
The spontaneous cascade optimization strategy of the double enrichment improves anion-derived solid electrolyte interphases to enable stable lithium-metal batteries†
Fengxu
Zhen‡
a,
Hong
Liu‡
a,
Yingbin
Wu‡
a,
Xinjia
Zhou
a,
Weiping
Li
 a,
Yuzhi
Chen
a,
Yuke
Zhou
a,
Haoyang
Wang
a,
Xiangkai
Yin
a,
Shujiang
Ding
*a,
Xiaodong
Chen
*b and
Wei
Yu
*a
a,
Yuzhi
Chen
a,
Yuke
Zhou
a,
Haoyang
Wang
a,
Xiangkai
Yin
a,
Shujiang
Ding
*a,
Xiaodong
Chen
*b and
Wei
Yu
*a
aSchool of Chemistry, Engineering Research Center of Energy Storage Materials and Devices, Ministry of Education, State Key Laboratory of Electrical Insulation and Power Equipment, Xi’an Jiaotong University, Xi’an, 710049, P. R. China. E-mail: dingsj@mail.xjtu.edu.cn; yuwei2019@mail.xjtu.edu.cn
bXi’an Institute of Optical Precision Machinery, Chinese Academy of Sciences, Xi’an Shanxi, 710000, P. R. China. E-mail: cxd_1979@163.com
First published on 5th April 2025
Abstract
Anion regulation represents a highly effective, convenient, and economical approach to generate LiF-rich solid electrolyte interfaces (SEIs). The anion decomposition process is influenced by charge density and anion concentration. However, current research primarily concentrates on increasing charge density to enhance anion decomposition. Herein, the spontaneous cascade optimization strategy driven by the double enrichment of anions and charges is proposed by utilizing NH2-MIL-101(Fe)@Copc (MOF@Copc). Specifically, NH2-MIL-101(Fe) functions as a TFSI− anion trap via the Lewis acid–base interactions and synergistic hydrogen bonding, thereby achieving primary optimization. Subsequently, the rich electronic structure of Copc facilitates charge delocalization and lowers the energy barrier for anion decomposition, allowing the C–F bonding to break more readily, thereby enabling further optimization. The π–π stacking interaction between the MOF and Copc facilitates the close association of adsorption and catalytic sites, allowing the continuous breakdown of the C–F series products in a chain reaction. The assembled LFP (19.26 mg cm−2) demonstrates a commercial-grade cathode area capacity, maintaining over 90% capacity retention across 350 cycles at 1C, with a capacity decay rate of only 0.02% per cycle. More importantly, this strategy enables the industrial-scale production of Ah-class anode-free lithium-metal pouch batteries exceeding 300 W h kg−1. Optimizing anion decomposition provides a novel perspective to advance the practical application of lithium-metal batteries.
Broader contextLithium metal batteries (LMBs), due to their exceptionally high energy density, are considered one of the most promising energy storage technologies. However, challenges such as dendrite formation and fragile solid electrolyte interphase (SEI) hinder the practical application of LMBs. Here, we present a multi-stage alternating arrangement of adsorption and catalytic sites—NH2-MIL-101(Fe)@Copc (MOF@Copc), which achieves a significant increase in the LiF content by regulating the mode of ‘enrichment-decomposition behavior’ of an anion. The content of LiF in the SEI is positively correlated with its cycle life. Therefore, under the lowest N/P conditions, the MOF@Copc/Cu anode-free lithium metal pouch battery demonstrates outstanding long-cycle stability. The protective layer contributes to simultaneously accelerating the development and deployment of high-energy-density LMBs and AFLMBs. |
Introduction
Lithium metal, recognized for its high theoretical capacity (3860 mA h g−1) and low redox potential (3.04 V lower than that of the standard hydrogen electrodes), has garnered significant attention.1,2 The highly reactive lithium metal interacts uncontrollably with the liquid electrolyte, inevitably leading to the formation of solid electrolyte interfaces (SEIs).3,4 The natural SEI is composed predominantly of organic substances (lithium alkyl carbonates) and contains lower amounts of inorganic substances (LiF, Li3N, etc.).5 However, the resulting SEI is structurally inhomogeneous and prone to fragmentation, leading to ruptures during continuous lithium stripping/deposition, and the rupture sites are highly prone to the formation of lithium dendrites, which in turn initiates a series of vicious cycles.6–11 LiF, possessing exceptional chemical and mechanical stability as well as a low Li+ diffusion barrier, is widely recognized as a key component in constructing a stable SEI.12,13Currently, the primary strategies to enhance the LiF content in SEIs involve the addition of fluorinated solvents, the construction of a high-concentration electrolyte, and the use of electrolyte additives. However, these strategies exhibit certain limitations in practice: most fluorinated solvents tend to generate corrosive HF and decomposition by-products (CH3OH), which accelerate the deterioration of the electrode interface.14,15 The high concentration of electrolyte is at the expense of a good solid–liquid contact interface,16 while fluorinated electrolyte additives incur high costs.17 The formation of LiF relies heavily on the decomposition of fluorine-containing compounds.18 Therefore, the modulation of fluorine-containing anions is expected to construct the in situ LiF-SEI without altering the electrolyte composition. However, the inability to generate an effective SEI layer on the lithium metal surface is attributable to two underlying reasons: (1) A limited number of anions at the electrode surface. The electric field, driven by the charging conditions, promotes anion migration toward the cathode electrode, thereby exacerbating the formation of an anion–deficient interface. (2) Slow electron transport kinetics. Lithium bis(trifluoromethanesulfonyl)imide (LiTFSI), characterized by its numerous C–F fragments and high ion conductivity, is considered a promising candidate for replacing LiPF6.19 However, since the decomposition of LiTFSI is electron-driven, the slow electron transfer kinetics hinder the anions from acquiring sufficient charge. This limitation leads to the formation of an SEI primarily composed of CF3−, which represents an incomplete reduction product of TFSI−.20–23 Therefore, the anionic decomposition process is constrained by a limited number of anions and a finite amount of electrical charge. Recent research has consistently demonstrated that optimizing electron-directed transport kinetics can effectively increase the degree of TFSI− decomposition. Gao et al. reported that Li–Au alloys produced by in situ lithiation possess significant electron-donating properties, thus facilitating the decomposition of LiTFSI within the electrolyte.24 Liu et al. enhanced the electron transfer kinetics using a Br-TPOM skeleton with high electron transfer capacity, thereby promoting the breakup of the C–F bond and leading to the formation of LiF.25 In parallel, Liu et al. constructed a “charge warehouse” using a porphyrin-organic framework (POF), serving as a charge source and ensuring sufficient charge transfer for the TFSI−, thereby facilitating the formation of a LiF-rich SEI layer.26 However, these studies primarily focus on the charge enrichment while overlooking the enrichment of anions. Addressing only one of these factors will not fully maximize anionic decomposition. Therefore, simultaneously enriching anions and enhancing electron transfer kinetics constitute effective methods for forming an excellent LiF-rich SEI.
Herein, a spontaneous cascade optimization strategy is proposed through the design of the NH2-MIL-101(Fe) @Copc composite material (Fig. 1). The central Fe3+ ion, featuring an empty orbital, participates in the Lewis acid–base interaction with TFSI−, which possesses lone pair electrons. Owing to the synergistic effects of Lewis acid–base interactions and the formation of hydrogen bonds with amino groups, precisely anchoring the wandering TFSI− ions to concentrate them on the surface of the modified layer, thus achieving initial optimization of anion-rich interfaces. Simultaneously, cobalt phthalocyanine (Copc) characterized by its electron-rich conjugated structure, creates an electron-rich environment through electron delocalization. An electron-rich environment is advantageous as it facilitates the formation of more coherent electron transport pathways, thus reducing the energy barriers faced by electrons during intermolecular transfer. The anion decomposition is facilitated by enhancing the electron transport kinetics. The two optimizations within the cascade strategy guarantee the efficient decomposition of the anions by the double enrichment of anions and charges.
 | ||
| Fig. 1 Schematic diagram of the principle of the modification layer NH2-MIL-101(Fe)@Copc. | ||
To validate the efficacy of the cascade optimization strategy, we constructed a MOF@Copc layer to serve as the artificial solid electrolyte interface (ASEI). Benefiting from the advancement of this strategy, the synergistic combination yields an increase in the LiF content from 30.2% to 41.7%. Impressively, Li–Cu half-cells modified with MOF@Copc exhibited stable cycling for more than 650 cycles, while maintaining an average coulombic efficiency (CE) of 98.85% under test conditions 1 mA cm−2. Meanwhile, MOF@Copc/Li‖LiFePO4 (approximately 11.05 mg cm−2), showed a capacity retention exceeding 90% after 1000 cycles at 1C, surpassing the performance reported in most literature reports on artificial SEI strategies. Also, the lifespan of anode-free Li metal pouch cells, with an energy density of 300 W h kg−1, can be extended by nearly four times. This study aims to significantly improve the cycle life of lithium metal batteries by maximizing anion decomposition to form LiF-rich SEIs.
Results and discussion
As shown in Fig. 2a, cobalt phthalocyanine(II) exhibits the strongest signals near the Fermi level, which theoretically suggests good conductivity. However, its tendency for spontaneous aggregation, combined with its poor film-forming properties, restricts its broader application as an artificial protective layer. Therefore, composite material NH2-Fe-MIL(101)@Copc can effectively regulate the dispersion of Copc clusters while ensuring great electrical conductivity. Additionally, as shown in Fig. 2b, the upward shift of the d-band center at the active sites strengthens the interaction with TFSI− anions, consistent with the adsorption energy results presented in Fig. 2d. Subsequently, (Fig. 2c) the calculation and analysis of differential charge densities reveal a significant charge difference between the two materials, with the directional transfer of electrons from cobalt phthalocyanine to the MOF. | ||
| Fig. 2 Mechanism of spontaneous cascade optimization strategy of MOF@Copc modified layer (a) total DOS(TDOS) for Copc, NH2-Fe-MIL(101) and NH2-Fe-MIL(101)@Copc. (b) εd of Copc, NH2-Fe-MIL(101) and NH2-Fe-MIL(101)@Copc. (c) Differential charge density diagram. (d) Binding energy of TFSI− with MOF and Li+ interactions. (e) LiTFSI decomposition mechanism. (f) The decomposition energy barrier of the rate-determining step of TFSI− under the conditions of MOF, Copc alone, and their synergistic collaboration. (g) DFT simulations with Bader charges (measured in units of |e|) illustrate the degradation dynamics of TFSI−, along with the corresponding ELF of TFSI− at various steady states. | ||
According to Fig. 2e, the decomposition reaction of LiTFSI is divided into four distinct stages, each of which is induced by electrons, ultimately resulting in the formation of LiF. This indicates that the degree of TFSI− decomposition can be optimized through electron-directed conduction. Cobalt phthalocyanine is characterized as a conjugated system with an 18 π electronic structure, where the two –NH– groups each contribute two electrons, while the remaining nitrogen and carbon atoms each donate one electron. We traced the four-step decomposition reaction of TFSI− using first-principles calculations (Fig. 2g and Fig. S1, ESI†). Bader charge analysis reveals that a 0.83 e− charge is transferred from the “electronic dark cloud” cobalt phthalocyanine to the TFSI− anion, resulting in N–S splitting. Subsequently, the CF3− group detaches from the SO2CF3− ionic fragments as the anion gains additional charges. The CF3− group then decomposes further into CF2− and F−. Ultimately, the F− ion combines with Li+ to form LiF. In addition, we investigated the effect of the cobalt phthalocyanine on the degree of TFSI−-dissociation using density-functional theory (DFT) (Fig. S2, ESI†) and detailed the changes in the C–F bond in Table S1 (ESI†). In the initial state, the C–F bonds of TFSI-anions are 1.343 Å, 1.344 Å, and 1.346 Å, respectively. Following electron transfer interactions with phthalocyanine, the bond lengths extended to 1.363 Å, 1.351 Å, and 1.350 Å, respectively. Cobalt phthalocyanine, due to its electron-rich nature, has been shown to weaken the interaction force between C and F atoms. The easier breakage of the C–F bond generates more LiF. Finally, the synergistic effect between Copc and MOF was further elucidated through the calculation of the decomposition energy barriers using DFT (Fig. 2f and Table S2 and Fig. S3a and b, ESI†). The calculation results demonstrate that Copc can lower the decomposition potential barrier of anions. Furthermore, TFSI− decomposition is further enhanced by the combined effects of charge transfer facilitated by Copc and anchoring by MOF.
NH2-MIL-101(Fe) was successfully synthesized using a straightforward hydrothermal synthesis strategy (Fig. 3a). Subsequently, NH2-MIL-101(Fe)@CoPc composites were synthesized via the impregnation method. The planar π-conjugated structure Copc causes molecular aggregation through π–π stacking.27 DFT calculations confirm that the binding energy between NH2-MIL-101(Fe) and Copc is lower than that between Copc molecules alone (Fig. S4, ESI†), suggesting that the stronger π–π interaction between NH2-MIL-101(Fe) and Copc is thermodynamically favorable. The NH2-MIL-101(Fe)@Copc composite material not only addresses the issue of spontaneous Copc aggregation but also ensures uniform and firm dispersion of Copc molecules on the iron-based metal–organic framework.28
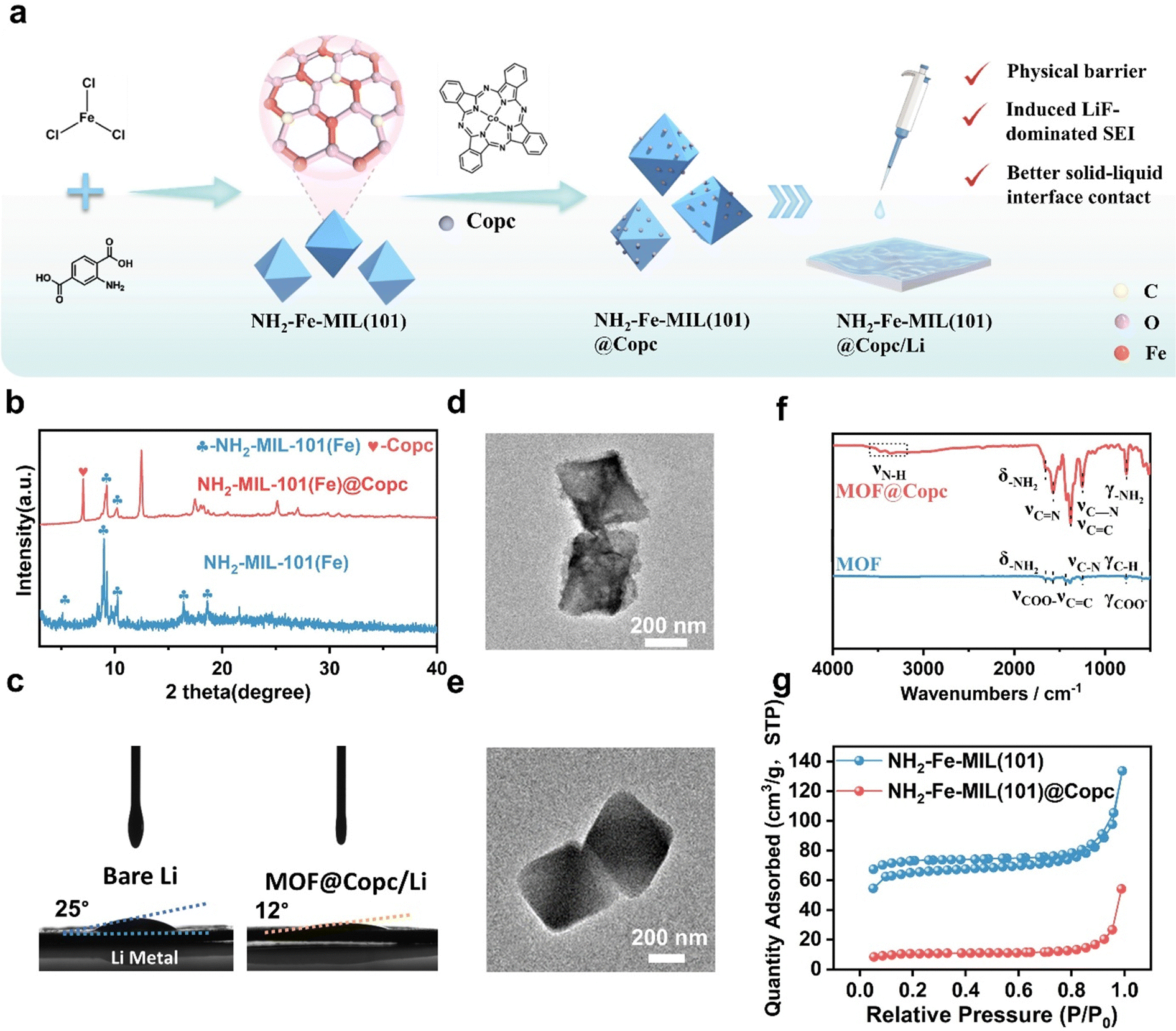 | ||
| Fig. 3 Characterization of the composite NH2-MIL-101(Fe)@Copc (a) schematic diagram of the preparation of NH2-MIL-101(Fe) and NH2-MIL-101(Fe)@CoPc nanocomposite. (b) XRD of NH2-MIL-101 (Fe) and NH2-MIL-101 (Fe)@CoPc. (c) Contact angle measurement of bare Li and MOF@Copc/Li surfaces. (d) and (e) TEM image of NH2-MIL-101(Fe) and NH2-MIL-101(Fe)@CoPc. (f) FTIR spectra of NH2-MIL-101(Fe)@CoPc. (g) N2 adsorption–desorption isotherm. | ||
The chemical structures of NH2-MIL-101(Fe), and NH2-MIL-101(Fe)@CoPc nanocomposites were analyzed using FT-IR spectroscopy (Fig. 3f). A broad absorption peak at 3368 cm−1 is identified as the characteristic absorption peak of cobalt phthalocyanine. Characteristic absorption peaks of NH2-MIL-101(Fe) at 768 cm−1 and 1658 cm−1 are attributed to the –NH2 distorted vibration and –NH2 variable angle vibration, respectively. Similarly, XRD spectra could confirm that Copc was successfully incorporated into NH2-MIL-101(Fe) (Fig. 3b). Contact angle measurements confirm that the presence of the MOF@Copc layer achieves a better solid–liquid interface (Fig. 3c). As depicted in the SEM and TEM images, NH2-MIL-101(Fe) exhibits a smooth surface with uniformly biconical-shaped blocks (Fig. 3d–e and Fig. S5 and S6, ESI†). Upon doping with Copc, the particles adhere to the surface of the MOF structure, which results in a roughened surface. Concurrently, the elemental distribution was analyzed using a transmission electron microscopy elemental map. NH2-MIL-101(Fe)@CoPc displays elements such as C, N, O, Fe, and Co, well-distributed throughout the selected TEM image areas, indicating that Copc was successfully dispersed on the surface of NH2-MIL-101(Fe). Finally, NH2-MIL-101(Fe) exhibits a specific surface area of 172.62 m2 g−1 (Fig. 3g), attributed to its larger surface area and porous structure. However, in the composite, the loaded Copc nanoparticles may obstruct the pores of NH2-MIL-101(Fe), leading to a reduction in the nitrogen adsorption capacity of the NH2-MIL-101(Fe)@CoPc nanocomposite, further proving that Copc was successfully loaded on the NH2-MIL-101(Fe).
The enrichment effect of the MOF on TFSI− was demonstrated through molecular dynamics simulations (Fig. 4a and Fig. S7–S9, ESI†). The radial distribution function shows that under the action of the MOF structure, the distance of the TFSI− anion from the lithium surface typically ranges between 0.18 nm and 0.4 nm. The result is primarily attributed to the coordinated effects involving the Lewis acid–base interaction of the central ion Fe3+ and the hydrogen bond formed by the H atom in the amino group, which effectively secures the TFSI− anion closer to the lithium surface.29–31 In the control group, the distance between the TFSI− anion and the lithium surface typically ranges from 0.24 nm to 0.8 nm, confirming that the strong repulsive force between the lithium metal and the anion pushes the anion away. The coordination numbers reveal that in the experimental MOF group, the coordination of TFSI− with the lithium surface is 0.42, compared to 0.13 in the control group. This suggests that the addition of MOF enables the lithium metal surface to more effectively absorb the TFSI−, facilitates the decomposition of TFSI−, and consequently boosts the formation of the LiF-SEI. To further demonstrate the enrichment effect of MOF on TFSI− anions, Li‖Li cells were assembled in the electrolytic cell for in operando Raman observations (Fig. 4d and e and Fig. S10, ESI†). The migration of TFSI− anions within electrolyte systems is evaluated by observing variations in anion strength between solid and liquid phases. After applying a constant current to the symmetric cell, TFSI− began to migrate, resulting in a gradual decrease in concentration on the upper side and an increase on the lower side. The presence of the modification layer mitigated the decrease in TFSI− concentration, limiting the movement of TFSI− and resulting in a slower decrease in concentration on the upper side. This provides strong support for the assertion that the modified MOF layer can enrich LiTFSI at the interface. Meanwhile, the red-shift phenomenon observed in both Raman and NMR spectra demonstrates that the MOF interacts with LiTFSI, leading to enrichment at the interface (Fig. 4b and c).
 | ||
| Fig. 4 Influence of MOF@Copc modified layer on SEI composition (a) schematic diagram of the distance between the TFSI− anion and the MOF/Li and Li surface. (b) and (c) The Raman and 7Li NMR spectra of MOF-LiTFSI and LiTFSI. (d) and (e) In operando Raman patterns of TFSI− characteristic peaks in the Li‖Li system and the MOF/Li‖Li/MOF system. (f) XPS spectra of C 1s for bare Li, MOF/Li, and MOF@Copc/Li electrodes. (g) TOF-SIMS spectra of the lithium surface after cycling in a Li‖Li and MOF@Copc/Li‖Li/MOF@Copc battery. Here LiF2− signal is chosen to represent LiF. (h) and (i) Microstructure of SEI under cryo-electron microscopy. | ||
To verify the effect of the modification layer on the kinetics, the redox behavior of LiTFSI was examined using cyclic voltammetry (CV), revealing a distinct cathodic peak at approximately 1.2 V (Fig. S11, ESI†). The observed increase in peak intensity is correlated with an enhanced reduction reaction of TFSI−. The substantial difference in peak intensities indicates that cobalt phthalocyanine fosters an electron-rich environment, thereby accelerating the reduction kinetics of TFSI−. Additionally, a notable peak around 0.3 V corresponds to the low potential deposition of Li.32 Around 1.6 V, the redox peak of LiNO3 is observed, with the variation in peak intensities suggesting that the electron-rich environment also enhances the electron-driven decomposition reaction of LiNO3.33
To elucidate the positive effects of the modification layer on SEI formation, X-ray photoelectron spectroscopy (XPS) was employed to analyze the composition of the SEIs after cycling. As shown in Fig. 4f, within the C 1s spectra, the content of product –CF2 predominates, while that of reactant –CF3 is significantly reduced. The positive effect of cobalt phthalocyanine on the electron transfer kinetics noticeably enhances the charge-induced decomposition reaction of LiTFSI. In the F 1s spectra, the MOF@Copc-modified layer yielded the highest LiF content (Fig. S12, ESI†). The same conclusion was reached through an analysis of the Li 1s data (Fig. S13, ESI†). At the same time, significant changes in Li3N content were observed, stemming from the reaction of the –NH2 group in the MOF with lithium metal,34–36 and from electron-rich environments that facilitate the decomposition of LiNO3. TOF-SIMS was utilized to analyze the differences in the distribution of LiF2−, LiF3−, and Li2F− ion fragments in SEI along the depth direction (Fig. 4g and Fig. S14, ESI†). The intensity of these fragments indicates that LiF produced by the modification layer is more uniformly distributed across the cross-section and vertical plane, completely exceeding the LiF content in the bare Li metal. The microstructure of the SEIs was examined using cryo-electron microscopy, revealing evident differences among the SEIs (Fig. 4h and i). The unmodified samples yielded a thick and inhomogeneous SEI, measuring 70 nm in thickness, whereas the modified samples formed a thin and homogeneous SEI with a thickness of just 50 nm. The thin and homogeneous SEI is crucial for regulating lithium deposition behavior and ion transport kinetics.37,38
To gain a deeper understanding of the significance of constructing MOF@Copc-ASEI, its electrochemical performance was assessed by assembling asymmetric Li–Cu half-cells. The MOF@Copc/Cu–Li cells exhibit satisfactory coulombic efficiency (CE) values and extended operating lives (Fig. 5a and Fig. S15, ESI†). Lithium modified with MOF@Copc/Li–Cu exhibits stable cycling for over 650 cycles at 1 mA cm−2 (1 mA h cm−2), achieving an average coulombic efficiency (CE) value of 98.85%. The MOF@Copc/Li-modified battery sustains more than three times the number of cycles compared to the other two batteries. Moreover, the half-cell MOF@Copc/Cu–Li demonstrated notably impressive cycling stability, even when the current density was increased to 3.0 mA cm−2 (Fig. 5b). The superior electrochemical performance demonstrated that MOF@Copc-induced construction of LiF-rich SEI not only enhances the stable cycling characteristics of the cell but also improves coulombic efficiency. Additionally, the nucleation barrier of Li was investigated using the time–voltage test curve (Fig. 5c), MOF@Copc exhibited a low lithium nucleation overpotential of 59.7 mV at 3 mA cm−2. However, the MOF/Cu–Li cells and bare Cu cells demonstrated high lithium nucleation overpotentials of 73.6 mV and 117.4 mV, respectively. The lowest nucleation and plateau overpotentials suggest that the ASEI-induced LiF interface effectively reduces the energy barrier to lithium nucleation and growth.12,39 To investigate the impact of the modified layer on Li-ion transfer kinetics, Li‖Cu half-cells and Li‖Li symmetric cells were assembled for subsequent measurements (Fig. S16, ESI†). Notably, the MOF@Copc-modified symmetric cell exhibits an exceptionally high Li+ transfer number (tLi+) of 0.765, compared to that of bare lithium. Higher tLi+ is achieved by anion immobilization.40 The Tafel curves indicate that the exchange current density, enhanced by MOF@Copc (I0 = 0.45 mA cm−2), is three times that of the unmodified one (I0 = 0.15 mA cm−2) (Fig. 5d). This enhancement suggests that the MOF@Copc modification layer significantly improves Li+ diffusion processes, thereby accelerating interfacial transfer kinetics.41 To further explore the effect of the MOF@Copc protective layer on ion transport, the activation energy (Ea) for lithium-ion migration at the interface was determined using the Arrhenius equation (Fig. 5e and Fig. S17, ESI†). The results indicated that bare Li (54.27 kJ mol−1) > MOF (45.97 kJ mol−1) > MOF@Copc (44.29 kJ mol−1). The spontaneous synergistic optimization strategy facilitates the enrichment and decomposition of TFSI−, resulting in the formation of a LiF-rich SEI that substantially enhances Li+ transport kinetics.42,43 Simulation results of the lithium-ion migration path reveal that the MOF@Copc interface layer displays a reduced lithium-ion diffusion barrier, promoting the rapid and uniform transport of Li ions (Fig. 5f)
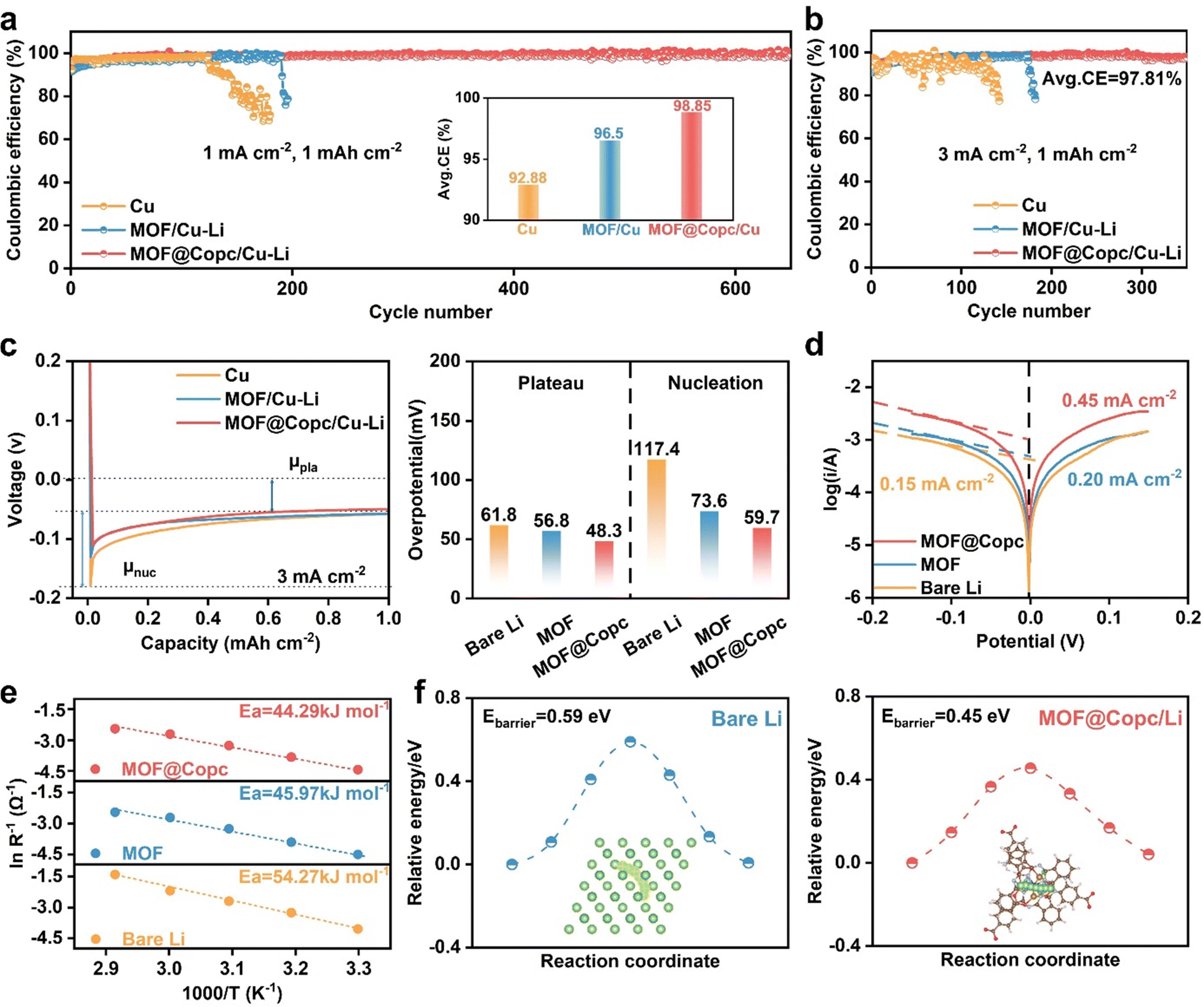 | ||
| Fig. 5 Electrochemical transport kinetics of modified lithium metal anodes (a) coulombic efficiencies of Li–Cu, MOF/Li–Cu, and MOF@Copc/Li–Cu cells at 1 mA cm−2 with a capacity of 1 mA h cm−2. (b) Coulombic efficiencies of Li–Cu, MOF/Li–Cu, and MOF@Copc/Li–Cu cells at 3 mA cm−2 with a capacity of 1 mA h cm−2. (c) The voltage–capacity curves of Li–Cu, MOF/Li–Cu, and MOF@Copc/Li–Cu cells at 3 mA cm−2 with a capacity of 1 mA h cm−2. (d) Tafel curves of symmetric cells. (e) Effect of temperature on the interfacial impedances of the modified and unmodified anode. (f) Li+ diffusion pathway and corresponding energy barriers of bare Li and MOF@Copc/Li. | ||
To demonstrate the enhanced cycling stability of MOF@Copc modification for lithium metal batteries (LMB), constant-current charge/discharge tests were conducted on symmetric cells (Fig. 6a). At a current density of 1 mA cm−2, bare Li cells experienced a sharp increase in overvoltage after just five cycles. For the MOF/Li symmetric cell, the voltage abruptly increased after 1000 hours. In contrast, the MOF@Copc-modified symmetric cell maintained continuous operation for 2000 hours. The amplified voltage curve illustrates that the MOF@Copc/Li batteries maintain a stable voltage plateau throughout the plating/stripping process.
 | ||
| Fig. 6 Electrochemical performance and characterization of lithium deposition layer (a) current density of 1 mA cm−2 with a capacity of 1 mA h cm−2. (b) The atomic force microscopy (AFM) testing of the surface roughness of lithium electrodes after cycling. (c) In situ optical microscopy observations of the Li deposition/stripping process on bare Li foil and MOF@Copc/Li. (d) Corresponding SEM images after plating for bare Li and MOF@Copc/Li. (e) Cryo-electron microscopic observation of the morphology of lithium deposition. | ||
To visualize the precise regulatory effect of the MOF@Copc layer on the excellent interface, the lithium electrode morphology of Li‖Li symmetric batteries after 1 mA cm−2 constant-current charging and discharging was examined using atomic force microscopy (AFM) (Fig. 6b). The surface roughness of the unmodified lithium anode, which measured 279.9 nm, is primarily attributable to the formation of numerous lithium dendrites and dead lithium after 100 cycles. The surface of the MOF@Copc-modified electrode remains smooth, with a roughness of 494.3 pm, which is mainly attributed to the fact that LiF plays an important role in inhibiting dendrite growth and promoting uniform deposition of lithium. Additionally, in operando optical microscopy (OM) was employed to observe Li plating behavior in real time. As shown in Fig. 6c and Fig. S18 (ESI†), both the bare Li and MOF@Copc/Li anodes exhibited smooth surfaces before lithium plating began. Subsequently, uneven Li plating emerged on the surface of bare Li after 15 minutes, leading to the formation of numerous lithium dendrites. In contrast, the MOF@Copc/Li anode gradually thickens during the entire lithium plating process while consistently maintaining a uniform and smooth surface. These results illustrate that the MOF@Copc/Li anode effectively inhibits the growth of lithium dendrites throughout the cycling process.
To more effectively demonstrate the morphological modulation of lithium deposition by the MOF@Copc protective layer during the cycling process, the morphological evolution during lithium deposition/exfoliation was investigated using non-in situ scanning electron microscopy (Fig. 6d and Fig. S19 and S20, ESI†). At a current density of 1 mA cm−2 and capacity of 1 mA h cm−2, numerous mossy lithium dendrites appeared on the surface of the bare lithium electrode, exhibiting a sparse and discrete deposition morphology. For the MOF/Li anode, the deposited lithium displayed a non-dense morphology with visible cracks. In contrast, the surface of the MOF@Copc-modified electrode exhibits a smooth and dense bulk deposition morphology. Further analysis of SEM images at the higher current density and capacity following the fiftieth cycle shows that MOF@Copc/Li exhibits uniform and flat lithium deposition morphology, which suggests that the formation of LiF-rich SEI induced by the protective layer of MOF@Copc can significantly inhibit the dendrites and improve the stability of the interface.44 Meanwhile, satisfactory spherical lithium deposition is more readily observable using cryo-electron microscopy (Fig. 6e and Fig. S21, ESI†). Owing to the minimal volume of the spherical units, the probability of interfacial side reactions in lithium metal is minimized, thereby enhancing the cycling reversibility of lithium metal batteries.45,46
A typical full cell was assembled to further explore the practical application potential of the MOF@Copc layer. The lithium metal anode, featuring a MOF@Copc-modified layer, was initially paired with an LFP cathode at a loading of 11.05 mg cm−2 (Fig. 7a). After enduring 1000 cycles, the cell demonstrated remarkable stability, with a capacity retention rate exceeding 90%. The electrochemical performance of MOF@Copc is notably superior compared to that reported in much of the existing literature (Fig. 7e).47–53 Meanwhile, under high current density (3C), the modified lithium anode maintains a capacity retention of up to 97.58% after 600 cycles (Fig. S22, ESI†). Surprisingly, the cell performance also remains outstanding when paired with higher loadings of LFP (Fig. 7c). Therefore, in situ electrochemical impedance spectroscopy (EIS) measurements and relaxation time distribution (DRT) of LFP‖Li cells were utilized for analysis (Fig. 7d and Fig. S23, ESI†). The change in the integrated area of the peak features associated with interfacial resistance after 100 cycles was significantly smaller for MOF@Copc/Li than for the cells with unmodified bare Li. The outcome is primarily attributed to the enhanced cycling stability resulting from an improved interface by the artificial SEI.54
 | ||
| Fig. 7 Battery performance of MOF@Copc protective layer (a) cycling performance of LFP-MOF@Copc/Li, LFP-MOF/Li, and LFP-bare Li cells at 1C rate. (b) Rate performance of LFP-MOF@Copc/Li, LFP-MOF/Li, and LFP-bare Li cells at 0.5C, 1C, 2C, 5C. (c) Cycling performances of high-loading LFP-MOF@Copc/Li and bare Li cells at 1C rate. (d) Distribution of relaxation times (DRT) results of LFP‖bare Li and LFP‖MOF@Copc/Li cells in the voltage range of 3.5–4.2 V. (e) Comparison of electrochemical performance of lithium metal anode modified by MOF@Copc with related work. (f) Cycling performance of the Cu‖NCM811 and MOF@Copc/Cu ‖ NCM811 pouch-type cell at 0.1C, Tests to light up commercially available mini-lamps. | ||
To further verify the rate capability of the full battery, charge–discharge rate tests were conducted at various current densities. As the charge–discharge current density increased, the discharge capacity of the battery was also increased to (0.5, 1, 2, and 5C, corresponding to 162.65, 149.81, 137.71, and 118.05 mA h g−1, respectively) (Fig. 7b). When the current density returned to 1C, a capacity of 149.57 mA h g−1 could be restored, exhibiting a satisfactory high magnification tolerance. Particularly at a current density of 5C, the capacity of MOF@Copc/Li was found to be 1.4 times that of bare lithium metal batteries. Voltage distribution diagrams at various rates demonstrate that, compared with LFP/bare Li cells, MOF@Copc/Li exhibits reduced voltage polarization (Fig. S24, ESI†). The MOF@Copc/Li cell exhibits excellent long-term cycling stability at a high rate of 20C, equivalent to one cycle every three minutes (Fig. S25, ESI†). After more than 9000 cycles, the capacity retention rate achieved an impressive 95.18%. The electrochemical cycling stability is attributed to the advanced spontaneous cascade optimization strategy. The synergistic effect of anion enrichment and excellent anion decomposition kinetics contributes to the SEI with a predominantly inorganic composition, which effectively regulates Li+ diffusion kinetics and lithium deposition behavior, facilitating fast charging of lithium-metal batteries.55
Subsequently, we further explore the compatibility of the modified MOF@Copc layer with high-voltage cathodes, we employed a high-loading LiNi0.8Co0.1Mn0.1O2 (NCM811) cathode (Fig. S26, ESI†). The cycling stability of MOF@Copc-modified layer batteries demonstrates that the proportion of inorganic components in the induced SEI is directly proportional to the cycle life of lithium metal batteries (LMB). The rate performance of MOF@Copc/Li-modified full cells is significantly superior to that of both bare Li and MOF/Li cells, particularly at high current densities (Fig. S27, ESI†). To achieve higher energy density, cycle stability tests were conducted under more challenging commercial pouch cell conditions. Notably, MOF@Copc/Cu demonstrates a longer cycle life compared to the unmodified copper foil. These pouch batteries are capable of powering light bulbs, thereby demonstrating their commercial potential in the field of flexible wearable electronics (Fig. 7f and Fig. S28, ESI†).
Conclusions
In summary, we propose the spontaneous cascade optimization strategy to modulate inorganic-rich SEIs. This strategy is used to achieve a two-step optimization by first enriching the anions at the interface and then modulating the electron transfer kinetics to promote the oxidative decomposition of the anions. Simulations and characterization studies reveal the critical role of Copc in the decomposition kinetics, and the electron-directed conduction of the electron-rich structure significantly lowers the decomposition energy barriers, thereby facilitating the decomposition of C–F bonds. It was demonstrated through in operando Raman spectroscopy and molecular dynamics simulations that NH2-MIL-101 (Fe) enriches anions at the interface. Therefore, the synergistic effect of these two composites, facilitated by the spontaneous cascade optimization strategy, maximizes the decomposition of anions. In situ construction of LiF-rich SEIs modulates Li+ transport kinetics and facilitates uniform lithium deposition, thereby resulting in excellent electrochemical performance of the modified anode. Notably, the MOF@Copc-modified Li‖Cu cell exhibited an exceptionally long cycle life. When paired with a highly loaded LFP, the performance exceeds that documented in the existing literature. Meanwhile, pouch batteries exhibit excellent electrochemical performance at 0.1C. We anticipate that the successful application of MOF@Copc ASEI will offer innovative insights to achieve uniform lithium deposition and enhance stable long-lifespan performance.Experimental section
Preparation of NH2-MIL-101(Fe)
As for the preparation of NH2-MIL-101(Fe), 2.75 g of FeCl3·6H2O and 1.105 g of 2-aminoterephthalic acid were each dissolved separately in 40 mL of DMF to prepare solution A and solution B. The two solutions were treated with ultrasonic for 15 min. Next, solution B was gradually added to solution A under ultrasonic to obtain a homogeneous solution. The mixture was then transferred to the Teflon-lined autoclave and heated at 110 °C for 24 h. After the product was cooled to room temperature, it was collected by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 5 minutes, followed by washing with DMF three times and ethanol twice. The product was then dried under vacuum at 60 °C for 12 hours. Finally, NH2-MIL-101(Fe) powder was obtained by grinding.56
000 rpm for 5 minutes, followed by washing with DMF three times and ethanol twice. The product was then dried under vacuum at 60 °C for 12 hours. Finally, NH2-MIL-101(Fe) powder was obtained by grinding.56
Preparation of NH2-MIL-101(Fe)@Copc
NH2-MIL-101(Fe)@CoPc composites were synthesized via the impregnation method. 5 mg of CoPc was weighed and transferred into a 50 mL single-necked flask, followed by the addition of 40 mL of DMF to ultrasonically dissolve the CoPc. 60 mg of NH2-MIL-101(Fe) was then added, and the mixture was macerated at 75 °C for 40 hours. The solvent was removed by distillation under reduced pressure. The residue was washed by centrifugation with ethanol several times and then dried at 120 °C for 12 hours. The reagents used during the experiment were Macklin reagents.Preparation of NH2-MIL-101(Fe)@Copc/Li
60 mg of MOF@Copc powder was dissolved in 1.64 mL of tetrahydrofuran and stirred for 4 hours. Subsequently, in the glove box, 50 μL of the solution was dropped on lithium tablets and dried for 2 hours to obtain MOF@Copc/Li.Electrode preparation and battery assembly
The cathode, Li metal anode, PP battery separator, and 40 μL electrolyte were assembled into a CR2025 button cell. All the CR2025 coin cells above and the pouch battery were assembled in an argon-filled glove box with oxygen content <0.1 ppm and H2O content <0.1 ppm.Battery testing
The cycling stability of Li‖Li, Li‖Cu, Li‖Li, and pouch cell were conducted using the Neware multichannel battery testing systems at 30 °C. The cycle performance of LiFePO4‖Li batteries are tested between 2.5 and 4.2 V (1.0 M LiTFSI in dioxolane (DOL)/1,2-dimethoxyethane (DME) = 1:1 (v/v) with 2.0% LiNO3, Duo Duo chem), while NCM811‖Li batteries were tested at charge–discharge ranges of 2.8–4.3 V. (1 M LiPF6 in DEC:EC = 1:1 vol% with 10%FEC, Duo Duo chem). The NCM811‖Cu anode-free lithium-metal pouch batteries were tested at charge–discharge ranges of 2.75–4.2 V. (1 M LiPF6 in DEC:EC = 1:1 Vol% with 10%FEC, Duo Duo chem). The C rates in all of the electrochemical measurements are defined based on 1C = 200 mA g−1 (NCM811) and 1C = 170 mA g−1 (LiFePO4). LFP, PVDF, and CB were mixed by ball milling in NMP in a mass ratio of 8:1:1, then made into a slurry, uniformly coated onto charcoal-coated aluminum foil, and dried in a vacuum oven at 120 °C for 12 hours. The mass loading of the active substance was about 2.1 mg cm−2.
Materials characterization
The Fourier transform infrared (FT-IR) spectra were tested on a Thermo Nicolet iS50 Fourier infrared spectrometer. The material MOF@Copc was determined using X-ray diffractometer (XRD, Bruker D8 ADVANCE). The SEM images were conducted by field-emission scanning electron microscopy (FESEM, Carl Zeiss Gemini 500), EDX spectroscope attached to SEM (EDX mapping). Atomic force microscope (AFM, Bruker Dimension ICON) was employed to observe surface morphology. Nuclear magnetic resonance (NMR) spectra were tested on a JEOL JNM-ECZ400S/L1 NMR spectrometer with d6-DMSO as solvent. The Raman spectroscopy measurement was performed using a Laser Raman Spectrometer (InVia Qontor, Renishaw) with a 785 nm excitation wavelength. X-ray photoelectron spectrometer (XPS, Thermo Fisher ESCALAB Xi+) was used to conduct surface elemental analysis. Time-of-flight secondary ion mass spectrometry (TOF-SIMS) analysis was performed using a PHI nano TOF‖time-of-flight SIMS equipped with a 30 kV Bi-cluster liquid metal ion gun. Cryo-transmission electron microscopy (Cryo-TEM) characterization was performed on a 200 kV F FEI Talos F200C. Changes in morphology of the Li electrode during the electrochemical deposition process were directly observed by using an in situ optical microscope. The surface topography of the lithium after the cycle was observed using a Laser confocal microscopy (CLSM, Leica DCM8).Electrochemical measurements
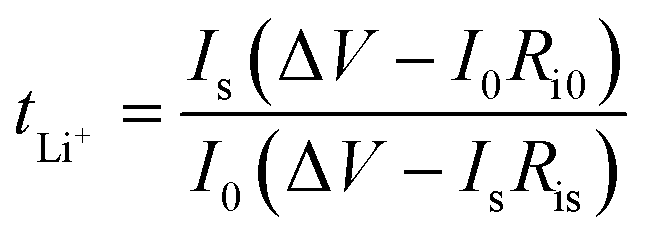 | (1) |
 | (2) |
| RCT−1 = Aexp(−Ea/RT) | (3) |
Calculation methods
The adsorption energy (Eads) can be calculated as follows:
| Eads (Li+ − TFSI−) = E(Li+ − TFSI−) − E(Li+) − E(TFSI−) | (4) |
| Eads (MOF − TFSI−) = E(MOF − TFSI−) − E(MOF) − E(TFSI−) | (5) |
| Eads (MOF − Copc) = E(MOF − Copc) − E(MOF) − E(Copc) | (6) |
Author contributions
F. Z. and H. L. and Y. W. contributed equally to this work, performed the experiments, and co-wrote the paper. X. Z., Y. C., and Y. Z. conceived the idea, planned the study, designed the experiment, and analyzed the data. F. Z. performed all of the experiments with the assistance of X. Y., H. W., and S. D. W. Y., and X. C. directed the revision of the article. All of the authors reviewed and commented on the manuscript.Data availability
All datasets generated or analyzed during the current study are included in the paper and its ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Key Research and Development Program of Shaanxi Province (2024PT-ZCK-82), and the Basic Strengthening Program (2022-JCJQ-JJ-0278). The Instrumental Analysis Center of Xi’an Jiaotong University is acknowledged for providing the test service. The authors deeply acknowledge the teachers at the Instrumental Analysis Center of Xi’an Jiaotong University, including Ms Jiamei Liu for the assistance with XPS, Ms Jia Liu for the assistance with in situ Raman test, and Ms Yan Liang for the assistance with the TOF-SIMS test.References
- H. Chen, Y. F. Yang, D. T. Boyle, Y. K. Jeong, R. Xu, L. S. de Vasconcelos, Z. J. Huang, H. S. Wang, H. X. Wang, W. X. Huang, H. Q. Li, J. Y. Wang, H. K. Gu, R. Matsumoto, K. Motohashi, Y. Nakayama, K. J. Zhao and Y. Cui, Nat. Energy, 2021, 6, 790–798 CrossRef CAS.
- X. He, D. Bresser, S. Passerini, F. Baakes, U. Krewer, J. Lopez, C. T. Mallia, Y. Shao-Horn, I. Cekic-Laskovic, S. Wiemers-Meyer, F. A. Soto, V. Ponce, J. M. Seminario, P. B. Balbuena, H. Jia, W. Xu, Y. B. Xu, C. M. Wang, B. Horstmann, R. Amine, C. C. Su, J. Y. Shi, K. Amine, M. Winter, A. Latz and R. Kostecki, Nat. Rev. Mater., 2021, 6, 1036–1052 Search PubMed.
- Y. Zhang, R. Qiao, Q. Nie, P. Zhao, Y. Li, Y. Hong, S. Chen, C. Li, B. Sun, H. Fan, J. Deng, J. Xie, F. Liu and J. Song, Nat. Commun., 2024, 15, 4454 CrossRef CAS PubMed.
- Z. J. Wang, Y. Y. Wang, Z. H. Zhang, X. W. Chen, W. Lie, Y. B. He, Z. Zhou, G. L. Xia and Z. P. Guo, Adv. Funct. Mater., 2020, 30, 2002414 CrossRef CAS.
- S. Y. Pan, J. W. Han, Y. Q. Wang, Z. S. Li, F. Q. Chen, Y. Guo, Z. S. Han, K. F. Xiao, Z. C. Yu, M. Y. Yu, S. C. Wu, D. W. Wang and Q. H. Yang, Adv. Mater., 2022, 34, 2203617 CrossRef CAS PubMed.
- Y. P. Gu, J. L. Hu, M. Lei, W. B. Li and C. L. Li, Adv. Energy Mater., 2024, 14, 2302174 CrossRef CAS.
- Y. P. Jiang, Q. Lv, C. Y. Bao, B. Wang, P. H. Ren, H. Y. Zhong, Y. Yang, X. M. Liu, Y. C. Dong, F. Jin, D. A. L. Wang, T. Xiong, H. K. Liu, S. X. Dou, J. Wang and J. M. Xue, Cell Rep. Phys. Sci., 2022, 3, 100785 CrossRef CAS.
- Y. Q. Liao, H. Y. Zhang, Y. F. Peng, Y. G. Hu, J. D. Liang, Z. L. Gong, Y. M. Wei and Y. Yang, Adv. Energy Mater., 2024, 14, 2304295 CrossRef CAS.
- X. Shen, R. Zhang, X. Chen, X. B. Cheng, X. Y. Li and Q. Zhang, Adv. Energy Mater., 2020, 10, 1903645 CrossRef CAS.
- P. B. Zhai, T. S. Wang, H. N. Jiang, J. Y. Wan, Y. Wei, L. Wang, W. Liu, Q. Chen, W. W. Yang, Y. Cui and Y. J. Gong, Adv. Mater., 2021, 33, 2006247 Search PubMed.
- Q. K. Zhang, X. Q. Zhang, J. Wan, N. Yao, T. L. Song, J. Xie, L. P. Hou, M. Y. Zhou, X. Chen, B. Q. Li, R. Wen, H. J. Peng, Q. Zhang and J. Q. Huang, Nat. Energy, 2023, 8, 725–735 Search PubMed.
- S. Z. Chang, J. B. Fang, K. Liu, Z. H. Shen, L. Zhu, X. Jin, X. J. Zhang, C. Q. Hu, H. G. Zhang and A. D. Li, Adv. Energy Mater., 2023, 13, 2204002 CrossRef CAS.
- Y. Z. Yang, J. Wang, Z. L. Li, Z. Yang, B. Wang and H. L. Zhao, ACS Nano, 2024, 18, 7666–7676 CrossRef CAS PubMed.
- H. Zhang, Z. Q. Zeng, F. F. Ma, X. L. Wang, Y. K. Wu, M. C. Liu, R. J. He, S. J. Cheng and J. Xie, Adv. Funct. Mater., 2023, 33, 2212000 CrossRef CAS.
- C. C. Su, M. N. He, M. Cai, J. Y. Shi, R. Amine, N. D. Rago, J. C. Guo, T. Rojas, A. T. Ngo and K. Amine, Nano Energy, 2022, 92, 106720 CrossRef CAS.
- H. K. Bergstrom and B. D. McCloskey, ACS Energy Lett., 2024, 9, 373–380 CrossRef CAS PubMed.
- C. Q. Jin, A. Xiang, Z. X. Wang, Q. Q. He, B. X. Li, X. K. Zhang, Y. Xiang, P. B. Zhai and Y. J. Gong, Adv. Energy Mater., 2025, 15, 2402811 CrossRef CAS.
- P. Zhao, Y. Zhang, B. Sun, R. Qiao, C. Li, P. Hai, Y. Wang, F. Liu and J. Song, Angew. Chem., Int. Ed., 2024, 63, e202317016 CrossRef CAS PubMed.
- Z. Li, L. Wang, X. D. Huang and X. M. He, Adv. Funct. Mater., 2024, 34, 2408319 CrossRef CAS.
- Y. J. Liu, X. Y. Tao, Y. Wang, C. Jiang, C. Ma, O. W. Sheng, G. X. Lu and X. W. Lou, Science, 2022, 375, 739–745 CrossRef CAS PubMed.
- Z. K. Ma, J. W. Chen, J. Vatamanu, O. Borodin, D. Bedrov, X. G. Zhou, W. G. Zhang, W. S. Li, K. Xu and L. D. Xing, Energy Storage Mater., 2022, 45, 903–910 CrossRef.
- H. A. Wang, H. Cheng, D. G. Li, F. Q. Li, Y. Wei, K. Huang, B. W. Jiang, H. H. Xu and Y. H. Huang, Adv. Energy Mater., 2023, 13, 2204425 CrossRef CAS.
- S. S. Yan, F. X. Liu, Y. Ou, H. Y. Zhou, Y. Lu, W. H. Hou, Q. B. Cao, H. Liu, P. Zhou and K. Liu, ACS Nano, 2023, 17, 19398–19409 CrossRef CAS PubMed.
- H. F. Zhuang, H. Xiao, T. F. Zhang, F. C. Zhang, P. Y. Han, M. Y. Xu, W. J. Dai, J. R. Jiao, L. Jiang and Q. M. Gao, Angew. Chem., Int. Ed., 2024, 63, e202407315 CrossRef CAS PubMed.
- X. Y. Zhou, F. F. Huang, X. D. Zhang, B. Zhang, Y. J. Cui, Z. H. Wang, Q. Yang, Z. S. Ma and J. Liu, Angew. Chem., Int. Ed., 2024, 63, e202401576 CrossRef CAS PubMed.
- W. Z. Liang, X. Y. Zhou, B. Zhang, Z. S. Zhao, X. Song, K. Chen, L. Wang, Z. S. Ma and J. Liu, Angew. Chem., Int. Ed., 2024, 63, e202320149 CrossRef CAS PubMed.
- J. Liu, D. W. Kang, Y. J. Fan, G. T. Nash, X. M. Jiang, R. R. Weichselbaum and W. B. Lin, J. Am. Chem. Soc., 2023, 146, 849–857 Search PubMed.
- Y. B. Song, S. Y. Hu, D. R. Cai, J. R. Xiao, S. F. Zhou and G. W. Zhan, ACS Appl. Mater. Interfaces, 2022, 14, 9151–9160 CrossRef CAS PubMed.
- L. Wei, X. Xu, K. Xi, Y. Lei, X. Cheng, X. B. Shi, H. H. Wu and Y. F. Gao, ACS Appl. Mater. Interfaces, 2024, 16, 33578–33589 CrossRef CAS PubMed.
- Y. H. Nie, T. Z. Yang, D. Luo, Y. Z. Liu, Q. Y. Ma, L. X. Yang, Y. Z. Yao, R. Huang, Z. Y. Li, E. M. Akinoglu, G. B. Wen, B. H. Ren, N. Zhu, M. Li, H. Liao, L. C. Tan, X. Wang and Z. W. Chen, Adv. Energy Mater., 2023, 13, 2204218 CrossRef CAS.
- L. Tu, Z. J. Zhang, Z. L. Zhao, X. Y. Xiang, B. H. Deng, D. Liu, D. Y. Qu, H. L. Tang, J. S. Li and J. P. Liu, Angew. Chem., Int. Ed., 2023, 62, e202306325 Search PubMed.
- C. Jiang, Q. Q. Jia, M. Tang, K. Fan, Y. Chen, M. X. Sun, S. F. Xu, Y. C. Wu, C. Y. Zhang, J. Ma, C. L. Wang and W. P. Hu, Angew. Chem., Int. Ed., 2021, 60, 10871–10879 CrossRef CAS PubMed.
- Q. C. Zhang, L. Xu, X. Y. Yue, J. J. Liu, X. Wang, X. Y. He, Z. D. Shi, S. Z. Niu, W. Gao, C. Cheng and Z. Liang, Adv. Energy Mater., 2023, 13, 2302620 CrossRef CAS.
- Z. Y. Han, H. R. Ren, Z. J. Huang, Y. B. Zhang, S. C. Gu, C. Zhang, W. H. Liu, J. L. Yang, G. M. Zhou, Q. H. Yang and W. Lv, ACS Nano, 2023, 17, 4453–4462 Search PubMed.
- Y. M. Y. Zhao, L. B. Li, H. Yang, S. B. Fan, S. Li and H. Tong, Energy Storage Mater., 2024, 65, 103126 CrossRef.
- Z. G. Chen, W. M. Chen, H. X. Wang, C. Zhang, X. Q. Qi, L. Qie, F. S. Wu, L. Wang and F. Q. Yu, Nano Energy, 2022, 93, 106836 CrossRef CAS.
- B. Jagger and M. Pasta, Joule, 2023, 7, 2228–2244 CrossRef CAS.
- S. N. Zhang, Y. H. Li, L. J. Bannenberg, M. Liu, S. Ganapathy and M. Wagemaker, Sci. Adv., 2024, 10, eadj8889 CrossRef CAS PubMed.
- Z. J. Ju, C. B. Jin, X. H. Cai, O. W. Sheng, J. C. Wang, J. M. Luo, H. D. Yuan, G. X. Lu, X. Y. Tao and Z. Liang, ACS Energy Lett., 2022, 8, 486–493 CrossRef.
- T. Duan, H. W. Cheng, Y. B. Liu, Q. C. Sun, W. Nie, X. G. Lu, P. P. Dong and M. K. Song, Energy Storage Mater., 2024, 65, 103091 CrossRef.
- L. G. Yue, X. Y. Wang, L. Chen, D. J. Shen, Z. H. Shao, H. Wu, S. F. Xiao, W. Q. Liang, Y. J. Yu and Y. Y. Li, Energy Environ. Sci., 2024, 17, 1117–1131 RSC.
- X. Z. Fan, J. H. Zhang, N. Yao, J. X. Chen, X. Chen and L. Kong, Adv. Energy Mater., 2024, 14, 2303336 CrossRef CAS.
- Z. H. Wu, C. Y. Wang, Z. Y. Hui, H. D. Liu, S. Wang, S. C. Yu, X. Xing, J. Holoubek, Q. S. Miao, H. L. Xin and P. Liu, Nat. Energy, 2023, 8, 340–350 CAS.
- G. X. Lu, Q. Q. Qiao, M. T. Zhang, J. S. Zhang, S. Li, C. B. Jin, H. D. Yuan, Z. J. Ju, R. Huang, Y. J. Liu, J. M. Luo, Y. Wang, G. M. Zhou, X. Y. Tao and J. Nai, Sci. Adv., 2024, 10, eado7348 CrossRef CAS PubMed.
- C. Y. Wang, C. P. Yang, Y. H. Du, Z. P. Guo and H. Ye, Adv. Funct. Mater., 2023, 33, 2303427 CrossRef CAS.
- X. R. Chen, Y. X. Yao, C. Yan, R. Zhang, X. B. Cheng and Q. Zhang, Angew. Chem., Int. Ed., 2020, 59, 7743–7747 CrossRef CAS PubMed.
- J. C. Guo, S. J. Tan, C. H. Zhang, W. P. Wang, Y. Zhao, F. Y. Wang, X. S. Zhang, R. Wen, Y. Zhang, M. Fan, S. Xin, J. Zhang and Y. G. Guo, Adv. Mater., 2023, 35, 2300350 CrossRef CAS PubMed.
- J. J. Pan, K. X. Shi, H. Wu, J. H. Li, R. Zhang, Q. B. Liu and Z. X. Liang, Adv. Energy Mater., 2024, 14, 2302862 CrossRef CAS.
- Z. Q. Shi, Y. M. Wang, X. Y. Yue, J. Zhao, M. M. Fang, J. J. Liu, Y. M. Chen, Y. T. Dong, X. Z. Yan and Z. Liang, Adv. Mater., 2024, 36, 2401711 CrossRef CAS PubMed.
- Y. Y. Zhang, Y. Guo, K. Yong, Q. Wang, M. Yao, Y. Zhang and H. Wu, Energy Environ. Sci., 2024, 17, 5819–5832 RSC.
- H. Liu, F. X. Zhen, X. K. Yin, Y. B. Wu, K. L. Yu, X. P. Kong, S. J. Ding and W. Yu, Angew. Chem., Int. Ed., 2025, 64, e202414599 CrossRef CAS PubMed.
- J. Chen, X. T. Deng, X. Jia, Y. Gao, H. Chen, Z. Q. Lin and S. J. Ding, J. Am. Chem. Soc., 2024, 146, 30836–30847 CrossRef CAS PubMed.
- Z. H. Sun, Y. K. Wang, S. Y. Shen, X. Y. Li, X. F. Hu, M. Y. Hu, Y. Q. Su, S. J. Ding and C. H. Xiao, Angew. Chem., Int. Ed., 2023, 62, e202309622 CrossRef CAS PubMed.
- G. X. Lu, X. R. Wu, M. F. Huang, M. T. Zhang, Z. H. Piao, X. W. Zhong, C. Li, Y. Z. Song, C. S. Chang, K. Yu and G. M. Zhou, Energy Environ. Sci., 2024, 17, 9555–9565 RSC.
- W. Wu, F. Niu, C. K. Sun, Q. R. Wang, M. Wang, J. Wang, Y. H. Deng, D. Ning, W. J. Li, J. Zhang, M. Chen, H. M. Cheng and C. L. Yang, Adv. Mater., 2024, 36, 2404630 CrossRef CAS PubMed.
- Y. P. Song, L. N. He, S. Zhang, X. Liu, K. Chen, Q. J. Jia, Z. H. Zhang and M. Du, Food Chem., 2021, 351, 129248 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 19 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee01219h |
| ‡ Co-first author. |
| This journal is © The Royal Society of Chemistry 2025 |