
Benzene layer-aligned electrochemical transformation of SWCNTs to redox-active macro-walled CNTs: enabling oxygen interference-free monitoring of ROS release from HeLa cancer cells†
Sakthivel
Srinivas
ab,
Nisha
Sivakumar
a,
Mouliganesh
Sekar
c,
Kavitha
Thirumurugan
 c and
Annamalai
Senthil Kumar
*ab
c and
Annamalai
Senthil Kumar
*ab
aNano and Bioelectrochemistry Research Laboratory, Carbon dioxide Research and Green Technology Centre, Vellore Institute of Technology, Vellore-632 014, India
bDepartment of Chemistry, School of Advanced Sciences, Vellore Institute of Technology, Vellore-632 014, India. E-mail: askumar@vit.ac.in; askumarchem@yahoo.com; Tel: +91-416-2202754
cStructural Biology Lab, Pearl Research Park, School of Biosciences & Technology, Vellore Institute of Technology, Vellore, Tamil Nadu, India
First published on 13th June 2024
Abstract
The search for novel carbon allotropes with unique electrochemical properties remains a key area of research in materials science. Here, we introduce a novel carbon nanotube material termed macro-walled CNTs, synthesized through an in situ benzene (BZ) electrochemical reaction on a glassy carbon electrode modified with single-walled carbon nanotubes (GCE/SWCNTs) in a pH 2 KCl–HCl electrolyte solution. The modified electrode, denoted as GCE/SWCNT@BZ-Redox (where BZ-Redox represents redox-active benzene species), exhibited a well-defined redox peak at E°′ = 150 ± 10 mV vs. Ag/AgCl, along with a surface excess value of 5.1 nmol cm−2. The physicochemical characterization of SWCNT@BZ-Redox, conducted using TEM, FTIR, Raman spectroscopy, and various electrochemical methods including scanning electrochemical microscopy (SECM) imaging, revealed a significant modification involving highly redox-active benzene multilayers with a diameter of approximately ∼200 nm, which is about 10 times larger than that of pristine SWCNTs (∼10–15 nm). Of particular interest is the stability of the redox peak under physiological pH conditions, as well as its ability to mediate hydrogen peroxide reduction reactions at a potential of −0.25 V vs. Ag/AgCl, akin to the reaction catalyzed by horseradish peroxidase enzymatic systems. For practical applications, continuous monitoring of reactive oxygen species (ROS), specifically H2O2 release kinetics from stressed HeLa cancer cells under simulated conditions, without any dissolved oxygen interference, was demonstrated using a home-made bath-injection analysis system coupled with a screen-printed electrode modified with SWCNT@BZ-Redox as a detector.
1. Introduction
Due to their unique electrical, chemical, and mechanical properties,1 carbon nanotubes (CNTs) have found diverse applications, including the development of chemical sensors,2,3 composite materials,4 semiconductors,5 energy materials,6 and hydrogen storage systems,7 among others. Recognized for their large specific surface area, high electronic conductivity, mechanical stability, micropore volume, and rapid adsorption equilibrium, CNTs are also acknowledged as effective adsorbents.8 Extensive research has been conducted to explore the adsorption capabilities of CNTs for various aromatic compounds such as benzene (BZ),9–13 polyaromatics,14 xylenes,15 chlorophenols,16 nitrobenzene,17 phenolic compounds,18 herbicides,19 and organic dyes.20,21 The adsorption mechanism typically involves π–π interactions between the sp2 carbons of the benzene ring in aromatic compounds and the sp2 carbon of the graphene units of CNTs, resulting in the formation of a few monolayer adsorbed species. It is important to note that high-pressure studies, conducted within the pressure range of 0.1 to 4 MPa, investigating the adsorption and removal of BZ from air using single-walled carbon nanotubes (SWCNTs), showed minor and predictable changes in the electronic and geometrical structures of SWCNTs.22 Similarly, chemisorption of BZ (BZchem) on CNTs was achieved by an in situ electrochemical generation of aryl radical ion species from aryldiazonium salts (CNT@BZchem), followed by a surface chemical reaction in an acid solution via C–C (covalent) bond formation.23–25 An interesting observation is the perpendicular orientation of covalently functionalized aryl radicals, contrasting the parallel alignment of physiosorbed BZ to the CNT plane. Furthermore, unzipping of CNTs using intercalation and oxidative conditions to form nanoribbons of graphene has been reported.26–30 Such modification procedures either slightly alter or reduce the dimensions of the graphene layers of the CNTs. In this study, we present a unique electrochemical reaction-assisted surface packing/alignment of BZ as a redox-active molecular species on SWCNTs, referred to as SWCNT@BZ-Redox. This modification results in approximately a ten-fold increase in the diameter (∼100 nm) of the SWCNTs and introduces redox functionality, diverging from previous literature on BZ adsorption onto and unzipping of carbon nanotube (CNT) systems.9,25–28 Our research findings provides new insights into the behavior of SWCNTs interacting with BZ in an electrochemical context, offering distinct perspectives from conventional findings.In the field of electrochemistry, owing to its high oxidation potential (>2 V vs. Ag/AgCl), BZ has gained attention as a solvent for the oxidation and reduction reactions of various organic molecules.30,31 Despite its prevalence in electrochemical studies, there are a limited number of studies in the literature exploring the electrochemical oxidation of BZ on solid electrodes like Pt,31 GCE,32 PbO2 (at ∼250 °C),33 VOx (at 50 °C),34 bifunctional Ni–(O–C2)4 sites,35 and Sb-SnO2/foam Ti nano-coating electrodes.36 Existing literature reports highlight the formation of oxidized products, including catechol,37 hydroquinone,38 polymeric BZ derivatives39 and CO2,40 underscoring the complexity of the oxidation process. In a recent development, our research group made a preliminary observation regarding the electrochemical-assisted π-stacking of BZ molecular species on multiwalled carbon nanotubes.41 This molecular system demonstrated a remarkably efficient redox signal, attributed to the formation of surface-confined BZ radical species. Notably, it exhibited profound electrocatalytic reduction of hydrogen peroxide. However, several questions remain unanswered in this context. These include the potential extensibility of the observed phenomena to other carbon nanomaterials, the influence of the surface features of BZ species packed CNTs, the impact of electrochemical properties on the orientation and dynamics of BZ adsorbed on CNTs, the applicability of this system to real-time applications, etc. Attaining a balance in comprehending these aspects is essential for advancing our understanding of the electrochemical behaviours of BZ on SWCNTs and its potential applications across different systems.
Reactive oxygen species (ROS), including the superoxide anion (O2˙−), hydrogen peroxide (H2O2) and the hydroxyl radical (HO˙), are typical byproducts of cellular metabolism. These ROS act as secondary messengers, influencing various normal physiological functions in the body. Additionally, growing evidence suggests a role for ROS in several diseases.42–44 For instance, an excessive release of ROS can trigger oxidative stress in HeLa cancer cells, resulting in cellular toxicity and ultimately leading to cell death.44,45 Research findings suggest that cancer cells are selectively killed by intracellular and extracellular ascorbic acid (AA, stress inducer) after certain hours of incubation.46,47 Considering the urgent need to develop selective and continuous monitoring methodologies for ROS in living cellular systems, this study employs HeLa cells as a model cellular system, along with micromolar concentrations of AA as a stress inducer to release and detect H2O2 (as a model ROS system).48,49
HeLa cells are a human cell-line derived from cervical cancer cells originating from the cancerous tumour of Henrietta Lacks in 1951.49–52 These cells have served as a fundamental research platform for medical researchers to study biology, cellular processes, potential treatments, and mechanisms of action.50–53 Traditionally, detection of H2O2 released from HeLa cells relied on enzyme-linked spectroscopy and chromatography techniques.54–56 However, electrochemical methods offer a more efficient and portable approach using disposable screen-printed electrodes, eliminating the need for a specialized person. For trace H2O2 detection in living HeLa cells, researchers have developed various chemically modified electrodes.56–59 These include enzyme-based hydrogels, boronic acid-functionalized frameworks, and graphene-blended metal oxide electrodes.57–59 However, interference from dissolved oxygen and limited stability under physiological conditions remain challenges for some of these methods, particularly those using metal oxides.60–62 This work presents a SWCNT@BZ-Redox system that addresses these concerns, offering highly selective H2O2 detection without dissolved oxygen interference at neutral pH. As a practical application, for the first time in the literature, a prototype batch injection analysis (BIA) system was developed for continuous monitoring of H2O2 from HeLa cells under simulated conditions. The overarching aims and objectives of this work are as follows: (i) to conduct electrochemical investigations of BZ oxidation reactions on SWCNT-modified GCE surfaces and analyse the surface-confined molecular species using various physicochemical and electrochemical techniques; (ii) to compare the electrochemical properties of BZ-functionalized SWCNTs prepared by covalent and π–π interaction methods; (iii) to visualize the surface features of SWCNT@BZ-Redox using the scanning electrochemical microscopy (SECM) technique with H2O2 as a molecular probe; (iv) to develop a highly efficient electroanalytical technique for oxygen interference-free sensing of H2O2; and (v) to demonstrate real sample analysis by studying H2O2 release from HeLa cells.
2. Experimental section
2.1. Materials and reagents
Benzene (HPLC grade, ≥99.9% pure), 4-nitrobenzenediazonium tetrafluoroborate (DZ), single-walled carbon nanotubes (SWCNTs; ≥95% purity on carbon basis, 0.84 nm average diameter), carbon nanofibers (CNF; >99.9% purity on a carbon basis, D × L: 100 nm × 20–200 μm), a graphitized mesoporous carbon-hydrophilic core (GMC, 99.95% purity, particle size: 5–50 μm) and activated charcoal (AC, 99.95% pure) were purchased from Sigma-Aldrich Chemicals (USA). Graphene nanoplatelets, 6–8 nm thick and 25 μm wide, were purchased from TCI Chemicals (India). All the other generic chemicals used in this work were of analytical standard. The pH 2 HCl–KCl supporting electrolyte and phosphate buffer solution of pH 7 prepared at 0.1 M ionic strength using deionized water (DIW) were used throughout the work. The DO concentration was measured using a commercial DO meter indicating [DO] = 3.1 ± 0.5 ppm.2.2. Instruments
For all electrochemical investigations, we employed a CHI760D electrochemical workstation (USA) in conjunction with a three-electrode setup. This setup consisted of a glassy carbon electrode (GCE) with a diameter of 3 mm as the working electrode, a 2 mm Pt-disc electrode as the counter electrode, and an Ag/AgCl (3 M KCl) electrode as the reference electrode. A portable dissolved oxygen meter (HI19142) from Hanna Instruments was utilized to measure the dissolved oxygen concentration. A high-resolution transmission electron microscope (HR-TEM, model: FEI-TECNAI G2-20 TWIN) was employed for imaging the morphology. SECM experiments were conducted using a Princeton Applied Research (PAR) VersaSCAN bipotentiostat instrument, with a 3 mm diameter chemically modified GCE and a 25 μm platinum (Pt) ultramicroelectrode (UME) as the probe electrode. The screen-printed electrode system coupled batch injection analysis system was custom-made and sourced from Metrohm (The Netherlands). An electronically robust pipette was procured from Glimson. Raman spectroscopic analysis was carried out using an i-Raman Plus spectrometer in the spectral range of 65–3400 cm−1 using a 532 nm laser wavelength. For FT-IR analysis, a SHIMADZU IRAffinity-1 instrument (Japan) was utilized employing the KBr method. Zensor disposable screen-printed electrodes (SPE) served as the base for Raman characterization.2.3. Preparation of the GCE/SWCNT@BZ-Redox modified electrode
Initially, the surface of the GCE was thoroughly cleaned using both mechanical and electrochemical methods. This process involved polishing with 0.5-micron alumina powder, subsequent rinsing with distilled water, and subjecting the electrode to ten continuous cycles of cyclic voltammetry within the potential range of −0.2 to 1 V vs. Ag/AgCl at a scan rate (v) of 50 mV s−1 in a pH 2 HCl/KCl solution. These steps were aimed at eliminating any physiosorbed impurities adhering to the electrode surface. The preparation of the GCE/SWCNT@BZ-Redox chemically modified electrode (CME) involved a three-step procedure. In step 1, the GCE was modified with SWCNTs by applying a 5 μL suspension of {SWCNT + ethanol} (10 mg mL−1), followed by drying at room temperature. The resulting GCE/SWCNT was then pretreated by subjecting it to twenty continuous CV cycles within the potential range of −0.4 to 1.2 V vs. Ag/AgCl in a pH 2 HCl/KCl solution. This pretreatment step facilitated the removal of trace metal impurities present in the SWCNTs via an oxidative dissolution method.63 In step 2, a 5 μL azeotropic mixture of {BZ + H2O}, prepared in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 volume ratio, was drop-casted onto the GCE/SWCNT surface (forming SWCNT@{BZ + H2O}ads) and left to dry in air at room temperature for approximately 5 minutes. In step 3, the SWCNT@{BZ + H2O}ads was subjected to either twenty continuous cycles of CV within an optimal potential window of −0.2 to 1.2 V vs. Ag/AgCl at a scan rate of 50 mV s−1 or potentiostatic polarization at an applied potential of 1.2 V vs. Ag/AgCl for 220 seconds in a pH 2 HCl–KCl solution (Scheme 1). Notably, both experimental procedures yielded quantitatively similar results. These steps led to the in situ generation of GCE/SWCNT@BZ-Redox on the electrode surface.
:1.5 volume ratio, was drop-casted onto the GCE/SWCNT surface (forming SWCNT@{BZ + H2O}ads) and left to dry in air at room temperature for approximately 5 minutes. In step 3, the SWCNT@{BZ + H2O}ads was subjected to either twenty continuous cycles of CV within an optimal potential window of −0.2 to 1.2 V vs. Ag/AgCl at a scan rate of 50 mV s−1 or potentiostatic polarization at an applied potential of 1.2 V vs. Ag/AgCl for 220 seconds in a pH 2 HCl–KCl solution (Scheme 1). Notably, both experimental procedures yielded quantitatively similar results. These steps led to the in situ generation of GCE/SWCNT@BZ-Redox on the electrode surface.
 | ||
| Scheme 1 Schematic representation of the electrochemical oxidation of benzene (BZ) on the SWCNT surface at about 1 V vs. Ag/AgCl in pH 2 KCl–HCl solution (A) and the surface confined redox-species formation of GCE/SWCNT@BZ-Redox and its selective hydrogen peroxide electrocatalytic reaction without interference from dissolved oxygen reduction in pH 7 phosphate buffer solution (B). Specific changes in the diameter of the SWCNTs before and after the electrochemical oxidation of BZ with the formation of a multilayered stack of redox active BZ-Redox. | ||
2.4. Preparation of the GCE/SWCNT@BZchem modified electrode
The preparation procedure was adapted from a previous study.64 The GCE/SWCNT modified electrode, prepared beforehand, was immersed in an acetonitrile (ACN) solvent containing 0.1 M tetrabutylammonium tetrafluoroborate as an electrolyte (N2 purged). To this solution, 5 mM 4-nitrobenzenediazonium tetrafluoroborate (n-Bu4NBF4) was added, and the electrode was subjected to CV potential cycling between 0.8 and −1.2 V vs. Ag/AgCl. Subsequently, the as-prepared GCE/SWCNT@BZchem electrode was rinsed with distilled water, dried, and subjected to electrochemical treatment as depicted in Fig. S1 (ESI†).2.5. HeLa cell culture maintenance and treatment
The human cervical carcinoma HeLa cell line was obtained from the National Centre for Cell Science (NCCS) in Pune, India. These cells were cultured in high-glucose Dulbecco's Modified Eagle Medium (DMEM, Invitrogen, USA), supplemented with 10% fetal bovine serum (FBS, Invitrogen, USA) and 1% antibiotic (penicillin–streptomycin, Invitrogen, USA).65 The cell cultures were maintained in a 5% CO2 incubator at 37 °C in a humidified environment, following established protocols. HeLa cells were grown in a T75 flask for expansion in preparation for the experiment. Once the cells reached 100% confluence, they were detached from the flask using trypsin. These cells were centrifuged at 1200 rpm for 4 min to obtain the pellet. This pellet was resuspended in PBS and centrifuged again. This washing step was repeated twice. Later, the cells were counted using a hemocytometer and 5 × 106 cells were taken for each group for the experiment in PBS buffer. Furthermore, the cells treated with 10 μM AA were incubated for 15, 30, 45, and 60 min, respectively, for the stimulation of ROS.65,66 To confirm the H2O2 generation, a conventional 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) assay was performed.67 The detailed experimental results and discussion are provided in the ESI† (Fig. S2).3. Results and discussion
3.1. Electrochemical response of the GCE/SWCNT@BZ-Redox CME
First, since BZ is partially soluble in water, the {BZ + H2O} azeotropic mixture was prepared, drop-casted on a bare GCE surface (i.e., GCE/{BZ + H2O}ads, where “ads” denotes adsorbed), and dried and its electrochemical response was examined by CV in a potential window of −0.2 to 1.2 V vs. Ag/AgCl at v = 50 mV s−1. No faradaic signal was observed in this setup, indicating the electrochemical inactivity and structural rigidity of BZ, which resisted oxidation under limited potential conditions (Fig. 1A, curves a and b). Intriguingly, repeating the same experiment on SWCNTs with the {BZ + H2O} mixture (i.e., GCE/SWCNT@{BZ + H2O}ads) revealed a growth-like redox response at an equilibrium potential, E1/2 = 150 ± 10 mV vs. Ag/AgCl (Fig. 1B, curve a). Subsequently, when the electrode was gently washed with blank electrolyte (pH 2 KCl–HCl) and subjected to CV in a fresh electrolytic solution, the redox peak was retained without any alteration in peak current response (Fig. 1C). The calculated relative standard deviation (RSD) of the redox peak at ten continuous CVs was 2.6%. Independently, repeating the experiment three times yielded appreciable reproducibility and good stability, with an error percentage of 4.7% (Fig. 1C). It is anticipated that the electrochemical treatment converted the electrochemically inactive BZ to redox-active species (BZ-Redox), which were stabilized via π–π bonding between the aromatic π-electrons of BZ-Redox and the sp2 carbons of SWCNTs (Scheme 1). Henceforth, this chemically modified electrode is designated as GCE/SWCNT@BZ-Redox, where BZ-Redox refers to the redox-active species of benzene.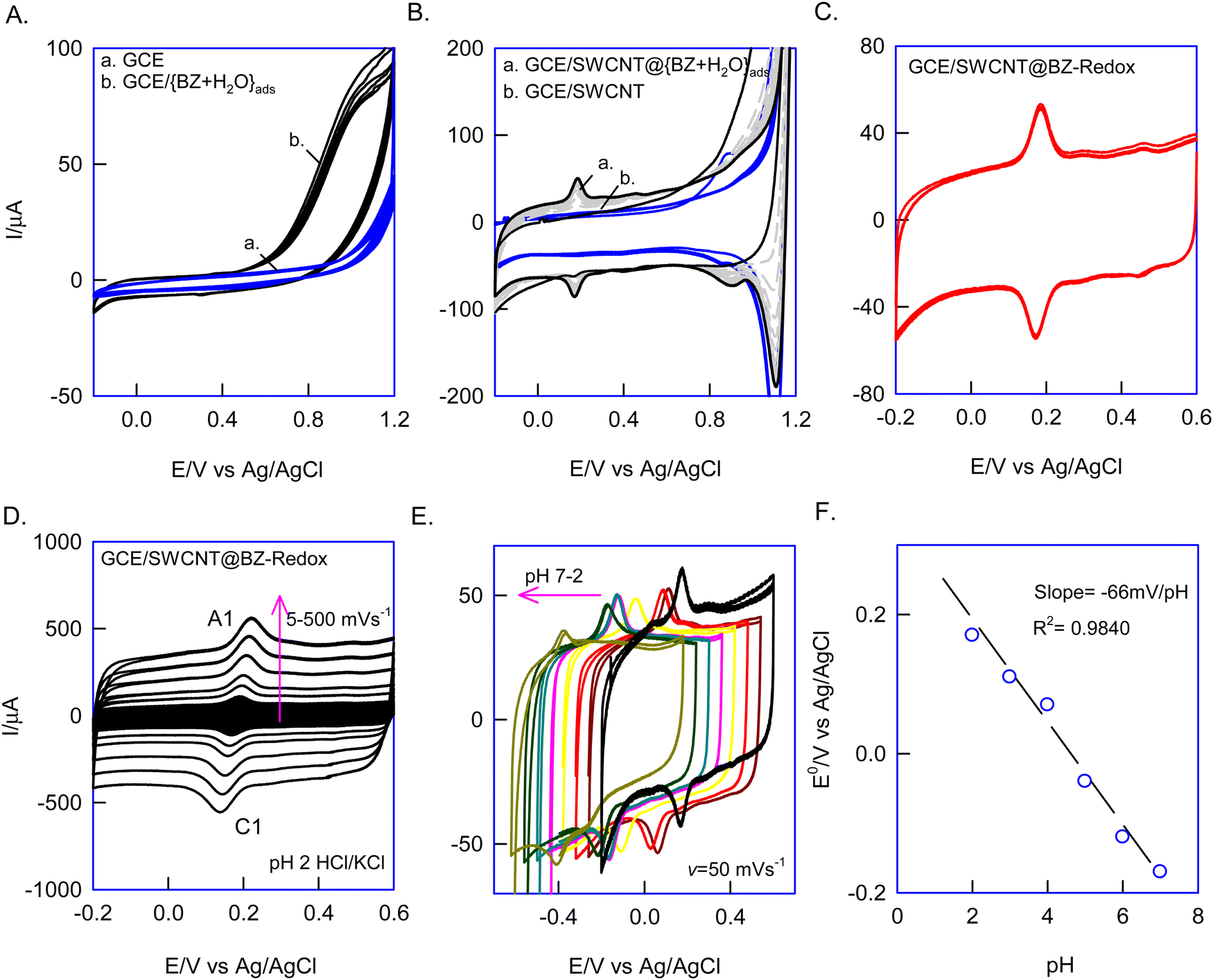 | ||
| Fig. 1 (A) Continuous CV responses of the bare GCE (curve a) and GCE/{BZ + H2O}ads (curve b). (B) CV responses recorded for GCE/SWCNT@{BZ + H2O}ads (curve a) and GCE/SWCNT (curve b) and (C) GCE/SWCNT@BZ-Redox in pH 2 HCl–KCl electrolytic solution at v = 50 mV s−1. Effects of the scan rate (D) and solution pH (E) on the CV responses of GCE/SWCNT@BZ-Redox. (F) Plot of E°/V vs. pH. | ||
Upon subjecting the GCE/SWCNT@BZ-Redox to the effect of scan rate experimentation in a pH 2 HCl–KCl solution, as illustrated in Fig. 1D, it was observed that the anodic and cathodic (A1/C1) peaks of the redox system increased in response to the scan rate. The plot of ipa and ipcversus v exhibited linearity, with a peak–peak potential difference of ∼0 V (ΔEp = Epa − Epc) observed at v = 10 mV s−1 (refer to the ESI,† Fig. S3). These findings suggest symmetrical redox behaviour attributable to facile electron transfer functionality, resulting in a calculated surface coverage, Γ, of 5.1 × 10−9 mol cm−2 for the GCE/SWCNT@BZ-Redox electrode. Furthermore, CV experiments were conducted in various pH solutions, as depicted in Fig. 1E. A systematic shift in the redox peak potential was observed relative to the solution pH. Plotting E°′/V against pH yielded a slope value of approximately −66 mV pH−1 within the pH range of 2 to 7 (see Fig. 1F), indicating a proton-coupled electron transfer reaction.
To understand the nature of the electron-transfer mechanism, we investigated the effect of an applied potential on the GCE/SWCNT@BZ-Redox formation. As part of this experiment, the precursor electrode GCE/SWCNT@{BZ + H2O}ads was freshly prepared and subjected to potentiation polarization and CV experiments. This experimental protocol involved initial potential polarization (Eapp) as step 1, followed by a CV response recorded as step 2, within a potential window of −0.4 to 1.2 V vs. Ag/AgCl. We calculated the ipa values from the CV graphs (step 2) and constructed a plot of ipavs. Eapp/V (Fig. 2A). The inset graphs in Fig. 2A are representative CV responses of the experiments. As shown in the plot in Fig. 2A, no redox signals were observed at negative potentials between −0.2 and −0.6 V (CV data not shown). Interestingly, a well-defined redox peak was observed at Eapp = 1.2 V with a maximum peak current (inset CV graph of Fig. 2A). This indicates that a positive potential is optimal for redox peak formation. To validate this, we conducted independent CV analyses by continuous potential sweeping at different potential windows (Fig. 2B). Notably, we only observed the A1/C1 redox peak at a positive potential window of −0.2 to 1.2 V vs. Ag/AgCl (Fig. 2B). These observations were consistent with the applied potential experiment. Overall, the experimental evidence showed that positive potential specifically influences BZ-Redox species formation. The plausible reason is likely the electrochemical oxidation reaction of water to dioxygen via the formation of hydroxyl radical-like intermediate species, which is favoured in aqueous solutions at 1.2 V vs. Ag/AgCl41 (Scheme 1). The experimental data suggest that hydroxyl radical-like intermediates facilitate the oxidation of surface-confined BZ molecules to redox-active CMEs. Regarding the nature of the redox species, one of the following may occur: (i) formation of dihydroxy derivatives of BZ (phenol and polyaromatic hydrocarbons such as catechol and quinone),34,39 (ii) production of polymeric products (polymerized benzene),39,68,69 or (iii) generation of cationic radical species such as BZ.41,70,71 Later physicochemical experiments with various characterization techniques will provide answers for the mechanism.
 | ||
| Fig. 2 Plot of (A) ipavs. applied potential (Eapp) of the SWCNT@{BZ + H2O}ads modified electrodes (freshly prepared) in pH 2 HCl/KCl (inset: representative CV responses of the modified electrode,GCE/SWCNT@BZ-Redox). (B) Effect of the potential window on the continuous CV responses of GCE/SWCNT@{BZ + H2O}ads (freshly prepared electrodes) in pH 2 HCl–KCl solution at v = 50 mV s−1. (C) Plot of surface excess ΓBZ-Redoxvs. different carbon nanomaterials@BZ-Redox. | ||
Furthermore, the effect of carbon materials on the formation of the BZ-Redox peak was investigated by modifying various carbon structures such as carbon nanofibers (CNF), graphitized mesoporous carbon (GMC), activated charcoal (AC), and graphene (Gr) on the GCE surface along with a {BZ + H2O} overlayer. From the resulting cyclic voltammetry (CV) responses, a bar chart comparing the surface excess ΓBZ-Redoxversus carbon nanomaterial was generated (Fig. 2C). Except for SWCNTs, all other carbon nanomaterials did not exhibit any significant faradaic responses (ESI,† Fig. S4). This observation may be attributed to differences in the characteristics of the edge plane and basal plane of the carbon surface.41 Furthermore, the molecular orientation and dynamics of benzene play an important role in the surface modification process. It is assumed that benzene preferentially interacts with the basal plane-rich SWCNTs. A plausible reason is that the stress created while aligning the benzene molecular species on CNTs is somewhat relieved, whereas on planar structures it is markedly demonstrated, resulting in unfavourable modification.
3.2. Physical characteristics of the GCE/SWCNT@BZ-Redox CME
To understand the surface features of the GCE/SWCNT@BZ-Redox electrode, it was subjected to TEM characterization with its control sample (Fig. 3A and B). The pristine SWCNTs showed a tubular graphitic structure with an average diameter of 15 ± 2 nm in agreement with the literature reports (Fig. 3A and B).25,26,72 In addition to this, impurities related to carbonaceous materials and metals were identified, which are due to the metal catalyst originating from the nature of the preparation method adopted under the preparation conditions. It is noteworthy that the intrinsic impurities cannot be removed completely from the CNT network structure. On the other side, after being subjected to surface modification with BZ-Redox, about a 10-fold enlargement in the diameter of SWCNTs (98 ± 5 nm) was observed (Fig. 3C and D). It is surprising to see such a macro-size modification of the SWCNT surface. It is noteworthy that the surface of modified SWCNTs is found to be smooth without any cracks and pores, highlighting the dense packing/alignment of the benzene species on the CNT. At this point, we suspected quinone, BZ-oligomers and polymers (polyphenylene)69,71 and other organic molecular (benzene radical species)70 systems immobilized on the SWCNT surface. In order to pinpoint the chemical functionalities, as a next step, the FT-IR response was recorded (Fig. 4A, curves a–d) for pristine SWCNTs, BZ (control) and SWCNT@BZ-Redox electrode systems. In the case of SWCNTs, IR signals due to the carbon–oxygen functional groups (C–O; 1000–1300 cm−1), sp2 units (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C; 1500–1700 cm−1) and fraction of impurities due to atmospheric CO2 adsorption (2200 cm−1) were noticed. Similarly, BZ showed characteristic signals of aromatic benzene units at 3060, 1977 and 1831 cm−1, ArCC bonding at 1438 cm−1 and C–H bonding at 681 cm−1. Interestingly, the FT-IR analysis of the SWCNT@BZ-Redox systems showed signals for the combination of SWCNTs and BZ without any other new peaks, but with a slight shift in the IR signals. It is important to note that there was no new signal observed in the regions ∼3500 and 750–820 cm−1 corresponding to the quinone and polyphenylene groups, respectively, which ruled out the formation of quinone and polymerized benzene derivative ions on the surface.38,69 It is suspected that redox-active benzene radical species may be formed and trapped on the surface of SWCNT@BZ-Redox. Further experiments were needed for the confirmation of the radical species on the surface. Fig. 4B (curves a–c) shows a comparative Raman spectroscopic response of the SWCNT and SWCNT@BZ-Redox samples. The SWCNT control sample showed characteristic Raman signals at 1334 cm−1 and 1574 cm−1 due to disordered graphitic (D, due to sp3 and oxygen functionalized carbon units) and ordered graphitic (G, due to sp2 carbon) bands of the carbon material. The calculated band intensity ratio, i.e., ID/IG value, for the SWCNT sample is 0.35. Interestingly, the Raman response of the SWCNT@BZ-Redox sample showed a qualitative similar pattern; indeed, the ID/IG value is increased to 0.86. This observation indicates the conversion of a fraction of the sp2 bonded graphitic structure to sp3 bonding non-graphitic carbon units after modification of BZ-Redox on the SWCNT surface.
C; 1500–1700 cm−1) and fraction of impurities due to atmospheric CO2 adsorption (2200 cm−1) were noticed. Similarly, BZ showed characteristic signals of aromatic benzene units at 3060, 1977 and 1831 cm−1, ArCC bonding at 1438 cm−1 and C–H bonding at 681 cm−1. Interestingly, the FT-IR analysis of the SWCNT@BZ-Redox systems showed signals for the combination of SWCNTs and BZ without any other new peaks, but with a slight shift in the IR signals. It is important to note that there was no new signal observed in the regions ∼3500 and 750–820 cm−1 corresponding to the quinone and polyphenylene groups, respectively, which ruled out the formation of quinone and polymerized benzene derivative ions on the surface.38,69 It is suspected that redox-active benzene radical species may be formed and trapped on the surface of SWCNT@BZ-Redox. Further experiments were needed for the confirmation of the radical species on the surface. Fig. 4B (curves a–c) shows a comparative Raman spectroscopic response of the SWCNT and SWCNT@BZ-Redox samples. The SWCNT control sample showed characteristic Raman signals at 1334 cm−1 and 1574 cm−1 due to disordered graphitic (D, due to sp3 and oxygen functionalized carbon units) and ordered graphitic (G, due to sp2 carbon) bands of the carbon material. The calculated band intensity ratio, i.e., ID/IG value, for the SWCNT sample is 0.35. Interestingly, the Raman response of the SWCNT@BZ-Redox sample showed a qualitative similar pattern; indeed, the ID/IG value is increased to 0.86. This observation indicates the conversion of a fraction of the sp2 bonded graphitic structure to sp3 bonding non-graphitic carbon units after modification of BZ-Redox on the SWCNT surface.
 | ||
| Fig. 3 TEM images of SWCNT (A) and (B) and SWCNT@BZ-Redox (C) and (D) electrodes under different magnifications. | ||
 | ||
| Fig. 4 (A) FTIR responses of (a) SWCNTs, (b) SWCNT@BZchem, (c) BZ and (d) SWCNT@BZ-Redox. (B) Raman spectra of (a) SWCNT@BZ-Redox, (b) SWCNT@BZchem and (c) SWCNTs. Note: chem = chemisorption. | ||
To validate the proposed BZ-Redox species, an independent control cyclic voltammetry experiment was conducted (ESI,† Fig. S5). This experiment compared the SWCNT@BZ-Redox electrode with SWCNTs in the presence of a 500 μM solution of 2,2,6,6-tetramethylpiperidin-1-yloxyl (TEMPO), an organic radical scavenging system, in a pH 2 HCl/KCl solution. Initially, a reversible redox response (A1/C1) was observed at E°′ = 0.528 V vs. Ag/AgCl, as well as an irreversible reduction peak (C1) at Epc = 0.3 V vs. Ag/AgCl corresponding to the aminooxy anion/TEMPO radical (I) and TEMPO radical/oxoammonium cation (II/III) electrochemical reactions of TEMPO on the GCE/SWCNT (curve a) modified electrode surface. Subsequent exposure of the GCE/SWCNT@BZ-Redox (curve b) electrode under the same experimental conditions resulted in the following observations: (i) disappearance of the original redox features of BZ-Redox with a 55% decrement in the current signal and (ii) a potential drift of ∼E°′ = 100 mV in the (A1/C1) redox peak. This outcome is plausibly attributed to a hydroxy radical quenching mechanism, confirming the existence of the BZ-redox peak.41
3.3. Chemisorption of BZ species on SWCNTs (SWCNT@BZchem)
The orientation and dynamics of adsorbed benzene species are crucial factors influencing redox activity. In this study, we prepared and tested a BZ molecule covalently linked to SWCNTs to investigate its electrochemical mechanism (Scheme 1A). Considering the geometric orientation, the covalently bonded BZ on SWCNTs may adopt a perpendicular arrangement,64 thus preventing the interaction between the aromatic units of BZ and the sp2 carbon of SWCNTs. Consequently, the absence of such interactions may hinder the generation of intermediate radical species, leading to negligible redox activity. To verify this hypothesis, we conducted CV studies on a freshly prepared GCE/SWCNT@BZchem system, similar to the GCE/SWCNT@BZ-Redox case, in a pH 2 KCl–HCl solution (ESI,† Fig. S1). Interestingly, we observed the absence of the CV response akin to the GCE/SWCNT@BZ-Redox scenario, indicating the absence of redox activity in this system. This experimental result concludes the specific influence of BZ adsorption on the redox activity of the molecular system.3.4. Electrocatalytic reduction of H2O2 by GCE/SWCNT@BZ-Redox
Fig. 5A depicts the CV response of the GCE/SWCNT@BZ-Redox system in the absence (curve a) and presence of a 500 μM H2O2 solution in pH 7 phosphate buffer (curve b). The SWCNT@BZ-Redox modified electrode exhibited a well-defined irreversible reduction peak starting at −140 mV vs. Ag/AgCl. As a control, bare SWCNTs were subjected to CV experiments with H2O2, revealing a significantly poorer electrochemical reduction signal beginning at approximately −0.4 V vs. Ag/AgCl, indicating a high overpotential of about 250 mV and a four-fold decrease in the current response. Additionally, the GCE/SWCNT@BZchem system was tested under similar conditions but failed to exhibit any noticeable faradaic signal (Fig. 5B, curves a–c). The overall results illustrate that an efficient electrocatalytic reduction response of H2O2 was observed in the GCE/SWCNT@BZ-Redox system. Subsequently, amperometry experiments were conducted with both GCE/SWCNT@BZ-Redox and GCE/SWCNT-control systems at an applied potential of Eapp = −0.25 V vs. Ag/AgCl, with continuous injections of 200 μM H2O2 in PBS of pH 7. The GCE/SWCNT@BZ-Redox system demonstrated a systematic increase in reduction current signals in response to the H2O2 injections (curve a), whereas the SWCNT control showed no such sensing current signals (Fig. 5C, curve b). The corresponding calibration plot of |ip/μA| vs. [H2O2/μM] displayed current linearity within the range of 0–1800 μM, with a current sensitivity of 60 nA μM−1 and a regression coefficient value of 0.9989. Seven continuous injections of 200 μM H2O2 yielded a RSD of 3.2%. Based on a signal-to-noise ratio value of 3, the calculated detection limit was 530 nM (Fig. 5D). Subsequent interference testing was performed on the optimized modified electrode by spiking 100 μM uric acid (UA), dopamine (DA), AA, cysteine (CySH) and glucose (Glu). Notably, there were no changes in the amperometry curve, except for H2O2 (Fig. 5E). In existing literature reports, there were few electrocatalysts for H2O2 sensing in HeLa cell systems. These include heteroatom-doped carbon systems, enzymes, metal–organic frameworks (MOFs), and Prussian blue analogues.73–77 Notably, many studies have achieved impressive detection limits in the nanomolar range. However, metal-based systems suffer due to DO interference in biological samples and Prussian blue analogues face surface instability at neutral pH.57,58,61,62 To address these limitations, in this study a DO interference experiment was conducted under CV and amperometry conditions (with N2 and O2 purging). The redox peak exhibits a minimal alternating current signal, regardless of the presence or absence of DO, and does not display any mediated reduction current response (ESI,† Fig. S6A and B). This highlights a primary advantage of this new modified electrode for the selective detection of H2O2, particularly in biological studies where oxygen species play a major role in biochemical reactions. The literature studies reporting H2O2 sensing systems in HeLa cells are provided in Table S1 (ESI†), with the present work and its limitations.57,58,61,62,73–77 | ||
| Fig. 5 (A) Comparative CV responses of SWCNT@BZ-Redox and (B) SWCNT@BZchem modified electrodes recorded at v = 10 mV s−1 without (a) and with (b) 500 μM H2O2 and the respective control experiments of bare SWCNTs (c) in PBS of pH 7. (C) Comparative amperometric i–t responses of GCE/SWCNT@BZ-Redox (a) and SWCNTs (b) upon continuous spikes of 200 μM H2O2 at Eapp = −0.25 V vs. Ag/AgCl and (D) its respective calibration plot of |ip/μA| vs. [H2O2]/μM. (E) Amperometric i–t responses of GCE/SWCNT@BZ-Redox with a 100 μM concentration of various interfering species: uric acid (UA), dopamine (DA), ascorbic acid (AA), cysteine (CySH) and glucose (Glu) under the same experimental conditions as those in Fig. 5C. Note: chem = chemisorption. | ||
3.5. SECM interrogation of the GCE/SWCNT@BZ-Redox CME
SECM is an advanced electrochemical technique used for investigating molecular reactions and conducting in situ surface morphological studies. Unlike conventional techniques such as AFM, SEM, and TEM, which provide the ex situ surface profiles of testing systems, SECM offers the advantage of providing the in situ morphology and active sites on the electrode surface during electrochemical reactions. We believe that SECM is a suitable technique for the morphological analysis of SWCNT@BZ-Redox in situ. In this study, a redox-active system, FeIII/II(CN)63−/4−, was chosen as a molecular probe to study the in situ surface of the modified electrode. The SECM imaging mechanism operates in a feedback mode, wherein FeIII(CN)63− is first reduced to FeII(CN)64− on the substrate and subsequently oxidized to FeIII(CN)63− on the probing tip (Pttip) continuously (Fig. 6A–F). The redox reaction occurs on the surface, and the measured tip current is imaged as the surface morphology. Prior to surface studies, CV experiments of GCE/SWCNT (Fig. 6A, curves a–c), GCE/SWCNT@BZchem (curve b) and GCE/SWCNT@BZ-Redox (curve c) were performed with 5 mM Fe(CN)63− in pH 2 KCl/HCl solution. A well-defined redox peak was observed in all electrodes at different magnitudes of current values, as depicted in Fig. 6A (curves a–c).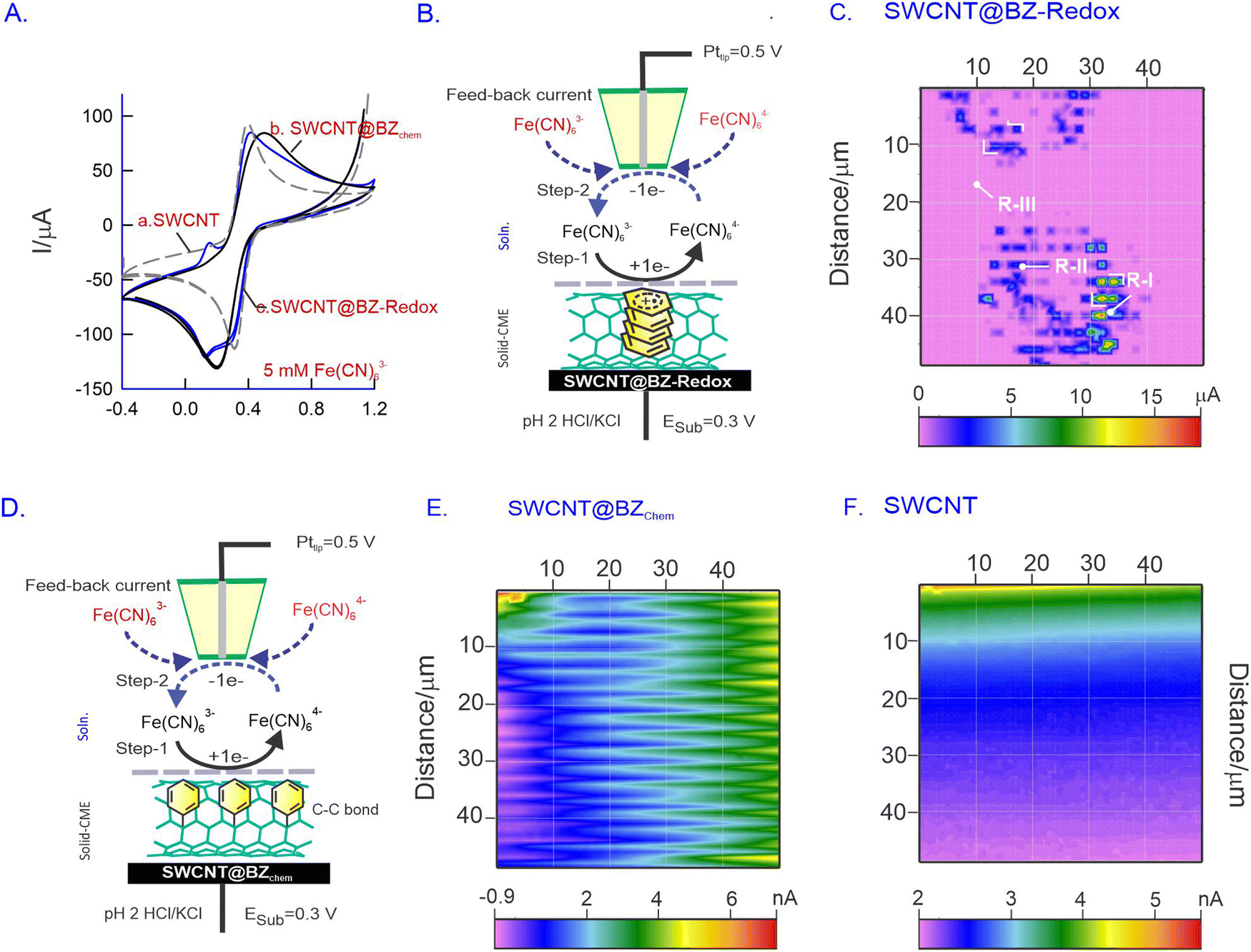 | ||
| Fig. 6 (A) Comparative CV responses of GCE/SWCNT (a), GCE/SWCNT@BZchem (b) and GCE/SWCNT@BZ-Redox (c) in pH 2 HCl/KCl containing 5 mM [Fe(CN)]63− at v = 50 mV s−1. Schematic representation of the feedback current based operation for GCE/SWCNT@BZ-Redox (B) and SWCNT@BZchem (D) electrodes and their respective surface mapping images (C) and (E) along with the control SWCNT electrode (F) using the SECM technique at Esub = 0.3 V and Pttip = 0.5 V vs. Ag/AgCl. (C), (E) and (F) Typical SECM surface morphology analysis results of GCE/SWCNT@BZ-Redox (D), SWCNT@BZchem (E) and SWCNTs (F). R-I–R-III = various electrode regions. Note: chem = chemisorption. | ||
Fig. 6B schematically explains the operational mechanism of the Fe(CN)63− redox reaction at the CMEs. In our study, the modified electrode served as the substrate electrode, while a platinum ultramicroelectrode (Pt-UME) with a 25 μm diameter functioned as the current collector tip. Initially, the potential of the substrate electrode was set to the reduction potential of Fe(CN)63− (0.3 V vs. Ag/AgCl; step 1), and the Pttip facilitated the counter oxidation reaction at a potential of 0.5 V vs. Ag/AgCl (step 2). Consequently, a continuous redox cycle was initiated, during which the Pttip collected current and generated a 50 μm × 50 μm image, revealing the locations of reactive hotspots on the surface (Fig. 6C, E and F). The proximity of the Pttip to the surface was achieved using the approach curve provided in the ESI,† Fig. S7.
Fig. 6C presents a typical SECM image of the SWCNT@BZ-Redox system showing microdots on a large pink background. The central part of the microdots has a maximum current signal of 15 μA, whereas the blue (5 μA) and pink coloured spots (1 μA) per background have relatively lower current values. Control SECM imaging experiments of SWCNTs and covalently anchored SWCNT@BZ systems showed about three orders of magnitude lower current signal (5 nA) than that of the SWCNT@BZ-Redox system. These results indicate the existence of inhomogeneous active sites on the modified electrode assembly (Fig. 6C). The observed hotspot dimensions were approximately 100 nm to 1 μm, resembling the thickness of SWCNT@BZ-Redox noticed in TEM measurements (Fig. 3C and D). This observation suggests the possible formation of basal plane sites which contain the active site BZ-Redox that enhances the electron transfer functionality. Additionally, we conducted a similar experiment using covalently anchored SWCNT@BZchem under identical operating conditions (Fig. 6E), and the current responses obtained exhibited wave-like patterns, affirming the formation of clustered arrays on the surface (Fig. 6E). In contrast, the bare SWCNT material showed a featureless response (Fig. 6F).
To visualize the active catalytic site, the H2O2 reduction reaction was also used as a molecular probe in the redox-competitive (RC) mode of the SECM operation, wherein the same electrochemical reactions are performed for imaging on both the substrate and the tip. Previously, our group implemented the RC mode of SECM imaging approach for visualizing the activities of a pre-anodized pencil graphite electrode (PGE) using DA,78 nickel hexacyanoferrate,79 and carbon nanofiber modified Binol redox species.80Fig. 7A is a schematic representation for redox-competitive (RC) mode SECM operation mechanism of the continuous reduction reaction of H2O2 using the GCE/SWCNT@BZ-Redox electrode (at open-circuit, step 1), and the Pt tip (step 1′) has been explored. Fig. 7B shows the SECM mapping of a 100 μm × 100 μm surface displaying the active hotspots with three different regions, distinguished as region I (∼400 nA), region II (∼200 nA), and region III (∼100 nA). The presence of metal impurity sites (in both inner and outer regions) on the SWCNT surface (as agglomerates) is likely responsible for the high current observed in this work. Region II, the ball-like feature observed in the mapping with a size of approximately 5 μm, is likely the area where SWCNTs are attached with BZ-Redox on the underlying electrode surface. Although specific morphological information was not obtained, these results indicate the surface heterogeneity of the modified electrode, whereas the control experiment of bare SWCNTs showed a featureless contour-like surface (Fig. 7C).
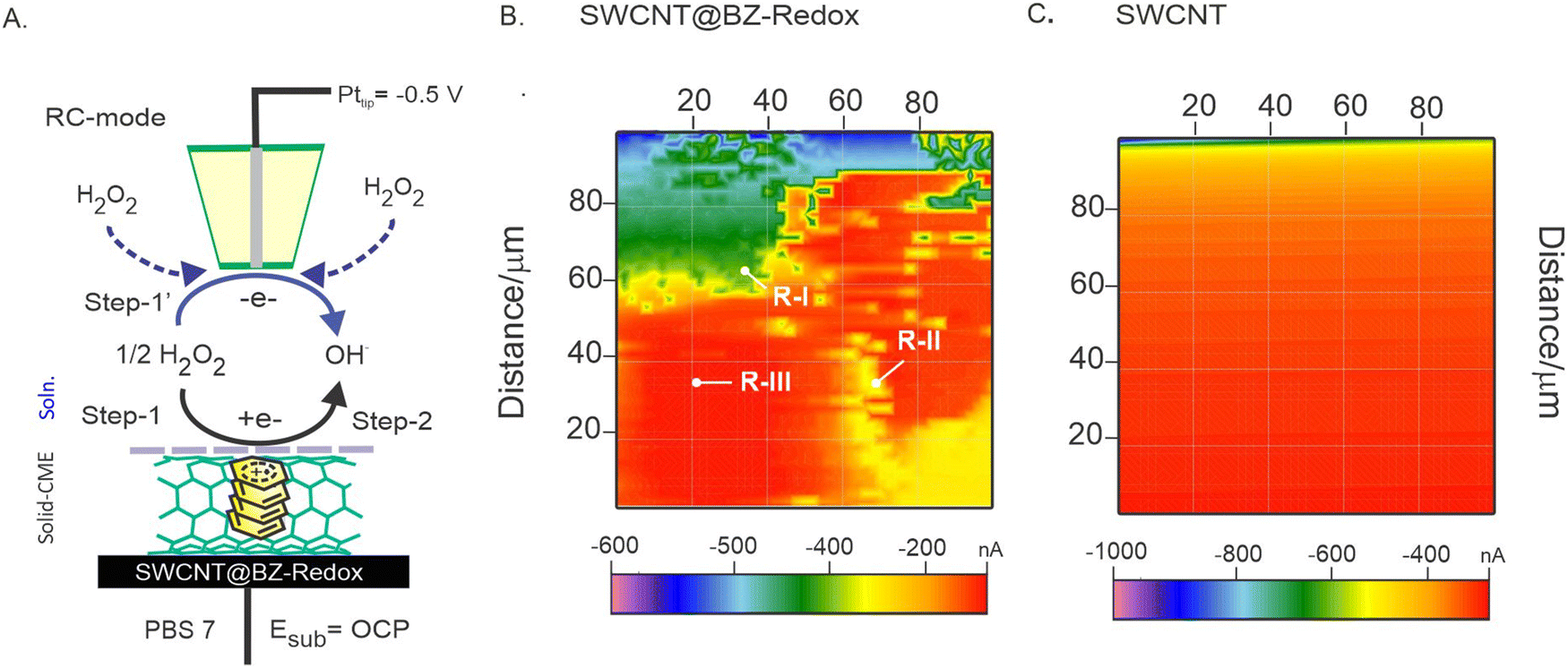 | ||
| Fig. 7 (A) Schematic illustration of the redox competitive mode operation mechanism of SECM. The respective SECM images of GCE/SWCNT@BZ-Redox (B) and SWCNTs (C) recorded in PBS of pH 7; Esub = OCP (open circuit potential) and Pttip = −0.5 V vs. Ag/AgCl. R-I–R-III = various surface regions. | ||
3.6. ROS monitoring in live HeLa cells
Since the biological system involves a lower sample volume (∼100 μL) and short-lived intermediate species, ROS, the use of conventional CV and amperometric i–t based electrochemical setups is not convenient and viable for HeLa cell monitoring. In this connection, we have adopted the BIA technique coupled with a three-in-one screen-printed electrode, consisting of SWCNT@BZ-Redox coated carbon as the working electrode, unmodified carbon as the counter electrode and Ag-ink modified electrode as the reference electrode, and a portable electrochemical workstation with 50 μL of sample injection for HeLa cell ROS monitoring. The software integrated with an electronic pipette was used for the sample injection (Fig. 9B). For this experiment, the amperometry i–t voltametric technique was adopted with an applied potential of Eapp = −0.25 V vs. Ag/AgCl. Fig. 8A shows the BIA responses of continuous spikes of H2O2 with increasing the concentration from 2 to 60 μM. The corresponding current signals exhibited a linear relationship with [H2O2] and the RSD value for the 10 spikes is 4.3% (Fig. 8B). The calculated current sensitivity and regression coefficient values are 1.2 μA μM−1 and 0.9970, respectively (Fig. 8C). The calculated detection limit is 530 nM. Comparing the analytical results with previous literature, we observed that our results were comparable and, in some cases, much better than those from previous literature (Table S1, ESI†).57,58,61,62,73–77 To assess the impact of interference, a study was conducted by introducing biochemical substances such as cysteine (CySH), hydrazine (Hyd), Cr3+, Cr6+, AA, UA, glucose (Glu) and H2O2 in a pH 7 PBS solution (Fig. 8D). Special attention was paid to the DO influence on the modified electrode, and continuous spikes of DO saturated PBS and repeated spikes did not show any changes in the current signals (ESI,† Fig. S6). This is the prime novelty of this work. Furthermore, the three-in-one screen printed electrode with the SWCNT@BZ-Redox working electrode and BIA were integrated as a programmable prototype device. We programmed the calibration plot obtained from our experiments into this device, which was then utilized to monitor the in situ release of H2O2 (one of the ROS species) in live HeLa cells. Subsequently, the cell culture, as prepared, underwent 15, 30, 45 and 60 min incubation (tincub) before being meticulously subjected to experimental analysis. Readings were acquired at various time points (tacquir) using the BIA-coupled prototype device (Fig. 9A). Fig. 9B shows the photographic documentation of the prototype coupled batch injection analysis system. When tincub = 15 and 30 minutes, no current signals were observed. However, at tincub = 45 and 60 minutes, a measurable amount of H2O2 was observed at an acquiring time (tacquir) of 80–140 s. Based on the calibration graph, the amounts of HeLa cells involved in the H2O2 release were calculated as 8 × 10−16 mol per cell and 1.3 × 10−15 mol per cell, respectively, at the 45th and 60th minutes of incubation (tincub). The amount of H2O2 produced by HeLa cells in the present work closely aligns with electrochemical analysis results with the range between ∼1.0 × 10−16 and 3 × 10−15 mol per cell.56,58,59 For the first time, we report a low sample volume based microfluidic analysis coupled with a prototype device system for continuous monitoring of H2O2 from the live cell system in this work. | ||
| Fig. 8 Batch injection analysis responses of (A) SPE/SWCNT@BZ-Redox upon continuous spiking of increasing H2O2 concentrations and (B) its respective calibration plot of |ip/μA| vs. [H2O2]/μM. BIA of (C) ten repetitive spikes of 2 μM H2O2 and (D) various interfering bio-analytes of 5 μM in a stirred 0.1 M PBS solution of pH 7 at Eapp = −0.25 V vs. Ag/AgCl. CySH = cysteine, Cr6+, Hyd = hydrazine, Cr3+, AA = ascorbic acid, Glu = glucose and UA = uric acid. | ||
 | ||
| Fig. 9 (A) BIA response of SPE/SWCNT@BZ-Redox upon continuous spikes of HeLa cells and HeLa cells + AA (10 μM) at various time points (a–d: 15–60 min) at an applied potential Eapp = −0.25 V vs. Ag/AgCl in PBS of pH 7. (B) Photograph of the BIA system coupled with a prototype model (inset: the cartoon of stressed HeLa cells). Note: acquir = acquiring and incub = incubation. | ||
For potential industrial application, we conducted a study on tannery wastewater. This wastewater contains a mixture of organic pollutants (phenols, sulphide, phosphate and other chemicals) and metals such as calcium, barium, chromium, manganese, iron, etc. Using the BIA coupled with SWCNT@BZ-Redox, we analysed this wastewater. The results of our experiment showed 98% recovery of the modified electrode after the spike of the complex real sample. This observation indicates that our new system can be used effectively to analyze various types of real-life samples (ESI,† Fig. S8).
4. Conclusion
In summary, we have successfully developed a SWCNT modified electrode (SWCNT@BZ-Redox) based on electrochemical oxidation of BZ on a SWCNT surface, which exhibits about a 10 times increase in the dimension of SWCNTs with a distinctive redox peak at E°′ = 150 ± 10 mV vs. Ag/AgCl in a pH 2 HCl/KCl solution. The observed redox peak demonstrates symmetrical characteristics, surface confinement, and efficient electron transfer behaviour. Infurther, the influence of the potential window on the modified electrode revealed a role of surface OH˙ radical formation at high potential, leading in turn to the in situ formation of BZ-Redox. To delve into the intricate mechanisms involved, we meticulously fabricated a vertically aligned nitrobenzyl diazonium compound and thoroughly investigated its electrochemical and physicochemical attributes through systematic experimental methodologies. Remarkably, the benzene π-stacked redox compound exhibited efficient electron transfer behaviour and bio-electrocatalytic activity towards the hydrogen peroxide reaction, while the covalently linked BZ system did not produce any significant current response. Additionally, as a part of an independent investigation, scanning electrochemical microscopy was employed to visualize surface hotspots utilizing feedback mechanisms and identifying electrocatalytic active sites. As a proof of concept, we developed a prototype device integrated with BIA technology for instantaneous H2O2 sensing in stressed HeLa cancer cells, showcasing continuous monitoring of H2O2 generation within a live HeLa cell system.Author contributions
Annamalai Senthil Kumar: conceptualization, validation, investigation, resources, data curation, writing – review and editing, supervision, project administration, funding acquisition and visualization. Sakthivel Srinivas: writing – review and editing, investigation, visualization, and device development. Nisha Sivakumar: first draft preparation, conceptualization, validation, investigation, resources, and data curation. Mouliganesh and Kavitha Thirumurugan: review, editing and HeLa cell part. All authors have given approval for the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge the financial support through the Department of Science and Technology – Science and Engineering Research Board (DST/CRG/2021/001048) scheme.References
- X. Hu, X. Bao, M. Zhang, S. Fang, K. Liu, J. Wang, R. Liu, S. H. Kim, R. H. Baughman and J. Ding, Adv. Mater., 2023, 35, 2303035 CrossRef CAS PubMed.
- V. Jain, T. Gurusamy, P. Gayathri and K. Ramanujam, New J. Chem., 2020, 44, 8849–8858 RSC.
- L. M. Peng, ACS Nano, 2023, 17, 22156–22166 CrossRef CAS PubMed.
- Y. Cai, H. Yu, L. Cheng, S. Guo, T. Liu, D. Chen, S. Huang, Z. Hu, Y. Wang and Y. Zhou, Adv. Sustainable Syst., 2023, 7, 2300272 CrossRef CAS.
- F. Zahoor, M. Hanif, U. Isyaku Bature, S. Bodapati, A. Chattopadhyay, F. Azmadi Hussin, H. Abbas, F. Merchant and F. Bashir, Phys. Scr., 2023, 98, 082003 CrossRef.
- M. Rani, M. Sehrawat, S. Sharma and B. P. Singh, J. Energy Storage, 2023, 73, 109063 CrossRef.
- R. Orinaková and A. Orinak, Fuel, 2011, 90, 3123–3140 CrossRef.
- F. Mashkoor, A. Nasar and Inamuddin, Environ. Chem. Lett., 2020, 18, 605–629 CrossRef CAS.
- V. V. Turov, G. P. Prikhod’ko, S. Y. Brichka and M. D. Tsapko, Russ. J. Phys. Chem., 2006, 80, 591–596 CrossRef CAS.
- C.-J. M. Chin, M.-W. Shih and H.-J. Tsai, Appl. Surf. Sci., 2010, 256, 6035–6039 CrossRef CAS.
- Y. Jiang and C. Wang, Int. J. Quantum Chem., 2019, 119, e25936 CrossRef.
- O. Y. Bakather, Ain Shams Eng. J., 2020, 11, 905–912 CrossRef.
- P. A. Gauden, A. P. Terzyk, G. Rychlicki, P. Kowalczyk, K. Lota, E. Raymundo-Pinero, E. Frackowiak and F. Beguin, Chem. Phys. Lett., 2006, 421, 409–414 CrossRef CAS.
- K. Yang, L. Zhu and B. Xing, Environ. Sci. Technol., 2006, 40, 1855–1861 CrossRef CAS PubMed.
- H. Anjum, K. Johari, N. Gnanasundaram, A. Appusamy and M. Thanabalan, J. Cleaner Prod., 2019, 221, 323–338 CrossRef CAS.
- H. Ding, X. Li, J. Wang, X. Zhang and C. Chen, J. Environ. Sci., 2016, 43, 187–198 CrossRef CAS PubMed.
- A. Dasgupta, J. Matos, H. Muramatsu, Y. Ono, V. Gonzalez, H. Liu, C. Rotella, K. Fujisawa, R. Cruz-Silva, Y. Hashimoto, M. Endo, K. Kaneko, L. R. Radovic and M. Terrones, Carbon, 2018, 139, 833–844 CrossRef CAS.
- D. Lin and B. Xing, Environ. Sci. Technol., 2008, 42, 7254–7259 CrossRef CAS PubMed.
- O. Duman, C. Ozcan, T. Gürkan Polat and S. Tunç, Environ. Pollut., 2019, 244, 723–732 CrossRef CAS PubMed.
- M. Athari, M. Fattahi, M. Khosravi-Nikou and A. Hajhariri, Sci. Rep., 2022, 12, 20415 CrossRef CAS PubMed.
- J. Luan, P.-X. Hou, C. Liu, C. Shi, G.-X. Li and H.-M. Cheng, J. Mater. Chem. A, 2016, 4, 1191–1194 RSC.
- X. Peng, Acta Phys.-Chim. Sin., 2014, 30, 2000–2008 CAS.
- Y. Zhang, Y. Shen, J. Li, L. Niu, S. Dong and A. Ivaska, Langmuir, 2005, 21, 4797–4800 CrossRef CAS PubMed.
- M. Knez, M. Sumser, A. M. Bittner, C. Wege, H. Jeske, S. Kooi, M. Burghard and K. Kern, J. Electroanal. Chem., 2002, 522, 70–74 CrossRef CAS.
- S. E. Kooi, U. Schlecht, M. Burghard and K. Kern, Angew. Chem., Int. Ed., 2002, 41, 1353–1355 CrossRef CAS PubMed.
- S. Mondal, S. Ghosh and C. R. Raj, ACS Omega, 2018, 3, 622–630 CrossRef CAS PubMed.
- D. V. Kosynkin, A. L. Higginbotham, A. Sinitskii, J. R. Lomeda, A. Dimiev, B. K. Price and J. M. Tour, Nature, 2009, 458, 872–876 CrossRef CAS PubMed.
- A. M. Dimiev, A. Khannanov, I. Vakhitov, A. Kiiamov, K. Shukhina and J. M. Tour, ACS Nano, 2018, 12, 3985–3993 CrossRef CAS PubMed.
- A. Maestro, E. Guzmán, F. Ortega and R. G. Rubio, Curr. Opin. Colloid Interface Sci., 2014, 19, 355–367 CrossRef CAS.
- M. Q. Xu, L. D. Xing, W. S. Li, X. X. Zuo, D. Shu and G. L. Li, J. Power Sources, 2008, 184, 427–431 CrossRef CAS.
- T. Osa, A. Yildiz and T. Kuwana, J. Am. Chem. Soc., 1969, 91, 3994–3995 CrossRef CAS.
- K. W. Kim, M. Kuppuswamy and R. F. Savinell, J. Appl. Electrochem., 2000, 30, 543–549 CrossRef CAS.
- A. J. Bard, W. M. Flarsheim and K. P. Johnston, J. Electrochem. Soc., 1988, 135, 1939 CrossRef CAS.
- B. Lee, H. Naito and T. Hibino, Angew. Chem., Int. Ed., 2012, 51, 440–444 CrossRef CAS PubMed.
- S. Zhang, M. Jin, H. Xu, W. Li, Y. Ye, T. Shi, H. Zhou, C. Chen, G. Wang, Y. Zhang, Y. Lin, L. Zheng, H. Zhang and H. Zhao, Adv. Sci., 2022, 9, 2204043 CrossRef CAS PubMed.
- B. Zhang, M. Chen, C. Zhang and H. He, Chemosphere, 2019, 217, 780–789 CrossRef CAS PubMed.
- M. P. Soriaga, J. L. Stickney and A. T. Hubbard, J. Electroanal. Chem. Interfacial Electrochem., 1983, 144, 207–215 CrossRef CAS.
- S. Ito, H. Okada, R. Katayama, A. Kunai and K. Sasaki, J. Electrochem. Soc., 1988, 135, 2996 CrossRef CAS.
- S. Zein El Abedin, N. Borissenko and F. Endres, Electrochem. Commun., 2004, 6, 422–426 CrossRef CAS.
- F. Montilla, E. Morallón and J. L. Vázquez, Electrochim. Acta, 2002, 47, 4399–4406 CrossRef CAS.
- S. Nisha, V. Lakshminarayanan and A. Senthil Kumar, Langmuir, 2020, 36, 9–19 CrossRef CAS PubMed.
- C. Lismont, I. Revenco and M. Fransen, Int. J. Mol. Sci., 2019, 20, 3673 CrossRef CAS PubMed.
- H. S. Wong, P. A. Dighe, V. Mezera, P.-A. Monternier and M. D. Brand, J. Biol. Chem., 2017, 292, 16804–16809 CrossRef CAS PubMed.
- C. Lennicke, J. Rahn, R. Lichtenfels, L. A. Wessjohann and B. Seliger, Cell Commun. Signaling, 2015, 13, 39 CrossRef PubMed.
- H. P. Woo, W. H. Yong, H. K. Suhn and Z. K. Sung, J. Cell. Biochem., 2007, 102, 98–109 CrossRef PubMed.
- B. V. Chernyak, D. S. Izyumov, K. G. Lyamzaev, A. A. Pashkovskaya, O. Y. Pletjushkina, Y. N. Antonenko, D. V. Sakharov, K. W. A. Wirtz and V. P. Skulachev, Biochim. Biophys. Acta, Bioenerg., 2006, 1757, 525–534 CrossRef CAS PubMed.
- B. M. Roberts, D. R. Fullerton and S. L. Elliott, Bios, 2015, 86, 134–143 CrossRef CAS.
- M. Uetaki, S. Tabata, F. Nakasuka, T. Soga and M. Tomita, Sci. Rep., 2015, 5, 13896 CrossRef PubMed.
- S. Morris, N. D. Geoghegan, J. B. A. Sadler, A. M. Koester, H. L. Black, M. Laub, L. Miller, L. Heffernan, J. C. Simpson, C. C. Mastick, J. Cooper, N. Gadegaard, N. J. Bryant and G. W. Gould, PeerJ, 2020, 8, e8751 CrossRef PubMed.
- S. L. Zhang, Y. S. Wang, T. Zhou, X. W. Yu, Z. T. Wei and Y. L. Li, Cytotechnology, 2012, 64, 477–484 CrossRef CAS PubMed.
- T. J. Preston, A. Abadi, L. Wilson and G. Singh, Adv. Drug Delivery Rev., 2001, 49, 45–61 CrossRef CAS PubMed.
- K. Maiese, Cells, 2023, 12, 2595 CrossRef CAS PubMed.
- Y. Feng, Y. Wang, C. Jiang, Z. Fang, Z. Zhang, X. Lin, L. Sun and W. Jiang, Life Sci., 2017, 181, 62–69 CrossRef CAS PubMed.
- F. Behrouzifar, S.-A. Shahidi, F. Chekin, S. Hosseini and A. Ghorbani-HasanSaraei, Spectrochim. Acta, Part A, 2021, 257, 119761 CrossRef CAS PubMed.
- F. Wen, Y. Dong, L. Feng, S. Wang, S. Zhang and X. Zhang, Anal. Chem., 2011, 83, 1193–1196 CrossRef CAS PubMed.
- S. M. Steinberg, Environ. Monit. Assess., 2013, 185, 3749–3757 CrossRef CAS PubMed.
- M. Lian, X. Chen, Y. Lu and W. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 25036–25042 CrossRef CAS PubMed.
- J. Peng, J. Zhao, Y. Zhao, P. Wu, L. Gou, S. Fu, P. Chen, Y. Lu and L. Yang, Int. J. Nanomed., 2029, 15, 6409–6420 CrossRef PubMed.
- Y. Fu, D. Huang, C. Li, L. Zou and B. Ye, Anal. Chim. Acta, 2018, 1014, 10–18 CrossRef CAS PubMed.
- A. Ganyecz and M. Kallay, J. Phys. Chem. C, 2021, 125, 8551–8561 CrossRef CAS PubMed.
- A. A. Karyakin, E. E. Karyakina and L. Gorton, Electrochem. Commun., 1999, 1, 78–82 CrossRef CAS.
- G. Chao, L. Zhang, J. Tian, W. Fan and T. Liu, Compos. Commun., 2021, 25, 100703 CrossRef.
- A. Heras, A. Colina, J. Lopez-Palacios, P. Ayala, J. Sainio, V. Ruiz and E. I. Kauppinen, Electrochem. Commun., 2009, 11, 1535–1538 CrossRef CAS.
- D. J. Guo and H. L. Li, Carbon, 2005, 43, 1259–1264 CrossRef CAS.
- Y. Zhang, Y. Wu, S. Tashiro, S. Onodera and T. Ikejima, Acta Pharmacol. Sin., 2011, 32, 1266–1275 CrossRef CAS PubMed.
- Q. Chen, M. G. Espey, M. C. Krishna, J. B. Mitchell, C. P. Corpe, G. R. Buettner, E. Shaded and M. Levine, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13604–13609 CrossRef CAS PubMed.
- N. S. Ng and L. Ooi, Bio. Protoc., 2021, 11, e3877 CAS.
- N. R. Lerner, J. Polym. Sci., 1974, 12, 2477–2495 CAS.
- P. Soubiran, S. Aeiyach, J. J. Aaron, M. Delamar and P. C. Lacaze, J. Electroanal. Chem. Interfacial Electrochem., 1988, 251, 89–102 CrossRef CAS.
- M. K. Eberhardt, J. Am. Chem. Soc., 1981, 103, 3876–3878 CrossRef CAS.
- S. Nisha and A. Senthil Kumar, J. Electroanal Chem., 2020, 878, 114680 CrossRef CAS.
- M. Trojanowicz, TrAC, Trends Anal. Chem., 2006, 25, 480–489 CrossRef CAS.
- W. Yang, X. Zhu, Q. Liu, Z. Lin, B. Qiu and G. Chen, Chem. Commun., 2011, 47, 3129–3131 RSC.
- B. Porstmann, T. Porstmann, D. Gaede, E. Nugel and E. Egger, Clin. Chim. Acta, 1981, 109, 175–181 CrossRef CAS PubMed.
- Y. Lin, Z. Tian, L. Zhang, J. Ma, Z. Jiang, B. J. Deibert, R. Ge and L. Chen, Nat. Commun., 2019, 10, 1–13 CrossRef PubMed.
- D. Rojas, J. F. Hernandez-Rodriguez, F. Della Pelle, M. Del Carlo, D. Compagnone and A. Escarpa, Biosens. Bioelectron., 2020, 170, 112669 CrossRef CAS PubMed.
- F. Ricci, A. Amine, G. Palleschi and D. Moscone, Biosens. Bioelectron., 2002, 18, 165–174 CrossRef PubMed.
- S. Srinivas, S. M. Senthil Kumar and A. Senthil Kumar, Langmuir, 2023, 39, 12563–12575 CrossRef CAS PubMed.
- S. Saikrithika, A. Shaju, B. Dinesh and A. S. Kumar, Electrochim. Acta, 2022, 405, 139806 CrossRef CAS.
- S. Srinivas and A. Senthil Kumar, J. Phys. Chem. C, 2022, 126, 8296–8311 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 2′,7′-Dichlorodihydrofluorescein diacetate assay procedure and results; effect of the scan rate on GCE/SWCNT@BZ-Redox; effect of the carbon nanomaterial towards BZ-Redox; TEMPO radical quenching experiment; effect of dissolved oxygen towards the GCE/SWCNT@BZ-Redox electrode; approach curves of SECM experiments; comparison of various chemically modified electrodes; BIA responses of real sample analysis using the standard addition method of the GCE/SWCNT@BZ-Redox electrode. See DOI: https://doi.org/10.1039/d4tc01653j |
| This journal is © The Royal Society of Chemistry 2024 |