
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Control of the molecular packing of chloroboron(III) and fluoroboron(III) subnaphthalocyanines by designing peripheral substituents†
Akuto Takagi and
Tadashi Mizutani *
*
Department of Molecular Chemistry and Biochemistry, Faculty of Science and Engineering, Doshisha University, Kyotanabe, Kyoto, 610-0321 Japan. E-mail: tmizutan@mail.doshisha.ac.jp
First published on 27th November 2017
Abstract
Chloroboron(III) and fluoroboron(III) hexa(1-alkynyl)- and hexa(2-arylethynyl)subnaphthalocyanines with a large dipole moment were prepared by Sonogashira coupling of hexaiodosubnaphthalocyanines with substituted acetylenes. Introduction of butyl or longer alkyl groups via ethynylene linkages on the periphery resulted in lower melting points and higher solubility in dichloromethane. X-ray diffraction (XRD) patterns of the cast film indicated that hexa(2-(4-hexylphenyl)ethynyl)- and hexa(2-(4-hexyloxyphenyl)ethynyl)subnaphthalocyanines with a B–F bond are packed in a discotic hexagonal columnar phase with lattice constants a = 59–64 Å and stacking distance c = 4.7–4.8 Å. The Q-band in visible spectra of the thin film of these B–F derivatives was blue-shifted, supporting the formation of H-aggregate in stacked columns. Polarized optical microscopy showed that these subnaphthalocyanines with a B–F bond exhibited a mesophase at 180 °C. XRD of these subnaphthalocyanines at 180 °C confirms a two dimensional hexagonal phase. XRD of the corresponding derivatives with a B–Cl bond showed that they were either amorphous or less crystalline. We suggest that the fluorine atom in the B–F group can fit into the cleft of cone-shaped subnaphthalocyanine, owing to the smaller van der Waals radius of F than Cl, to stabilize the columnar packing structure.
Introduction
Cyclic oligopyrroles such as porphyrins and phthalocyanines are important dyes whose optical, redox, and semi-conductive properties have been employed in active materials for diverse optoelectric systems, such as photosynthesis, solar cells,1 and electroluminescence devices.2 Much effort has been directed toward the control of the molecular packing of these dyes,3 which would affect the electrical conductivity of their films.4 Charge carrier mobilities of phthalocyanines with eight alkoxy groups were reported to be dependent on the molecular packing, i.e., crystalline, mesoscopic and isotropic phases.5 Among cyclic oligopyrroles, subphthalocyanines have a unique cone shape,6 and π-conjugation occurs on the cone-shaped framework.7 Verreet et al.8 reported that subnaphthalocyanine showed better energy conversion efficiency than subphthalocyanine in an organic solar cell combined with C60 due to the red shifted absorption. Subnaphthalocyanine with a larger core size than subphthalocyanine also has advantages as a semi-conductive material since Warman et al.9 reported that the charge mobilities increase with increasing core sizes of the discotic molecules. In addition to the visible light absorption, redox properties, and semi-conductive properties, subphthalocyanines and subnaphthalocyanines have a large dipole moment nearly perpendicular to the π-conjugated electronic systems. Therefore, control of molecular packing would lead to unique dielectric functions that other cyclic oligo tetrapyrroles cannot exhibit. There have been several studies on the control of assembly of subphthalocyanines. Torres and co-workers reported that subphthalocyanines with nine dodecyloxy groups showed a liquid-crystalline phase which shows permanent polarization in the presence of electric fields.10We have reported synthesis of subnaphthalocyanines with six halogen atoms on the naphthalene rings to tune the HOMO/LUMO energy levels as an active layer of photovoltaic cells.12 Compared to subphthalocyanine, subnaphthalocyanine has a smaller HOMO–LUMO gap, and adsorbs visible light in the 600–700 nm range. Halogenation of subnaphthalocyanine resulted in a better orbital energy matching with fullerenes or fluorosubphthalocyanines at the p–n junction.11 Introduction of six halogens in the periphery of subnaphthalocyanines cancelled the original dipole moment of subnaphthalocyanines because the dipole moment of a B–Cl bond and the combined dipole moment of C–halogen bonds are anti-parallel. As shown in Table 1, dipole moments of subnaphthalocyanines increases as the peripheral substituents are Cl < F < I < H < 2-phenylethynyl. Therefore, substitution of alkynyl groups on the naphthalene rings is an attractive strategy to obtain highly dipolar subnaphthalocyanines. The B–Cl derivatives showed a larger dipole moment than the B–F derivatives, but the B–Cl derivatives are more labile than the B–F derivatives. In this paper we report that hexaiodosubnaphthalocyanines were converted to hexa(1-alkynyl)subnaphthalocyanines and hexa(2-arylethynyl)subnaphthalocyanines, and changes in the peripheral substituents as well as the axial ligand from Cl to F can effectively control the molecular packing of these subnaphthalocyanine derivatives.
| Peripheral substituent (R) | Dipole moment/D | |
|---|---|---|
| X = Cl | X = F | |
| a B3LYP/LAN2DZ for R = I, B3LYP/6-31G(D) for others. | ||
| Cl | 1.51 | 0.22 |
| F | 2.61 | 1.08 |
| I | 3.78 | 2.76 |
| H | 5.24 | 3.91 |
PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– C– |
6.32 | 4.88 |
Results and discussion
Synthesis of chloroboron(III) and fluoroboron(III) hexa(1-alkynyl)subnaphthalocyanines and hexa(2-arylethynyl)subnaphthalocyanines
Chart 2 shows structures of subnaphthalocyanines studied in this paper. Synthetic routes to chloroboron(III) subnaphthalo-cyanines 1a–f and fluoroboron(III) subnaphthalocyanines 2a–g are shown in Scheme 1. Chloroboron(III) 3,4,12,13,21,22-hexaiodosubnaphthalocyanine was prepared according to the literature.12,13 Bender and coworkers reported that cyclotrimerization of unsubstituted 2,3-naphthalenedicarbonitrile in the presence of BCl3 gave by-products with naphthalene rings chlorinated,14 while cyclotrimerization of 6,7-diiodo-2,3-naphthalenedicarbonitrile 11 did not give any chlorinated by-products owing to the inactivation of naphthalene ring toward electrophilic substitution by iodine groups. Therefore, isolation of 3 was facile. Sonogashira coupling was employed to substitute iodines of hexaiodosubnaphthalocyanine 3 with alkynyl groups.15 B–Cl derivatives 1a–f were isolated with silica gel column chromatography eluted with toluene.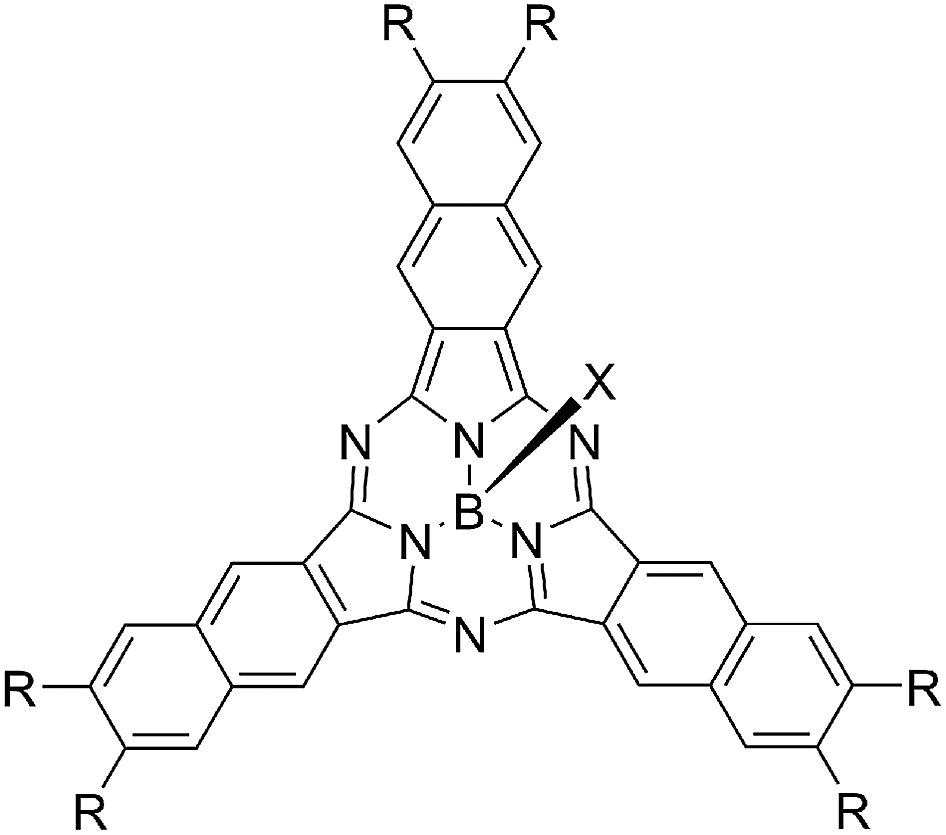 | ||
| Chart 1 Structures of subnaphthalocyanines. | ||
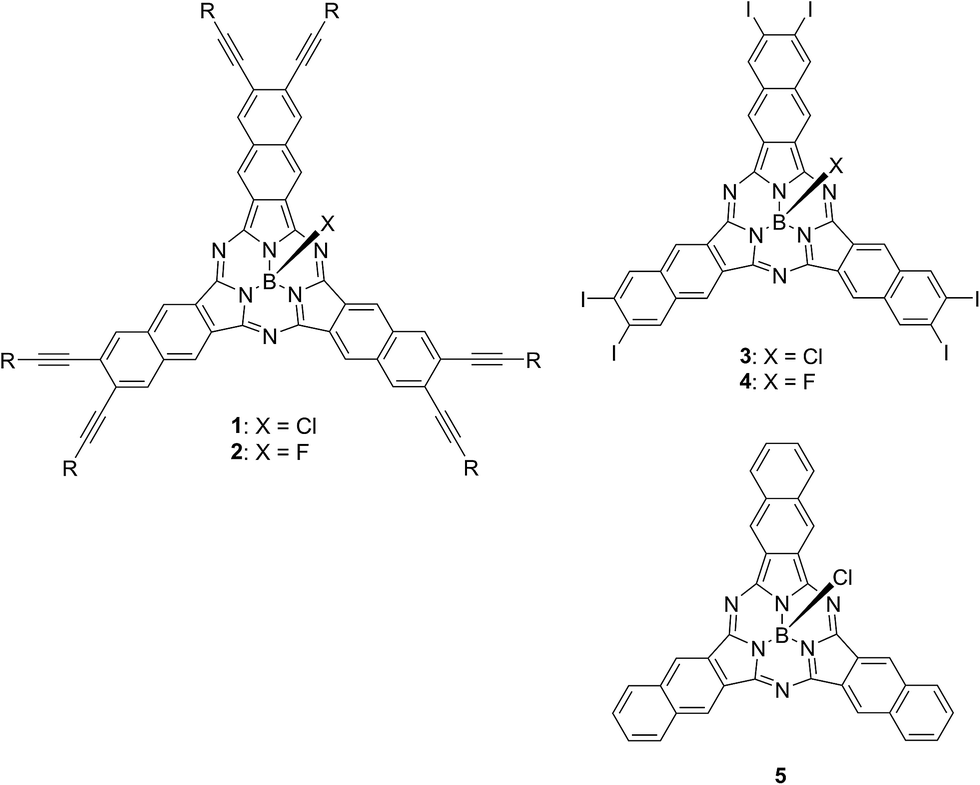 | ||
| Chart 2 Structures of subnaphthalocyanines 1–5. | ||
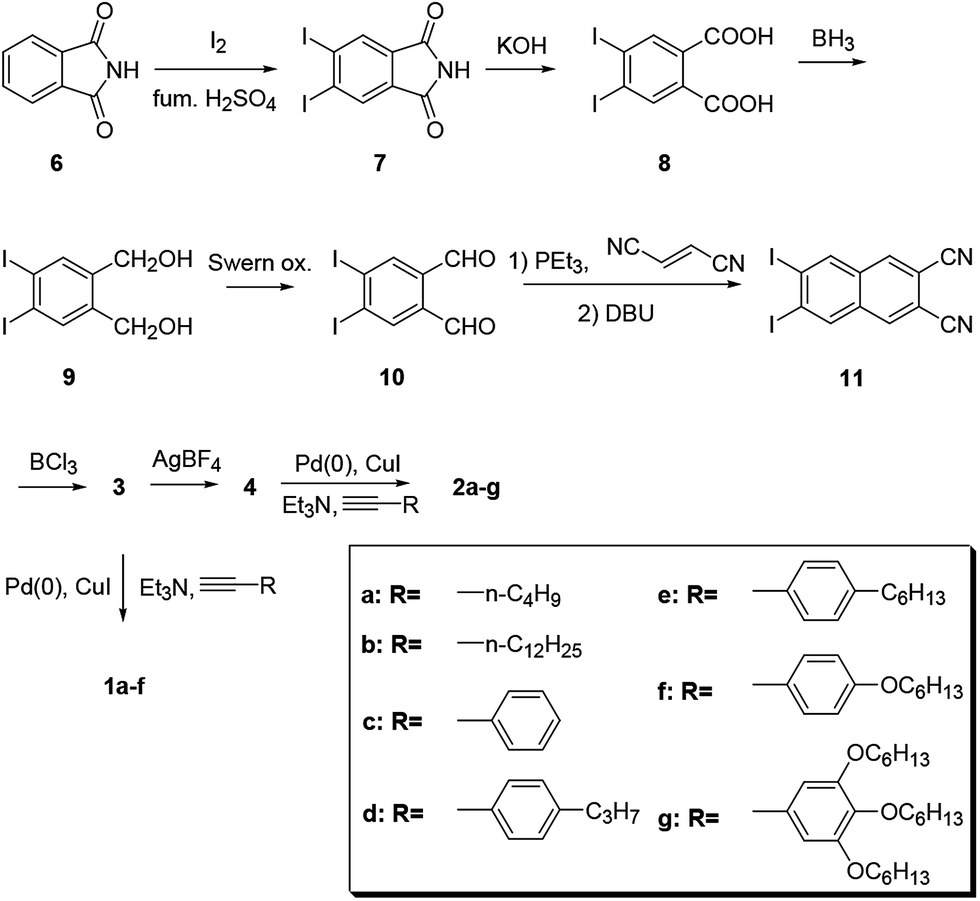 | ||
| Scheme 1 Synthesis of substituted subnaphthalocyanines 1 and 2. | ||
Substitution of axial Cl with F by Torres's procedure16 gave fluoroboron(III) 3,4,12,13,21,22-hexaiodosubnaphthalocyanine 4. B–F derivatives 2a–g were prepared similarly from fluoroboron(III) hexaiodosubnaphthalocyanine 4 by Sonogashira coupling, and isolated using silica gel column chromatography, except for 2c. For 2c, owing to low solubility in organic solvents, the product was crystallized by adding hexane to the reaction mixture, and the crystal was washed with organic solvents, and dried in vacuo. The precursor 15, a coupling component for 2g, was prepared according to the literature (Scheme 2).17 These subnaphthalocyanines were identified by 1H NMR, 13C NMR, MALDI-TOF mass spectroscopy, high resolution FAB mass spectroscopy and UV-visible spectroscopy. Their purities were checked by gel-permeation HPLC, which clearly detected the presence of penta- or tetrasubstituted by-products before silica gel column purification. Isolated yields of Sonogashira coupling are listed in Table 2. The yields of Sonogashira coupling were 3–8% for B–Cl derivatives 1, while they were 10–27% for B–F derivatives 2. Lower yields of B–Cl derivatives 1 can be ascribed to the labile B–Cl bond. The B–F derivatives 2 were also prepared from the B–Cl derivatives 1 by substitution of Cl with F, but the overall yields were lower in this route, due to the labile B–Cl bond.
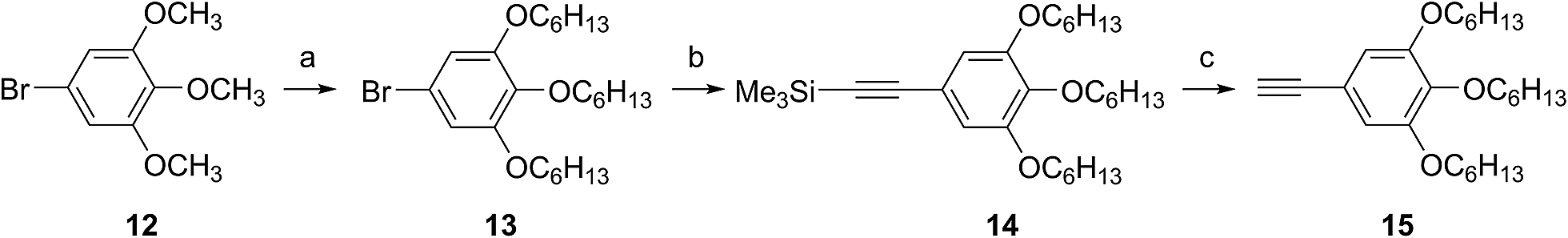 | ||
| Scheme 2 Synthesis of arylethyne 15. Reagents and conditions. (a) BBr3; C6H13Br, K2CO3, 65%; (b) trimethylsilylacetylene, Pd(PPh3)4–CuI, piperidine, 80 °C, 98%; (c) K2CO3, DCM/MeOH, 86%. | ||
| Yield (%) | Yield (%) | ||
|---|---|---|---|
| 1a | 7 | 2a | 19 |
| 1b | 6 | 2b | 16 |
| 1c | 8 | 2c | 10 |
| 1d | 8 | 2d | 20 |
| 1e | 6 | 2e | 22 |
| 1f | 3 | 2f | 27 |
| 2g | 10 |
Solubilities and melting points
Solubilities and melting points of subnaphthalocyanines 1a–f and 2a–g are listed in Table 3. Solubilities were determined by measuring the absorbances of saturated solutions with UV-visible spectrophotometry. The molar extinction coefficients at the Q-band peak were determined using 2 × 10−6 M CH2Cl2 solutions. Compared to unsubstituted chloroboron(III) subnaphthalocyanine 5, 1a–f were 40 to 310 times more soluble in CH2Cl2. Solubilities increased with longer alkyl groups for homologs 1a/1b and 1c/1d/1e. By substitution of axial Cl with F, solubilities were much lower, showing that a smaller fluorine atom would stabilize molecular packing in the solid state. This observation is contrary to the previous report that fluoroboron(III) subphthalocyanine showed higher solubility than chloroboron(III) subphthalocyanine and bromoboron(III) subphthalocyanine.18 Melting points of 1-alkynyl derivatives 2a and 2b were lower than 120 °C, while those of 2-phenylethynyl and 2-(4-propylphenyl)ethynyl derivative (2c and 2d) were higher than 375 °C. Thus, melting points were lowered by introduction of alkyl groups in the periphery, and raised by introduction of phenyl groups. Polarized optical microscope (POM) observation under cross-Nicole conditions displayed that the thin film of chloroboron(III) derivatives 1a–f were optically isotropic in the solid state, indicating that these are in a glassy state. Therefore, the temperatures reported for 1a–f listed in Table 3 are glass transition temperatures. For fluoroboron(III) derivatives 2a–f, the thin film was crystalline, as confirmed by the bright image observed with a POM under cross-Nicole conditions. The thin film of 2g was not crystalline as judged by POM observations.| Solubility (g L−1) | Melting point or glass transition temperature (°C) | |
|---|---|---|
| a Glass transition temperature. | ||
| 1a | 5.02 | 77–80a |
| 1b | 9.37 | 70–73a |
| 1c | 1.19 | >375 |
| 1d | 1.52 | >375 |
| 1e | 2.13 | 102–108a |
| 1f | 3.70 | 98–106a |
| 2a | 1.35 | 114–118 |
| 2b | 2.30 | 100–105 |
| 2c | 6.48 × 10−4 | >375 |
| 2d | 0.90 | >375 |
| 2e | 1.62 | 170–178 |
| 2f | 2.01 | 168–176 |
| 2g | 10.62 | 80–86a |
| 5 | 0.03 | 270–276 |
UV-visible spectra in solutions and of cast films
Chloroboron(III) subnaphthalocyanine 5 in toluene shows a strong absorption at 660 nm and an absorption at 299 nm in the near UV with almost equal absorbances.19 The former band is called a Q-band, and the latter band is called a B-band or a Soret band. Fig. 1 shows UV-visible spectra of B–Cl derivatives 1 in CH2Cl2. The absorption maxima in the Q-band were shifted to the longer wavelength in the order: 5 (H) < 1a, b (1-alkynyl) < 1c–f (2-arylethynyl). The molar absorption coefficients of 1a–f were larger than that of the parent 5. The substituent effects are more obvious in the B-band in the 300–500 nm region: 1a and 1b with 1-alkynyl groups showed peaks at 318 and 369 nm, while 1c–f with 2-arylethynyl groups showed a more intense absorption in the B-band. Fig. 2 shows the visible spectra of B–Cl derivatives of the cast film on silicate glass. A solution of 1 in dichloromethane was dropped on silicate glass, and the film was kept at room temperature in Ar for 2 days. The absorption maxima in the Q-band were shifted to the longer wavelength in the order: 5 (H) < 1a, 1b (1-alkynyl) < 1d–f (4-alkyl or 4-alkoxyphenylethynyl) < 1c (phenyl).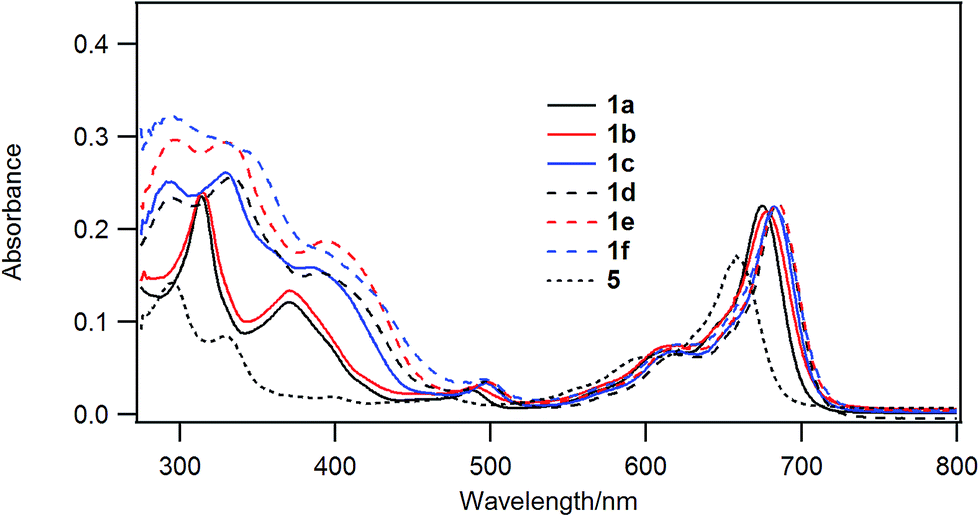 | ||
| Fig. 1 UV-visible spectra of 1a (black solid line), 1b (red solid line), 1c (blue solid line), 1d (black dashed line), 1e (red dashed line), and 1f (blue dashed line) and the parent chloroboron(III) subnaphthalocyanine 5 (black dotted line). The concentration was 2 × 10−6 M in dichloromethane. | ||
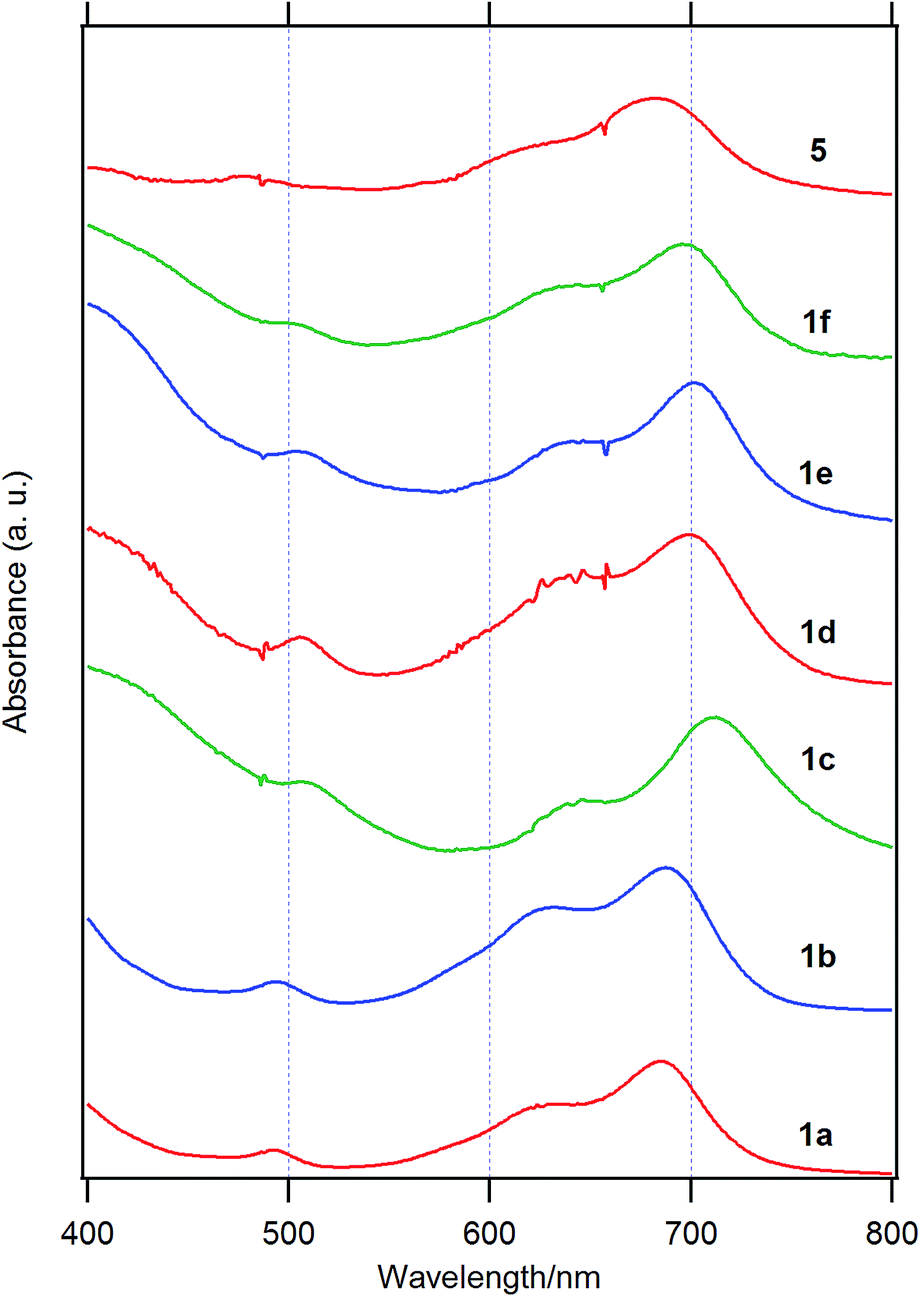 | ||
| Fig. 2 Visible spectra of 1a, 1b, 1c, 1d, 1e, and 1f and the parent chloroboron(III) subnaphthalocyanine 5 on silicate glass. | ||
In Table 4 are listed the absorption maxima in solution and of the cast film. The shifts of the Q-band of B–Cl derivatives of the cast film to the longer wavelength compared to that in solutions indicate that J-aggregate forms in the cast film. The displacements are 9–30 nm, and increased in the order: 1a, 1b (alkyl) < 1d–f (alkylphenyl or alkoxyphenyl) < 5 (H) < 1c (phenyl).
| λabs (nm) | |||
|---|---|---|---|
| In CH2Cl2 | On silicate glass | Shift | |
| 1a | 675 | 685 | 10 |
| 1b | 678 | 687 | 9 |
| 1c | 682 | 712 | 30 |
| 1d | 685 | 698 | 13 |
| 1e | 685 | 703 | 18 |
| 1f | 683 | 696 | 13 |
| 5 | 658 | 680 | 22 |
Fig. 3 shows the UV-visible spectra of the B–F derivatives 2 in CH2Cl2. The absorption maxima were shifted to the longer wavelength in the order: 2a, b (alkyl) < 2c–f (phenyl) in CH2Cl2. The B-band was also affected by substituents in a similar way as the B–Cl derivatives. The molar absorption coefficients of all of alkynyl substituted derivatives were similar. Fig. 4 shows visible spectra of B–F derivatives 2 of the cast film on silicate glass. Solutions of 2a–b and 2d–g in dichloromethane and 2c in pyridine were dropped on silicate glass, the solvent was evaporated, and the resulting film was kept in Ar for 2 days before recording visible spectra. The spectra were almost identical with those of as-prepared film. In Table 5 are listed the absorption maxima in solution and of the cast film. It is noteworthy that the Q-band absorption envelope of 2b–f is different from that in CH2Cl2, that of 1 in CH2Cl2, and that of 1 of the cast film. The Q-band of 2b–f of the cast film exhibited larger absorption in the 600–650 nm range than the absorption band in the 650–700 nm range. The shifts to the shorter wavelength indicate that 2b–f form H-aggregate in the cast film.20 Phthalocyanines with oligo(ethylene oxide) units showed a blue-shifted Q-band, and it was ascribed to formation of H-aggregate.21 The Q-band spectral shapes are similar to those reported for μ-oxo dimers of iron phthalocyanines22,23 and subphthalocyanines,24 supporting the formation of a stacked dimer or a stacked column in the cast film. In contrast, the Q-band of 2a and 2g of the cast film was similar to those observed for B–Cl derivatives 1: 10–14 nm red-shift relative to the peak maximum in solution indicated that 2a and 2g do not form H-aggregate in the cast film.
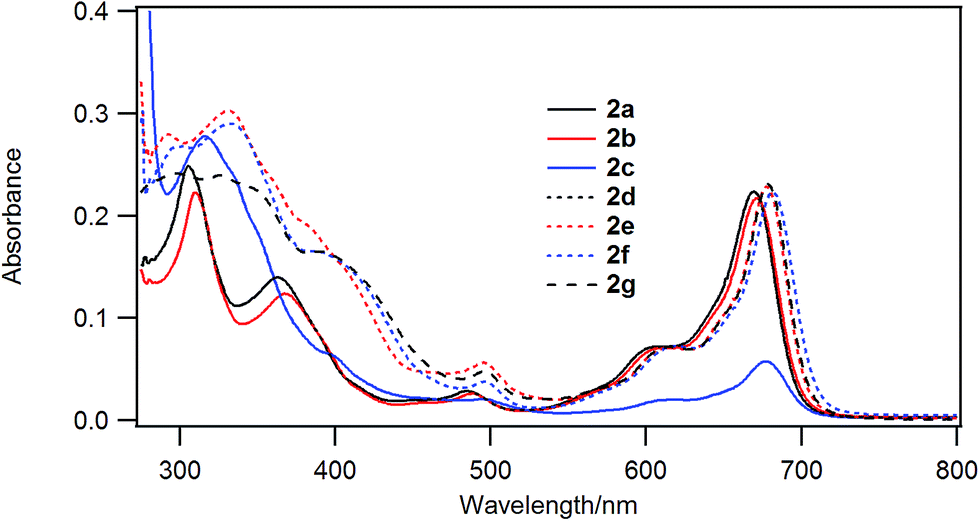 | ||
| Fig. 3 UV-visible spectra of 2a (black solid line), 2b (red solid line), 2c (blue solid line), 2d (black dotted line), 2e (red dotted line), 2f (blue dotted line), and 2g (black dashed line). The concentration of 2a, 2b and 2d–g was 2 × 10−6 M in dichloromethane. Visible spectra of 2c was the spectra of saturated dichloromethane solution. | ||
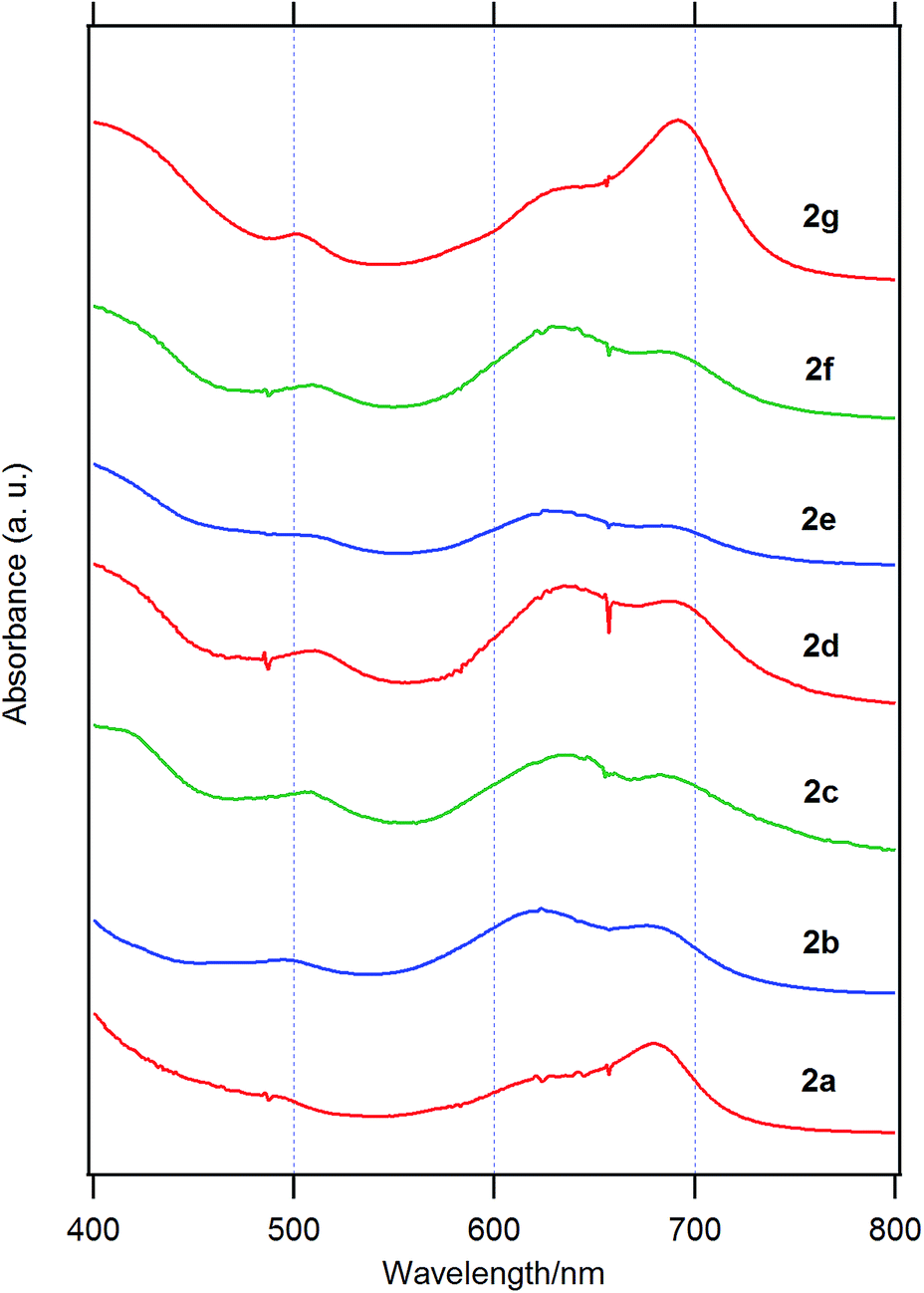 | ||
| Fig. 4 Visible spectra of 2a, 2b, 2c, 2d, 2e, 2f, and 2g on silicate glass. | ||
| λabs (nm) | |||
|---|---|---|---|
| In CH2Cl2 | On silicate glass | Shift | |
| 2a | 669 | 679 | 10 |
| 2b | 671 | 624 | −47 |
| 2c | 677 | 632 | −45 |
| 2d | 678 | 642 | −36 |
| 2e | 678 | 625 | −53 |
| 2f | 681 | 629 | −52 |
| 2g | 678 | 692 | 14 |
Fluorescence spectra
Fig. 5 shows the fluorescence emission spectra of the B–F derivatives in CH2Cl2. In Table 6 are listed the emission maxima, Stokes shifts, and fluorescence quantum yields. The fluorescence quantum yields were determined based on the reported quantum yield of unsubstituted derivative 5 (0.22).25 The fluorescence quantum yields decreased in the order: 5 (H) > 2a (–C4H9) > 2b (–C12H25) > 2d (–PhC3H7) > 2e (–PhC6H13) > 2f (–PhOC6H13), 2g (–Ph(OC6H13)3) > 2c (–Ph).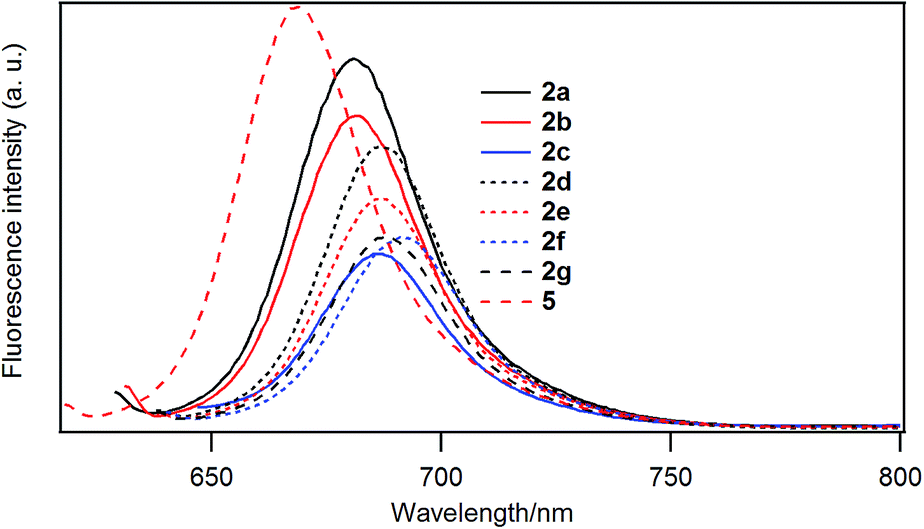 | ||
| Fig. 5 Fluorescence spectra of 2a (black solid line), 2b (red solid line), 2c (blue solid line), 2d (black dotted line), 2e (red dotted line), 2f (blue dotted line), and 2g (black dashed line), and 5 (red dashed line) at 298 K in dichloromethane where the samples were excited at (λmax abs – 50) nm. Each sample has absorbance of 0.02 at the excitation wavelength. | ||
| λabs/nm | λem/nm | Stokes shift (nm) | Fluorescence quantum yield | |
|---|---|---|---|---|
| 2a | 669 | 681 | 12 | 0.19 |
| 2b | 671 | 682 | 11 | 0.16 |
| 2c | 677 | 687 | 10 | 0.09 |
| 2d | 678 | 687 | 9 | 0.14 |
| 2e | 678 | 686 | 8 | 0.11 |
| 2f | 680 | 691 | 11 | 0.10 |
| 2g | 678 | 688 | 10 | 0.10 |
| 5 | 658 | 669 | 11 | 0.22 |
Fluorescence emission spectra of 2b, 2d, 2e and 2f in CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMSO (1:1, v/v) are shown in Fig. S51.† In addition to the emission band peaked at 685–689 nm, 2b and 2e exhibited a new band around 800 nm. Excitation spectra for the new band are shown in Fig. S52 and S53,† and they were similar to the UV-visible spectra of the thin film shown in Fig. 4. Therefore, 2b and 2e form H-aggregate in CH2Cl2–DMSO (1:1), whose structures are similar to those formed in the cast film.
:DMSO (1:1, v/v) are shown in Fig. S51.† In addition to the emission band peaked at 685–689 nm, 2b and 2e exhibited a new band around 800 nm. Excitation spectra for the new band are shown in Fig. S52 and S53,† and they were similar to the UV-visible spectra of the thin film shown in Fig. 4. Therefore, 2b and 2e form H-aggregate in CH2Cl2–DMSO (1:1), whose structures are similar to those formed in the cast film.
Polarized optical microscope
Fig. 6 shows the polarized optical microscopic images of the cast film of 2e and 2f under crossed-Nicols conditions at 180 °C above the melting point. The film was sandwiched between two glasses and the glasses were pressed above the melting point. These images clearly showed that these films are optically anisotropic above the melting point. We also attempted to detect phase transition by differential scanning calorimetry, while no clear enthalpy changes were observed. Slow kinetics due to highly viscous nature and/or low degree of crystallinity may be responsible for the lack of DSC peaks. Serrano and coworkers reported that no DSC peak was observed for the liquid crystalline porphyrin derivatives.26 | ||
| Fig. 6 POM microphotographs of 2e and 2f at 180 °C under crossed-Nicols conditions. | ||
Powder X-ray diffraction
A dichloromethane solution of 2d, 2e, and 2f was dropped on silicate glass, and the glass plate was dried at room temperature for 2 weeks in dark in Ar to allow crystallization. A thin film of 2d, 2e, and 2f prepared in this way was subjected to powder X-ray diffraction studies (Fig. 7). All films showed a diffraction peak in the low angle region around 2θ of 2.8°, showing a long distance periodicity. The diffraction peaks were indexed based on a two-dimensional hexagonal lattice as shown in Tables 7–9. The hexagonal lattice parameter a ranged from 34.1 to 37.3 Å. We also assigned some of the peaks to the stacking distance along the c direction. It should be noted that the peaks originating from the stacking distance at 2θ = 29.45° and 31.69° for 2e as well as the peaks at 28.34° and 31.70° for 2f were not observed for the as-prepared cast film before aging. This observation suggests that the hexagonal column are formed quickly, while the periodic stacking of the cone-shaped molecules occurs slowly. The lattice constant c ranged from 4.7 to 4.8 Å. These X-ray diffraction studies confirm the hexagonal columnar structure of fluoroboron(III) subnaphthalocyanines bearing 2-(4-alkylphenyl)ethynyl groups in the cast film. In contrast to the B–F derivatives, XRD of B–Cl derivatives 1d, 1e, and 1f indicates that they are poorly crystalline (see Fig. S78–S80 in ESI†). Clays, Kim and coworkers27 reported that hexakis(hexadecylthio)subphthalocyanine exhibited hexagonal columnar mesophase with a lattice constant a = 33.02 Å. The molecular size of this compound is similar to 2f, if the alkyl chains adopt anti conformer. Torres and coworkers reported that dodecafluorosubphthalocyanine with a B–F bond formed a one-dimensional column in the crystal, where the B–B distance, i.e., the stacking distance, was 4.5 Å.28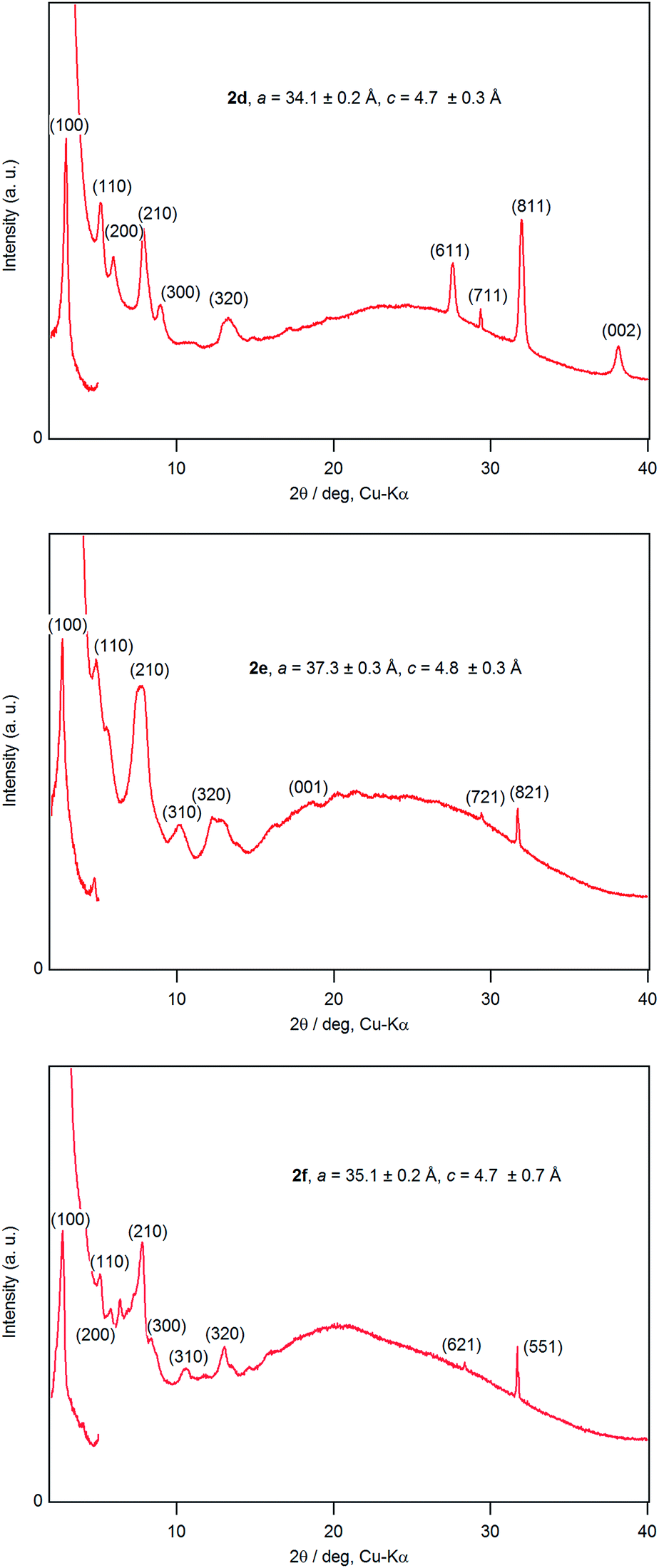 | ||
Fig. 7 Powder X-ray diffraction patterns of 2d–f. Lattice constants, a and c, were determined by least-squares fitting based on the hexagonal lattice: 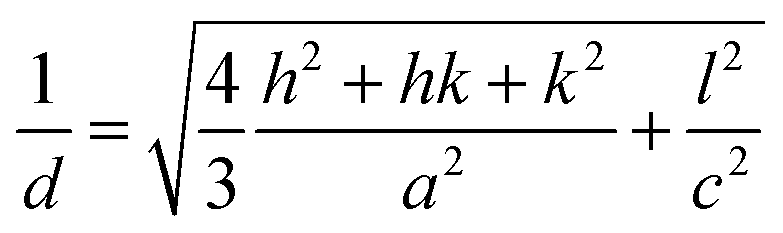 . While the XRD patterns were recorded with a 1D silicon strip detector for a wide angle region, 2θ > 3°, they were recorded with a 0D detector for the low angle region, 2θ = 2–5°. . While the XRD patterns were recorded with a 1D silicon strip detector for a wide angle region, 2θ > 3°, they were recorded with a 0D detector for the low angle region, 2θ = 2–5°. | ||
| 2θ (deg) | d (Å) | (hkl) | 2θ (deg) | d (Å) | (hkl) |
|---|---|---|---|---|---|
| 2.95 | 29.88 | (100) | 13.19 | 6.71 | (320) |
| 5.32 | 16.59 | (110) | 27.54 | 3.24 | (611) |
| 6.08 | 14.52 | (200) | 29.34 | 3.04 | (711) |
| 7.84 | 11.27 | (210) | 31.93 | 2.80 | (811) |
| 8.95 | 9.87 | (300) | 38.10 | 2.36 | (002) |
| 2θ (deg) | d (Å) | (hkl) | 2θ (deg) | d (Å) | (hkl) |
|---|---|---|---|---|---|
| 2.70 | 32.65 | (100) | 13.02 | 6.79 | (410) |
| 4.84 | 18.24 | (110) | 18.41 | 4.82 | (001) |
| 7.52 | 11.75 | (210) | 29.45 | 3.03 | (721) |
| 7.83 | 11.28 | (300) | 31.69 | 2.82 | (821) |
| 10.13 | 8.73 | (310) | 42.62 | 2.12 | (622) |
| 12.22 | 7.23 | (320) |
| 2θ (deg) | d (Å) | (hkl) | 2θ (deg) | d (Å) | (hkl) |
|---|---|---|---|---|---|
| 2.90 | 30.45 | (100) | 10.51 | 8.41 | (310) |
| 5.10 | 17.31 | (110) | 12.98 | 6.81 | (320) |
| 5.78 | 15.28 | (200) | 28.34 | 3.15 | (621) |
| 7.81 | 11.31 | (210) | 31.70 | 2.82 | (551) |
| 8.26 | 10.70 | (300) |
Fig. 8 shows XRD patterns of films of 2e and 2f at 25 °C and 180 °C. Diffraction peaks owing to two dimensional hexagonal lattice (hk0) appeared at 180 °C, indicating that the films retain the hexagonal columnar structure at 180 °C. The diffraction peaks in the range 28–32° due to the stacking ordering along the c-axis direction disappeared at 180 °C. Combined with the POM observations described above, 2e and 2f were in a discotic hexagonal columnar mesophase at 180 °C.
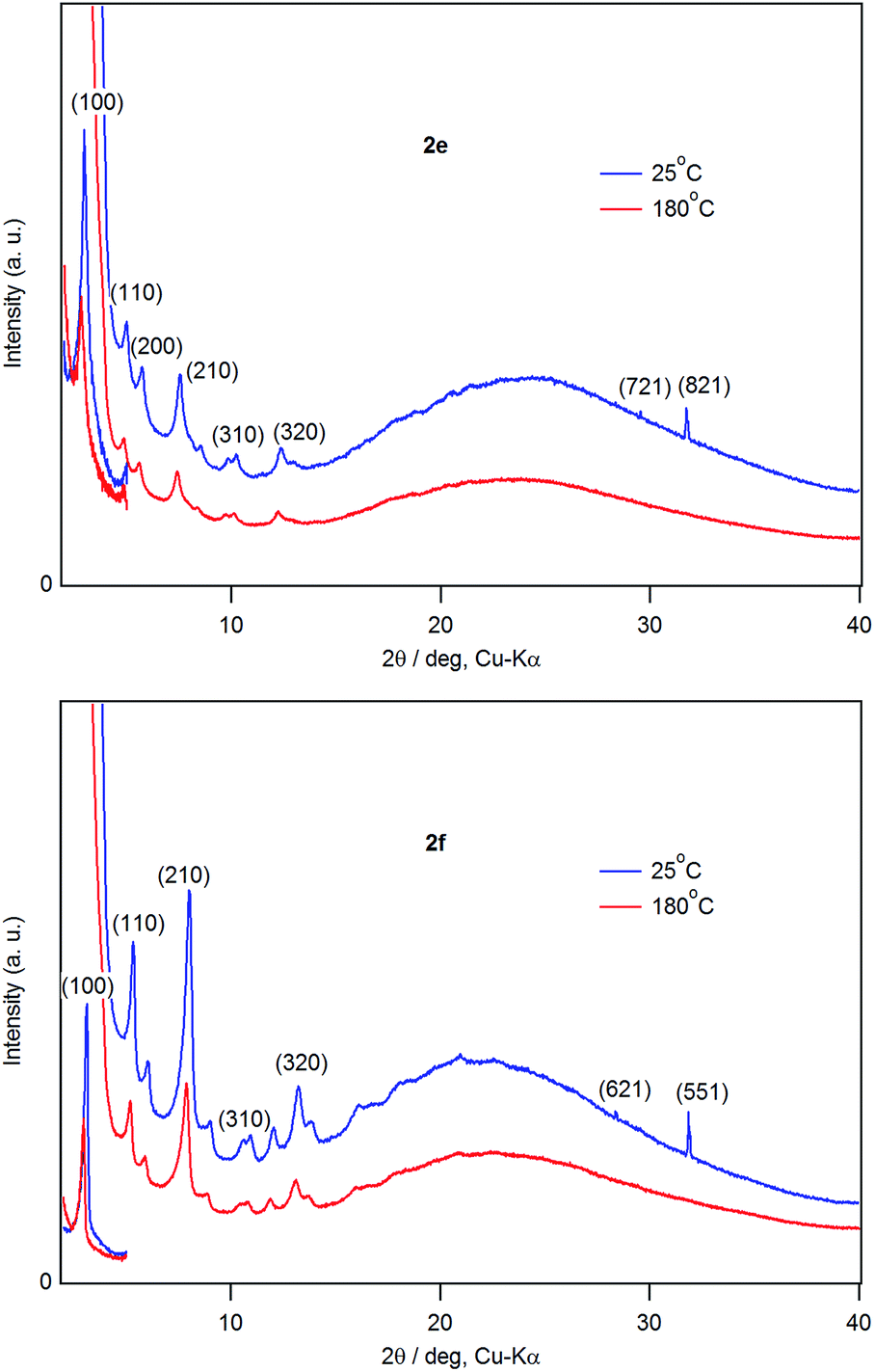 | ||
| Fig. 8 Variable-temperature XRD of 2e and 2f. | ||
Molecular orbital calculations of the stacked dimer of 1c and 2c
The energy of stacked dimers of 1c and 2c relative to monomeric 1c and 2c was calculated at the B3LYP/6-31G(D) level with a fixed distance between the two B atoms of the dimer. The energies were plotted against the B–B distance in Fig. 9. The energy of the dimeric B–Cl derivative 1c decreased as the B–B distance was longer, while the energy of the dimeric B–F derivative 2c showed a minimum at about 4.7 Å of the B–B distance. The geometry optimized structures are shown in Fig. 10. At the B–B distance of 5.5 Å, the dimeric 1c showed a bent structure due to a large van der Waals radius of Cl, while the dimeric 2c showed a linear structure even at a B–B distance of 3.9 Å. The van der Waals radii of B,29 C, F, and Cl30 are 1.92, 1.7, 1.47, and 1.75 Å. Therefore, sum of the van der Waals radii, B + F, are shorter than that of C + C, while sum of the van der Waals radii, B + Cl, are larger than that of C + C. These molecular modelling studies indicate that the smaller van der Waals radius of F atom can fit to the cleft of cone-shaped subnaphthalocyanine and lead to a stable stacked structure.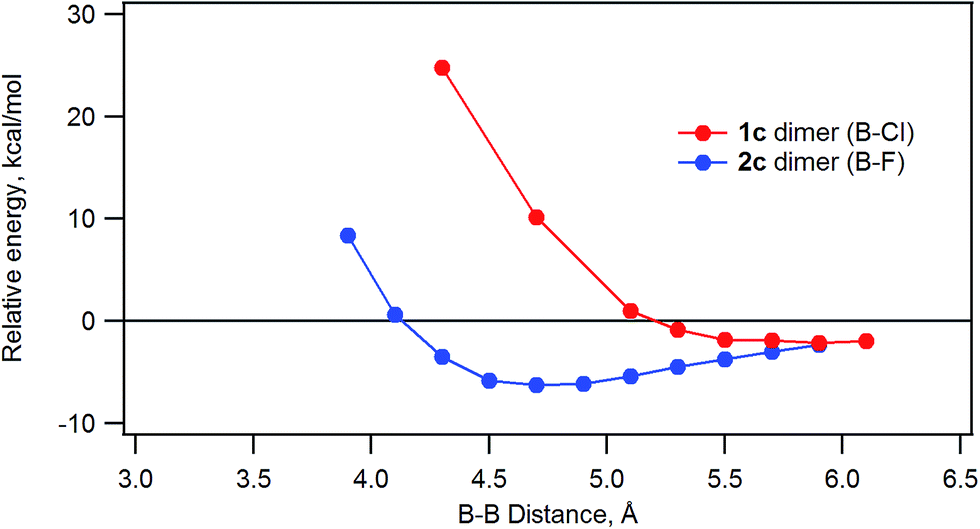 | ||
| Fig. 9 Plot of the relative energy of dimeric 1c or 2c against the B–B distance calculated by ab initio B3LYP/6-31G(D). | ||
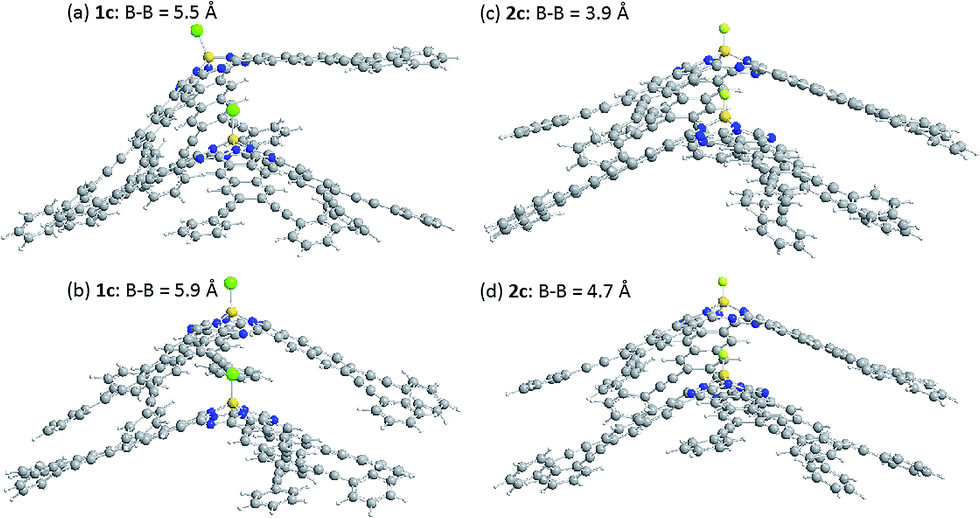 | ||
| Fig. 10 Optimized structures of dimeric subnaphthalocyanines 1c and 2c. (a) 1c, B–B distance of 5.5 Å, (b) 1c, B–B distance of 5.9 Å, (c) 2c, B–B distance of 3.9 Å, and (d) 2c, B–B distance of 4.7 Å. | ||
Experimental
1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a JEOL JNM-ECX DELTA spectrometer using tetramethylsilane as an internal reference (0 ppm). Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectra were obtained with a Brucker Autoflex Speed mass spectrometer. High resolution FAB MS spectra were recorded with a JEOL JMS-700 MStation. Gel-permeation HPLC was performed with a Shimadzu liquid chromatography LC-6AD using Tosoh TSKgel G2000H as a stationary phase and chloroform as eluant. Powder X-ray diffraction was recorded with a Rigaku SmartLab diffractometer with Cu-Kα radiation. The low angle region, 2θ of 2 to 5°, was recorded with the 0D detector, while the wide angle region, 2θ of 3 to 40°, was recorded with the 1D silicon strip detector. Variable temperature XRD was recorded for a thin film of subnaphthalocyanines on silicate glass placed in a thermostated Pt holder. Melting points were determined by observing changes in the sample sandwiched between two glasses heated at a rate of 2 °C min on Mettler-Toredo FP82HT hot stage with a polarized optical microscope Nikon Eclipse LV100 POL. Molecular orbital calculations were performed with Gaussian 09.31Synthesis of subnaphthalocyanines
1H-NMR (CDCl3, 500 MHz): δ (ppm) = 9.25 (s, 6H), 8.36 (s, 6H), 2.58 (t, 12H), 1.76–1.66 (m, 12H), 1.65–1.57 (m, 12H), 1.02 (t, 18H). 13C-NMR (CDCl3, 125 MHz): δ (ppm) 148.74, 132.97, 131.94, 128.88, 124.98, 121.64, 95.97, 79.61, 30.87, 22.11, 19.57, 13.77. MALDI-TOF mass: m/z = 1061.3 [(M + H)+], calcd for C72H67BN6Cl m/z = 1061.5. HRMS (FAB): m/z = 1060.5144, calcd for 12C721H6614N635Cl 11B m/z = 1060.5131. Visible (dichloromethane) λmax/nm (logε): 675 (5.05).
1H-NMR (CDCl3, 500 MHz): δ (ppm) = 9.26 (s, 6H), 8.35 (s, 6H), 2.57 (t, 12H), 1.74–1.70 (m, 12H), 1.42–1.22 (m, 108H), 0.88 (t, 18H). 13C-NMR (CDCl3, 125 MHz): δ (ppm) 148.86, 132.94, 131.96, 128.93, 125.05, 121.66, 96.11, 79.57, 31.95, 29.75, 29.71, 29.66, 29.40, 29.35, 29.10, 28.87, 22.71, 19.91, 14.14. MALDI-TOF mass: m/z = 1734.4 [(M + H)+], calcd for C120H163BN6Cl m/z = 1734.3. Visible (dichloromethane) λmax/nm (logε): 678 (5.04).
1H-NMR (CDCl3, 500 MHz): δ (ppm) = 9.25 (s, 6H), 8.51 (s, 6H), 7.73–7.60 (m, 12H), 7.45–7.33 (m, 18H). MALDI-TOF mass: m/z = 1181.2 [(M + H)+], calcd for C84H43BN6Cl m/z = 1181.3. HRMS (FAB): m/z = 1180.3259, calcd for 12C841H4214N635Cl 11B m/z = 1180.3253. Visible (dichloromethane) λmax/nm (logε): 682 (5.05).
1H-NMR (CDCl3, 500 MHz): δ (ppm) = 9.25 (s, 6H), 8.49 (s, 6H), 7.58 (d, 12H, 5 Hz), 7.21 (d, 12H, 5 Hz), 2.64 (t, 12H), 1.72–1.65 (m, 12H), 0.98 (t, 18H). MALDI-TOF mass: m/z = 1432.8 [M+], calcd for C102H78BN6Cl m/z = 1432.6. Visible (dichloromethane) λmax/nm (logε): 685 (5.06).
1H-NMR (CDCl3, 500 MHz): δ (ppm) = 9.26 (s, 6H), 8.50 (s, 6H), 7.58 (d, 12H, 5 Hz), 7.21 (d, 12H, 5 Hz), 2.66 (t, 12H), 1.66–1.62 (m, 12H), 1.40–1.29 (m, 36H), 0.91 (t, 18H). MALDI-TOF mass: m/z = 1686.1 [(M + H)+], calcd for C120H115BN6Cl m/z = 1685.9. Visible (dichloromethane) λmax/nm (logε): 685 (5.06).
1H-NMR (CDCl3, 500 MHz): δ (ppm) = 9.29 (s, 6H), 8.49 (s, 6H), 7.59 (d, 12H, 10 Hz), 6.92 (d, 12H, 10 Hz), 4.01 (t, 12H), 1.84–1.79 (m, 12H), 1.51–1.47 (m, 12H), 1.42–1.32 (m, 24H), 0.93 (t, 18H). MALDI-TOF mass: m/z = 1781.1 [M+], calcd for C120H114BN6O6Cl m/z = 1780.9. Visible (dichloromethane) λmax/nm (logε): 683 (5.04).
1H-NMR (CDCl3, 500 MHz): δ (ppm) = 9.22 (s, 6H), 8.34 (s, 6H), 2.58 (t, 12H), 1.73–1.68 (m, 12H), 1.63–1.58 (m, 12H), 1.01 (t, 18H). MALDI-TOF mass: m/z = 1044.7 [M+], calcd for C72H66BN6F m/z = 1044.5. HRMS (FAB): m/z = 1044.5413, calcd for 12C721H6614N619F 11B m/z = 1044.5426. Visible (dichloromethane) λmax/nm (logε): 669 (5.05).
1H-NMR (CDCl3, 500 MHz): δ (ppm) = 9.21 (s, 6H), 8.33 (s, 6H), 2.57 (t, 12H), 1.75–1.69 (m, 12H), 1.42–1.25 (m, 108H), 0.87 (t, 18H). MALDI-TOF mass: m/z = 1717.5 [M+], calcd for C120H162BN6F m/z = 1717.3. Visible (dichloromethane) λmax/nm (logε): 671 (5.04).
MALDI-TOF mass: m/z = 1164.3 [M+], calcd for C84H42BN6F m/z = 1164.4. Visible (dichloromethane) λmax/nm (logε): 677 (5.01).
1H-NMR (CDCl3, 500 MHz): δ (ppm) = 9.11 (s, 6H), 8.42 (s, 6H), 7.54 (d, 12H, 5 Hz), 7.15 (m, 12H, 10 Hz), 2.62 (t, 12H), 1.69–1.64 (m, 12H), 0.98 (t, 18H). MALDI-TOF mass: m/z = 1416.6 [M+], calcd for C102H78BN6F m/z = 1416.6. Visible (dichloromethane) λmax/nm (logε): 678 (5.06).
1H-NMR (CDCl3, 500 MHz): δ (ppm) = 9.04 (s, 6H), 8.39 (s, 6H), 7.53 (d, 12H, 10 Hz), 7.11 (d, 12H, 5 Hz), 2.63 (t, 12H), 1.65–1.61 (m, 12H), 1.42–1.25 (m, 36H), 0.92 (t, 18H). MALDI-TOF mass: m/z = 1668.9 [M+], calcd for C120H114BN6F m/z = 1668.9. Visible (dichloromethane) λmax/nm (logε): 678 (5.06).
1H-NMR (CDCl3, 500 MHz): δ (ppm) = 8.88 (s, 6H), 8.28 (s, 6H), 7.51 (d, 12H, 10 Hz), 6.81 (d, 12H, 10 Hz), 3.95 (t, 12H), 1.85–1.78 (m, 12H), 1.53–1.45 (m, 12H), 1.43–1.30 (m, 24H), 0.95 (t, 18H). MALDI-TOF mass: m/z = 1765.0 [M+], calcd for C120H114BN6O6F m/z = 1764.9. Visible (dichloromethane) λmax/nm (logε): 681 (5.05).
1H-NMR (CDCl3, 500 MHz): δ (ppm) = 9.33 (s, 6H), 8.51 (s, 6H), 6.58 (s, 12H), 3.98 (t, 12H), 3.89 (t, 24H), 1.82–1.68 (m, 36H), 1.53–1.40 (m, 36H), 1.39–1.27 (m, 72H), 0.97–0.80 (m, 54H). 13C-NMR (CDCl3, 125 MHz): δ (ppm) 153.14, 150.29, 139.54, 132.74, 132.18, 129.34, 124.27, 121.96, 117.28, 110.19, 95.67, 87.08, 73.54, 69.10, 31.78, 31.62, 30.33, 29.33, 25.82, 25.77, 22.71, 22.65, 14.10, 14.05. MALDI-TOF mass: m/z = 2966.0 [M+], calcd for C192H258BN6O18F m/z = 2966.0. Visible (dichloromethane) λmax/nm (logε): 678 (5.06).
:1) to obtain 6.2 g (65%) of 13 as a white powder.1H-NMR (CDCl3, 500 MHz): δ (ppm) = 6.67 (s, 2H), 3.92 (m, 6H), 1.84–1.67 (m, 6H), 1.57–1.23 (m, 18H), 0.94–0.85 (m, 9H). MALDI-TOF mass: m/z = 456.2 [M+], calcd for C24H41BrO3 m/z = 456.2.
:1) to obtain 5.1 g (98%) of 14.1H-NMR (CDCl3, 500 MHz): δ (ppm) = 6.66 (s, 2H), 3.97–3.93 (m, 6H), 1.81–1.69 (m, 6H), 1.49–1.28 (m, 18H), 0.92–0.87 (m, 9H). MALDI-TOF mass: m/z = 497.4 [(M + Na)+], calcd for C29H50O3SiNa m/z = 497.3.
:1) to give 2.9 g of title product 15 (86%). 1H NMR spectrum was identical to that reported in the literature.321H-NMR (CDCl3, 500 MHz): δ (ppm) = 6.69 (s, 2H), 3.98–3.93 (m, 6H), 2.99 (s, 1H), 1.81–1.70 (m, 6H), 1.49–1.30 (m, 18H), 0.92–0.88 (m, 9H). MALDI-TOF mass: m/z = 425.3 [(M + Na)+], calcd for C26H42O3Na m/z = 425.3.
Conclusions
Chloroboron(III) and fluoroboron(III) hexaethynyl-subnaphthalocyanines were prepared by Sonogashira coupling. The B–F derivatives showed higher melting points and lower solubilities than the corresponding B–Cl derivatives. XRD, POM, visible spectroscopic studies indicated that the B–F derivatives were packed in the hexagonal columnar structure, while the B–Cl derivatives were poorly crystalline. The B–F derivatives with hexylphenyl or hexyloxyphenyl groups showed mesophase at 180 °C. We demonstrated that design of the peripheral substituents and the axial halogen is efficient strategy to control molecular packing of highly polar subnaphthalocyanines.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Drs Toshinobu Ohno and Yo Shimizu for valuable discussions. We also thank Koji Ohoka for his help in high resolution mass spectroscopic studies.Notes and references
- (a) G. E. Morse and T. P. Bender, ACS Appl. Mater. Interfaces, 2012, 4, 5055 CrossRef CAS PubMedG. de la Torre, G. Bottari and T. Torres, Adv. Energy Mater., 2017, 7, 1601700 CrossRef; (b) K. Cnops, G. Zango, J. Genoe, P. Heremans, M. V. Martinez-Diaz, T. Torres and D. Cheyns, J. Am. Chem. Soc., 2015, 137, 8991 CrossRef CAS PubMed.
- D. Hohnholz, S. Steinbrecher and M. Hanack, J. Mol. Struct., 2000, 521, 231 CrossRef CAS.
- (a) C. Piechocki, J. Simon, A. Skoulios, D. Guillon and P. Weber, J. Am. Chem. Soc., 1982, 104, 5245 CrossRef CAS; (b) B. A. Gregg, M. A. Fox and A. J. Bard, J. Am. Chem. Soc., 1989, 111, 3024 CrossRef CAS; (c) B. A. Gregg, M. A. Fox and A. J. Bard, J. Phys. Chem., 1990, 94, 1586 CrossRef CAS; (d) P. G. Schouten, J. M. Warman, M. P. de Haas, M. A. Fox and H.-L. Pan, Nature, 1991, 353, 736 CrossRef CAS; (e) K. Hatsusaka, K. Ohta, I. Yamamoto and H. Shirai, J. Mater. Chem., 2001, 11, 423 RSC.
- (a) H. E. Katz, Z. Bao and S. L. Gilat, Acc. Chem. Res., 2001, 34, 359 CrossRef CAS PubMed; (b) C. D. Sheraw, T. N. Jackson, D. L. Eaton and J. E. Anthony, Adv. Mater., 2003, 15, 2009 CrossRef CAS; (c) C. R. Newman, C. D. Frisbie, D. A. da Silva Filho, J.-L. Bredas, P. C. Ewbank and K. R. Mann, Chem. Mater., 2004, 16, 4436 CrossRef CAS; (d) S. Sergeyev, W. Pisula and Y. H. Geerts, Chem. Soc. Rev., 2007, 36, 1902 RSC; (e) Q. Ye, J. Chang, K.-W. Huang and C. Chi, Org. Lett., 2011, 13, 5960 CrossRef CAS PubMed.
- P. G. Schouten, J. M. Warman, M. P. de Haas, C. F. van Nostrum, G. H. Gelinck, R. J. M. Nolte, M. J. Copyn, J. W. Zwikker, M. K. Engel, M. Hanack, Y. H. Chang and W. T. Ford, J. Am. Chem. Soc., 1994, 116, 6880 CrossRef CAS.
- H. Kietaibl, Monatsh. Chem., 1974, 105, 405 CrossRef CAS.
- C. G. Claessens, D. Gonzalez-Rodriguez, M. S. Rodriguez-Morgade, A. Medina and T. Torres, Chem. Rev., 2014, 114, 2192 CrossRef CAS PubMed.
- B. Verreet, S. Schols, D. Cheyns, B. P. Rand, H. Gommans, T. Aernouts, P. Heremans and J. Genoe, J. Mater. Chem., 2009, 19, 5295 RSC.
- A. M. van der Craats and J. M. Warman, Adv. Mater., 2001, 13, 130 CrossRef.
- J. Guilleme, J. Arago, E. Orti, E. Cavero, T. Sierra, J. Ortega, C. L. Folcia, J. Etxebarria, D. Gonzalez-Rodriguez and T. Torres, J. Mater. Chem. C, 2015, 3, 985 RSC.
- (a) K. L. Mutolo, E. I. Mayo, B. P. Rand, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2006, 128, 8108 CrossRef CAS PubMed; (b) H. Gommans, T. Aernouts, B. Verreet, P. Heremans, A. Medina, C. G. Claessens and T. Torres, Adv. Funct. Mater., 2009, 19, 3435 CrossRef CAS; (c) H. Gommans, T. Aernouts, B. Verreet, P. Heremans, A. Medina, C. G. Claessens and T. Torres, Adv. Funct. Mater., 2009, 19, 3435 CrossRef CAS; (d) B. Ma, C. H. Woo, Y. Miyamoto and J. M. J. Frechet, Chem. Mater., 2009, 21, 1413 CrossRef CAS.
- (a) Y. Takao, T. Masuoka, K. Yamamoto, T. Mizutani, F. Matsumoto, K. Moriwaki, K. Hida, T. Iwai, T. Ito, T. Mizuno and T. Ohno, Tetrahedron Lett., 2014, 55, 4564 CrossRef CAS; (b) K. Yamamoto, A. Takagi, M. Hada, R. Taniwaki, T. Mizutani, Y. Kimura, Y. Takao, K. Moriwaki, F. Matsumoto, T. Ito, T. Iwai, K. Hida, T. Mizuno and T. Ohno, Tetrahedron, 2016, 72, 4918 CrossRef CAS.
- D. S. Terekhov, K. J. M. Nolan, C. R. McArthur and C. C. Leznoff, J. Org. Chem., 1996, 61, 3034 CrossRef CAS PubMed.
- J. D. Dang, D. S. Josey, A. J. Lough, Y. Li, A. Sifate, Z.-H. Luc and T. P. Bender, J. Mater. Chem. A, 2016, 4, 9566 CAS.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 4467 CrossRef CAS.
- J. Guilleme, D. Gonzalez-Rodriguez and T. Torres, Angew. Chem., Int. Ed., 2011, 50, 3506 CrossRef CAS PubMed.
- D. Velasco, V. Jankauskas, J. Stumbraite, J. V. Grazulevicius and V. Getautis, Synth. Met., 2009, 159, 654 CrossRef CAS.
- M. V. Fulford, D. Jaidka, A. S. Paton, G. E. Morse, E. R. L. Brisson, A. J. Lough and T. P. Bender, J. Chem. Eng. Data, 2012, 57, 2756 CrossRef CAS.
- G. Martin, G. Rojo, F. Agullo-Lopez, V. R. Ferro, J. M. G. de la Vega, M. V. Martinez-Diaz, T. Torres, I. Ledoux and J. Zyss, J. Phys. Chem. B, 2002, 106, 13139 CrossRef CAS.
- M. Kasha, H. R. Rawls, M. Ashraf and A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371 CrossRef CAS.
- J. M. Kroon, R. B. M. Koehorst, M. van Dijk, G. M. Sanders and E. J. R. Sudholter, J. Mater. Chem., 1997, 7, 615 RSC.
- C. Colomban, E. V. Kudrik, V. Briois, J. C. Shwarbrick, A. B. Sorokin and P. Afanasiev, Inorg. Chem., 2014, 53, 11517 CrossRef CAS PubMed.
- B. Koksoy, O. Soyer, E. B. Orman, A. R. Ozkaya and M. Bulut, Dyes Pigm., 2015, 118, 166 CrossRef CAS.
- N. Kobayashi, T. Ishizaki, K. Ishii and H. Konami, J. Am. Chem. Soc., 1999, 121, 9096 CrossRef CAS.
- S. Nonell, N. Rubio, B. del Rey and T. Torres, J. Chem. Soc., Perkin Trans. 2, 2000, 1091 RSC.
- A. Concellon, M. Marcos, P. Romero, J. L. Serrano, R. Termine and A. Golemme, Angew. Chem., Int. Ed., 2017, 56, 1259 CrossRef CAS PubMed.
- S. H. Kang, Y.-S. Kang, W.-C. Zin, G. Olbrechts, K. Wostyn, K. Clays, A. Persoons and K. Kim, Chem. Commun., 1999, 1661 RSC.
- (a) M. S. Rodriguez-Morgade, C. G. Claessens, A. Medina, D. Gonzalez-Rodriguez, E. Gutierrez-Puebla, A. Monge, I. Alkorta, J. Elguero and T. Torres, Chem.–Eur. J., 2008, 14, 1342 CrossRef CAS PubMed; (b) J. Guilleme, M. J. Mayoral, J. Calbo, J. Arago, P. M. Viruela, E. Orti, T. Torres and D. Gonzalez-Rodriguez, Angew. Chem., Int. Ed., 2015, 54, 2543 CrossRef CAS PubMed.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, in Gaussian 09, Revision A.02, 2009 Search PubMed; M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
- Y. Long, H. Chen, Y. Yang, H. Wang, Y. Yang, N. Li, K. Li, J. Pei and F. Liu, Macromolecules, 2009, 42, 6501 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra11104e |
| This journal is © The Royal Society of Chemistry 2017 |