
Cu(I)-catalyzed double C–H amination: synthesis of 2-iodo-imidazo[1,2-a]pyridines†
Divya
Dheer
ab,
K. Ranjith
Reddy
ac,
Santosh K.
Rath
ab,
P. L.
Sangwan
ab,
Parthasarthi
Das
abc and
Ravi
Shankar
*ab
aAcademy of Scientific and Innovative Research (AcSIR), Jammu Campus, India. E-mail: rshankar@iiim.ac.in
bBio-Organic Chemistry Division, Indian Institute of Integrative Medicine (CSIR), Jammu 180001, India
cMedicinal Chemistry Division, Indian Institute of Integrative Medicine (CSIR), Jammu 180001, India
First published on 8th April 2016
Abstract
An iodine/CuI mediated double oxidative C–H amination reaction has been developed for the synthesis of 2-iodo-imidazo[1,2-a]pyridines. The salient features of this methodology are mild reaction conditions and high regioselectivity. The designed compounds e.g. 2-iodo-imidazo[1,2-a]pyridines (3e) may serve as active pharmaceutical ingredients (APIs) of marketed drugs like saripidem and nicopidem. Further saripidem was synthesised by using 3e as an intermediate.
Heteroaromatic scaffolds containing bridgehead nitrogen atoms such as imidazo[1,2-a]pyridine, imidazo[1,2-a]pyrazine, imidazo[1,2-a]pyrimidine are found in many biologically active molecules.1 A number of imidazo[1,2-a]pyridine derived analogues have been successfully used for various biological activities such as antiviral, antibacterial and anti-inflammatory.2 Top selling drugs like alpidem (as an anxiolytic agents),3 zolpidem (used in the treatment of insomnia),4 nicopidem & saripidem (as an anxiolytic agents),5 minodronic acid (used for the treatment of osteoporosis),6 and optically active GSK812397 drug (HIV infection)7 are derived from imidazo[1,2-a]pyridine scaffold (Fig. 1).
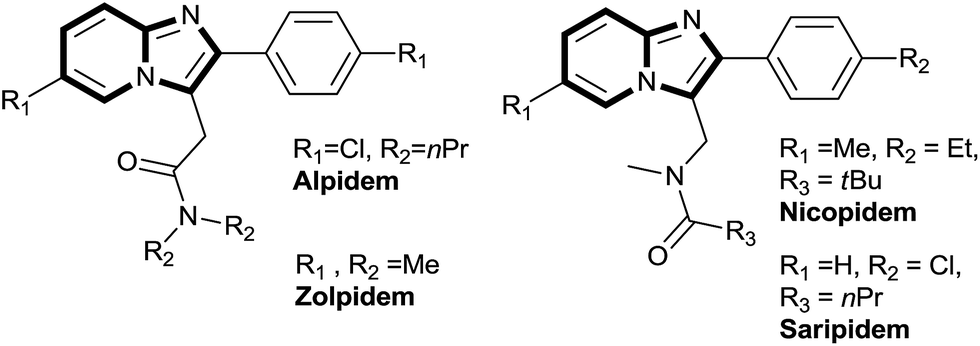 | ||
| Fig. 1 Imidazo[1,2-a]pyridine derived drugs. | ||
Therefore, synthesis of imidazo[1,2-a]pyridine derivatives remain an active area of research in organic synthesis because of above said biological importance and scaffold of marketed drug. In the recent years, various metal-catalyzed8 and metal free9 synthetic methods for synthesis of imidazo[1,2-a]pyridine have been reported, the general approach used for the synthesis of imidazo[1,2-a]pyridine is based on the intermolecular cyclization of 2-aminopyridine and other precursor such as phenylacetaldehyde, α-bromoaldehyde, bromo-ketones, iodonium salt, and bromoalkyne.1,10,11 In addition to above halogenated hetero-aromatic scaffolds have always been a centre of attraction in medicinal chemistry because they can be easily modified by classical coupling reaction such as Suzuki,12 Heck13 and Sonogashira reaction14 during structure activity relationship (SAR) study.
A careful survey of the literature revealed that the synthesis of 2-halo-substituted imidazo[1,2-a]pyrimidines has been rarely explored. Zhao's group15 designed a catalyst copper supported on manganese oxide-based octahedral molecular sieves OMS-2 (CuOx/OMS-2) for the synthesis of 3-iodoimidazo[1,2-a]pyridines from acetophenones whereas Jiang's group16 developed copper-catalyzed method for the synthesis of 2-haloimidazopyridines with aminopyridines and haloalkynes via intermolecular oxidative cyclization (Scheme 1). The substrate 1-haloalkyne used in the Jiang's method, the main disadvantage of this method is that synthesis of 1-haloalkyne requires special halogenating reagents and tedious workup processes. To overcome the drawback, we developed an efficient and simple one-pot Cu-catalyzed protocol for the synthesis of 2-iodo-imidazo[1,2-a]pyridine by using alkynes, 2-aminopyridine, and iodine in presence of O2.
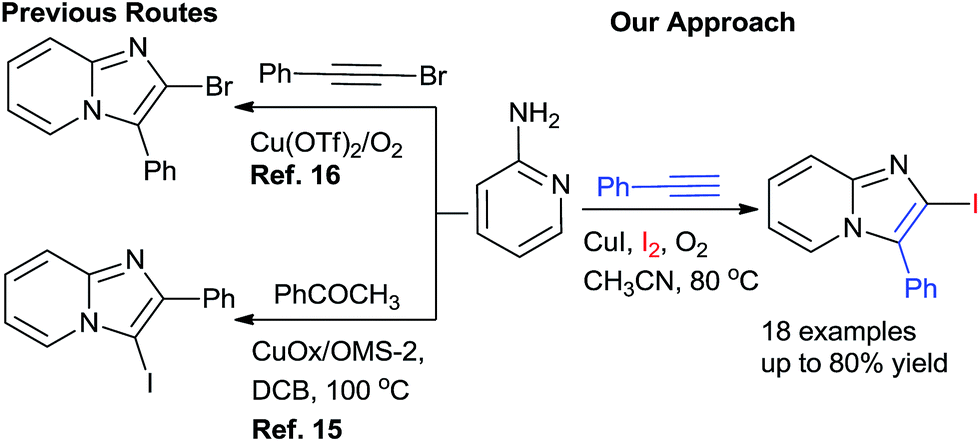 | ||
| Scheme 1 Synthesis of 2/3-haloimidazo[1,2-a]pyridine. | ||
Initially, reaction was performed between 2-aminopyridine (1a, 1 eq.) and phenylacetylene (2a, 1.5 eq.) in optimizing the reaction conditions (Table 1) by using Cu(OTf)2 and I2 (1 eq.) in CH3CN at 80 °C in open air condition (Table 1, entry 1), but product formation was not observed. However, the desired 2-iodo-3-phenylimidazo[1,2-a]pyridine (3a) was observed in 50% yield (Table 1, entry 2) under O2 atmosphere. The spectral data for 3a was found in accordance with literature.16,17
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Iodine source | Yield |
| a n.r.: no reaction. | ||||
| 1 | Cu(OTf)2 | CH3CN | I2 | n.r. |
| 2 | Cu(OTf)2 | CH3CN | I2 | 50 |
| 3 | Cu(OTf)2 | DMF | I2 | 25 |
| 4 | Cu(OTf)2 | Dioxane | I2 | 40 |
| 5 | Cu(OTf)2 | Toluene | I2 | Trace |
| 6 | CuI | CH3CN | I2(1) | 60 |
| 7 | CuI | CH 3 CN | I 2 (1.5) | 80 |
| 8 | CuI | CH3CN | I2(2) | 70 |
| 9 | CuI | CH3CN | NIS | Trace |
| 10 | CuI | CH3CN | I2 | Trace |
| 11 | CuI | DMSO | I2 | Trace |
| 12 | CuBr | CH3CN | I2 | 45 |
| 13 | CuBr2 | CH3CN | I2 | 55 |
| 14 | CuCl | CH3CN | I2 | 50 |
| 15 | CuCl2 | CH3CN | I2 | 60 |
| 16 | Cu(OAc)2 | CH3CN | I2 | 35 |
| 17 | CuI | CH3CN | — | n.r. |
| 18 | — | CH3CN | I2 | n.r. |
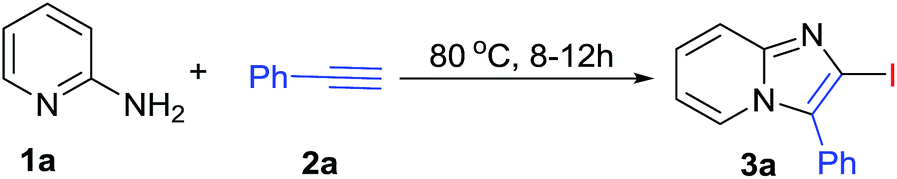
Subsequently, a number of solvents such as DMF, 1,4-dioxane, toluene were investigated but CH3CN was found most suitable reaction medium in terms of yield (Table 1, entries 2–5). A series of other copper catalyst such as CuI, CuBr, CuBr2, CuCl, CuCl2 and Cu(OAc)2 were also examined for improving the yield (Table 1, entries 6–16). The higher yield was obtained when the reaction was carried out with CuI (10 mol%) and I2 (1.5 equiv.) in CH3CN under O2 atmosphere (Table 1, entry 7) at 80 °C. The yield of 3a was slightly decreased when increasing the ratio of I2 (2.0 equiv.) (Table 1, entry 8), which may be due to oxidative behaviour of I2.18 The other source of iodine like NIS was also examined (Table 1, entry 9), the result revealed that I2 is better source than NIS in this reaction condition. The control experiments, were also carried out without CuI or I2 (Table 1, entries 18 and 17) under same reaction conditions but no reaction happened in both case. To avoid unstability of iodoalkyne & minimization of homocouple formation, initially all reaction were carried out at room temperature by stirring for 10 min. There after the temperature was raised gradiently upto 80 °C. In this optimised reaction condition better results were obtained. The synthesis of 2-iodo-imidazo[1,2-a]pyridines via 1-iodoalkyne was carried out with another controlled reaction. 1-Iodoalkyne & 2-aminopyridine were reacted in which with I2 or without I2. In result we observed same desired product in both conditions with dimmer of terminal alkyne as expected this prove I2 is just playing a role for synthesis of iodoalkyne and also observed homocoupled of 1-iodoalkyne in the presence of O2 (Scheme 2).
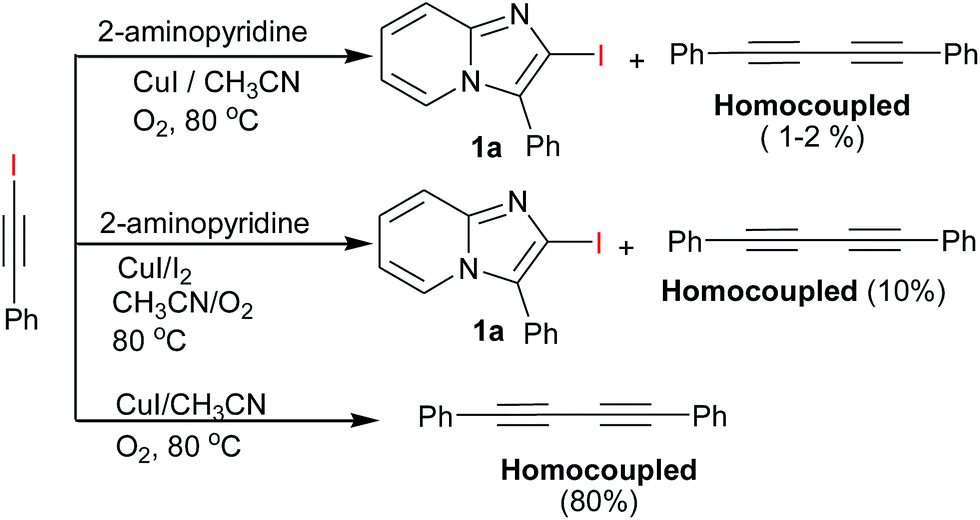 | ||
| Scheme 2 Controlled experiment. | ||
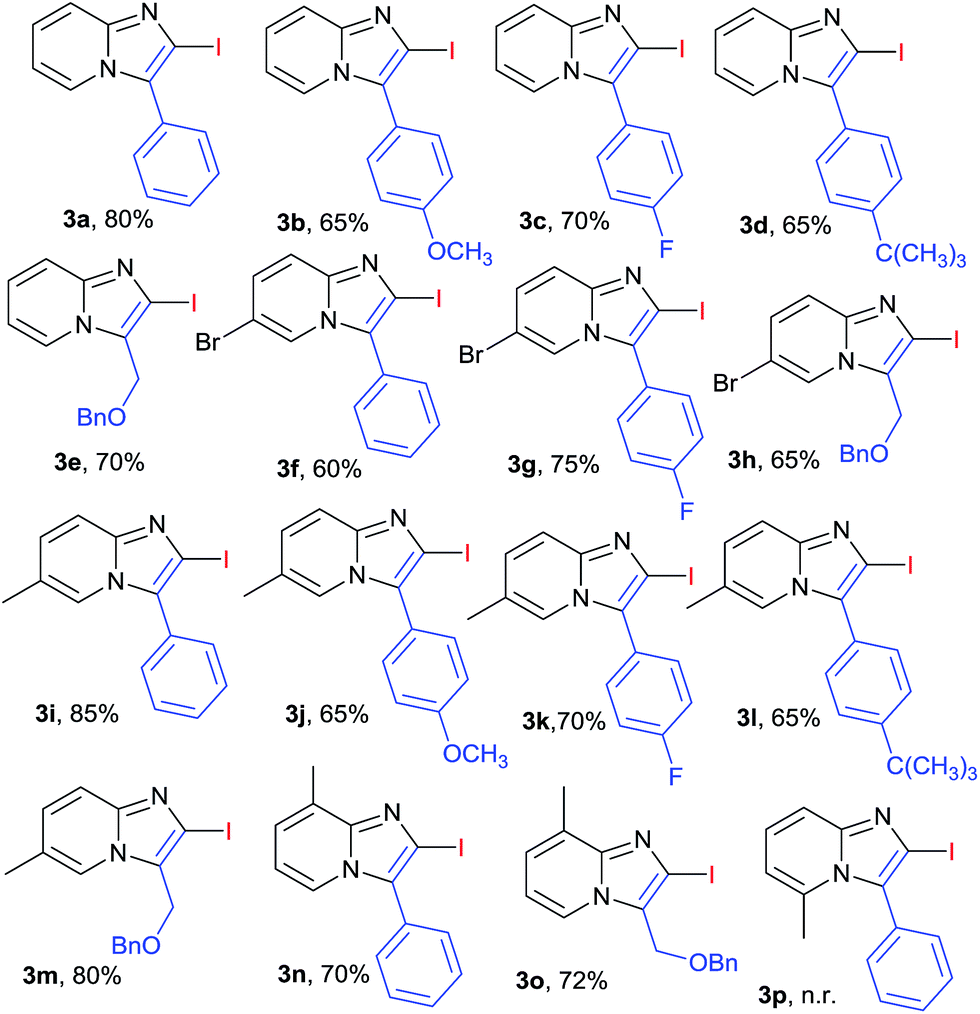
The scope of the reaction was further explored by using various substituted 2-amino pyridines and substituted acetylenes (involving 10 mol% of CuI and 1.5 equiv. of I2 in CH3CN under O2 at 80 °C). The designed 2-iodoimidazo[1,2-a]pyridines (3a–3p) were obtained in moderate to good yields (Table 2). In general, substituted phenylacetylene that contained either electron-donating or electron-withdrawing group reacted smoothly with 2-aminopyridine in 60–80% yield. The electron deficient alkynes were more favourable such as 1-ethynyl-4-fluorobenzene yielded product (3c, 70%) comparatively alkynes with electron donating group such as OMe or t-butyl yielded (3b, 65%) and (3d, 65%) respectively. Various 2-aminopyridines with different substituent's were found suitable partner for the synthesis of 2-halo-substituted-imidazo[1,2-a]pyridines. 2-Aminopyridines with methyl or halo group at different position (3- and 5- position) were successfully transformed into corresponding products in good yield (3f–3o, 60–85%). In case of 2-amino-6-methylpyridine no reaction proceed may be due to steric hindrance could play the role in the formation of desired product (3p). It was observed 2-aminopyridine with electron donating group at various position is favourable for the synthesis of 2-halo-substituted-imidazo[1,2-a]pyridines (3k–3o, 65–85%), compare to 2-amino-pyridine with halo substitution (3f–3h, 60–75%). A halo substituted imidazo[1,2-a]pyridine (3f–3j) at 2 and 6 position can open a window for further modification. Recently Chioua19 and Zhu20 reported synthesis of the anxiolytic saripidem and nicopidem by imidazo[1,2-a]pyridin-3-yl-methanol. With these background, the transformation of benzyl protected propargyl alcohol under same reaction condition with substituted 2-aminopyridine, gave the corresponding compound (3e, 3h, 3m and 3o) in good yield that can be convert to active pharmaceutical ingredients (APIs) of marketed drugs like saripidem and nicopidem.
|
|
|---|
| a All reaction were carried out using 1a (1.0 eq.) and 2a (1.5 eq.),with CuI (10 mol%) catalyst, in CH3CN 80 °C under O2 atmosphere, reaction time 8–12 h. |
|
On the basis of controlled experimental and literature evidence,19–21 a postulated reaction mechanism for the synthesis of 2-iodo-imidazo[1,2-a]pyridine analogue via CuI/I2 promoted oxidative cyclization is displayed in Scheme 3. As initially reaction of substituted acetylene with CuI/I2 gives crude adduct 1-iodoalkynes 5via Cu(I) phenylethyne 4, characterized by mass and 1H NMR spectroscopic analysis.16 Then the triple bond of 5 is activated by the coordination with Cu(I) and react with 2-aminopyridine could initially form complex 6. Then migratory insertion of haloalkyne occurred and provide reactive Cu(III) intermediate 7 after deprotonation and oxidation.21 Finally the reductive elimination of 7 give desired cyclised compound 3a.
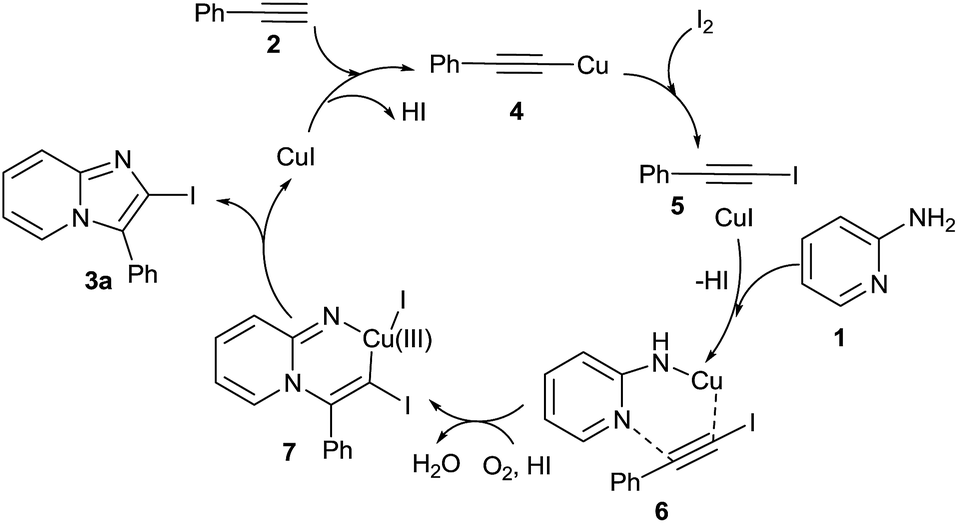 | ||
| Scheme 3 Proposed mechanism. | ||
Halogenated aromatics are known to convert into the corresponding aryl, alkenyl or alkynyl group under cross-coupling condition. To investigate the scope in coupling reaction, we further functionalized 2-iodo-imidazo[1,2-a]pyridines in some important analogues 8, 9, and 10 in excellent yields through Suzuki, Heck, and Sonogashira reaction respectively (Scheme 4).
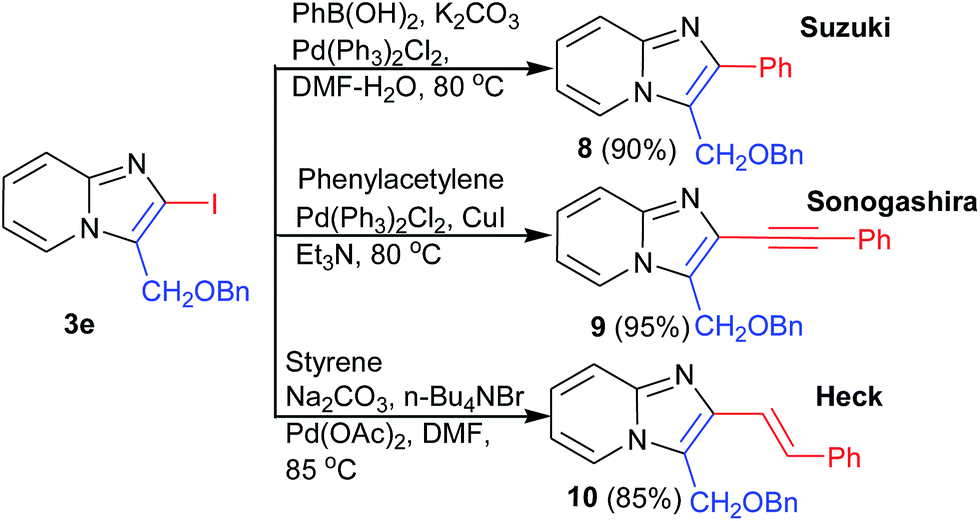 | ||
| Scheme 4 Synthetic utility of the 2-iodoimidazo[1,2-a]pyridine. | ||
The utility of synthesised APIs, was confirmed by using 2-iodo-imidazo[1,2-a]pyridines (3e) in the synthesis of anxioltic saripidem (Scheme 5). Suzuki reaction of intermediate 3e with p-chlorophenylboronic acid gave the intermediate 11 in 90% yield. Debenzylation of 11 with BCl3 at −78 °C afforded alcohol in good yield that was further converted in intermediate 12 by reaction of butyronitrile in the presence of H2SO4. Finally saripidem was obtained by methylation of 12 in the presence of NaH via reported method in excellent yield.19
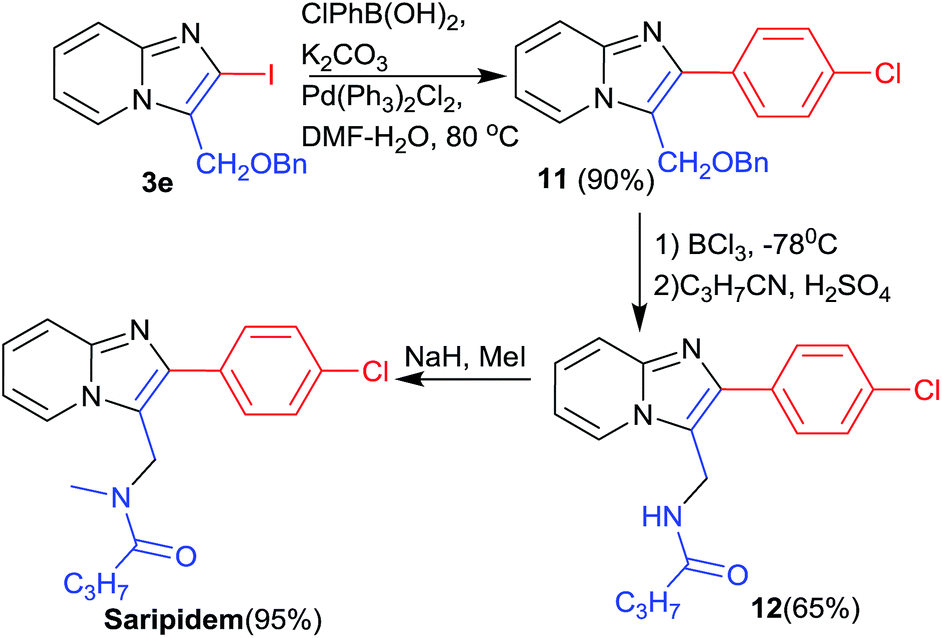 | ||
| Scheme 5 Total synthesis of saripidem from 3e. | ||
In summary, we have developed an efficient one-pot process for synthesis of 2-iodo-imidazo[1,2-a]pyridine analogues via CuI/I2 promoted oxidative cyclization of 2-aminopyridine. This procedure is general and diverse array of designed 2-iodo-imidazo[1,2-a]pyridine derivatives were synthesized with challenging substitution. Further saripidem was synthesised successfully by using the key intermediate 3e obtained through our develop method. This prove that important heterocycles can be used as advanced intermediate for marketed drugs like nicopidem and saripidem.
Acknowledgements
D. D.; K. R. R. And S. K. R thank to CSIR and ICMR, New Delhi, India for their research fellowship, respectively. This research work was financially supported by CSIR-New Delhi (BSC 0108) IIIM communication no. IIIM/1814/2015.Notes and references
- (a) A. K. Bagdi, S. Santra, K. Monir and A. Hajra, Chem. Commun., 2015, 51, 1555 RSC; (b) R. Goel, V. Luxami and K. Paul, Org. Biomol. Chem., 2015, 13, 3525 RSC; (c) K. Pericherla, P. Kaswan, K. Pandey and A. Kumar, Synthesis, 2015, 47, 887 CrossRef CAS; (d) K. R. Reddy, A. S. Reddy, R. Shankar, R. Kant and P. Das, Asian J. Org. Chem., 2015, 4, 573 CrossRef CAS.
- (a) F. Couty and G. Evano, in Comprehensive Heterocyclic Chemistry III, ed. A. R. Katritzky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, Oxford, 2008, vol. 11, p. 409 Search PubMed; (b) E. C. Gueiffier and A. Gueiffier, Mini-Rev. Med. Chem., 2007, 7, 888 CrossRef.
- S. Z. Langer, S. Arbilla, J. Benavides and B. Scatton, Adv. Biochem. Psychopharmacol., 1990, 46, 61 CAS.
- L. Almirante, L. Polo, A. Mugnaini, E. Provinciali, P. Rugarli, A. Biancotti, A. Gamba and W. Murmann, J. Med. Chem., 1965, 8, 305 CrossRef CAS.
- R. J. Boerner and H. J. Moller, Psychopharmacology, 1997, 4, 145 Search PubMed.
- H. Mori, M. Tanaka, R. Kayasuga, T. Masuda, Y. Ochi, H. Yamada, K. Kishikawa, M. Ito and T. Nakamura, Bone, 2008, 43, 840 CrossRef CAS.
- Y. Abe, H. Kayakiri, S. Satoh, T. Inoue, Y. Sawada, N. Inamura, M. Asano, I. Aramori, C. Hatori, H. Sawai, T. Oku and H. Tanaka, J. Med. Chem., 1998, 41, 4587 CrossRef CAS.
- (a) P. Y. Choy, K. C Luk, Y. Wu, C. M. So, L. Wang and F. Y. Kwong, J. Org. Chem., 2015, 80, 1457 CrossRef CAS; (b) L. Zhao, H. Zhan, J. Liao, J. Huang, Q. Chen, H. Qiu and H. Cao, Catal. Commun., 2014, 56, 65 CrossRef CAS; (c) M. Lesieur, F. Lazreg and C. S. J. Cazin, Chem. Commun., 2014, 50, 8927 RSC; (d) H. Yang, L. Yang, Y. Li, F. Zhang, H. Liu and B. Yi, Catal. Commun., 2012, 26, 11 CrossRef CAS.
- (a) A. Kamal, C. N. Reddy, M. Satyaveni, D. Chandrasekhar, J. B. Nanubolu, K. K. Singarapu and R. A. Maurya, Chem. Commun., 2015, 51, 10475 RSC; (b) R. Adams and J. S. Dix, J. Am. Chem. Soc., 1958, 80, 4618 CrossRef CAS; (c) G. Liu, X. Cong, J. He, S. Luo, D. Wu and J. Lan, J. Chem. Res., 2012, 36, 687 CrossRef CAS.
- S. K. Lee and J. K. Park, J. Org. Chem., 2015, 80, 3723 CrossRef CAS.
- Z. Wu, Y. Pan and X. Zhou, Synthesis, 2011, 14, 2255 Search PubMed.
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (b) J. Han, Y. Liu and R. Guo, J. Am. Chem. Soc., 2009, 131, 2060 CrossRef CAS; (c) L. Liu, Y. Zhang and Y. Wang, J. Org. Chem., 2005, 70, 6122 CrossRef CAS.
- (a) C. Wu and J. Zhou, J. Am. Chem. Soc., 2014, 136, 650 CrossRef CAS; (b) M. L. Kantam, P. Srinivas, J. Yadav, P. R. Likhar and S. Bhargava, J. Org. Chem., 2009, 74, 4882 CrossRef CAS; (c) A. Fayol, Y. Q. Fang and M. Lautens, Org. Lett., 2006, 8, 4203 CrossRef CAS.
- (a) R. Severin, J. Reimer and S. Doye, J. Org. Chem., 2010, 75, 3518 CrossRef CAS; (b) R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084 RSC.
- X. Meng, C. Yu, G. Chen and P. Zhao, Catal. Sci. Technol., 2015, 5, 372 RSC.
- Y. Gao, M. Yin, W. Wu, H. Huang and H. Jiang, Adv. Synth. Catal., 2013, 355, 2263 CrossRef CAS.
- (a) G. Pelletier, S. Lie, J. J. Mousseau and A. B. Charette, Org. Lett., 2012, 14, 5464 CrossRef CAS; (b) W. Chen, J. Zhang, B. Wang, Z. Zhao, X. Wang and Y. Hu, J. Org. Chem., 2015, 80, 2413 CrossRef CAS.
- (a) S. C. Lu, P. R. Zheng and G. Liu, J. Org. Chem., 2012, 77, 7711 CrossRef CAS; (b) A. S. Lopez, J. M. C. Plane, A. R. Baker, L. J. Carpenter, R. V. Glasow, J. C. G. O. Martín, G. McFiggans and R. W. Saunders, Chem. Rev., 2012, 112, 1773 CrossRef.
- M. Chioua, E. Soriano, L. Infantes, M. L. Jimeno, J. M. Contelles and A. Samadi, Eur. J. Org. Chem., 2013, 35 CrossRef CAS.
- H. Wang, Y. Wang, D. Liang, L. Liu, J. Zhang and Q. Zhu, Angew. Chem., Int. Ed., 2011, 50, 5678 CrossRef CAS.
- Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra02953a |
| This journal is © The Royal Society of Chemistry 2016 |