
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Raising the (metastable) bar: 100% photo-switching in [Pd(Bu4dien)(η1-![[N with combining low line]](https://www.rsc.org/images/entities/h2_char_004e_0332.gif) O2)]+ approaches ambient temperature†
O2)]+ approaches ambient temperature†
Lauren E.
Hatcher
Department of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: l.e.hatcher@bath.ac.uk
First published on 11th February 2016
Abstract
100% nitro–nitrito linkage isomerism is reported in single-crystals of the Pd(II)–nitrite system [Pd(Bu4dien)(η1-![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) O2)]BPh4 (Bu4dien = N,N,N′,N′-tetrabutyldiethylenetriamine, BPh4 = tetraphenylborate). Complete conversion to a metastable endo-nitrito-(η1-
O2)]BPh4 (Bu4dien = N,N,N′,N′-tetrabutyldiethylenetriamine, BPh4 = tetraphenylborate). Complete conversion to a metastable endo-nitrito-(η1-![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) NO) isomer is achieved after just 15 min irradiation with 400 nm LED light. The system is entirely metastable below 240 K, while pseudo-steady-state photocrystallographic experiments confirm that the excited state isomer is retained, at substantial conversion levels, under continuous illumination until 260 K. These results show promise for new linkage isomer systems, based on heavier transition metal centres, which display full functionality under near-ambient conditions.
NO) isomer is achieved after just 15 min irradiation with 400 nm LED light. The system is entirely metastable below 240 K, while pseudo-steady-state photocrystallographic experiments confirm that the excited state isomer is retained, at substantial conversion levels, under continuous illumination until 260 K. These results show promise for new linkage isomer systems, based on heavier transition metal centres, which display full functionality under near-ambient conditions.
Introduction
Photo-switchable molecular compounds, based on organic and organometallic chemistries, continue to be important in the search for new functional materials that will address a number of real-world challenges, including renewable energy solutions, sensors and data storage.1 Light-induced reactions are tunable, site-specific and are often efficient and clean, all of which increases their popularity for a number of applications. In particular, molecules whose photo-switching behaviour is retained in the solid-state can be advantageous as these are often more easily developed into devices. Where possible, single-crystal X-ray diffraction (SC-XRD) continues to be the preferred method to study these solid-state systems, providing highly-accurate, 3-D structural parameters at all stages of the reaction. The detailed study of photochemical switching in crystalline materials was pioneered by Schmidt, Cohen et al. in the 1960s,2–4 whose work with trans-cinnamic acid derivatives developed an understanding of the conditions necessary for [2 + 2] photo-dimerisation in single-crystals. While this early work relied primarily on SC-XRD experiments conducted on a crystal in its ground state (GS), dramatic advancements in the SC-XRD method since this time now allow the determination of GS, metastable state (MS) and entirely transient excited state species over a range of time-scales and temperatures.5,6 Such experiments are conducted via so-called photocrystallographic methods7 and require in situ irradiation of a crystal either before or during the X-ray experiment.The earliest photocrystallographic studies were conducted by Coppens et al. in the mid-1990s and were the first experiments to unambiguously determine the presence of MS linkage isomers at low temperature in the Fe–nitrosyl complex sodium nitroprusside.8 Metastable linkage isomers have the potential to be exploited as functional materials for applications where a long-lived excited state is desirable. In the last 20 years, single-crystal-to-single-crystal linkage isomerism has been reported for a number of small di- and tri-atomic ligands including nitrosyl,9–12 di-nitrogen,13 sulfur dioxide14–18 and nitrite groups.19–24 Despite these successes, there remain some key factors that limit the functionality of metastable linkage isomer systems and must be overcome before these can be considered viable candidates for real devices. One such limitation is the maximum level of photo-activation that can be achieved in the crystal. Ideally, 100% activation that is reversible, controllable and reproducible over many repeat cycles should be achieved. The first fully-reversible, 100% photo-activation in a linkage isomer crystal was reported by Warren et al. in 2009, in [Ni(dppe)(NO2)Cl] (dppe = bis-diphenylphosphinoethane).19 100% excitation is rationalised by the inclusion of the bulky di-phosphine ligand as this photo-inert fragment can dominate the packing, providing a “reaction cavity”25–27 in which the smaller nitrite ligand can rearrange without imparting undue strain to the surrounding lattice. Since that time, this crystal engineering principle has been utilised by Raithby et al. in designing several Ni(II)–nitrite species with bulky co-ligands, which are capable of very high nitro–nitrito conversion in the single-crystal.20,22–24 A second factor limiting the efficacy of metastable linkage isomers as functional molecular switches is the relatively low critical temperature, or “MS limit”, above which the system regains its GS arrangement. Currently, MS limits for the high-converting Ni(II)–nitrite complexes reported to-date do not exceed 200 K. A few Pd(II) and Pt(II)–nitrite systems boast MS limits between 200 and 240 K,21,28 while some Ru–SO2 compounds reach limits as high as 250 K.14,29 However, these systems do not achieve high levels of GS to MS conversion. MS linkage isomers capable of 100% switching at near-ambient temperature would be most desirable for practical applications. The current study reports here-in a novel Pd(II)–nitrite species capable of 100% photo-activation, with a long-lived nitrito-(η1-NO) isomer still detectable by SC-XRD methods at 250 K.
Experimental section
Synthetic preparations
All manipulations were carried out in air. All reagents were used as purchased, without the need for further purification.Single-crystals of the tetrahydrofuran (THF) solvate (1) were obtained by recrystallization from THF/diethyl ether.
UV/vis diffuse reflectance spectroscopy
UV/vis diffuse reflectance spectra were collected on a Shimadzu UV-2600 spectrophotometer with an integrating sphere. Data were collected on a powdered crystalline sample of (1) pelleted with KBr, between 800 and 200 nm at ambient temperature.Single-crystal X-ray crystallography
Standard single-crystal X-ray diffraction (SC-XRD) experiments were conducted at the University of Bath using a Rigaku–Oxford Diffraction Gemini A Ultra diffractometer equipped with an Atlas CCD detector. The crystal temperature was controlled by an Oxford Instruments CryojetXL liquid nitrogen flow device, between 100–300 K. Data collection, indexing and integration procedures were completed with CrysAlis Pro.31 Structures were solved by dual-space methods using SHELXT32 and refined by full-matrix least squares on F2 using SHELXL-2014.33Photocrystallographic experiments
In situ irradiation of the crystal was achieved using a specifically designed LED ring array.34 Six 400 nm LEDs (Bivar Opto™ LED5-UV-400-30 Series, 350 mcd, 3.8 V, 12 mW) are positioned 1 cm from the sample in a uniform circle. SC-XRD data collection, indexing and reduction were completed as outlined previously. Metastable state (MS) X-ray data were refined against ground state (GS) coordinates and a Fourier electron density difference map (photodifference map) used to identify new features corresponding to the MS. In the final refinement, partially-activated systems were treated as a disorder model with linkage isomer occupancies refined using standard free variables.Results
Ground state (GS) crystal structure
A single-crystal of (1) was mounted in oil at ambient temperature and cooled to 100 K in the dark, before a standard SC-XRD dataset was obtained (Table 1). Fig. 1 shows the structure of the asymmetric unit (the full atomic numbering scheme for the palladium cation is given in Fig. S1†). With this data it is possible to conduct a detailed analysis at the molecular level and, in terms of the crystal packing, establish whether a switch of coordination mode is likely in (1).| Ground state (GS) | 400 nm metastable state (MS) | |
|---|---|---|
| Photo-conversion | 0% | 100% |
| Temperature | 100(2) K | 100(2) K |
| Wavelength | 0.71073 Å | 0.71073 Å |
| Empirical formula | C48H73B1N4O3Pd1 | C48H73B1N4O3Pd1 |
| Formula weight | 871.31 g mol−1 | 871.31 g mol−1 |
| Crystal size | 0.17 × 0.05 × 0.03 mm | 0.17 × 0.05 × 0.03 mm |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/n | P21/n |
| Unit cell parameters (constrained) | a = 11.5457(4) Å, α = 90° | a = 11.4775(4) Å, α = 90° |
| b = 13.4021(5) Å, β = 95.335(4)° | b = 13.3647(5) Å, β = 94.569(3)° | |
| c = 29.7173(12) Å, γ = 90° | c = 29.9849(8) Å, γ = 90° | |
| Volume | 4578.4(3) Å3 | 4584.9(3) Å3 |
| Z | 4 | 4 |
| Density (calculated) | 1.264 M g m−3 | 1.262 M g m−3 |
| Absorption coefficient μ | 0.451 mm−1 | 0.448 mm−1 |
| F(000) | 1856 | 1856 |
| R(int) | 0.0459 | 0.0661 |
| Completeness (to θ = 25.00°) | 0.998 | 0.998 |
| R 1 (observed data) | 0.0428 | 0.0428 |
| wR2 (all data) | 0.0863 | 0.0901 |
| Reflections (independent) | 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 627 (9335) 627 (9335) |
31062 (9355) |
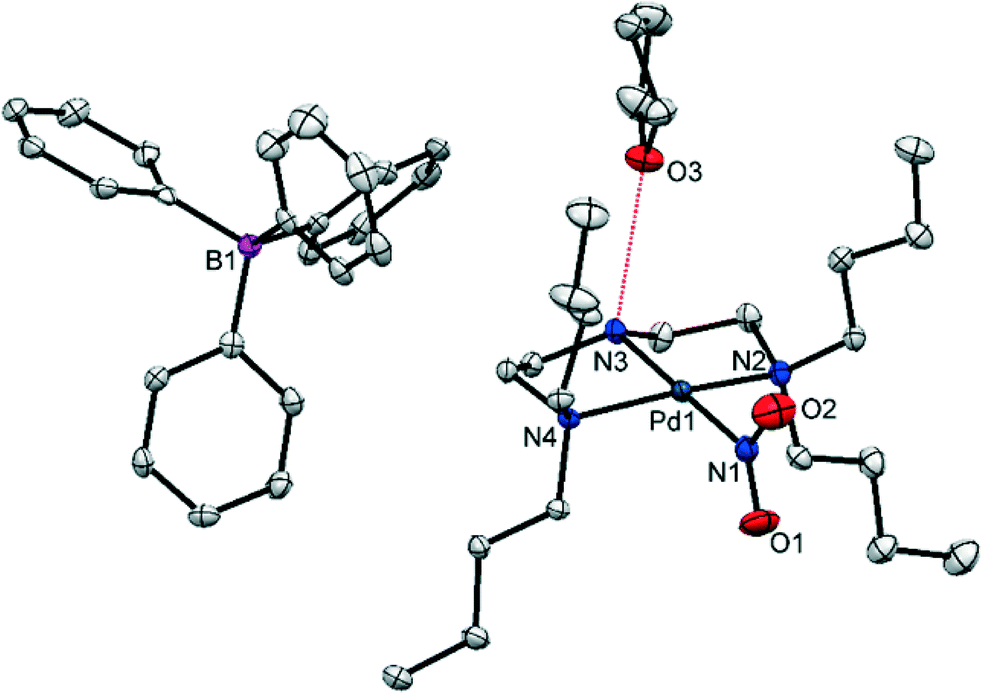 | ||
| Fig. 1 Single-crystal X-ray structure for the asymmetric unit of (1) showing the atomic arrangement in the ground state (GS), before any irradiation. Ellipsoids are set at 50% probability and hydrogen atoms are omitted for clarity (red dashed line = hydrogen bonding to THF solvent molecule). | ||
(1) crystallises in the monoclinic space group P21/c with one complex cation, one BPh4 anion and one THF solvent molecule in the asymmetric unit. The Pd(II) cation is square planar, with three coordination sites occupied by the chelating amine. The fourth site is filled by the nitrite ligand, which is monodentate and adopts solely nitro-(η1-O2) coordination in the GS. The N(3)–H(3)⋯O(3) hydrogen bond, highlighted in Fig. 1, is the only strong intermolecular interaction in the asymmetric unit and does not involve the potentially isomerisable nitro-(η1-O2) group.
Details of the packing arrangement in the GS structure of (1) are outlined in Fig. 2. Pd(II) cations are aligned along the crystallographic b-axis, with adjacent cations related by the 21 screw axis symmetry. Although the view in Fig. 2 suggests that adjacent nitro-(η1-O2) ligands point towards one another, they are not close in 3-D space due to the ½ translation along the b-direction. Each NO2 group is surrounded on the opposite side by butyl chains within the same cation. The BPh4 anions occupy positions near to the backbone of the triamine ligands, away from the bulky butyl groups. Finally, solvent THF molecules exist in discrete pockets that might reasonably be considered the result of inefficient packing between the other, bulkier components. Void space calculations conducted in Mercury,35 with the THF solvent molecules removed, provide representations of these cavities (Fig. S2†). Using a contact surface calculation (probe radius 1.2 Å, grid spacing 0.1 Å) a total of 13.2% of the unit cell volume is found to comprise THF solvent. A solvent-accessible void calculation (with the same parameters) reduces this percentage to just 2.2%. Void space calculations conducted on the crystal structure as a whole, including THF, suggest no cavities are present in the GS and indicate a close-packed structure. Further quantification of the packing efficiency is possible by calculating a packing coefficient (see ESI† for method details). Using molecular volumes calculated by Hirschfeld Stockholder Partitioning in CrystalExplorer,36,37 a packing coefficient of 0.98 is determined. Finally, three intermolecular close-contact interactions exist between nitro-(η1-O2) and the surrounding crystal structure (Fig. S3†). Any intermolecular interactions involving nitro-(η1-O2) in the GS must necessarily be overcome for linkage isomerism to occur in (1) and literature precedent suggests that strong intermolecular interactions can limit photo-activation.23 However, given that these are all relatively weak intermolecular C–H⋯O contacts, they are not expected to cause a significant barrier to excitation.
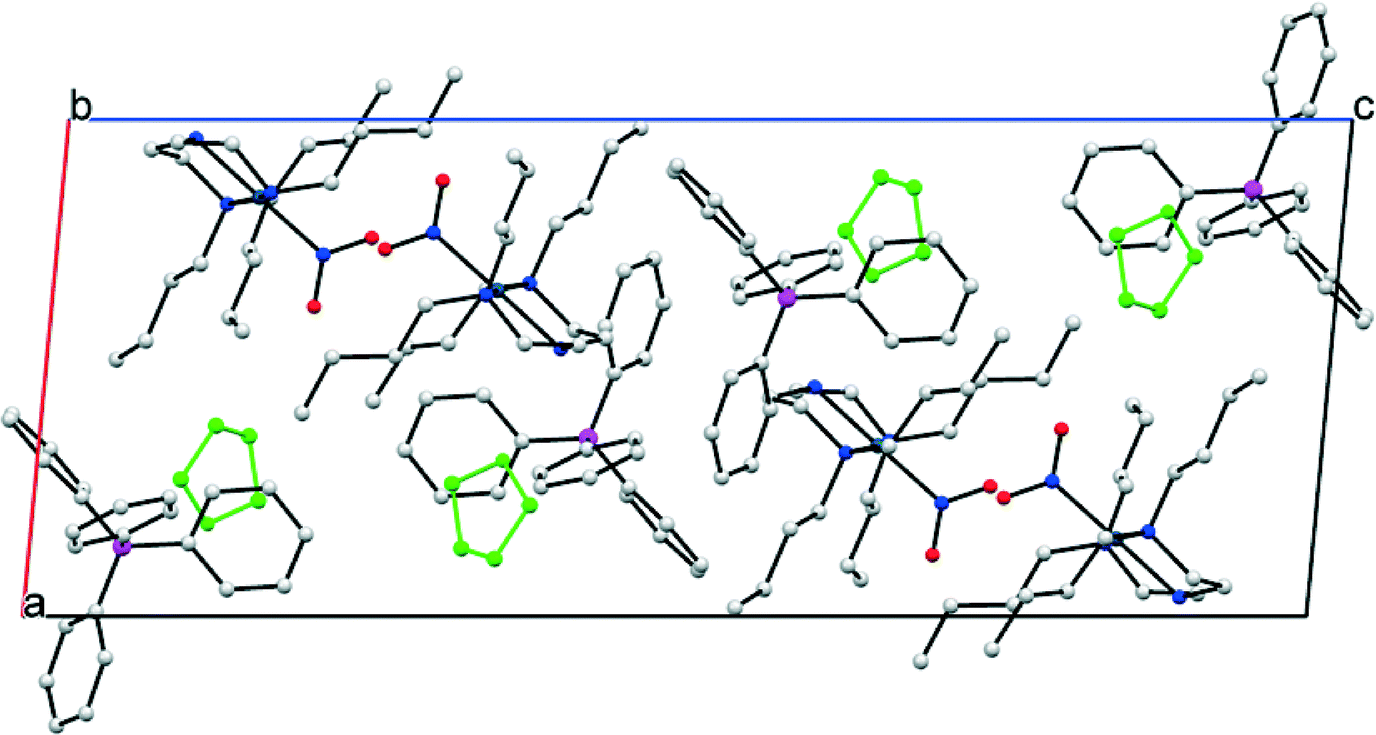 | ||
| Fig. 2 Crystal packing diagram (one unit cell) for the GS structure of (1), before any irradiation. The packing diagram is viewed along the crystallographic b-axis, c-axis horizontal (hydrogen atoms omitted and THF solvent molecules highlighted in green for clarity). | ||
UV/vis spectroscopy
The UV/vis diffuse reflectance spectrum for (1) contains a single, broad maximum centred at approximately 300 nm, with a tail that extends to approximately 470 nm (Fig. S4†). The spectrum indicates that irradiation in the 200–470 nm range might induce a response. A wavelength in the tail of the UV/vis maxima was considered most suitable for excitation in the solid-state, in line with work by Enkelmann, Abdelmoty et al.38,39 As such, a peak LED wavelength of 400 nm was used for all photocrystallographic experiments with (1).Steady-state photocrystallography
NO) isomer. Similar data collections were then conducted for further irradiation times at intervals between 1 and 60 min. The refined GS and MS occupancies are provided in Table S1.†
Fig. 3 shows the plot of endo-nitrito-(η1-NO) occupancy vs. irradiation time, for 0–15 min illumination. Remarkably, 100% photo-conversion is achieved in (1) after just 15 min. To the author's knowledge, this is the fastest reported example of a fully-complete single-crystal-to-single-crystal nitro–nitrito isomerisation reaction. Kinetic parameters for the photo-reaction can be obtained by application of the JMAK theory for solid-state kinetics22,40–45 to the data points in Fig. 3:
| α(t) = 1 − e−ktn | (1) |
NO) occupancy] at a time t, k is the rate constant and n is the Avrami constant. Least-squares fitting of eqn (1) to the data gives values of k = 0.30(2) min−1 (0.0049(3) s−1) and n = 0.98(7).
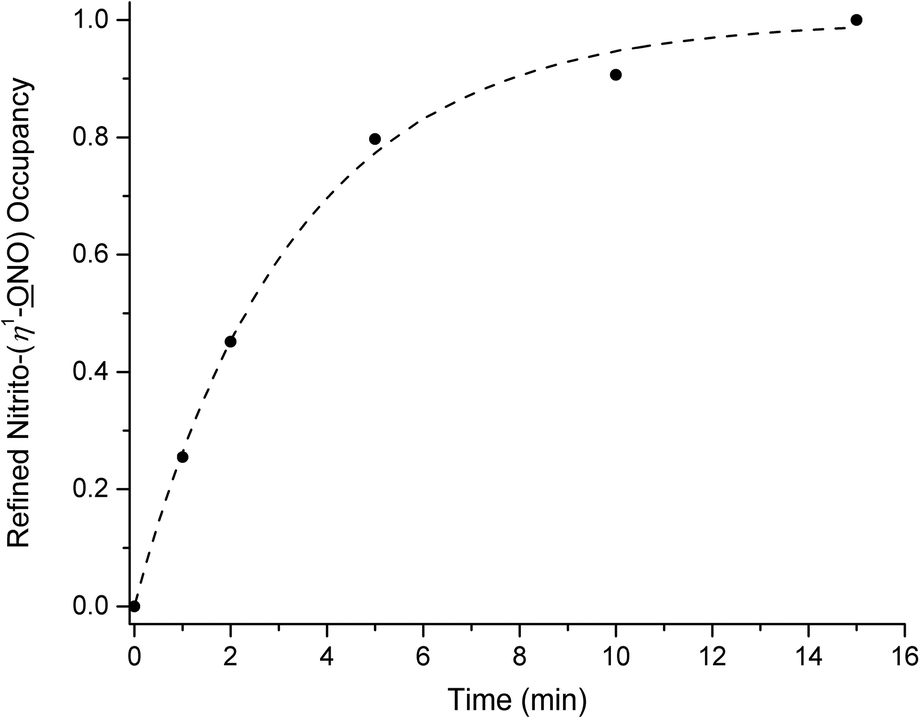 | ||
| Fig. 3 Plot of crystallographically-refined nitrito-(η1-NO) occupancy vs. irradiation time (excitation at λ = 400 nm) for steady-state photocrystallographic experiments with (1) between 0 and 15 min illumination. JMAK kinetic analysis: k = 0.30(2) min−1, (0.0049(3) s−1), n = 0.98(7), R2 = 0.99. | ||
A value of n ≈ 1 indicates a homogeneous spread of nucleation sites for MS growth occurring throughout the crystal lattice.
Extended illumination of the crystal for a total of 60 min induced no further change, indicating that a photostationary state had been reached after 15 min. The photo-excited crystal was held in the dark for a further 1 h and a subsequent data collection showed still no change, proving the excited state is metastable at 100 K. SC-XRD data for the MS structure are given in Table 1.
Variable temperature (VT) studies were then conducted on the same crystal, to determine the temperature range over which the system is metastable. The crystal was heated in situ and paused at intervals to allow for SC-XRD data collections. Full occupancy data for VT studies are provided in Table S2,† with selected data given in Table 2. (1) remains metastable between 100 and 200 K (Table S2†). The onset of MS decay occurs on warming to 220 K, with complete recovery of the GS by 240 K.
O2) and nitrito-(η1-NO) occupancies for variable temperature studies conducted under steady-state and pseudo-steady-state conditions
| Temp/K | Steady-state | Pseudo-steady-state | ||
|---|---|---|---|---|
| (η1-O2) |
(η1-NO) |
(η1-O2) |
(η1-NO) |
|
| 100 | 0.00 | 1.00 | — | — |
| 200 | 0.00 | 1.00 | 0.00 | 1.00 |
| 220 | 0.71 | 0.29 | 0.00 | 1.00 |
| 240 | 1.00 | 0.00 | 0.00 | 1.00 |
| 250 | 1.00 | 0.00 | 0.53 | 0.47 |
| 260 | 1.00 | 0.00 | 1.00 | 0.00 |
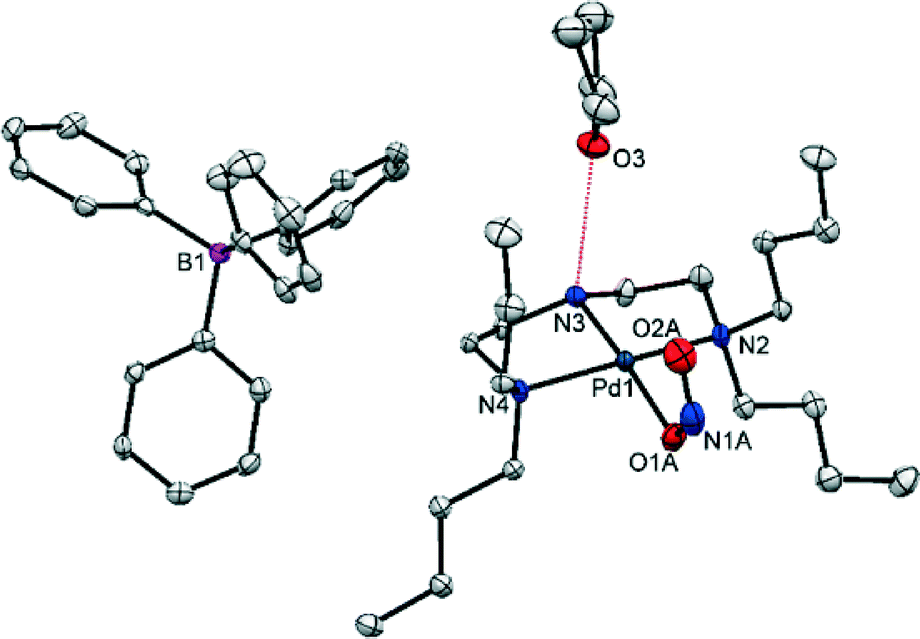 | ||
| Fig. 4 Single-crystal X-ray structure for the asymmetric unit of (1) showing the atomic arrangement in the metastable state (MS) after 1 h irradiation at λ = 400 nm. Ellipsoids set at 50% probability and hydrogen atoms omitted for clarity (red dashed line = hydrogen bonding to THF solvent molecule). | ||
 | ||
| Fig. 5 Overlay of unit cell packing diagrams for the GS (red) and MS (blue) of (1), showing subtle changes in the unit cell as a result of excitation at λ = 400 nm. | ||
There is a small increase in the unit cell volume following 100% excitation, comprising 6.5(4) Å3 or 0.14% of the GS volume. The changes are anisotropic: while the c-axis is found to lengthen by 0.268(1) Å, the a and b axes contract by a smaller amount (Δa = −0.068(1) Å and Δb = −0.037(1) Å). This asymmetry may suggest the changes are a result of the isomerisation, as nitro–nitrito rearrangement occurs approximately along c. However, the reduction in β-angle is the most significant, with Δβ = −0.766(5)°.
Pseudo-steady-state photocrystallography
Pseudo-steady-state experiments were next conducted to ascertain the response in (1) to continuous illumination at temperatures close to the MS limit (200–260 K). As the crystal is “pumped” by LEDs throughout the experiment, the pseudo-steady-state isomer ratio determined at temperatures above the MS limit describes an equilibrium situation between the forward nitro–nitrito photo-excitation and the reverse nitrito–nitro decay processes. A summary of the refined nitro-(η1-O2) and nitrito-(η1-NO) occupancies under continuous LED irradiation (λ = 400 nm) are given in Table 2. In contrast to the VT experiments conducted in the dark, under continuous illumination 100% excitation is maintained up to 240 K and 47% excitation is still determined at 250 K. By 260 K no evidence of the excited state nitrito-(η1-NO) isomer could be determined and the SC-XRD refinement indicated 100% occupancy of the GS nitro-(η1-O2) isomer, in the time-frame of the experiment.
Discussion
Photo-activation level
(1) is the first reported example of complete, 0–100% MS photo-activation in the single-crystal for a Pd(II)–nitrite linkage isomer. Previously, nitrito-(η1-NO) occupancies of 46% and 39%, in [Pd(PPh3)2(NO2)2] and [Pd(AsPh3)2(NO2)2] respectively, were the highest reported MS photo-conversion levels for Pd(II) systems starting from a clean, 100% nitro-(η1-O2) GS.21 A photo-induced, 100% nitrito-(η1-NO) isomer is also reported in [Pd(NO2)(C^N)(PPh3)] (C^N = papaverine) in 2012,28 however this MS arrangement was produced from a GS structure that already contained 77% of the nitrito-(η1-NO) ligand, meaning the maximum GS–MS conversion in this case was 23%.
In light of previous work, the Bu4dien ancillary ligand and BPh4 counterion are chosen specifically for their high steric demands. Analysis of the crystal packing arrangement (Fig. 2, above) confirms that these bulky, photo-inert fragments dominate the structure and outline the reaction cavity within which nitro–nitrito isomerisation can occur. Fig. 6 provides a pictorial representation of the reaction cavity space available for the nitrite ligand in the GS and MS structures. Reaction cavity volumes were determined in Mercury35 by removing the atoms of the nitrite ligand and conducting a contact surface void space calculation. The results of these reaction cavity calculations are given in Table 3. Visually, there is little change in reaction cavity size between the GS and MS arrangements, while the calculations indicate only a very small change in the reaction cavity volume, ΔVc, of +1.46 Å3 (0.05% unit cell volume) per molecule. In line with the Topochemical Postulate outlined by Schmidt and Cohen,2–4 it is expected that solid-state reactions requiring the least change in the crystalline environment will proceed to highest completeness. As such, the requirement for only small ΔVc on excitation is likely to facilitate the high level of photo-activation achieved in (1). Additionally, the nitrite ligand is not involved in strong hydrogen bonds in the GS or MS and only weaker C–H⋯O interactions must be broken and re-formed on excitation (Fig. S3†). As stronger intermolecular contacts are expected to raise the energy barrier to linkage isomerism, this also rationalises the high photo-conversion level in (1).
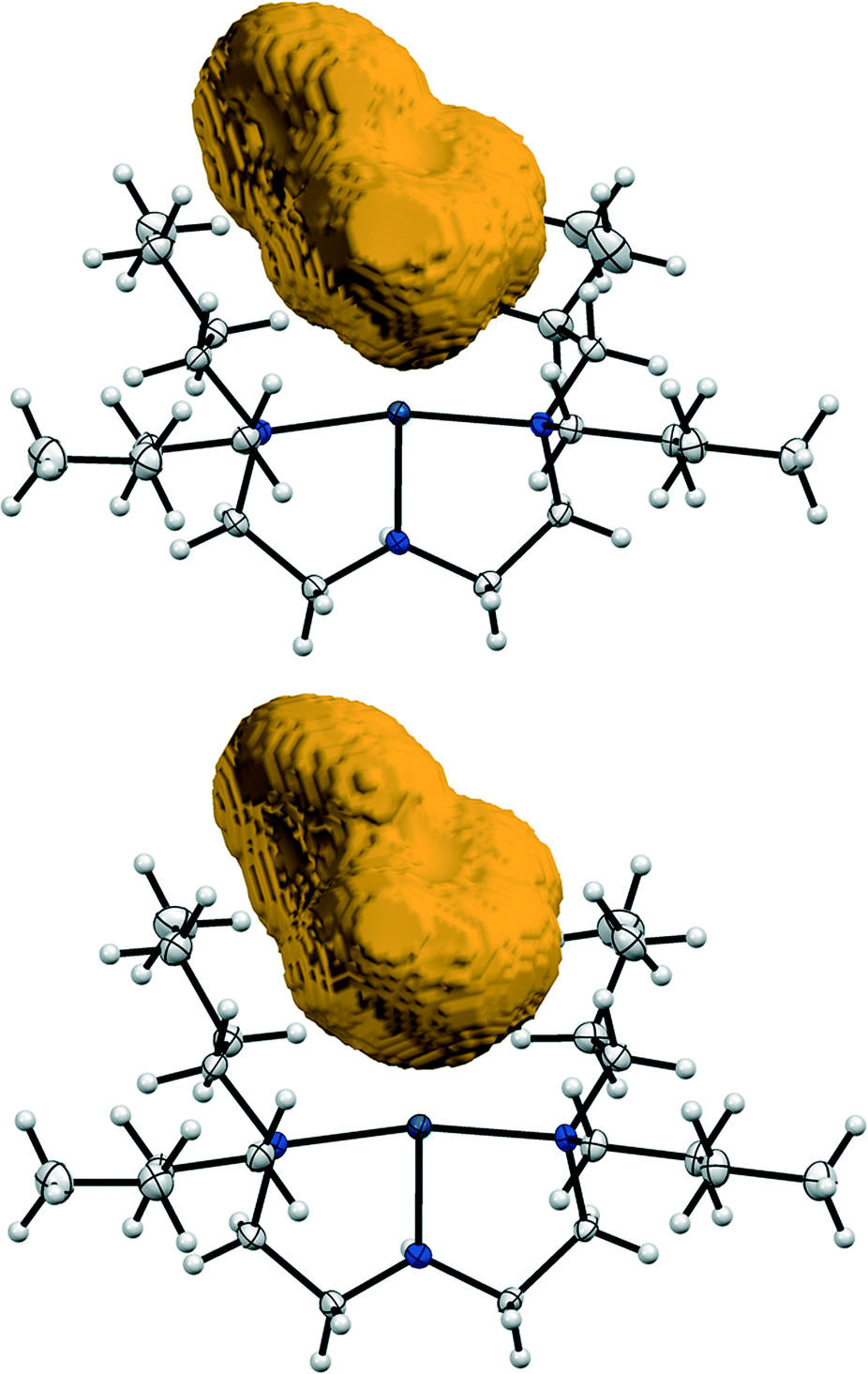 | ||
| Fig. 6 Reaction cavity volumes: top = GS cavity, bottom = MS cavity. Reaction cavity volumes are determined by removing the atoms of the nitrite ligand before conducting a contact surface void calculation in Mercury35 (probe radius 1.2 Å, grid spacing 0.1 Å). | ||
| Cavity size (per unit cell, u.c) | Cavity size/Z (per molecule) | ΔVc (per molecule) | ||||
|---|---|---|---|---|---|---|
| Vol (Å3) | % u.c | Vol (Å3) | % u.c | Vol (Å3) | % u.c | |
| GS | 198.36 | 4.30 | 49.59 | 1.08 | ||
| MS | 204.20 | 4.50 | 51.05 | 1.13 | 1.46 | 0.05 |
In addition to steric factors, the absorption properties of (1) will likely contribute to the overall activation level. The choice of an excitation wavelength in the tail of the UV/vis profile is expected to help maximise conversion.38,39 400 nm light should have sufficient energy to promote a response, but at the same time not be too strongly absorbed in outer layers of the crystal, allowing light to penetrate more evenly throughout the bulk.
Reaction rate and speed of response
The photo-induced linkage isomerisation reaction in (1) reaches completion after just 15 min irradiation. This is remarkably fast, in comparison to the reaction rates reported for similar photo-reactions in single-crystals. Other systems displaying 100% nitro–nitrito conversion, including [Ni(dppe)(NO2)Cl],19 [Ni(dppe)(NO2)2],24 [Ni(dcpe)(NO2)2] (dcpe = bis-dicyclohexyl-phosphinoethane)24 and [Ni(Et4dien)(NO2)2] (Et4dien = N,N,N′,N′-tetraethyldiethylenetriamine)22 achieve maximum conversion in 90 min, 60 min, 60 min and 120 min, respectively. It is possible that the butyl substituents in the triamine co-ligand may facilitate faster linkage isomer switching in (1). These butyl chains are expected to be relatively flexible, and so may help to disappate any strain induced in the lattice as a result of linkage isomerism. This should reduce the nitro–nitrito energy barrier and so promote a faster reaction. Such a hypothesis is difficult to quantify by crystallographic data alone, although qualitative support is gained on considering the shape of the GS and MS reaction cavities (Fig. 6). Although there is little overall change in reaction cavity size between the GS and MS, close inspection shows some subtle variation in the shape of each cavity, particularly in the regions surrounded by the butyl substituents, indicating some degree of flexibility.Metastable limit and near ambient-temperature functionality
Under steady-state conditions, the onset of MS decay in (1) occurs around 220 K and this MS limit is comparable to other reported Pd(II)–nitrite linkage isomers,21 although these do not achieve 100% activation. Other high-converting Ni(II)–nitrite systems generally display lower MS limits. Previous reports highlight the trend for complexes with heavier transition metal (TM) centres to display higher MS limits, suggesting a link between the reduced kinetic lability of the TM(II)–ligand bond with heavier metals and the stability of the MS isomer.21More interestingly, SC-XRD experiments conducted under pseudo-steady-state conditions allow full characterisation of the excited state nitrito-(η1-NO) isomer with 100% occupancy at 240 K (−33 °C) and 47% occupancy as high as 250 K (−23 °C). These temperatures are approaching ambient conditions and are the highest reported operating temperatures for a Pd(II)–nitrite linkage isomer switch. In contrast to the response under continuous illumination, no evidence of the excited state endo-nitrito isomer is found at 240 K and above when the excitation source is removed. As such, complex (1) shows potential as a light-on, functionality-on/light-off, functionality-off molecular switch in the 240–250 K region. Given that the largest difference occurs at 240 K (light on = 100%, light off = 0%) this would likely be the most suitable operating temperature. Before any such device could be considered practicable however, it is important to ascertain the speed of excitation and decay in the crystal at 240 K. Unfortunately, given that the photo-reaction is essentially dynamic at this temperature, it was not possible to conduct full SC-XRD experiments to determine the change in nitro:nitrito ratio with time in this case. Instead, an estimation of the isomerisation reaction progress was made by following the change in unit cell parameters, as this approach required only a few frames of X-ray data per time-point. These kinetic experiments were completed at 240 K for both the excitation (light on) and decay (light off) processes. The results of these experiments are given in Fig. 7. Only the β-angle showed a clear trend under both conditions, which might be expected considering that this is the parameter which changes the most on photo-activation under steady-state conditions (Table 1). As expected, a clear reduction in the β-angle is observed on photo-excitation and a corresponding increase can be seen with excited state decay. Unfortunately, it was not possible to obtain reliable kinetic parameters from these data by least-squares fitting of eqn (1), given the low level of accuracy in the measurements. However, it does appear that both the excitation and decay processes reach completion in approximately 15 min, a similar time to the excitation process followed at 100 K. These experiments provide a strong starting point for time-resolved synchrotron powder X-ray diffraction (PXRD) experiments with (1) at 240 K, for which very fast data collection times would allow many and more accurate data points to be determined for each reaction profile.
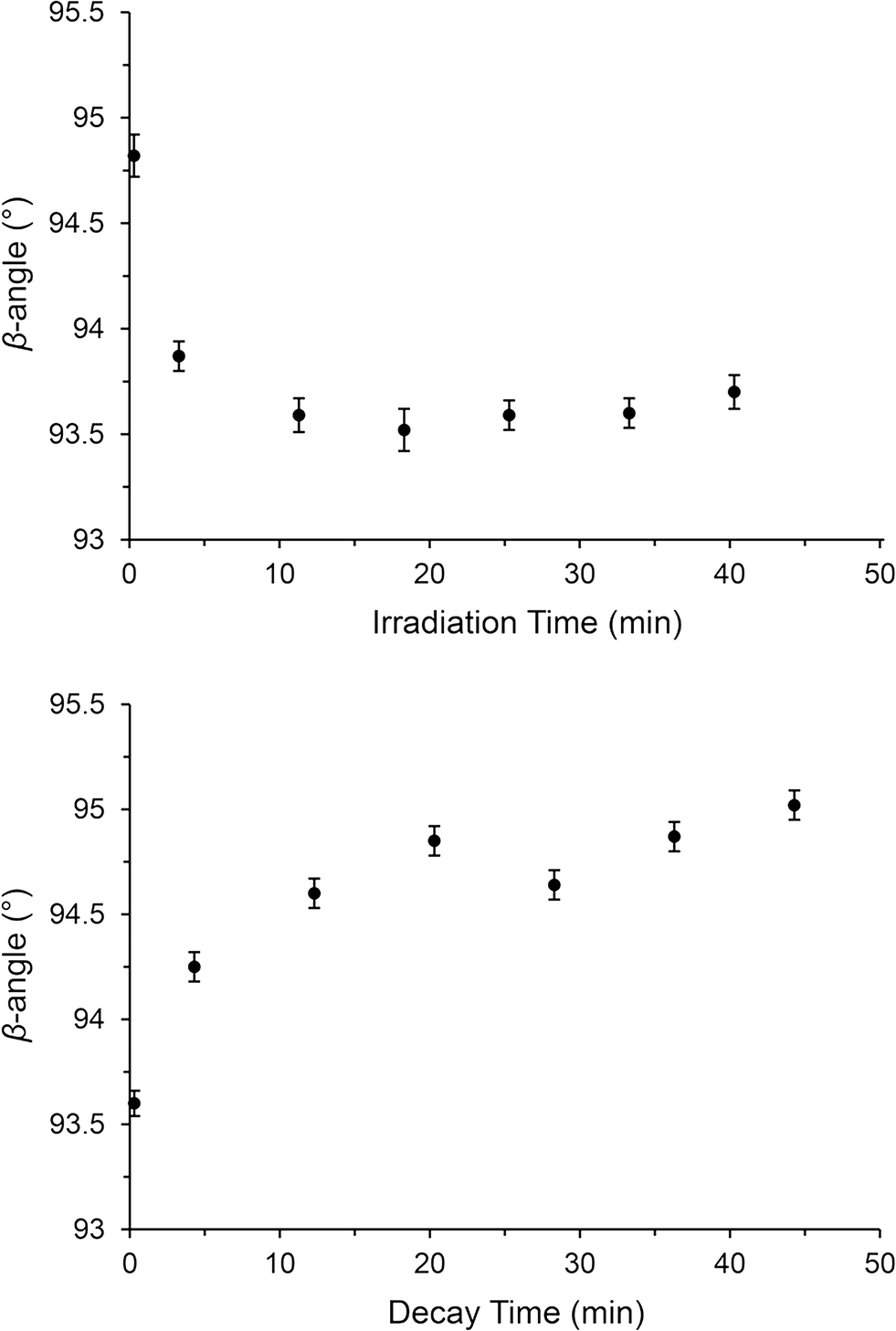 | ||
| Fig. 7 SC-XRD kinetics experiments at 240 K, following the change in β-angle as a result of nitrite linkage isomerism. Top: Plot of β-angle vs. irradiation time under continuous illumination (λ = 400 nm). Bottom: Plot of β-angle vs. decay time in the dark. | ||
Conclusions
The observation of 100%, fully-reversible linkage isomerism in the Pd(II)–nitrite complex (1) is encouraging, as it proves that complete nitro–nitrito conversion in the single-crystal is not limited to complexes containing first-row TM centres. Detailed analysis of the packing effects in both GS and MS structures once again validates the approach of including sterically-demanding ancillary fragments into the crystal system, to facilitate high levels of photo-conversion. In addition, analysis of the size and shape of the reaction cavity provides both a qualitative and quantitative indication of changes in the immediate location of the nitrite ligand as a result of linkage isomer switching, which also supports the hypothesis that bulky photo-inert fragments help the single-crystal-to-single-crystal isomerisation reaction to proceed topotactically.While steady-state photocrystallographic experiments provide important, accurate and detailed information on the MS structure, pseudo-steady-state SC-XRD experiments are equally important to understand the progress of linkage isomer switching at temperatures close to the MS limit. The determination of a photo-excited endo-nitrito-(η1-NO) isomer by SC-XRD as high as 240–250 K in (1) is promising evidence that linkage isomer switching is approaching viable operating temperatures. The result is suggestive that the design of linkage isomer systems containing either heavier TM centres, or less kinetically labile ambidentate ligands, might offer a new approach to maximising MS limits, and further investigation in this area would be profitable.
There remain a number of key challenges in the development of viable solid-state linkage isomer devices and future work with (1) will focus on obtaining more detailed understanding of both the rate of GS–MS switching at relevant temperatures and the reproducibility of this process over extended operating periods.
Acknowledgements
LEH is grateful to the EPSRC for financial support for this project (EP/K004956) and to Prof. Paul Raithby at the University of Bath for continued support and guidance. Additional thanks go to the DySS team at the Research Complex at Harwell, UK for access to UV/vis diffuse reflectance equipment.Notes and references
- J. Zhang, Q. Zou and H. Tian, Adv. Mater., 2013, 25, 378–399 CrossRef CAS PubMed.
- M. D. Cohen and G. M. J. Schmidt, J. Chem. Soc., 1964, 1996–2000 RSC.
- M. D. Cohen, G. M. J. Schmidt and F. I. Sonntag, J. Chem. Soc., 1964, 2000–2013 RSC.
- G. M. J. Schmidt, J. Chem. Soc., 1964, 2014–2021 RSC.
- P. Coppens, I. I. Vorontsov and T. Graber, Acta Crystallogr., Sect. A: Found. Crystallogr., 2005, 61, 162–173 CrossRef PubMed.
- L. E. Hatcher and P. R. Raithby, Coord. Chem. Rev., 2014, 277–278, 69–79 CrossRef CAS.
- P. Coppens, Angew. Chem., Int. Ed., 2009, 48, 4280–4281 CrossRef CAS PubMed.
- M. D. Carducci, M. R. Pressprich and P. Coppens, J. Am. Chem. Soc., 1997, 119, 2669–2678 CrossRef CAS.
- B. Cormary, S. Ladeira, K. Jacob, P. G. Lacroix, T. Woike, D. Schaniel and I. Malfant, Inorg. Chem., 2012, 51, 7492–7501 CrossRef CAS PubMed.
- D. V. Fomitchev, T. R. Furlani and P. Coppens, Inorg. Chem., 1998, 37, 1519–1526 CrossRef CAS.
- P. Coppens, I. Novozhilova and A. Kovalevsky, Chem. Rev., 2002, 102, 861–884 CrossRef CAS PubMed.
- D. V. Fomitchev, P. Coppens, T. Li, K. A. Bagley, L. Chen and G. B. Richter-Addo, Chem. Commun., 1999, 2013–2014 RSC.
- D. V. Fomitchev, K. A. Bagley and P. Coppens, J. Am. Chem. Soc., 2000, 122, 532–533 CrossRef CAS.
- A. Y. Kovalevsky, K. A. Bagley, J. M. Cole and P. Coppens, Inorg. Chem., 2002, 42, 140–147 CrossRef PubMed.
- K. F. Bowes, J. M. Cole, S. L. G. Husheer, P. R. Raithby, T. L. Savarese, H. A. Sparkes, S. J. Teat and J. E. Warren, Chem. Commun., 2006, 2448–2450 RSC.
- S. O. Sylvester, J. M. Cole and P. G. Waddell, J. Am. Chem. Soc., 2012, 134, 11860–11863 CrossRef CAS PubMed.
- S. O. Sylvester and J. M. Cole, J. Phys. Chem. Lett., 2013, 4, 3221–3226 CrossRef CAS.
- S. O. Sylvester and J. M. Cole, Adv. Mater., 2013, 25, 3388 CrossRef CAS.
- M. Warren, S. Brayshaw, A. Johnson, S. Schiffers, P. Raithby, T. Easun, M. George, J. Warren and S. Teat, Angew. Chem., Int. Ed., 2009, 5821–5824 CrossRef.
- L. E. Hatcher, M. R. Warren, D. R. Allan, S. K. Brayshaw, A. L. Johnson, S. Fuertes, S. Schiffers, A. J. Stevenson, S. J. Teat, C. H. Woodall and P. R. Raithby, Angew. Chem., Int. Ed., 2011, 50, 8371–8374 CrossRef CAS PubMed.
- M. R. Warren, S. K. Brayshaw, L. E. Hatcher, A. L. Johnson, S. Schiffers, A. J. Warren, S. J. Teat, J. E. Warren, C. H. Woodall and P. R. Raithby, Dalton Trans., 2012, 41, 13173–13179 RSC.
- L. E. Hatcher, J. Christensen, M. L. Hamilton, J. Trincao, D. R. Allan, M. R. Warren, I. P. Clarke, M. Towrie, D. S. Fuertes, C. C. Wilson, C. H. Woodall and P. R. Raithby, Chem. – Eur. J., 2014, 20, 3128–3134 CrossRef CAS PubMed.
- L. E. Hatcher, E. J. Bigos, M. J. Bryant, E. M. MacCready, T. P. Robinson, L. K. Saunders, L. H. Thomas, C. M. Beavers, S. J. Teat, J. Christensen and P. R. Raithby, CrystEngComm, 2014, 16, 8263–8271 RSC.
- M. R. Warren, T. L. Easun, S. K. Brayshaw, R. J. Deeth, M. W. George, A. L. Johnson, S. Schiffers, S. J. Teat, A. J. Warren, J. E. Warren, C. C. Wilson, C. H. Woodall and P. R. Raithby, Chem. – Eur. J., 2014, 20, 5468–5477 CrossRef CAS PubMed.
- E. V. Boldyreva, Solid State Ionics, 1997, 101–103(Part 2), 843–849 CrossRef CAS.
- Y. Ohashi, Crystallogr. Rev., 2013, 19, 2–146 CrossRef CAS.
- A. J. Blake, N. R. Champness, T. L. Easun, D. R. Allan, H. Nowell, M. W. George, J. Jia and X.-Z. Sun, Nat. Chem., 2010, 2, 688–694 CrossRef CAS PubMed.
- S. E. Bajwa, T. E. Storr, L. E. Hatcher, T. J. Williams, C. G. Baumann, A. C. Whitwood, D. R. Allan, S. J. Teat, P. R. Raithby and I. J. S. Fairlamb, Chem. Sci., 2012, 3, 1656–1661 RSC.
- J. M. Cole, K. Y. M. Yeung, G. Pace, S. O. Sylvester, D. Mersch and R. H. Friend, CrystEngComm, 2015, 17, 5026–5031 RSC.
- R. D. Feltham, G. Elbaze, R. Ortega, C. Eck and J. Dubrawski, Inorg. Chem., 1985, 24, 1503–1510 CrossRef CAS.
- Rigaku Oxford Diffraction, CrysAlisPro Software system, version 1.171.37.31, Rigaku Corporation, Oxford, UK, 2015 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- S. K. Brayshaw, J. W. Knight, P. R. Raithby, T. L. Savarese, S. Schiffers, S. J. Teat, J. E. Warren and M. R. Warren, J. Appl. Crystallogr., 2010, 43, 337–340 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- J. J. McKinnon, A. S. Mitchell and M. A. Spackman, Chem. – Eur. J., 1998, 4, 2136–2141 CrossRef CAS.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627–668 CrossRef PubMed.
- V. Enkelmann, G. Wegner, K. Novak and K. B. Wagener, J. Am. Chem. Soc., 1993, 115, 10390–10391 CrossRef CAS.
- I. Abdelmoty, V. Buchhoz, L. Di, C. Guo, K. Kowitz, V. Enkelmann, G. Wegner and B. M. Foxman, Cryst. Growth Des., 2005, 5, 2210–2217 CAS.
- M. Avrami, J. Chem. Phys., 1939, 7, 1103–1112 CrossRef CAS.
- M. Avrami, J. Chem. Phys., 1940, 8, 212–224 CrossRef CAS.
- J. B. Benedict and P. Coppens, J. Phys. Chem. A, 2009, 113, 3116–3120 CrossRef CAS PubMed.
- R. More, G. Busse, J. Hallmann, C. Paulmann, M. Scholz and S. Techert, J. Phys. Chem. C, 2010, 114, 4142–4148 CAS.
- D.-K. Cao, T. V. Sreevidya, M. Botoshansky, G. Golden, J. B. Benedict and M. Kaftory, J. Phys. Chem. A, 2010, 114, 7377–7381 CrossRef CAS PubMed.
- A. G. Jarvis, H. A. Sparkes, S. E. Tallentire, L. E. Hatcher, M. R. Warren, P. R. Raithby, D. R. Allan, A. C. Whitwood, M. C. R. Cockett, S. B. Duckett, J. L. Clark and I. J. S. Fairlamb, CrystEngComm, 2012, 14, 5564–5571 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1441774-1441792 contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce02434j |
| This journal is © The Royal Society of Chemistry 2016 |