
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence2-(Anthracenyl)-4,5-bis(2,5-dimethyl(3-thienyl))-1H-imidazole: regulatable stacking structures, reversible grinding- and heating-induced emission switching, and solid-state photodimerization behavior†
Jun-Feng
Chen
a,
Dan-Ping
Gong
a,
Jing
Wen
b,
Haibo
Ma
b and
Deng-Ke
Cao
*a
aState Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, P. R. China. E-mail: dkcao@nju.edu.cn
bKey Laboratory of Mesoscopic Chemistry of MOE, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, P. R. China
First published on 8th October 2015
Abstract
2-(Anthracenyl)-4,5-bis(2,5-dimethyl(3-thienyl))-1H-imidazole (anbdtiH) has been synthesized. Its three solid-state structures, anbdtiH·CH3Cl (1), anbdtiH·2CH3OH (2) and anbdtiH2·CF3COO·CH3OH·H2O (3), have been constructed by skillfully choosing CHCl3 or CH3OH as the solvent and using or not using CF3COOH, with the aim of modifying the intermolecular hydrogen bonds and/or π⋯π stacking interactions. The three distinct structures show significantly different solid-state luminescence behaviors, an orange-red emission at 603 nm for 1, a blue emission at 453 nm for 2, and a green emission at 533 nm for 3. Upon grinding, these emission wavelengths exhibit evident variations, a blue-shift of Δλ = 83 nm for 1, a red-shift of Δλ = 20 nm for 2, and a blue-shift of Δλ = 54 nm for 3. The emission color of 1 can be reversibly switched between orange-red and green upon regulation of the intermolecular (N–H)imidazole⋯Nimidazole hydrogen bonds by a grinding–heating process. Moreover, compound 3 can undergo a solid-state [4π + 4π] photodimerization reaction upon irradiation with sunlight, forming 3-dimer. Based on the crystal structures of 1–3, this work discusses the relationship between the molecular stacking mode and the luminescence behavior/photochemical reactivity.
Introduction
In solid-state organic molecular materials, the molecular stacking mode plays a key role in obtaining the desired physical and chemical properties.1 Intermolecular hydrogen bonds and π⋯π stacking are two kinds of main supramolecular interactions, which could regulate the molecular stacking mode, thus leading to different functional properties, such as stimuli-responsive luminescence,2 and photochemical reactivity.3 Considerable attention has been focused on anthracene derivatives, mainly because of their intriguing luminescent properties4–8 and interesting [4π + 4π] photodimerization behaviors.9 Several anthracene derivatives were found to show reversible emission changes induced by grinding and heating.6–8 For example, Wang, Zhang et al. have reported 3(5)-(9-anthryl)pyrazole and its one derivative,6 which show different emission-color changes upon grinding, from blue to green and from green to blue, respectively. Tian et al. synthesized 9,10-bis((E)-2-(pyridin-2-yl)vinyl)anthracene (BP2VA) and 9,10-bis((E)-2-(pyridin-4-yl)vinyl)anthracene (BP4VA), each of which revealed a variety of emission colors from green to yellow upon grinding.7 Xiao, Yi et al. recently described four crystal structures of 2-(anthracen-9-yl)-4,5-diphenyl-1H-imidazole (ADPI) and their grinding-induced emission changes.8 Interestingly, the ground samples of these compounds could restore their initial emission colors upon heating at a temperature in the range of 90–170 °C.6–8 These stimuli-responsive luminescent materials have potential applications in rewritable optical media, information storage, imaging, sensors, switches,10etc. Crystal-structure analyses indicate that the emission-color changes of these anthracene derivatives are mainly due to the regulation of the intermolecular π⋯π stacking interactions between the anthracene moieties by grinding and heating.6–8 This finding provides a guiding principle for the design and fabrication of anthracene-based molecular materials with grinding-/heating-responsive emissions. However, to this day, it is unclear whether we could use a grinding–heating process to regulate the intermolecular hydrogen bonds in an anthracene-based molecular material, thus resulting in grinding-/heating-responsive luminescence behavior. So far, no document has answered this question. Therefore, more anthracene derivatives need to be designed and synthesized to understand the relationship between the molecular stacking mode and the grinding-/heating-responsive emission, and also to explore their photochemical reactivity.In this paper, we designed and synthesized an anthracene derivative, namely 2-(anthracenyl)-4,5-bis(2,5-dimethyl(3-thienyl))-1H-imidazole (anbdtiH, Scheme 1). In this compound, the imidazole unit could be involved in the formation of intermolecular hydrogen bonds. The two thiophene groups could provide a certain steric hindrance to promote non-parallel packing, and facilitate visible luminescence in the solid state. Under the different crystallization conditions of using CHCl3 or CH3OH as the solvent and adding or not adding CF3COOH, we obtained three kinds of solid-state structures of anbdtiH, including anbdtiH·CH3Cl (1), anbdtiH·2CH3OH (2), and anbdtiH2·CF3COO·CH3OH·H2O (3), which reveal different intermolecular hydrogen bonds and/or π⋯π stacking interactions. Herein, we discuss the relationship between the distinct molecular stacking modes in 1–3 and the related grinding-/heating-responsive luminescence behaviors. Moreover, we report the photodimerization behavior of 3, and the structural characterization of the resultant compound 3-dimer.
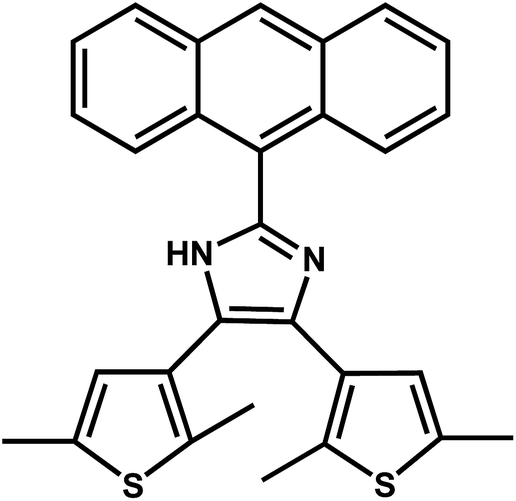 | ||
| Scheme 1 The structure of anbdtiH. | ||
Results and discussion
For the preparation of 1–3, different solvents were used, CHCl3 for 1, and CH3OH for 2 and 3. In addition, CF3COOH was used for preparing 3. The choice of these crystallization conditions is based on the following considerations. (1) CHCl3 molecules generally do not become involved in the formation of hydrogen bonds, which would allow neighboring anbdtiH molecules to connect to each other through (N–H)imidazole⋯Nimidazole hydrogen bonds. (2) Compared to CHCl3, CH3OH molecules have a stronger capability for forming hydrogen bonds between a CH3OH molecule and the imidazole unit of anbdtiH. (3) CF3COOH can not only protonate the imidazole unit of anbdtiH, but can also connect to anbdtiH through a hydrogen bond. Our experimental results indicate that these different crystallization conditions can lead to the distinct packing structures of 1–3.Compound 1 crystallizes in the hexagonal non-centrosymmetric space group P65, while the monoclinic space group P21/c is observed for 2 and 3. Their crystallographic details and selected bond lengths are listed in Tables 1 and S1,† respectively. Both 1 and 2 consist of neutral anbdtiH molecules and solvent molecules (CHCl3 and CH3OH molecules, respectively) (Fig. S6†), while 3 is an organic salt, which is made up of an anbdtiH2+ cation, CF3COO− anion, and CH3OH and H2O molecules (Fig. S7†). The anbdtiH/anbdtiH2+ molecules in the three compounds show similar structural features, where the two thiophene groups of each molecule adopt an antiparallel conformation, and the imidazole unit is nearly perpendicular to the anthracene moiety, exhibiting a dihedral angle of 85.7(1)° in 1, 86.5(1)° in 2, and 84.4(1)° in 3.
| 1 | 2 | 3 | 3-dimer·2DMF | |
|---|---|---|---|---|
| a R 1 = ∑||Fo| − |Fc||/∑|Fo|. wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2. | ||||
| Formula | C30H25C13N2S2 | C31H32O2N2S2 | C32H31O4N2F3S2 | C64H62O2N6S4 |
| M | 583.99 | 528.71 | 628.71 | 1075.44 |
| Crystal system | Hexagonal | Monoclinic | Monoclinic | Orthorhombic |
| Space group | P65 | P21/c | P21/c | Pbca |
| a/Å | 13.8888(10) | 22.178(6) | 20.1561(15) | 18.220(5) |
| b/Å | 13.8888(10) | 7.454(2) | 9.0577(7) | 12.964(4) |
| c/Å | 25.754(4) | 18.052(5) | 17.1173(13) | 24.265(7) |
| α/° | 90 | 90 | 90 | 90 |
| β/° | 90 | 104.184(5) | 93.2870(10) | 90 |
| γ/° | 120 | 90 | 90 | 90 |
| V/Å−3 | 4302.3(8) | 2893.5(14) | 3119.9(4) | 5731(3) |
| Z | 6 | 4 | 4 | 4 |
| D c/g cm−3 | 1.352 | 1.214 | 1.338 | 1.246 |
| F(000) | 1812 | 1120 | 1312 | 2272 |
| GooF on F2 | 1.032 | 1.036 | 1.021 | 1.032 |
| R 1, wR2 [I > 2σ(I)]a | 0.0631, 0.1294 | 0.0554, 0.1224 | 0.0467, 0.1601 | 0.0698, 0.1470 |
| R 1, wR2 (all data)a | 0.1269, 0.1637 | 0.1234, 0.1468 | 0.0587, 0.1742 | 0.1672, 0.1789 |
| (Δρ)max, (Δρ)min (e Å−3) | 0.275, −0.284 | 0.267, −0.274 | 0.429, −0.276 | 0.302, −0.270 |
As shown in Fig. 1 and S8,† compound 1 shows a supramolecular left-handed chiral chain structure, where neighboring anbdtiH molecules connect to each other through hydrogen bonds N2–H⋯N1a = 2.858(1) Å [symmetry code a = y, −x + y, z + 1/6], i.e. (N–H)imidazole⋯Nimidazole hydrogen bonds. These chiral chains stack through van der Waals interactions, and the interchain space is filled with CHCl3 molecules. It should be noted that no CD signal was observed for the bulk sample, indicating that 1 is a racemic mixture. In compound 2, neighboring anbdtiH molecules are linked by two CH3OH molecules in a chain structure (Fig. 2) through hydrogen bonds O2⋯N1 = 2.753(1) Å, N2⋯O1a = 2.856(1) Å, and O1⋯O2b = 2.672(1) Å [symmetry codes a = x, y, z + 1; b = x, −y + 1/2, z − 1/2]. Compound 3 exhibits a supramolecular layer structure (Fig. 3), where neighboring anbdtiH2+ cations are connected by CF3COO−, H2O and CH3OH through hydrogen bonds N2⋯O1W = 2.697(1) Å, O3⋯O2 = 2.792(1) Å, N1⋯O2a = 2.745(1) Å, O1W⋯O1b = 2.852(1) Å, and O1W⋯O3c = 2.734(1) Å [symmetry codes a = x, y, z − 1; b = x, y + 1, z − 1; c = x, −y + 3/2, z − 1/2]. It is interesting that the anthracene moieties from neighboring layers show face-to-face π⋯π stacking interactions, with a centroid–centroid distance of 3.684(1) Å (Fig. 4).11 This distance is below 4.2 Å, suggesting the possible occurrence of a solid-state [4π + 4π] photodimerization reaction.12 Clearly, compounds 1–3 reveal distinct packing structures, which would result in different luminescence behaviors and photochemical reactivities.
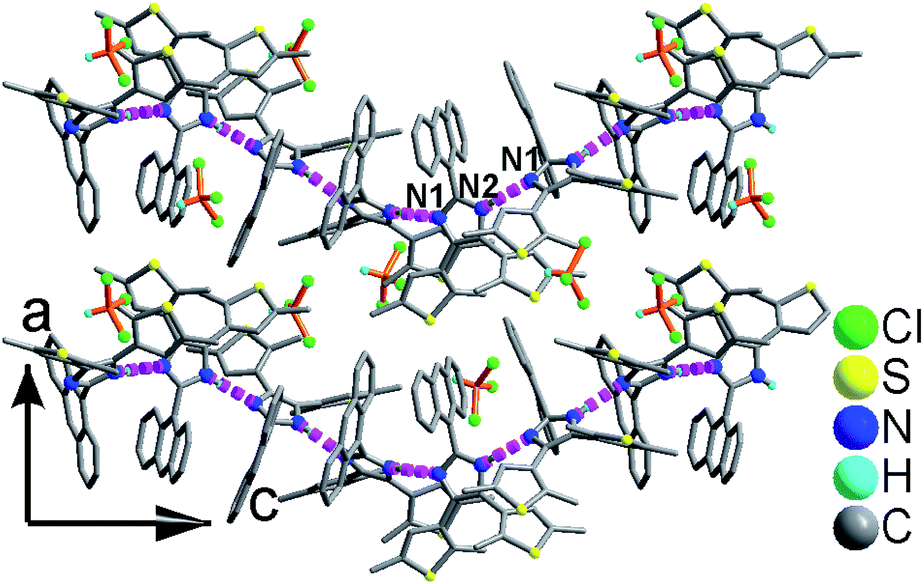 | ||
| Fig. 1 Supramolecular left-handed chiral chain structure in 1. | ||
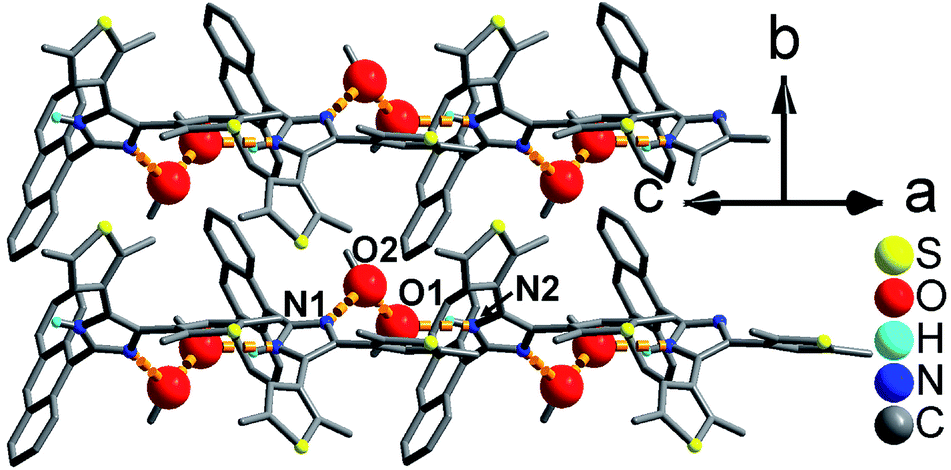 | ||
| Fig. 2 Supramolecular chain structure in 2. The red balls are the O atoms from the CH3OH molecules. | ||
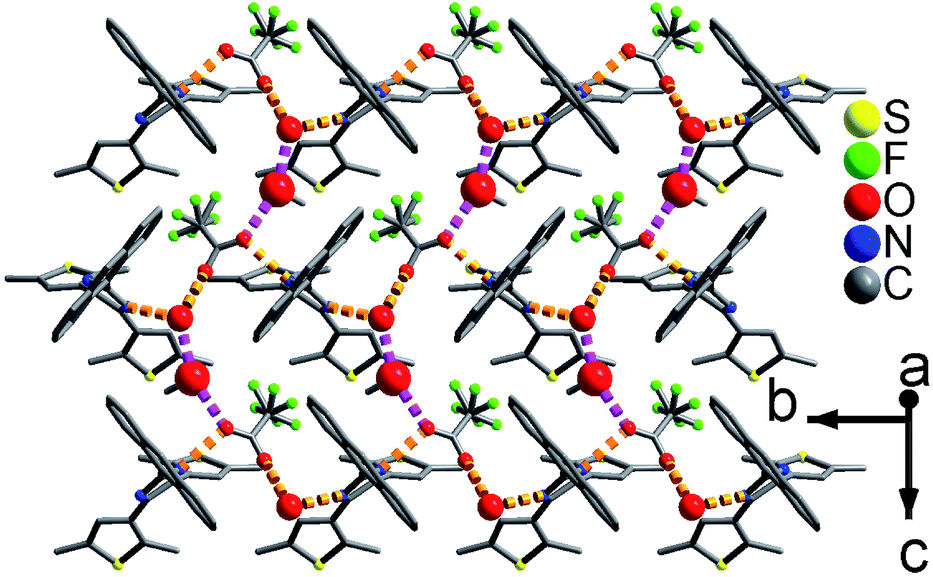 | ||
| Fig. 3 Supramolecular layer in 3. The large and medium red balls are the O atoms from the CH3OH and H2O molecules, respectively. | ||
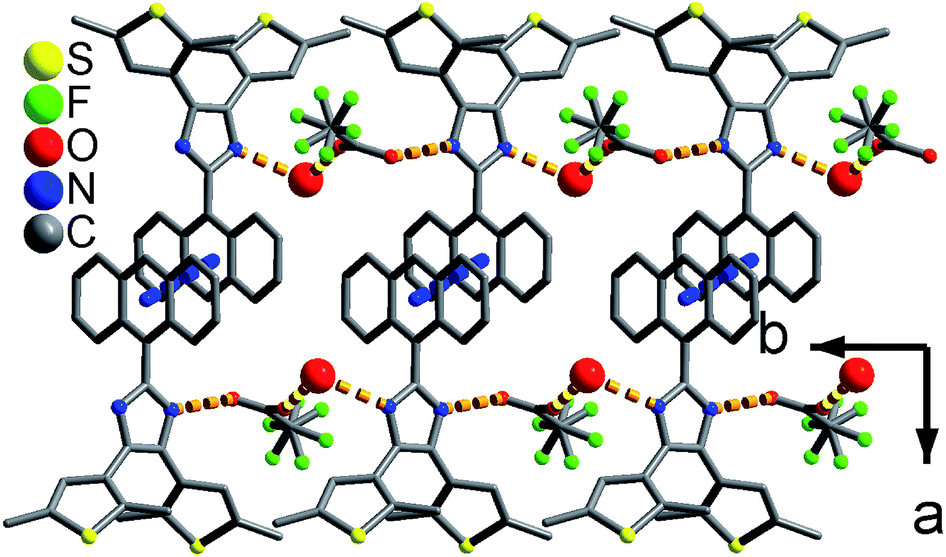 | ||
| Fig. 4 The π⋯π stacking interactions between the anthracene moieties in 3. The red balls are the O atoms from the H2O molecules. | ||
The solid-state luminescence spectrum of 1 shows three main emissions at 488, 539 and 603 nm (Fig. 5). In contrast, 2 and 3 exhibit shorter emission wavelengths (maxima at 453 and 533 nm, respectively). Thus, the three compounds reveal different emission colors upon irradiation with 365 nm light – orange-red for 1, blue for 2, and green for 3 (Fig. 6). The orange-red emission of 1 could be related to the intermolecular (N–H)imidazole⋯Nimidazole hydrogen bonds (Fig. 1), which could result in direct electronic coupling between neighboring anbdtiH molecules.13 This is supported by the following grinding experiment (see the paragraph after the quantum chemical calculations). Compound 2 reveals a blue emission mainly because of the following two reasons. (1) Compared to 1, neighboring anbdtiH molecules are separated by two CH3OH molecules, thus resulting in an absence of electronic coupling between neighboring anbdtiH molecules. (2) The hydrogen bond interactions between the CH3OH molecule and imidizole nitrogen atom N1/N2 (Fig. 2) could result in a blue-shifted emission, which is confirmed by the emission spectra of anbdtiH in different solvents (Fig. S10†). The emission in CH3OH (498 nm) occurs at a shorter wavelength than the emission in toluene (505 nm), CH2Cl2 (516 nm), and CH3CN (521 nm). Compound 3 has a longer emission wavelength than that of 2, mainly due to the π⋯π stacking interactions between the anthracene moieties (Fig. 4).14
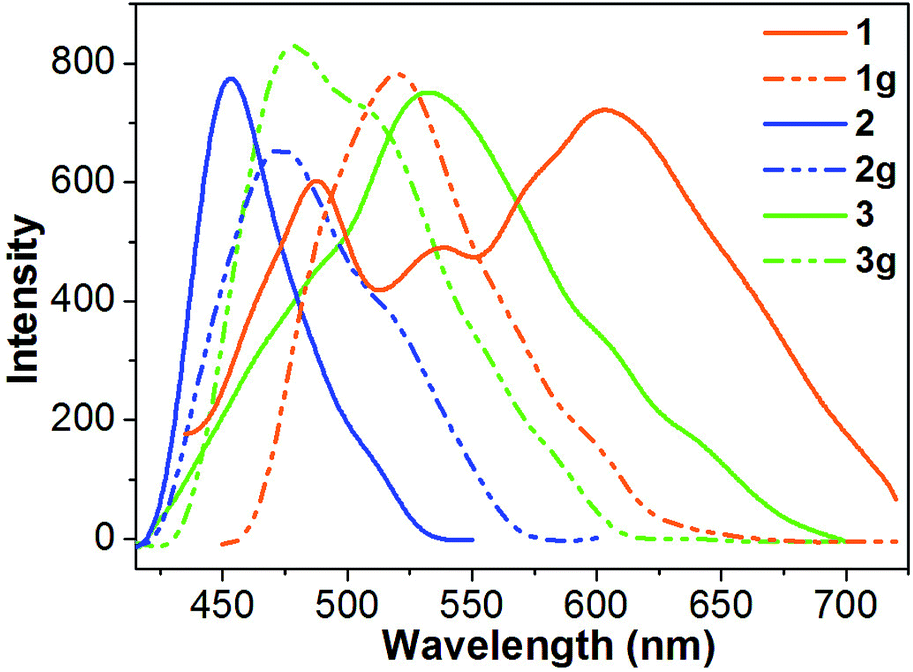 | ||
| Fig. 5 Solid-state fluorescence spectra (λ = 380 nm) of 1–3 and their ground samples 1g–3g at room temperature. | ||
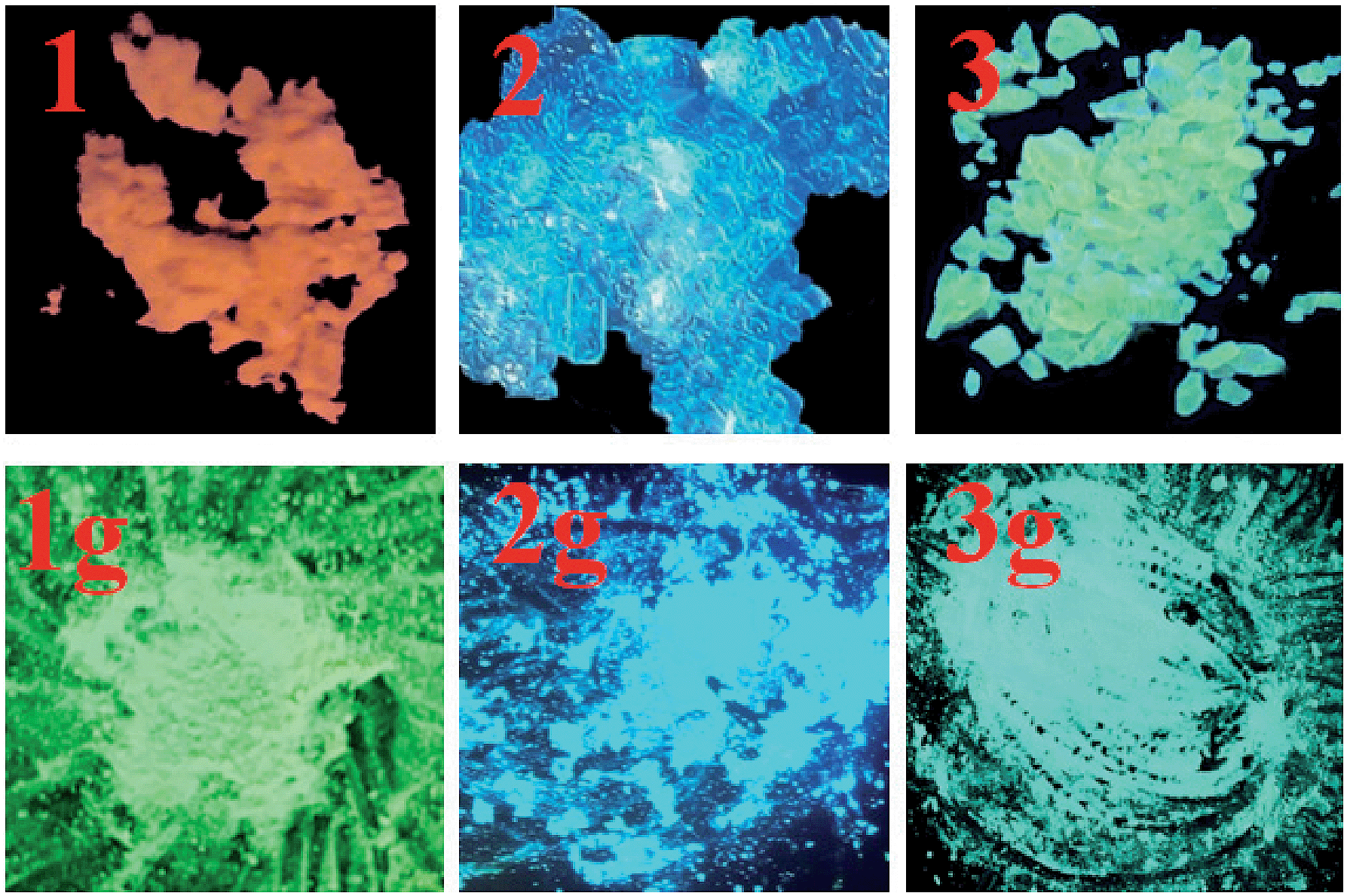 | ||
| Fig. 6 Emission colors of 1–3 and their ground samples 1g–3g at room temperature, under a lamp with 365 nm wavelength light. | ||
In order to understand the different emission behaviors of 1–3, we performed quantum chemical calculations for the first dipole-allowed excited states in these compounds at the CAM-B3LYP/6-31G(d,p) level (the computational details are provided in the ESI†). The dominant natural transition orbitals (NTOs, Fig. S11†) show that the emission chromophore in each compound is mainly located at the anthracene moiety of the anbdtiH/anbdtiH2+ molecule. In compound 2, the emission localizes at the central anbdtiH molecule because of the too weak π–π electronic coupling with other molecules, and, accordingly, this compound has the largest emission energy. On the contrary, the central molecules in both 1 and 3 show remarkable π–π electronic coupling with neighboring molecules, which may result in a red-shifted emission. It should be noted that the computational result of the intermolecular π–π electronic coupling in 1 is in agreement with the supramolecular chain structure showing a centroid–centroid distance of 3.765(1) Å between the two benzene rings from neighboring anthracene moieties (Fig. S9†). The NTOs for compound 1 are shown to be well distributed at the anthracene moieties of the central molecule and its two neighbors, while the NTOs for 3 can only delocalize over the central anbdtiH2+ molecule and its one nearest-neighbor molecule because the distance between the central molecule and its other neighbor is too large [the plane⋯plane distance between the two anthracene moieties is ≥6.991(1) Å in Fig. 3] to produce effective π–π electronic coupling. This may explain why compound 1 has a more significant red shift than 3.
After grinding, the stacking structure of 1 was destroyed, obtaining sample 1g. This was confirmed by their different XRD patterns (Fig. S12†). Compared to 1, sample 1g shows only a few broad and weak signals. As shown in Fig. 5 and 6, 1g reveals a green emission at 520 nm, which is very similar to the emission of the anbdtiH monomolecule in CH2Cl2 (516 nm) (Fig. S10†). This suggests that neighboring anbdtiH molecules in 1g have no obvious intermolecular (N–H)imidazole⋯Nimidazole hydrogen bond interactions, such as those in 1. The destruction of the hydrogen bonds in 1 by grinding was further confirmed by the ATR IR spectra of 1 and 1g (Fig. S13†). The spectra show obvious differences in the peaks around 840, 1140, 1382, 1431, 1560, 1974, and 2030 cm−1. The different emission wavelengths of 1 and 1g (603 nm vs. 520 nm) indicate that the connection of neighboring anbdtiH molecules through (N–H)imidazole⋯Nimidazole hydrogen bonds could increase the intermolecular electronic coupling, thus resulting in the orange-red emission of 1. We measured the lifetimes of compound 1, and obtained τ1 = 5.2 ns (45.2% contribution) and τ2 = 0.6 ns (54.8% contribution) for the emission at 488 nm, τ1 = 6.3 ns (77.5% contribution) and τ2 = 0.5 ns (22.5% contribution) for the emission at 539 nm, and τ = 6.1 ns for the emission at 603 nm. Compared to 1, the ground sample 1g exhibits lifetimes of τ1 = 5.1 ns (49.9% contribution) and τ2 = 1.3 ns (50.1% contribution) for its emission at 520 nm. In addition, compounds 2 and 3 also exhibit emission-color changes upon grinding (Fig. 6) due to grinding-induced changes of the hydrogen bonds and/or π⋯π stacking interactions. Their emission-wavelength changes (red-shift of Δλ = 20 nm for 2, and blue-shift of Δλ = 54 nm for 3) are clearly smaller than that for 1 (blue-shift of Δλ = 83 nm) (Fig. 5, Table S3,† Δλ calculation based on the maximum emission).
As shown in Fig. S14,† after heating at 110 °C for 10 minutes, sample 1g could change its emission color from green to orange-red, indicating the formation of a new structure, namely 1gh. A similar heating-induced structural transformation was also observed in the other anthracene derivative.6–8 The XRD pattern of 1gh is different from that of 1 (Fig. S12†), which excludes the possibility of a heating-induced structural transformation from 1g to 1. It should be noted that the emission bands of 1gh (484, 533, and 604 nm) are very similar to those of 1 (488, 539, and 603 nm) (Fig. S15†). This suggests that neighboring anbdtiH molecules in 1gh connect to each other through (N–H)imidazole⋯Nimidazole hydrogen bonds. After grinding, 1gh could switch its emission color from orange-red to green (Fig. S14†). We have not studied the influence of heating on the emission colors of 2g and 3g because they easily lost lattice CH3OH and/or H2O molecules upon heating. These lattice molecules are involved in hydrogen bond connections among anbdtiH/anbdtiH2+ molecules. Their losses would complicate the structures and luminescence behaviors of the samples.
Clearly, the emission color of compound 1 could be reversibly switched between orange-red and green due to grinding-induced destruction and heating-induced formation of intermolecular (N–H)imidazole⋯Nimidazole hydrogen bonds. It should be noted that compound 1 is significantly different to reported anthracene derivatives showing grinding- and heating-responsive luminescence behaviors,6–8 which have been described in the introduction. The emission-color switching in those compounds is mainly due to the regulation of intermolecular π⋯π stacking interactions between the anthracene moieties by grinding and heating. To the best of our knowledge, compared to those compounds, 1 is the first anthracene derivative where the intermolecular hydrogen bonds can be regulated by grinding and heating, thus resulting in reversible emission-color switching.
Considering the perfect face-to-face π⋯π stacking interactions between the anthracene moieties in compound 3 (Fig. 4), we studied its solid-state [4π + 4π] photodimerization behavior. Since compound 3 has strong UV-vis absorption in the range of 200–450 nm (Fig. S16†), sunlight was used to irradiate its crystalline-phase sample (see the detail in the ESI†). After removing the unreacted sample, a white powder was obtained, namely 3-dimer. Compounds 3 and 3-dimer show significantly different UV-vis spectra (Fig. S17†). The former exhibits an intense absorption band at 255 nm, and the typical anthracene absorption bands at 351, 367 and 386 nm.15 The latter reveals a strong absorption band at 228 nm and a weak absorption band at 290 nm. In order to clarify the solid-state photodimerization behavior of 3, we measured the crystal structure of 3-dimer·2DMF. It crystallizes in the orthorhombic space group Pbca, and has a centrosymmetric molecular structure (Fig. 7). Two anbdtiH molecules connect to each other by C1–C8A and C8–C1A bonds [1.624(5) Å]. These distances are comparable to those in the dimer compound 1-(9-anthracenylmethyl)-3-ethylimidazolium iodide [1.636(4) Å].16 The central rings of the two original anthracene moieties lose their aromaticity and bend along the C1, C8 and C1A, C8A axes, respectively, forming the shape of flying wings. The dihedral angle between the two wings is 132.1(1)°. The two imidazole–thiophene moieties reach out roughly along the C1, C8 and C1A, C8A axes, respectively, forming a C8–C1–C15 angle of 164.755 (1)°. The two thiophene groups attached to each imidazole unit adopt an antiparallel conformation (Fig. S18†). In addition, the structure of 3-dimer was also confirmed by its 1H NMR spectrum (Fig. S2†). Our experimental results indicate that compound 3 could undergo a solid-state [4π + 4π] photodimerization reaction upon irradiation with sunlight, forming 3-dimer. In addition, compounds 1 and 2 have not shown solid-state [4π + 4π] photodimerization behavior, which is in agreement with their packing structures not having face-to-face π⋯π stacking interactions between neighboring anthracene moieties.
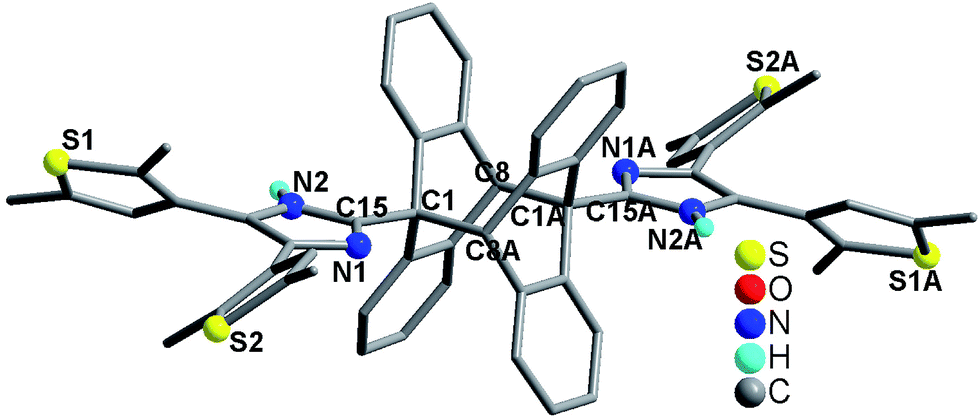 | ||
| Fig. 7 Molecular structure of 3-dimer·2DMF. All H atoms attached to C atoms and the two lattice DMF molecules are omitted for clarity. | ||
Conclusions
We have synthesized a new anthracene derivative containing an imidazole unit, namely anbdtiH. Based on the molecular assembly of anbdtiH through intermolecular hydrogen bonds and/or π⋯π stacking interactions, compounds anbdtiH·CH3Cl (1), anbdtiH·2CH3OH (2) and anbdtiH2·CF3COO·CH3OH·H2O (3) have been prepared, which show distinct packing structures, and different luminescence and photodimerization behaviors. Compound 1 shows a supramolecular left-handed chiral chain structure, where neighboring anbdtiH molecules connect to each other through (N–H)imidazole⋯Nimidazole hydrogen bonds. In contrast, neighboring anbdtiH/anbdtiH2+ molecules in 2 and 3 are separated by solvent molecules and counter ions (CH3OH, H2O, and CF3COO−), and are linked through hydrogen bonds to form a supramolecular chain structure and layer structure, respectively. In addition, compound 3 shows face-to-face π⋯π stacking interactions between interlayer anthracene moieties. Compounds 1–3 reveal an orange-red emission at 603 nm, a blue emission at 453 nm, and a green emission at 533 nm, respectively. These emissions can be evidently changed by grinding, resulting in a blue-shift of Δλ = 83 nm for 1, a red-shift of Δλ = 20 nm for 2, and a blue-shift of Δλ = 54 nm for 3, because of the variation in intermolecular hydrogen bonds and/or π⋯π stacking interactions. Compared to the regulation of π⋯π stacking interactions between anthracene moieties by a grinding–heating process,6–8 compound 1 is the first anthracene derivative where the intermolecular (N–H)imidazole⋯Nimidazole hydrogen bonds can be reversibly changed by grinding and heating, thus resulting in emission-color switching between orange-red and green. In addition, compound 3 is photoactive due to the perfect face-to-face π⋯π stacking between anthracene moieties. Thus, its solid-state [4π + 4π] photodimerization results in compound 3-dimer. Our experimental results demonstrate that the molecular stacking mode of anbdtiH can be purposefully modified by hydrogen bonds and/or π⋯π stacking interactions, thus obtaining the expected luminescence and/or photodimerization behaviors.Acknowledgements
We thank the financial support from the NSF of Jiangsu province China (BK 20141314), the NSF of China (No. 21171090), the NSF of China-Royal Society of UK International Exchanges Scheme-Cost Share Program (No. 21411130119) and the National Science Fund for Talent Training in Basic Science (No. J1103310).Notes and references
- (a) S.-J. Yoon and S. Y. Park, J. Mater. Chem., 2011, 21, 8388 Search PubMed; (b) F. Li, N. Gao, H. Xu, W. Liu, H. Shang, W. Yang and M. Zhang, Chem.–Eur. J., 2014, 20, 9991 CrossRef CAS PubMed and references therein.
- (a) S. Yagai, S. Okamura, Y. Nakano, M. Yamauchi, K. Kishikawa, T. Karatsu, A. Kitamura, A. Ueno, D. Kuzuhara, H. Yamada, T. Seki and H. Ito, Nat. Commun., 2014, 5, 5013 CrossRef PubMed; (b) G. Fan and D. Yan, Sci. Rep., 2014, 4, 4933 CAS; (c) M.-S. Yuan, D.-E. Wang, P. Xue, W. Wang, J.-C. Wang, Q. Tu, Z. Liu, Y. Liu, Y. Zhang and J. Wang, Chem. Mater., 2014, 26, 2467 CrossRef CAS.
- (a) L. R. MacGillivray, G. S. Papaefstathiou, T. Friscic, T. D. Hamilton, D.-K. Bucar, Q. Chu, D. B. Varshney and I. Georgiev, Acc. Chem. Res., 2008, 41, 280 CrossRef CAS PubMed; (b) Y. Ishida, A. S. Achalkumar, S. Kato, Y. Kai, A. Misawa, Y. Hayashi, K. Yamada, Y. Matsuoka, M. Shiro and K. Saigo, J. Am. Chem. Soc., 2010, 132, 17435 CrossRef CAS PubMed; (c) K. M. Hutchins, J. C. Sumrak and L. R. MacGillivray, Org. Lett., 2014, 16, 1052 CrossRef CAS PubMed.
- (a) Z. Fei, N. Kocher, C. J. Mohrschladt, H. Ihmels and D. Stalke, Angew. Chem., Int. Ed., 2003, 42, 783 CrossRef CAS PubMed; (b) A. N. Sokolov, T. Friscic and L. R. MacGillivray, J. Am. Chem. Soc., 2006, 128, 2806 CrossRef CAS PubMed; (c) X.-F. Fu, Y.-F. Yue, R. Guo, L.-L. Li, W. Sun, C.-J. Fang, C.-H. Xu and C.-H. Yan, CrystEngComm, 2009, 11, 2268 RSC; (d) Z. Zhang, Y. Zhang, D. Yao, H. Bi, I. Javed, Y. Fan, H. Zhang and Y. Wang, Cryst. Growth Des., 2009, 9, 5069 CrossRef CAS; (e) Y. Mizobe, T. Hinoue, A. Yamamoto, I. Hisaki, M. Miyata, Y. Hasegawa and N. Tohnai, Chem.–Eur. J., 2009, 15, 8175 CrossRef CAS PubMed; (f) T. Hinoue, M. Miyata, I. Hisaki and N. Tohnai, Angew. Chem., Int. Ed., 2012, 51, 155 CrossRef CAS PubMed; (g) Y.-X. Li, H.-B. Zhou, J.-L. Miao, G.-X. Sun, G.-B. Li, Y. Nie, C.-L. Chen, Z. Chen and X.-T. Tao, CrystEngComm, 2012, 14, 8286 RSC; (h) J. Zhang, J. Chen, B. Xu, L. Wang, S. Ma, Y. Dong, B. Li, L. Ye and W. Tian, Chem. Commun., 2013, 49, 3878 RSC; (i) M. Sugino, K. Hatanaka, Y. Araki, I. Hisaki, M. Miyata and N. Tohnai, Chem.–Eur. J., 2014, 20, 3069 CrossRef CAS PubMed.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973 CrossRef CAS PubMed.
- Z. Zhang, D. Yao, T. Zhou, H. Zhang and Y. Wang, Chem. Commun., 2011, 47, 7782 RSC.
- (a) Y. Dong, B. Xu, J. Zhang, X. Tan, L. Wang, J. Chen, H. Lv, S. Wen, B. Li, L. Ye, B. Zou and W. Tian, Angew. Chem., Int. Ed., 2012, 51, 10782 CrossRef CAS PubMed; (b) Y. Dong, J. Zhang, X. Tan, L. Wang, J. Chen, B. Li, L. Ye, B. Xu, B. Zou and W. Tian, J. Mater. Chem. C, 2013, 1, 7554 RSC.
- R. Li, S. Xiao, Y. Li, Q. Lin, R. Zhang, J. Zhao, C. Yang, K. Zou, D. Li and T. Yi, Chem. Sci., 2014, 5, 3922 RSC.
- (a) F. Cicogna, G. Ingrosso, F. Lodato, F. Marchetti and M. Zandomeneghi, Tetrahedron, 2004, 60, 11959 CrossRef CAS PubMed; (b) I. Zouev, T. Lavy and M. Kaftory, Eur. J. Org. Chem., 2006, 4164 CrossRef CAS PubMed; (c) M. Horiguchi and Y. Ito, J. Org. Chem., 2006, 71, 3608 CrossRef CAS PubMed; (d) I. Zouev, D.-K. Cao, T. V. Sreevidya, M. Telzhensky, M. Botoshansky and M. Kaftory, CrystEngComm, 2011, 13, 4376 RSC; (e) L. Zhu, A. Agarwal, J. Lai, R. O. Al-Kaysi, F. S. Tham, T. Ghaddar, L. Mueller and C. J. Bardeen, J. Mater. Chem., 2011, 21, 6258 RSC.
- (a) M. Irie, T. Fukaminato, T. Sasaki, N. Tamai and T. Kawai, Nature, 2002, 420, 759 CrossRef CAS PubMed; (b) S. Kobatake, S. Takami, H. Muto, T. Ishikawa and M. Irie, Nature, 2007, 446, 778 CrossRef CAS PubMed; (c) T. Mutai, H. Satou and K. Araki, Nat. Mater., 2005, 4, 685 CrossRef CAS PubMed; (d) A. Kishimura, T. Yamashita, K. Yamaguchi and T. Aida, Nat. Mater., 2005, 4, 546 CrossRef CAS PubMed; (e) S. Srinivasan, P. A. Babu, S. Mahesh and A. Ajayaghosh, J. Am. Chem. Soc., 2009, 131, 15122 CrossRef CAS PubMed.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885 RSC.
- (a) M. O'Donnell, Nature, 1968, 218, 460 CrossRef PubMed; (b) H. Bouas-Laurent, A. Castellan, J.-P. Desvergne and R. Lapouyade, Chem. Soc. Rev., 2000, 29, 43 RSC.
- (a) J. Yang, D. Seneviratne, G. Arbatin, A. M. Andersson and J. C. Curtis, J. Am. Chem. Soc., 1997, 119, 5329 CrossRef CAS; (b) D. A. Williamson and B. E. Bowler, J. Am. Chem. Soc., 1998, 120, 10902 CrossRef CAS; (c) G. Canzi, J. C. Goeltz, J. S. Henderson, R. E. Park, C. Maruggi and C. P. Kubiak, J. Am. Chem. Soc., 2014, 136, 1710 CrossRef CAS PubMed.
- H. Zhang, Z. Zhang, K. Ye, J. Zhang and Y. Wang, Adv. Mater., 2006, 18, 2369 CrossRef CAS PubMed.
- (a) H. Bouas-Laurent, A. Castellan, J.-P. Desvergne and R. Lapouyade, Chem. Soc. Rev., 2000, 29, 43 RSC; (b) M. Kercher, B. Konig, H. Zieg and L. D. de Cola, J. Am. Chem. Soc., 2002, 124, 11541 CrossRef CAS PubMed.
- Q.-X. Liu, H.-B. Song, F.-B. Xu, Q.-S. Li and Z.-Z. Zhang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, o674 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Physical measurements, syntheses, quantum chemical calculations, selected bond lengths, 1H NMR spectra, XRD patterns, ATR IR spectra, diffuse reflectance spectra, luminescence spectra, photograph of the emission color, and crystal structures. CCDC 1415320–1415323. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03201f |
| This journal is © The Royal Society of Chemistry 2016 |