
Iron–sulfur cluster biosynthesis and trafficking – impact on human disease conditions
C.
Wachnowsky†
ab,
I.
Fidai†
ac and
J. A.
Cowan
 *abc
*abc
aDepartment of Chemistry and Biochemistry, The Ohio State University, 100 West 18th Avenue, Columbus, Ohio 43210, USA. E-mail: cowan@chemistry.ohio-state.edu
bThe Ohio State Biochemistry Program, The Ohio State University, USA
cThe Biophysics Graduate Program, The Ohio State University, USA
First published on 26th September 2017
Abstract
Iron–sulfur clusters (Fe–S) are one of the most ancient, ubiquitous and versatile classes of metal cofactors found in nature. Proteins that contain Fe–S clusters constitute one of the largest families of proteins, with varied functions that include electron transport, regulation of gene expression, substrate binding and activation, radical generation, and, more recently discovered, DNA repair. Research during the past two decades has shown that mitochondria are central to the biogenesis of Fe–S clusters in eukaryotic cells via a conserved cluster assembly machinery (ISC assembly machinery) that also controls the synthesis of Fe–S clusters of cytosolic and nuclear proteins. Several key steps for synthesis and trafficking have been determined for mitochondrial Fe–S clusters, as well as the cytosol (CIA – cytosolic iron–sulfur protein assembly), but detailed mechanisms of cluster biosynthesis, transport, and exchange are not well established. Genetic mutations and the instability of certain steps in the biosynthesis and maturation of mitochondrial, cytosolic and nuclear Fe–S cluster proteins affects overall cellular iron homeostasis and can lead to severe metabolic, systemic, neurological and hematological diseases, often resulting in fatality. In this review we briefly summarize the current molecular understanding of both mitochondrial ISC and CIA assembly machineries, and present a comprehensive overview of various associated inborn human disease states.
 C. Wachnowsky | Christine Wachnowsky first studied biochemistry at Allegheny College in the lab of Dr Margaret Nelson, focusing on cell differentiation in Dictyostelium discoideum. She earned her PhD at the Ohio State University as part of the Ohio State Biochemistry Program. Christine conducted research in the laboratory of Dr James A. Cowan on the topic of iron–sulfur cluster biosynthesis and trafficking in relation to disease states. |
 I. Fidai | Insiya Fidai completed her Bachelor's and Master' in Biotechnology from the University of Mumbai, India. During her Masters, she worked on biocompatible synthesis of noble metal nanoparticles using various reducing sugars under the guidance of Dr Pankaj Poddar at the National Chemical Laboratory, Pune, India. She then earned her PhD at The Ohio State University as part of the Ohio State Biophysics Graduate Program. In Dr Cowan's laboratory, Insiya worked on ATCUN complexes as potential catalytic metallodrugs and role of iron–sulfur clusters in biogenesis, trafficking and storage, and implications for disease states. |
 J. A. Cowan | James A. Cowan graduated with first class honours in Chemistry from the University of Glasgow, before moving to Cambridge University to pursue research in synthetic organic chemistry and chemical physics. Subsequently he took up a postdoctoral appointment at Caltech to pursue studies in bioinorganic chemistry before taking up a faculty position at The Ohio State University. Dr Cowan's research interests cover metal catalysis, metals in medicine, and the cellular chemistry of iron and metal cofactors. |
Significance to metallomicsHerein, we summarize current models for iron–sulfur cluster biogenesis, which reflects the complex and intricate series of cellular exchange pathways required for proper iron homeostasis and metal cofactor trafficking. Given the complexity of iron–sulfur cluster assembly and delivery, which begins in the mitochondria and includes a mechanism for nuclear delivery, malfunctions in these trafficking processes can lead to a number of disease conditions. In this review we report on Fe–S cluster-based diseases that have been attributed to defects in cluster trafficking proteins and pathways and can lead to lethal consequences. |
Introduction
Iron–sulfur (Fe–S) clusters are small inorganic cofactors, composed of iron and sulfur. Protein-bound Fe–S clusters are among the most structurally and functionally versatile cofactors across all kingdoms of life.1 These cofactors are evolutionarily conserved and ubiquitous in nature,2 with the ability to be formed spontaneously in solution from Fe2+/Fe3+, RS−, and inorganic S2−, or from Fe2+, R-SH and sulfur under reducing conditions. However, Fe–S clusters are susceptible to oxidation, presenting challenges for cluster synthesis and binding to proteins due to atmospheric oxygen. In addition, the iron-mediated Fenton reaction poses a severe threat to the genome due to cytotoxicity via DNA damage.2a,3 As such, highly conserved and controlled mechanisms are in place for cluster production and trafficking to target proteins to allow for their wide range of function as protein cofactors.Over the last decade, several new roles associated with Fe–S clusters have emerged, both as a result of progress in identifying and purifying proteins with intact clusters, and also due to the higher level of sophistication of the biophysical techniques used in their characterization. Yet the presence of these clusters remains puzzling, given the challenges present with oxygen and the potential for cellular toxicity. This raises the fundamental question of why Fe–S clusters have been retained as a vital component of essential macromolecules ranging from proteins to nucleic acids, and highlights the need for mechanistic and biochemical studies to promote understanding of the role of these versatile cofactors. With recent advances, a variety of disease conditions have been associated with Fe–S cluster proteins that impact overall cluster trafficking. Herein, we briefly review the cluster biogenesis process and describe the associated inborn human disease phenotypes.
Mitochondrial Fe–S cluster biogenesis
Elucidation of the molecular and cellular basis of Fe–S cluster biogenesis and transfer to putative Fe–S cluster proteins has been an area of intense investigation since it was discovered that biogenesis is not spontaneous, but rather catalyzed.4 Three distinct pathways have been identified for bacterial Fe–S cluster biosynthesis.5 The ISC system is considered the major Fe–S cluster assembly system in humans,1a,2b required not only for the maturation of mitochondrial but also cytosolic and nuclear Fe/S proteins. This machinery primarily mediates three steps of Fe–S cluster biosynthesis; namely, (1) cluster assembly on the scaffold protein, (2) chaperone-assisted release of the Fe–S cluster and a currently unclear mechanism of mitochondrial export, if necessary, and (3) subsequent transfer to putative target apo-proteins.1b,c Much of the research concerning mitochondrial Fe–S cluster biosynthesis has been carried out in yeast cells, human cells, and with human, yeast, or bacterial proteins in vitro. The overview presented here summarizes a compilation of this research, with the goal of primarily describing the current model for the human system.1. Cluster assembly on the scaffold protein
The first step in Fe–S cluster biogenesis is the assembly of iron and sulfur on the scaffold protein IscU. Sulfur is provided by the eukaryotic cysteine desulfurase complex of NFS1–LYRM4 (human ortholog of the Nfs1–Isd11 complex), which converts cysteine to alanine through a persulfide intermediate (Fig. 1 and Table 1).6 The precise details of the assembly complex of Iscu–Isd11–Nfs are somewhat unclear, as larger complexes have been observed in yeast and bacteria, but the physiological significance of these oligomers is unknown.7 Redox chemistry ensues, although the details are poorly understood, possibly with the assistance of a redox-active ferredoxin.7d,8 Mitochondria import ferrous iron in a membrane potential-dependent manner via the mammalian carrier proteins mitoferrin 1 and 2.9 This iron is then transferred to IscU through an iron donor protein called frataxin (Fxn),10 for complete assembly of a [2Fe–2S] cluster. Frataxin can also function as a regulatory partner in the Nfs1–Isd11–IscU complex to control not only sulfur production, but also iron entry during Fe–S cluster assembly, to accelerate the rate of Fe–S cluster biogenesis.11,12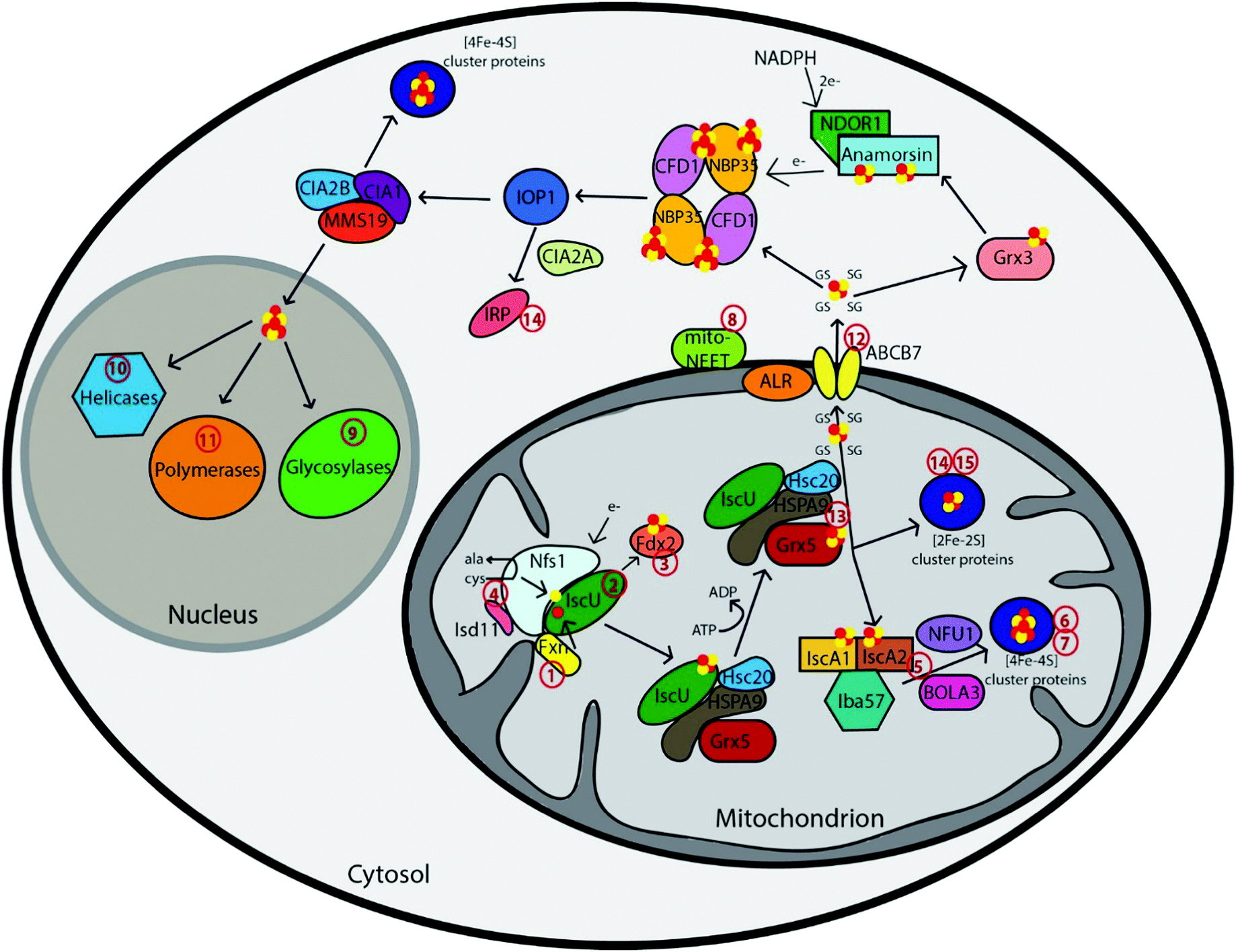 | ||
| Fig. 1 Schematic representation summarizing current models of mammalian Fe–S cluster biogenesis based on disease conditions. Nfs1, the cysteine desulfurase, with its cofactor protein Isd11 donates sulfur from L-cysteine for nascent Fe–S cluster assembly. Together, they bind to the primary scaffold protein IscU, the regulator protein frataxin (Fxn), which controls cysteine desulfurase activity and iron delivery, and a source of electrons. The newly formed IscU-bound [2Fe–2S] is subsequently transferred via a dedicated chaperone-co-chaperone system (Hsc20–HSPA9) to Grx5. From Grx5, the Fe–S clusters are either directly inserted into mitochondrial [2Fe–2S] proteins or transferred via specific carrier systems such as the one composed of IscA1, IscA2 and Iba57 to mitochondrial [4Fe–4S] proteins. To date, the presence of another cluster type (such as a glutathione-complexed [2Fe–2S] cluster) under physiological conditions cannot be excluded. For extra-mitochondrial Fe–S cluster, an uncharacterized compound (X) is exported for cytosolic and nuclear cluster trafficking via the Fe–S cluster export machinery consisting of ALR, glutathione and ABCB7. Species X has been proposed to be a glutathione-complexed [2Fe–2S] cluster and is depicted as the [2Fe–2S](GS)4 complex.17 Cytosolic cluster trafficking has been studied primarily in yeast. Fe–S clusters can be trafficked through Grx3 to anamorsin, which provides electrons to the cytosolic scaffold CFD1–NBP35. From there, [4Fe–4S] clusters are transferred via IOP1 to the CIA proteins and final target proteins, such as IRP. The CIA proteins can complex with MMS19 for cluster delivery to nuclear targets, like polymerases, helicases, and glycosylases. The red numbers in the figure correspond to the disease numbers in the review and in Table 1. | ||
| Number | Protein | Function | Disease | Most common cause |
|---|---|---|---|---|
| 1 | Frataxin | Iron donor; allosteric effector of NFS1 | Friedreich's ataxia (FRDA) | GAA-triplet expansion causing transcriptional silencing |
| 2 | IscU | Fe–S cluster scaffold | IscU myopathy | Splice mutation in intron 4 resulting in decreased protein levels |
| 3 | Fdx2 | Electron transport | Fdx2 myopathy | c. 1A > T mutation disrupting the initiation codon |
| 4 | Nfs1 | Sulfur donor for cluster assembly | Infantile mitochondrial complex II/III | p. R72Q decreasing transcripts and protein levels |
| 4 | ISD11 | Stability factor | Combined oxidative phosphorylation deficiency 19 (COXPD19) | p. R68L resulting in undetectable ISD11 protein levels |
| 5 | Nfu | Late acting [Fe–S] targeting factor | MMDS1 | p. G208C resulting in insufficient downstream holo targets |
| 5 | BOLA3 | Late acting [Fe–S] targeting factor | MMDS2 | Truncation mutation resulting in insufficient downstream holo targets |
| 5 | Iba57 | [4Fe–4S] cluster assembly factor | MMDS3 | p. Q314P causing protein misfolding and degradation |
| 5 | IscA1/2 | [4Fe–4S] cluster assembly | MMDS4 | p. G77S resulting in insufficient downstream holo targets |
| 6 | Ind1 | Assembly factor for complex I | Complex I deficiency | p. G56R and a c. 815-27T > C branch site mutation leading to aberrant splicing |
| 6 | SDHB | Respiratory complex II | Neurodegeneration and cancer | Mutations at Fe–S cluster ligating cysteines preventing cluster binding |
| 7 | SDHAF1 | Assembly factor for complex II | leukoencephalopathy | Mutations that disrupt binding interface for either Hsc20 or SDHB prevent cluster maturation on SDHB |
| 8 | MitoNEET | Maintenance of iron homeostasis | Association with cystic fibrosis, Parkinson's disease, and cancer | Related to decrease in mRNA levels |
| 9 | MUTYH | Base excision repair of 8-oxoguanine: adenine mismatches | Colorectal polyposis | p. P281L resulting in low protein that cannot bind DNA |
| 10 | FANCJ | Genome maintenance and double-strand break repair | Fanconi anemia and breast cancer | p. M299I upregulates helicase activity or p. A349P that impairs DNA–protein interactions |
| 10 | XPD | Nucleotide excision repair and transcription | Xeroderma pigmentosum (XP), Cockayne's syndrome (CS), trichothiodystrophy (TTD), and Cerebro-oculo-facial-skeletal (COFS) syndrome | p. R112H that impairs transcriptional function and inactivates helicase activity |
| 10 | ChlR1 | Genome maintenance | Warsaw breakage syndrome | p. R263Q inhibits DNA binding and DNA-dependent hydrolysis |
| 10 | RTEL1 | Maintenance of telomeres | Cancer, dyskeratosis congenita (DC), and Hoyeraal-Hreidarsson syndrome (HHS) | Protein mutations that prevent telomere maintenance |
| 11 | POL δ and ε | DNA polymerases | Cancer | Unclear connection |
| 12 | ABCB7 | Fe–S cluster export from the mitochondria | X-linked sideroblastic anemia (XLSA) | Protein mutations that result in mitochondrial iron overload |
| 13 | Grx5 | [2Fe–2S] cluster carrier | Sideroblastic-like anemia | Aberrant splicing leading to decreased Fe–S cluster transfer and impaired iron homeostasis |
| 14 | IRP | Iron homeostasis | Parkinson's disease | Erroneous regulation generating reactive oxygen species |
| 14 | Grx2 | Cluster carrier and redox regulation | Parkinson's disease | Erroneous regulation of GSH levels |
| 15 | FECH | Heme biosynthesis | Erythropoietic protoporphyria | Over 130 different mutations that impair function |
2. Chaperone assisted release of the Fe–S cluster and mitochondrial export, if necessary
Cluster release from IscU to target proteins is mediated by a dedicated chaperone system comprising the Hsp70-type protein HSPA9 (homolog of yeast Ssq1 and bacterial HscA) and the co-chaperone Hsc20 (homolog of yeast Jac1 and bacterial HscB) (Fig. 1 and Table 1).13 The chaperone complex binds and induces a conformational change in IscU, which enables release of the IscU-bound Fe–S cluster either to selected target proteins or to specific Fe–S cluster carrier proteins, with the proposed major target being the monothiol glutaredoxin, Grx5, a homodimeric protein, that binds a [2Fe–2S] cluster with two exogenous glutathione ligands for downstream cluster delivery and trafficking (Fig. 1).14From Grx5, the [2Fe–2S] cluster can also be targeted to other downstream proteins within the mitochondria, or for export from the mitochondria to the cytosol via the ABC transporter ABCB7 (yeast Atm1) (Fig. 1).15 The crystal structures of yeast Atm1 and a related bacterial ABC transporter that confers heavy metal tolerance16 indicate that the transporter exports a sulfur containing molecule from the mitochondria to the cytosol, which is critical for the CIA machinery.17 A role for glutathione in the ISC export process has been established in yeast cells, where a depletion of glutathione results in a phenotype similar to that of Atm1 deficiency that produces iron overload in mitochondria.18 Hence, glutathione is required for extra-mitochondrial Fe–S cluster biogenesis and therefore impacts cellular iron homeostasis.19 Prior reports have provided evidence for a tetrameric GSH-coordinated [2Fe–2S] cluster as a possible substrate for the exporter protein.17a In fact, mixing GSH-coordinated [2Fe–2S] complex with purified Atm1 stimulated its ATPase activity up to 1.8-fold.17b Structures were solved with potential glutathione and oxidized glutathione (GSSG) binding sites about 5 Å apart, which could readily accommodate a [2Fe–2S] cluster and provide support for a glutathione complexed Fe–S cluster.16In vivo determination of these glutathione complexed [2Fe–2S] clusters can further strengthen the physiological existence of these small molecules in the cell. Another study has suggested the molecule GSSSG, i.e., the oxidized form of GSSH (glutathione persulfide) and GSH, as the Atm1 substrate and potential sulfur carrier.20 Nevertheless, an as yet ill-defined species X must be exported from the mitochondria in order to support proper Fe–S cluster trafficking in other cellular compartments.
3. Transfer to putative target apo-proteins
Recent studies have shown that a number of other proteins are further involved in Fe–S cluster biogenesis. Following release of the Fe–S cluster from the scaffold protein, it can either be directly delivered to [2Fe–2S] target proteins or converted to a [4Fe–4S] cluster.1b,c A current model suggests that [4Fe–4S] cluster maturation requires additional ISC proteins: IscA1 and IscA2, and the folate binding protein Iba57,21 in a process that requires sequential delivery of a [2Fe–2S], postulated to be from Grx5, with reductive coupling to form a [4Fe–4S] cluster (Fig. 1).22 However, knockdown of these IscA proteins does not eliminate production of [4Fe–4S] clusters, so other proteins and complexes may utilize a similar reductive coupling mechanism on target Fe–S cluster proteins.21b Other more specialized targeting factors have evolved to assist the maturation of dedicated subsets of [4Fe–4S] proteins and also for transfer of [2Fe–2S] clusters downstream of the scaffold assembly to final target proteins, such as Nfu, BolA3, Ind1, and SDHAF (Fig. 1 and Table 1).1a However, the precise mapping of these pathways and all of the potential functions of these proteins remain unclear. For example, Nfu may act as an alternative scaffold protein to IscU, because Nfu can accommodate both a [2Fe–2S] cluster as well as a labile [4Fe–4S] cluster in vitro and pass these on to apo-proteins.23Cytosolic and nuclear Fe–S cluster protein biogenesis
The biogenesis of cytosolic and nuclear Fe–S proteins is heavily dependent on the conserved mitochondrial Fe–S cluster (ISC) assembly machinery in addition to the ABC transporter protein and cytosolic Fe–S cluster (CIA) assembly machinery. To date, nine components of the CIA machinery have been identified that play a role in mobilization of Fe–S cluster and its insertion into extra-mitochondrial apo-proteins. The assembly process has been primarily studied in yeast and comprises two mains steps:1a,4c (1) [4Fe–4S] cluster assembly on the Cfd1–Nbp35 scaffold complex and (2) transfer of Fe–S clusters to target apo-proteins from the CIA scaffold complex.1b,c Yeast nomenclature has been used throughout this section and describes the proposed models based on the work in yeast. Similar pathways are likely functional in humans too.1. [4Fe–4S] cluster assembly on the CFD1–NBP35 scaffold complex
An initial step in cytosolic Fe–S protein biogenesis is the assembly and binding of a transient [4Fe–4S] cluster on the cytosolic scaffold protein complex, a heterotetramer comprised of the two P-loop NTPases Cfd1 and Nbp35, which bind two different kinds of [4Fe–4S] clusters (Fig. 1).4c,24 A loosely bound [4Fe–4S] cluster at the C-terminus of both proteins bridges the two complex subunits together, whereas the other [4Fe–4S] cluster is tightly coordinated at the N-terminal domain of the Nbp35 protein. The lability of the C-terminus cluster is considered to be important for the second step, involving transfer of the cluster to target apo-proteins. Generation of the functionally essential N-terminal Fe–S cluster (N) of Nbp35 depends on the flavoprotein Tah18 (yeast homolog of NDOR1) and the Fe–S protein Dre2 (yeast homolog of CIAPIN1), which serve as an NADPH-dependent electron (e−) transfer chain (Fig. 1).25 Cytosolic–nuclear Fe–S protein biogenesis additionally requires the cytosolic multidomain glutaredoxin, Grx3, yet its precise function and site of action in the pathway remains to be determined,26 although studies from the yeast system suggest roles in iron regulation and maturation of other proteins such as ribonucleotide reductase.26b,27 Accordingly, it is speculated that these cytosolic monothiol glutaredoxins play a general role in iron trafficking.2. Transfer of Fe–S clusters to target apo-proteins from the CIA scaffold complex
The bridging Fe–S clusters are released from Cfd1–Nbp35 and transferred to apo-proteins in a reaction mediated by the Fe–S protein Nar1 and the CIA targeting complex Cia1–Cia2–Mms19 (Fig. 1).1a,4c Nar1 coordinates two [4Fe–4S] cofactors that are similar to those in hydrogenases and bridges the early and late stage CIA machinery.61,62 Cia1, Cia2 and Mms19 form dimeric and tetrameric complexes in both yeast and mammalian cells, and interact with target apo-proteins and assure specific Fe–S cluster insertion,28 including the crucial function of maturation of nuclear Fe–S proteins (Fig. 1). Several of the proteins involved in mitochondrial Fe–S cluster biogenesis have also been reported to be localized in the cytosol such as IscU, Nfu, Nfs1 and Isd11.29 However, their functional relevance is yet to be established.Diseases associated with Fe–S cluster biogenesis proteins
A number of inborn human diseases have been identified with mutations associated either with the gene or the protein involved in Fe–S cluster biogenesis. In recent years, additional diseases have been identified with the increasing availability of information concerning the function and properties of Fe–S cluster proteins.1b,c Several of these disease states are discussed below (Table 1). Additional disease states related to Fe–S cluster maturation have been discussed in terms of infectious agents, but those will only be described briefly in this work.Diseases associated with mitochondrial Fe−S cluster biogenesis
The requirement for ISD11 in stabilizing Nfs1 has been confirmed in patients with mutations on ISD11, who demonstrated a similar phenotype in the form of respiratory complex deficiency, termed combined oxidative phosphorylation deficiency 19 (COXPD19).40 Interestingly, these patients also exhibited further downstream complications with deficiencies in complexes I, II and III, in addition to other mitochondrial and cytosolic Fe–S proteins.40a Both patients were homozygous for a missense mutation (p. Arg68Leu),40a which caused the levels of ISD11 to be undetectable and a decrease in respiratory chain complexes to below 40%. This mutation also impacted the interaction with Nfs1 by promoting aggregation and inhibiting desulfurase activity.40a Furthermore, IscU levels were also observed to decrease when the R68L mutation was present, suggesting that the complex is also essential for promoting the stability of IscU for overall Fe–S cluster biogenesis and trafficking.40b
For MMDS1, five patients have been described with a homozygous mutation in Nfu (p. Arg182Gln), where the mutation is close to a splice site and results in skipping of exon 6 and no detectable protein levels.46 Most other patients were described with a homozygous mutation (p. Gly208Cys) that affects an amino acid close to a highly conserved Fe–S cluster binding domain of the Nfu protein. This mutation does not affect protein levels but does impair Fe–S cluster binding and transfer, most likely due to a perturbed monomer–dimer equilibrium that limits cellular reconstitution of Nfu.41f,47 Most recently, a study of patients with the heterozygous gene for Nfu, one with the altered splice site or with the Gly208Cys missense mutation and the native allele, has revealed that low levels of the wild type Nfu mRNA were produced, which yielded sufficient functional LIAS to avoid lipoic acid deficiency, but the wild type transcripts were insufficient to support functional respiratory complex II.41d
In the case of MMDS2, 6 different patients have been identified with mutations on BOLA3.41b,42 The first patient demonstrated a single base pair duplication that produced a frameshift with a premature stop codon and therefore loss of the full length protein.41b Two other patients have been characterized as having a missense mutation at a highly conserved residue (p. Ile67Asn)42b that could affect the overall protein fold based on the solution NMR structure of human BOLA3.48 The remaining three patients all demonstrated a truncating mutation of eight amino acids upstream of the first truncating mutation.42a To date, the molecular role of BOLA3 remains to be elucidated; however the availability of an NMR structure48,49 with additional biochemical characterization will provide increasing understanding of its role in MMDS.
Similar to BOLA3 in MMDS2, a wide variety of mutations on Iba57 have been identified for MMDS3. Initial reports described different patients with a range of phenotypes. The first case resulted in the most severe condition caused by a missense mutation (p. Gln314Pro) that appeared to result in protein misfolding with subsequent degradation to concentrations below critical levels.43a The mildest phenotype was observed in a patient with an Iba57 mutation that resulted in reduced mRNA levels because of aberrant splicing and a frameshift mutation that yielded a truncated form of Iba57.43c The intermediate phenotype of MMDS3 was due to a mutation on Iba57 that drastically reduced its ability to function in Fe–S biogenesis with diminished levels of [4Fe–4S] clusters.43b Most recently, four additional mutations on Iba57 have been observed with generally the same symptoms and phenotypes.43d The prevalence of such diverse mutations on Iba57 that all result in MMDS3 makes prediction of the disease difficult, based on genetic information, and strengthens the need for further biochemical characterization of Iba57 to understand why so many possible causes of the disease exist.
MMDS4 was the most recent class to be discovered, with six patients exhibiting a missense mutation (p. Gly77Ser) on IscA2, which is located in a highly conserved cluster binding domain. However, the exact effect of the mutation is unclear, since mRNA levels were not significantly impacted.44 Interestingly, patient fibroblasts also showed lower levels of IscA1 and Iba57 than control fibroblasts.44 How this mutation results in lower protein levels of these partner proteins remains unclear. Similarly, an IscA1 mutation (p. Glu87Lys) has been implicated in causing MMDS-like symptoms, which may result in a new class of the disorder, as one patient has been found with this mutation that is expected to lead to protein instability due to loss of a conserved salt-bridge.45
The class of MMDS disorders describes a newly discovered disease condition and further details on how mutations in key Fe–S biogenesis proteins influence cellular function, and their roles that result in disease, need to be determined in order to fully aid patients.
In particular, complex I of the respiratory complex, also known as NADH-CoQ oxidoreductase, is composed of 44 subunits, utilizes at least 10 assembly factors for correct complex formation, and accounts for one-third to one-quarter of mitochondrial disease, which occurs in about 1 out of every 5000 births.51 One such assembly factor, Ind1 or NUBPL, has been implicated as an Fe–S cluster delivery protein that is specifically required for complex I assembly and activity in yeast,51 and mutations or perturbations of the Ind1 gene have resulted in a specific deficiency of complex I, while other respiratory complexes remain intact.52 Genetic sequencing has found two mutations in patients: namely, a p. Gly56Arg missense mutation and a c. 815-27T > C branch site mutation. Of the two, pathogenicity has been more attributed to the branch site mutation, since it results in aberrant splicing, very low levels of the NUBPL protein, and reduced levels of fully assembled complex I, as little as 20%.52,53 However, there remains the possibility that the missense mutation contributes to the pathogenicity of the branch site mutation. Furthermore, recent work in Arabidopsis thaliana suggests that Ind1 may also play a role in translational control,54 which may contribute to phenotypes observed in the pathogenic state. However, no work has been done in human cell lines to either evaluate this additional functional role or examine the possible role of Ind1 in Fe–S cluster delivery.
In terms of the neurological impact, one patient has been identified with a missense mutation at an evolutionarily conserved residue (p. Asp48Val) near an iron–sulfur cluster binding site, which is hypothesized to be crucial for efficient electron transport through the respiratory chain and resulted in an almost complete loss of SDHB and decreased levels of SDHA.56 As with many mitochondrial disorders, there is no cure for SDH deficiency; however, the patient in this study was supplied with an oral treatment of coenzyme Q10 to yield a subjective improvement in strength.56
The remainder of patients with mutations on SDHB have been linked to some form of cancer. A recent analysis of the Leiden Open Variation Database showed that 37% of all the reported cancer cases linked to SDHB are due to mutations to the cluster cysteinyl ligands that prevented cluster binding, or in the L(I)YR motif, which blocks chaperone recognition by Hsc20 and the HSPA9 chaperone to prevent binding interactions with either of these proteins.58
As is the case for respiratory complex I, maturation factors for SDH have also been implicated in causing disease. In the case of SDHB, two assembly factors exist: SDHAF1 and SDHAF3.55b,59 Both factors are believed to help protect SDHB from oxidative damage during the assembly of the complete SDH complex by shielding the nascent SDHB Fe–S clusters from damage by reactive oxygen species.55b,59 SDHAF1 also functions in concert with Fe–S cluster chaperones in recruiting the chaperone complex to SDHB via the L(I)YR motif,60 and was found to co-immunoprecipitate with Hsc20.58b As such, mutations on SDHAF1 that disrupt the binding interface for either SDHB or Hsc2060 have also been implicated in causing leukoencephalopathy,61 where accumulation of succinate in the white matter of the brain is caused by a loss of SDH activity. The accumulation of succinate was due to degradation of SDHB, when non-functional SDHAF1 was present; however, succinate levels could be reduced following treatment with riboflavin, which serves to increase the flavinylation of SDHA and allows it to convert succinate to fumarate to restore the activity of the Krebs cycle.60
Given its broad role in metabolism, mitoNEET has also been associated with cystic fibrosis due to a decrease in mitoNEET mRNA,68 Parkinson's disease due to an interaction with PARKIN,69 and cancer and tumorogenesis,62,70 where mitoNEET was found to be upregulated in breast cancer cells.71 Despite the association of mitoNEET with broad cellular processes and diseases, many questions remain unanswered concerning its specific cellular mechanism and the connection between its role in Fe–S cluster biosynthesis and overall mitochondrial regulation. Furthermore, mitoNEET is still under consideration as a target for drug development for the treatment of cancer and diabetes based on overall mitochondrial dysfunction,62,63 with additional possibilities considering its likely roles in Parkinson's and cystic fibrosis. As more is learned concerning mitoNEET and its physiological roles, better therapeutics may be developed to compensate for disruptions in its function.
Diseases associated with cytosolic Fe–S cluster proteins
Although a number of proteins have been identified as components of the cytosolic Fe–S cluster machinery,4b,15,72 many have only been discovered or characterized in depth over the last 5 years. For this reason, many of these proteins have not yet been linked to disease conditions. However, given their required roles in the biogenesis of essential Fe–S clusters and maintenance of iron homeostasis, there is the possibility of disease association as these proteins become better understood and disease states are examined in greater depth. For example, examination of proteins in the NAR family in Arabidopsis thaliana73 and yeast74 have indicated conserved roles in maintaining a response to oxidative stress, which may have downstream implications for genome maintenance,72 due to the connection to DNA damage repair pathways and corresponding involvement in aging disorders.Likewise, the cytosolic Fe–S protein MMS19, has not specifically been associated with any diseases. However, this protein has been characterized as required for maturation of many nuclear Fe–S cluster containing proteins that are particularly involved in DNA replication and repair, such as FANCJ, XPD, RTEL, and POLD (discussed below).75 The essential nature of MMS19 in maturation of key nuclear proteins may link it to cancer and neurodegenerative phenotypes in the future.75b
Diseases associated with nuclear Fe–S cluster containing proteins
With oxygen sensitivity and possible toxicity associated with Fe–S clusters, it was assumed that most nucleic acid enzymes did not contain Fe–S clusters. However, this view changed with the discovery of an Fe–S cluster in the DNA helicase XPD and associated protein members that are involved in DNA repair,76 and the possibility that Fe–S cluster containing enzymes find DNA damage by probing DNA integrity electronically.77 Today, all DNA polymerases involved in replication and also certain helicases that participate in Okazaki fragment processing contain some form of Fe–S clusters.2a,78FANCJ, in conjunction with 15 other genes, is implicated in causing Fanconi anemia; a condition characterized by congenital defects, progressive bone marrow failure, susceptibility to cancer with chromosome breaks, and sensitivity to DNA damaging agents that result in intra-strand cross-links.84 Furthermore, FANCJ plays a role in genome stability by unwinding G-quadruplexes85 and by conducting double-strand break repair.86 Of the numerous mutations identified in the gene for FANCJ that are linked to various disease states, two of them (M299I and A349P) occur in the iron–sulfur cluster binding domain, where the M299I mutation has been found in breast cancer patients,83a,87,88 and the A349P mutation causes Fanconi anemia.89 These mutational analyses have demonstrated the importance of the Fe–S cluster in the function of FANCJ and its role in downstream DNA processing activities in disease.83
A second Fe–S cluster-containing helicase is XPD, which serves key roles in nucleotide excision repair to remove photo-damaged DNA bases, and in transcription as a member of the TFIIH complex.83 Mutations in XPD have been linked to xeroderma pigmentosum (XP), Cockayne's syndrome (CS), trichothiodystrophy (TTD), and Cerebro-oculo-facial-skeletal (COFS) syndrome.83 To date, only one of these disorders has been linked to a mutation in the Fe–S cluster binding domain. In patients with TTD, a R112H mutation is present at a highly conserved residue that removes a hydrogen bond to a cluster-ligating cysteine,90 resulting in impaired interactions with proteins in the TFIIH complex and involvement in helicase inactivation.83a,90a
The last two Fe–S cluster helicases that we will describe have been implicated in disease much more recently. The helicase ChlR1, or DDX11, is less understood than those already discussed. Its specific role in DNA processing and genome integrity is unclear; however, it may serve primarily to unwind intermediate DNA structures present during replication and recombination,91 with additional potential roles in sister chromatid cohesion, heterochromatin organization, viral genome maintenance, DNA repair, and G-quadruplex unwinding, via its helicase and ATPase activities.92 Although its precise function is unknown, the first human patient with a defect in ChlR1 was identified in 2010 in a condition termed Warsaw breakage syndrome, due to either a splice site mutation93 or p. Arg263Gln mutation in ChlR1, which perturbs a conserved arginine residue in the Fe–S cluster binding domain.94
The last Fe–S cluster helicase that we will discuss is the regulator of telomere length 1 (RTEL1), which functions to maintain telomere integrity and unwinds various DNA structures that are assembled in the process of replication, recombination, and repair.95 Thus far, a number of RTEL1 variants have been found that exhibit single nucleotide polymorphisms (SNPs) and increased susceptibility for brain tumors or hepatocellular carcinomas, suggesting a key role for the protein in tumorigenesis.95 Mutations of RTEL1 in coding regions have instead been implicated in premature aging disorders,95 such as dyskeratosis congenita (DC)96 and Hoyeraal-Hreidarsson syndrome (HHS),97 which is considered a more severe form of DC. Both diseases are characterized by multi-system bone marrow failure, due to impaired telomere maintenance.95 One mutation (p. Glu275Lys) has been found in the Fe–S cluster domain,96b but the biochemical characterization and impact of this residue on the cluster has not yet been examined.
Further biochemical characterization is required to understand the Fe–S cluster helicase family, particularly to determine the role of the cluster in relation to disease phenotypes. Efforts in examining the molecular mechanisms will aid current potential therapies that use chemical rescue by small molecules to restore function to mutated helicases and treat disease conditions.2a
Conditions resulting in mitochondrial iron overload
Although the disorders below are linked primarily to the misregulation of iron, they occur specifically in Fe–S cluster proteins. Whether this link is due to a simple abundance of iron or to a more direct connection between Fe–S clusters and iron regulation, perhaps via the iron regulatory protein (IRP), remains uncertain and requires further research.Most recently, a phenotype similar to that observed for sideroblastic anemia has been linked to mutations in the mitochondrial Fe–S cluster biosynthesis chaperone, HSPA9, which has also been implicated in key roles in erythroid differentiation.109 Mutations to HSPA9 result in lower mRNA expression levels with incomplete loss of function leading to the anemia,110 a phenotype which may be under a process of complex regulation.
Fe–S cluster proteins linked to neurological disorders
Similar to the diseases related to iron overload, neurological disorders are often related to iron misregulation; however, it remains to be seen if there is a more direct connection between Fe–S clusters and these conditions. Recent work has indicated a link between mitochondrial dysfunction and neurodegenerative disorders, such as Parkinson's disease, Alzheimer's disease, and Huntington's disease.111 These neurodegenerative conditions are thought to exist as a feedback loop, where mitochondrial dysfunction causes increased production of reactive oxygen species, which decreases Fe–S cluster and heme biosynthesis and leads to IRP activation and iron overload in mitochondria.111,112 This accumulated iron in turn generates additional reactive oxygen species via Fenton chemistry. In the case of Alzheimer's disease, these factors promote oligomerization of amyloid-β to form plaques113 and the formation of Fe3+-paired helical filament aggregates114 to further drive iron accumulation and decrease the levels of the soluble Tau protein.115 Similarly, in Huntington's disease the Huntington protein (Htt) is believed to play a role in maintaining iron homeostasis via the transferrin receptor;111b,116 when mutated in the disease state, Htt likely perturbs iron uptake and contributes to mitochondrial dysfunction.111b In the class of the neurodegenerative disorders, Parkinson's disease has most been associated with iron–sulfur cluster proteins.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :800 individuals with a survival rate of about 15 years after diagnosis. The degenerative process affects dopaminergic neurons leading to motor function deficits, such as resting tremor, rigidity, and postural instability.111,112 Age is considered to be the main risk factor for Parkinson's-however, environmental toxins or genetic susceptibility are also implicated.117 Data suggests that patients with Parkinson's also have a defect in ISC synthesis, partial inhibition of complex I of the respiratory chain, and increased iron levels in dopaminergic neurons (in the substantia nigra),112,118 again due to erroneous regulation of iron homeostasis by IRP.111,112 In addition to this, the pathway which delivers transferrin-bound iron to mitochondria and to complex I of the respiratory chain is found in the substantia nigra dopaminergic neurons, but also consists of transferrin receptor 2.119 In Parkinson's, there is accumulation of oxidized transferrin inside mitochondria, which results in the release of ferrous iron from transferrin and the generation of hydroxyl radicals via Fenton chemistry.119a,120 Furthermore, glutaredoxin 2 (Grx2) has been implicated in promoting the iron accumulation feedback loop in Parkinson's disease based on its ability to regulate cellular redox status by acting on its substrate glutathione (GSH) to impact overall iron homeostasis.112,121 As our understanding of these neurodegenerative disorders is expanded, therapeutic approaches are being explored as well. Thus far, some success has been observed in patients with Parkinson's and other diseases in this area through the use of iron-chelation therapy.111b However, only one of the chelators has been successful in clinical trials,122 since many of them cause adverse side effects due to lack of targeting to specific tissues, resulting in systemic iron depletion.111b,112 New compounds or approaches that include targeting with fewer broad effects could provide a successful treatment option.
:800 individuals with a survival rate of about 15 years after diagnosis. The degenerative process affects dopaminergic neurons leading to motor function deficits, such as resting tremor, rigidity, and postural instability.111,112 Age is considered to be the main risk factor for Parkinson's-however, environmental toxins or genetic susceptibility are also implicated.117 Data suggests that patients with Parkinson's also have a defect in ISC synthesis, partial inhibition of complex I of the respiratory chain, and increased iron levels in dopaminergic neurons (in the substantia nigra),112,118 again due to erroneous regulation of iron homeostasis by IRP.111,112 In addition to this, the pathway which delivers transferrin-bound iron to mitochondria and to complex I of the respiratory chain is found in the substantia nigra dopaminergic neurons, but also consists of transferrin receptor 2.119 In Parkinson's, there is accumulation of oxidized transferrin inside mitochondria, which results in the release of ferrous iron from transferrin and the generation of hydroxyl radicals via Fenton chemistry.119a,120 Furthermore, glutaredoxin 2 (Grx2) has been implicated in promoting the iron accumulation feedback loop in Parkinson's disease based on its ability to regulate cellular redox status by acting on its substrate glutathione (GSH) to impact overall iron homeostasis.112,121 As our understanding of these neurodegenerative disorders is expanded, therapeutic approaches are being explored as well. Thus far, some success has been observed in patients with Parkinson's and other diseases in this area through the use of iron-chelation therapy.111b However, only one of the chelators has been successful in clinical trials,122 since many of them cause adverse side effects due to lack of targeting to specific tissues, resulting in systemic iron depletion.111b,112 New compounds or approaches that include targeting with fewer broad effects could provide a successful treatment option.
Fe–S cluster protein maturation in human infectious agents
Iron–sulfur clusters also play a vital role in prokaryotic metabolism, as catalysts, redox sensors or electron trafficking agents in a variety of important proteins. For example, across different strains of E. coli there are over 150 proteins that contain Fe–S clusters and over 50 different proteins in Mycobacterium tuberculosis.131 Fe–S clusters have consequently been exploited for bactericidal toxicity, where antibiotics promote cell respiration, forming endogenous superoxide and peroxide species, which degrade Fe–S clusters that serve important roles in bacterial cell cycles, and DNA and protein biogenesis.132 Fe–S clusters have also been targeted in terms of human pathogens in Pseudomonas aeruginosa. Deletion of the transcriptional regulator iscR leads to increased susceptibility to peroxides and severely decreased virulence in mouse and Drosophila models.133 Much work has been done in studying Fe–S clusters in Mycobacterium tuberculosis, a facultative pathogen that can live inside macrophages and lead to tuberculosis. These bacteria contain a family of WhiB-like 4Fe–4S cluster-containing transcriptional regulators that could be exploited as an attractive potential antibacterial target, since these regulators play a vital role in mycobacterial cell division, lipid metabolism, oxidative stress, and antibiotic resistance.134Summary and perspectives
Since Fe–S clusters are required in a number of essential proteins or as necessary cofactors for a multitude of enzymatic reactions and physiological processes, conditions that perturb the process of cluster biogenesis and trafficking often lead to disease states. Additionally, a number of these perturbations lead to lethal consequences, making them challenging to investigate, which highlights the importance of understanding the molecular details of Fe–S cluster biosynthesis. Furthermore, recent work in bacteria, utilizing various double mutants and media supplementation, has demonstrated that these genetically engineered forms can grow, albeit slowly, without Fe–S clusters.135 The implications of this work in relation to human Fe–S cluster-based diseases are currently unclear, but may provide alternative treatment strategies in the future.To date, the majority of inborn human diseases related to biogenesis or Fe–S cluster binding proteins are caused by mitochondrial proteins.1a,2b No specific diseases have been associated with the cytosolic system, even though they have demonstrated roles in yeast viability (Table 1).1a,4c,15,75,98,136 Furthermore, diseases have been linked to nuclear proteins that bind Fe–S clusters to function in DNA replication and repair;2a,80,82–84,86,90a,93,95,100c,d however, the presence of the cluster and some of the specific cellular functions remain unclear, presenting challenges when trying to understand the role of the cluster in disease conditions. Aside from diseases associated with specific Fe–S cluster proteins, a subset of neurological disorders, such as Alzheimer's and Parkinson's, have been linked to the Fe–S cluster biogenesis pathway based on the presence of free iron and overall iron dysregulation in mitochondria.111 In addition, Fe–S cluster proteins from all of the cellular compartments have been implicated in a variety of cancers.3 The essential nature and necessary function of these metal cofactors dictates the need for a better understanding of their biogenesis and trafficking mechanisms to clarify the molecular basis of disease states and lead to potential therapeutic approaches.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by a grant from the National Institutes of Health [AI072443]. Christine Wachnowsky was supported by an NIH Chemistry/Biology Interface training grant (T32 GM095450) and an Ohio State University Presidential Fellowship.References
-
(a) N. Maio and T. A. Rouault, Iron–sulfur cluster biogenesis in mammalian cells: new insights into the molecular mechanisms of cluster delivery, Biochim. Biophys. Acta, 2015, 1853, 1493–1512 CrossRef CAS PubMed
; (b) J. J. Braymer and R. Lill, Iron–sulfur cluster biogenesis and trafficking in mitochondria, J. Biol. Chem., 2017, 292, 12754–12763 CrossRef CAS PubMed
-
(a) J. O. Fuss, C.-L. Tsai, J. P. Ishida and J. A. Tainer, Emerging critical roles of Fe–S clusters in DNA replication and repair, Biochim. Biophys. Acta, 2015, 1853, 1253–1271 CrossRef CAS PubMed
- S. Toyokuni, F. Ito, K. Yamashita, Y. Okazaki and S. Akatsuka, Iron and thiol redox signaling in cancer: an exquisite balance to escape ferroptosis, Free Radical Biol. Med., 2017, 108, 610–626 CrossRef CAS PubMed
-
(a) R. Lill, Function and biogenesis of iron–sulphur proteins, Nature, 2009, 460, 831–838 CrossRef CAS PubMed
-
(a) D. C. Johnson, D. R. Dean, A. D. Smith and M. K. Johnson, Structure, function, and formation of biological iron–sulfur clusters, Annu. Rev. Biochem., 2005, 74, 247–281 CrossRef CAS PubMed
-
(a) A. Pandey, H. Yoon, E. R. Lyver, A. Dancis and D. Pain, Isd11p protein activates the mitochondrial cysteine desulfurase Nfs1p protein, J. Biol. Chem., 2011, 286, 38242–38252 CrossRef CAS PubMed
-
(a) O. Gakh, W. Ranatunga, D. Y. Smith IV, E.-C. Ahlgren, S. Al-Karadaghi, J. R. Thompson and G. Isaya, Architecture of the human mitochondrial iron–sulfur cluster assembly machinery, J. Biol. Chem., 2016, 291, 21296–29321 CrossRef CAS PubMed
-
(a) Y. Shi, M. Ghosh, G. Kovtunovych, D. R. Crooks and T. A. Rouault, Both human ferredoxins 1 and 2 and ferredoxin reductase are important for iron–sulfur cluster biogenesis, Biochim. Biophys. Acta, 2012, 1823, 484–492 CrossRef CAS PubMed
-
(a) G. Shaw, J. Cope, L. Li, K. Corson, C. Hersey, G. Ackermann, B. Gwynn, A. Lambert, R. Wingert, D. Traver, N. Trede, B. Barut, Y. Zhou, E. Minet, A. Donovan, A. Brownlie, R. Balzan, M. Weiss, L. Peters, J. Kaplan, L. Zon and B. Paw, Mitoferrin is essential for erythroid iron assimilation, Nature, 2006, 440, 96–100 CrossRef CAS PubMed
-
(a) G. Isaya, H. O’Neill, O. Gakh, S. Park, R. Mantcheva and S. Mooney, Functional studies of frataxin, Acta Paediatr., 2004, 93, 68–72 CrossRef CAS
- F. Colin, A. Martelli, M. Clemancey, J. Latour, S. Gambarelli, L. Zeppieri, C. Birck, A. Page, H. Puccio and S. O. S. Choudens, Mammalian frataxin controls sulfur production and iron entry during de novo Fe4S4 cluster assembly, J. Am. Chem. Soc., 2013, 135, 733–740 CrossRef CAS PubMed
-
(a) C. Tsai and D. Barondeau, Human frataxin is an allosteric switch that activates the Fe–S cluster biosynthetic complex, Biochemistry, 2010, 49, 9132–9139 CrossRef CAS PubMed
-
(a) A. D. Sheftel, O. Stehling, A. J. Pierik, H.-P. Elsässer, U. Mühlenhoff, H. Webert, A. Hobler, F. Hannemann, R. Bernhardt and R. Lill, Humans possess two mitochondrial ferredoxins, Fdx1 and Fdx2, with distinct roles in steroidogenesis, heme, and Fe/S cluster biosynthesis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11775–11780 CrossRef CAS PubMed
-
(a) M. Uzarska, R. Dutkiewicz, S. Freibert, R. Lill and U. Muhlenhoff, The mitochondrial Hsp70 chaperone Ssq1 facilitates Fe/S cluster transfer from Isu1 to Grx5 by complex formation, Mol. Biol. Cell, 2013, 24, 1830–1841 CrossRef CAS PubMed
- R. Lill, R. Dutkiewicz, S. A. Freibert, T. Heidenreich, J. Mascarenhas, D. J. Netz, V. D. Paul, A. J. Pierik, N. Richter, M. Stümpfig, V. Srinivasan, O. Stehling and U. Mühlenhoff, The role of mitochondria and the CIA machinery in the maturation of cytosolic and nuclear iron–sulfur proteins, Eur. J. Cell Biol., 2015, 94, 280–291 CrossRef CAS PubMed
-
(a) J. Y. Lee, J. G. Yang, D. Zhitnitsky, O. Lewinson and D. C. Rees, Structural basis for heavy metal detoxification by an Atm1-type ABC exporter, Science, 2014, 343, 1133–1136 CrossRef CAS PubMed
-
(a) W. Qi, J. Li, C. Y. Chain, G. A. Pasquevich, A. F. Pasquevich and J. A. Cowan, Glutathione complexed Fe–S centers, J. Am. Chem. Soc., 2012, 134, 10745–10748 CrossRef CAS PubMed
-
(a) P. Cavadini, G. Biasiotto, M. Poli, S. Levi, R. Verardi, I. Zanella, M. Derosas, R. Ingrassia, M. Corrado and P. Arosio, RNA silencing of the mitochondrial ABCB7 transporter in HeLa cells causes an iron-deficient phenotype with mitochondrial iron overload, Blood, 2007, 109, 3552–3559 CrossRef CAS PubMed
- C. Kumar, A. Igbaria, B. D’Autreaux, A. G. Planson, C. Junot, E. Godat, A. K. Bachhawat, A. Delaunay-Moisan and M. B. Toledano, Glutathione revisited: a vitalfunction in iron metabolism and ancillary role in thiol-redox control, EMBO J., 2011, 30, 2044–2056 CrossRef CAS PubMed
- T. A. Schaedler, J. D. Thornton, I. Kruse, M. Schwarzlander, A. J. Meyer, H. W. van Veen and J. Balk, A conserved mitochondrial ATP-binding cassette transporter exports glutathione polysulfide for cytosolic metal cofactor assembly, J. Biol. Chem., 2014, 289, 23264–23274 CrossRef CAS PubMed
-
(a) C. Gelling, I. W. Dawes, N. Richhardt, R. Lill and U. Muhlenhoff, Mitochondrial Iba57p is required for Fe/S cluster formation on aconitase and activation of radical SAM enzymes, Mol. Cell. Biol., 2008, 28, 1851–1861 CrossRef CAS PubMed
-
(a) K. D. Kim, W. H. Chung, H. J. Kim, K. C. Lee and J. H. Roe, Monothiol glutaredoxin Grx5 interacts with Fe–S scaffold proteins Isa1 and Isa2 and supports Fe–S assembly and DNA integrity in mitochondria of fission yeast, Biochem. Biophys. Res. Commun., 2010, 392, 467–472 CrossRef CAS PubMed
-
(a) W. H. Tong, G. N. L. Jameson, B. H. Huynh and T. A. Rouault, Subcellular compartmentalization of human Nfu, an iron–sulfur cluster scaffold protein, and its ability to assemble a [4Fe–4S] cluster, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9762–9767 CrossRef CAS PubMed
-
(a) A. Hausmann, D. J. A. Netz, J. Balk, A. J. Pierik, U. Muhlenhoff and R. Lill, The eukaryotic P loopNTPaseNbp35: an essential component of the cytosolic and nuclear iron–sulfur protein assembly machinery, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3266–3271 CrossRef CAS PubMed
-
(a) L. Banci, I. Bertini, V. Calderone, S. Ciofi-Baffoni, A. Giachetti, D. Jaiswal, M. Mikolajczyk, M. Piccioli and J. Winkelmann, Molecular view of an electron transfer process essential for iron–sulfur protein biogenesis, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7136–7141 CrossRef CAS PubMed
-
(a) P. Haunhorst, E. M. Hanschmann, L. Brautigam, O. Stehling, B. Hoffmann, U. Muhlenhoff, R. Lill, C. Berndt and C. H. Lillig, Crucial function of vertebrate glutaredoxin 3 (PICOT) in iron homeostasis and hemoglobin maturation, Mol. Biol. Cell, 2013, 24, 1895–1903 CrossRef CAS PubMed
-
(a) C. E. Outten and A. N. Albetel, Iron sensing and regulation in Saccharomyces cerevisiae: ironing out the mechanistic details, Curr. Opin. Microbiol., 2013, 16, 662–668 CrossRef CAS PubMed
-
(a) M. Seki, Y. Takeda, K. Iwai and K. Tanaka, IOP1 protein is an external component of the human cytosolic iron–sulfur cluster assembly (CIA) machinery and functions in the MMS19 protein-dependent CIA pathway, J. Biol. Chem., 2013, 288, 16680–16689 CrossRef CAS PubMed
-
(a) W. H. Tong, G. N. Jameson, B. H. Huynh and T. A. Rouault, Subcellular compartmentalization of human Nfu, an iron–sulfur cluster scaffold protein, and its ability to assemble a [4Fe–4S] cluster, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9762–9767 CrossRef CAS PubMed
-
(a) A. H. Koeppen, Friedreich's ataxia: pathology, pathogenesis, and molecular genetics, J. Neurol. Sci., 2011, 303, 1–12 CrossRef CAS PubMed
- S. Schmucker and H. Puccio, Understanding the molecular mechanisms of Friedreich's ataxia to develop therapeutic approaches, Hum. Mol. Genet., 2010, 19, 103–110 CrossRef PubMed
- M. G. Cotticelli, L. Rasmussen, N. L. Kushner, S. McKellip, M. I. Sosa, A. Manouvakhova, S. Feng, E. L. White, J. A. Maddry, J. Heemskerk, R. J. Oldt, L. F. Surrey, R. Ochs and R. B. Wilson, Primary and secondary drug screening assays for Friedreich ataxia, J. Biomol. Screening, 2012, 17, 303–313 CrossRef CAS PubMed
- F. Mochel, M. A. Knight, W. H. Tong, D. Hernandez, K. Ayyad, T. Taivassalo, P. M. Andersen, A. Singleton, T. A. Rouault, K. H. Fischbeck and R. G. Haller, Splice mutation in the ironesulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance, Am. J. Hum. Genet., 2008, 82, 652–660 CrossRef CAS PubMed
- A. Olsson, L. Lind, L. E. Thornell and M. Holmberg, Myopathy with lactic acidosis is linked to chromosome 12q23.3-24.11 and caused by an intron mutation in the ISCU gene resulting in a splicing defect, Hum. Mol. Genet., 2008, 17, 1666–1672 CrossRef CAS PubMed
-
(a) P. S. Sanaker, M. Toompuu, V. E. Hogan, L. He, C. Tzoulis, Z. M. C. Lightowlers, R. W. Taylor and L. A. Bindoff, Differences in RNA processing underlie the tissue specific phenotype of ISCU myopathy, Biochim. Biophys. Acta, 2010, 1802, 539–544 CrossRef CAS PubMed
- K. White, Y. Lu, S. Annis, A. E. Hale, B. N. Chau, J. E. Dahlman, C. Hemann, A. R. Opotowsky, S. O. Vargas, I. Rosas, M. A. Perrella, J. C. Osorio, K. J. Haley, B. B. Graham, R. Kumar, R. Saggar, W. D. Wallace, D. J. Ross, O. F. Khan, A. Bader, B. R. Gochuico, M. Matar, K. Polach, N. M. Johannessen, H. M. Prosser, D. G. Anderson, R. Langer, J. L. Zweier, L. A. Bindoff, D. Systrom, A. B. Waxman, R. C. Jin and S. Y. Chan, Genetic and hypoxic alterations of the microRNA-210-ISCU1/2 axis promote iron–sulfur deficiency and pulmonary hypertension, EMBO Mol. Med., 2015, 7, 695–713 CrossRef CAS PubMed
- Y. Funauchi, C. Tanikawa, P. H. Yi Lo, J. Mori, Y. Daigo, A. Takano, Y. Miyagi, A. Okawa, Y. Nakamura and K. Matsuda, Regulation of iron homeostasis by the p53-ISCU pathway, Sci. Rep., 2015, 5, 16497 CrossRef CAS PubMed
- R. Spiegel, A. Saada, J. Halvardson, D. Soiferman, A. Shaag, S. Edvardson, Y. Horovitz, M. Khayat, S. A. Shalev, L. Feuk and O. Elpeleg, Deleterious mutation in FDX1L gene is associated with a novel mitochondrial muscle myopathy, Eur. J. Hum. Genet., 2013, 22, 902–906 Search PubMed
- S. M. K. Farhan, J. Wang, J. F. Robinson, P. Lahiry, V. M. Siu, C. Prasad, J. B. Kronick, D. A. Ramsay, C. A. Rupar and R. A. Hegele, Exome sequencing identifies NFS1 deficiency in a novel Fe–S cluster disease, infantile mitochondrial complex II/III deficiency, Mol. Genet. Genomic Med., 2014, 2, 73–80 CrossRef CAS PubMed
-
(a) S. C. Lim, M. Friemel, J. E. Marum, E. J. Tucker, D. L. Bruno, L. G. Riley, J. Christodoulou, E. P. Kirk, A. Boneh, C. M. DeGennaro, M. Springer, V. K. Mootha, T. A. Rouault, S. Leimkuhler, D. R. Thorburn and A. G. Compton, Mutations in LYRM4, encoding iron–sulfur cluster biogenesis factor ISD11, cause deficiency of multiple respiratory chain complexes, Hum. Mol. Genet., 2013, 22, 4460–4473 CrossRef CAS PubMed
-
(a) U. Ahting, J. A. Mayr, A. V. Vanlander, S. A. Hardy, S. Santra, C. Makowski, C. L. Alston, F. A. Zimmermann, L. Abela, B. Plecko, M. Rohrbach, S. Spranger, S. Seneca, B. Rolinski, A. Hagendorff, M. Hempel, W. Sperl, T. Meitinger, J. Smet, R. W. Taylor, R. Van Coster, P. Freisinger, H. Prokisch and T. B. Haack, Clinical, biochemical, and genetic spectrum of seven patients with NFU1 deficiency, Front. Genet., 2015, 6, 123 Search PubMed
-
(a) P. R. Baker, M. W. Friederich, M. A. Swanson, T. Shaikh, K. Bhattacharya, G. H. Scharer, J. Aicher, G. Creadon-Swindell, E. Geiger, K. N. MacLean, W. T. Lee, C. Deshpande, M. L. Freckmann, L. Y. Shih, M. Wasserstein, M. B. Rasmussen, A. M. Lund, P. Procopis, J. M. Cameron, B. H. Robinson, G. K. Brown, R. M. Brown, A. G. Compton, C. L. Dieckmann, R. Collard, C. R. Coughlin, E. Spector, M. F. Wempe and J. L. K. Van Hove, Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5, Brain, 2013, 137, 366–379 CrossRef PubMed
-
(a) N. Ajit Bolar, A. V. Vanlander, C. Wilbrecht, N. Van der Aa, J. Smet, B. De Paepe, G. Vandeweyer, F. Kooy, F. Eyskens, E. De Latter, G. Delanghe, P. Govaert, J. G. Leroy, B. Loeys, R. Lill, L. Van Laer and R. Van Coster, Mutation of the iron–sulfur cluster assembly gene IBA57 causes severe myopathy and encephalopathy, Hum. Mol. Genet., 2013, 22, 2590–2602 CrossRef CAS PubMed
- Z. N. Al-Hassnan, M. Al-Dosary, M. Alfadhel, E. A. Faqeih, M. Alsagob, R. Kenana, R. Almass, O. S. Al-Harazi, H. Al-Hindi, O. I. Malibari, F. B. Almutari, S. Tulbah, F. Alhadeq, T. Al-Sheddi, R. Alamro, A. AlAsmari, M. Almuntashri, H. Alshaalan, F. A. Al-Mohanna, D. Colak and N. Kaya, ISCA2 mutation causes infantile neurodegenerative mitochondrial disorder, J. Med. Genet., 2015, 52, 186–194 CrossRef CAS PubMed
- A. Shukla, M. Hebbar, A. Srivastava, R. Kadavigere, P. Upadhyai, A. Kanthi, O. Brandau, S. Bielas and K. M. Girisha, Homozygous p.(Glu87Lys) variant in ISCA1 is associated with a multiple mitochondrial dysfunctions syndrome, J. Hum. Genet., 2017, 62, 723–727 CrossRef CAS PubMed
- J. M. Cameron, A. Janer, V. Levandovskiy, N. MacKay, T. A. Rouault, W.-H. Tong, I. Ogilvie, E. A. Shoubridge and B. H. Robinson, Mutations in iron–sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes, Am. J. Hum. Genet., 2011, 89, 486–495 CrossRef CAS PubMed
- C. Wachnowsky, N. A. Wesley, I. Fidai and J. A. Cowan, Understanding the molecular basis for Multiple Mitochondrial Dysfunctions Syndrome 1 (MMDS1) – Impact of a disease-causing Gly208Cys substitution on structure and activity of NFU1 in the Fe/S cluster biosynthetic pathway, J. Mol. Biol., 2017, 429, 790–807 CrossRef CAS PubMed
- M. A. Uzarska, V. Nasta, B. D. Weiler, F. Spantgar, S. Ciofi-Baffoni, M. R. Saviello, L. Gonnelli, U. Muehlenhoff, L. Banci and R. Lill, Mitochondrial Bol1 and Bol3 function as assembly factors for specific iron–sulfur proteins, eLife, 2016, 5, e15991 Search PubMed
- A. Melber, U. Na, A. Vashisht, B. D. Weiler, R. Lill, J. A. Wohlschlegel and D. R. Winge, Role of Nfu1 and Bol3 in Iiron–sulfur cluster transfer to mitochondrial clients, eLife, 2016, 5, e15991 Search PubMed
- N. Maio, K. S. Kim, A. Singh and T. A. Rouault, A single adaptable cochaperone-scaffold complex delivers nascent iron–sulfur clusters to mammalian respiratory chain complexes I–III, Cell Metab., 2017, 25, 945–953 CrossRef CAS PubMed
- I. E. Scheffler, Mitochondrial disease associated with complex I (NADH-CoQ oxidoreductase) deficiency, J. Inherited Metab. Dis., 2014, 38, 405–415 CrossRef PubMed
- S. H. Kevelam, R. J. Rodenburg, N. I. Wolf, P. Ferreira, R. J. Lunsing, L. G. Nijtmans, A. Mitchell, H. A. Arroyo, D. Rating, A. Vanderver, C. G. M. van Berkel, T. E. M. Abbink, P. Heutink and M. S. van der Knapp, NUBPL mutations in patients with complex I deficiency and a distinct MRI pattern, Neurology, 2013, 80, 1577–15783 CrossRef CAS PubMed
-
(a) E. J. Tucker, M. Mimaki, A. G. Compton, M. McKenzie, M. T. Ryan and D. R. Thorburn, Next-generation sequencing in molecular diagnosis: NUBPL mutations highlight the challenges of variant detection and interpretation, Hum. Mutat., 2012, 33, 411–418 CrossRef CAS PubMed
- M. M. Wydro, P. Sharma, J. M. Foster, K. Bych, E. H. Meyer and J. Balk, The evolutionarily conserved iron–sulfur protein INDH is required for complex I assembly and mitochondrial translation in Arabidopsis, Plant Cell, 2013, 25, 4014–4027 CrossRef CAS PubMed
-
(a) A. S. Hoekstra and J.-P. Bayley, The role of complex II in disease, Biochim. Biophys. Acta, 2013, 1827, 543–551 CrossRef CAS PubMed
- C. L. Alston, J. E. Davison, F. Meloni, F. H. van der Westhuizen, L. He, H.-T. Hornig-Do, A. C. Peet, P. Gissen, P. Goffrini, I. Ferrero, E. Wassmer, R. McFarland and R. W. Taylor, Recessive germline SDHA and SDHB mutations causing leukodystrophy and isolated mitochondrial complex II deficiency, J. Med. Genet., 2012, 49, 569–577 CrossRef CAS PubMed
- M. A. Pantaleo, A. Astolfi, M. Urbini, M. Nannini, P. Paterini, V. Indio, M. Saponara, S. Formica, C. Ceccarelli, R. Casadio, G. Rossi, F. Bertolini, D. Santini, M. G. Pirini, M. Fiorentino, U. Basso and G. Biasco, Analysis of all subunits, SDHA, SDHB, SDHC, SDHD, of the succinate dehydrogenase complex in KIT/PDGFRA wild-type GIST, Eur. J. Hum. Genet., 2013, 22, 32–39 Search PubMed
-
(a) N. Saxena, N. Maio, D. R. Crooks, C. J. Ricketts, Y. Yang, M.-H. Wei, T. W. M. Fan, A. N. Lane, C. Sourbier, A. Singh, J. K. Killian, P. S. Meltzer, C. D. Vocke, T. A. Rouault and W. M. Linehan, SDHB-deficient cancers: the role of mutations that impair iron sulfur cluster delivery, J. Natl. Cancer Inst., 2016, 108, djv287 CrossRef PubMed
- U. Na, W. Yu, J. Cox, D. K. Bricker, K. Brockmann, J. Rutter, C. S. Thummel and D. R. Winge, The LYR factors SDHAF1 and SDHAF3 mediate maturation of the iron–sulfur subunit of succinate dehydrogenase, Cell Metab., 2014, 20, 253–266 CrossRef CAS PubMed
- N. Maio, D. Ghezzi, D. Verrigni, T. Rizza, E. Bertini, D. Martinelli, M. Zeviani, A. Singh and R. Carrozzo, Tracey A. Rouault, Disease-causing SDHAF1 mutations impair transfer of Fe–S clusters to SDHB, Cell Metab., 2016, 23, 292–302 CrossRef CAS PubMed
- A. Ohlenbusch, S. Edvardson, J. Skorpen, A. Bjornstad, A. Saada, O. Elpeleg, J. Gärtner and K. Brockmann, Leukoencephalopathy with accumulated succinate is indicative of SDHAF1 related complex II deficiency, Orphanet J. Rare Dis., 2012, 7, 69 CrossRef PubMed
- S. Tamir, M. L. Paddock, M. Darash-Yahana-Baram, S. H. Holt, Y. S. Sohn, L. Agranat, D. Michaeli, J. T. Stofleth, C. H. Lipper, F. Morcos, I. Z. Cabantchik, J. N. Onuchic, P. A. Jennings, R. Mittler and R. Nechushtai, Structure–function analysis of NEET proteins uncovers their role as key regulators of iron and ROS homeostasis in health and disease, Biochim. Biophys. Acta, 2015, 1853, 1294–1315 CrossRef CAS PubMed
- W. J. Geldenhuys, T. C. Leeper and R. T. Carroll, mitoNEET as a novel drug target for mitochondrial dysfunction, Drug Discovery Today, 2014, 19, 1601–1606 CrossRef CAS PubMed
- C. M. Kusminski, W. L. Holland, K. Sun, J. Park, S. B. Spurgin, Y. Lin, G. R. Askew, J. A. Simcox, D. A. McClain, C. Li and P. E. Scherer, MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity, Nat. Med., 2012, 18, 1539–1549 CrossRef CAS PubMed
- R. Nechushtai, A. R. Conlan, Y. Harir, L. Song, O. Yogev, Y. Eisenberg-Domovich, O. Livnah, D. Michaeli, R. Rosen, V. Ma, Y. Luo, J. A. Zuris, M. L. Paddock, Z. I. Cabantchik, P. A. Jennings and R. Mittler, Characterization of Arabidopsis NEET reveals an ancient role for NEET proteins in iron metabolism, Plant Cell, 2012, 24, 2139–2154 CrossRef CAS PubMed
- I. Ferecatu, S. Goncalves, M. P. Golinelli-Cohen, M. Clemancey, A. Martelli, S. Riquier, E. Guittet, J. M. Latour, H. Puccio, J. C. Drapier, E. Lescop and C. Bouton, The diabetes drug target mitoNEET governs a novel trafficking pathway to rebuild an Fe–S cluster into cytosolic aconitase/iron regulatory protein 1, J. Biol. Chem., 2014, 289, 28070–28086 CrossRef CAS PubMed
- M.-P. Golinelli-Cohen, E. Lescop, C. Mons, S. Gonçalves, M. Clémancey, J. Santolini, E. Guittet, G. Blondin, J.-M. Latour and C. Bouton, Redox control of the human iron–sulfur repair protein mitoNEET activity via its iron–sulfur cluster, J. Biol. Chem., 2016, 291, 7583–7593 CrossRef CAS PubMed
- G. L. Taminelli, V. Sotomayor, A. G. Valdivieso, M. L. Teiber, M. A. C. Marın and T. A. Santa-Coloma, CISD1 codifies a mitochondrial protein upregulated by the CFTR channel, Biochem. Biophys. Res. Commun., 2008, 365, 856–862 CrossRef CAS PubMed
- K. Okatsu, S.-i. Iemura, F. Koyano, E. Go, M. Kimura, T. Natsume, K. Tanaka and N. Matsuda, Mitochondrial hexokinase HKI is a novel substrate of the Parkin ubiquitin ligase, Biochem. Biophys. Res. Commun., 2012, 428, 197–202 CrossRef CAS
-
(a) A. F. Salem, D. Whitaker-Menezes, A. Howell, F. Sotgia and M. P. Lisanti, Mitochondrial biogenesis in epithelial cancer cells promotes breast cancer tumor growth and confers autophagy resistance, Cell Cycle, 2012, 11, 4174–4180 CrossRef CAS PubMed
- Y.-S. Sohn, S. Tamir, L. Song, D. Michaeli, I. Matouk, A. R. Conlan, Y. Harir, S. H. Holt, V. Shulaev, M. L. Paddock, A. Hochberg, I. Z. Cabanchick, J. N. Onuchic, P. A. Jennings, R. Nechushtaia and R. Mittler, NAF-1 and mitoNEET are central to human breast cancer proliferation by maintaining mitochondrial homeostasis and promoting tumor growth, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14676–14681 CrossRef CAS PubMed
- V. D. Paul and R. Lill, Biogenesis of cytosolic and nuclear iron–sulfur proteins and their role in genome stability, Biochim. Biophys. Acta, 2015, 1853, 1528–1539 CrossRef CAS PubMed
- M. Nakamura, D. M. Buzas, A. Kato, M. Fujita, N. Kurata and T. Kinoshita, The role of Arabidopsis thaliana NAR1, a cytosolic iron–sulfur cluster assembly component, in gametophytic gene expression and oxidative stress responses in vegetative tissue, New Phytol., 2013, 199, 925–935 CrossRef CAS PubMed
- M. V. Corbin, D. A. P. Rockx, A. B. Oostra, H. Joenje and J. C. Dorsman, The iron–sulfur cluster assembly network component NARFL is a key element in the cellular defense against oxidative stress, Free Radical Biol. Med., 2015, 89, 863–872 CrossRef CAS PubMed
-
(a) K. Gari, A. M. L. Ortiz, V. Borel, H. Flynn, J. M. Skehel and S. J. Boulton, MMS19 links cytoplasmic iron–sulfur cluster assembly to DNA metabolism, Science, 2012, 337, 243–245 CrossRef CAS PubMed
- J. Rudolf, V. Makrantoni, W. Ingledew, M. Stark and M. White, The DNA repair helicases XPD and FancJ have essential iron–sulfur domains, Mol. Cell, 2006, 23, 801–808 CrossRef CAS PubMed
- R. Service, Science, 2014, 346, 1284–1287 CrossRef CAS PubMed
-
(a) S. Pokharel and J. Campbell, Cross talk between the nuclease and helicase activities of Dna2: role of an essential iron–sulfur cluster domain, Nucleic Acids Res., 2012, 40, 7821–7830 CrossRef CAS PubMed
- A. H. S. de Oliveira, A. E. da Silva, I. M. de Oliveira, J. A. P. Henriques and L. F. Agnez-Lima, MutY-glycosylase: an overview on mutagenesis and activities beyond the GO system, Mutat. Res., 2014, 769, 119–131 CrossRef CAS PubMed
- F. Mazzei, A. Viel and M. Bignami, Role of MUTYH in human cancer, Mutat. Res., 2013, 743-744, 33–43 CrossRef CAS PubMed
- A. A. Out, C. M. J. Tops, M. Nielsen, M. M. Weiss, I. J. H. M. van Minderhout, I. F. A. C. Fokkema, M.-P. Buisine, K. Claes, C. Colas, R. Fodde, F. Fostira, P. F. Franken, M. Gaustadnes, K. Heinimann, S. V. Hodgson, F. B. L. Hogervorst, E. Holinski-Feder, K. Lagerstedt-Robinson, S. Olschwang, A. M. W. van den Ouweland, E. J. W. Redeker, R. J. Scott, B. Vankeirsbilck, R. V. Grønlund, J. T. Wijnen, F. P. Wikman, S. Aretz, J. R. Sampson, P. Devilee, J. T. den Dunnen and F. J. Hes, Leiden open variation database of the MUTYH gene, Hum. Mutat., 2010, 31, 1205–1215 CrossRef CAS PubMed
- M. K. Brinkmeyer and S. S. David, Distinct functional consequences of MUTYH variants associated with colorectal cancer: damaged DNA affinity, glycosylase activity and interaction with PCNA and Hus1, DNA Repair, 2015, 34, 39–51 CrossRef CAS PubMed
-
(a) A. N. Suhasini and R. M. Brosh, Disease-causing missense mutations in human DNA helicase disorders, Mutat. Res., 2013, 752, 138–152 CrossRef CAS PubMed
- H. Walden and A. J. Deans, The Fanconi anemia DNA repair pathway: structural and functional insights into a complex disorder, Annu. Rev. Biophys., 2014, 43, 257–278 CrossRef CAS PubMed
- S. K. Bharti, J. A. Sommers, F. George, J. Kuper, F. Hamon, K. Shin-ya, M. P. Teulade-Fichou, C. Kisker and R. M. Brosh, Specialization among iron–sulfur cluster helicases to resolve G-quadruplex DNA structures that threaten genomic stability, J. Biol. Chem., 2013, 288, 28217–28229 CrossRef CAS PubMed
-
(a) S. B. Cantor and S. Guillemette, Hereditary breast cancer and the BRCA1-associated FANCJ/BACH1/BRIP1, Future Oncol., 2011, 7, 253–261 CrossRef CAS PubMed
- R. Gupta, S. Sharma, K. M. Doherty, J. A. Sommers, S. B. Cantor and R. M. Brosh, Inhibition of BACH1 (FANCJ) helicase by backbone discontinuity is overcome by increased motor ATPase or length of loading strand, Nucleic Acids Res., 2006, 34, 6673–6683 CrossRef CAS PubMed
- J. A. Sommers, N. Rawtani, R. Gupta, D. V. Bugreev, A. V. Mazin, S.
B. Cantor and R. M. Brosh, FANCJ uses its motor ATPase to destabilize protein–DNA complexes, unwind triplexes, and inhibit RAD51 strand exchange, J. Biol. Chem., 2009, 284, 7505–7517 CrossRef CAS PubMed
-
(a) O. Levran, C. Attwooll, R. T. Henry, K. L. Milton, K. Neveling, P. Rio, S. D. Batish, R. Kalb, E. Velleuer, S. Barral, J. Ott, J. Petrini, D. Schindler, H. Hanenberg and A. D. Auerbach, The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia, Nat. Genet., 2005, 37, 931–933 CrossRef CAS PubMed
-
(a) L. Fan, J. O. Fuss, Q. J. Cheng, A. S. Arvai, M. Hammel, V. A. Roberts, P. K. Cooper and J. A. Tainer, XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations, Cell, 2008, 133, 789–800 CrossRef CAS PubMed
- Y. Wu, J. A. Sommers, I. Khan, J. P. de Winter, J. Brosh and M. Robert, Biochemical characterization of Warsaw breakage syndrome helicase, J. Biol. Chem., 2011, 287, 1007–1021 CrossRef PubMed
- S. K. Bharti, I. Khan, T. Banerjee, J. A. Sommers, Y. Wu and R. M. Brosh, Molecular functions and cellular roles of the ChlR1 (DDX11) helicase defective in the rare cohesinopathy Warsaw breakage syndrome, Cell. Mol. Life Sci., 2014, 71, 2625–2639 CrossRef CAS PubMed
- P. van der Lelij, K. H. Chrzanowska, B. C. Godthelp, M. A. Rooimans, A. B. Oostra, M. Stumm, M. Z. Zdzienicka, H. Joenje and J. P. de Winter, Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1, Am J. Hum. Genet., 2010, 86, 262–266 CrossRef CAS PubMed
- J.-M. Capo-Chichi, S. K. Bharti, J. A. Sommers, T. Yammine, E. Chouery, L. Patry, G. A. Rouleau, M. E. Samuels, F. F. Hamdan, J. L. Michaud, R. M. Brosh Jr, A. Mégarbane and Z. Kibar, Identification and biochemical characterization of a novel mutation in DDX11 causing Warsaw breakage syndrome, Hum. Mutat., 2013, 34, 103–107 CrossRef CAS PubMed
- J.-B. Vannier, G. Sarek and S. J. Boulton, RTEL1: functions of a disease-associated helicase, Trends Cell Biol., 2014, 24, 416–425 CrossRef CAS PubMed
-
(a) B. J. Ballew, M. Yeager, K. Jacobs, N. Giri, J. Boland, L. Burdett, B. P. Alter and S. A. Savage, Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in Dyskeratosis congenita, Hum. Genet., 2013, 132, 473–480 CrossRef CAS PubMed
-
(a) Z. Deng, G. Glousker, A. Molczan, A. J. Fox, N. Lamm, J. Dheekollu, O.-E. Weizman, M. Schertzer, Z. Wang, O. Vladimirova, J. Schug, M. Aker, A. Londoño-Vallejo, K. H. Kaestner, P. M. Lieberman and Y. Tzfati, Inherited mutations in the helicase RTEL1 cause telomere dysfunction and Hoyeraal–Hreidarsson syndrome, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3408–E3416 CrossRef CAS PubMed
- D. J. A. Netz, C. M. Stith, M. Stümpfig, G. Köpf, D. Vogel, H. M. Genau, J. L. Stodola, R. Lill, P. M. J. Burgers and A. J. Pierik, Eukaryotic DNA polymerases require an iron–sulfur cluster for the formation of active complexes, Nat. Chem. Biol., 2011, 8, 125–132 CrossRef PubMed
- T. N. Moiseeva, A. M. Gamper, B. L. Hood, T. P. Conrads and C. J. Bakkenist, Human DNA polymerase ε is phosphorylated at serine-1940 after DNA damage and interacts with the iron–sulfur complex chaperones CIAO1 and MMS19, DNA Repair, 2016, 43, 9–17 CrossRef CAS PubMed
-
(a) E. Heitzer and I. Tomlinson, Replicative DNA polymerase mutations in cancer, Curr. Opin. Genet. Dev., 2014, 24, 107–113 CrossRef CAS PubMed
- T. Fujiwara and H. Harigae, Pathophysiology and genetic mutations in congenital sideroblastic anemia, Pediatr. Int., 2013, 55, 675–679 CrossRef CAS PubMed
- M. S. Protasova, A. P. Grigorenko, T. V. Tyazhelova, T. V. Andreeva, D. A. Reshetov, F. E. Gusev, A. E. Laptenko, I. L. Kuznetsova, A. Y. Goltsov, S. A. Klyushnikov, S. N. Illarioshkin and E. I. Rogaev, Whole-genome sequencing identifies a novel ABCB7 gene mutation for X-linked congenital cerebellar ataxia in a large family of Mongolian ancestry, Eur. J. Hum. Genet., 2015, 24, 550–555 Search PubMed
- K. Sato, Y. Torimoto, T. Hosoki, K. Ikuta, H. Takahashi, M. Yamamoto, S. Ito, N. Okamura, K. Ichiki, H. Tanaka, M. Shindo, K. Hirai, Y. Mizukami, T. Otake, M. Fujiya, K. Sasaki and Y. Kohgo, Loss of ABCB7 gene: pathogenesis of mitochondrial iron accumulation in erythroblasts in refractory anemia with ringed sideroblast with isodicentric (X)(q13), Int. J. Hematol., 2011, 93, 311–318 CrossRef PubMed
-
(a) C. Camaschella, A. Campanella, L. De Falco, L. Boschetto, R. Merlini, L. Silvestri, S. Levi and A. Iolascon, The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload, Blood, 2007, 110, 1353–1358 CrossRef CAS PubMed
- G. Liu, S. Guo, G. J. Anderson, C. Camaschella, B. Han and G. Nie, Heterozygous missense mutations in the GLRX5 gene cause sideroblastic anemia in a Chinese patient, Blood, 2014, 124, 2750–2751 CrossRef CAS PubMed
- G. Liu, Y. Wang, G. J. Anderson, C. Camaschella, Y. Chang and G. Nie, Functional analysis of GLRX5 mutants reveals distinct functionalities of GLRX5 protein, J. Cell. Biochem., 2016, 117, 207–217 CrossRef CAS PubMed
- C. Johansson, A. K. Roos, S. J. Montano, R. Sengupta, P. Filippakopoulos, K. Guo, F. von Delft, A. Holmgren, U. Oppermann and K. L. Kavanagh, The crystal structure of human GLRX5: iron–sulfur cluster co-ordination, tetrameric assembly and monomer activity, Biochem. J., 2011, 433, 303–311 CrossRef CAS PubMed
- R. A. Wingert, J. L. Galloway, B. Barut, H. Foott, P. Fraenkel, J. L. Axe, G. J. Weber, K. Dooley, A. J. Davidson, B. Schmidt, B. H. Paw, G. C. Shaw, P. Kingsley, J. Palis, H. Schubert, O. Chen, J. Kaplan, C. The Tübingen Screen and L. I. Zon, Deficiency of glutaredoxin 5 reveals Fe–S clusters are required for vertebrate haem synthesis, Nature, 2005, 436, 1035–1039 CrossRef CAS PubMed
- Y. Shan and G. Cortopassi, Mitochondrial Hspa9/Mortalin regulates erythroid differentiation via iron–sulfur cluster assembly, Mitochondrion, 2016, 26, 94–103 CrossRef CAS PubMed
- K. Schmitz-Abe, S. J. Ciesielski, P. J. Schmidt, D. R. Campagna, F. Rahimov, B. A. Schilke, M. Cuijpers, K. Rieneck, B. Lausen, M. L. Linenberger, A. K. Sendamarai, C. Guo, I. Hofmann, P. E. Newburger, D. Matthews, A. Shimamura, P. J. L. M. Snijders, M. C. Towne, C. M. Niemeyer, H. G. Watson, M. H. Dziegiel, M. M. Heeney, A. May, S. S. Bottomley, D. W. Swinkels, K. Markianos, E. A. Craig and M. D. Fleming, Congenital sideroblastic anemia due to mutations in the mitochondrial HSP70 homologue HSPA9, Blood, 2015, 126, 2734–2738 CrossRef CAS PubMed
-
(a) P. J. Urrutia, N. P. Mena and M. T. Nunez, The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders, Front. Pharmacol., 2014, 5, 38 Search PubMed
- Y. Muñoz, C. M. Carrasco, J. D. Campos, P. Aguirre and M. T. Núñez, Parkinson's disease: the mitochondria-iron link, Parkinson's Dis., 2016, 1–21 Search PubMed
-
(a) F. Hefti, W. F. Goure, J. Jerecic, K. S. Iverson, P. A. Walicke and G. A. Krafft, The case for soluble Aβ oligomers as a drug target in Alzheimer's disease, Trends Pharmacol. Sci., 2013, 34, 261–266 CrossRef CAS PubMed
- C. Guo, P. Wang, M.-L. Zhong, T. Wang, X.-S. Huang, J.-Y. Li and Z.-Y. Wang, Deferoxamine inhibits iron induced hippocampal tau phosphorylation in the Alzheimer transgenic mouse brain, Neurochem. Int., 2013, 62, 165–172 CrossRef CAS PubMed
- P. Lei, S. Ayton, D. I. Finkelstein, L. Spoerri, G. D. Ciccotosto, D. K. Wright, B. X. W. Wong, P. A. Adlard, R. A. Cherny, L. Q. Lam, B. R. Roberts, I. Volitakis, G. F. Egan, C. A. McLean, R. Cappai, J. A. Duce and A. I. Bush, Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export, Nat. Med., 2012, 18, 291–295 CrossRef CAS PubMed
-
(a) A. L. Lumsden, T. L. Henshall, S. Dayan, M. T. Lardelli and R. I. Richards, Huntingtin-deficient zebrafish exhibit defects in iron utilization and development, Hum. Mol. Genet., 2007, 16, 1905–1920 CrossRef CAS PubMed
- A. J. Lees, J. Hardy and T. Revesz, Parkinson's disease, Lancet, 2009, 373, 2055–2066 CrossRef CAS
-
(a) D. Berg and H. Hochstrasser, Iron metabolism in Parkinsonian syndromes, Mov. Disord., 2006, 21, 1299–1310 CrossRef PubMed
-
(a) P. G. Mastroberardino, E. K. Hoffman, M. P. Horowitz, R. Betarbet, G. Taylor and D. Cheng,
et al., A novel transferrin/TfR2-mediated mitochondrial iron transport system is disrupted in Parkinson's disease, Neurobiol. Dis., 2009, 34, 417–431 CrossRef CAS PubMed
- M. P. Horowitz and J. T. Greenamyre, Mitochondrial iron metabolism and its role in neurodegeneration, J. Alzheimer's Dis., 2010, 20, 551–568 CrossRef PubMed
- D. W. Lee, D. Kaur, S. J. Chinta, S. Rajagopalan and J. K. Andersen, A disruption in iron–sulfur center biogenesis via inhibition of mitochondrial dithiol glutaredoxin 2 may contribute to mitochondrial and cellular iron dysregulation in mammalian glutathione-depleted dopaminergic cells: implications for Parkinson's disease, Antioxid. Redox Signaling, 2009, 11, 2083–2094 CrossRef CAS PubMed
-
(a) M. Katsuyama, K. Iwata, M. Ibi, K. Matsuno, M. Matsumoto and C. Yabe-Nishimura, Clioquinol induces DNA double-strand breaks, activation of ATM, and subsequent activation of p53 signaling, Toxicology, 2012, 299, 55–59 CrossRef CAS PubMed
- D. P. Barupala, S. P. Dzul, P. J. Riggs-Gelasco and T. L. Stemmler, Synthesis, delivery and regulation of eukaryotic heme and Fe–S cluster cofactors, Arch. Biochem. Biophys., 2016, 592, 60–75 CrossRef CAS PubMed
- M. Lecha, H. Puy and J.-C. Deybach, Erythropoietic protoporphyria, Orphanet J. Rare Dis., 2009, 4, 19 CrossRef PubMed
- H. Nakano, A. Nakano, Y. Toyomaki, S. Ohashi, K. Harada, R. Moritsugu, H. Takeda, A. Kawada, Y. Mitsuhashi and K. Hanada, Novel ferrochelatase mutations in Japanese patients with erythropoietic protoporphyria: high frequency of the splice site modulator IVS3-48C polymorphism in the Japanese population, J. Invest. Dermatol., 2006, 126, 2717–2719 CrossRef CAS PubMed
- X.-F. Kong, J. Ye, D.-Y. Gao, Q.-M. Gong, D.-H. Zhang, Z.-M. Lu, Y.-M. Lu and X.-X. Zhang, Identification of a ferrochelatase mutation in a Chinese family with erythropoietic protoporphyria, J. Hepatol., 2008, 48, 375–379 CrossRef CAS PubMed
- M. Balwani and D. Doheny, Loss-of-function ferrochelatase and gain-of-function erythroid-specific 5-aminolevulinate synthase mutations causing erythropoietic protoporphyria and X-linked protoporphyria in North American patients reveal novel mutations and a high prevalence of X-linked protoporphyria, Mol. Med., 2013, 19, 1 Search PubMed
- M. S. Farrag, J. Kucerova, L. Šlachtova, O. Šeda, J. Šperl and P. Martasek, A novel mutation in the FECH Gene in a Czech family with erythropoietic protoporphyria and a population study of IVS3-48C variant contributing to the disease, Folia Biol., 2015, 61, 227–232 CAS
- Y. Wang, N. B. Langer, G. C. Shaw, G. Yang, L. Li, J. Kaplan, B. H. Paw and J. R. Bloomer, Abnormal mitoferrin-1 expression in patients with erythropoietic protoporphyria, Exp. Hematol., 2011, 39, 784–794 CrossRef CAS PubMed
-
(a) W. Chen, H. A. Dailey and B. H. Paw, Ferrochelatase forms an oligomeric complex with mitoferrin-1 and Abcb10 for erythroid heme biosynthesis, Blood, 2010, 116, 628–630 CrossRef CAS PubMed
- H. Beinert, Iron–sulfur proteins: ancient structures, still full of surprises, J. Biol. Inorg. Chem., 2000, 5, 2–15 CrossRef CAS PubMed
-
(a) M. A. Kohanski, D. J. Dwyer and J. J. Collins, How antibiotics kill bacteria: from targets to networks, Nat. Rev. Microbiol., 2010, 423–435 CrossRef CAS PubMed
-
(a) Y. S. Choi, D. H. Shin, I. Y. Chung, S. H. Kim, Y. J. Heo and Y. H. Cho, Identification of Pseudomonas aeruginosa genes crucial for hydrogen peroxide resistance, J. Microbiol. Biotechnol., 2007, 17, 1344–1352 Search PubMed
-
(a) R. P. Morris, L. Nguyen, J. Gatfield, K. Visconti, K. Nguyen, D. Schnappinger, S. Ehrt, Y. Liu, L. Heifets, J. Pieters, G. Schoolnik and C. J. Thompson, Ancestral antibiotic resistance in Mycobacterium tuberculosis, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12200–12205 CrossRef CAS PubMed
- A. G. Rocha and A. Dancis, Life without Fe–S clusters, Mol. Microbiol., 2016, 99, 821–826 CrossRef CAS PubMed
-
(a) D. J. A. Netz, A. J. Pierik, M. Stumpfig, E. Bill, A. K. Sharma, L. J. Pallesen, W. E. Walden and R. Lill, A bridging [4Fe–4S] cluster and nucleotide binding are essential for function of the Cfd1–Nbp35 complex as a scaffold in iron–sulfur protein maturation, J. Biol. Chem., 2012, 287, 12365–12378 CrossRef CAS PubMed
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |