vapour on membrane permeability and selectivity is stronger than that of other minor components regarding competitive sorption, plasticization and ageing.2 Besides, this influence depends on the hydrophilic or hydrophobic character of the membrane material or the affinity of water with the different gases.22,23 In spite of the amount of works on high free volume glassy membranes,16,24,25 to the best of our knowledge, only a few have dealt with the water effect on the gas permeability through such polymer membranes as PTMSP, PIM-1 and thermally rearranged (TR) materials.21,26,27 In all such membranes, the presence of humidity reduced the gas permeability affecting to a lesser extent the selectivity. Therefore, there is still limited information on the effect of water vapour in these high free volume polymer membranes,17,28 and even less in MMMs.
In this work, we studied the water vapour effect on N2, CH4 and CO2 gas sorption and permeation through the 20 wt% zeolite A/PTMSP MMM developed in our laboratory19 in the range 0–75% relative humidity (R.H.). This MMM is comprised of a hydrophilic porous filler in a hydrophobic high free volume matrix. The performance of the MMM has been compared to that of pure PTMSP membranes. These experimental results have been described by a simple but effective model that describes the permeation process under humid conditions in the framework of the solution-diffusion mechanism. In particular, the free volume theory was considered to account for the gas diffusivity29 while the solubility, taking into account the multicomponent competitive sorption of gas and water in the membrane,30 was modelled by the NELF model. For this purpose, the N2, CH4 and CO2 sorption curves of this MMM have been also experimentally obtained by means of the pressure-decay technique to be introduced in the model applied in this work.
2. Results and discussion
In Fig. 1, the N2, CH4 and CO2 permeabilities obtained at 35 °C and 1 bar of upstream pressure, in the range of 0–75% R.H. are plotted as a function of water vapour activity. The dry gas permeability of pure PTMSP membranes agrees with the range reported for other PTMSP membranes in literature.16,24,25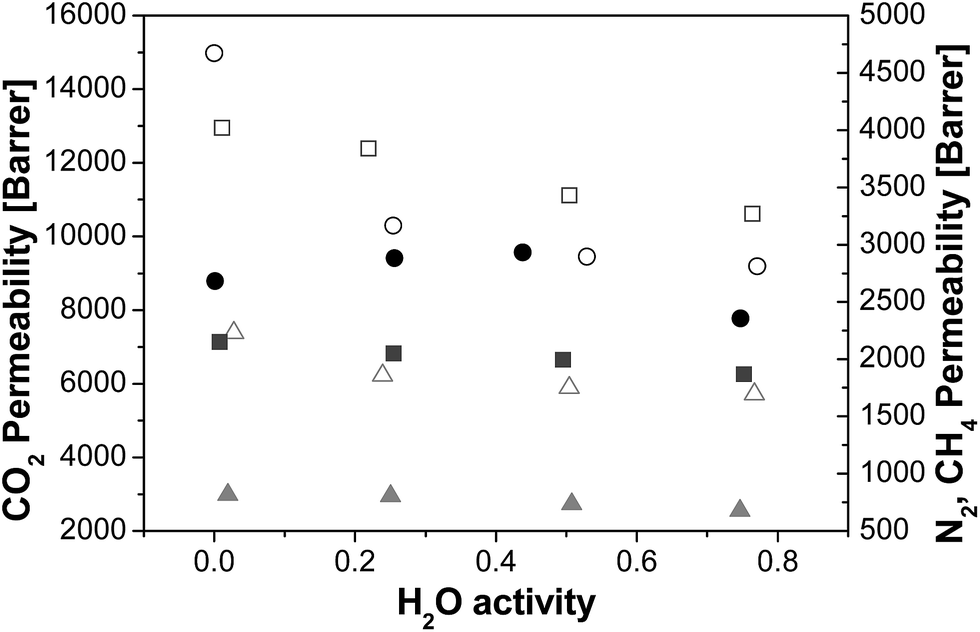 | ||
| Fig. 1 Permeability (barrer) at different relative humidity (T = 35 °C p = 1 bar) for the zeolite A/PTMSP MMM (full symbols) and PTMSP membranes (void symbols). N2 (grey triangles), CH4 (dark grey squares) and CO2 (black circles). | ||
Interestingly, the addition of zeolite A to PTMSP in the MMM caused a decrease of permeability in the order of magnitude of 50% for N2 and CH4, and to a lesser extent for CO2. This has been previously attributed to the MMM dual-layer morphology, as already reported in a previous work.19 This MMM is comprised of two layers, one almost pure PTMSP layer at the top, and a zeolite layer at the bottom where the PTMSP acts as binder and the zeolite particles partly occupy the free volume of the final MMM. This contributes to the rigidification and densification of the membrane matrix, by reducing further the flexibility of the polymer chains and somewhat inhibiting the gas diffusion.31 In that work, we observed that the CO2 permeability decreased with increasing temperature up to a maximum zeolite loading that depended on the composition of the zeolite particles, thus revealing the molecular sieving effect of the zeolite A in the PTMSP matrix.
Regarding the water effect, the gas permeability through PTMSP membranes generally decreased with increasing water activity for all gases investigated, in agreement with other polyimide membranes made of PIM-1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21,32 or Matrimid,29 due to the reduction in diffusivity when water molecules occupy part of the free volume of the polymer. The gas permeability through the zeolite A/PTMSP MMM also decreased for N2 and CH4 with increasing water activity, while the CO2 permeability showed a slight increase to about 50% RH, followed by a slight decrease. At average relative humidity, the CO2 permeability of the MMM is similar to the value measured in pure PTMSP membranes. The behaviour of the humid gas permeability in zeolite A/PTMSP MMM can be correlated qualitatively with the gas sorption in the pure zeolite A particles, at two different hydration levels. It is well known that zeolite A is extremely hydrophilic, so that it contains some water adsorbed from the environment, at ambient conditions.33,34 Such water can be removed by treating the system at high temperatures. The exact quantification of the amount of water adsorbed by the zeolite as a function of relative humidity and temperature was beyond the scope of the present paper, although we checked by thermal gravimetric analysis (shown in Fig. S1 of the ESI†), the water content in the undried and dried zeolite, obtaining a 12.6% and 5.7% content of water in the undried, “as received”, and dried at 105 °C zeolite, respectively, to account for the effect of the residual water content in the zeolite sample on the sorption of the different gases with different contents of humidity (Fig. S2 of the ESI†). Since the residual water content in the MMM is in the range 0.5 to 3.1 wt%, with increasing zeolite A loading, which accounts for the dispersion of the hydrophilic zeolite in the hydrophobic polymer matrix, these data can help interpreting the humid permeability results.
21,32 or Matrimid,29 due to the reduction in diffusivity when water molecules occupy part of the free volume of the polymer. The gas permeability through the zeolite A/PTMSP MMM also decreased for N2 and CH4 with increasing water activity, while the CO2 permeability showed a slight increase to about 50% RH, followed by a slight decrease. At average relative humidity, the CO2 permeability of the MMM is similar to the value measured in pure PTMSP membranes. The behaviour of the humid gas permeability in zeolite A/PTMSP MMM can be correlated qualitatively with the gas sorption in the pure zeolite A particles, at two different hydration levels. It is well known that zeolite A is extremely hydrophilic, so that it contains some water adsorbed from the environment, at ambient conditions.33,34 Such water can be removed by treating the system at high temperatures. The exact quantification of the amount of water adsorbed by the zeolite as a function of relative humidity and temperature was beyond the scope of the present paper, although we checked by thermal gravimetric analysis (shown in Fig. S1 of the ESI†), the water content in the undried and dried zeolite, obtaining a 12.6% and 5.7% content of water in the undried, “as received”, and dried at 105 °C zeolite, respectively, to account for the effect of the residual water content in the zeolite sample on the sorption of the different gases with different contents of humidity (Fig. S2 of the ESI†). Since the residual water content in the MMM is in the range 0.5 to 3.1 wt%, with increasing zeolite A loading, which accounts for the dispersion of the hydrophilic zeolite in the hydrophobic polymer matrix, these data can help interpreting the humid permeability results.
On one hand, from Fig. S2a,† it seems that the CO2 adsorption in the zeolite is not inhibited by the presence of water, possibly due to the higher affinity of CO2 for water molecules.29,35 On the other hand, we can see in Fig. S2b and S2c† that the uptake of CH4 and N2 in the dried zeolite sample is much higher than that measured on the undried one, which contains water molecules adsorbed from the atmosphere. This means that the water present in zeolite A does inhibit the sorption of gases like CH4 and N2 due to competitive effects. This behaviour is consistent with that observed in the humid permeability of the zeolite A/PTMSP MMM, where the relative humidity was detrimental for the permeation of N2 and CH4, but not for the transport of CO2 molecules. Besides, the shape of the isotherms is similar to those obtained for the zeolite A at the same conditions of this work, i.e. low pressures, by Palomino et al.36
The effect of water on permeability is clarified using the normalized gas permeability, defined as the ratio between the permeability at a given R.H. and the dry gas permeability, and plotted on Fig. 2. Indeed, the normalised gas permeability of pure PTMSP membranes shows a similar decrease for all three gases with increasing water activity, being somewhat stronger for CO2. In particular, the CO2 permeability through the PTMSP membranes is decreased by 40% from the dry value at a water activity of 0.75, whereas the permeability of N2 and CH4 is only reduced by 20%. The data agree with the reduction of N2 permeability reported by Scholes et al.21 in mixed gas separation experiments and water activity below 0.40. The reduction in CO2 permeability reported by those authors was lower than 12%, thus smaller than the reduction observed here.
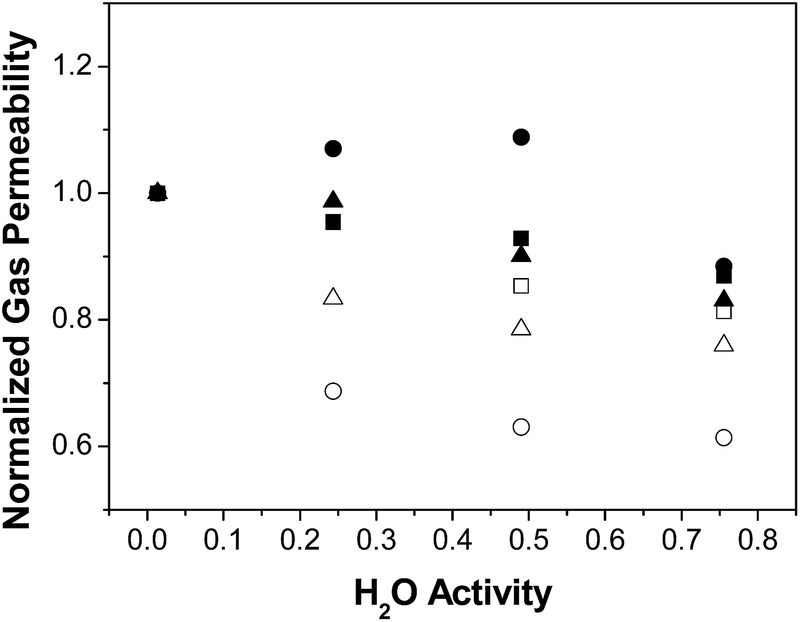 | ||
| Fig. 2 Normalised gas permeability P/Pdry through the zeolite A/PTMSP MMM, (full symbols) PTMSP membranes (void symbols): N2 (triangles), CH4 (squares), CO2 (circles). | ||
The large decrease of the CO2 permeability through the PTMSP membrane is related to a combination of competitive sorption of N2 and CH4 with water in the micro cavities,32 water clustering and pore blockage, which reduce diffusivity and CO2 solubility, thus compensating the effect of CO2 dissolution in water and the expected increase in CO2 permeation through the membrane.37 The permeability reduction with increasing water activity will be evaluated in the next section when discussing the modelling.38 Such decreasing trend in permeability was also observed for N2 and CH4 through the zeolite A/PTMSP MMM, though less pronounced than that of pure PTMSP membranes.
The deviations are attributed to the different experimental set-ups. The influence of water activity on gas permeability visible in Fig. 2 was definitively lower in the case of the zeolite A/PTMSP MMM than in the pristine PTMSP membrane. On average, the gas permeability of the MMM decreased by 9% and 15% from the dry permeability at 0.5 and 0.75 water activity, respectively. The decrease in gas permeability never exceeded 15 ± 2.6% in the R. H. range investigated. The error of experimental permeabilities, calculated by experiments repetition, is always below 3%. In contrast, the CO2 permeability increases up to 10% at a water activity of 0.50, due to the higher affinity of CO2 with water,29 the hydrophilic character of zeolite A33,34 and the higher sorption at higher water activities.39 The reduction of CO2 permeability through MMM observed in Fig. 2 may be attributed to the combination of competitive sorption between water and gas molecules and the plasticization by water of the high free volume polymer, as observed also by Nakamura et al.28 for PTMSP-related membranes in dry and wet states.
Fig. 3 shows the effect of the R.H. on the membrane separation performance in terms of the distance from the Robeson upper bound.9 In general, the CO2/N2 and CO2/CH4 selectivities of pure PTMSP are not strongly affected by the presence of water in the feed, with a slight increment in the distance from the upper bound upon increasing R.H., as observed in Matrimid.29 In contrast, it is clear that the overall permselectivity of the zeolite A/PTMSP MMM approaches the upper bound with increasing R.H., up to 50%. The difference in membrane performance in the presence of water vapour is attributed to the presence of the zeolite, which increased the membrane selectivity in humid conditions. This is due to the enhancement in the CO2 permeability of the MMM with the water content in the gas feed, which leads to an additional improvement in the CO2 selectivity in the presence of water, with respect to dry conditions.
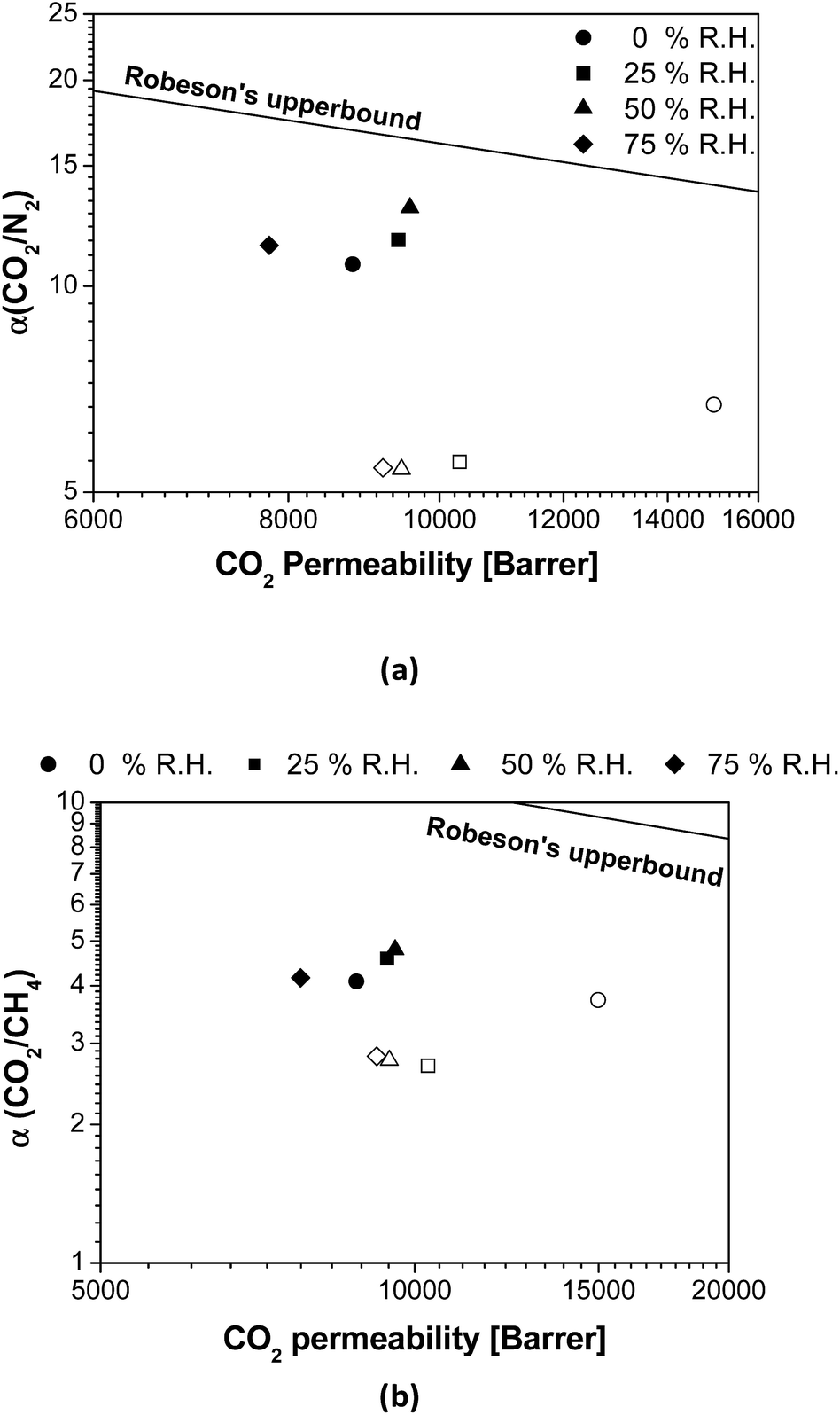 | ||
| Fig. 3 Comparison of (a) CO2/N2 and (b) CO2/CH4 selectivity as a function of R.H. with the Robeson's upper bound.9 Open symbols: PTMSP. Filled symbols: zeolite A/PTMSP MMM. The symbols indicate the direction towards increasing relative humidity values as follows: 0% (circles) < 25% (squares) < 50% (triangles) < 75% (rhombuses). | ||
The CO2 permeability of the MMM is slightly lower than that of the pristine PTMSP membrane and the increase in CO2/CH4 selectivity is not high enough to counteract the decrease in CO2 permeability, thus the separation performance is maintained at about the same distance from the upper bound. This agrees with other MMM formed by zeolites and glassy polymers, because of the combined effects of CH4 and water vapour on CO2 permeation.37 This is also attributed to the reduction of free volume by the space occupied by the zeolite particles and the rigidity imparted to the polymer matrix thereby,19 which has a larger effect than plasticization or competitive sorption.40 This issue requires some research effort in the materials knowledge, but as a starting point it can be noticed that the CO2 solubility of the hydrated zeolite is almost the same as the dried zeolite, in opposition to the solubility of N2 or CH4, as discussed above regarding Fig. S2.†
2.1. Modelling of permeability in the presence of water
| Component | T* (K) | p* (MPa) | ρ* (g cm−3) |
|---|---|---|---|
| PTMSP | 416 | 405 | 1.250 |
| CO2 | 300 | 630 | 1.515 |
| N2 | 145 | 160 | 0.943 |
| CH4 | 215 | 250 | 0.500 |
| H2O | 670 | 2400 | 1.050 |
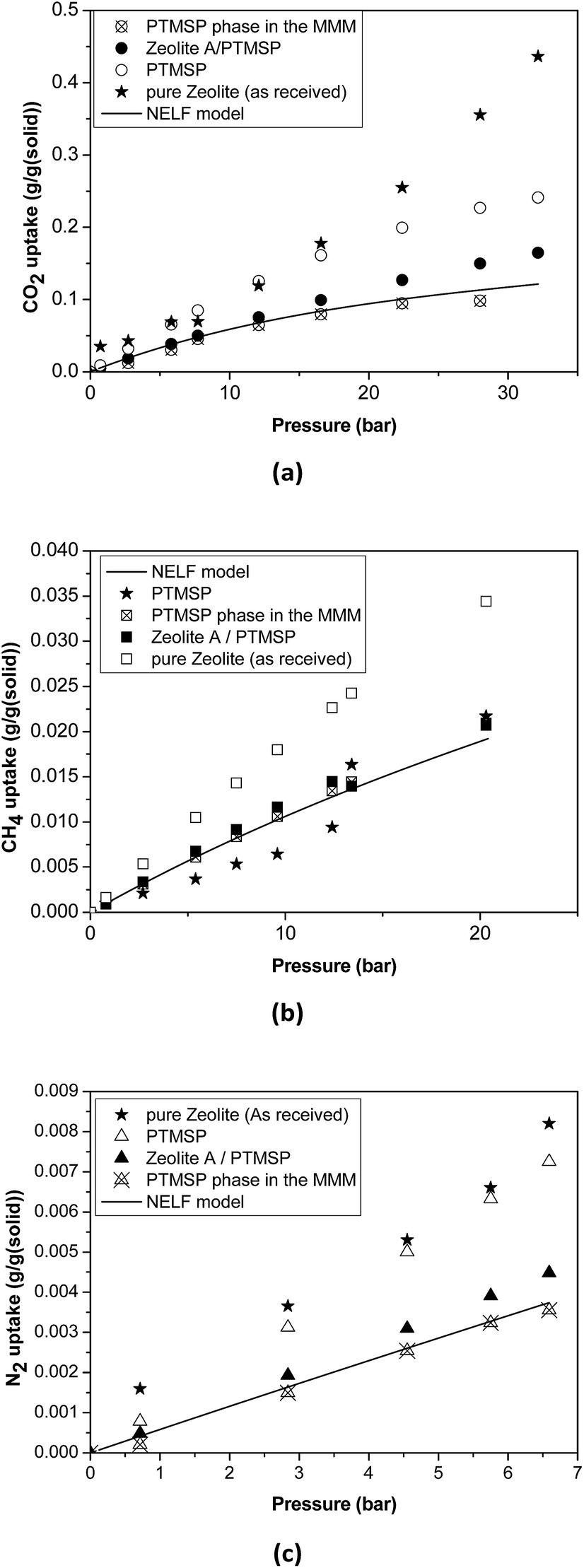 | ||
| Fig. 4 Sorption isotherms in the pure zeolite, pure PTMSP, zeolite A/PTMSP MMM and the PTMSP phase in the MMM. The solid line represents the adjustment with the NELF model. | ||
The kij, gas–polymer interaction parameter and ksw, swelling parameter calculated for PTMSP are reported in Table 2. Such parameters were adjusted with the pure gas solubility isotherms in pristine PTMSP membranes, as represented in Fig. S3 in the ESI.† For all gases, kij is slightly positive, in agreement with the values reported for other glassy polymers as PIM-147 and HAB-6FDA polyimides.48 For water vapour sorption, the binary parameters reported in Table 2 were adjusted using the isotherm reported by Scholes et al.21
| Components | PTMSP | |
|---|---|---|
| kij | ksw (MPa−1) | |
| CO2 | 0.075 | 0.009 |
| CH4 | 0.020 | 0.007 |
| N2 | 0.10 | 0 |
| H2O | −0.17 | 0 |
In addition, the model has been modified to take into account the gas sorption into MMMs.46,49 The mass of penetrant adsorbed in the MMM per unit mass of total solid, ΩM, is evaluated from the mass adsorbed on the pure filler per unit mass of filler, ΩF, and that adsorbed in the polymeric phase of the MMM per unit mass of polymer, ΩP,MM:
| ΩM = wF × ΩF + (1 − wF) × ΩP,MM | (1) |
For “ideal” MMMs, the sorption capacities of the polymer and the filler in the composite material are considered equal to the pure component values so a simple additive rule is used to determine the sorption capacity of the composite. However, mixed matrices of high free volume glassy polymers, such as PTMSP, may not show this additive behaviour because of how the inorganic rigid phase affects the volumetric space of the polymer.50 In a first approximation, however, we can assume that the adsorption capacity of the filler remains the same as in pure state:
| ΩF = Ω0F | (2) |
Eqn (2) becomes:
| ΩM = wFΩ0F + (1 − wF) × ΩP,MM | (3) |
| ΩP,MM ≠ Ω0P, ρP,MM ≠ ρ0P and FFVP,MM ≠ FFV0P |
By applying the NELF model,41–44 the value of ΩP,MM is univocally related to the MM polymer density ρP,MM:
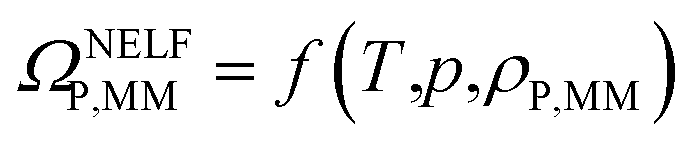 | (4) |
Eqn (4) was used to estimate the density of the polymer phase in the MMM, ρP,MM. This density value can be associated to the fractional free volume value in the MMM, FFVP,MM by
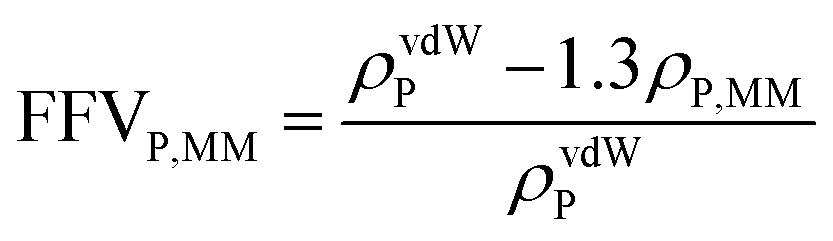 | (5) |
Thus, the sorption isotherm in the polymeric phase of the MMM, ΩP,MM, was adjusted by NELF model and eqn (4), looking at the MMM, pure zeolite and PTMSP membrane sorption data, and allowing ρP,MM and ksw to iterate, while keeping kij equal to the value obtained for pure PTMSP (Table 2). The sorption isotherms thus obtained are represented in Fig. 4 for CO2, CH4 and N2, respectively. Here, it is observed that the polymer phase in the MMM adsorbs less gas than in the pure state. This is attributed to the fact that the polymeric matrix is constrained by the presence of the zeolite.
The density of the polymer in the MMM can be lower or higher than the pure polymer density in the same operation conditions, depending on the interaction with the filler. For instance, the polymer density decreased, resulting in lower FFV and transport parameters in MMM where silica domains were generated in situ in a PTMSP solution via sol–gel.51 Other studies31,52 revealed that when pre-formed hydrophobic fumed non-porous silica particles were added to PTMSP, lower density of the MMM than the pure PTMSP membranes was achieved, resulting in additional free volume mainly due to poor adhesion. The porous zeolite particles are larger than those non-porous silica particles so they are not supposed to disrupt the chain packing and the changes in density and permeability are caused by the flow through the zeolite particles, which depends on the size and morphology, influencing the compatibility with the polymer matrix.53 In particular, the value of ρP,MM that best adjusts the data of all the gases is equal to 0.885 g cm−3, which is higher than the value for pure PTMSP membranes, and corresponds to a lower fractional free volume FFVP,MM of 0.162. The swelling coefficient ksw diminishes from 0.007 to 0.005 MPa−1, for CH4, and from 0.005 MPa−1 to zero for CO2. This indicates a rigidification of the polymer matrix in the MMM.
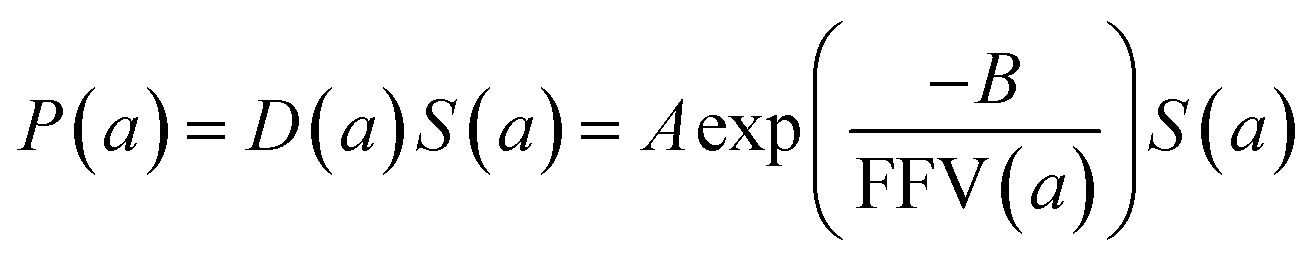 | (6) |
Since for rigid glassy polymers the diffusivity is known to dominate the transport of small substances,55 in this work, two approaches have been undertaken to model the influence of water activity on permeability. The first one considers that the predominant effect of water in the membrane matrix is due to the kinetic part of permeability in eqn (6), that is, the diffusivity. The humid gas permeability is thus expressed in terms of the humid fractional free volume, FFV, as function of the water activity a, with two adjustable parameters that depend on the gas penetrant i,29,56 using a simple but effective model reported recently by Ansaloni et al.,20 as
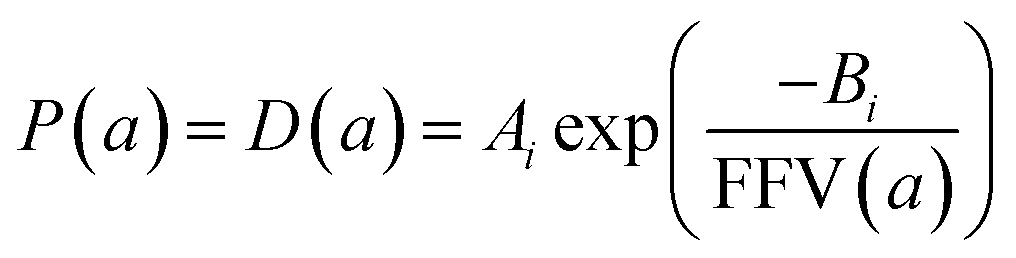 | (7) |
This approach estimates that the permeability reduction observed in the PTMSP-based membranes in the presence of water is only due to the reduction of the free volume because of the partial occupation by the absorbed water molecules and the zeolite particles embedded in the polymer matrix. No swelling is induced by the water vapour, due to the high rigidity of the membrane matrix and the low quantity of absorbed water by the polymer.21 When water is present, the FFV is estimated by considering both the occupied volume of the polymer chains, the zeolite particles and the water molecules adsorbed in the membrane matrix.57,58 The volume of a water molecule is assumed to be constant and proportional to its van der Waals volume over the whole water activity range. The expression that links the FFV with the water activity in this approach is given by eqn (8),
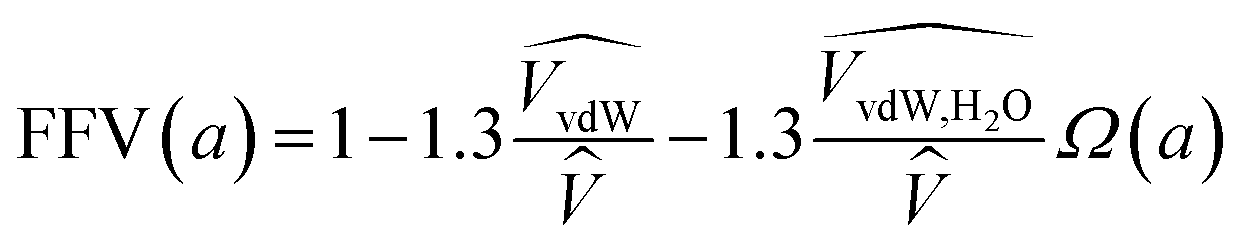 | (8) |
![[V with combining circumflex]](https://www.rsc.org/images/entities/i_char_0056_0302.gif) and
and 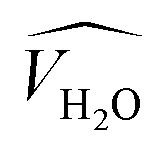 are the occupied volumes of the membrane and water,
are the occupied volumes of the membrane and water, 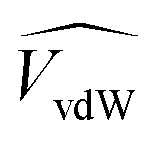 and
and 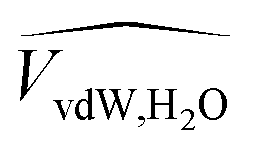 are the membrane and water van der Waals volume, respectively, estimated by Bondi's method.59 Ω(a) is the amount of water vapour in the membrane, expressed as g H2O/g membrane. In this work, this value for the pristine PTMSP membrane has been taken from water solubility isotherms reported in literature,21 while that of the zeolite A/PTMSP MMM has been calculated by the additive rule from water sorption in PTMSP and zeolite A,60 respectively. All the parameters used to calculate the FFV in eqn (8) are given in Table 3.
are the membrane and water van der Waals volume, respectively, estimated by Bondi's method.59 Ω(a) is the amount of water vapour in the membrane, expressed as g H2O/g membrane. In this work, this value for the pristine PTMSP membrane has been taken from water solubility isotherms reported in literature,21 while that of the zeolite A/PTMSP MMM has been calculated by the additive rule from water sorption in PTMSP and zeolite A,60 respectively. All the parameters used to calculate the FFV in eqn (8) are given in Table 3.
| Parameter | Value | Unit | Reference |
|---|---|---|---|
 (PTMSP) (PTMSP) |
0.728 | cm3 g−1 | 51 |
| ρ(PTMSP) | 0.750 | g cm−3 | 51 |
| ρ(zeolite A) | 0.720 | g cm−3 | Commercial data from Sigma-Aldrich |
| ρ(PTMSP phase in the MMM) | 0.885 | g cm−3 | For the diffusivity + solubility model. From the NELF model in Section 2.1.1 |
| ρ(zeolite A/PTMSP) | 0.744 | g cm−3 | For the diffusivity model. Calculated by the additive rule, for the 20 wt% zeolite A/PTMSP MMM, from the ρ(PTMSP) |
| ρ(zeolite A/PTMSP) | 0.846 | g cm−3 | For the diffusivity + solubility model. Calculated by the additive rule, with the ρ(PTMSP phase in the MMM) for the 20 wt% zeolite A/PTMSP MMM |
 (H2O) (H2O) |
0.602 | cm3 g−1 | 29 |
When calculating the effect of water on the gas solubility, S(a), the multicomponent sorption has to be considered. Thus, a gas–water interaction parameter kij (Table 4), was added to the NELF characteristic parameters in Table 1 and the gas-polymer interaction and swelling parameters in PTMSP and MMM, kij and ksw, calculated above, in order to model the solubility of CO2, CH4 and N2 in zeolite A/PTMSP MMM and pristine PTMSP membranes, in the presence of relative humidity.
| Components | kij |
|---|---|
| CO2–H2O | −0.117 |
| CH4–H2O | 0 |
| N2–H2O | 0 |
The calculated humid gas solubilities S(a) are plotted in Fig. 5 as a function of the water activity. It is worth remarking that the solubility of CO2 increases with increasing water content while the solubility of the other gases decreases, because of their general lower solubility.30 These observations explain some of the experimental observations discussed above, by the affinity of CO2–water, and the kij in Table 4.
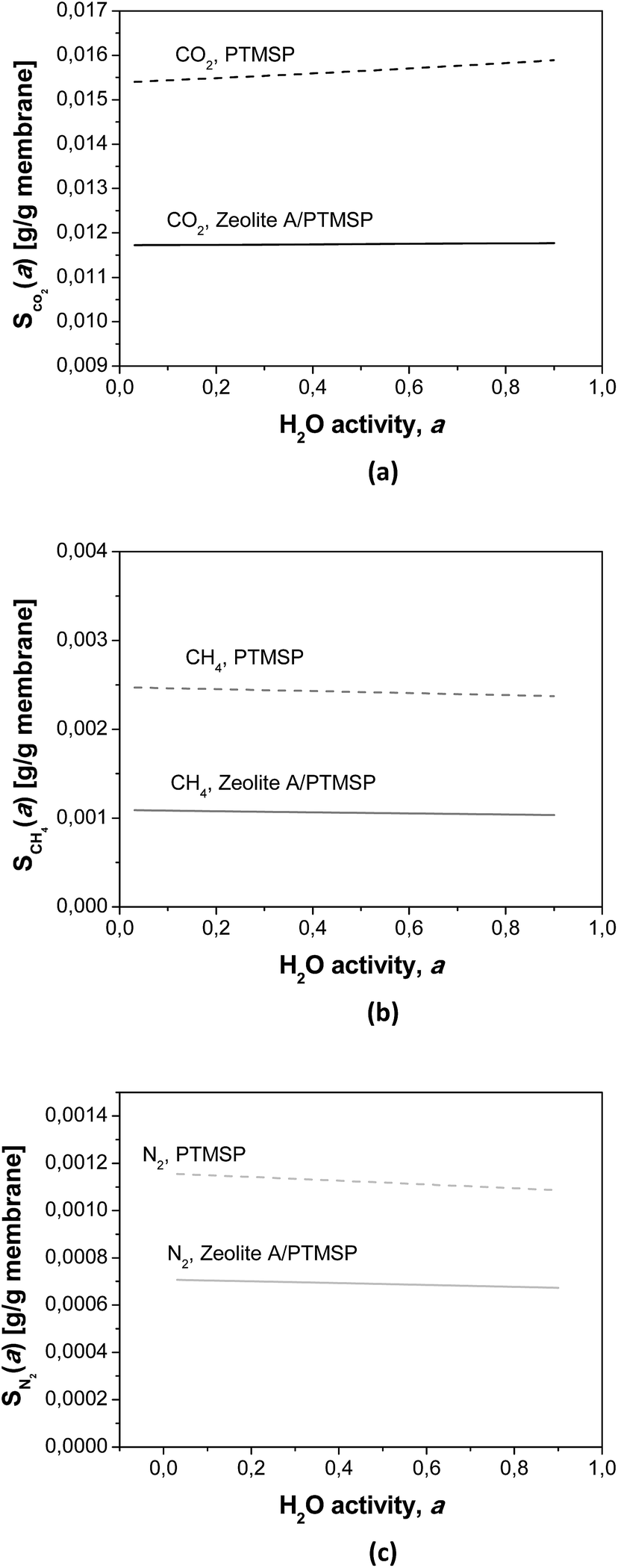 | ||
| Fig. 5 Calculated humid gas solubility S(a) as a function of water activity of (a) CO2, (b) CH4 and (c) N2, and in the PTMSP membrane (dotted lines) and the zeolite A/PTMSP MMM (continuous lines) calculated by the NELF model, at the same pressure and temperature as the experimental isotherms. | ||
The effect of water activity in diffusivity has been estimated using the experimental data of humid gas permeability and the humid gas solubility of the membranes obtained by NELF model shown in Fig. 5, by means of the solution diffusion model. The humid gas diffusivities calculated this way are represented in Fig. 6. It can be seen that the expected strong influence of the decrease in gas diffusivity in the permeability with increasing water activity through the pristine PTMSP membranes is somehow softened by the effect of the zeolite particles in the MMM, because of the combining effects of competitive gas sorption and water clustering and membrane matrix rigidification mentioned above.
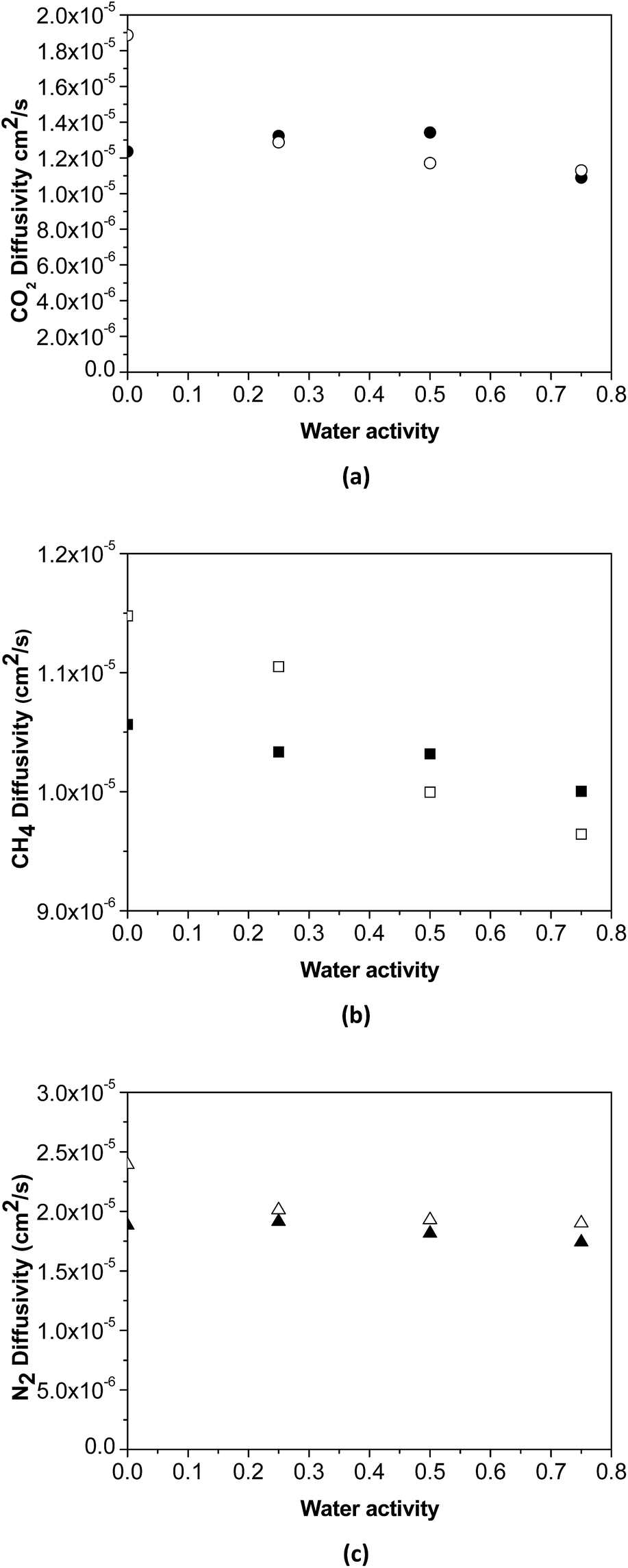 | ||
| Fig. 6 Calculated humid gas diffusivity D(a) as a function of water activity of (a) CO2, (b) CH4 and (c) N2, in the PTMSP membrane (void symbols) and the zeolite A/PTMSP MMM (filled symbols). | ||
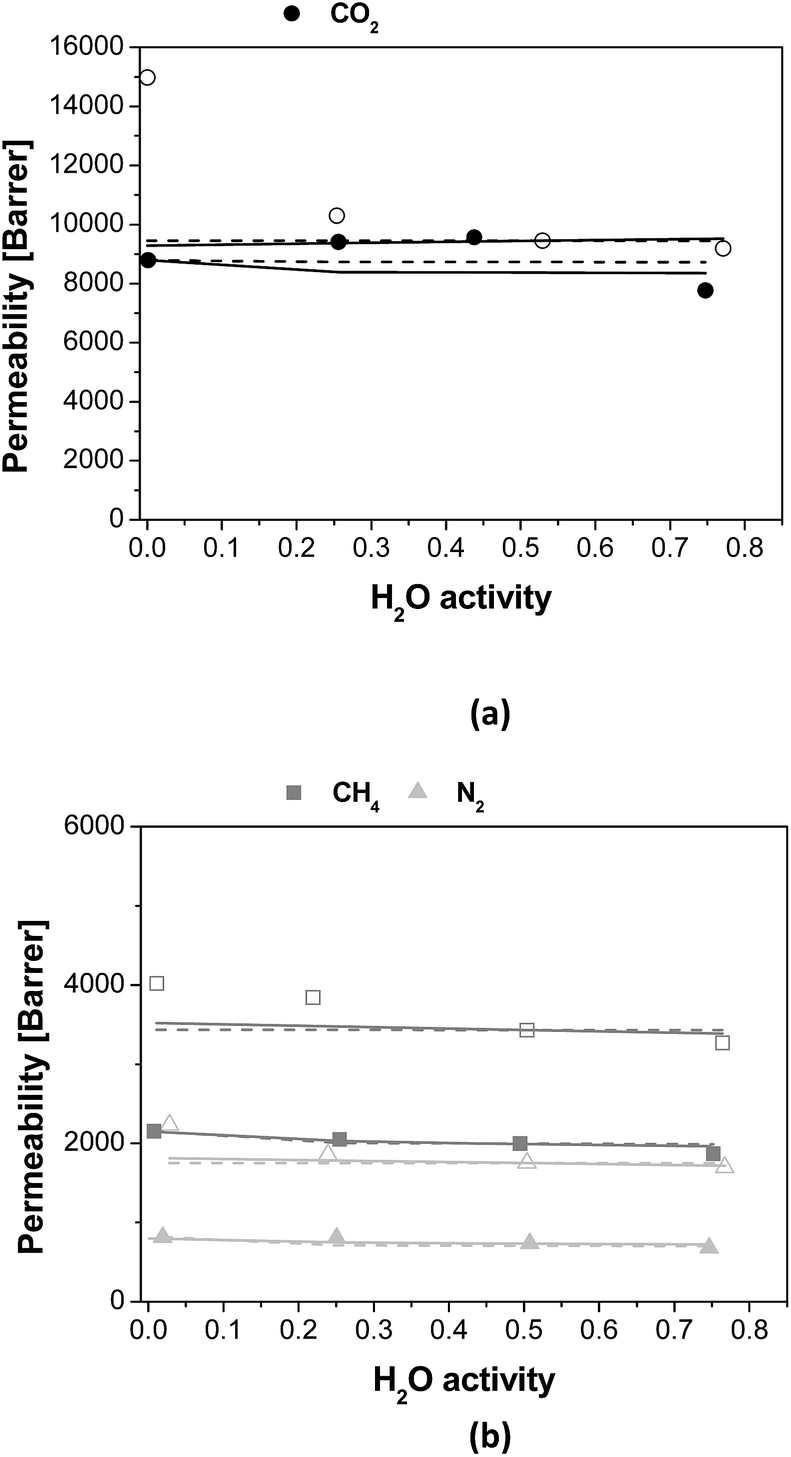 | ||
| Fig. 7 Permeability of (a) CO2 (black circles) and (b) N2 (light grey triangles) and CH4 (grey squares) as function of water activity for the PTMSP membrane (void symbols) and the zeolite A/PTMSP MMM (full symbols). Dashed lines correspond to the diffusivity-based model and continuous lines to the (diffusivity + solubility)-based model. | ||
The adjustable parameters A and B obtained from both approaches are given in Table 5, as a function of membrane material and gas penetrant. The deviations from the experimental data for both model approaches are also included in Table 5. The parameter A calculated considering only the effect on humid gas diffusivity on humid gas permeability, by eqn (7), is higher for the PTMSP membrane than the zeolite A/PTMSP MMM, being the highest that of CO2 and the lowest that of N2. This observation is consistent with the order or magnitude of permeability reported by other authors for other polyimide membranes, like Matrimid,29 PSf62 or 6FDA-6FpDA.38 When the model equation takes both solubility and diffusivity on the description of humid gas permeability, as in eqn (6), the A parameter of CH4 is lower for the PTMSP membrane than the MMM, while that of N2 is higher for PTMSP than MMM and that of CO2 does not vary. This is also consistent with the experimental behaviour of the humid permeability in the MMM observed in this work. The parameter B predicted by the complete solubility and diffusivity based model is the same in the pristine PTMSP membrane and MMM, for all gases, in agreement with reported data for co-polyetherimides.30
| Modelling approach | Parameter | N2 | CH4 | CO2 |
|---|---|---|---|---|
| Diffusivity model | A (105 cm2 s−1) | 2.70 (PTMSP) | 3.35 (PTMSP) | 9.05 (PTMSP) |
| 2.10 (MMM) | 3.21 (MMM) | 7.70 (MMM) | ||
| B (−) | 0.180 (PTMSP) | 0.047 (PTMSP) | 0.042 (PTMSP) | |
| 0.319 (MMM) | 0.165 (MMM) | 0.015 (MMM) | ||
| Error (%) | 7.58 (PTMSP) | 7.54 (PTMSP) | 12.70 (PTMSP) | |
| 4.68 (MMM) | 2.14 (MMM) | 7.38 (MMM) | ||
| Diffusivity + solubility model | A (105 cm2 s−1) | 2.24 (PTMSP) | 1.19 (PTMSP) | 1.36 (PTMSP) |
| 1.79 (MMM) | 3.05 (MMM) | 1.18 (MMM) | ||
| B (−) | 0.040 (PTMSP) | 0.046 (PTMSP) | 0.040 (PTMSP) | |
| 0.049 (MMM) | 0.043 (MMM) | 0.047 (MMM) | ||
| Error (%) | 6.06 (PTMSP) | 6.45 (PTMSP) | 11.96 (PTMSP) | |
| 4.03 (MMM) | 1.49 (MMM) | 7.69 (MMM) |
It is important to remark that both model approaches employed in this work are able to predict the experimental performance of the PTMSP membrane under humid conditions in a very accurate manner, using only two adjustable parameters depending on the permeating gas and membrane composition. However, the model accounting for solubility and diffusivity estimates better the experimental performance of the MMM. This is attributed to the reduction of free volume by the space occupied by the zeolite particles and the rigidity imparted to the polymer matrix thereby,19 which has a larger effect than plasticization or competitive sorption,40 which is accentuated by the presence of the water molecules. As hinted above, this issue requires some research effort in the materials knowledge regarding water sorption and diffusivity, however, it should be remarked that the CO2 solubility of the hydrated zeolite is almost the same as the dried zeolite, in opposition to the solubility of N2 or CH4 (Fig. S2 in the ESI†).
3. Experimental
3.1. Materials
Membranes were prepared by the solution casting method as reported elsewhere.19 Pure PTMSP membranes were prepared from a 1.5 wt% toluene solution of PTMSP (ABCR GmbH, Karlsruhe, Germany) and cast on a glass Petri dish. The membranes were then covered by a Petri dish at ambient conditions, to ensure the slow evaporation of the solvent. Before the permeation tests, membranes were immersed in liquid methanol for 5 min, because this was the method that ensured that all the PTMSP-based membranes were in similar initial conditions and presented reproducible permeation performance,19 since this is a common procedure for high fractional free volume polymers.63,64 The MMM were prepared in a similar way, except that the zeolite particles were previously dispersed in the solvent for 2 h and added to the polymer solution and stirred for other 24 h prior to the casting on the glass plate. The nominal zeolite loading in the PTMSP matrix for the MMM studied in this work was 20 wt%, which revealed the highest permselectivity performance in CO2/N2 separation from a group of PTMSP- based MMM prepared with small-pore zeolites of different Si/Al and topology, due to the dual-layer morphology of the zeolite 4A/PTMSP MMM, due to the different densities between zeolite particles and PTMSP and the slow evaporation procedure, which has already been discussed in our previous publications.19,65 This MMM consists of a top layer of almost pure PTMSP and a bottom layer containing the zeolite A particles, where the PTMSP acts as a binder, and thanks to the good compatibility between the zeolite 4 A and the PTMSP, this layer provides thermal and mechanical resistance as well as selectivity to the membrane.The thickness of the membranes was measured using a digital micrometre (Mitutoyo, precision of ±1 μm). The average thickness of the membranes tested in this work was 60 ± 6 μm.
The membrane density was obtained by the buoyancy method (Sartorius Density Kit YDK01), weighing the samples in air and water.
3.2. Humid gas permeation experiments
The effect of the water vapour content on the transport performance of zeolite A/PTMSP MMM and pristine PTMSP membranes has been studied at 35 °C in the relative humidity (R.H.) range from 0 to 75%. Humid gas permeability tests were carried out in a modified constant volume variable pressure permeometer. The system is equipped with a manometric apparatus that determines the amount of gas diffusing across the membrane by the variation of the gas pressure in a calibrated downstream volume, described in previous works66,67 and schematized in Fig. S4 in the ESI.† There is a humidifying section on the upstream side of the membrane, a water vapour reservoir and a purge flow. After placing the membrane into the sample holder, a vacuum test was performed to ensure the absence of leakage. At the beginning of the experiment, the membrane was equilibrated to the same level of water activity (i.e. the ratio between partial pressure and vapour pressure of water29) on both sides. When equilibrium was reached, the downstream volume of the apparatus was closed, and the upstream side of the membrane was fed by a gas stream at the same R. H. of the equilibrated membrane. As a consequence, the water activity at both sides of the membrane remained constant along the measurement, so that only the gas species was diffusing across the membrane23 at each level of R. H.3.3. Sorption experiments
The sorption of N2, CH4 and CO2 pure gases in the membranes and zeolite powders were measured using a pressure decay equipment reported elsewhere40 (drawn in Fig. S5 in the ESI†) with an average precision of the data higher than 93%. Before every pure gas sorption experiment, the whole equipment was kept under vacuum overnight. The membrane sample chamber D2 was then isolated from the rest of the equipment by closing valve V1. The volume separated from the surroundings with closed valves V1, V2 and V3 was then pressurized with the penetrant gas. Once the pressure was stable, the experiment was started by opening V1 and waiting several minutes to reach the equilibrium. Then, V1 was closed to set the post-equilibrium pressure. Sequential filling stages were carried out by increasing the pressure stepwise in order to obtain a complete sorption isotherm. The volumes of the membrane sample and pre-chambers, were calibrated by helium expansion experiments using a metal cylinder of known volume as a volume standard. The sorption isotherms were obtained in the order: N2, CH4 and CO2 at 35 °C. The sorption isotherms for CH4 and CO2 were taken up to 30 bar, while those of N2 only to 10–12 bar, because of the pressure of the N2 bottle. The zeolite samples were measured both as received and dried at 105 °C for 24 h in a vacuum oven.4. Conclusions
In this work, the effect of humidity on the gas permeability of N2, CH4 and CO2 has been studied in the range of relative humidity from 0% to 75% at 35 °C in the new zeolite A/PTMSP MMM whose performance in dry conditions improves that of the pure PTMSP membranes. At a 50% R. H., while the gas permeability of all the studied gases decreases for pristine PTMSP membranes, the CO2 permeability through the zeolite A/PTMSP MMM increases up to 10% from the dry value, and N2 and CH4 permeabilities decrease with water activity.This behaviour can be related with the reduction of free volume and increased rigidity in the MMM compared to PTMSP membranes. Furthermore, it has been proven that the adsorption of CO2 in pure zeolite A is not impaired by the presence of water, while that of CH4 and of N2 is. Therefore, the presence of zeolite A in PTMSP, under humid conditions, may be generally favourable to the permeation of CO2, while not helping that of N2 or CH4. Thus, the selectivity of zeolite A/PTMSP MMM is enhanced with increasing relative humidity in the feed in all the water activity range studied.
The sorption of gases in PTMSP and MMM has been measured experimentally and adjusted accurately by the NELF model, for all gases considered, using an approach for composite structures.
The permeability of gases in humid conditions in both pure PTMSP membranes and MMM is also modelled with a tool based on the free volume theory for diffusion, and NELF for sorption, with only two adjustable parameters. While the diffusivity only –based model equation predicts accurately the humid gas permeability through pristine PTMSP glassy polyimide membranes, the consideration of both solubility and diffusivity is necessary to approach the experimental results obtained in the laboratory for the novel zeolite A/PTMSP MMM under humid conditions in a very accurate way. This gives scope to the relevance of considering the multicomponent competitive sorption and the free volume reduction by occupation of the hydrophobic polymer matrix by water molecules and the hydrophilic character of zeolite fillers in the model prediction of new membranes with potential application in the treatment of humid gas streams.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support is gratefully acknowledged to the Spanish Ministry of Economy and Competitiveness (MINECO) under projects CTQ2016-76231-C2-1-R and CTQ2012-31229 at the University of Cantabria. A. F. B. also thanks the MINECO for the Early Stage Researcher (BES2013-064266) contract and the short stay grant to work at the University of Bologna for 3 months (EEBB-I-17-12097).References
- R. S. Haszeldine, Science, 2009, 325, 1647–1652 CrossRef CAS PubMed.
- J. C. Abanades, B. Arias, A. Lyngfelt, T. Mattisson, D. E. Wiley, H. Li, M. T. Ho, E. Mangano and S. Brandani, Int. J. Greenhouse Gas Control, 2015, 40, 126–166 CrossRef CAS.
- E. S. Rubin, H. Mantripragada, A. Marks, P. Versteeg and J. Kitchin, Prog. Energy Combust. Sci., 2012, 38, 630–671 CrossRef CAS.
- P. Luis, T. Van Gerven and B. Van der Bruggen, Prog. Energy Combust. Sci., 2012, 38, 419–448 CrossRef CAS.
- R. Khalilpour, K. Mumford, H. Zhai, A. Abbas, G. Stevens and E. S. Rubin, J. Cleaner Prod., 2015, 103, 286–300 CrossRef CAS.
- L. Giordano, D. Roizard, R. Bounaceur and E. Favre, Energy, 2016, 116, 517–525 CrossRef CAS.
- K. Ramasubramanian, Y. Zhao and W. S. Winston Ho, AIChE J., 2013, 59, 1033–1045 CrossRef CAS.
- S. Wang, X. Li, H. Wu, Z. Tian, Q. Xin, G. He, D. Peng, S. Chen, Y. Yin, Z. Jiang, M. D. Guiver, S. Wang, X. Li, H. Wu, Z. Tian, Q. Xin, G. He, D. Peng, S. Chen, Y. Yin, Z. Jiang and M. D. Guiver, Energy Environ. Sci., 2016, 9, 1863–1890 CAS.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400 CrossRef CAS.
- S. Roussanaly, R. Anantharaman, K. Lindqvist, H. Zhai and E. Rubin, J. Membr. Sci., 2016, 511, 250–264 CrossRef CAS.
- M. Rezakazemi, A. Ebadi Amooghin, M. M. Montazer-Rahmati, A. F. Ismail and T. Matsuura, Prog. Polym. Sci., 2014, 39, 817–861 CrossRef CAS.
- D. Bastani, N. Esmaeili and M. Asadollahi, J. Ind. Eng. Chem., 2013, 19, 375–393 CrossRef CAS.
- N. Jusoh, Y. Fong Yeong, T. Leng Chew, K. Keong Lau and A. Mohd Shariff, Sep. Purif. Technol., 2016, 454, 321–344 CrossRef.
- A. Morisato, H. C. Shen, S. S. Sankar, B. D. Freeman, I. Pinnau and C. G. Casillas, J. Polym. Sci., Part B: Polym. Phys., 1996, 34, 2209–2222 CrossRef CAS.
- K. Nagai, T. Masuda, T. Nakagawa, B. D. Freeman and I. Pinnau, Prog. Polym. Sci., 2001, 26, 721–798 CrossRef CAS.
- T. C. Merkel, R. P. Gupta, B. S. Turk and B. D. Freeman, J. Membr. Sci., 2001, 191, 85–94 CrossRef CAS.
- X. Y. Wang, A. J. Hill, B. D. Freeman and I. C. Sanchez, J. Membr. Sci., 2008, 314, 15–23 CrossRef CAS.
- S. D. Kelman, B. W. Rowe, C. W. Bielawski, S. J. Pas, A. J. Hill, D. R. Paul and B. D. Freeman, J. Membr. Sci., 2008, 320, 123–134 CrossRef CAS.
- A. Fernández-Barquín, C. Casado-Coterillo, M. Palomino, S. Valencia and A. Irabien, Chem. Eng. Technol., 2015, 38, 658–666 CrossRef.
- L. Ansaloni, J. R. Nykaza, Y. Ye, Y. A. Elabd and M. Giacinti Baschetti, J. Membr. Sci., 2015, 487, 199–208 CrossRef CAS.
- C. A. Scholes, J. Jin, G. W. Stevens and S. E. Kentish, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 719–728 CrossRef CAS.
- G. Q. Chen, S. Kanehashi, C. M. Doherty, A. J. Hill and S. E. Kentish, J. Membr. Sci., 2015, 487, 249–255 CrossRef CAS.
- M. Giacinti Baschetti, M. Minelli, J. Catalano and G. C. Sarti, Int. J. Hydrogen Energy, 2013, 38, 11973–11982 CrossRef CAS.
- T. Mizumoto, T. Masuda and T. Higashimura, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 255–2561 CrossRef.
- W. Yave, K.-V. Peinemann, S. Shishatskiy, V. Khotimskiy, M. Chirkova, S. Matson, E. Litvinova and N. Lecerf, Macromolecules, 2007, 40, 8991–8998 CrossRef CAS.
- C. A. Scholes, B. D. Freeman and S. E. Kentish, J. Membr. Sci., 2014, 470, 132–137 CrossRef CAS.
- J. H. Lee, J. Lee, H. J. Jo, J. G. Seong, J. S. Kim, W. H. Lee, J. Moon, D. Lee, W. J. Oh, J. gu Yeo and Y. M. Lee, J. Membr. Sci., 2017, 539, 412–420 CrossRef CAS.
- K. Nakamura, T. Kitagawa, S. Nara, T. Wakamatsu, Y. Ishiba, S. Kanehashi, S. Sato and K. Nagai, Ind. Eng. Chem. Res., 2013, 52, 1133–1140 CrossRef CAS.
- L. Ansaloni, M. Minelli, M. Giacinti Baschetti and G. C. Sarti, J. Membr. Sci., 2014, 471, 392–401 CrossRef CAS.
- L. Olivieri, A. Tena, M. G. De Angelis, A. Hernández Giménez, A. E. Lozano and G. C. Sarti, Green Energy Environment, 2016, 1, 201–210 CrossRef.
- S. J. D. Smith, H. Lau, J. I. Mardel, M. Kitchin, K. Konstas, P. Ladewig and M. R. Hill, J. Mater. Chem. A, 2016, 4, 10627–10634 CAS.
- E. Lasseuguette, M. Carta, S. Brandani and M. C. Ferrari, Int. J. Greenhouse Gas Control, 2016, 50, 93–99 CrossRef CAS.
- T. T. Moore and W. J. Koros, J. Appl. Polym. Sci., 2007, 104, 4053–4059 CrossRef CAS.
- R. Y. Yanagida, A. A. Amaro and K. Seff, J. Phys. Chem., 1973, 77, 805–809 CrossRef CAS.
- X. Ren, K. Nishimoto, M. Kanezashi, H. Nagasawa, T. Yoshioka and T. Tsuru, Ind. Eng. Chem. Res., 2014, 53, 6113–6120 CrossRef CAS.
- M. Palomino, A. Corma, F. Rey and S. Valencia, Langmuir, 2010, 26, 1910–1917 CrossRef CAS PubMed.
- B. Ozturk and F. Demirciyeva, Chem. Eng. J., 2013, 222, 209–217 CrossRef CAS.
- C. Tsvigu, E. Pavesi, M. G. G. De Angelis and M. Giacinti Baschetti, J. Membr. Sci., 2015, 485, 60–68 CrossRef CAS.
- H. Azher, C. Scholes, S. Kanehashi, G. Stevens and S. Kentish, J. Membr. Sci., 2016, 519, 55–63 CrossRef CAS.
- O. Vopička, M. G. De Angelis and G. C. Sarti, J. Membr. Sci., 2014, 449, 97–108 CrossRef.
- M. Minelli and G. C. Sarti, J. Membr. Sci., 2013, 435, 176–185 CrossRef CAS.
- M. Minelli and G. C. Sarti, Fluid Phase Equilib., 2016, 424, 44–51 CrossRef CAS.
- M. Minelli and G. C. Sarti, J. Membr. Sci., 2017, 521, 73–83 CrossRef CAS.
- M. G. De Angelis and G. C. Sarti, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 97–120 CrossRef PubMed.
- K. Nagai, S. Kanehashi, S. Tabei and T. Nakagawa, J. Membr. Sci., 2005, 251, 101–110 CrossRef CAS.
- M. G. De Angelis and G. C. Sarti, Ind. Eng. Chem. Res., 2008, 47, 5214–5226 CrossRef CAS.
- M. Minelli, G. Coochi, L. Ansaloni, M. Giacinti Baschetti, M. G. De Angelis and F. Doghieri, Ind. Eng. Chem. Res., 2013, 52, 8936–8945 CrossRef CAS.
- M. Galizia, K. A. Stevens, Z. P. Smith, D. R. Paul and B. D. Freeman, Macromolecules, 2016, 49, 8768–8779 CrossRef.
- M. C. Ferrari, M. Galizia, M. G. De Angelis and G. C. Sarti, Ind. Eng. Chem. Res., 2010, 49, 11920–11935 CrossRef CAS.
- A. J. Hill, B. D. Freeman, M. Jaffe, T. C. Merkel and I. Pinnau, J. Mol. Struct., 2005, 739, 173–178 CrossRef CAS.
- M. Galizia, M. G. De Angelis, M. Messori and G. C. Sarti, Ind. Eng. Chem. Res., 2014, 53, 9243–9255 CrossRef CAS.
- M. C. Ferrari, M. Galizia, M. G. De Angelis and G. C. Sarti, Ind. Eng. Chem. Res., 2010, 49, 11920–11935 CrossRef CAS.
- M. Woo, J. Choi and M. Tsapatsis, Microporous Mesoporous Mater., 2008, 110, 330–338 CrossRef CAS.
- A. L. Ahmad, J. K. Adewole, C. P. Leo, S. Ismail, A. S. Sultan and S. O. Olatunji, J. Membr. Sci., 2015, 480, 39–46 CrossRef CAS.
- J. Park and D. R. Paul, J. Membr. Sci., 1997, 125, 23–39 CrossRef CAS.
- J. S. S. Vrentas, J. L. L. Duda and H. C. C. Ling, J. Membr. Sci., 1989, 40, 101–107 CrossRef CAS.
- Y. Huang, X. Wang and D. R. Paul, J. Membr. Sci., 2006, 277, 219–229 CrossRef CAS.
- G. Consolati, I. Genco, M. Pegoraro and L. Zanderighi, J. Polym. Sci., Part B: Polym. Phys., 1996, 34, 357–367 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- K. Okamoto, H. Kita and K. Horii, Ind. Eng. Chem. Res., 2001, 40, 163–175 CrossRef CAS.
- R. H. Perry, D. W. Green and J. O. Maloney, Perry's Chemical Engineer's Handbook, McGraw-Hill Professional, 8th edn, 2007, vol. 27 Search PubMed.
- B. W. Rowe, B. D. Freeman and D. R. Paul, Polymer (Guildf), 2009, 50, 5565–5575 CrossRef CAS.
- S. D. Kelman, S. Matteucci, C. W. Bielawski and B. D. Freeman, Polymer (Guildf), 2007, 48, 6881–6892 CrossRef CAS.
- A. J. Hill, S. J. Pas, T. J. Bastow, M. I. Burgar, K. Nagai, L. G. Toy and B. D. Freeman, J. Membr. Sci., 2004, 243, 37–44 CrossRef CAS.
- A. Fernández-Barquín, C. Casado-Coterillo, M. Palomino, S. Valencia and A. Irabien, Sep. Purif. Technol., 2016, 157, 102–111 CrossRef.
- M. Minelli, M. G. Baschetti, F. Doghieri, M. Ankerfors, T. Lindström, I. Siró and D. Plackett, J. Membr. Sci., 2010, 358, 67–75 CrossRef CAS.
- J. Catalano, T. Myezwa, M. G. De Angelis, M. G. Baschetti and G. C. Sarti, Int. J. Hydrogen Energy, 2012, 37, 6308–6316 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra13039b |
| This journal is © The Royal Society of Chemistry 2018 |