
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tailoring the optical properties of rubrene films through epitaxy-induced amorphous-to-crystal transition†
Silvia
Trabattoni
 ,
Luisa
Raimondo
*,
Alessandro
Minotto
*,
Angelo
Monguzzi
,
Francesco
Meinardi
and
Adele
Sassella
,
Luisa
Raimondo
*,
Alessandro
Minotto
*,
Angelo
Monguzzi
,
Francesco
Meinardi
and
Adele
Sassella
Department of Materials Science, University of Milano-Bicocca, via Cozzi 55, I-20125, Milano, Italy. E-mail: luisa.raimondo@unimib.it; alessandro.minotto@unimib.it
First published on 20th June 2024
Abstract
Crystalline rubrene (RUB) with the orthorhombic structure can be regarded as a workhorse in organic optoelectronics. So far, however, its great potential for device integration has been held back by the struggle to obtain high-quality and photo-oxidation-resistant RUB thin films. Here, we propose an effective strategy to obtain homogeneous, highly crystalline, and oriented RUB thin films, which relies on the spontaneous amorphous-to-crystal transition driven by organic epitaxy occurring at room temperature in vacuum; this crucial process dictates the final morphological, structural, and optoelectronic properties of the film. To probe the kinetics of the transition, we combine ex situ analysis via polarized optical spectroscopy and atomic force microscopy with a photoluminescence investigation carried out in situ, based on monitoring the efficiency of the singlet fission process typical of crystalline RUB. Building on the insights gained, we tune the thin film growth and post-growth parameters to obtain centimeter-scale, highly homogeneous and crystalline RUB thin films, consisting of several μm-sized and coherently oriented domains, featuring oxidation resistance.
Introduction
Rubrene (RUB) is an extensively studied organic semiconductor that, in its orthorhombic polymorphic structure, exhibits very high mobility of charge carriers,1 record exciton diffusion length,2 extremely efficient triplet fusion (TF) and singlet fission (SF).3–5 This combination of properties makes RUB a versatile platform for a wide range of applications, spanning from transistors to solar cells and organic light-emitting diodes (OLEDs), also featuring RUB-based photon-upconverting (via TF) or -downconverting (via SF) layers.6–11It is now well established that, for most of these applications in devices, a high degree of crystallinity of RUB is necessary. This is indeed particularly crucial for improving charge transport and exciton diffusion and, notably, the SF efficiency.1,2,12 Obtaining highly crystalline and homogeneous RUB thin films with extended domains is, therefore, the fundamental goal that has long been pursued by many research groups in view of boosting device performance. However, for combining the exceptional semiconducting properties of orthorhombic bulk single crystals with the processability and versatility of thin films, two main hurdles are yet to be overcome: the first and most important one is the tendency of RUB to grow naturally amorphous or as spherulites13,14 on all substrates relevant for device applications;15–18 the latter forms are nonetheless poorly controlled and, when crystalline, they display just a radial orientation that strongly limits their use.19–21 The second problem is photo-oxidation, which is extremely efficient in amorphous RUB,22–24i.e., it occurs fast enough to prevent RUB use under ambient conditions without a suitable capping layer. In contrast, photo-oxidation is virtually suppressed in RUB crystals.24,25 This is afforded by a few-monolayer-thick epitaxial native RUB oxide (RUBOX) known to form on crystalline RUB films;26 such a RUBOX layer acts as a passivation layer25 suppressing the diffusion of oxygen in the underlying crystalline films.27 The oxidation issue thus further supports the importance of maximizing the degree of crystallinity in RUB films.
To obtain crystalline RUB films, researchers adopted different methods, such as incorporating buffer layers,15,28–31 post-growth thermal annealing of amorphous RUB films,32–37 or via appropriate modifications of solution-based processes.38,39 When choosing vacuum deposition, which offers the highest level of control, RUB crystallinity can be promoted by a suitable choice of the substrate, leveraging epitaxy to obtain large crystalline domains with coherent orientation directly onto the substrates, both inorganic40–43 and organic ones,44–47 and/or by tuning the growth parameters, for example, the substrate temperature,24,48 deposition rate,45 duration of exposure to air49 or storage time in vacuum.45 Nonetheless, most of these approaches often lead to crystalline RUB films displaying poor homogeneity, a limited size domain and a limited degree of long-range order and orientation.
In this work, we delve into the growth process of RUB thin films on β-alanine (β-ala) single-crystal substrates via organic molecular beam epitaxy (OMBE), which yields RUB films with the orthorhombic structure, unparalleled high crystallinity, and single-crystal-like properties. Specifically, by investigating the evolution of the morphology of RUB films and concurrently monitoring its effect on both the film optical response and photo-physics, we demonstrate that the growth process of crystalline RUB films originates from the spontaneous amorphous-to-crystal transition occurring at room temperature during storage in a ultra-high vacuum (UHV) environment, triggered and driven by organic epitaxy. We then built on the rationalization of the amorphous-to-crystal transition to show how suitable tuning of the growth and post-growth parameters allows fine control of the transition itself. Ultimately, this enables the tailoring of the degree of film crystallinity and the crystalline domain size, and, in turn, the overall properties of RUB films.
Experimental section
Materials
β-ala was purchased from Sigma-Aldrich. Single crystals were grown from supersaturated water solutions at 30 °C following the procedure described elsewhere;50 they were cleaved in air just before their use as substrates for film deposition. The area of β-ala substrates is up to 0.5 cm2. RUB powder was purchased from Acros Organics (99%).Growth
The deposition of RUB was carried out on freshly cleaved (010)-oriented β-ala substrates using OMBE (base pressure ≈1 × 10−9 torr) with a Knudsen-type effusion cell by setting the source temperature to 185 °C. Under these conditions, the growth rate R was lower than 1 Å min−1, except for the rate optimization experiments in which the source temperature was increased up to 200 °C for testing rates up to 3 Å min−1. The nominal thickness d of the film was monitored in situ using a quartz crystal microbalance ranging from 1 Å to 3 nm. The substrate temperature was varied from room temperature to 45 °C. The storage time of the sample (ts) in UHV after deposition was varied from about 15 min to 72 h.Atomic force microscopy
AFM images were acquired in air using a Bruker Multimode Nanoscope V in intermittent contact mode with silicon tips (spring constant = 40 N m−1; resonance frequency ≈ 340 kHz; tip curvature radius < 10 nm) and with an image resolution of 512 × 512 pixels.Optical spectroscopy
Normal incidence optical transmission measurements were carried out in the spectral range of 1.9 to 5.2 eV using a PerkinElmer Lambda 900 spectrometer, equipped with a depolarizer and Glan–Taylor calcite polarizers. The films were analysed on a macroscopic scale, defined by a beam spot size of a few mm2.Photoluminescence characterization
In situ steady-state photoluminescence was recorded using a defocused in situ 355 nm laser excitation (Laser Export, 1.5 mW) and a nitrogen-cooled charge-coupled device (CCD) detector Spex 2000 coupled to a monochromator Triax 190 (HORIBA Jobin-Yvon), with a spectral resolution of 0.5 nm. The laser beam excited the sample at non-normal incidence (about 20° to the sample surface normal) through a BOMCO quartz window and the photoluminescence was collimated and collected by a lens placed about 50 cm from the sample. All spectra were corrected for the optical response of the setup.Results and discussion
Analysis of the growth process
To investigate the growth process of crystalline RUB thin films on β-ala substrates, a combined study of the morphology and optical response of 3-nm-thick films grown at room temperature was carried out ex situ as a function of the storage time ts under UHV conditions in the OMBE chamber after growth at room temperature. A nominal thickness of 3 nm was selected here to afford full coverage of the substrate. Fig. 1 shows the AFM height and phase-contrast images and optical absorption spectra of films stored up to ts = 72 h: in particular, Fig. 1a–c, d–f and g–i refer to samples stored for ts = 24, 48, and 72 h, respectively. First, the morphology of different samples should be analyzed. The film stored for ts = 24 h (Fig. 1a) is composed of coalescent round-shaped structures and a few flat islands with heights of about 6–9 nm. The different components are more clearly detectable in the phase-contrast AFM image (Fig. 1b), which unveils the presence of phases with different chemical–physical properties,51 identified here as the amorphous and crystalline RUB phases, respectively. By increasing ts up to 48 h (Fig. 1d and e), the amount of the crystalline phase (molecularly flat islands) increases at the expense of the amorphous phase; however, at the same time, the morphology becomes highly heterogeneous. Namely, by observing the height image (Fig. 1d), two regions displaying completely different morphologies can be identified. In the left part of the image, the crystalline phase consists of terraces with 1.3-nm-high steps and flat-bottomed holes that are 12–18 nm deep and 120–150 nm wide. In the right part of the image, some round-shaped structures with heights and widths of about 20 nm and 150 nm, respectively, are detected, surrounded by few-nm-thick (up to 9 nm) crystalline islands. A further increase of ts up to 72 h (Fig. 1g and h) leads to a highly homogeneous morphology, now displaying a flat phase, characterized by some terraces with steps of 1.31 ± 0.09 nm. This height fairly matches the spacing between the monomolecular layers enclosed between (200) planes in the orthorhombic RUB polymorph (a = 26.86 Å, b = 7.19 Å, c = 14.43 Å).8,52 The flat phase covers the whole substrate, except for few small holes (8–9 nm deep and 80–100 nm wide) with a smaller average size and lower density than those observed in the samples stored for a shorter ts. In the phase-contrast image (Fig. 1h), the boundaries between the individual domains are evident, indicating that, after nucleation, the nuclei grow independently until they coalesce to form a flat film. Hence, the AFM analysis shows that, if stored in UHV for a suitably long ts, the initially amorphous RUB film spontaneously transforms into a highly homogeneous, flat, and crystalline film. | ||
| Fig. 1 AFM height images and corresponding phase-contrast images of 3-nm-thick RUB films grown on (010)-β-ala with (a,b) ts = 24 h, (d) and (e) ts = 48 h and (g) and (h) ts = 72 h. All AFM images are (5 × 5) μm2. For the films in each row, the normal incidence optical absorption spectra are also reported in (c), (f), and (i), as collected with linearly polarized incident light with an electric field E parallel to [100]β-ala (black curves) and parallel to [001]β-ala (red curves). The spectra were collected in air about 24 h after sample extraction from the UHV chamber. A constant background was subtracted from the spectra for a better comparison. | ||
The same sample evolution with ts can be revealed on a macroscopic scale by analyzing the polarized optical absorption spectra. In Fig. 1c, the optical response of the film stored for ts = 24 h reveals the response of different phases and slight optical anisotropy. Specifically, the strongly polarized sharp peak at 3.72 eV, detected for light polarization with E parallel to [100]β-ala, is characteristic of the orthorhombic crystalline RUB phase;8,53,54 instead, the broad and slightly structured band in the range of 3.9 to 4.2 eV detected for the perpendicular light polarization (E parallel to [001]β-ala) can be attributed to both crystalline and amorphous RUB.8,23,53,54 However, since all spectra in Fig. 1 were collected after the AFM analysis (i.e., about 24 hours after extraction from the UHV chamber), the contribution to the overall optical response from amorphous RUB is expected to be negligible due to photo-oxidation. Indeed, as shown in Fig. S1 (ESI†), the optical fingerprints of amorphous RUB, namely the characteristic band at about 4 eV and the complete vibronic progression of the RUB S0 → S1 transition in the 2–3 eV range,55 disappear within few hours of exposure to ambient conditions. The legacy of the disappeared amorphous phase in Fig. 1c is the prominent RUBOX band at 4.9 eV, which is indeed the mark of complete photo-oxidation of amorphous RUB.23 As ts increases, so does optical anisotropy, and the optical fingerprints of crystalline RUB in the range 3.5–4.5 eV become more evident; in particular, the 3.72 eV sharp peak in the //[100]β-ala-polarized spectra (Fig. 1f and i). This evolution of the optical response can be readily attributed to an increase of crystallinity and order in the RUB film, which is in agreement with the conclusions inferred from the corresponding AFM data and previous report.8 Notably, for ts = 72 h, the highly anisotropic optical response of fully crystalline (100)-oriented orthorhombic RUB on the (010)-oriented β-ala substrate is found,8,53 in the whole spectral range, including the 2–3 eV range (inset in Fig. 1i), where the weak S0 → S1 vibronic progression, which is now clearly visible, lacks the 0–0 component at about 2.35 eV, which is not detectable at normal incidence in (100)-oriented crystalline RUB.8,53 Remarkably, the increase of the oxidation resistant crystalline phase with ts leads to a concomitant suppression of the contribution of RUBOX to the whole optical response.
The marked evolution of the morphological and optical properties, shown in Fig. 1 as a function of ts, is the hallmark of the amorphous-to-crystal transition taking place spontaneously in the UHV chamber. In the early growth stages, RUB films on (010)-β-ala are composed of mainly an amorphous phase and few crystalline islands (Fig. 1a). The storage in UHV allows the amorphous phase to undergo a spontaneous and complete transition to the crystalline phase (Fig. 1g) provided that ts = 72 h or longer, which can therefore be considered as the critical storage time for the occurrence of the complete transition (hereafter labeled tc) for the growth conditions under use. Therefore, Fig. 1a–f (ts < tc) show the intermediate stages of the transition. Looking more closely at these stages of the transition offers the opportunity to probe the actual process taking place in a vacuum and, ultimately, the mechanisms triggering the amorphous-to-crystal transition. For this purpose, the post-growth evolution of the morphology of a 3-nm-thick sample grown under the same conditions and stored for ts < tc is analyzed in more detail. Fig. 2 shows the morphology of the sample surface monitored by AFM for several hours, right after extraction from the UHV growth chamber. The overall inhomogeneous morphology in the full (5 × 5) μm2 image closely resembles that of the sample shown in Fig. 1d. Four regions with different and characteristic morphologies, labelled with letters A to D in Fig. 2, were selected for the analysis.
 | ||
| Fig. 2 AFM height image (5 × 5) μm2 of a 3-nm-thick RUB film grown on (010)-β-ala collected immediately after air exposure. The letters A to D identify four (1.2 × 1.2) μm2 areas of the image, whose evolution with time is reported below as a function of air exposure time (columns). The arrows in the images are to highlight the evolution of specific features. | ||
In region A, the round-shaped amorphous structures observed at t = 0 h (i.e., right after extraction from UHV) shrink within 2 h and eventually disappear for a longer time (see, for example, the structures marked by the black arrows), leaving a shallow hollow in its original position. Concurrently, the height of the flat and initially few-nm-thick crystalline islands surrounding the amorphous structures increases (see also the profiles in Fig. S2, ESI†). A completely different evolution of the morphology can be observed in region B, which is mainly characterized by a holed crystalline network with a height of 10–15 nm. The white arrow in the t = 0 h image marks an ∼11-nm-high round-shaped structure made of amorphous RUB inside a hole in the flat crystalline network; the same structure vanishes after t = 2 h, leaving a hole with a flat bottom. At the same time, the height of the surrounding crystalline network increases, but the difference in height from the bottom of the hole remains constant over time (see also the profile in Fig. S2, ESI†), thus suggesting that the bottom is also rising. No major changes in morphology are observed after 15 h of evolution, aside from a slight enlargement of the hole. The evolution of region C shows the rising of the bottom even more clearly. The round-shaped structures depicted with light blue arrows at t = 0 h disappear after t = 2 h while an island emerges from the flat bottom, with the thickness increasing from one to two monolayers over time. In contrast, at the center of region D, a uniform crystalline island is visible already at t = 0 h, with monomolecular terraces at its edges. As the morphology evolves, the surface of such a concave island levels off with time (i.e., terraces disappear), reaching an almost uniform thickness after 15 h. Note that in all regions of the sample, no further evolution was observed after 15 h.
The post-growth evolution of regions A to C, despite apparently dissimilar, hints at a common amorphous-to-crystal transition mechanism acting spontaneously in vacuum at room temperature. Namely, amorphous RUB is a metastable phase that slowly undergoes the transition induced by organic epitaxy, starting from the interface with a crystalline phase, corresponding to either the β-ala substrate or the surrounding crystalline RUB network (via a diffusion-limited process). By increasing ts up to tc, the flat bottom of the holes in the crystalline network slowly fills up, the holes close up, and the monomolecular terraces in the crystalline islands progressively level off (see region D). At the end of the transition, i.e., for ts ≥ tc, a fully crystalline and homogeneous film consisting of flat and large crystalline domains originating from the coalescence of the islands is obtained. Interestingly, a similar mechanism occurs with inorganic materials and is known as solid-phase epitaxy,56 where a metastable amorphous layer grown on a crystalline templating substrate undergoes the amorphous-to-crystal transition via epitaxy, starting from the interface with the crystalline substrate. The fundamental difference is that, while in solid-phase epitaxy the transition is induced by external factors, such as heating or ion bombardment, in the case of RUB films the amorphous-to-crystal transition occurs spontaneously at room temperature.
The presence of crystalline phases is, therefore, clearly identified as the trigger for the observed amorphous-to-crystal transition in RUB films. The same phenomenon was also previously observed in the case of RUB films deposited on tetracene single crystals45 or via organic droplet epitaxy,49 where crystalline RUB itself or a different crystalline material, respectively, induces the transition of metastable amorphous RUB. On the contrary, in the absence of crystalline phases forming epitaxial interfaces with crystalline RUB, the direct transformation of amorphous RUB into crystalline RUB is hindered, as in the case of RUB deposited onto potassium acid phthalate (KAP) single crystals.23
Finally, it is worth highlighting that the morphology of all samples, in particular those stored for ts < tc, is characterized by the presence of deep holes, with lateral size and density decreasing with increasing ts. From the above description, it is safe to interpret such holes as the original sites of the round-shaped amorphous structures composing the films in the early stages of growth and then supplying molecules to the (already present) crystalline RUB network. Similar holes were previously observed in RUB films deposited on several different substrates,36,41,45,46,57 where nonetheless the amorphous-to-crystal transition leads to crystalline films with different degrees of long-range order and orientation.
The above ex situ analysis carried out in air effectively unveils the mechanisms behind the amorphous-to-crystal transition, despite the concomitant photo-oxidation of the amorphous RUB phase, complicating the interpretation. We made here a further step by probing the amorphous-to-crystal transition by monitoring the photoluminescence (PL) of the films during storage in UHV (in situ) to prevent any interaction with oxygen and thus photo-oxidation. Fig. 3 shows the evolution in time of the PL spectrum of a 3-nm-thick RUB film, grown on β-ala using the same protocol as the films discussed in Fig. 1. The lineshape of the PL spectra in Fig. 3a resembles that of amorphous RUB films,58 with a vibronic progression dominated by the 0–0 and 0–1 components peaking at 2.18 eV and 2.06 eV, respectively. With increasing ts, the PL intensity drops dramatically and the extent of such a drop is quantitatively illustrated in Fig. 3b, which shows the integrated PL versus ts. The PL integrals can be fitted to a single exponential function of the form A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) exp(−ts/τ) + B, where A and B are constants and τ can be interpreted as a phenomenological decay time constant, here of approximately 18 h. Note that τ can be directly linked to the evolution of the film morphology and the optical response shown in Fig. 1, which is ascribed to the amorphous-to-crystal transition. Furthermore, the single-exponential nature of the PL drop suggests that it is caused by a single quenching mechanism.
exp(−ts/τ) + B, where A and B are constants and τ can be interpreted as a phenomenological decay time constant, here of approximately 18 h. Note that τ can be directly linked to the evolution of the film morphology and the optical response shown in Fig. 1, which is ascribed to the amorphous-to-crystal transition. Furthermore, the single-exponential nature of the PL drop suggests that it is caused by a single quenching mechanism.
 | ||
| Fig. 3 (a) PL spectra of a 3-nm RUB film deposited on (010)-β-ala measured in situ in the OMBE chamber at different ts. The spectra are collected at room temperature by exciting the film at 3.5 eV; note that at this excitation energy the absorption of the RUB film does not change with time and polarization (Fig. 1). (b) Evolution of integrated PL (normalized at ts = 0 h). The red line represents a single exponential fit of the experimental data (black dots). | ||
Importantly, since the RUB film was grown in a UHV environment and kept in the UHV environment for the whole duration of the PL experiment, we can rule out that the PL drop is due to RUB oxidation or, in general, to an increased density of exciton quenching sites with ts.59,60 Instead, the dominant source of PL quenching can be identified as the spontaneous amorphous-to-crystal transition discussed above since an increase of the crystallinity of RUB should translate into enhanced singlet fission.12,61 In RUB crystals and polycrystalline films, SF occurs with near-unit efficiency, leading to a relatively low PL quantum yield (of the order of a few percent)61–63 with a substantial fraction (>90%)4 of the PL originating from triplet fusion, which enables the partial conversion/recycling of dark triplets to bright singlets. In contrast, an order of magnitude higher PL quantum yield is obtained in amorphous RUB films, where the molecules are randomly oriented, or in matrices with diluted RUB molecules,12,63 where SF is suppressed.
In summary, the combination of the above ex situ and in situ analyses is a powerful tool to monitor the evolution of the structure and morphology of RUB films grown on β-ala on a timescale ranging from tens of minutes to days. It would also be extremely instructive to follow the growth kinetics on a faster timescale, as previously demonstrated by other groups.64–66 However, the characterization of such early stages of growth is beyond the scope of the present work. Notably, the methodology reported here allowed us to demonstrate and probe, with a proper temporal resolution, the occurrence of a spontaneous amorphous-to-crystal transition driven by organic epitaxy, unveiling the mechanisms behind this transition and showing how the properties of RUB films evolve when the contribution of the crystalline phase increases.
Improving the quality of crystalline RUB films
The unveiling of a spontaneous amorphous-to-crystal transition driven by epitaxy involved in the growth of crystalline RUB thin films gives an opportunity to control it by properly tuning the growth parameters in view of two main purposes. The first is to speed up the amorphous-to-crystal transition (i.e., shortening tc) while maintaining the final crystallinity of the RUB films, given that the process described above requires tens of hours for its completion even at nominal film thicknesses as low as a few nm. The second goal, even more important, is to obtain crystalline RUB films with the highest quality, i.e., highly crystalline and homogeneous thin films with extended domains in view of device integration.The shortening of tc can be obtained by either increasing the deposition rate R or lowering the thickness d. In both cases, given the adjusted growth parameters, the time necessary for full amorphous-to-crystal transition (tRc and tdc, respectively) needs to be carefully controlled. As discussed before, 72 h is the tc required for the complete crystallization of a 3-nm-thick RUB film if the growth rate is set at R1 ∼ 1 Å min−1 ( , hereafter). If the rate is increased, 72 h is not enough to reach the full crystallinity of the film, as demonstrated in Fig. 4, where the AFM images and absorption spectra of a 3-nm-thick RUB film deposited with R2 ∼ 3 Å min−1 and stored in UHV for 72 h are reported. The observed film morphology, with a mix of flat islands, holes, and a few round-shaped structures, is closer to that of the sample grown at rate R1 and stored in UHV for
, hereafter). If the rate is increased, 72 h is not enough to reach the full crystallinity of the film, as demonstrated in Fig. 4, where the AFM images and absorption spectra of a 3-nm-thick RUB film deposited with R2 ∼ 3 Å min−1 and stored in UHV for 72 h are reported. The observed film morphology, with a mix of flat islands, holes, and a few round-shaped structures, is closer to that of the sample grown at rate R1 and stored in UHV for  (Fig. 1d and e). Looking at the polarized absorption spectra, despite the resemblance to those of the sample stored for
(Fig. 1d and e). Looking at the polarized absorption spectra, despite the resemblance to those of the sample stored for  (Fig. 1i), the evident RUBOX shoulder at 4.9 eV23 suggests that the amorphous-to-crystal transition was incomplete upon air exposure since photo-oxidation strongly affects the amorphous RUB phase. Thus, in general, when the growth rate is increased (R2 > R1), the OMBE process shifts more out of equilibrium,67 so that a longer ts is required to complete the amorphous-to-crystal transition, i.e.
(Fig. 1i), the evident RUBOX shoulder at 4.9 eV23 suggests that the amorphous-to-crystal transition was incomplete upon air exposure since photo-oxidation strongly affects the amorphous RUB phase. Thus, in general, when the growth rate is increased (R2 > R1), the OMBE process shifts more out of equilibrium,67 so that a longer ts is required to complete the amorphous-to-crystal transition, i.e. .
.
 | ||
| Fig. 4 (a) AFM height image (5 × 5) μm2 of a 3 nm RUB film grown on (010)-β-ala with a 3 Å min−1 deposition rate and ts = 72 h. (b) Normal incidence optical absorption spectra of the same film. A constant background was subtracted from the spectra for a better comparison. | ||
When considering the second approach, that is shortening tc by reducing the film thickness, it is reasonable to expect that tdc drops, assuming that the storage time needed for complete amorphous-to-crystal transition is strictly related to the RUB mass deposited initially in the amorphous structures. Fig. 5 shows the morphology of the two films grown under the same conditions as the films in Fig. 1 (thickness d1 = 3 nm), with lower thicknesses, namely d2 = 0.1 nm (Fig. 5a) and d3 = 1 nm (Fig. 5b). These samples were stored in UHV for ts = 15 min and 2 h, respectively.
 | ||
| Fig. 5 AFM height images of (a) a 1-Å-thick RUB film and (b) a 1-nm-thick RUB film on (010)-β-ala. In (a), the signal profile along the white line is also reported. | ||
A uniform distribution of 1 monolayer-high islands (1.41 ± 0.07 nm, see the profile) is observed in the thinnest film (Fig. 5a), with irregular borders and some small holes. Although it is one order of magnitude shorter than that in previous experiments, ts is long enough to afford complete amorphous-to-crystal transition for such a low nominal thickness 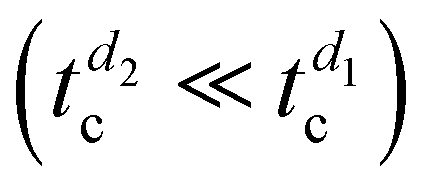 . However, only a partial substrate coverage of about 75% is reached, suggesting that, without modifying other parameters, reducing too much of the initial RUB thickness may not be a viable solution to speed up the whole growth process. Looking at the 1-nm-thick film (Fig. 5b), the surface presents crystalline islands fully covering the substrate, with steps that are one or a few molecular layers high, which is indicative of a successful amorphous-to-crystal transition with satisfactory coverage. However, the morphology is not the desired one since molecularly flat films over a larger scale and without holes are highly preferred in view of device integration. Therefore, limiting the nominal thickness of the RUB film (d2,3 < d1) allows shortening
. However, only a partial substrate coverage of about 75% is reached, suggesting that, without modifying other parameters, reducing too much of the initial RUB thickness may not be a viable solution to speed up the whole growth process. Looking at the 1-nm-thick film (Fig. 5b), the surface presents crystalline islands fully covering the substrate, with steps that are one or a few molecular layers high, which is indicative of a successful amorphous-to-crystal transition with satisfactory coverage. However, the morphology is not the desired one since molecularly flat films over a larger scale and without holes are highly preferred in view of device integration. Therefore, limiting the nominal thickness of the RUB film (d2,3 < d1) allows shortening 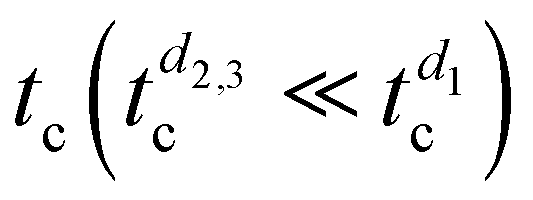 but is not a definitive solution for improving the overall quality of crystalline RUB films.
but is not a definitive solution for improving the overall quality of crystalline RUB films.
To achieve the second and most important goal, i.e., to improve the degree of crystallinity and the homogeneity of the film and to concomitantly increase the size of crystalline domains, another approach should be used, namely, increasing the substrate temperature (Tsub) during growth so as to favor the surface diffusion of the RUB molecules giving crystalline islands.23,48 In Fig. 6, the AFM images collected on the surface of four RUB films grown with the same growth parameters and different Tsub, ranging from 20 to 45 °C, are reported. Note that for the sample grown at Tsub = 45 °C, ts was reduced from 2 h to 1 h, indicating that the amorphous-to-crystal transition at 45 °C is at least ten times faster than that at room temperature. All samples consist of monomolecular-high and flat islands, which become larger with increasing Tsub. At Tsub ≤ 35 °C, some holes were still visible, and the density decreased with Tsub. At Tsub = 45 °C (i.e., the highest temperature investigated here, Fig. 6d), no holes can be observed and the crystalline islands are so wide that, within the (5 × 5) μm2 scanned area, only two levels can be detected: one corresponding to a RUB layer covering the whole substrate, and the other consisting of islands with lateral size > 1 μm and thickness of one monolayer (1.38 ± 0.11 nm). The increasing size of crystalline islands in the different samples in Fig. 6 is a clear effect of increasing Tsub, which provides the adsorbed RUB molecules with additional kinetic energy, thereby favoring surface diffusion.68
 | ||
| Fig. 6 AFM height images (5 × 5) μm2 of 1-nm-thick RUB films deposited at different temperatures of the (010)-β-ala substrate: (a) Tsub = 20 °C (same as in Fig. 5a), (b) Tsub = 30 °C, (c) Tsub = 35 °C, and (d) Tsub = 45 °C. | ||
In view of the improvement of the homogeneity of crystalline RUB films, it can be concluded that increasing Tsub is the best approach, offering two key improvements in one fell swoop: the tc required to allow the completion of the amorphous-to-crystal transition is significantly reduced, and the homogeneity of the films is greatly improved.
Conclusions
In this study, we report the successful growth of crystalline RUB thin films with a molecularly flat surface made of > 1 μm-wide coherently oriented domains displaying the orthorhombic structure with considerably limited grain boundary density. Notably, the morphology of such crystalline films is homogeneous on a macroscopic scale of the order of tens of mm2, limited only by the size and defects of the substrate.We achieved this by rationalizing the physical process controlling the growth and quality of crystalline RUB films on β-ala, which is an amorphous-to-crystal transition. Through a combined morphology and optical response study, we demonstrate that the amorphous-to-crystal transition in RUB/β-ala systems occurs spontaneously at room temperature and is driven by organic epitaxy, triggering the crystallization of amorphous RUB at the interface with the β-ala substrate, yielding films with single-crystal-like properties.
To enable this, however, it is of paramount importance to prevent photo-oxidation, which is highly efficient in amorphous RUB and can outcompete the amorphous-to-crystal transition. We thus demonstrated that, to maximize film quality and crystallinity, the film must be stored in a vacuum chamber after growth until the amorphous-to-crystal transition is complete.
We also directly assessed the completion of the amorphous-to-crystal transition in UHV (in situ) in a non-invasive way by examining the evolution of the RUB film PL during storage. Namely, we tracked the amorphous-to-crystal transition by observing the rate at which the PL intensity drops, i.e., by monitoring the efficiency of the SF process, which is a distinctive characteristic of the RUB crystalline phase and contributes to making it a particularly attractive organic semiconductor.
In view of device integration, we show that the growth of RUB crystalline films can be substantially expedited (from tens of hours to 1 h or less for completion) by increasing the substrate temperature from room temperature to just 45 °C. Concurrently, the morphology improved significantly, giving the most homogenous films among those reported here, with the largest crystalline domains and complete substrate coverage. Importantly, such an optimal morphology is fundamental when thick RUB films may be necessary since they can be grown by exploiting homoepitaxy at a high rate without compromising film quality. Finally, because of the water solubility of the β-ala substrate, the crystalline RUB film could be readily transferred to virtually any device-relevant substrate, as demonstrated in a previous work,8 while retaining substantially unaffected film properties.
Author contributions
Silvia Trabattoni: investigation and writing – original draft. Luisa Raimondo: conceptualization, investigation, methodology, and writing – original draft. Alessandro Minotto: funding acquisition and writing – original draft. Angelo Monguzzi: investigation and writing – review & editing. Francesco Meinardi: writing – review & editing. Adele Sassella: conceptualization, methodology, supervision, and writing – original draft.Data availability
The data supporting the findings of this work are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Prof. C. Antonini for helpful discussions and Dr G. Vaccaro for preliminary PL measurements. The Italian Ministero dell’Università e della Ricerca (MUR) is acknowledged for funding through the PRIN project 2022SCWMT2 (SCINTILLA).References
- E. Menard, V. Podzorov, S.-H. Hur, A. Gaur, M. E. Gershenson and J. A. Rogers, Adv. Mater., 2004, 16, 2097 CrossRef CAS
.
- H. Najafov, B. Lee, Q. Zhou, L. C. Feldman and V. Podzorov, Nat. Mater., 2010, 9, 938–943 CrossRef CAS PubMed
- A. Ryasnyanskiy and I. Biaggio, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 193203 CrossRef
- I. Biaggio and P. Irkhin, Appl. Phys. Lett., 2013, 103, 263301 CrossRef
- G.-H. Deng, J. B. Brown, H. Fisher, Z.-C. Huang-Fu, Y. Qian, T. Zhang, A. Harutyunyan, H. Chen, G. Chen and Y. Rao, Chem. Phys. Rev., 2023, 4, 041313 CrossRef CAS
- V. C. Sundar, J. Zaumseil, V. Podzorov, E. Menard, R. L. Willett, T. Someya, M. E. Gershenson and J. A. Rogers, Science, 2004, 303, 1644 CrossRef CAS PubMed
- V. Podzorov, E. Menard, A. Borissov, V. Kiryukhin, J. A. Rogers and M. E. Gershenson, Phys. Rev. Lett., 2004, 93, 086602 CrossRef CAS PubMed
- S. Trabattoni, L. Raimondo, M. Campione, D. Braga, V. C. Holmberg, D. J. Norris, M. Moret, A. Ciavatti, B. Fraboni and A. Sassella, Adv. Mater. Interfaces, 2015, 2, 1500423 CrossRef
- A. Rao and R. H. Friend, Nat. Rev. Mater., 2017, 2, 17063 CrossRef CAS
- R. Nagata, H. Nakanotani, W. J. Potscavage Jr. and C. Adachi, Adv. Mater., 2018, 30, 1801484 CrossRef PubMed
- W. Sun, A. Ronchi, T. Zhao, J. Han, A. Monguzzi and P. Duan, J. Mater. Chem. C, 2021, 9, 14201 RSC
- D. M. Finton, E. A. Wolf, V. S. Zoutenbier, K. A. Ward and I. Biaggio, AIP Adv., 2019, 9, 095027 CrossRef
- L. Wang, Y. Li, F. Zou, H. Du, L. Sun, J. Zhang, X. Song and G. Song, RSC Adv., 2016, 6, 3532 RSC
- L. Gránásy, T. Pusztai, G. Tegze, J. A. Warren and J. F. Douglas, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 72, 011605 CrossRef PubMed
- J. H. Seo, D. S. Park, S. W. Cho, C. Y. Kim, W. C. Jang, C. N. Whang, K.-H. Yoo, G. S. Chang, T. Pedersen, A. Moewes, K. H. Chae and S. J. Cho, Appl. Phys. Lett., 2006, 89, 163505 CrossRef
- J. J. Kim, H. M. Lee, J. W. Park and S. O. Cho, J. Mater. Chem. C, 2015, 3, 2650 RSC
- D. Käfer, L. Ruppel, G. Witte and C. Wöll, Phys. Rev. Lett., 2005, 95, 166602 CrossRef PubMed
- H. H. Fong, S. K. So, W. Y. Sham, C. F. Lo, Y. S. Wu and C. H. Chen, Chem. Phys., 2004, 298, 119 CrossRef CAS
- S.-W. Park, J.-M. Choi, K. H. Lee, H. W. Yeom, S. Im and Y. K. Lee, J. Phys. Chem. B, 2010, 114, 5661 CrossRef CAS PubMed
- A. L. Foggiatto, Y. Takeichi, K. Ono, H. Suga, Y. Takahashi, M. A. Fusella, J. T. Dull, B. P. Rand, K. Kutsukake and T. Sakurai, Org. Electron., 2019, 74, 315 CrossRef CAS
- J. A. Tan, J. T. Dull, S. E. Zeltmann, J. A. Tulyagankhodjaev, H. M. Johnson, A. Liebman-Peláez, B. D. Folie, S. A. Dönges, O. Khatib, J. G. Raybin, T. D. Roberts, L. M. Hamerlynck, C. P. N. Tanner, J. Lee, C. Ophus, K. C. Bustillo, M. B. Raschke, H. Ohldag, A. M. Minor, B. P. Rand and N. S. Ginsberg, Adv. Funct. Mater., 2023, 33, 2207867 CrossRef CAS
- M. Kytka, A. Gerlach, F. Schreiber and J. Kováč, Appl. Phys. Lett., 2007, 90, 131911 CrossRef
- S. Uttiya, L. Raimondo, M. Campione, L. Miozzo, A. Yassar, M. Moret, E. Fumagalli, A. Borghesi and A. Sassella, Synth. Met., 2012, 161, 2603 CrossRef
- D. Käfer and G. Witte, Phys. Chem. Chem. Phys., 2005, 7, 2850 RSC
- L. Raimondo, S. Trabattoni, M. Moret, N. Masciocchi, M. Masino and A. Sassella, Adv. Mater. Interfaces, 2017, 4, 1700670 CrossRef
- E. Fumagalli, L. Raimondo, L. Silvestri, M. Moret, A. Sassella and M. Campione, Chem. Mater., 2011, 23, 3246 CrossRef CAS
- D. D. T. Mastrogiovanni, J. Mayer, A. S. Wan, A. Vishnyakov, A. V. Neimark, V. Podzorov, L. C. Feldman and E. Garfunkel, Sci. Rep., 2014, 4, 4753 CrossRef CAS PubMed
- M. Haemori, J. Yamaguchi, S. Yaginuma, K. Itaka and H. Koinuma, Jpn. J. Appl. Phys., 2005, 44, 3740 CrossRef CAS
- X. Qian, T. Wang and D. Yan, Org. Electron., 2013, 14, 1052 CrossRef CAS
- M. A. Fusella, S. Yang, K. Abbasi, H. H. Choi, Z. Yao, V. Podzorov, A. Avishai and B. P. Rand, Chem. Mater., 2017, 29, 6666 CrossRef CAS
- Z. Li, J. Du, Q. Tang, F. Wang, J.-B. Xu, J. C. Yu and Q. Miao, Adv. Mater., 2010, 22, 3242 CrossRef CAS PubMed
- H. M. Lee, H. Moon, H.-S. Kim, Y. N. Kim, S.-M. Choi, S. Yoo and S. O. Cho, Org. Electron., 2011, 12, 1446 CrossRef CAS
- T. R. Fielitz and R. J. Holmes, Cryst. Growth Des., 2016, 16, 4720 CrossRef CAS
- M. Nothaft and J. Pflaum, Phys. Status Solidi B, 2008, 245, 788 CrossRef CAS
- B. Verreet, P. Heremans, A. Stesmans and B. P. Rand, Adv. Mater., 2013, 25, 5504 CrossRef CAS PubMed
- C. Du, W. Wang, L. Li, H. Fuchs and L. Chi, Org. Electron., 2013, 14, 2534 CrossRef CAS
- S.-W. Park, S. H. Jeong, J.-M. Choi, J. M. Hwang, J. H. Kim and S. Im, Appl. Phys.
Lett., 2007, 91, 033506 CrossRef
- N. Stingelin-Stutzmann, E. Smits, H. Wondergem, C. Tanase, P. Blom, P. Smith and D. De Leeuw, Nat. Mater., 2005, 4, 601 CrossRef CAS PubMed
- L. Wang, L. Wang, P. Zhang, L. Zhang, Q. Xie and Y. Liu, Synth. Metals, 2019, 248, 68 CrossRef CAS
- C.-H. Lee, T. Schiros, E. J. G. Santos, B. Kim, K. G. Yager, S. J. Kang, S. Lee, J. Yu, K. Watanabe, T. Taniguchi, J. Hone, E. Kaxiras, C. Nuckolls and P. Kim, Adv. Mater., 2014, 26, 2812 CrossRef CAS PubMed
- Y. Wei, D. Xue, L. Ji, Q. Wang, X. Jiang, Y. Sun, Z. Wang, L. Huang and L. Chi, Chin. J. Chem., 2022, 40, 1298 CrossRef CAS
- M. Lan, Z.-H. Xiong, G.-Q. Li, T.-N. Shao, J.-L. Xie, X.-F. Yang, J.-Z. Wang and Y. Liu, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195322 CrossRef
- S. Wali, Q. Yin, J. Li, G. Si, M. Shafi, J. Ren and H. Zhang, J. Mater. Chem. C, 2022, 10, 1289 RSC
- W.-S. Hu, S.-Z. Weng, Y.-T. Tao, H.-J. Liu and H.-Y. Lee, Org. Electron., 2008, 9, 385 CrossRef CAS
- M. Campione, M. Moret, L. Raimondo and A. Sassella, J. Phys. Chem. C, 2009, 113, 20927 CrossRef CAS
- L. Raimondo, E. Fumagalli, M. Moret, M. Campione, A. Borghesi and A. Sassella, J. Phys. Chem. C, 2013, 117, 13981 CrossRef CAS
- H. Chang, W. Li, H. Tian, Y. Geng, H. Wang, D. Yan and T. Wang, Org. Electron., 2015, 20, 43 CrossRef CAS
- C. H. Hsu, J. Deng, C. R. Staddon and P. H. Beton, Appl. Phys. Lett., 2007, 91, 193505 CrossRef
- A. Sassella, L. Raimondo, M. Campione and A. Borghesi, Adv. Mater., 2013, 25, 2804 CrossRef CAS PubMed
- S. Trabattoni, M. Moret, M. Campione, L. Raimondo and A. Sassella, Cryst. Growth Des., 2013, 13, 4268 CrossRef CAS
- J. P. Cleveland, B. Anczykowski, A. E. Schmid and V. B. Elings, Appl. Phys. Lett., 1998, 72, 2613 CrossRef CAS
- O. D. Jurchescu, A. Meetsma and T. T. M. Palstra, Acta Crystallogr., Sect. B: Struct. Sci., 2006, 62, 330 CrossRef PubMed
- S. Tavazzi, L. Silvestri, M. Campione, A. Borghesi, A. Papagni, P. Spearman, A. Yassar, A. Camposeo and D. Pisignano, J. Appl. Phys., 2007, 102, 023107 CrossRef
- M. Campione, L. Raimondo, M. Moret, P. Campiglio, E. Fumagalli and A. Sassella, Chem. Mater., 2009, 21, 4859–4867 CrossRef CAS
- M. Kaschke, N. P. Ernsting and F. P. Schafer, Opt. Commun., 1988, 66, 211 CrossRef CAS
-
M. A. Herman, W. Richter and H. Sitter, Epitaxy, Springer-Verlag Berlin Heidelberg, Germany, 2004 Search PubMed
- M. A. Fusella, F. Schreiber, K. Abbasi, J. J. Kim, A. L. Briseno and B. P. Rand, Nano Lett., 2017, 17, 3040 CrossRef CAS PubMed
- P. Irkhin, A. Ryasnyanskiy, M. Koehler and I. Biaggio, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 085143 CrossRef
- P. Irkhin, I. Biaggio, T. Zimmerling, M. Döbeli and B. Batlogg, Appl. Phys. Lett., 2016, 108, 063302 CrossRef
- D. G. Bossanyi, Y. Sasaki, S. Wang, D. Chekulaev, N. Kimizuka, N. Yanai and J. Clark, J. Mater. Chem. C, 2022, 10, 4684 RSC
- P. Baronas, G. Kreiza, L. Naimovičius, E. Radiunas, K. Kazlauskas, E. Orentas and S. Juršėnas, J. Phys. Chem. C, 2022, 126, 15327 CrossRef CAS PubMed
- E. Radiunas, M. Dapkevičius, S. Raišys and K. Kazlauskas, Phys. Chem. Chem. Phys., 2022, 24, 24345 RSC
- E. Radiunas, L. Naimovičius, S. Raišys, A. Jozeliūnaitė, E. Orentas and K. Kazlauskas, J. Mater. Chem. C, 2022, 10, 6314 RSC
- A. Winkler, Surf. Sci., 2016, 643, 124 CrossRef CAS PubMed
- F. Balzer, C. Röthel, H.-G. Rubahn, A. Lützen, J. Parisi, R. Resel and M. Schiek, J. Phys. Chem. C, 2016, 120, 7653 CrossRef CAS
- A. J. Fleming, F. P. Netzer and M. G. Ramsey, J. Phys.: Condens. Matter, 2009, 21, 445003 CrossRef CAS PubMed
- A. Sassella, M. Campione, A. Papagni, C. Goletti, G. Bussetti, P. Chiaradia, V. Marcon and G. Raos, Chem. Phys., 2006, 325, 193 CrossRef CAS
- B. Krause, F. Schreiber, H. Dosch, A. Pimpinelli and O. H. Seeck, Europhys. Lett., 2004, 65, 372 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Post-growth evolution in air of the polarized absorption spectra and the morphology after air exposure. See DOI: https://doi.org/10.1039/d4tc01618a |
| This journal is © The Royal Society of Chemistry 2024 |