
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbon nanotube graphene multilevel network based phase change fibers and their energy storage properties†
Xiaoyu
Yang
ab,
Jingna
Zhao
 *b,
Tanqian
Liao
c,
Wenya
Li
c,
Yongyi
Zhang
b,
Chengyong
Xu
a,
Xiaohua
Zhang
*d and
Qingwen
Li
b
*b,
Tanqian
Liao
c,
Wenya
Li
c,
Yongyi
Zhang
b,
Chengyong
Xu
a,
Xiaohua
Zhang
*d and
Qingwen
Li
b
aSchool of Science, Nanchang Institute of Technology, Nanchang 330099, China
bKey Laboratory of Multifunctional Nanomaterials and Smart Systems, Suzhou Institute of Nano-Tech and Nano-Bionics, Chinese Academy of Sciences, Suzhou 215123, China. E-mail: jnzhao2008@sinano.ac.cn
cSchool of Textile Science and Engineering, Xi'an Polytechnic University, Xi'an 710048, China
dInnovation Center for Textile Science and Technology, Donghua University, Shanghai 201620, China. E-mail: zhangxh@dhu.edu.cn
First published on 11th September 2024
Abstract
Phase change fibers with abilities to store/release thermal energy and responsiveness to multiple stimuli are of high interest for wearable thermal management textiles. However, it is still a challenge to prepare phase change fibers with superior comprehensive properties, especially proper thermal conductivity. Here, we report a cooperative in situ impregnation strategy to introduce graphene oxide (GO) and polyethylene glycol (PEG) together into the carbon nanotube (CNT) network during the expansion process and construct a 1D–2D multilevel skeleton, resulting in a CNT/GO/PEG composite phase change fiber. The presence of GO plays a more important role in increasing the interfacial contact and space volume, resulting in the characteristics of high loading (up to 96.8–98.4%), phase change enthalpy, and relatively lower thermal conductivity. Therefore, the CNT/GO/PEG phase change fiber demonstrates higher thermal efficiency during the exothermic process, showing good thermal management characteristics.
1 Introduction
Flexible multifunctional fabrics have attracted wide attention because of their intelligent sensing and interaction properties.1–3 Fibers, as the basic building blocks of smart fabrics, should not only meet the basic performance of forming fabrics but also be endowed with more intelligent and functional properties, such as sensing, energy storage, temperature regulation, etc.4–7 People's pursuit of temperature freedom greatly promotes the development of high-tech thermal functional fibers, while the phase change latent heat properties of phase change materials (PCM) perfectly match this point and drive a new attractive field. However, PCMs suffer from issues such as low thermal conductivity, liquid leakage, poor mechanical properties, etc., and difficulty in fiber formation.8–16 These drawbacks of individual phase change materials need to be overcome by compositing with reinforcements to form composite phase change fibers.Phase change fibers are primarily prepared by utilizing various spinning techniques to fabricate phase change materials into fibers, including melt spinning,17,18 electrospinning,19 and wet spinning,20,21 in which polymers play the roles of supporting structure. These methods solved the problem of fiber formation from phase change materials, but there are still challenges of rigorous requirements for the preparation of the spinning solution, poor continuity of fiber formation, low mechanical strength, and poor cyclic thermal stability. Therefore, it is necessary to find higher-performance ESI.†
Nanocarbon materials,22–24 due to their unique characteristics such as low density, high electrical, and thermal conductivity, high mechanical strength and microporous confinement, are considered ideal reinforcements. There are mainly two ways to realize the combination. One is directly mixing CNT or graphene powders with PCMs which is effective in improving the thermal properties, stability, and electrical conductivity of PCMs, but still suffers from the disadvantages of agglomeration, poor orientation, and low content. The other way is to combine nanocarbon assemblies (e.g., fibers,25–27 foams,28,29 aerogels,30–32etc.) into PCMs, among which fibers show more promising prospects due to their flexible adaptability to various anisotropic surfaces. Li et al.27 filled PCM into graphene aerogel fibers and developed a variety of multiresponsive smart fibers which showed excellent responsivity and reversible energy storage and conversion. Chen et al.33 loaded lauric acid into hollow carbon fibers, resulting in temperature-regulating supports with strong photothermal conversion ability and high latent heat. These methods overcome the issues of poor thermal stability and liquid leakage compared with single-phase change materials. However, considering the urgent demand of wearable thermal management textiles, challenges still remain in terms of mechanical properties, electrical properties, and poor thermal conductivity. Carbon nanotube (CNT) fibers, due to their excellent mechanical, electrical, and thermal properties, have become one of the best choices for reinforcing composite phase change fibers. In our previous work,26 an expansion-based in situ impregnation strategy was developed to fabricate CNT/PEG phase change fibers with superior comprehensive performance, whereby CNTs formed an interconnected 3 dimensional (3D) nano network and provided efficient heat transport pathways and sufficient microvoids to confine polymeric PCMs. Yet, the relatively high thermal conductivity (27 W m−1 K−1) while increasing the rate of heat absorption also leads to a faster heat release rate, limiting the efficiency of heat utilization. So, it is of great importance to develop composite phase change fibers simultaneously exhibiting high heat absorption and low heat release characteristics.
Herein, we report an efficient method for simultaneously impregnating GO and PEG into a CNT network during expansion, resulting in a 3D nanocarbon multi-layer structure leading to the preparation of a CNT/GO/PEG composite fiber. The presence of GO can further increase the interface contacts, specific surface area, and volumetric efficiency within the CNT network, resulting in a decrease in the overall thermal conductivity of the system due to the increase of interfacial thermal resistance, while increasing the loading capacity of phase change materials within the network structure. The structure can effectively inhibit the excessive thermal conductivity of the phase change fibers and improve the efficiency of the use of latent heat of phase change.
2 Experimental
2.1 Materials
The carbon nanotubes were prepared by floating catalytic chemical vapor deposition (FCCVD). For the fiber growth, acetone was used as the carbon source, ferrocene (0.5 wt%) as the catalyst, and thiophene (1.0 wt%) as the growth promoter, all of these were mixed together as the reaction solution. A mixture of hydrogen and argon gases in a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was used as the carrier gas. The reaction solution was injected into the reaction tube with a temperature of 1300 °C together with the carrier gas, then the carbon nanotubes were catalytically grown to form sock-like aerogels, pulled out from the furnace tube, and densified into continuous carbon nanotube fibers.34,35 The carbon nanotubes in the fiber were mainly double-walled with a diameter of ∼6 Å and bundles with a size of 30–50 nm.
:1 was used as the carrier gas. The reaction solution was injected into the reaction tube with a temperature of 1300 °C together with the carrier gas, then the carbon nanotubes were catalytically grown to form sock-like aerogels, pulled out from the furnace tube, and densified into continuous carbon nanotube fibers.34,35 The carbon nanotubes in the fiber were mainly double-walled with a diameter of ∼6 Å and bundles with a size of 30–50 nm.
The PCM used in this study was a PEG purchased by Macklin Company with an average molecular weight of 6000, which was chosen mainly due to its hydrophilicity, high thermal stability, chemical stability, non-toxic toxicity, and so on. The graphene oxide was bought from Hangzhou Gaoxi Technology with sizes of 20–30 μm, and was ultrasonically broken by an ultrasonic cell grinder at a power ratio of 30% for 5 s, 10 s, 20 s, 30 s, 1 min, 2 min, and 10 min, resulting in GO sizes of 0.5 μm, 2.5 μm, 4.5 μm, 6.5 μm, 8.5 μm, and 10.5 μm, respectively.
2.2 In situ cooperative impregnation
The electrolysis-induced expansion of carbon nanotube networks was carried out in 0.01 mol L−1 H2SO4 electrolyte. To achieve in situ impregnation, PEG and GO were also dissolved and dispersed in the electrolyte at concentrations of 20 wt% and 0.02–0.11 wt%, respectively.The CNT fiber was connected to the negative electrode of a DC power, by using a conductive rod, and a platinum plate to the positive electrode. The current passing through the CNT fiber was set at 50 mA. As shown in Fig. 1a, H2 is precipitated on the surface of the carbon nanotube fiber, leading to the super high volumetric expansion CNT network. During the expansion process, GO and PEG are in situ incorporated into the CNT network. Then the wet CNT/GO/PEG fiber is drawn out of the solution by a winder at a draw rate of approximately 1.3 cm s−1. The coiled fiber is vacuum dried at 40 °C for 24 hours to obtain a composite phase change fiber.
 | ||
| Fig. 1 Expansion-based impregnation to prepare CNT/GO/PEG fibers. (a) Experimental setup. (b) Optical picture of CNT fiber. (c) Optical picture of expanded CNT network. (d) Surface morphology of CNT/GO/PEG composite fiber. (e) Knotting morphology of in situ composite CNT/GO/PEG fiber. (f) Internal microstructure of CNT/GO/PEG composite fiber. Both GO and PEG enter the CNT fiber network. (g) Surface morphology of CNT/GO/PEG fiber fabric. (h) TGA curves of pure PEG and different composite fibers. (i) XRD curves of pure PEG and composite PCM of CNT/PEG, CNT/GO/PEG. | ||
2.3 Characterizations
The fibers surface morphology and microstructures were characterized by S-4800 High-Resolution SEM (Hitachi High-Tech Corp., Tokyo, Japan). The thermal conductivity of composite phase change fibers was evaluated using the 3ω harmonic method. Where the harmonic 3ω term U3ω was extracted and thus used to evaluate the conductivity based on the relationship between U3ω, κ, R, and temperature coefficient of resistance αCR, see ref. 36 and 37. The endothermic and exothermic processes were obtained with a 200-F3 Differential Scanning Calorimeter (DSC, NETZSCH-Geratebau GmbH, Selb, Germany) at a heating and cooling rate of 10 K min−1 with N2 as a shielding gas. The latent heat was the integral of the DSC signal over time. The PEG mass fraction was evaluated according to TGA tests by using a thermogravimetric analyzer (TGA) (NETZSCH-Geratebau GmbH, Selb, Germany), with N2 as the shielding gas and a heating rate of 10 K min−1. The crystalline properties of the fibers were tested by an X-ray diffractometer (D8 Advance, Bruker AXS, Germany), with a scanning speed of 5° min−1 and a scanning range from 10° to 60°. The infrared photographs were taken with a Fluke Ti480 PRO infrared camera (Shanghai Fluke Test Instruments Co., Ltd).3 Results and disscussion
3.1 Preparation of CNT/GO/PEG composite fiber
The preparation process and structure of CNT/GO/PEG composite phase change fibers were shown in Fig. 1a. During electrolysis, H2 was generated from the CNT network surface and the bubbles led to a volume expansion. PEG and GO were in situ impregnated into the expanded CNT network to form a three-dimensional CNT/GO/PEG composite structure. Then the CNT/GO/PEG composite was pulled out of the bath and collected by a winding bobbin, shrinking due to the effect of fiber surface tension. After being dried in an oven at 40 °C for 24 hours, a continuous CNT/GO/PEG composite phase change fiber was obtained.The original CNT fiber morphology and the expanded CNT network were shown in Fig. 1b and c, respectively. It was evident that the width of the ribbon was increased by nearly one order of magnitude from 40–50 μm to 2–3 mm, resulting in a volume expansion of thousands of times, which providing enough spaces and channels for PEG and GO impregnating. Fig. 1d showed the smooth surface of the composite fiber, which has a diameter of 135 μm, increasing by more than 300% compared with the pure CNT original 40 μm, indicating that the CNT/GO multilevel expansion structure certainly provided a high volume for the PEG loading. Moreover, due to the properties of CNT, the composite fiber could be bent, knotted flexibly, and even woven into fabrics, as seen in Fig. 1e and f. The distribution of GO within the CNT/GO/PEG fiber was characterized by tearing, as shown in Fig. 1g, substantiating that GO had already entered into the CNT network. The PEG mass fraction was characterized by TGA (Fig. 1h), demonstrating that the expanded CNT network allowed for more PEG loading.26,38 In the CNT/GO/PEG structure, the presence of GO further enhanced the PEG mass fraction in this work, see Fig. 1h. The weight loss of the CNT/PEG and CNT/GO/PEG composite fiber was 95.1% and 98.4% respectively. Compared with our previous work, the relatively lower PEG loading was mainly due to the larger and longer molecular weight of PEG 6000 limiting its entrance into the inner network. In addition, as discussed above, the CNT/GO multilevel network also affected the thermal diffusion of the composite fiber, it can be seen from Fig. 1h the thermal decomposition temperatures decreased from 340 °C for pure PEG to 260 °C for the CNT/GO/PEG composite fiber, still significantly much higher than that of PEG, indicating that the thermal stability of the composite fiber was not adversely affected.
The addition of GO as a support material inevitably affected the crystallization properties of PEG and thus influenced the crystalline morphology of CNT/GO/PEG composite fiber. In order to analyze the crystallization behavior, the PEG, CNT/PEG, and CNT/GO/PEG composite PCMs were tested by XRD at room temperature and the results were shown in Fig. 1i. It can be seen that pure PEG had two prominent obvious diffraction peaks near 2θ = 19.2° and 2θ = 23.3°, and there were almost no peak shifts for the CNT/PEG and CNT/GO/PEG. On the other hand, it was worth noting the peak value of 2θ = 19.2° of CNT/PEG and CNT/GO/PEG increased and became sharper in comparison to the pure PEG, and meanwhile, the CNT/GO/PEG exhibited a lower value than CNT/PEG. All of these indicated that the introduction of nanocarbon improved the PEG nucleation on CNT surfaces due to their unique sp2 and sp3 structure, resulting in much larger grain size, modified pattern distribution, and higher crystallinity. The lower peak value of CNT/GO/PEG may be mainly due to the GO further increasing the CNT network volume, which raises the PEG loading as well as weakening the PEG/nanocarbon interfacial interaction.
In addition, other physical properties of the composite phase change fiber were also characterized. The FTIR results shown in Fig. S1 (ESI†) indicated that the peak positions of the CNT/GO/PEG composite fiber were almost identical to those of pure PEG. Similarly, the Raman spectra in Fig. S2 (ESI†) revealed no peak shifts, and the decrease of IG/ID was significant due to the composition of PEG. All of these illustrated that there was no new bond among CNT, GO, and PEG, and no mutual damage to each other. On the other hand, PEG is known for its poor mechanical strength and non-conductive, which were greatly improved by the presence of CNT network and GO, consistent with our previous work,26,38 as shown in Fig. S3 and S4 (ESI†). The CNT/GO/PEG fiber exhibited a tensile strength of 142 MPa and electrical conductivity from 6.8 to 7.3 × 103 S m−1, and also showed typical electrical-thermal properties (Fig. S5, ESI†).
To better show the GO effect on PEG loading, GO with different sizes and concentrations were introduced into the compounding structure. SEM was used to characterize the GO morphology inside the PEG-impregnated CNT network, as shown in Fig. 2a, it was found that GO with different sizes (including 0.5 μm, 2.5 μm, 4.5 μm, 6.5 μm, 8.5 μm, 10.5 μm) could all be infiltrated into the interior of the CNT network, constructing a multilevel structure and freely distributing with almost no aggregation. The size distributions were also shown in Fig. 2b. The PEG mass fraction was characterized by using TGA tests and the results demonstrated high PEG loading with the GO size and concentration changing (Fig. 2c). With the variation of GO size from 0.5 μm to 10.5 μm, the PEG mass fraction was 96.8%, 97.5%, 97.6%, 98.4%, 97.8%, and 97.5%, respectively. Maintaining the GO size at 6.5 μm, PEG mass fraction was all above 97%, exhibiting higher PEG loading effectiveness. This is mainly attributed to the introduction of GO further enlarged the volume space of carbon nanotube networks. As shown in Fig. S6 (ESI†), the pores of the network structure could reach up to 1 μm in size, while PEG molecular chains are tens of nanometres in size, thus providing more sufficient channels and regions for the loading of PEG, and thereby increasing the PEG loading within the network. Another notable phenomenon was the highest PEG loading (98.4%) occurred at the GO size of 6.5 μm with 0.05 wt%. This is because small-sized GO sheets would enter into relatively large quantities but can not support a larger network space, whereas over-sized GO had a very limited ability to enter the interior of the network. In other words, an excess of GO incorporation may result in closed pores which hindering PEG loading, while too little GO also cannot effectively expand the 3D CNT network.
 | ||
| Fig. 2 Effect of GO on PEG loading. (a) Microscopic morphology of GO with different radial sizes in the interior fiber. (b) GO size distribution with a concentration of 0.5 wt%. (c) PEG loading with the changes of GO size from 0.5 m to 10.5 m and GO concentration from 0.02 wt% to 0.11 wt%. | ||
3.2 Thermal properties
Thermal property is one of the most critical factors in evaluating phase change materials. Here, the 3ω method was used to measure the thermal conductivity of composite fibers, as shown in Fig. 3a. A single fiber was suspended on a printed circuit board, and silver paste was used to connect the fiber to four Cu pads. Due to the conductive feature of the composite fiber, the third harmonic voltage U3ω can be accurately obtained by applying a small sinusoidal current Iω to the fiber. Combined with the length of the fiber, the electrical resistance, and the temperature coefficient of resistance, the value of the fiber's thermal conductivity can be calculated. More details can be referred to ref. 36 and 37. It has been well known that electrolysis-induced expansion is a non-damaging process for the CNT network,26,38,39 allowing which to maintain excellent thermal properties within the composite structure. Therefore, the tested thermal conductivity of CNT/PEG reached up to 27.3 W m−1 K−1 (Fig. 3b), in agreement with our previous works.26 | ||
| Fig. 3 Thermal conductivity of composite fibers. (a) Schematic diagram of 3ω measurement circuit connection. (b) Thermal conductivity of CNT/GO/PEG fibers with different concentrations and sizes. | ||
The modification of poor thermal conductivity is important for pure PCMs, however, excessively high thermal conductivity is also a disadvantage due to the rapid heat dissipation during the exothermic process. The insertion of two-dimensional GO into the one-dimensional CNT network increased the interior interface contacts of the network and introduced higher thermal resistance, resulting in a decreased lower thermal conductivity from 12.5 W m−1 K−1 to 13.5 W m−1 K−1, which was only half of CNT/PEG (Fig. 3b), yet still much higher than the pure PEG (0.4 W m−1 K−1). Furthermore, it was found that with the variation of GO size and concentration, the thermal conductivity remained at essentially the same level. Together with the PEG loading discussed above, it is clear that a small amount of GO insertion could be sufficient to achieve the purpose of thermal conductivity suppression in the CNT/GO/PEG composite structure.
3.3 Phase change properties
The energy storage capacity, phase change temperature, and degree of supercooling are important characteristics of PCMs. Meanwhile, the high PEG loading can guarantee a high storage capacity of CNT/GO/PEG fiber. DSC testing was used to investigate the details of the phase change process, Fig. 4a and b showed the typical DSC curve of pure PEG, CNT/PEG fiber, and CNT/GO/PEG fiber. For pure PEG, the peak temperature of the melting and crystallization were measured Tmelt = 70.6 °C and Tcrys = 37.5 °C. When the CNT network skeleton was introduced, the Tmelt and Tcrys shifted to 63.1 °C and 40.1 °C, respectively. The presence of GO had little effect on the Tmelt, but a strong one on Tcrys. When the GO size increased from 0.5 μm to 10.5 μm, Tcrys rightly shifted to 40.0 °C, 39.9 °C, 39.9 °C, 39.0 °C, 38.6 °C, 36.9 °C, comparing with the pure PEG. Furthermore, it is worth noting that the smaller the size, the farther the Tcrys to that of the pure PEG, and the smaller the degree of supercooling. In terms of concentration, there was also a similar Tcrys right shift and smaller supercooling degree with the concentration increasing (Fig. 4c). For phase change materials, the crystallization process directly affected the phase change behavior of the efficiency of thermal energy utilization. Here, GO played an important role in regulating heat transfer in the CNT/GO/PEG structure and thus influenced the crystallization behavior during the phase change process, which was also reflected in the thermal conductivity above. | ||
| Fig. 4 Phase change properties of CNT/GO/PEG composite fibers. (a) Typical DSC curves of pure PEG, CNT/PEG fibers, and CNT/GO/PEG fibers. (b) Typical DSC curves of pure PEG, CNT/PEG fibers, and CNT/GO/PEG fibers. (c) The melting and crystallization temperatures. (d) Evolution of the fusion enthalpy. | ||
As it was difficult to measure the GO mass fraction in the CNT/GO/PEG composite fiber from the GO size and concentration analysis. Here, the effect of GO size and concentration on the phase change enthalpy was conducted. The solid–liquid melting peak and liquid–solid freezing peak were used to calculate the melting and freezing latent heat values, respectively, see Fig. 4d and Table 1. The corresponding melting and freezing enthalpies were achieved by integrating the peak area of the melting and crystallization peaks. It seemed that Hmelt = 141.8, 143.6, 149.5, 154.4, 150.9, 151.4 J g−1 and Hcrys = 140.6, 142.9, 147.2, 152.5, 148.7, 149.2 J g−1 with the GO size increasing, the optimal value occurred at 6.5 μm. When the GO size was fixed and the concentration was varied, it was found that the peak of Hmelt = 154.4 J g−1 and Hcrys = 150.6 J g−1 still remained at 0.05 wt%, which was 97.6% of pure PEG 6000. Therefore, for the overall phase change performance, the size of 6.5 μm and 0.05 wt% was the optimal choice. Clearly, the variation of GO in the composite would not obviously change the thermal properties, yet could affect the crystalline behavior and thus the phase change enthalpy.
| Samples | PEG loading (wt%) | Thermal conductivity (W m−1 K−1) | T melt (°C) | H melt (J g−1) | T crys (°C) | H crys (J g−1) | R (%) | φ (%) | |
|---|---|---|---|---|---|---|---|---|---|
| Pure PEG 6000 | 99.2 | 0.4 | 70.6 | 158.2 | 37.5 | 156.0 | |||
| CNT/PEG | 95.1 | 27.4 | 63.1 | 132.7 | 40.1 | 131.4 | 83.9 | 84.1 | |
| GO size (μm) | 0.5 | 96.8 | 12.9 | 65.7 | 141.8 | 40.0 | 140.6 | 89.6 | 89.9 |
| 2.5 | 97.5 | 12.3 | 64.2 | 143.6 | 39.9 | 142.9 | 90.8 | 91.2 | |
| 4.5 | 97.6 | 12.1 | 64.0 | 149.5 | 39.9 | 147.2 | 94.5 | 94.4 | |
| 6.5 | 98.4 | 12.7 | 63.9 | 154.4 | 39.0 | 152.5 | 97.6 | 97.7 | |
| 8.5 | 97.8 | 13.0 | 64.3 | 150.9 | 38.6 | 148.7 | 95.4 | 95.4 | |
| GO concentration (wt%) | 10.5 | 97.5 | 12.7 | 64.8 | 151.4 | 37.9 | 149.2 | 95.7 | 95.7 |
| 0.02 | 97.5 | 13.5 | 64.2 | 153.3 | 37.9 | 151.6 | 96.9 | 97.0 | |
| 0.05 | 98.4 | 12.7 | 63.8 | 154.4 | 38.6 | 152.5 | 97.6 | 97.7 | |
| 0.08 | 97.9 | 12.4 | 64.7 | 151.8 | 40.1 | 150.6 | 96.0 | 96.2 | |
| 0.11 | 97.4 | 13.5 | 64.2 | 146.4 | 40.2 | 144.6 | 92.5 | 92.6 | |
In order to more visually represent the effective encapsulation of PCM in the CNT/GO/PEG composite structure, the encapsulation ratio (R) was determined by phase change enthalpy. The R is calculated by the enthalpies of the DSC measurements and eqn (1).40–42
 | (1) |
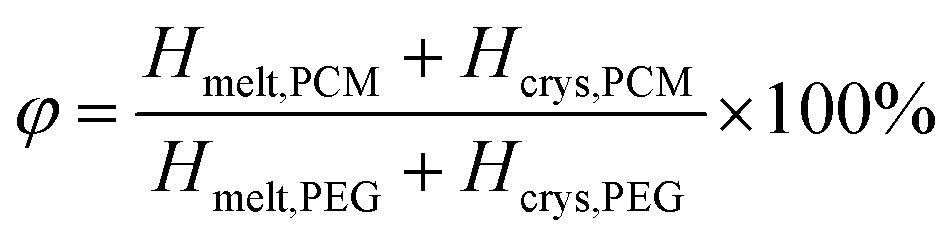 | (2) |
According to eqn (2), the φ of the CNT/GO/PEG and CNT/PEG fiber were 84.1% and 97.7% respectively, illustrating the addition of GO can improve the enthalpy of CNT/GO/PEG composite. This increase was mainly due to the PEG high loading effect of GOs space-expanding role. More details of the influence of GO size and concentration were summarised in Table 1.
3.4 Photothermal energy storage
It is well known that nanocarbon materials (including carbon nanotubes, graphene, fullerenes, etc.) were excellent light-absorbing materials. However, pure carbon materials cannot store the absorbed heat energy, an effective way is to introduce them into PCMs to simultaneously realize both light absorption and thermal storage. In order to evaluate the photothermal characteristics, CNT/PEG and CNT/GO/PEG composite fibers were woven into small pieces of fabric, see Fig. 5a. Diffuse reflectance spectroscopy was used to obtain the absorption spectroscopy for different samples. As shown in Fig. 5b, the CNT/PEG and CNT/GO/PEG fabrics exhibited significantly higher light absorbability than pure PEG, and the presence of GO had little influence on light absorption. | ||
| Fig. 5 Photothermal properties of different fabrics. (a) The tested fabrics based on CNT, CNT/PEG, and CNT/GO/PEG fibers. (b) Absorption spectroscopy of the different samples. (c) The temperature-time curves of photothermal performance. (d) The thermal management demonstration. (e) The corresponding infrared thermographic photos. | ||
The photothermal energy storage was characterized by a home-made set-up, a light irradiation of 150 mW cm−2 was used to warm the fabric samples, and the temperature was recorded via thermocouples. Fig. 5c showed the temperature-time curves for different samples, and the corresponding infrared thermographic photos at different times were provided in Fig. 5e. The pure PEG film could just get warmed to 32 °C in 180 s, far below Tmelt, and then rapidly cooled down to room temperature after the light-off, not exhibiting any phase change performance. The CNT fabric showed a high ability to absorb photo energy and could be warmed up to 84.1 °C in 170 s, yet did not store the energy and caused a rapid temperature decrease due to the high emissivity after the light-off. For the CNT/PEG and CNT/GO/PEG fabrics, there were clear stages of heat storage and release. The CNT/PEG fabric grew up to a plateau that set around 52 °C and then reached 93.2 °C. The CNT/GO/PEG fabric demonstrated a similar on-set but a lower warming temperature of 86.3 °C due to the presence of GO. There were clear melting plateaus that could be used for energy storage and temperature regulation. When the light was off, the CNT/PEG and CNT/GO/PEG fabrics also showed an extended solidification plateau. Especially, the CNT/GO/PEG fabric showed a much higher maintained temperature than the CNT/PEG, such as 44.9 °C and 41.1 °C in 300 s, 44.2 °C and 38.6 °C in 400 s, 29.4 °C and 25.9 °C in 400 s, respectively. The corresponding infrared thermographic photos for the CNT, CNT/PEG, and CNT/GO/PEG fabrics (Fig. 5c) further visually demonstrated the superior insulation characteristic of CNT/GO/PEG fabric.
In order to more straightforwardly validate the thermal management effectiveness of CNT/GO/PEG fabric, a scenario (see Fig. 5d, inside) was designed to demonstrate the internal room environment. The fabric was wrapped around a glass bottle and a black cotton was also chosen as a comparison sample. The interior temperature inside the room was recorded during the light-on and light-off, as shown in Fig. 5d. Due to the high blackness of the cotton and CNT fabrics, the interior temperature could grow up to above 60 °C, but without any heat storage. Thus after the light-off, the temperature decreased very rapidly to ambient one. As for the CNT/PEG and CNT/GO/PEG fabrics, the temperature–time curves exhibited clear heat storage and release in a wider range of time, and the CNT/GO/PEG fabric showed a much gentler and smoother cooling process, and also reflected a better thermal insulation effectiveness. All of these suggested that CNT/GO/PEG fabric exhibited better thermal management characteristics.
3.5 Thermal stability
The CNT/GO network structure also provided excellent thermal stability, as well as superior anti-liquid leakage and cyclic stability. Fig. 6a showed a cyclic DSC test on the CNT/GO/PEG composite fiber, where the curves did not show any evident changes in the endothermic and exothermic peaks during the 200 cycles of heating and cooling process. The corresponding melting enthalpy exhibited Hmelt consistently around 154.4 J g−1 (see Fig. 6b), indicating full retention of the phase changeability. This means the thermal cycles did not cause any degradation or deterioration. Meanwhile, when heating the PEG film and CNT/GO/PEG fabric with an alcohol lamp, the former melted and lost its original shape in a very short time, while there was no change in the CNT/GO/PEG fabric due to the strong confinement of the CNT/GO network, as shown in Fig. 6d, which exhibited excellent shape stability upon heating. | ||
| Fig. 6 Cycling test of CNT/GO/PEG composite fiber. (a) A cyclic DSC curves. (b) The corresponding crystalizing enthalpy as a function of cycle number. (c) A cyclic photothermal test for the CNT/GO/PEG composite fiber, one cycle of 15 min. (d) The photos of pure PEG film, CNT/PEG, and CNT/GO/PEG composite fabric of heating by a flame. | ||
Similarly, the photothermal cyclic performance was also tested after 200 cycles. The temperature–time curves shown in Fig. 6c display distinct plateaus in each cycle, indicating the composite fibers could gradually release the absorbed photothermal energy for over 15 minutes. In addition, the time stability was also evaluated by testing the composite fibers after 1 week, 1 month and, 3 months respectively, as shown in Fig. S7 (ESI†), the DSC curves demonstrated perfect overlap across these different time points. All of this is strong evidence that the PEG was effectively confined inside the CNT/GO network structure.
4 Conclusion
A cooperative in situ impregnation strategy was adopted to introduce GO and PEG into the CNT network, resulting in the preparation of CNT/GO/PEG phase change fibers. The obtained CNT/GO/PEG fibers exhibited high PEG loading (from 96.7 wt% to 98.4 wt%), excellent thermal conductivities, and favorable phase change enthalpies. The presence of GO constructed a 1D–2D multilevel network skeleton structure, which was crucial for enhancing interfacial contact, leading to decreased thermal conductivity (12.5–13.5 W m−1 K−1). This also significantly improved photothermal utilization efficiency and performed a more favorable thermal insulation effectiveness. Additionally, due to the rich interface and strong confinement of the network, the CNT/GO/PEG fiber demonstrated high polymer loading ability and superior thermal stability, including anti-leakage and consistent phase change abilities. These typical characteristics present a promising opportunity for the development of thermal management applications.Data availability
Data will be made available on request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors acknowledge financial supports by the National Natural Science Foundation of China (52373031), the Key Research Program Industrial Textiles Collaborative Innovation Center Project of Shaanxi Provincial Department of education (No. 20JY026), Natural Science Foundation of Jiangsu Province (K20221247).References
- X. Chen, P. Cheng, Z. Tang, X. Xu, H. Gao and G. Wang, Carbon-based composite phase change materials for thermal energy storage, transfer, and conversion, Adv. Sci., 2021, 8(9), 2001274 CrossRef CAS PubMed.
- X. Chen, H. Gao, Z. Tang, W. Dong, A. Li and G. Wang, Optimization strategies of composite phase change materials for thermal energy storage, transfer, conversion and utilization, Energy Environ. Sci., 2020, 13(12), 4498–4535 RSC.
- J. Xu, X. Zhang and L. Zou, A review: Progress and perspectives of research on the functionalities of phase change materials, J. Energy Storage, 2022, 54, 105341 CrossRef.
- Z. Tang, H. Gao, X. Chen, Y. Zhang, A. Li and G. Wang, Advanced multifunctional composite phase change materials based on photo-responsive materials, Nano Energy, 2021, 80, 105454 CrossRef CAS.
- F. Mahdavian, A. Allahbakhsh, D. Rodrigue and A. Reza Bahramian, An analytical model for the energy storage potential of phase change materials supported by polymeric colloidal aerogels, J. Energy Storage, 2023, 72, 108568 CrossRef.
- X. Li, Y. Zhao, X. Min, J. Xiao, X. Wu, R. Mi, Y. Liu, Z. Huang and M. Fang, Carbon nanotubes modified graphene hybrid aerogel-based composite phase change materials for efficient thermal storage, Energy Build., 2022, 273, 112384 CrossRef.
- G. Wang, Z. Tang, Y. Gao, P. Liu, Y. Li, A. Li and X. Chen, Phase change thermal storage materials for interdisciplinary applications, Chem. Rev., 2023, 123(11), 6953–7024 CrossRef CAS PubMed.
- A. Sar, A. Bicer, F. A. Al-Sulaiman, A. Karaipekli and V. V. Tyagi, Diatomite/cnts/peg composite pcms with shape-stabilized and improved thermal conductivity: preparation and thermal energy storage properties, Energy Build., 2018, 164, 166–175 CrossRef.
- X. Zhao, D. Zou and S. Wang, Flexible phase change materials: Preparation, properties and application, Chem. Eng. J., 2022, 431, 134231 CrossRef CAS.
- Y. Lu, D. Yu, H. Dong, J. Lv, L. Wang, H. Zhou, Z. Li, J. Liu and Z. He, Magnetically tightened form-stable phase change materials with modular assembly and geometric conformality features, Nat. Commun., 2022, 13(1), 1397 CrossRef CAS PubMed.
- J. Bao, D. Zou, S. Zhu, Q. Ma, Y. Wang and Y. Hu, A medium-temperature, metal-based, microencapsulated phase change material with a void for thermal expansion, Chem. Eng. J., 2021, 415, 128965 CrossRef CAS.
- K. Yuen Leong, M. Rosdzimin Abdul Rahman and B. A. Gurunathan, Nano-enhanced phase change materials: a review of thermo-physical properties, applications and challenges, J. Energy Storage, 2019, 21, 18–31 CrossRef.
- X. Kong, Y. Fu and J. Yuan, Novel flexible phase change materials with high emissivity, low thermal conductivity and mechanically robust for thermal management in outdoor environment, Appl. Energy, 2023, 348, 121556 CrossRef CAS.
- J. Cheng, S. Niu, M. Kang, Y. Liu, F. Zhang, W. Qu, Y. Guan and S. Li, The thermal behavior and flame retardant performance of phase change material microcapsules with modified carbon nanotubes, Energy, 2022, 240, 122821 CrossRef CAS.
- D. Zhang, M. Chen, S. Wu, Q. Liu and J. Wan, Preparation of expanded graphite/polyethylene glycol composite phase change material for thermoregulation of asphalt binder, Constr. Build. Mater., 2018, 169, 513–521 CrossRef CAS.
- N. Sheng, Z. Rao, C. Zhu and H. Habazaki, Honeycomb carbon fibers strengthened composite phase change materials for superior thermal energy storage, Appl. Therm. Eng., 2020, 164, 114493 CrossRef CAS.
- W. Xia, H. Xiang, Z. Zhou, X. Fei and M. Zhu, Hybridizing rational designed hydrophobic peg-based derivatives into nanoporous f-sio2 as form-stable phase change materials for melt-spun pa6 phase change fibers with a superior washing durability, Composites Commun., 2021, 24, 100633 CrossRef.
- W. Xia, X. Fei, Q. Wang, Y. Lu, M. Tendo Innocent, J. Zhou, S. Yu, H. Xiang and M. Zhu, Nano-hybridized form-stable ester@ f-SiO2 phase change materials for melt-spun pa6 fibers engineered towards smart thermal management fabrics, Chem. Eng. J., 2021, 403, 126369 CrossRef CAS.
- Y. Cai, C. Gao, X. Xu, Z. Fu, X. Fei, Y. Zhao, Q. Chen, X. Liu, Q. Wei and G. He, et al., Electrospun ultrafine composite fibers consisting of lauric acid and polyamide 6 as form-stable phase change materials for storage and retrieval of solar thermal energy, Sol. Energy Mater. Sol. Cells, 2012, 103, 53–61 CrossRef CAS.
- M. Li, F. Gan, J. Dong, Y. Fang, X. Zhao and Q. Zhang, Facile preparation of continuous and porous polyimide aerogel fibers for multifunctional applications, ACS Appl. Mater. Interfaces, 2021, 13(8), 10416–10427 CrossRef CAS PubMed.
- W. Li, L. Xu, X. Wang, R. Zhu and Y. Yan, Phase change energy storage elastic fiber: a simple route to personal thermal management, Polymers, 2021, 14(1), 53 CrossRef PubMed.
- M. Tehrani, Advanced electrical conductors: an overview and prospects of metal nanocomposite and nanocarbon based conductors, Phys. Status Solidi A, 2021, 218(8), 2000704 CrossRef CAS.
- G. Abdeali, A. Reza Bahramian and M. Abdollahi, Review on nanostructure supporting material strategies in shape-stabilized phase change materials, J. Energy Storage, 2020, 29, 101299 CrossRef.
- C. Li, B. Zhang, B. Xie, X. Zhao, J. Chen, Z. Chen and Y. Long, Stearic acid/expanded graphite as a composite phase change thermal energy storage material for tankless solar water heater, Sustainable Cities Soc., 2019, 44, 458–464 CrossRef.
- Y. Qian, N. Han, X. Gao, X. Gao, W. Li and X. Zhang, Cellulose-based phase change fibres for thermal energy storage and management applications, Chem. Eng. J., 2021, 412, 128596 CrossRef CAS.
- J. Zhao, W. Zhang, Q. Lu, T. Liao, W. Li, X. Zhang and Q. Li, High energy-capacity and multiresponsive phase change fibers via in situ polymer composition with expanded carbon nanotube networks, Chem. Eng. J., 2024, 481, 148262 CrossRef CAS.
- G. Li, G. Hong, D. Dong, W. Song and X. Zhang, Multiresponsive graphene-aerogel-directed phase-change smart fibers, Adv. Mater., 2018, 30(30), 1801754 CrossRef PubMed.
- S. Liu, W. Wang, S. Xin, X. Meng and Z. Zhang, Preparation and characterization of oligomeric thermal phase change polyurethane foam, J. Energy Storage, 2023, 72, 108703 CrossRef.
- J. Shi, H. Du, Z. Chen and S. Lei, Review of phase change heat transfer enhancement by metal foam, Appl. Therm. Eng., 2023, 219, 119427 CrossRef CAS.
- K. Sun, Y. Kou, H. Dong, S. Ye, D. Zhao, J. Liu and Q. Shi, The design of phase change materials with carbon aerogel composites for multi-responsive thermal energy capture and storage, J. Mater. Chem. A, 2021, 9(2), 1213–1220 RSC.
- G. Li, X. Zhang, J. Wang and J. Fang, From anisotropic graphene aerogels to electron-and photo-driven phase change composites, J. Mater. Chem. A, 2016, 4(43), 17042–17049 RSC.
- Z. Zheng, H. Liu, D. Wu and X. Wang, Polyimide/mxene hybrid aerogel-based phase-change composites for solar-driven seawater desalination, Chem. Eng. J., 2022, 440, 135862 CrossRef CAS.
- L. Chen, X. Sun, K. Cheng, P. D. Topham, M. Xu, Y. Jia, D. Dong, S. Wang, Y. Liu and L. Wang, et al., Temperature-regulating phase change fiber scaffold toward mild photothermal-chemotherapy, Adv. Fiber Mater., 2022, 4(6), 1669–1684 CrossRef CAS.
- K. Wu, B. Wang, Y. Niu, W. Wang, C. Wu, T. Zhou, L. Chen, X. Zhan, Z. Wan and S. Wang, et al., Carbon nanotube fibers with excellent mechanical and electrical properties by structural realigning and densification, Nano Res., 2023, 16(11), 12762–12771 CrossRef CAS.
- T. Zhou, Y. Niu, Z. Li, H. Li, Z. Yong, K. Wu, Y. Zhang and Q. Li, The synergetic relationship between the length and orientation of carbon nanotubes in direct spinning of high-strength carbon nanotube fibers, Mater. Des., 2021, 203, 109557 CrossRef CAS.
- L. Qiu, P. Guo, X. Yang, Y. Ouyang, Y. Feng, X. Zhang, J. Zhao, X. Zhang and Q. Li, Electro curing of oriented bismaleimide between aligned carbon nanotubes for high mechanical and thermal performances, Carbon, 2019, 145, 650–657 CrossRef CAS.
- L. Qiu, H. Zou, X. Wang, Y. Feng, X. Zhang, J. Zhao, X. Zhang and Q. Li, Enhancing the interfacial interaction of carbon nanotubes fibers by au nanoparticles with improved performance of the electrical and thermal conductivity, Carbon, 2019, 141, 497–505 CrossRef CAS.
- T. Liao, W. Li, J. Zhao, X. Yang, W. Zhang, J. Zou, B. Zhao, X. Zhang and Q. Li, Cooperative in situ impregnation of peg and au nanoparticles into expanded cnt network toward composite phase change fibers with high storage capacity and photothermal conversion, Adv. Mater. Technol., 2024, 2400259 CrossRef.
- J. Wang, J. Zhao, L. Zhao, Q. Lu, T. Zhou, Z. Yong, P. Wang, X. Zhang and Q. Li, Interfacial-bubbling-induced nondestructive expansion to reconstruct superstrong and multifunctional carbon nanotube fibers, Carbon, 2021, 184, 24–33 CrossRef CAS.
- H. Zhang, X. Wang and D. Wu, Silica encapsulation of n-octadecane via sol-gel process: a novel microencapsulated phase-change material with enhanced thermal conductivity and performance, J. Colloid Interface Sci., 2010, 343(1), 246–255 CrossRef CAS PubMed.
- P. Cheng, H. Gao, X. Chen, Y. Chen, M. Han, L. Xing, P. Liu and G. Wang, Flexible monolithic phase change material based on carbon nanotubes/chitosan/poly(vinyl alcohol), Chem. Eng. J., 2020, 397, 125330 CrossRef CAS.
- S. Yu, X. Wang and D. Wu, Microencapsulation of n-octadecane phase change material with calcium carbonate shell for enhancement of thermal conductivity and serving durability: synthesis, microstructure, and performance evaluation, Appl. Energy, 2014, 114, 632–643 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc03006k |
| This journal is © The Royal Society of Chemistry 2024 |