
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRu(IV)–Ru(IV), Ru(III)–Ru(IV), and Ru(III)–Ru(III) complexes having a sulfato bridging ligand on the doubly oxido-/hydroxido-bridged core, and their crystallographic and electronic structures†
Tomoyo
Misawa-Suzuki
 *a,
Yusuke
Kataoka
b,
Sota
Mafune
a,
Teresa
Niwa
a and
Hirotaka
Nagao
a
*a,
Yusuke
Kataoka
b,
Sota
Mafune
a,
Teresa
Niwa
a and
Hirotaka
Nagao
a
aDepartment of Materials and Life Sciences, Faculty of Science and Technology, Sophia University, 7-1 Kioicho, Chiyoda-ku, Tokyo 102-8554, Japan. E-mail: t_misawa@sophia.ac.jp
bDepartment of Chemistry, Graduate School of Natural Science and Technology, Shimane University, 1060, Nishikawatsu, Matsue 690-8504, Shimane, Japan
First published on 27th December 2024
Abstract
The Ru2(IV,IV), Ru2(III,IV), and Ru2(III,III) complexes with the doubly oxido- and/or hydroxido-bridged diamond core {Ru2(μ-O(H))2}, bridged by an η1:η1:μ2-type bidentate sulfato ligand, [{Ru(L)}2(μ-O)2(μ-O2SO2)]m+ (m = 1: [III,IV]+; m = 2: [IV,IV]2+), [{RuIII,IV(L)}2(μ-O)(μ-OH)(μ-O2SO2)]2+ ([III,IV_1H]2+), and [{RuIII,III(L)}2(μ-OH)2(μ-O2SO2)]2+ ([III,III_2H]2+) (L = ethylbis(2-pyridylmethyl)amine), were synthesised as ClO4−-salts, and their crystal and electronic structures investigated. The corresponding hydrogencarbonato-bridged Ru2(III,III) complex, [{RuIII,III(L)}2(μ-OH)2(μ-O2COH)]3+ ([III,III(HCO3)_2H]3+), was also prepared and its crystallographic and electronic structures compared to those of the sulfato-bridged system, [III,III_2H]2+. All the sulfato-bridged complexes isolated were confirmed in the Pourbaix diagram, wherein the redox potential was plotted as a function of pH. The pKas were determined using UV-vis-NIR spectroscopic measurements and incorporated into the Pourbaix diagram. This is the first sequential study, both experimentally and theoretically, on the various oxidation states of Ru centres and the number of protons on the {Ru2(μ-O(H))2} core in the same tridentate ligand system for complexes with the {Ru2(μ-O(H))2(μ-O2XY)}-type dinuclear framework, which are analogous to the intermediate structures of the diiron-containing soluble methane monooxygenase.
Introduction
Multi-nuclear transition metal complexes having a doubly oxido- and/or hydroxido-bridged core, {M2(μ-O(H))2}, have attracted much attention as reaction centres in a number of natural metalloenzymes. For instance, the oxidation states of the active site of soluble methane monooxygenase (sMMO) range from Fe2(II,II) to Fe2(IV,IV) during the course of the catalytic reaction.1 Studies on the structural changes of/around the reaction centres, namely, the {M2(μ-O(H))n} (n = 1, 2) core, accompanied by redox reactions coupled with proton transfers, are important for discussions on reaction mechanisms. For example, the doubly oxido- and/or hydroxido-bridged metal core, {FeIV,IV2(μ-O)2}, is the potential structure of the active species Q, but the debate over whether the core structure is the “open-type” or “closed-type” has long been continued regarding Q. Furthermore, the resting state (MMOHox) has been reported to have the {FeIII,III2(μ-OH)2} core. A number of studies on the electronic structures, catalytic oxidation reactions, and model reactions by sMMO analogues have been reported using diiron2 or dinickel centres.3 As structural model compounds of such diiron-centred reaction sites, ruthenium complexes as the same 8th group element-containing compounds have also been intensively investigated; however, the number of studies is still limited. Although Ru2(V,V),6a,b Ru2(IV,IV),6 Ru2(III,IV),4 and Ru2(III,III)6b,5 complexes with the {Ru2(μ-O(H))2} diamond core have been reported in different supporting ligand systems, only a few in the same supporting ligand system have been reported to date, making direct comparisons of the properties and reactivities of the reaction centres difficult. Bercaw et al. reported the isovalent species, Ru2(V,V), Ru2(IV,IV), and Ru2(III,III) with the phosphate-donor oxygen tripod ligand [CoIII(Cp)(PO(OR)2)3]− (Cp = cyclopentadienyl, R = Me, Et) system;6a,b however, the mixed-valent species have not been isolated and characterised in their ligand system.We reported diruthenium complexes with {Ru2(μ-O(H))n} (n = 1, 2) cores using the ethylbis(2-pyridylmethyl)amine (ebpma) tridentate ligand.7b–f For the {Ru2(μ-O(H))2} core, we synthesised carbonato-,7e hydrogencarbonato-,7e acetato-,7c,d and nitrato-bridged7c complexes in the Ru2(III,IV) states, in which S = 1/2 at 298 K, and compared their structures and properties. In addition, we recently reported the Ru2(IV,IV) complexes using carbonato and hydrogencarbonato bridging ligands, and their reactivity in solution and application toward the oxidation of organic substrates were demonstrated.7f
In this study, sulfato-bridged complexes in the Ru2(IV,IV), Ru2(III,IV), and Ru2(III,III) states and a hydrogencarbonato-bridged Ru2(III,III) complex, for which only the higher-valent Ru2(IV,IV) and Ru2(III,IV) species with the corresponding frameworks have been identified,7e,f were synthesised. The crystal structures and electronic structures of these complexes in one ligand system were investigated and directly compared. These complexes are significant as structural models for the series of reaction intermediates of sMMO.
Results and discussion
Synthesis of sulfato-bridged complexes in the Ru2(IV,IV), Ru2(III,IV), and Ru2(III,III) states and a hydrogencarbonato-bridged Ru2(III,III) complex having the {Ru2(μ-O(H))2} cores
The synthetic procedure is summarised in Scheme 1. Sulfato-bridged Ru2(III,IV) complexes having the {Ru2(μ-O)2} or {Ru2(μ-O)(μ-OH)} core, [{RuIII,IV(ebpma)}2(μ-O)2(μ-O2SO2)]ClO4·0.25(CH3)2CO·4H2O ([III,IV]ClO4) and [{RuIII,IV(ebpma)}2(μ-O)(μ-OH)(μ-O2SO2)](ClO4)2·3H2O ([III,IV_1H](ClO4)2), respectively, were synthesised through reactions of a nitrato-bridged complex, [{RuIII,IV(ebpma)}2(μ-O)2(μ-O2NO)](ClO4)2·1.5H2O ([III,IV(NO3)](ClO4)2), which was prepared by the reaction of a singly oxido-bridged complex ([{RuIII,IVCl2(ebpma)}2(μ-O)]ClO4·0.75(CH3)2CO)7b with four molar equivalents of AgNO3.7c Above pH 3 in the presence of sulfate sources such as Na2SO4, the doubly oxido-bridged complex ([III,IV]ClO4) was synthesised, and at approximately pH 1, the hydroxido- and oxido-bridged complex ([III,IV_1H](ClO4)2) was isolated. [III,IV_1H](ClO4)2 could also be formed in a reaction of [{RuIII,IVCl2(ebpma)}2(μ-O)]+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7b with ten-fold Na2SO4 in water/acetone if four molar equivalents of AgTfO (TfO− = trifluoromethanesulfonate) were present. The corresponding Ru2(IV,IV) complex, [{RuIV,IV(ebpma)}2(μ-O)2(μ-O2SO2)](ClO4)2·0.5H2O ([IV,IV](ClO4)2), was isolated through the one-electron oxidation of [III,IV(NO3)]2+ by cerium(IV) ammonium sulfate (CAS) at pH 2.3, owing to nitrato ligand substitution to sulfate in CAS. [IV,IV]2+ was also afforded by the one-electron oxidation of [III,IV]+ by cerium(IV) ammonium nitrate. The Ru2(III,III) complex having the doubly hydroxido-bridged core {Ru2(μ-OH)2}, [{RuIII,III(ebpma)}2(μ-OH)2(μ-O2SO2)](ClO4)2·3H2O ([III,III_2H](ClO4)2), was synthesised via a one-electron reduction reaction of [III,IV_1H]2+ by SnCl2·2H2O, coupled with a protonation, at approximately pH 2. [III,III_2H]2+ was also afforded directly from [III,IV(NO3)]2+ under similar reaction conditions via the formation of [III,IV_1H]2+ in solution. A corresponding Ru2(III,III) complex having a hydrogencarbonato bridging ligand, [III,III(HCO3)_2H](ClO4)3·H2O, was newly prepared in the reaction of the corresponding Ru2(III,IV) complex, [III,IV(CO3)_2H]3+.7e
7b with ten-fold Na2SO4 in water/acetone if four molar equivalents of AgTfO (TfO− = trifluoromethanesulfonate) were present. The corresponding Ru2(IV,IV) complex, [{RuIV,IV(ebpma)}2(μ-O)2(μ-O2SO2)](ClO4)2·0.5H2O ([IV,IV](ClO4)2), was isolated through the one-electron oxidation of [III,IV(NO3)]2+ by cerium(IV) ammonium sulfate (CAS) at pH 2.3, owing to nitrato ligand substitution to sulfate in CAS. [IV,IV]2+ was also afforded by the one-electron oxidation of [III,IV]+ by cerium(IV) ammonium nitrate. The Ru2(III,III) complex having the doubly hydroxido-bridged core {Ru2(μ-OH)2}, [{RuIII,III(ebpma)}2(μ-OH)2(μ-O2SO2)](ClO4)2·3H2O ([III,III_2H](ClO4)2), was synthesised via a one-electron reduction reaction of [III,IV_1H]2+ by SnCl2·2H2O, coupled with a protonation, at approximately pH 2. [III,III_2H]2+ was also afforded directly from [III,IV(NO3)]2+ under similar reaction conditions via the formation of [III,IV_1H]2+ in solution. A corresponding Ru2(III,III) complex having a hydrogencarbonato bridging ligand, [III,III(HCO3)_2H](ClO4)3·H2O, was newly prepared in the reaction of the corresponding Ru2(III,IV) complex, [III,IV(CO3)_2H]3+.7e
 | ||
| Scheme 1 Synthesis of a series of sulfato-bridged diruthenium complexes and a hydrogencarbonato-bridged complex. | ||
The sulfato ligand functions as a multidentate bridging ligand with versatile coordination modes to form multinuclear complexes (for example, with first,8–16 second,17–24 and third row25–29 d-block elements and lanthanoids30–33 and actinoids34,35), and it has been investigated as one of the building blocks of multinuclear frameworks or self-assembled structures. Among the coordination modes, there have been a limited number of Ru dimers,20 and to the best of our knowledge, only a small number of crystal structures have been characterised or finely proposed in the η1:η1:η1:μ2-mode20f or in the η1:η1:μ2-mode20a–c,g which is the same type as the complexes described herein. Therefore, reporting the η1:η1:μ2-O2SO22−-bridged complexes in the Ru2(IV,IV), Ru2(III,IV), and Ru2(III,III) states significantly expands the available structural data across a wide range of oxidation states.
X-ray crystallography
The crystal structures of all the species, [IV,IV]2+, [III,IV]+, [III,IV_1H]2+, [III,III_2H]2+, and [III,III(HCO3)_2H]3+, were successfully characterised. Crystallographic data are summarised in Table S1,† and selected metric parameters are listed in Table 1. The crystal structures are shown in Fig. 1–3 and Fig. S1 and S2.† Each ruthenium centre had a distorted hexacoordinate octahedral geometry and was coordinated by tridentate ebpma in the facial manner; the other three coordination sites were occupied by two bridging oxido or hydroxido ligands and one of the sulfato oxygens. The sulfato ligand bridged the {Ru2(μ-O(H))2} core through two oxygens as a bidentate ligand. The hydrogencarbonato in [III,III(HCO3)_2H]3+ bridged the {Ru2(μ-OH)2} core in a similar manner to a bidentate ligand. Both the bridging oxido or hydroxido ligands were located trans to the pyridyl (py) and amine nitrogen atoms, which was the same geometrical configuration as that of previously reported systems.7c–f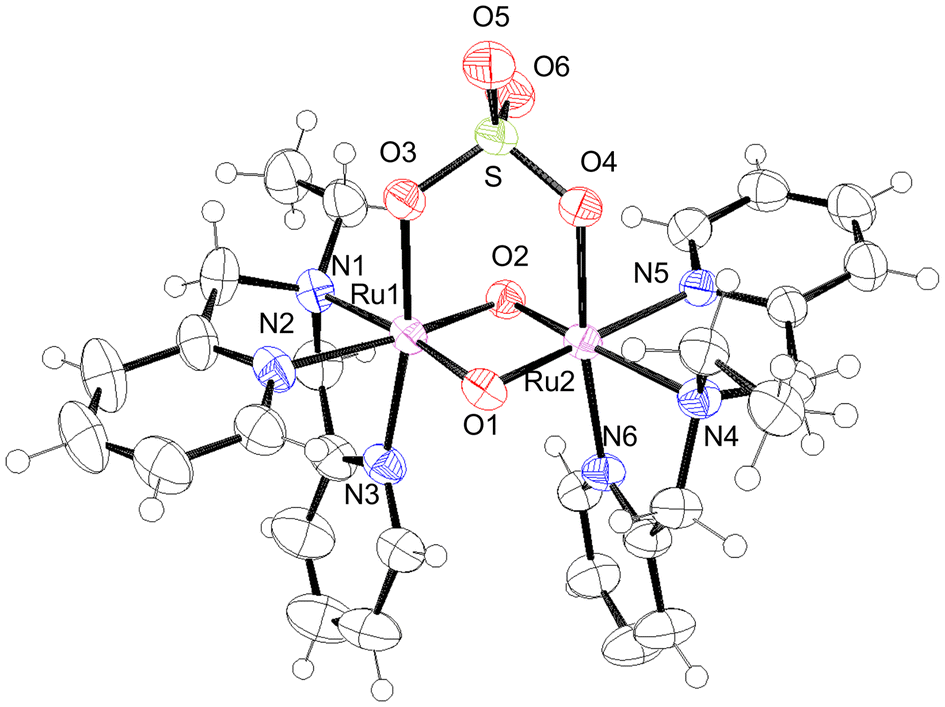 | ||
| Fig. 1 ORTEP drawing of [IV,IV]2+ shown at the 50% thermal probability. | ||
 | ||
| Fig. 2 ORTEP drawing of [III,IV_1H]2+ shown at the 50% thermal probability (notation of H is added to the bridging oxygen where the Ru–O bond lengths were elongated). | ||
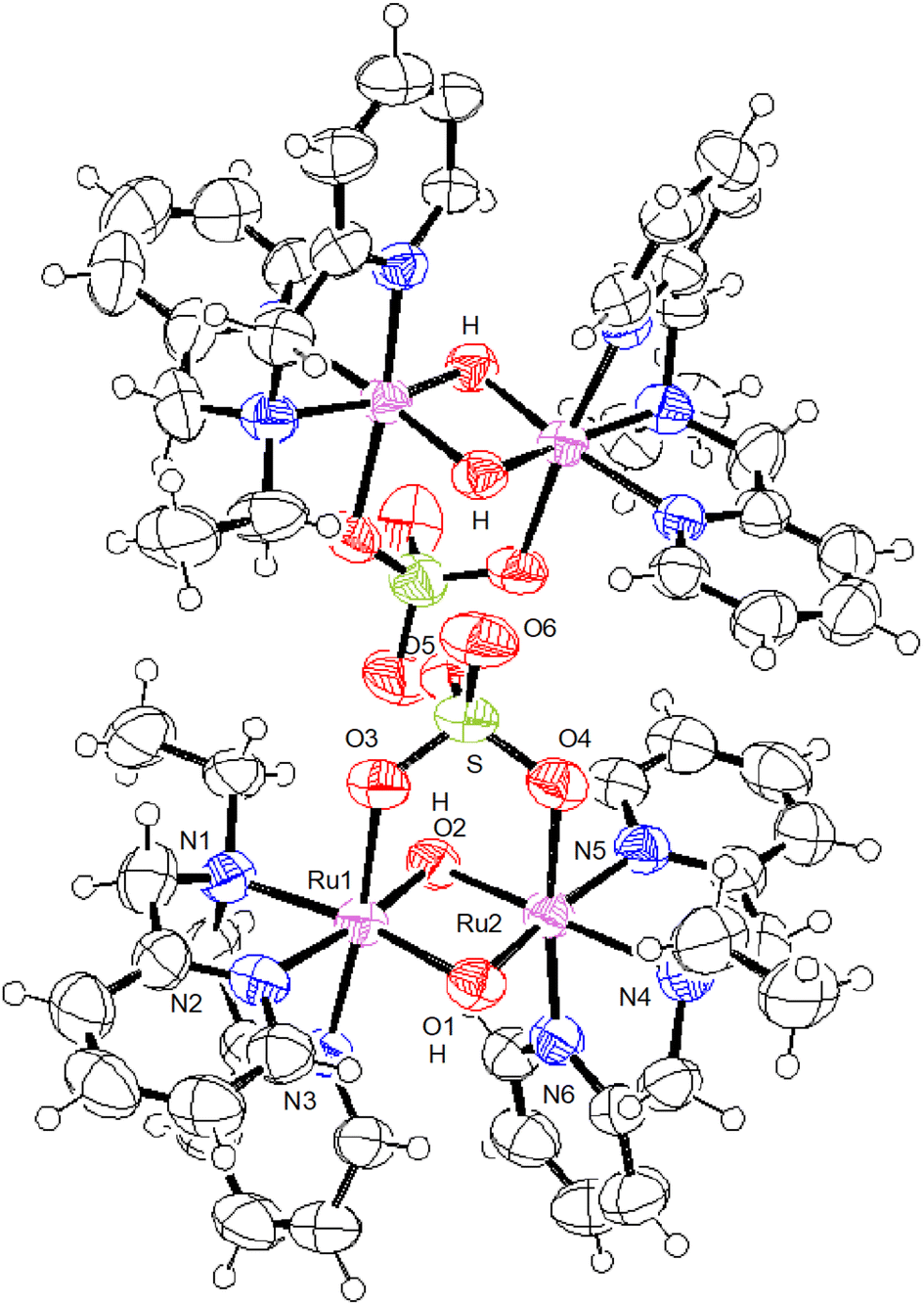 | ||
| Fig. 3 ORTEP drawing of [III,III_2H]2+ shown at the 50% thermal probability (notation of H is added for the bridging oxygen). | ||
| Ru(III)–Ru(III) | Ru(III)–Ru(IV) | Ru(IV)–Ru(IV) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| [III,III(HCO3)_2H](ClO4)3·0.25H2O | [III,III_2H](ClO4)2·1.5H2O | [III,IV]ClO4·3.5H2O | [III,IV_1H](ClO4)2·0.75H2O | [IV,IV](ClO4)2 ·H2O | ||||||
| Bond distances/Å | ||||||||||
| Ru–O1 | 2.028(3) | 2.026(2) | 2.023(6) | 2.023(6) | 1.930(2) | 1.945(2) | 1.921(2) | 1.913(2) | 1.9080(19) | 1.9042(19) |
| Ru–O2 | 2.014(3) | 2.024(3) | 2.010(6) | 2.020(6) | 1.938(3) | 1.937(3) | 2.030(2) | 2.028(2) | 1.9173(19) | 1.926(2) |
| Ru1⋯Ru2 | 2.5657(4) | 2.5695(7) | 2.4705(4) | 2.5342(3) | 2.4470(3) | |||||
| Ru–O(bidentate ligand) | 2.095(3) | 2.070(3) | 2.121(5) | 2.109(5) | 2.093(3) | 2.089(3) | 2.080(2) | 2.086(3) | 2.0520 (18) | 2.0419(18) |
| Bond angles/° | ||||||||||
| Ru1–O1,2—Ru2 | 78.52(9) | 78.88(10) | 78.9(2) | 79.2(2) | 79.22(9) | 79.22(10) | O1O2− 82.77(9) | O2OH–: 77.28(8) | 79.87(7) | 79.08(7) |
| O1–Ru1,2–O2 | 101.03(10) | 100.74(10) | 101.0(2) | 100.6(2) | 100.92(10) | 100.46(10) | 99.51(10) | 99.87(10) | 100.49(8) | 100.30 (8) |
| Ru–O(bidentate ligand)-S/C | 120.0(2) | 121.7(3) | 119.7(3) | 117.9(3) | 122.89(16) | 123.53(15) | 119.93(15) | 118.31(14) | 121.76(11) | 121.71(11) |
The metric parameters around the {Ru2(μ-O(H))2} core differed depending on the electronic structure of the Ru centres and the number of protons on the core, whereas those around the {μ-O2SO2}2− ligand were similar among the complexes (Table 1). Between [IV,IV]2+ and [III,IV]+ having the {Ru2(μ-O)2} core, the Ru–O and Ru⋯Ru distances were shorter for [IV,IV]2+ (average: 1.917 and 2.4470(3) Å) than those of [III,IV]+ (average: 1.939 and 2.4705(4) Å) because of the more cationic Ru(IV) centres, while the bond angles around the core were similar. The shorter Ru⋯Ru distance in [IV,IV]2+ indicated stronger interactions between the Ru centres. In [III,IV_1H]2+ having the {Ru2(μ-O)(μ-OH)} core, the Ru–O2 distances in [III,IV_1H]2+ (average: 2.029 Å) was elongated by 0.05–0.10 Å compared to the other Ru–O1 bond lengths in [III,IV_1H]2+ (average: 1.917 Å) and [III,IV]+ having the {Ru2(μ-O)2} core (average: 1.939 Å), suggesting that one of the bridging oxido ligands (O2), which was away from one of the terminal sulfato oxygens (O6), was singly protonated. Accordingly, the Ru⋯Ru distance was elongated (2.5342(3) Å) compared to that of [III,IV]+ (2.4705(3) Å), and the Ru–OH–Ru angle (77.28(8)°) was slightly smaller than the Ru–O–Ru angle (82.77(9)° in [III,IV_1H]2+). It is noteworthy that, according to the theoretical calculations, a geometrical isomer of [III,IV_1H]2+, in which O1 is singly protonated instead of O2 (Table S10† Model-B), could slightly be stabilized by 3.13 kcal mol−1 in free energy (Table S11†) owing to the intramolecular hydrogen bonding interactions between O1–H and O6–SO32− (1.677 Å). However, it was rather challenging to separate the isomers in the synthetic procedures.
For [III,III_2H]2+, the two Ru–OH bond lengths (average: 2.019 Å) were almost identical to those of [III,IV_1H]2+ (average: 2.019 Å); however, because both oxygen atoms were singly protonated, the Ru⋯Ru distance (2.5695(7) Å) was longer than that in [III,IV_1H]2+ (2.5342(3) Å) but comparable to that in [III,III(HCO3)_2H]3+ (2.5657(4) Å). The angles around the {Ru2(μ-OH)2} core differ slightly from those of [III,IV_1H]2+. The longer Ru⋯Ru distances in [III,III_2H]2+ and [III,III(HCO3)_2H]3+ suggest weaker interactions between the dπ(Ru) centres. The closer orbital energies of the LUMO and HOMO may contribute to unpairing the electrons in the HOMO, resulting in the observation of a certain extent of a paramagnetic character of [III,III_2H]2+, as discussed later.
The Ru–O3,4(sulfato) bond lengths were longer in [III,III_2H]2+ (average: 2.115 Å), followed by [III,IV]+ (average: 2.091 Å) and [III,IV_1H]2+ (average: 2.083 Å), and [IV,IV]2+ (average: 2.047 Å), indicating that the sulfato ligand coordinated more strongly to the more cationic {Ru2(μ-O(H))2} core. Furthermore, the Ru–N2,5(py, trans-O(oxido/hydroxido)) bond lengths were longer in [III,IV]+ and [IV,IV]2+ having the {Ru2(μ-O)2} core (2.0912.103 Å) compared to those in [III,IV_1H]2+ and [III,III_2H]2+ having the {Ru2(μ-O)(μ-OH)} or {Ru2(μ-OH)2} cores (2.062–2.078 Å), respectively. Upon protonation to the bridging oxido ligand, the pπ(O)–dπ(Ru) interactions became stronger by decreasing the number of lone pair electrons on oxygen.
The metric parameters were also compared with those of complexes having different bridging ligands, {μ-O2CO}2− ([IV,IV(CO3)]2+7f or [III,IV(CO3)]+7e), {μ-O2COH}− ([IV,IV(HCO3)]3+,7f [III,IV(HCO3)]2+,7e [III,IV(HCO3)_1H]3+,7e or [III,III(HCO3)_2H]3+), {μ-O2CCH3}− ([III,IV(CH3O2)]2+7c or [III,IV(CH3O2)_1H]3+7d), and {μ-O2NO}− ([III,IV(NO3)]2+7c). The order of the Ru⋯Ru distances was consistent among the Ru2(IV,IV) and Ru2(III,IV) complexes; [IV,IV]2+ (2.4470(3) Å) > [IV,IV(HCO3)]3+ (2.4389(3) Å) > [IV,IV(CO3)]2+ (2.4261(3) Å) ≈ [III,IV]+ (2.4705(4) Å) > [III,IV(HCO3)]2+ (2.4547(2) Å) > [III,IV(CH3O2)]2+ (2.4463(10) Å) > [III,IV(CO3)]+ (2.4434(8) Å) > [III,IV(NO3)]2+ (2.4334(4) Å). These results were in accordance with the order of the angles of Ru–O–X (X = S(sp3) > C(sp2) > N(sp2)) of the respective {μ-O2XOn}m− (n = 1, 2), in which the central X atom has a different geometry and flexibility upon coordinating to the Ru centres. Notably, the electrostatic nature of the {μ-O2XOn}m− ligand does not seem to influence the metric parameters. The same reasoning regarding the Ru⋯Ru distance is applicable to complexes having the asymmetric {Ru2(μ-O)(μ-OH)} core and the Ru2(III,III) complexes having the {Ru2(μ-OH)2} core; [III,IV_1H]2+ (2.5342(3) Å) > [III,IV(HCO3)_1H]3+ (2.5332(5) Å) > [III,IV(CH3CO2)_1H]2+ (2.5201(6) Å) and [III,III_2H]2+ (2.5695(7) Å) > [III,III(HCO3)_2H]3+ (2.5657(4) Å).
Electronic structures
The 1H NMR spectra of [IV,IV]2+, [III,IV]+, and [III,IV_1H]2+ showed some broad signals, which were observed only in the diamagnetic region and were not assignable (Fig. S4 and S5†). Therefore, magnetic measurements were performed at 298 K. The effective magnetic moment (μeff) of [IV,IV](ClO4)2·0.5H2O was 0.94μB, indicating the paramagnetic character of the d4–d4 system, almost consistent with those of the corresponding carbonato- and hydrogencarbonato-bridged complexes, [IV,IV(CO3)]2+ and [IV,IV(HCO3)]3+ (1.04 and 0.85μB, respectively).7f We newly performed the theoretical calculations of [IV,IV]2+ for the quintet, open-shell singlet, and closed-shell singlet states to testify which best describes the electronic structure of [IV,IV]2+. The free energy was lower for the open-shell singlet state than those of other states (Table S8†). Specifically, the paramagnetic features experimentally studied could be supported by the theoretical result that the free energy of the open-shell singlet was lower than that of the closed-shell singlet state by −6.27 kcal mol−1. The metric parameters of the optimised structure for the open-shell singlet state were closer to those observed in the single crystal XRD analysis than those obtained from the calculation for the quintet state (Table S7†). Therefore the MO energy diagram of the open-shell singlet state is shown in Fig. S35.† The spin density was clearly localised on the {Ru2(μ-O)2} core (Fig. S36†), showing the stronger anti-ferromagnetic interactions between the Ru (S = 1) centers.The μeff values of [III,IV]ClO4·0.25(CH3)2CO·4H2O and [III,IV_1H](ClO4)2·3H2O were 1.79 (296 K) and 1.69μB (298 K), respectively, suggesting one electronic spin on the {RuIII,IV2(μ-O)2} core (S = 1/2). The theoretical calculations were performed assuming the doublet state for both [III,IV]+ and [III,IV_1H]2+ and the optimised structures (Fig. S37 and S39†) were well-consistent with the crystal structures (Tables S9 and S10†). The spin density plot supported the paramagnetic character of the {Ru2(μ-O(H))2} core (Fig. S38 and S40†). ESR signals centred at g = 2.07 − 2.09 were observed for [III,IV]+ and [III,IV_1H]2+ at both 298 (powder samples) and 93 K (frozen acetonitrile) (Fig. S8, S9 and Table S3†), which supported the paramagnetic characters of the {Ru2(μ-O(H))2} core.
For [III,III_2H]2+ and [III,III(HCO3)_2H]3+, the 1H and 13C NMR spectra confirmed the diamagnetic character of the {RuIII,III2(μ-OH)2} core (Fig. S6 and S7†). The proton or carbon atoms of the ebpma ligand could be assigned in the respective spectrum. The protons at the bridging oxido ligands were observed at 16.60 and 15.75 ppm for [III,III_2H]2+ and [III,III(HCO3)_2H]3+, respectively, and were reasonably quantified as 2H (Fig. S6†). The more downshifted hydroxido proton signals for [III,III_2H]2+ suggested higher electron density, which was consistent with the pKa values of the {Ru2(μ-OH)2} (5.7) and {Ru2(μ-O)(μ-OH)} cores (4.2) (eqn (3)), determined from spectroscopic investigations (Fig. S14 and S15†). Although the NMR spectra of [III,III_2H](ClO4)2·3H2O were reasonably assigned, its paramagnetic character was also suggested in the magnetic measurements (μeff = 1.15μB at 298 K). Based on the theoretical calculations, which were performed for both closed-shell singlet and triplet states, the singlet state was stabilised by only −0.168 kcal mol−1 in free energy (298 K) compared to the triplet state (Table S13†). The spin state transition from the closed-shell singlet to triplet state could occur by such a lower thermal barrier. These computational results could explain the magnetic characters of [III,III_2H]2+.
The MO energy diagram of the closed-shell singlet state is shown in Fig. S41†. The metric parameters of the optimized structure obtained for a singlet state calculation perfectly agreed with those experimentally confirmed (Table S12†). Based on the theoretical results for [III,III_2H]2+, the calculations were performed for [III,III(HCO3)_2H]3+ as the closed-shell singlet state (Fig. S42 and Table S14†).
UV-vis-NIR spectroscopic properties and electrochemical behaviours in CH3CN
The electron transition spectra in acetonitrile of [IV,IV]2+, [III,IV]+, and [III,IV_1H]2+ (Fig. 4 and Table 2) exhibited maximum absorption wavelengths (λmax) of 699, 945, and 1077 nm, respectively, which could be assigned to the characteristic electron transitions from and to the {Ru2(μ-O(H))2} diamond core. These bands indicated that the SOMO of the {Ru2(μ-O)2} core was more stabilised in the order of [IV,IV]2+ > [III,IV]+ > [III,IV_1H]2+. [III,III_2H]2+ showed an intense band at 328 nm, assigned to the metal-to-ligand charge transfer (MLCT) band from the {RuIII,III2(μ-OH)2} core to the π*(py) of the ebpma ligand. On comparing the spectrum of [IV,IV]2+ (699 nm) with those of [IV,IV(CO3)]2+ (762 nm) and [IV,IV(HCO3)]3+ (669 nm),7f it is found that the order of the λmax corresponds to that of the electron-donating character of the bidentate ligand on the {Ru2(μ-O)2} core ({μ-O2CO}2−7f > {μ-O2SO2}2− > {μ-O2COH}−;7fTable 2 and Fig. S10†), indicating the destabilisation of the core due to the coordination of a more electron-donating ligand. Time-dependent DFT calculations were performed for all the complexes and the wavelengths of characteristic absorption bands in the visible and near-IR regions relatively matched the experimentally derived spectra (Fig. S43–S46 and Table S15†).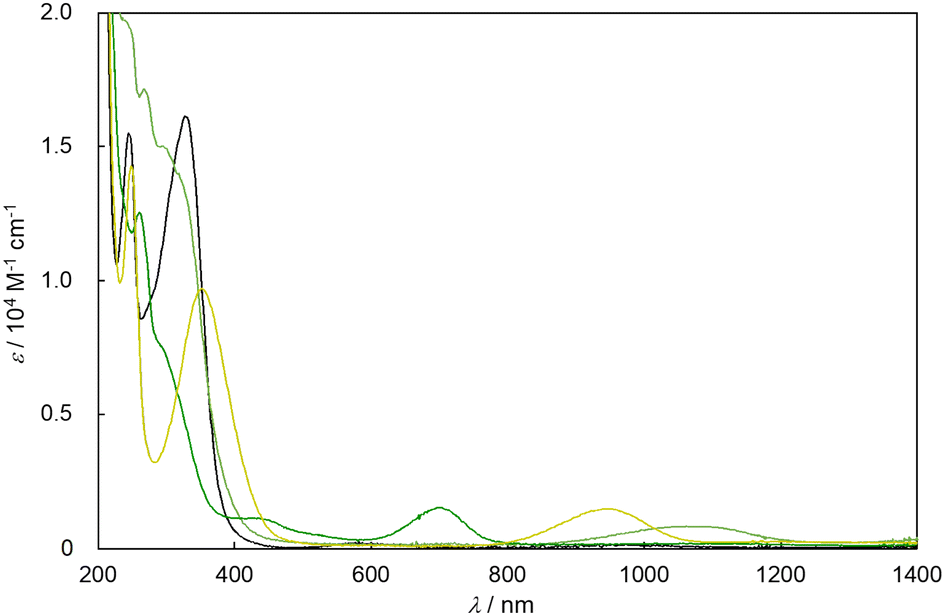 | ||
| Fig. 4 UV-vis-NIR spectra of [IV,IV]2+ (green line), [III,IV]+ (olive line), [III,IV_1H]2+ (yellow-green line), and [III,III_2H]2+ (black line) in CH3CN. | ||
| Bridging bidentate ligand | (E vs. Ag|0.01 M AgNO3)/V | λ max/nm (ε/M−1 cm−1) | ||
|---|---|---|---|---|
| Ru(IV)–Ru(IV)/Ru(III)–Ru(IV) | Ru(III)–Ru(IV)/Ru(III)–Ru(III) | {Ru2(μ-O)2}-{Ru2(μ-O)2}* | ||
| Ru(IV)–Ru(IV) | Ru(III)-Ru(IV) | |||
| a Not isolated, confirmed only in solution. | ||||
| CO32−7e |
0.15 | −1.10 | 762 (1440) | 1067 (660) |
| SO42−this work |
0.31 | −1.02 | 699 (1710) | 945 (1440) |
| CH3CO2−7c |
0.57 | −0.75 | 677a | 977 (1440) |
| HCO3−7f |
0.60 | 669 (1970) | 972 (1490) | |
| NO3−7c |
0.93 | −0.52 | 586a | 762 (1300) |
Furthermore, a relationship was found between λmax and the redox potentials, which will be discussed in the following section. These results were consistent with those previously reported for Ru2(III,IV) complexes (Table 2).7c–e The effect of the electron-donating character of the bidentate bridging ligands in complexes having the asymmetric {RuIII,IV2(μ-O)(μ-OH)} core was evident from their electrochemical properties ({μ-O2SO2}2− > {μ-O2CCH3}−7d > {μ-O2COH}−;7e,fTable 3 and Fig. S11†). However, the effect was much smaller than that on the corresponding complexes with the {RuIII,IV2(μ-O)2} core. This would be because the impact of protonation of the {Ru2(μ-O)2} core on the destabilization of MO energies of frontier orbitals was much larger than that of the electrostatic differences of bidentate bridging ligands {μ-O2XY}n−.
| (E vs. Ag|0.01 M AgNO3 (CH3CN))/V | λ max/nm (ε/M−1 cm−1) | |||
|---|---|---|---|---|
| Ru(IV)–Ru(IV)/Ru(III)–Ru(IV) | Ru(III)–Ru(IV)/Ru(III)–Ru(III) | Ru(III)–Ru(III)/Ru(II)–Ru(III) | ||
| [III,IV_1H]2+this work |
1.07 | −0.29 | −0.98 | 294 (15000), 1077 (840) |
| [III,IV(CH3CO2)_1H]3+7c |
1.33 | −0.22 | −0.90 | 269 (16400), 1161 (841) |
| [III,IV(HCO3)_1H]3+7f |
1.32 | −0.20 | −0.86 | 336 (11600), 964 (1330) |
| [III,III_2H]2+this work |
1.14 | −0.30 | −0.98 | 328 (16100) |
| [III,III(HCO3)_2H]3+this work |
1.25 | −0.22 | −0.88 | 331 (15200) (in CH3OH) |
The absorption wavelengths of [III,III_2H]2+ (331 nm) and [III,III(HCO3)_2H]2+ (328 nm), which could be attributed to the MLCT bands, and molar coefficients (ε = 1.61 × 104 and 1.52 × 104 M−1 cm−1, respectively) were comparable (Fig. 4 and Table 3), indicating that the electron transition energies and probabilities are approximately independent of the type of bidentate bridging ligand.
Cyclic voltammetry (CV) was performed in acetonitrile containing tetra-n-butylammonium perchlorate (TBAP) at 298 K (Fig. 5) and electrochemical analyses were carried out (Fig. S16–S33†). [IV,IV]2+ and [III,IV]+ showed four distinct redox waves: a quasi-reversible oxidation wave at 2.03 V, reversible one-electron oxidation and reduction waves at 0.31 and −1.02 V, respectively, and an irreversible reduction wave at −2.16 V. Based on the hydrodynamic voltammetry (HDV) analysis of [III,IV]+ (Fig. S19–S21†), the waves at 0.31 and −1.02 V were attributed to the one-electron couples of Ru2(IV,IV)/Ru2(III,IV) and Ru2(III,IV)/Ru2(III,III), and the reversibility was confirmed using different scan rates (Fig. S16, S17 and Table S4†). The irreversible waves at 2.03 and −2.16 V were attributed to the one-electron couples of Ru2(IV,V)/Ru2(IV,IV) and Ru2(III,III)/Ru2(II,III), respectively, in line with our previous reports.7b,c For the reduction wave at −2.16 V, the electrode reaction could be assigned to a one-electron process involving an electrode reaction–chemical reaction–electrode reaction (ECE) according to our previous reports.7b–e In the cyclic voltammogram of [III,IV]+, after the second oxidation of Ru2(IV,V)/Ru2(IV,IV), no cathodic peak current corresponding to the Ru2(IV,IV)/Ru2(III,IV) redox couple was observed, suggesting the occurrence of a structural change upon the second oxidation and a high reactivity for the Ru2(IV,V) species. The variations in the redox potentials of complexes having different bridging ligands indicated that the electron-donating nature is in the order of {μ-O2CO}2−7f > {μ-O2SO2}2− > {μ-O2CCH3}−7c ≈ {μ-O2COH}−7e > {μ-O2NO}−.7c The λmax values of the Ru2(IV,IV) complexes were related to the order of their redox potentials, which reflect the SOMO, namely, the {Ru2(μ-O)2} core energies. The cyclic voltammogram of [III,IV_1H]2+ exhibited irreversible oxidation waves at 1.07 and 2.02 V and reversible reduction waves at −0.29 and −0.98 V. The reversible waves were assigned to the Ru2(III,IV)/Ru2(III,III) (−0.29 V) and Ru2(III,III)/Ru2(II,III) couples (−0.98 V), which were confirmed by HDV analysis (Fig. S25–S27†), and the reversibility was confirmed using different scan rates (Fig. S22, S23 and Table S5†). The irreversible waves were attributed to the Ru2(IV,IV)/Ru2(III,IV) (1.07 V) and Ru2(IV,V)/Ru2(IV,IV) couples (2.02 V). The oxidation wave at 2.02 V is identical to those of [III,IV]+ and [IV,IV]2+, indicating that deprotonation from the {Ru2(μ-O)(μ-OH)} core was accompanied by the one-electron oxidation of [III,IV_1H]2+. The redox waves of [III,IV_1H]2+ having the {Ru2(μ-O)(μ-OH)} core were more positive than those of [III,IV]+ having the {Ru2(μ-O)2} core.
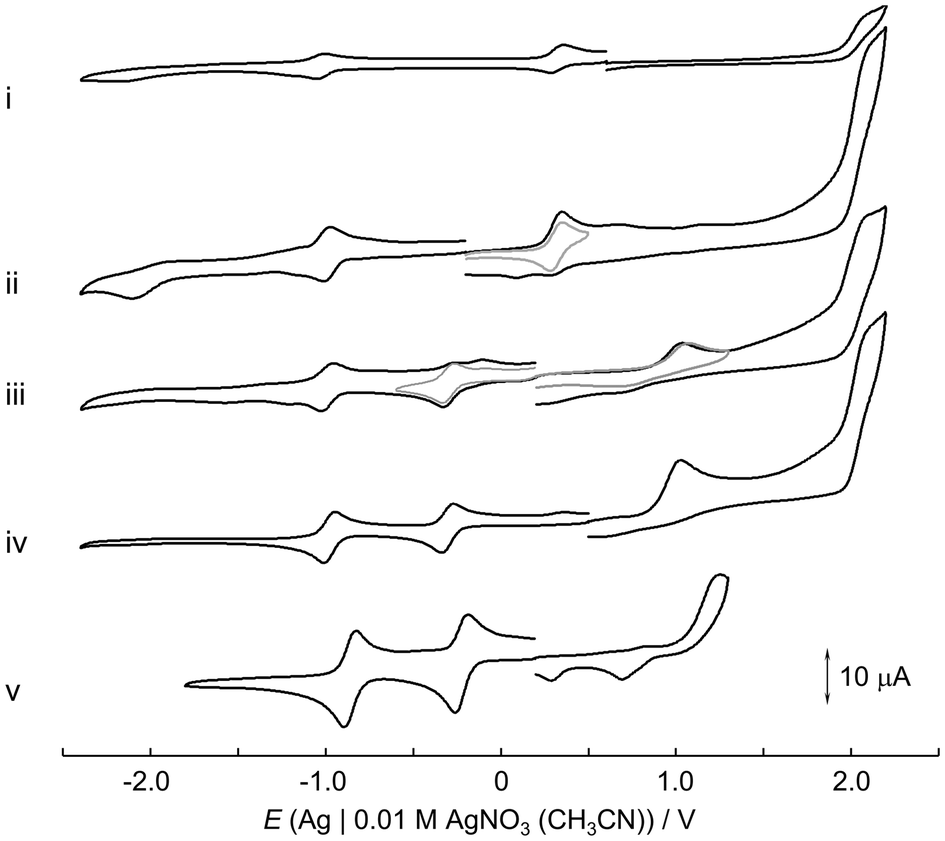 | ||
| Fig. 5 Cyclic voltammograms of (i) [IV,IV]2+ (0.66 mM), (ii) [III,IV]+, (iii) [III,IV_1H]2+, (iv) [III,III_2H]2+, and (v) [III,III_2H(HCO3)]3+ in CH3CN (c = 1.0 mM unless otherwise noted; scan rate, 100 mV s−1; 298 K). | ||
The comproportionation constants (Kc) of the mixed-valence state in [III,IV]+ and [III,IV_1H]2+ were calculated to be 3.1 × 1022 and 4.5 × 1022, respectively, according to the Nernst equation (eqn (S4)–(S6)†), corresponding to Class III of the Robin–Day classification.36 The evaluation on the mixed-valency revealed a stable mixed-valent state of Ru3.5−Ru3.5, which is described as Ru2(III,IV) for the clarity of redox reactions. These values were in line with those of other Ru2(III,IV) complexes in our Rbpma ligand system (R = ethyl or benzyl; 1022–1024).7d
[III,III_2H]2+ exhibited two irreversible oxidation waves at 1.14 and 2.05 V, corresponding to the Ru2(IV,IV)/Ru2(III,IV) and Ru2(III,IV)/Ru2(III,III) couples, respectively. The reversible redox waves at −0.30 and −0.98 V were assigned to the one-electron reduction couples of Ru2(III,III)/Ru2(II,III) and Ru2(II,III)/Ru2(II,II), respectively (Fig. S31–S33†). These redox behaviours indicated that upon the one-electron oxidation of [III,III_2H]2+, one-deprotonation occurs to form [III,IV_1H]2+, and the second oxidation involves further one-deprotonation to eventually form [IV,IV]2+. The reversible reduction processes (Fig. S28, S29 and Table S6†) indicated that both {RuII,III2(μ-OH)2} and {RuII,II2(μ-OH)2} cores were formed without structural changes under the measurement conditions. [III,III(HCO3)_2H]3+ exhibited similar redox behaviours with potentials at 1.25 (Epa), −0.22 (E1/2) and −0.88 V (E1/2), which were slightly more positive than those of [III,III_2H]2+.
Electrochemical and spectroscopic behaviours in aqueous solutions
The electrochemical behaviours of [III,IV_1H]2+ were investigated in aqueous solution (Fig. S34†) and a Pourbaix diagram was obtained by plotting the redox potentials as a function of pH between pH 0.5 and 8.3 (Fig. 6). The UV-vis-NIR spectrum measured at pH 10.95 showed much lower intensity compared to those obtained at lower pH (Fig. S13†), indicating that [III,IV_1H]2+ underwent a structural change, and was thus excluded from the Pourbaix diagram. In all pH regions, one reversible oxidation wave, corresponding to the Ru2(IV,IV)/Ru2(III,IV) couple, and multiple reversible or irreversible reduction waves were observed. | ||
| Fig. 6 Pourbaix diagram of sulfato-bridged complexes (the {μ-O2SO2}2− moiety is omitted for clarity in the figure). | ||
The protonation/deprotonation equilibria and electrode reactions are summarised in eqn (1)–(15). In the Ru2(III,IV) region, the pKa between [III,IV_1H]2+ and [III,IV]+ was at pH 2.1 (eqn (1)), consistent with the spectroscopic investigations (Fig. S13†). A pKa was not observed in the Ru2(IV,IV) region between pH 0.5 and 8.3, indicating the presence of only [IV,IV]2+. The spectroscopic data suggested that the pKa between {[IV,IV_1H]3+} and [IV,IV]2+ was as low as −0.3 (eqn (2) and Fig. S12†), although it was out of scope of the accurate pH evaluation with the glass electrode, therefore, {[IV,IV_1H]3+} is only present under strongly acidic conditions. [IV,IV]2+ seemed to be unstable above pH 5, as evidenced by the UV-vis-NIR spectra above pH 5.03, likely changing into [III,IV_1H]2+ (Fig. S12 and S13†). In the Ru2(III,III) region, the isolated species [III,III_2H]2+ is present below pH 5.7 (pKa), consistent with the spectroscopic results (Fig. S14†). The correponding pKa of [III,III(HCO3)_2H]3+was observed at 4.2 (Fig. S15,†eqn (3), eqn (S1)†). Above the pKa and below approximately pH 8, a Ru2(III,III) species having the {Ru2(μ-O)(μ-OH)} core, {[III,III_1H]+}, was present, but it started to decompose at pH 11 (grey line in Fig. S14†). The unstable character of {[III,III_1H]+} in strongly basic aqueous media was similar to that of {[III,III(CO3)]}. Finally, in the Ru2(II,II) region, complexes having the {Ru2(μ-OH)2}, {Ru2(μ-O)(μ-OH)}, or {Ru2(μ-O)2} cores would be present owing to the two pKas approximately at 4.1 and 6.6, although they were not isolated.
Protonation/deprotonation equilibria:
 | (1) |
 | (2) |
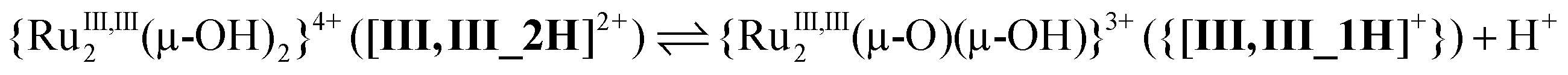 | (3) |
 | (4) |
| {RuII,II2(μ-OH2)(μ-OH)}3+⇌{RuII,II2(μ-OH)2}2+ + H+ | (5) |
Redox reactions:
between Ru2(III,IV) and Ru2(IV,IV)
above pH 2.1:
| {RuIII,IV2(μ-O)2}3+⇌{RuIV,IV2(μ-O)2}4+ + e− | (6) |
below pH 2.1:
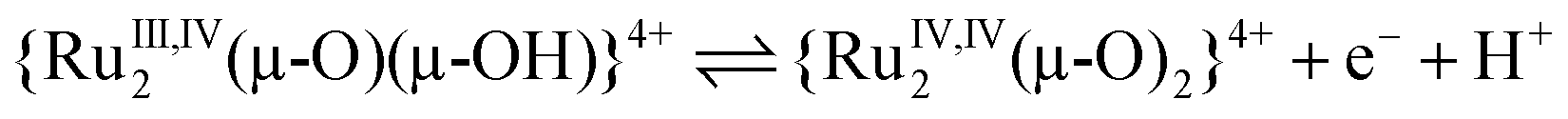 | (7) |
between Ru2(III,IV) and Ru2(III,III)
above pH 5.7:
 | (8) |
between pH 2.1 and 5.7:
| {RuIII,IV2(μ-O)2}3+ + e− + 2H+⇌{RuIII,III2(μ-OH)2}4+ | (9) |
below pH 2.1:
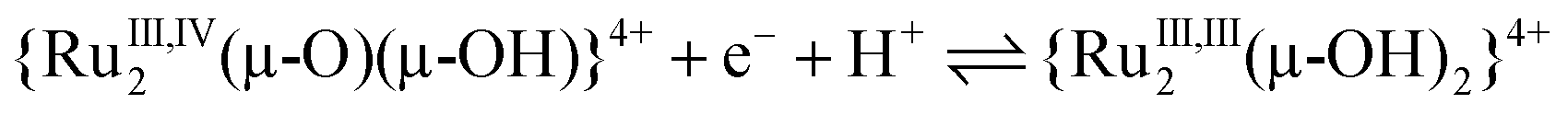 | (10) |
between Ru2(III,III) and Ru2(II,III)
above pH 5.7:
 | (11) |
between pH 2.0 and 5.7:
| {RuIII,III2(μ-OH)2}4+ + e− ⇌{RuII,III2(μ-OH)2}3+ | (12) |
between Ru2(III,III) and Ru2(II,II)
below pH 2.0:
 | (13) |
between Ru2(II,III) and Ru2(II,II)
above pH 6.6:
| {RuII,III2(μ-OH)2}3+ + e− ⇌{RuII,II2(μ-OH)2}2+ | (14) |
between pH 4.1 and 6.6:
 | (15) |
The pKa(s) in every oxidation state was(were) observed at a higher pH for the sulfato-bridged complex than that of the acetato-bridged complex in the Pourbaix diagram, indicating the higher basicity of the (μ-O(H))2 core of the present system: pKa = 1.5 between [III,IV(CH3CO2)_1H]3+ and [III,IV(CH3CO2)]2+ (eqn (1)); 5.6 between {[III,III(CH3CO2)_2H]3+} and [III,III(CH3CO2)_1H]2+ (eqn (3)); and 3.7 and 6.6 between {[II,II(CH3CO2)_4H]3+} and {[II,II(CH3CO2)_3H]2+} (eqn (4)) and {[II,II(CH3CO2)_3H]2+} and {[II,II(CH3CO2)_2H]+} (eqn (5)), respectively.7c The pKas of singly protonated species in the Ru2(III,IV) state were 2.4, 2.1, and 1.5 for [III,IV(HCO3)]2+,7e [III,IV]+, and [III,IV(CH3CO2)]2+,7c respectively, indicating that the order of increasing acidity is hydrogencarbonato > sulfato > acetato, which is not necessarily correlated with the electrostatic nature of the bridging ligand. For the Ru2(III,III) state, pKa = 5.7 and 4.2 for [III,III_2H]2+ and [III,III(HCO3)_2H]3+, respectively, indicating that the basicity of the bridging oxygen is higher in [III,III(HCO3)_2H]3+ owing to the weaker electron-donating hydrogencarbonato compared to sulfato.
Conclusions
A series of Ru(IV)–Ru(IV), Ru(III)–Ru(IV), and Ru(III)–Ru(III) complexes, in which the bidentate sulfato ligand bridged the doubly oxido- and/or hydroxido-bridged diruthenium core {Ru2(μ-O(H))2}, were synthesised all using the same tridentate ligand, ebpma. The hydrogencarbonato-bridged Ru2(III,III) complex was also synthesised. The electronic structures of these complexes were systematically investigated experimentally and theoretically as structural analogues of the reaction intermediates proposed in iron-containing metalloenzymes such as sMMO.The single-crystal X-ray analyses of all complexes revealed the structural differences among the {Ru2(μ-O)2}, {Ru2(μ-O)(μ-OH)} and {Ru2(μ-OH)2} cores, which depended on the oxidation state of the Ru centres and the number of protons on the oxido ligand(s). Interesting paramagnetic characteristics were revealed for [IV,IV]2+ and [III,III_2H]2+ in addition to those of [III,IV]+ and [III,IV_1H]2+ with S = ½ through magnetic measurements and NMR spectroscopy, which could be rationalised by the quantum mechanical calculations that the open-shell singlet state was more stabilized for [IV,IV]2+ and a possible spin transition from a closed-shell singlet state to triplet state could occur for [III,III_2H]2+. Notably, all the complexes isolated could be confirmed in the Pourbaix diagram. The basicity of the bridging oxido ligand(s) of the {Ru2(μ-O(H))2} core were discussed in each electronic state; the pKa values of {[IV,IV_1H]3+}, [III,IV_1H]2+, and [III,III_2H]2+ were −0.3, 2.1, and 5.7, respectively. Under more strongly acidic conditions, the sulfato ligand may be protonated and utilised as a proton accepting site in future applications. In addition to the Ru2(IV,IV), Ru2(III,IV), and Ru2(III,III) states, the Ru2(II,III) or Ru2(II,II) state can be accessed using the ebpma ligand system. These oxidation states are analogous to the intermediate or transient species expected in the sMMO cycle.
Experimental
Materials
All chemical reagents were used as purchased without further purification. RuCl3·nH2O (Ru content, 40.11 wt%) was purchased from Furuya Metal Inc. Co., Ltd Japan or Tanaka Holdings Co., Ltd Japan. The ebpma ligand and fac-[RuIIICl3(ebpma)] were prepared according to the literature procedures.7a [{RuIII,IVCl2(ebpma)}2(μ-O)]ClO4·0.75(CH3)2CO was prepared according to the procedure reported in ref. 7f and the synthesis of [{RuIII,IV(ebpma)}2(μ-O)2(μ-O2NO)](ClO4)2·1.5H2O ([III,IV(NO3)](ClO4)2·1.5H2O) followed the procedure of the corresponding PF6-salt reported in ref. 7c.Measurements
The μeff was measured at 296–298 K using a Sherwood Scientific Magway MSB MK1 balance (UK) with Hg[Co(SCN)4] as the calibrant. X-band ESR spectra were recorded using a JEOL (Japan) JESFA300 ESR spectrometer at 93 and 298 K. 1H and 13C NMR spectra were measured using a JEOL JNM-ECA (500 MHz) spectrometer. IR spectra were recorded using a Shimadzu (Japan) IR Affinity-1 spectrophotometer for samples prepared as KBr disks. Elemental analyses were carried out with a PerkinElmer (US) 2400-II or at the Institute of Physical and Chemical Research (RIKEN, Japan). Fast atom bombardment mass spectrometry (FAB MS) was conducted using a JEOL JMS-SX700 V with an m-nitrobenzyl alcohol matrix. UV-vis-NIR spectra were measured using a Shimadzu (Japan) UV-3600 Plus spectrophotometer in acetonitrile, methanol, or aqueous solutions using a quartz cell with a 1 cm path length. Cyclic voltammetry, Osteryoung square wave voltammetry, and differential pulse voltammetry were performed in acetonitrile containing 0.1 M TBAP (Nakarai Tesque Ltd, Japan) as a supporting electrolyte with a glassy carbon disk working electrode (ϕ = 1.0 or 1.6 mm), Ag|0.01 M AgNO3(CH3CN) reference electrode, and platinum counter electrode using an ALS Model 630E electrochemical analyser (BAS Inc., Japan.). At the end of each measurement, ferrocene ([FeIIICp2]+/[FeIICp2] (Cp = cyclopentadienyl), 0.074 V in CH3CN vs. Ag | 0.01 M AgNO3(CH3CN)) was added as the internal standard to scale the redox potentials. For the measurements in 0.1 M NaOH–0.1 M H3PO4 aqueous buffer solutions, a glassy carbon working electrode (ϕ = 1.0 mm), Ag | 3.0 M AgCl reference electrode, and platinum wire counter electrode were used. Hydrodynamic voltammetry (HDV) was carried out with a rotating platinum disk or a rotating glassy carbon disk working electrode (ϕ = 3.0 mm) and a BAS RDE-2 rotating electrode.Theoretical calculations
The computational calculations were performed with (U)B3LYP-D3BJ37 as a functional and LANL08(f)(Ru), aug-cc-pVDZ(N, O, S), and cc-pVDZ(H, C) as basis sets after the self-consistent field (SCF) calculations, using the Gaussian 16 program38 for [IV,IV]2+, [III,IV]+, [III,IV_1H]2+, and [III,III_2H]2+. The Grimme dispersion correction (D3BJ) was employed in addition to B3LYP as a better solution in our system in comparison with the ωB97XD and the sole B3LYP functions. The initial electronic structures were estimated by the fragment method, dividing the complex cations into four moieties; the {Ru2(μ-O(H))2} core, μ-O2SO22− ligand, and two ebpma ligands. The structures were optimized with full-optimization (no symmetric restrains) and the polarizable continuum model (PCM) was applied, in which acetonitrile was selected as a representative solvent used in experiments reported herein. For the assignment of electron transition spectra, time-dependent (TD)-DFT calculations were performed in similar methods. For [III,III(HCO3)_2H]3+, costless simpler calculations were performed with B3LYP/LANL2DZ levels.Single-crystal X-ray diffraction analysis
Single crystals suitable for X-ray diffraction analysis were obtained at 298 K by slow evaporation of a water/acetone reaction solution, with pH 2.7 for [IV,IV](ClO4)2·H2O, pH 5.4 for [III,IV]ClO4·3.5H2O, pH 2.3 for [III,IV_1H](ClO4)2·0.75H2O, pH 1.9 for [III,III_2H](ClO4)2·1.5H2O, and pH 1.9 for [III,III(HCO3)_2H](ClO4)3·0.25H2O. The intensity data were collected on a Rigaku (Japan) Model Synergy-S system, using multilayer mirror monochromated CuKα radiation (1.54184 Å). All the calculations were performed using CrysAlisPro data collection and processing software (Rigaku Corporation (2015)). Structures were solved using direct methods, expanded using Fourier techniques, and refined using full-matrix least-squares techniques on F2 using the SHELXT39 implementation in Olex2 1.5.40 For the disorder modelling of perchlorate and solvent water, parts 1 and 2 were used, restraints (DFIX, DANG, RIGU, SIMU, and ISOR) were applied where necessary to make more sensible models, and constraint (EADP) was utilized only if the split atoms were located too close to each other. The structures are represented by ORTEP-3 for windows41 (CCDC 2355836, 2355837, 2355838, 2355839 and 2355840† for [IV,IV](ClO4)2·H2O, [III,IV]ClO4·3.5H2O, [III,IV_1H](ClO4)2·0.75H2O, [III,III_2H](ClO4)2·1.5H2O, and [III,III(HCO3)_2H](ClO4)3·0.25H2O, respectively).Author contributions
T. M.-S.: conceptualization, supervision, data curation, visualization, writing – original draft preparation, and writing – reviewing and editing; Y. K.: data curation, visualization, and writing – reviewing and editing. S. M. and T. N.: data curation and visualization. H. N.: supervision and writing – reviewing and editing.Data availability
The data supporting this article have been included as part of the ESI.†Crystallographic data for [[IV,IV](ClO4)2·H2O, [III,IV]ClO4·3.5H2O, [III,IV_1H](ClO4)2·0.75H2O, [III,III_2H](ClO4)2·1.5H2O, and [III,III(HCO3)_2H](ClO4)3·0.25H2O] have been deposited at the CCDC under 2355836, 2355837, 23558382355839, and 2355840,† respectively, and can be obtained from [https://doi.org/10.1039/D4DT02833C].
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate Dr Hiroyoshi Ohtsu (Fujitsu Ltd) for the SCXRD analysis. We are grateful for the financial support from Japan Society for the Promotion of Science Grant-in-aid for young scientists (22K14696) (T.M.-S.) and The Joint Usage/Research Center for Catalysis (23AY0076, 23AY0358, and 23AY0449) (T.M.-S.).References
- (a) C. E. Tinberg and S. J. Lippard, Acc. Chem. Res., 2011, 44, 280 CrossRef CAS PubMed; (b) R. Banerjee, Y. Proshlyakov, J. D. Lipscomb and D. A. Proshlyakov, Nature, 2015, 518, 431 CrossRef CAS.
- (a) A. B. Jacobs, R. Banerjee, D. E. Deweese, A. Braun, J. T. Babicz, L. B. Gee, K. D. Sutherlin, L. H. Böttger, Y. Yoda, M. Saito, S. Kitao, Y. Kobayashi, M. Seto, K. Tamasaku, J. D. Lipscomb, K. Park and E. I. Solomon, J. Am. Chem. Soc., 2021, 143, 16007 CrossRef CAS; (b) M. Martinho, G. Xue, A. T. Fiedler, L. Que, E. L. Bominaar and E. Münck, J. Am. Chem. Soc., 2009, 131, 5823 CrossRef CAS PubMed; (c) G. Xue, R. D. Hont, E. Münck and L. Que, Nat. Chem., 2010, 2, 400 CrossRef CAS; (d) E. Y. Tshuva and S. J. Lippard, Chem. Rev., 2004, 104, 987 CrossRef CAS PubMed.
- Y. Morimoto, Y. Takagi, T. Saito, T. Ohta, T. Ogura, N. Tohnai, M. Nakano and S. Itoh, Angew. Chem., Int. Ed., 2018, 57, 7640 CrossRef CAS PubMed.
- A. Geilenkirchen, P. Neubold, R. Schneider, K. Wieghardt, U. Florke, H.-J. Haupt and B. Nuber, J. Chem. Soc., Dalton Trans., 1994, 457 RSC.
- K. Wieghardt, W. Herrmann and M. Koppen, Z. Naturforsch., 1984, 39b, 1335 CrossRef CAS.
- (a) J. M. Power, K. Evertz, L. Henling, R. Marsh, W. P. Schaefer, J. A. Labinger and J. E. Bercaw, Inorg. Chem., 1990, 29, 5058 CrossRef CAS; (b) E. P. Kelson, L. M. Healing, W. P. Schaefer, J. A. Labinger and J. E. Bercaw, Inorg. Chem., 1993, 32, 2863 CrossRef CAS; (c) T. Nakano, T. Abe, T. Matsumoto, K. Kimura, G. Nakamura, S. Hayami, Y. Shiota, K. Yoshizawa and S. Ogo, RSC Adv., 2022, 12, 12253 RSC; (d) Y.-R. He, M. Zhang, W.-H. Huang, X.-X. Xue, Z.-Q. Zhou, L.-N. Feng, J.-H. Yang and B. Liu, Dalton Trans., 2024, 53, 4598 RSC.
- (a) Y. Shimizu, S. Fukui, T. Oi and H. Nagao, Bull. Chem. Soc. Jpn., 2008, 81, 1285 CrossRef CAS; (b) T. Suzuki, K. Matsuya, T. Kawamoto and H. Nagao, Eur. J. Inorg. Chem., 2014, 722 CrossRef CAS; (c) T. Suzuki, Y. Suzuki, T. Kawamoto, R. Miyamoto, S. Nanbu and H. Nagao, Inorg. Chem., 2016, 55, 6830 CrossRef CAS; (d) T. Misawa-Suzuki, T. Watanabe, M. Okamura, S. Nanbu and H. Nagao, Inorg. Chem., 2020, 59, 612 CrossRef CAS; (e) T. Misawa-Suzuki, S. Mafune and H. Nagao, Inorg. Chem., 2021, 60, 9996 CrossRef CAS; (f) T. Misawa-Suzuki and H. Nagao, Dalton Trans., 2023, 52, 2863 RSC.
- (a) A. Abarca, A. Martín and M. Mena, Inorg. Chem., 1995, 34, 5437 CrossRef CAS; (b) Q.-F. Zhang, T. C. H. Lam, X.-Y. Yi, E. Y. Y. Chan, W.-Y. Wong, H. H. Y. Sung, I. D. Williams and W.-H. Leung, Chem. – Eur. J., 2005, 11, 101 CrossRef.
- (a) S. Srivastava, O. P. Pandey and S. K. Sengupta, Transition Met. Chem., 1996, 21, 262 CrossRef CAS; (b) M. I. Khan, S. Cevik, D. Powell, S. Li and C. J. Óconnor, Inorg. Chem., 1998, 37, 81 CrossRef CAS PubMed; (c) C. H. Ng, C. W. Lim, S. G. Teoh, H.-K. Fun, A. Usman and S. W. Ng, Inorg. Chem., 2002, 41, 2 CrossRef CAS; (d) S. B. Rasmussen, R. M. Rasmussen, R. Fehrmann and K. Nielsen, Inorg. Chem., 2003, 42, 7123 CrossRef CAS; (e) K. Kanamori, E. Kameda and K. Okamoto, Bull. Chem. Soc. Jpn., 2006, 79, 1881 CrossRef CAS; (f) M. R. Maurya, A. Kumar, M. Ebel and D. Rehder, Inorg. Chem., 2006, 45, 5924 CrossRef CAS.
- (a) S. Indubla and D. Ramaswamy, J. Inorg. Nucl. Chem., 1973, 35, 2055 CrossRef; (b) S. A. Sadeek, M. S. Refat and S. M. Teleb, J. Korean Chem. Soc., 2004, 48, 358 CrossRef CAS; (c) J. Glerup, P. A. Goodson, D. J. Hodgson, M. A. Masood and K. Michelsen, Inorg. Chim. Acta, 2005, 358, 295 CrossRef CAS.
- Y. Bi, Y. Chen and X. Lu, Chin. J. Chem., 2011, 29, 2034 CrossRef CAS.
- (a) N. Arulsamy, P. A. Goodson, D. J. Hodgson, J. Glerup and K. Michelsen, Inorg. Chim. Acta, 1994, 216, 21 CrossRef CAS; (b) A. C. Sudik, A. R. Millward, N. W. Ockwig, A. P. Côté, J. Kim and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 7110 CrossRef CAS PubMed; (c) H.-Z. Shi, Z.-L. Li and Y.-K. Shan, Z. Kristallogr. —New Crystal Structures, 2005, 220, 245 CAS; (d) A. Horn, I. Vencato, A. J. Bortoluzzi, V. Drago, M. A. Novakd and A. Neves, J. Braz. Chem. Soc., 2006, 17, 1584 CrossRef CAS; (e) N. Loganathan and A. C. Roodt, Z. Kristallogr. —New Crystal Structures, 2013, 228, 476 CAS; (f) N. Xu, E. G. Abucayon, D. R. Powell and G. B. Richter-Addo, Nitric Oxide, 2016, 52, 16 CrossRef CAS PubMed.
- (a) E. P. Hertzenberg and J. C. Bailar, Inorg. Chem., 1971, 10, 2371 CrossRef CAS; (b) V. V. Ponomarova, V. V. Komarchuk, I. Boldog, A. N. Chernega, J. Sielerc and K. V. Domasevitch, Chem. Commun., 2002, 436 RSC; (c) M. Bera, A. B. S. Curtiss, G. T. Musie and D. R. Powell, Inorg. Chem. Commun., 2008, 11, 1033 CrossRef CAS; (d) H. Zasłona and P. Drożdżewski, J. Coord. Chem., 2011, 64, 2262 CrossRef; (e) F. Setifi, Z. Setifi, P. Konieczny, C. Glidewell, S. Benmansour, C. J. Gómez-García, F. Grandjean, G. J. Long, R. Pelka and J. Reedijk, Polyhedron, 2019, 157, 558 CrossRef CAS.
- (a) N. M. Karayannis, C. M. Mikulski, L. L. Pytlewski and M. M. Labes, Inorg. Chim. Acta, 1974, 10, 97 CrossRef CAS; (b) C. Papatriantafyllopoulou, G. Aromi, A. J. Tasiopoulos, V. Nastopoulos, C. P. Raptopoulou, S. J. Teat, S. J. A. Escuer and S. P. Perlepes, Eur. J. Inorg. Chem., 2007, 2761 CrossRef CAS; A. Cagnolini, B. Ballard, H. P. Engelbrecht, T. L. Rold, C. Barnes, C. Cutler, T. J. Hoffman, R. Kannan, K. Katti and S. S. Jurisson, Nucl. Med. Biol., 2011, 38, 63 Search PubMed; (c) L.-H. Jia, R.-Y. Li, Z.-M. Duan, S.-D. Jiang, B.-W. Wang, Z.-M. Wang and S. Gao, Inorg. Chem., 2011, 50, 144 CrossRef CAS PubMed; (d) C. M. Nagaraja, T. K. Maji and C. N. R. Rao, Inorg. Chim. Acta, 2012, 389, 85 CrossRef CAS.
- (a) J. J. Girerd, S. Jeannin, Y. Jeannin and O. Kahn, Inorg. Chem., 1978, 17, 3034 CrossRef CAS; (b) F. Cariati, G. Micera, A. Scozzafava, G. Minghetti and G. Banditelli, Inorg. Chem., 1982, 21, 3843 CrossRef CAS; (c) L. K. Thompson, A. W. Hanson and B. S. Ramaswamy, Inorg. Chem., 1984, 23, 2459 CrossRef CAS; (d) H. Enders, D. Nöthe, E. Rossato and W. E. Hatfield, Inorg. Chem., 1984, 23, 3467 CrossRef; (e) P. J. van Koningsbruggen, D. Gatteschi, R. A. G. de Graaff, J. G. Haasnoot, J. Reedijk and C. Zanchini, Inorg. Chem., 1995, 34, 5175 CrossRef CAS; (f) S. Dhar, M. Nethaji and A. R. Chakravarty, Dalton Trans., 2004, 4180 RSC; (g) S. Chowdhury, P. Mal, C. Basu, H. Stoeckli-Evans and S. Mukherjee, Polyhedron, 2009, 28, 3863 CrossRef CAS; (h) T. S. Lobana, R. Sultana, G. Hundala and R. J. Butcher, Dalton Trans., 2010, 39, 7870 RSC; (i) R. Vafazadeh, R. Esteghamat-Panah, A. C. Willis and A. F. Hill, Polyhedron, 2012, 48, 51 CrossRef CAS; (j) A. D. Burrows, M. F. Mahon, V. M. Sebestyen, Y. Lan and A. K. Powell, Inorg. Chem., 2012, 51, 10983 CrossRef CAS PubMed; (k) K. Hchicha, M. Korb, R. Badraouicd and H. Naïli, New J. Chem., 2021, 45, 13775 RSC.
- (a) S. A. Sadeek, J. Phys. Chem. Solids, 1993, 54, 919 CrossRef CAS; (b) B. Ali, I. G. Dance, D. C. Craig and M. L. Scudder, J. Chem. Soc., Dalton Trans., 1998, 1661 RSC; (c) C. Papatriantafyllopoulou, C. P. Raptopoulou, A. Terzis, J. F. Janssens, E. Manessi-Zoupa, S. P. Perlepes and J. C. Plakatouras, Polyhedron, 2007, 26, 4053 CrossRef CAS; (d) A. B. S. Curtiss, M. Bera, G. T. Musie and D. R. Powell, Dalton Trans., 2008, 2717 RSC; (e) Z. Chen, X. Li and F. Liang, J. Solid State Chem., 2008, 181, 2078 CrossRef CAS; (f) F. Wang, X.-Y. Wu, Z.-G. Zhao, Q.-S. Zhang, Y.-M. Xie, R. Yu and C.-Z. Lu, Inorg. Chim. Acta, 2010, 363, 1320 CrossRef CAS; (g) L. Wan, C. Zhang, Y. Xing, Z. Li, N. Xing, L. Wan and H. Shan, Inorg. Chem., 2012, 51, 6517 CrossRef CAS PubMed; (h) R. Tsukamoto, R. Aoki, R. Sakamoto, R. Toyoda, M. Shimada, Y. Hattori, M. Asaoka, Y. Kitagawa, E. Nishibori, M. Nakano and H. Nishihara, Chem. Commun., 2017, 53, 3657 RSC.
- C. Gonźalez-García, C. García-Pascual, R. Buŕon, D. G. Calatayud, J. Perle, M. A. Mendiola and E. López-Torres, Polyhedron, 2022, 223, 115945 CrossRef.
- D. F. DeTar, J. Am. Chem. Soc., 1980, 102, 7991 CrossRef.
- (a) C. L. Angell, F. A. Cotton, B. A. Frenz and T. R. Webb, J. Chem. Soc., Chem. Commun., 1973, 399 RSC; (b) M. Cindric, D. Matkovic-Calogovic, V. Vrdoljak and B. Kamenar, Struct. Chem., 1998, 9, 353 CrossRef CAS.
- (a) T. A. Fomina, Zh. Neorg. Khim., 1989, 34, 1552 CAS; (b) I. V. Kuz'menko, A. N. Zhilyaev, T. A. Fomina, M. A. Poraj-Koshits and I. B. Baranovskij, Zh. Neorg. Khim., 1989, 34, 2548 Search PubMed; (c) A. N. Zhilyaev, I. V. Kuz'menko, T. A. Fomina, S. B. Katser and I. B. Baranovskii, Zh. Neorg. Khim., 1993, 38, 847 CAS; (d) F. A. Cotton, T. Datta, L. Labella and M. Shang, Inorg. Chim. Acta, 1993, 203, 55 CrossRef CAS; (e) L. Heerman, H. V. Nyen and W. D'Olieslager, Inorg. Chim. Acta, 1996, 244, 191 CrossRef CAS; (f) W.-H. Leung, H. Zheng, J. L. C. Chim, J. Chan, W.-T. Wong and I. D. Williams, J. Chem. Soc., Dalton Trans., 2000, 423 RSC; (g) E. Nakatani, Y. Takai and H. Kurosawa, J. Organomet. Chem., 2007, 692, 278 CrossRef CAS.
- (a) I. B. Baranovskii, A. N. Zhilyaev and L. M. Dikareva, Zh. Neorg. Khim., 1985, 30, 1785 CAS; (b) L. M. Dikareva, Y. V. Zefirov, A. N. Zhilyaev, I. B. Baranovskii and M. A. Porai-Koshits, Zh. Neorg. Khim., 1987, 32, 118 CAS.
- C. A. Kruithof, A. Berger, H. P. Dijkstra, F. Soulimani, T. Visser, M. Lutz, A. L. Spek, R. J. M. K. Gebbink and G. van Koten, Dalton Trans., 2009, 3306 RSC.
- L.-L. Song, Q.-H. Jin, L.-N. Cui and C.-L. Zhang, Inorg. Chim. Acta, 2010, 363, 2425 CrossRef CAS.
- (a) R. Tamasi and R. Cini, Dalton Trans., 2003, 2928 RSC; (b) Y. B. Ding, Y. Cheng, Z.-L. Zhang, J. Zhang, Y.-G. Yin and W.-H. Gao, Inorg. Chem. Commun., 2009, 12, 45 CrossRef CAS; (c) Y.-K. Gu, X. Li, H. Liang and X.-B. Deng, Inorg. Chem. Commun., 2011, 14, 52 CrossRef CAS; Y.-J. Chang, D. Liu, Z.-G. Ren and J.-P. Lang, Inorg. Chem. Commun., 2011, 14, 403 Search PubMed.
- A. Falber, L. Todaro, I. Goldberg, M. V. Favilla and C. M. Drain, Inorg. Chem., 2008, 47, 454 CrossRef CAS PubMed.
- R. W. Berg, M. I. Ferré and S. J. C. Schäffer, Vib. Spectrosc., 2006, 42, 346 CrossRef CAS.
- (a) F. A. Cotton, B. A. Frenz and L. W. Shive, Inorg. Chem., 1975, 14, 649 CrossRef CAS; (b) M. Irmler and G. S. Meyer, Naturwissenschaften, 1988, 75, 308 CrossRef CAS; (c) I. S. Gonçalves, A. D. Lopes, T. R. Amarante, F. A. A. Paz, N. J. O. Silva, M. Pillinger, S. Gago, F. Palacio, F. E. Kühn and C. C. Romão, Dalton Trans., 2009, 10199 RSC.
- (a) E. F. Hills, D. T. Richens and A. G. Sykes, Inorg. Chem., 1986, 25, 3144 CrossRef CAS; (b) C. A. Miller, T. S. Janik, M. R. Churchill and J. D. Atwood, Inorg. Chem., 1996, 35, 3683 CrossRef CAS.
- (a) T. G. Appleton, J. R. Hall, D. W. Neale and S. F. Ralph, Inorg. Chim. Acta, 1983, 77, L149 CrossRef CAS; (b) J. Terheijden, G. van Koten, W. P. Mul and D. J. Stufkens, Organometallics, 1986, 5, 519 CrossRef CAS; (c) S. Camadanli, N. Deveci, G. Gokagac and H. Isci, Inorg. Chim. Acta, 2003, 351, 1 CrossRef CAS.
- (a) Y. Xing, Z. Shi, G. Li and W. Pang, Dalton Trans., 2003, 940 RSC; (b) H. Akkari, P. Bénard-Rocherullé, H. Mérazig, T. Roisnel and J. Rocherullé, Solid State Sci., 2006, 8, 704 CrossRef CAS.
- B. M. Casaria and V. Langer, Z. Anorg. Allg. Chem., 2007, 633, 1055 CrossRef.
- N. Xu, W. Shi, D.-Z. Liao, S.-P. Yan and P. Cheng, Inorg. Chem., 2008, 47, 8748 CrossRef CAS.
- (a) K.-L. Wonga, Y.-M. Zhu, Y.-Y. Yang, G.-L. Law, H.-H. Fan, P. A. Tanner and W.-T. Wong, Inorg. Chem. Commun., 2009, 12, 52 CrossRef; (b) X. Xiao, Y. Wei, W. Zheng and K. Wu, Z. Anorg. Allg. Chem., 2011, 637, 741 CrossRef CAS.
- (a) P. Thuéry, N. Keller, M. Lance, J.-D. Vigner and M. Nierlich, Acta Crystallogr., Sect. C:Cryst. Struct. Commun., 1995, 51, 1526 CrossRef; (b) M. B. Doran, A. J. Norquist, C. L. Stuarta and D. Óhare, Acta Crystallogr., Sect. E:Struct. Rep. Online, 2004, 60, m996 CrossRef CAS.
- J. Lin, G. B. Jin and L. Soderholm, Inorg. Chem., 2016, 55, 10098 CrossRef CAS PubMed.
- Advances in Inorganic Chemistry, ed. H. J. Emeleus and A. G. Sharpe, Academic Press, New York, 1960, vol. 2 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.02, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., A., 2015, 71, 3 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., C, 2015, 71, 3 CrossRef PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data and spectroscopic and electrochemical measurements are described. CCDC 2355836–2355840. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02833c |
| This journal is © The Royal Society of Chemistry 2025 |