 Open Access Article
Open Access ArticleBiomimetic zinc chlorin–poly(4-vinylpyridine) assemblies: doping level dependent emission–absorption regimes†
Ville
Pale‡
a,
Taru
Nikkonen‡
b,
Jaana
Vapaavuori
c,
Mauri
Kostiainen
c,
Jari
Kavakka
b,
Jorma
Selin
a,
Ilkka
Tittonen
*a and
Juho
Helaja
*b
aDepartment of Micro and Nanosciences, Aalto University, P.O. Box 13500, FI-00076 Aalto, Finland. E-mail: ilkka.tittonen@aalto.fi; Fax: +35 8207227012; Tel: +35 8405437564
bLaboratory of Organic Chemistry, Department of Chemistry, University of Helsinki, P.O. Box 55, FI-00014, Finland. E-mail: juho.helaja@helsinki.fi; Fax: +35 8919150466; Tel: +35 8919150430
cDepartment of Applied Physics, Aalto University, P.O. Box 15100, FI-00076 Aalto, Finland
First published on 29th January 2013
Abstract
To develop biomimetic dye–polymers for photonics, two different types of Zn chlorin–poly(4-vinylpyridine) (P4VP) assemblies were prepared by varying Zn pyro-pheophorbide a methylester (ZnPPME) and Zn 31-OH-pyro-pheophorbide a methylester (Zn-31-OH-PPME) doping levels. 1H NMR spectroscopy and diffusion ordered NMR spectroscopy (DOSY) studies revealed that a coordinative interaction between Zn chlorin and P4VP was predominant in solution (d5-nitrobenzene). Small angle X-ray scattering (SAXS) and transmission electron microscopy (TEM) characterization of bulk samples of polystyrene-block-poly(4-vinylpyridine) (PS-b-P4VP) doped with variable amounts of Zn chlorin showed that the pigment doping transformed the native cylindrical block copolymer nanostructures to lamellar morphologies. The result indicates that the pyridine moiety–Zn chlorin coordination is stronger than the aggregation tendency between the pigment molecules even in the solid state. UV-Vis absorption spectroscopy studies of a Zn chlorin–P4VP thin film showed characteristic monomeric chlorin spectra, while steady-state fluorescence spectroscopy displayed quenching of fluorescence and time-resolved studies indicated shortening of fluorescence lifetimes with an increasing chlorin doping level. Notably, time-resolved fluorescence spectroscopy revealed that the lifetime decay changed from monoexponential to biexponential above 0.5 wt% (ca. 0.001 equiv.) loadings. The Förster analysis implies that excitonic chlorin–chlorin interactions are observed in the thin films when the distance between the pigment molecules is approximately 50 Å. The Zn chlorin–P4VP solid films emit strongly up to 1 wt% (ca. 0.002 equiv.) doping level above which the chlorin–chlorin interactions start to linearly dominate with an increase of doping level, while with 10 wt% (ca. 0.02 equiv.) loading less than 10% of fluorescence remains. Doping levels up to 300 wt% (0.5 equiv.) can be used in absorbing materials without the formation of chlorin aggregates. These defined optical response regions pave the way for photonic materials based on biopigment assemblies.
Introduction
Dye-doped organic polymeric materials have significant applications in functional materials and optoelectronics, such as organic electronics, organic light-emitting diodes (OLEDS) and organic solar cells, sensors, optical storage media and organic lasers.1–9 The advantages of these materials are their structural versatility, low cost and ease of fabrication, as well as a strong and fast response to light stimuli.4 Various polymers have been used as hosts in non-covalent anchoring of the guest dye molecules into the polymer matrix.8 The main advantages in the supramolecular approach are the avoidance of synthetic steps required in covalent matrix build-up as well as easily adjustable doping levels. However, the typical strong aggregation tendency of dyes is a major drawback in a non-covalent approach, which in practice often sets upper limits for usable doping levels. In turn, especially for thin films, low optical density reduces the efficiency of the systems, which is critical, e.g., for organic lasers.10 Therefore, supramolecular materials in which the host (e.g. polymer)–guest assembly overcomes aggregative dye–dye interactions are highly desirable. Existing doping dyes cover a wide range of conjugated molecules, aromatics and heterocycles.11 Nevertheless, biological chromophores integrated in polymers are rare in photonic and optoelectronic applications, even though biological systems have been extensively studied in non-polymeric solar cell applications.12 We were interested in preparing and studying chlorophyll based supramolecular dye materials, which would have functional similarities with their biological counterparts.In the photosynthetic systems of green plants and algae, chlorophylls (Chl) function in light absorption, excitation energy transfer and charge separation processes.13 A single Chl pigment can act in any of these roles depending on the molecular environment and the mutual distance between the chromophores. In photosynthetic protein complexes the Chl’s are precisely organized by a protein scaffold to fulfill these tasks. One crucial interaction is the coordination between the central metal (Mg) of Chl and protein amino acid residue. Fig. 1a depicts a fragment of light-harvesting complex II (LHC-II)14 associated with Photosystem II, in which the Mg's of the Chl's are coordinated by various electron pair donors of different amino acid residues. Overall, the pigment assembly is seemingly random; the distances and angles between the Chl planes vary substantially, and as a whole the system has a high degree of complexity. Nevertheless, one distinctive feature of LHC-II is the lack of any intermolecular coplanar plane to plane interactions.
![(a) Fragment (amino acid residue numbers 215–163 from left to right) of Spinach light-harvesting complex (LHC II). Chl's bound to Mg atoms are coordinated by various electron pair donors such as the imidazole nitrogen of histidine (His or H, residue 212), amide oxygens of glutamine (Gln or Q, residue 197) and asparagine (Asn or N, residue 183) and carboxylate oxygen of glutamic acid (Glu or E, residue 180). The figure was produced with a Discovery Studio protein modeling program using the Protein Data Bank coordinates [PDB accession number 1RWT]. (b) Sketch of self-assembled aggregates formed by Mg–oxygen electron pair coordination and hydrogen bonding in BChl. (c) Illustration of Zn–pyridine interaction between Zn chlorin and P4VP.](/image/article/2013/TC/c3tc00499f/c3tc00499f-f1.gif) | ||
| Fig. 1 (a) Fragment (amino acid residue numbers 215–163 from left to right) of Spinach light-harvesting complex (LHC II). Chl's bound to Mg atoms are coordinated by various electron pair donors such as the imidazole nitrogen of histidine (His or H, residue 212), amide oxygens of glutamine (Gln or Q, residue 197) and asparagine (Asn or N, residue 183) and carboxylate oxygen of glutamic acid (Glu or E, residue 180). The figure was produced with a Discovery Studio protein modeling program using the Protein Data Bank coordinates [PDB accession number 1RWT]. (b) Sketch of self-assembled aggregates formed by Mg–oxygen electron pair coordination and hydrogen bonding in BChl. (c) Illustration of Zn–pyridine interaction between Zn chlorin and P4VP. | ||
In contrast to that mentioned above, in green sulphur bacteria the chlorosomal antennae structures are formed by self-assembly of chlorosomal chlorophylls without any protein guidance.15,16 While the former protein complexes are beyond artificial assembly, there are several examples of how to mimic a chlorosomal J-aggregate assembly by chlorophyll derivatives or even with porphyrins.17,18 It has been established that suitably functionalized metallochlorins and porphyrins exhibit a tendency to form self-assembled aggregates essentially by metal–oxygen electron pair coordination (Fig. 1b).19 In these tightly packed aggregates the distances of the chromophores are small, and hence the excitonic couplings are strong between the molecules. In this strong coupling regime excitation energy is delocalized over multiple chromophores resulting in an efficient coherent energy transport that can enable close to unity quantum efficiencies in energy transfer processes.20–22
In a pioneering chlorophyll–P4VP investigation, Seely has demonstrated that chlorophylls (a and b) remain in a monomeric form in solution with the aid of the polymer. This finding was evidenced by the measured absorbance and fluorescence spectra in nitromethane solution.23 Recently, we found out that Zn chlorins also tend to bind tightly with pyridine hosts in solution.24,25 It has been shown in several solution studies that metalloporphyrins have an affinity to P4VP moieties26 as well as to other pyridines including supramolecular hosts.27–30 Yet this topic has not been extensively studied in the solid state until very recently; Krishnamoorthy and coworkers31 have prepared Zn porphyrin–P4VP thin films for field effect transistors (FETs) demonstrating that the porphyrin pigment becomes evenly distributed into the polymer matrix even with high porphyrin–pyridine moiety ratios.
In this work, we investigate the structural and optical properties of non-covalent supramolecular Zn chlorin–P4VP assemblies in solution and solid state thin films as schematically illustrated in Fig. 1c. For this purpose we have chosen Zn pyro-pheophorbide a methylester (ZnPPME) and Zn 31-OH-pyro-pheophorbide a methylester (Zn-31-OH-PPME), which are chemically rather stable and straightforward to derivatise from algae isolated pigments.
Experimental section
General
All the reactions were performed under argon in the dark using standard Schlenk techniques. 1H and DOSY NMR spectra were recorded at 27 °C using a Varian INOVA 500 MHz Spectrometer. The DOSY spectra were measured using a Bipolar Pulse Pair Stimulated Echo (BPPSTE) pulse sequence.32 The small angle X-ray scattering (SAXS) measurements were performed at the Dutch–Belgian beamline (BM26) of the European Synchrotron Radiation Facility in Grenoble using a beam of 10 keV with an area of 0.35 × 0.5 mm2 at the sample position. One-dimensional SAXS data were obtained by azimuthally averaging the 2D scattering data. The magnitude of the scattering vector was given by: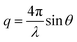 , where 2θ is the scattering angle. Bright-field TEM was performed on a FEI Tecnai 12 transmission electron microscope operating at an accelerating voltage of 120 kV. The absorption and fluorescence spectra of the polymer–dye thin films were measured with a Lambda 950 (Perkin-Elmer) UV-Vis spectrometer and a Cary Eclipse (Varian) fluorescence spectrometer, respectively. For the fluorometer, the used photomultiplier voltage was 800 V and the monochromator entrance and exit slits were 5 nm in all the measurements. In the time-resolved measurements, a frequency-doubled mode-locked Ti:sapphire laser (Coherent, Mira; FWHM = 150 ps, pulse repetition rate 76 MHz, λexc = 410 nm, average excitation intensity 5 W cm−2) was used to probe the samples. For the detection, a Peltier-cooled microchannel plate photomultiplier tube (Hamamatsu: Vbias = 2.7 kV, acquisition time 45 s) along with single-photon counting electronics was used. The used scanning electron microscope (SEM) was Helios Nanolab 600 (FEI).
, where 2θ is the scattering angle. Bright-field TEM was performed on a FEI Tecnai 12 transmission electron microscope operating at an accelerating voltage of 120 kV. The absorption and fluorescence spectra of the polymer–dye thin films were measured with a Lambda 950 (Perkin-Elmer) UV-Vis spectrometer and a Cary Eclipse (Varian) fluorescence spectrometer, respectively. For the fluorometer, the used photomultiplier voltage was 800 V and the monochromator entrance and exit slits were 5 nm in all the measurements. In the time-resolved measurements, a frequency-doubled mode-locked Ti:sapphire laser (Coherent, Mira; FWHM = 150 ps, pulse repetition rate 76 MHz, λexc = 410 nm, average excitation intensity 5 W cm−2) was used to probe the samples. For the detection, a Peltier-cooled microchannel plate photomultiplier tube (Hamamatsu: Vbias = 2.7 kV, acquisition time 45 s) along with single-photon counting electronics was used. The used scanning electron microscope (SEM) was Helios Nanolab 600 (FEI).
Materials
All the solvents used in the syntheses, sample preparation and measurements were obtained as HPLC quality and used as received. All commercial polymers were used without further purification. Poly(4-vinylpyridine) (P4VP, Mw ∼ 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) used in NMR measurements was purchased from Sigma Aldrich. Polystyrene-block-poly(4-vinylpyridine) (PS-b-P4VP, Mw = 47600–20900 g mol−1) used for TEM and SAXS measurements and poly(4-vinylpyridine) (P4VP Mw = 5100 g mol−1) used for the optical measurements were supplied by Polymer Source, Inc. The glass wafers which were used as substrates in spin coating were purchased from Menzel-Gläser.
000 g mol−1) used in NMR measurements was purchased from Sigma Aldrich. Polystyrene-block-poly(4-vinylpyridine) (PS-b-P4VP, Mw = 47600–20900 g mol−1) used for TEM and SAXS measurements and poly(4-vinylpyridine) (P4VP Mw = 5100 g mol−1) used for the optical measurements were supplied by Polymer Source, Inc. The glass wafers which were used as substrates in spin coating were purchased from Menzel-Gläser.
Synthesis
Chlorophyll a was extracted and purified from Spirulina pacifica and converted into pyro-pheophorbide a methylester (1),33 which was used as a starting material for the syntheses. Zn pyro-pheophorbide a methylester24,25 and Zn 31-OH-pyro-pheophorbide a methylester (31-epimeric mixture, (31R)/(31S) = 1/1)34 (Fig. 2) were synthesized according to literature procedures and their NMR spectra were in agreement with the published data.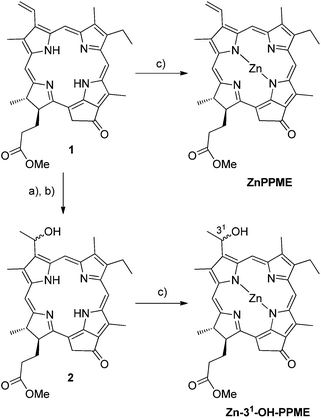 | ||
| Fig. 2 (a) HBr/AcOH, (b) H2SO4/MeOH and (c) Zn(OAc)2/MeOH-DCM. | ||
Samples for small angle X-ray scattering (SAXS)
Samples were prepared by dissolving the polymer and chlorin in chloroform yielding homogeneous mixtures with varying compositions. The solutions were stirred for 24 h, after which the solvent was slowly evaporated at room temperature. The samples were then dried under vacuum at 30 °C and high-vacuum annealed for several days.Samples for transmission electron microscopy (TEM)
The bulk samples prepared as described above were embedded in epoxy and thin sections (∼60 to 70 nm) were microtomed at room temperature using a Leica Ultracut UCT-ultramicrotome and a Diatome diamond knife. Samples were stained in I2 vapor for several hours in order to improve contrast.Samples for optical measurements
Standard solutions of P4VP (5 wt%) and Zn chlorin (0.1 wt%) were prepared using THF as the solvent. The required amounts of the P4VP and Zn chlorin standard solutions were mixed together in order to obtain polymer–dye assemblies with variable doping values (from 0.1 wt% up to 12 wt% for fluorescence measurements and higher doping levels for absorption measurements). These solutions were left in the dark to stir for 24 h. The Zn chlorin–P4VP solutions were then spin-coated on a glass substrate that was cleaned using acetone, isopropanol and DI water, after which the samples were heated for 2 h at 80 °C in an oven to remove any residual solvent.Results and discussion
The nature of the interaction between ZnPPME and P4VP was studied in solution using 1H NMR spectroscopy. The measurements were performed in a weakly coordinating solvent, d5-nitrobenzene, to make coordination favourable between Zn and the pyridine electron pair. Fig. 3 shows that in the 1H NMR spectrum, chlorin ring methine proton resonances are shifted downfield and clearly broadened after the addition of ∼3 equiv. P4VP (molar amount calculated from the amount of pyridine units in P4VP). Correspondingly, the signals for pyridine protons are shifted upfield due to being exposed to the aromatic ring current of the chlorin ring. Together these effects indicate that the chlorin units are in close interaction with the polymer in a similar manner to the one we have recently measured for tightly bound pyridine derivative–Zn chlorin complexes.24,25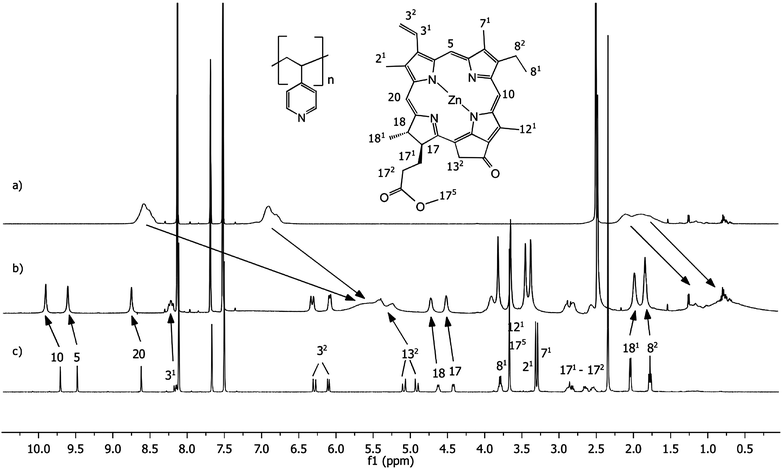 | ||
| Fig. 3
1H NMR spectra of (a) P4VP (polymer 52.6 μM; the molarity of pyridine units in the polymer backbone is 30 mM), (b) 1:3 mixture of ZnPPME (10 mM) and P4VP (polymer 52.6 μM; the molarity of pyridine units in the polymer backbone is 30 mM), (c) ZnPPME (10 mM) measured in d5-nitrobenzene. | ||
In addition to the 1H NMR studies, diffusion ordered NMR spectroscopy (DOSY) provided us with a convenient tool to probe the formation of the ZnPPME–P4VP assembly, since the method provides information about molecular mobility in solution.35 In DOSY experiments, spectra of compounds in the sample are separated according to their diffusion coefficients, which are proportional to their hydrodynamic volumes. Hence, any monomeric small molecule, when binding to a macromolecule, will experience a significant decrease in diffusion rate. The separate DOSY measurements of ZnPPME and P4VP in d5-nitrobenzene yielded translation diffusion coefficients (D values) of 2.1 × 10−9 m2 s−1 and 0.3 × 10−9 m2 s−1, respectively.† The obtained values indicate high mobility for the chlorin and sluggish motion for the polymer. The addition of ⅓ equivalents (with respect to pyridine units) of ZnPPME into the polymer mixture gave approximately the same D value, 0.3 × 10−9 m2 s−1, for the ZnPPME–P4VP assembly as for P4VP alone. This result shows that in solution, P4VP is capable of tight coordinative Zn chlorin binding even with rather high pigment loading.
In order to probe how pyridine coordination can be utilized in the construction of solid material assemblies, we used a PS-b-P4VP diblock copolymer to direct the assembly of ZnPPME. Our main motivation herein was to find out how well the pyridine coordination can compete with the Zn chlorin self-aggregation tendency with high pigment loadings and whether the amorphous material becomes organized and has a homogeneous character. The pure block copolymer without ZnPPME self-assembles into a cylindrical nanostructure, where P4VP alone forms hexagonally packed cylinders in a PS matrix (Fig. 4a). The small angle X-ray (SAXS) scattering curve from a bulk sample shows the most prominent Bragg peak at q* = 0.0132 Å−1 corresponding to the reflection from the (hk) (10) plane and a calculated (2π/q*) long period (Lp) of 48 nm. Multiple less intense reflections with peak position ratios qn/q* ≈ √3:√7:√9 are also observed indicating a hexagonally packed cylindrical structure, which is also clearly visible in the transmission electron microscopy (TEM) image. A cylindrical nanostructure is to be expected as the P4VP weight fraction is 0.31.36 When Zn chlorin (0.5 equiv. per pyridine) is added to the polymer, a clear change in the structure morphology from cylindrical to lamellar is observed (Fig. 4b). The ZnPPMEs can be selectively coordinated by the pyridine moieties, thus increasing the volume fraction of the P4VP block to yield a lamellar morphology. The SAXS curve shows the first peak at a magnitude of the scattering vector q* = 0.0148 Å−1 and multiple reflections at 2q*, 3q* and 4q* corresponding to a lamellar morphology with Lp = 42 nm. The change in morphology can also be clearly observed by TEM, which now shows a lamellar morphology.
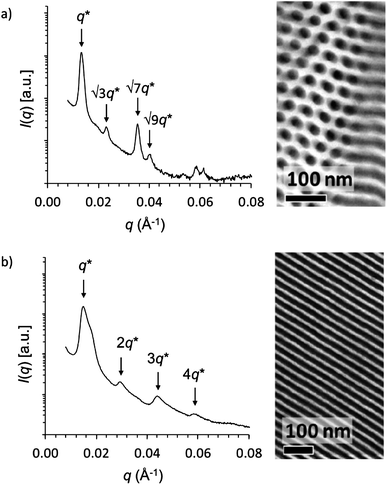 | ||
| Fig. 4 Small angle X-ray scattering and transmission electron microscopy data of (a) PS-b-P4VP(ZnPPME)0 and (b) PS-b-P4VP(ZnPPME)0.5 showing cylindrical and lamellar morphologies, respectively. | ||
Even with 1.0 equiv. loading of Zn chlorin (with respect to pyridine units), the SAXS data indicate a dominant lamellar morphology.† Semiempirical molecular modelling of a P4VP fragment with the PM6 method confirms that a full, 1.0 equiv., loading level would be spatially favourable, i.e., geometry optimized systems show that each pyridine unit coordinates to ZnPPME.†
With the knowledge of tight coordinative binding at hand, the next stage was to study the influence of doping level on the optical properties. The Zn chlorin–P4VP assemblies (ZnPPME–P4VP and Zn-31-OH-PPME–P4VP) were spun on glass substrates. Scanning electron microscope (SEM) and ellipsometer measurements showed comparatively smooth surfaces, with the average thickness being ca. 200 nm for the spun films.† The UV-vis absorption spectra of Zn chlorin aggregates and Zn chlorin–P4VP complex films are presented in Fig. 5a and b. Distinctively, the Zn chlorins alone form aggregates, the UV-vis spectra of which display characteristic line broadening and strong bathochromic shifts for the Qy band. For ZnPPME and Zn-31-OH-PPME, the Qy band absorption shifts from the monomer to aggregate are 666 → 677 and 655 → 699 nm, respectively. In the presence of P4VP the aggregation is prevented with 0.5 equiv. doping of ZnPPME, while the increase of the chlorin doping level induced slight bathochromic shifts for the Soret and Qy bands. Similarly, the corresponding doping of P4VP with Zn-31-OH-PPME resists aggregation up to 0.5 equiv. doping level, while with 1 equiv. the spectrum shows clearly some characteristics of aggregation. Overall, the absorption spectra prove that with the assistance of P4VP, Zn chlorins display characteristic monomeric absorption spectra up to 0.5 equiv. chlorin loadings, i.e., spectra without notable aggregation characteristics.
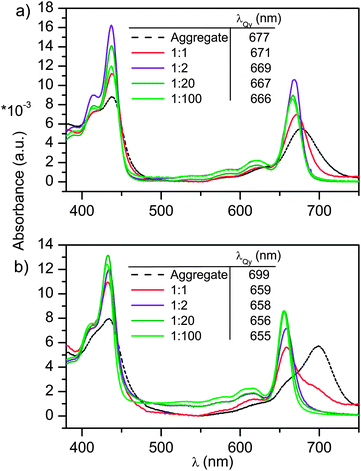 | ||
| Fig. 5 The normalized absorption spectra of (a) P4VP(ZnPPME) and (b) P4VP(Zn-31-OH-PPME) with different dye loadings. The black broken line shows the reference spectra of polymer-free chlorin aggregates. The doping level is given as equiv. (with respect to the pyridine units) of the dye in the polymer. | ||
Fluorescence properties of the spin-coated thin films were studied to find out how solid-state chlorin emission depends on the doping level. As a default in the solid state, plain chlorophyll compounds have a strong tendency to form aggregates which exhibit very short excited state lifetimes, and therefore negligible fluorescence.37 The steady-state fluorescence spectra of spin coated Zn chlorin–P4VP thin films studied with variable chlorin doping levels show expectedly that higher dye concentrations lead to stronger excitonic chlorin–chlorin interactions and thus to the quenching of fluorescence (Fig. 6a and b). Additionally, for both dyes the emission maximum wavelength displays a slight bathochromic shift (ca. 2 nm) with increasing pigment concentration. The quenching strength for ZnPPME and Zn-31-OH-PPME in P4VP assemblies was quantified from Stern–Volmer type plots; the fluorescence intensity normalized with the initial fluorescence intensity F0 was plotted as a function of dye loading. The Stern–Volmer plot in Fig. 6c shows that fluorescence quenches linearly with increasing dye loading; roughly 50% of the emission is quenched at 4 wt% loading, while 10 wt% quenches 80% of the emission. Nevertheless, at high doping levels of 6–10 wt% the quenching becomes increasingly more efficient for Zn-31-OH-PPME–P4VP, indicating that the 31-hydroxyl group starts to compete with P4VP, leading to enhanced excitonic chlorin–chlorin interactions.
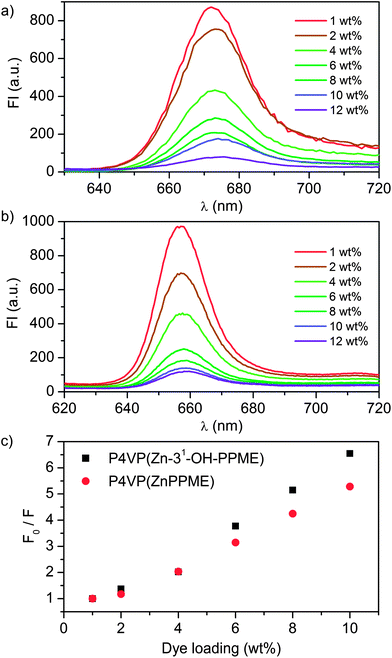 | ||
| Fig. 6 The fluorescence spectra of (a) P4VP(ZnPPME) (λexc = 437 nm) and (b) P4VP(Zn-31-OH-PPME) (λexc = 432 nm) with variable dye loadings. (c) Stern–Volmer type plots of P4VP(ZnPPME) and P4VP(Zn-31-OH-PPME) assemblies display quenching of fluorescence upon increase of dye loading. Values obtained for P4VP(ZnPPME) at λem = 671 nm and for P4VP(Zn-31-OH-PPME) at λem = 660 nm. | ||
Additionally, time-resolved fluorescence measurements were carried out to study the connection between the fluorescence lifetime and the doping level. The measured decay of the fluorescence intensity was fitted using a double-exponential function in eqn (1)
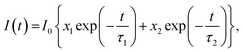 | (1) |
| <τ> = x1τ1 + x2τ2, | (2) |
The results of the calculated fluorescence lifetimes are presented in Table 1 and plotted in Fig. 7 as a function of dye loading. In accordance with the fluorescence intensity data, averaged fluorescence lifetimes 〈τ〉 decrease when the dye loading increases. This, together with the observed fluorescence intensity quenching, indicates that the quenching mechanism is purely dynamic. The quenching results from the energy transfer between identical molecules, also referred to as homo-FRET. Analysis of the fluorescence lifetimes with low dye loadings (up to 0.5 wt%) reveals that the fluorescence intensity decay exhibits essentially monoexponential decay (Table 1). This is explained by the large relative separation of the chromophores, low loadings resulting in a weak coupling between the molecules. Interestingly, when the dye loading is increased, the fluorescence intensity starts to show a biexponential decay, indicating a pronounced intermolecular interaction between the chromophores. The change from mono- to biexponential decay takes place between 0.5 and 1.0 wt% doping levels, indicating that above this region the chlorin–chlorin interaction based quenching mechanism starts to dominate.
| Wt% | τ 1, ns (x1) | τ 2, ns (x2) | 〈τ〉, ns |
|---|---|---|---|
| P4VP(ZnPPME) | |||
| 0.1 | 3.68 (98%) | 0.45 (2%) | 3.62 ± 0.12 |
| 0.25 | 3.63 (99%) | 0.72 (1%) | 3.61 ± 0.16 |
| 0.5 | 3.14 (94%) | 0.85 (6%) | 3.00 ± 0.26 |
| 1 | 1.78 (67%) | 0.69 (33%) | 1.42 ± 0.07 |
| 2 | 1.31 (84%) | 0.46 (16%) | 1.17 ± 0.16 |
| 4 | 0.36 (60%) | 0.13 (40%) | 0.27 ± 0.01 |
| 6 | 0.37 (80%) | 0.12 (20%) | 0.32 ± 0.02 |
| 8 | 0.30 (69%) | 0.08 (31%) | 0.23 ± 0.03 |
| 10 | 0.10 (97%) | 0.50 (3%) | 0.11 ± 0.01 |
| P4VP(Zn-3 1 -OH-PPME) | |||
| 0.1 | 3.66 (99%) | 0.24 (1%) | 3.61 ± 0.07 |
| 0.25 | 3.51 (98%) | 0.25 (2%) | 3.45 ± 0.09 |
| 0.5 | 3.04 (90%) | 0.78 (10%) | 2.9 ± 0.05 |
| 1 | 2.31 (79%) | 0.76 (21%) | 1.99 ± 0.19 |
| 2 | 1.13 (84%) | 0.39 (16%) | 1.01 ± 0.11 |
| 4 | 0.50 (75%) | 0.20 (25%) | 0.43 ± 0.02 |
| 6 | 0.40 (80%) | 0.12 (20%) | 0.37 ± 0.02 |
| 8 | 0.37 (78%) | 0.12 (22%) | 0.32 ± 0.01 |
| 10 | 0.08 (74%) | 0.24 (26%) | 0.12 ± 0.01 |
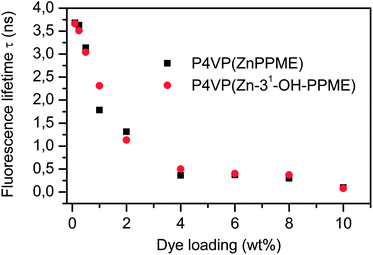 | ||
| Fig. 7 The fluorescence lifetimes for P4VP(ZnPPME) and P4VP(Zn-31-OH-PPME) assemblies given as a function of dye weight percent. | ||
Seely has shown in the classic solution studies of P4VP–chlorin, with several chlorin derivatives, that with low pigment loadings, i.e. pyridine/Chl > 500, the quantum yields of fluorescence become constant.23,38,39 We presume that in our case the situation is essentially similar; below 1 wt% chlorin loading levels fluorescence lifetimes remain rather constant (Table 1 and Fig. 7), implying that below this level the quantum yields resemble that of the monomeric pigment. Recently, it has been shown in a zinc tetraphenylporphyrin study that the excited state dynamics changed slightly upon the ligation of pyridine as the fluorescence lifetime shortened from 1.95 ns to 1.58 ns in toluene and from 2.05 ns to 1.61 ns in polystyrene/toluene mixtures respectively.40 Yet these fluorescence lifetime changes are rather small and thus have only a minor effect on the quantum yield. We expect that for our chlorin system the behaviour is similar.
Finally, to gain further insight into Zn chlorin–P4VP assemblies, we calculated intermolecular chlorin–chlorin distances at different doping average values (from 0.1 to 10 wt%) using both microscopic and macroscopic approximations assuming homogeneous chlorin distribution in P4VP.† The microscopic approximation was obtained using the Connolly solvent excluded volumes for the pigment and the polymer 4-VP unit. The macroscopic approximation was calculated using the material density to obtain the chromophore number density in a square lattice. The fluorescence lifetimes of ZnPPME–P4VP and Zn-31-OH-PPME–P4VP assemblies were plotted as a function of intermolecular distances to reveal a shortening of the fluorescence lifetimes upon decrease of chlorin–chlorin distances.† The calculated Förster distance (R0) using the microscopic approximation for ZnPPMEs was 47.8 Å and for Zn-31-OH-PPMEs it was 48.3 Å, whereas the corresponding values for the macroscopic approximation were 44.7 Å and 45.2 Å for ZnPPMEs and Zn-31-OH-PPMEs, respectively. Noticeably, both approximations yielded similar results. These values are in good accordance with Förster distances, 47 Å and 57 Å, reported between Chl b–Chl a and Chl b–Chl b, in a light-harvesting a/b complex (LHCIIb).41
For comparison, van Zandvoort et al. have successfully prepared nitrocellulose–Chl a assemblies on films with relatively high doping levels by casting the films from DMSO solutions.42 Consistently, photophysical studies of these films showed similar optical properties for the studied chlorins including monomeric absorption and emission spectra of Chl a at low concentrations. Additionally, the measured fluorescence lifetime decay was monoexponential at low concentrations and biexponential at higher concentrations. Also, the calculated Förster radius in the matrix was similar, being 59 ± 5 Å. Distinctly, for the benefit of the P4VP assemblies in our study we can state that P4VP has no overlapping signals in the visible spectral region, homogeneous films are easy to prepare, and moreover, unlike nitrocellulose, P4VP is chemically stable in material applications.
Conclusions
In the visible spectral region, the polymer host (P4VP) was found to be an optically transparent scaffold for Zn chlorins which are analogous to chlorophyll–protein complexes. The spin coated Zn chlorin–P4VP thin film assemblies indicated that coordinative pyridine moiety–Zn chlorin bonding was dominating over chlorin–chlorin assembling tendency up to 0.5 equiv. doping levels. Upon further increase of Zn chlorin doping, linear quenching was observed for steady-state fluorescence. The quenching process was identified to be dynamic quenching resulting from the energy transfer (FRET) between identical molecules. The approximated Förster distance R0 correlated well with the literature values for related dyes. Doping levels for absorbing materials lie at ca. 0.5 equiv. (ca. 300 wt%) and for emitting materials at ca. 0.002 equiv. (ca. 1 wt%).The photonic application potential of the studied assemblies lies in the light absorbing–harvesting realm that was initially anticipated from the antennae biomimicry. The small Stokes shift (∼6 nm) makes these dye assemblies alone less attractive for emission applications. To overcome this limitation, Tamiaki et al. showed in chlorin aggregate studies that a bacteriochlorin, i.e., a more red-shifted chlorin, integrated into an aggregate was able to receive and emit the light harvested by the chlorin assemblies.43 We propose that a similar strategy would also be viable for the studied assembly to strengthen its emission efficiency, e.g., for solid state lasers, single photon emitters, optical switches and OLEDs. Another material development option would be in the high doping absorption regime; Zn chlorin–P4VP assemblies could be built in contact with electron acceptors to construct optoelectronic devices such as solar cells and FETs.
Acknowledgements
Dr Marco Mattila is kindly acknowledged for his assistance with the time-resolved fluorescence measurements. We gratefully acknowledge the Academy of Finland [project no. (I.T.) 129043 and (J.H.) 135113], the Graduate School of Organic Chemistry and Chemical Biology (GSOCCB) and the Finnish National Graduate School for Materials Physics for financial support. The National Centre for Scientific Computing (CSC) is acknowledged for computational resources.Notes and references
- B. R. Crenshaw and C. Weder, Adv. Mater., 2005, 17, 1471–1476 CrossRef CAS.
- M. Berggren, A. Dodabalapur, R. E. Slusher and Z. Bao, Nature, 1997, 389, 466–469 CrossRef CAS.
- M. Maeda, H. Ishitobi, Z. Sekkat and S. Kawata, Appl. Phys. Lett., 2004, 85, 351–353 CrossRef CAS.
- O. Ostroverkhova and W. E. Moerner, Chem. Rev., 2004, 104, 3267–3314 CrossRef CAS.
- A. Shundo, Y. Okada, F. Ito and K. Tanaka, Macromolecules, 2012, 45, 329–335 CrossRef CAS.
- R. N. Dsouza, U. Pischel and W. M. Nau, Chem. Rev., 2011, 111, 7941–7980 CrossRef CAS.
- L. Cerdán, A. Costela and I. García-Moreno, Org. Electron., 2012, 13, 1463–1469 CrossRef.
- A. Priimagi, S. Cattaneo, R. H. A. Ras, S. Valkama, O. Ikkala and M. Kauranen, Chem. Mater., 2005, 17, 5798–5802 CrossRef CAS.
- A. Priimagi, M. Kaivola, F. J. Rodriguez and M. Kauranen, Appl. Phys. Lett., 2007, 90, 121103 CrossRef.
- I. D. W. Samuel and G. A. Turnbull, Chem. Rev., 2007, 107, 1272–1295 CrossRef CAS.
- A. A. Ishchenko, Pure Appl. Chem., 2008, 80, 1525–1538 CrossRef CAS.
- M. R. Narayan, Renewable Sustainable Energy Rev., 2012, 16, 208–215 CAS.
- V. Balzani, A. Credi and M. Venturi, ChemSusChem, 2008, 1, 26–58 CrossRef CAS.
- Z. Liu, H. Yan, K. Wang, T. Kuang, J. Zhang, L. Gui, X. An and W. Chang, Nature, 2004, 428, 287–292 CrossRef CAS.
- T. S. Balaban, Photosynth. Res., 2005, 86, 251–262 CrossRef CAS.
- N. Nelson and A. Ben-Shem, Nat. Rev. Mol. Cell Biol., 2004, 5, 971–982 CrossRef CAS.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef.
- S. Patwardhan, S. Sengupta, L. D. A. Siebbeles, F. Würthner and F. C. Grozema, J. Am. Chem. Soc., 2012, 134, 16147–16150 CrossRef CAS.
- T. S. Balaban, H. Tamiaki and A. R. Holzwarth, in Top. Curr. Chem., Supramolecular Dye Chemistry, ed. F. Würthner, Springer-Verlag, Berlin Heidelberg, 2005, vol. 258, pp. 1–38 Search PubMed.
- R. E. Blankenship, in Molecular Mechanisms of Photosynthesis, Blackwell Science Ltd, United Kingdom, 2002, pp. 61–94 Search PubMed.
- G. S. Engel, T. R. Calhoun, E. L. Read, T. Ahn, T. Mancal, Y. Cheng, R. E. Blankenship and G. R. Fleming, Nature, 2007, 446, 782–786 CrossRef CAS.
- M. Orrit, Science, 1999, 285, 349–350 CrossRef CAS.
- G. R. Seely, J. Phys. Chem., 1976, 80, 447–451 CrossRef CAS.
- J. S. Kavakka, S. Heikkinen and J. Helaja, Eur. J. Org. Chem., 2008, 4932–4937 CrossRef CAS.
- J. S. Kavakka, S. Heikkinen, I. Kilpeläinen, N. V. Tkachenko and J. Helaja, Chem. Commun., 2009, 758–760 RSC.
- D. M. Guldi, G. M. A. Rahman, S. Qin, M. Tchoul, W. T. Ford, M. Marcaccio, D. Paolucci, F. Paolucci, S. Campidelli and M. Prato, Chem.–Eur. J., 2006, 12, 2152–2161 CrossRef CAS.
- G. Bottari, O. Trukhina, M. Ince and T. Torres, Coord. Chem. Rev., 2012, 256, 2453–2477 CrossRef CAS.
- I. Beletskaya, V. S. Tyurin, A. Y. Tsivadze, R. Guilard and C. Stern, Chem. Rev., 2009, 109, 1659–1713 CrossRef CAS.
- F. D'Souza, R. Chitta, S. Gadde, M. E. Zandler, A. L. McCarty, A. S. D. Sandanayaka, Y. Araki and O. Ito, Chem.–Eur. J., 2005, 11, 4416–4428 CrossRef CAS.
- R. F. Kelley, R. H. Goldsmith and M. R. Wasielewski, J. Am. Chem. Soc., 2007, 129, 6384–6385 CrossRef CAS.
- A. Arulkashmir, R. Y. Mahale, S. S. Dharmapurikar, M. K. Jangid and K. Krishnamoorthy, Polym. Chem., 2012, 3, 1641–1646 RSC.
- M. D. Pelta, H. Barjat, G. A. Morris, A. L. Davis and S. J. Hammond, Magn. Reson. Chem., 1998, 36, 706–714 CrossRef CAS.
- K. M. Smith, D. A. Goff and D. J. Simpson, J. Am. Chem. Soc., 1985, 107, 4946–4954 CrossRef CAS.
- H. Tamiaki, S. Takeuchi, S. Tsudzuki, T. Miyatake and R. Tanikaga, Tetrahedron, 1998, 54, 6699–6718 CrossRef CAS.
- Y. Cohen, L. Avram, T. Evan-Salem and L. Frish, in Analytical Methods in Supramolecular Chemistry, ed. C. A. Schalley, Wiley-VCH, Weinheim, 2007, pp. 163–219 Search PubMed.
- A. Laiho, R. H. A. Ras, S. Valkama, J. Ruokolainen, R. Österbacka and O. Ikkala, Macromolecules, 2006, 39, 7648–7653 CrossRef CAS.
- H. Scheer, in Chlorophylls and Bacteriochlorophylls (Advances in Photosynthesis and Respiration), ed. B. Grimm, R. J. Porra, W. Rüdiger and H. Scheer, Springer, Dordrecht, 2006, vol. 25, pp. 1–26 Search PubMed.
- G. R. Seely, J. Phys. Chem., 1970, 74, 219–227 CrossRef CAS.
- G. R. Seely, J. Phys. Chem., 1976, 80, 441–446 CrossRef CAS.
- M. M. Yatskou, R. B. M. Koehorst, A. van Hoek, H. Donker and T. J. Schaafsma, J. Phys. Chem. A, 2001, 105, 11432–11440 CrossRef CAS.
- R. Horn and H. Paulsen, J. Biol. Chem., 2004, 279, 44400–44406 CrossRef CAS.
- M. A. M. J. van Zandvoort, D. Wróbel, P. Lettinga, G. van Ginkel and Y. K. Levine, Photochem. Photobiol., 1995, 62, 279–289 CrossRef CAS.
- H. Tamiaki, T. Miyatake, R. Tanikaga, A. R. Holzwarth and K. Schaffner, Angew. Chem., Int. Ed. Engl., 1996, 35, 772–774 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Molecular modelling; DOSY, SAXS, TEM and SEM measurement data; fluorescence lifetime decays; intermolecular distance and Förster radius calculations. See DOI: 10.1039/c3tc00499f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2013 |