
Mitigating the volume expansion and enhancing the cycling stability of ferrous fluorosilicate-modified silicon-based composite anodes for lithium-ion batteries†
Jichang
Sun
ac,
Xiaoyi
Liu
a,
Penglun
Zheng
*c,
Yang
Zhao
d,
Yun
Zheng
 *a,
Jingchao
Chai
a and
Zhihong
Liu
*b
*a,
Jingchao
Chai
a and
Zhihong
Liu
*b
aSchool of Optoelectronic Materials and Technology, Jianghan University, Wuhan, 430056, P. R. China. E-mail: zhengyun@jhun.edu.cn
bState Key Laboratory of Precision Blasting, Jianghan University, Wuhan, 430056, P. R. China. E-mail: liuzh@jhun.edu.cn
cCollege of Civil Aviation Safety Engineering, Civil Aviation Flight University of China, Guanghan, 618307, P.R. China. E-mail: zhengpenglun@cafuc.edu.cn
dChina Institute of Ocean Engineering (Tsing Tao), Qingdao, 266555, P. R. China
First published on 30th August 2024
Abstract
Silicon has emerged as a prominent candidate for anodes in advanced lithium-ion batteries due to its exceptional theoretical capacity and low operational potential. Despite its advantages, silicon-based anodes face significant challenges, including substantial volume changes, formation of an unstable solid-electrolyte interphase (SEI) film, and voltage hysteresis during lithium alloying/dealloying, which compromise their cycling stability. This study introduces a novel ferrous fluorosilicate (FeSiF6)-modified silicon-based composite anode. FeSiF6 is prepared via a simple reaction between Fe–Si alloys and hydrofluoric acid (HF). Various treatment methods are employed to create modified silicon-based composites with different compositions and morphologies. This innovative composite material prevents the formation of crystalline Li15Si4 and facilitates the formation of a stable SEI film, thereby markedly improving the cycling stability of the silicon-based anodes. Among these, the composite material Fe–Si@F@C (consisting of Fe–Si alloy@FeSiF6@graphite) demonstrates a stable discharge capacity of 975 mA h g−1 after 200 cycles at 1 A g−1, with ∼94% capacity retention, and outstanding rate performance (664.4 mA h g−1 at 4 A g−1). In comparison, the Fe–Si alloy/graphite anode without FeSiF6 shows a much lower discharge capacity of 458 mA h g−1 at 1 A g−1 after 200 cycles and 291.8 mA h g−1 at 4 A g−1. These findings underscore the critical role of FeSiF6 in modifying silicon-based anodes and enhancing their cycling stability, significantly increasing their potential for commercial application in next-generation lithium-ion batteries.
1. Introduction
Silicon has gained widespread attention as a potential anode material for next-generation lithium-ion batteries (LIBs), owing to its superior theoretical capacity (3589 mA h g−1 based on the alloying reaction 4Si + 15Li+ + 15e− → Li15Si4 at room temperature), abundant availability (constituting about 28% of the Earth's crust), and cost-effectiveness (approximately 5 US$ per kg).1,2 Despite these advantages, the extensive volume change (nearly 383% expansion/contraction) experienced by silicon during the charge/discharge cycles results in electrode cracking/fracturing, formation of an unstable solid electrolyte interphase (SEI) film, loss of electrical contact, and considerable capacity fade.3–5 Consequently, the research community has dedicated efforts towards microstructural/compositional optimization, binder enhancement, and electrolyte engineering to address these issues.4,6–8 Nanostructuring of silicon into forms such as nanoparticles, nanowires, and nanosheets has become a focal point of investigation,9,10 aimed at reducing stress from the formation of crystalline Li15Si4 (c-Li15Si4) during full lithiation11 and improving the diffusion rates of lithium ions within these nanoarchitectures.12,13 In particular, silicon nanoparticles with a critical size of approximately 150 nm have shown resilience against cracking or fracturing in their initial lithiation cycle.14 However, the large specific surface area of these nanostructures predisposes them to aggregation, leading to excessive lithium/electrolyte consumption (which reduces the initial coulombic efficiency), and impedes electron transport at the electrode/electrolyte interface, thus adversely affecting battery performance.15,16To tackle these issues, one can employ encapsulation technology and surface modification, incorporate electrolyte additives, or combine silicon with other materials (i.e., compositing).15,17,18 Encapsulation involves enveloping silicon in the highly conductive and buffering matrix (for example, carbon layer, carbon nanotubes, and graphene), mitigating the adverse effects of the electrode's expansion and improving cycling stability.19 Surface modification primarily entails functionalizing the surface of silicon particles through specific techniques (for instance, atomic/molecular layer deposition20) to enhance their interface stability with electrolytes.21 Furthermore, the use of electrolyte additives like organic fluoroethylene carbonate (FEC) and vinylene carbonate (VC) has been proven to facilitate the formation of an SEI film, predominantly comprising LiF and cross-linked polyethylene oxide.18,22 This enhancement in SEI film stability reduces electrolyte decomposition, boosts coulombic efficiency, and thereby aids in better capacity retention. However, it is important to highlight that the FEC additive is fully consumed when the delithiation cutoff voltage exceeds 0.7 V, utilizing lithium metal as the counter electrode, which prompts a rapid decline in capacity.23 In addition, the presence of Lewis acid PF5 in LiPF6-based electrolytes triggers the defluorination of FEC at higher temperatures,24 leading to the emission of gaseous species (such as CO2 and H2) and corrosive HF.25 This corrosive HF can breach the SEI layer, reacting with the silicon and resulting in the degradation of the active silicon material.5
Additionally, the formation of composites through the combination of silicon with other materials, such as carbonaceous materials, metal oxides and intermetallic compounds, boosts both its structural stability and electrochemical performance.26,27 Composite materials typically provide benefits such as straightforward preparation, increased mechanical stability,4 improved electron transport/Li-ion diffusion,28 increased surface reaction activity,29 and stabilized SEI film.30,31 These benefits have turned silicon-based composite anode materials into a hotspot for research and development, offering new possibilities for the application of next-generation lithium-ion batteries. For example, silicon-based nanocomposites with FeSi2 and carbon exhibit a reversible charge capacity of 1045 mA h g−1 and an initial coulombic efficiency of 87%.32 This benefits from the buffer effect of the highly elastic/lithium-inactive FeSi2 phase and conductive carbon layers. Furthermore, the study demonstrates that different carbon-based conductive modifiers are crucial for enhancing the conductivity and electrochemical performance of silicon-based anodes in lithium batteries.33–35 Liu et al. found that fluorine-containing precursor carbon materials significantly improve the performance of pure-phase nano-silicon particles.36 The porous nanocomposites they prepared exhibited a specific capacity of approximately 660 mA h g−1 after 50 cycles, maintaining 75% of the capacity retention rate. Interestingly, compared to other commonly used carbon sources, such as polyvinyl chloride, pitch, and sucrose, the Si/C composites made with fluorine-containing precursors also showed improved capacity retention rates.
While conventional additives partially stabilize the SEI film and mitigate continuous side reactions between silicon and the electrolyte, their effectiveness is often compromised by their inability to uniformly withstand silicon's substantial volumetric changes, resulting in ongoing lithium consumption. Fluorine-containing compounds possess strong antioxidative properties, which help mitigate the issue of Li–Si alloying by enhancing the SEI film on the anode surface.37 In addition to additives, composite materials also play a crucial role in suppressing silicon lithiation and enhancing its stability. Electrochemical performance analysis indicates that the presence of ferrous fluorosilicate (FeSiF6) can prevent the irreversible formation of Li2SiF6 to some extent, especially after combining silicon with graphite, further stabilizing the anode. Moreover, these additives/composites play a crucial role in suppressing the lithiation of silicon, enhancing its stability. It is also hypothesized that the presence of FeSiF6 can to some extent prevent the irreversible formation of Li2SiF6, especially following the integration of silicon with graphite, which further stabilizes the anode.38
This study proposes a straightforward synthesis method for FeSiF6 and investigates its effectiveness in improving the performance of silicon-based anodes. We characterized the electrochemical performance of the materials and explored the impact of varying FeSiF6 concentrations on anode performance. Differential capacity analysis indicates that the FeSiF6 modifier plays a critical role in inhibiting the formation of c-Li15Si4 during lithiation, thereby significantly reducing electrode expansion. The Fe–Si alloy@FeSiF6@graphite (Fe–Si@F@C) composite anodes exhibited robust performance, maintaining a stable discharge capacity of 975 mA h g−1 after 200 cycles at a current density of 1 A g−1, which corresponds to approximately 94% capacity retention. The composite anode also demonstrated impressive rate performance (664.4 mA h g−1 at 4 A g−1). The findings from this study are anticipated to significantly advance research on silicon-based anodes and underscore their potential for commercial applications, marking a notable contribution toward the development of more resilient and efficient LIBs.
2. Experimental section
2.1 Material preparation
Phase composition analysis by X-ray diffraction (XRD) revealed the presence of FeSi and FeSi2 phases in the initial Fe–Si alloy powders, as shown in Fig. S2.† The XRD pattern of the synthesized FeSiF6 crystals, presented in Fig. S3,† matches closely with the reference pattern for FeSiF6·6H2O (PDF#26-0799), demonstrating that the Fe–Si alloy (FeSix) reacted with HF to produce FeSiF6, as per the reaction: FeSix + HF → FeSiF6 + H2 (gas). The morphology and structure of the FeSiF6 crystals are depicted in Fig. S3a and b,† illustrating a hexagonal columnar formation with a width of around 2 µm and relatively smooth side surfaces. A high-resolution image in Fig. S3c† reveals stacked structures at the top, presumed to form during the crystallization phase, indicative of the FeSiF6 crystals' continuous growth through evaporation and crystallization. In contrast, scanning electron microscopy (SEM) images (Fig. S4†) of the initial Fe45Si55 alloy show irregular particles with an uneven size distribution, highlighting the synthesis method's effectiveness in producing a uniform FeSiF6 morphology.
Transmission electron microscopy (TEM) images (Fig. S3d†) depict the columnar structure of FeSiF6, encased in a transparent thin outer layer. The high-magnification TEM image in Fig. S3e† reveals hollow structures within the crystals and their uneven distribution, suggesting that these voids may serve as insertion sites for Li+ ions in electrode materials, potentially enhancing stability. The high-resolution TEM (HRTEM) images in Fig. S3f† display distinct lattice fringes, with measurements at three different locations yielding lattice spacings of 0.42 nm, 0.38 nm, and 0.33 nm, corresponding to the (012), (021), and (202) planes of FeSiF6 (PDF#26-0799), respectively. Furthermore, elemental mapping in Fig. S3g† showcases the even distribution of F, Fe, and Si elements within the FeSiF6 structure, affirming its compositional uniformity.
| Abbreviations for materials | Differences | Preparation method | Composition (wt%) |
|---|---|---|---|
| Fe–Si–T@C | Employing an excess of FeSi alloy (incomplete reaction) | (a) Add 10 mL of 1 M hydrofluoric acid to a beaker; (b) mix in 1 g of Fe25Si75 alloy powder (500 mesh, 99% purity, Benyu Metal Materials) and seal the beaker; (c) heat at 40 °C for 12 hours to react; (d) dry the mixture at 80 °C under vacuum for 24 hours; (e) mix 0.6 g of the resultant powder with 0.4 g of graphite (from Dongguan KELIDA) in a ball mill; (f) mill with stainless steel balls for 30 minutes in a SPEX 8000M mill (with a fixed clamp speed of 875 cycles per minute); (g) rinse with ethanol, filter, and dry at 80 °C under vacuum for 6 hours | Graphite 40%, silicon 21.2%, FeSiF6 22.8%, and FeSi2 16% |
| Fe–Si@C–T | Utilizing an excess of HF (the Fe–Si alloy fully reacted) | (a) Combine 0.6 g of Fe25Si75 alloy powder with 0.4 g of graphite in a ball milling jar; (b) add 40 g of 3 mm and 10 g of 1.5 mm stainless steel balls to the jar and mill for 30 minutes using a SPEX 8000M mill; (c) wash the milled powder with ethanol, filter, and dry at 80 °C under vacuum for 6 hours; (d) soak the dry powder in 10 mL of 1 M hydrofluoric acid, seal with plastic wrap, heat to 40 °C on a heating plate for 12 hours to ensure complete reaction; (e) finally, transfer the reacted mixture to a Petri dish and dry under vacuum at 80 °C for 24 hours | Graphite 40%, silicon 12.2%, FeSiF6 41.3%, and FeSi2 6.5% |
| Fe–Si@F@C | Applying an equal mixture of both Fe–Si alloy and FeSiF6 | (a) Measure 0.3 g each of Fe25Si75 alloy powder and FeSiF6 powder, along with 0.4 g of graphite, and place them into a ball milling jar; (b) add 40 g of 3 mm and 10 g of 1.5 mm stainless steel balls to the jar, then mill for 30 minutes using a high-energy ball mill (SPEX 8000M); (c) rinse the milled composite with ethanol, filter out the powder, and dry it under vacuum at 80 °C for 6 hours | Graphite 40%, silicon 21.8%, FeSiF6 29.1%, and FeSi2 9.1% |
2.2 Morphology and composition characterization
The crystal structure of the resulting samples was characterized by X-ray diffraction (XRD) using a multifunctional powder X-ray diffractometer (X'Pert Powder, PANalytical, The Netherland) with a Cu Kα radiation source. The morphology and microstructure of the samples were characterized by field-emission scanning electron microscopy (FE-SEM, Hitachi SU8010, Japan) and high-resolution transmission electron microscopy (TEM, FEI Talos F200X, USA). X-ray photoelectron spectroscopy (XPS, Axis Supra+, Kratos Shimadzu, Japan) was used to identify the chemical states of Si, C, F, O, Li, and Fe in the samples.2.3 Electrochemical measurements
The electrochemical performance was characterized by assembling a CR2032 button cell with lithium cells serving as the counter/reference electrode. The anode consists of 70 wt% active materials, 20 wt% acetylene black, and 10 wt% polyvinylidene fluoride (PVDF). These powders were mixed with N-methyl-2-pyrrolidone and stirred on a magnetic stirrer for 4 hours to obtain a homogeneous slurry. The slurry was evenly coated onto a copper foil and then dried in an oven at 60 °C for 8 h. The weight of the active material on the electrode is about 0.5–1.0 mg cm−2, with a thickness of ∼15 µm, as measured using a micrometer. The weight of the active material is calculated based on the weight of the composite material. The electrolyte is LB-064 (Do Do Chem) comprising 1.0 M LiPF6 in ethylene carbonate (EC) and dimethyl carbonate (DMC) (EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMC = 1:1 by volume). A microporous polypropylene film (Celgard 2400) was used as the separator for the button cell. Cyclic voltammetry (CV) was performed on an electrochemical workstation (model VSP-300, BioLogic, France) with a voltage range of 0.005–3 V and sweep rates of 0.1 and 0.5 mV s−1. Electrochemical impedance spectroscopy (EIS) tests were conducted on the same electrochemical workstation at open circuit potentials with frequencies ranging from 100 kHz to 0.01 Hz and an AC amplitude of 5 mV. A multi-channel battery test system (LAND CT2001A, Wuhan LAND Electronics Co., Ltd., China) was used for galvanostatic charge–discharge (GCD) measurements in the range of 0.005–3 V (vs. Li+/Li).
:DMC = 1:1 by volume). A microporous polypropylene film (Celgard 2400) was used as the separator for the button cell. Cyclic voltammetry (CV) was performed on an electrochemical workstation (model VSP-300, BioLogic, France) with a voltage range of 0.005–3 V and sweep rates of 0.1 and 0.5 mV s−1. Electrochemical impedance spectroscopy (EIS) tests were conducted on the same electrochemical workstation at open circuit potentials with frequencies ranging from 100 kHz to 0.01 Hz and an AC amplitude of 5 mV. A multi-channel battery test system (LAND CT2001A, Wuhan LAND Electronics Co., Ltd., China) was used for galvanostatic charge–discharge (GCD) measurements in the range of 0.005–3 V (vs. Li+/Li).
3. Results and discussion
Pure-phase crystalline FeSiF6 is efficiently synthesized from Fe–Si alloys (comprising FeSi and FeSi2) using a straightforward reaction with HF, followed by vacuum filtration and drying. This method is simpler and more resource-efficient than traditional approaches, offering environmental benefits.39,40 The material's transformation was evaluated through SEM images and the reaction process is illustrated in Fig. 1a–c. XRD analyses for Fe–Si@C–T, Fe–Si–T@C, and Fe–Si@F@C composites, presented in Fig. 1d, confirm the formation of FeSiF6 in all samples post-HF treatment. The Fe–Si@C–T sample shows a complete disappearance of FeSi2 peaks, leaving only the silicon phase. Meanwhile, Fe–Si–T@C reveals a mix of Si, FeSi2, and FeSiF6, with Si being the predominant phase due to the partial conversion of FeSi2 into FeSiF6, thereby reducing the presence of both FeSi and FeSi2. | ||
| Fig. 1 (a–c) Schematic diagram for the synthesis of Fe–Si@C–T, Fe–Si–T@C, and Fe–Si@F@C. (d) XRD results of Fe–Si@C–T, Fe–Si–T@C, and Fe–Si@F@C samples. | ||
SEM analyses were conducted on the three materials to examine the effects of various preparation methods on morphology, as shown in Fig. 2. The analysis highlights notable differences in morphology: Fe–Si@C–T prepared by ball milling and acid treatment (Fig. 2a) differs significantly from those prepared by other methods. Specifically, Fe–Si@C–T (Fig. 2b) and Fe–Si@F@C (Fig. 2c) show a layered graphite structure, offering a buffer for silicon anode volume expansion and improving stability.15,41 Conversely, the Fe–Si@C–T processed through ball milling and acid treatment lacks this layered structure, presenting smoother surfaces with graphite well integrated with FeSiF6 and Si. The TEM images (Fig. 2d) corroborate these observations, displaying a uniform graphite distribution in Fe–Si@C–T and distinct FeSiF6 particles. Fig. 2e and f clearly depict the graphite layers enveloping other components in a sandwich-like manner. Elemental analysis of Fe–Si@C–T (Fig. 2g) indicates a core–shell structure, with Fe, F, and C forming an outer layer around Si, suggesting that this method effectively creates core–shell morphologies.
 | ||
| Fig. 2 SEM images of (a) Fe–Si@C–T, (b) Fe–Si–T@C, and (c) Fe–Si@F@C. TEM images of (d) Fe–Si@C–T, (e) Fe–Si–T@C, and (f) Fe–Si@F@C. (g) HAADF-STEM image and corresponding EDX mapping images of Fe–Si@C–T. | ||
Research shows FeSi2 enhances silicon anodes' cycling stability through a buffering effect,15,42 while graphite creates a conductive network, improving electrical conductivity.15,28,43 CV tests on Fe–Si@C–T, Fe–Si–T@C, and Fe–Si@F@C (Fig. S5†) revealed similar oxidation–reduction peak positions for the materials, with variations in intensity due to different compositions and morphologies. The initial scans show peaks below 0.1 V indicating Li+ insertion into crystalline silicon to form amorphous Li–Si (a-LiSix) alloys,44,45 with further lithiation (at 30–60 mV) transforming these into crystalline phase c-Li15Si4.46 Peaks at 0.34 V and 0.53 V are linked to the reconversion of a-LiSix and c-Li15Si4 to amorphous silicon.47 Despite FeSi2 being present, it shows no distinct redox activity with Li.48 However, FeSiF6's CV curve (Fig. S6†) displays reversible peaks at 0.92 V and 1.4 V for oxidation, and 2.02 V and 1.62 V for reduction, similar to FeOF's Li+ storage reactions,49 suggesting these peaks represent Fe(III) ↔ Fe(II) intercalation and conversion processes, respectively.
During the initial lithiation, Li-ions intercalate into FeSiF6, causing a partial oxidation of Fe2+ to Fe3+ without altering the FeSiF6 structure. In subsequent lithiation, the accumulation of Li-ions continues to reduce Fe3+ back to Fe2+ until all Fe3+ is fully converted. The oxidation states of other elements remain stable within the tested voltage range, indicating no significant changes. The CV curve for the Fe–Si@F@C material shows a slight shift in the Fe oxidation–reduction voltage in FeSiF6, possibly due to energy absorption by other components, leading to a minor voltage increase.50,51 A comparison of the CV curves of the three materials suggests that a higher presence of the FeSi2 phase correlates with an elevated oxidation peak voltage. The similarity of curves between 1.0 V and 3.0 V in Fig. S5a and c,† along with XRD data, implies a relationship with FeSiF6 content. Additionally, with more charging and discharging cycles, the redox peaks' intensity grows, indicating enhanced electrolyte penetration and gradual activation of the anode material.52
The GCD curves for the three composite anodes at 0.1 A g−1 reveal distinct characteristics in Fig. S7a and b,† and 3a. An extended voltage plateau below 0.1 V during the first discharge (Fig. 3a and S7b†) signifies the alloying of lithium with silicon, forming LixSi alloys, while a plateau from 0.3 V to 0.6 V during charging indicates de-alloying.53 Comparing the GCD curves of the three composite materials reveals that all three exhibit a distinct lithiation reaction plateau around 1.5 V, which disappears in subsequent cycles. The initial discharge curves of Fe–Si@F@C and Fe–Si@C–T are more similar, likely due to the sufficient content of FeSiF6 in both materials. During discharge, the presence of FeSiF6 promotes the formation of a different SEI film, consuming some Li+ and increasing the Li storage capacity of the anode. Consequently, this reaction does not occur during subsequent charge–discharge cycles, leading to a capacity decrease and lower initial coulombic efficiency.54–56 Despite similar electrochemical behavior across Fe–Si@C–T, Fe–Si–T@C, and Fe–Si@F@C samples, their specific capacities differ. Fe–Si@C–T displays a first-cycle discharge/charge capacity of 1291.5/907.5 mA h g−1, achieving an initial coulombic efficiency (CE) of 70.2%, which improves to 92.7% in the second cycle. Fe–Si–T@C shows initial capacities of 1828.8/1414.8 mA h g−1 with an initial CE of 77.36%. Fe–Si@F@C records initial capacities of 1924.9/1497.7 mA h g−1, with the CE increasing to 94.6% in the second cycle and stabilizing at approximately 96.8% in later cycles. FeSiF6 alone has an initial CE of 49.68%, suggesting FeSiF6 and graphite significantly enhance the reversible lithium storage capacity of silicon-based anodes. Fe–Si@C–T demonstrates the most consistent rate performance, delivering capacities of 1187.5 to 534.4 mA h g−1 across current densities from 0.1 to 4 A g−1 (Fig. 3b). Fe–Si@F@C shows competitive rate performance at higher current densities, with capacities of 789 and 664.4 mA h g−1 at 2 and 4 A g−1, respectively. Fig. 3c showcases the cycling performance of the three anodes, with Fe–Si@C–T and Fe–Si@F@C demonstrating remarkable stability and minimal capacity loss after 200 cycles at 1 A g−1, maintaining discharge capacities of 685.7 and 974.9 mA h g−1, respectively, for a ∼94% capacity retention. By comparing the composition and electrochemical performance of the three composite materials, it is evident that the higher the FeSiF6 content, the more stable the electrochemical performance of the material. Both Fe–Si–T@C and Fe–Si@F@C composites exhibit a slight increase in capacity, primarily due to structural defects introduced during ball milling.57 The initial capacity decline in Fe–Si–T@C is attributed to the volumetric expansion of silicon. XRD analysis shows that the silicon and graphite content ratios are almost identical in both composites, with the difference being the varying amounts of FeSiF6 and FeSi2. It is apparent that FeSiF6 stabilizes the silicon anode's performance better than FeSi2.
 | ||
| Fig. 3 (a) GCD curves of Fe–Si@F@C in the 1st, 2nd and 5th cycles measured at 0.1 A g−1. (b) Rate performance of Fe–Si@C–T, Fe–Si–T@C, and Fe–Si@F@C at different current densities. (c) Cycling performance of Fe–Si@C–T, Fe–Si–T@C, and Fe–Si@F@C at 1 A g−1. (d) EIS spectra of Fe–Si@C–T, Fe–Si–T@C, and Fe–Si@F@C at a scan rate of 0.1 mV s−1. The inset shows the equivalent circuit model used for EIS fitting. (e) Linear fitting of the Z′–ω−1/2 (ω = 2πf) relationship for Fe–Si@C–T, Fe–Si–T@C, and Fe–Si@F@C. | ||
To more intuitively analyze the material's performance and the reasonableness of its capacity, we compiled the data into Table 2 for comparison. The purchased high-energy-density graphite has a relatively high specific capacity. According to Wei et al.'s report,58 the capacity of amorphous graphite can reach up to 810 mA h g−1 in the voltage range of 0–1.2 V. Based on the study conducted by Zhao et al.,59 which employed calculations to determine the theoretical capacity of materials, we can similarly use the data from the figure and the previously analyzed material composition to estimate the theoretical capacity of the composite material. The calculation formula is as follows:

| Electrode | Si | Graphite | FeSiF6 | Fe–Si@C–T | Fe–Si–T@C | Fe–Si@F@C |
|---|---|---|---|---|---|---|
| IDSC (mA h g−1) | 3854.2 | 1313.4 | 1330.0 | 1291.5 | 1828.9 | 1924.9 |
| ICSC (mA h g−1) | 2759.1 | 937.7 | 660.7 | 907.5 | 1414.9 | 1497.7 |
| SDSC (mA h g−1) | 2718.3 | 896.7 | 653.0 | 1258.4 | 1439.6 | 1474.6 |
| SCSC (mA h g−1) | 2565.3 | 827.3 | 602.5 | 1166.6 | 1521.2 | 1395.5 |
| TSC (mA h g−1) | — | — | — | 1544.9 | 1645.5 | 1752.7 |
| ICE (%) | 71.58 | 71.32 | 49.68 | 70.2 | 77.36 | 78.84 |
| C200 (mA h g−1) | 128.0 | 274.4 | 427.9 | 679.8 | 917.5 | 971.9 |
| RPID (%) | 23.75 | 135.5 | 93.6 | 96.45 | 104.17 | 104.25 |
Therefore, the theoretical capacity of the composite materials is:
| Fe–Si@C–T: 3854.2 × 12.2% + 1330 × 41.3% + 1313.4 × 40% = 1544.9 mA h g−1 |
| Fe–Si–T@C: 3854.2 × 21.2% + 1330 × 22.8% + 1313.4 × 40% = 1645.5 mA h g−1 |
| Fe–Si@F@C: 3854.2 × 21.8% + 1330 × 29.1% + 1313.4 × 40% = 1752.7 mA h g−1 |
From the calculation results, it can be seen that the test results of Fe–Si–T@C and Fe–Si@F@C are higher than the calculated values. This may be due to the modification effects of the ball-milled composite materials on the material interface, which significantly enhance the cycling stability of the materials. However, more Li+ is consumed during the SEI formation in the initial cycles.60 As the composite materials are more stable, the capacity retention rate of the composite materials also improves significantly, resulting in an increase in capacity in subsequent cycles compared to the initial material capacity.57
Furthermore, capacity calculations based on the material composition revealed that the initial cycle capacity is somewhat high. This is primarily due to two factors: first, the formation of the SEI film during the initial discharge consumes additional Li+ ions.60 Second, the structure of the material itself contributes to this phenomenon. Characterization of the material revealed the formation of a unique sandwich structure, with FeSiF6 containing some void structures. These structural features increase the material's defect density, allowing it to store more Li+.57 By comparing the initial capacities of the composite material Fe25Si75@C and the pristine Fe25Si75 material (Fig. S9†), it is evident that the initial capacity of Fe25Si75@C is only ∼40% of the pristine material, whereas Fe25Si75–T exhibits an initial capacity of 93.6% of the pristine material. This indicates that although graphite improves the capacity stability of the material, it sacrifices a certain amount of capacity, possibly due to incomplete graphite encapsulation, leading to the reaction of silicon with Li2PF6 to form Li2SiF6, consuming lithium ions.38 On the other hand, the addition of FeSiF6 results in a smaller loss of capacity. The composite of the two materials achieves an improvement beyond the sum of their individual effects. Furthermore, from the analysis above, it can be inferred that the presence of FeSiF6 can to some extent transform Li2PF6, rendering it a reversible process, thereby reducing Li+ consumption and increasing specific capacity.
EIS analysis of the three iron–silicon composite anodes before cycling provided insights into their electrochemical reaction processes, using equivalent circuit models to interpret Nyquist plots (Fig. 3d). The high-frequency semi-circle represents the SEI film resistance (Rf), and the medium-frequency one indicates charge-transfer impedance (Rct), with a low-frequency straight line denoting Li+ diffusion within the material (Warburg impedance, Zw). In the model, RΩ is the cell's internal resistance, and CPE is the constant phase element. Fe–Si–T@C showed the lowest Rct at approximately 40 Ω, suggesting excellent stability. Fe–Si@F@C and Fe–Si@C–T presented Rct values of about 88 Ω and 245 Ω, respectively. Compared to the base material Fe25Si75 (Fig. S10†), which has an Rct of 462 Ω, surface modifications like graphite coating improved conductivity, reducing Rct to 273 Ω. With FeSiF6, Rct decreased further to 164 Ω, indicating FeSiF6's significant role in enhancing charge transfer kinetics, especially when combined with graphite. Analyzing the slope of the low-frequency Z′–ω−1/2 curve (Fig. 3e) showed the Fe–Si@F@C anode with the lowest slope (306.3), implying a higher Li+ diffusion coefficient than the others.
Differential capacity (dQ/dV) curves after various cycles at 1 A g−1 for the composite anodes (Fig. S11a–c†) highlight initial lithiation peaks for c-Li15Si4 (∼0.45–0.51 V) and a-LixSi (∼0.27–0.34 V), with shifts attributed to restricted Li-ion diffusion61 and polarization effects under high current density.62 As cycling progresses, the anodic peaks for Fe–Si@C–T and Fe–Si@F@C shift towards lower voltages, suggesting enhanced lithium insertion/extraction kinetics and reduced polarization.63 In contrast, Fe–Si–T@C shows variable peak shifts, indicating a lower initial polarization that increases over cycles but improves versus the original material, possibly due to FeSiF6's effect on polarization. Notably, the sharp peaks at 0.15 V for Fe–Si–T@C and Fe–Si@F@C, versus a broader peak for Fe–Si@C–T, reflect different carbon structures from varied synthesis methods, linked to lithium deintercalation from lithiated graphite. Increased FeSiF6 content in composite anodes correlates with higher anodic peak values after 100 cycles, suggesting FeSiF6's role in enhancing cycling stability. Higher FeSiF6 content in silicon-based composite anodes correlates with increased anodic peak values after 100 cycles, suggesting FeSiF6's role in enhancing cycling stability. During delithiation, two cathodic peaks for amorphous silicon phases, L1(a-Si) and L2(a-Si), are noted, with L2(a-Si) at 0.25 V becoming more pronounced and stable in capacity as FeSiF6 content increases. Over cycles, both cathodic peaks diminish, with L1(a-Si)'s decrease highlighting the challenge of lithiating amorphous silicon without affecting crystalline silicon, as per Obrovac's findings.64 In contrast, L2(a-Si) provides a transition region, stabilizing the cyclic performance of the material.
To elucidate FeSiF6's impact on composite material performance, dQ/dV plots for the initial and fifth cycles at 0.1 A g−1 were analyzed, highlighting changes in c-Li15Si4 peak (at ∼0.45 V) intensities. Initially, Fe25Si75 showed a c-Li15Si4 peak intensity of ∼10.71 mA h g−1 V−1 (Fig. 4a), which reduced to ∼3.04 mA h g−1 V−1 after five cycles (Fig. 4b). In contrast, Fe–Si–T@C (Fig. S12a†), Fe–Si@C–T (Fig. S12b†), and Fe–Si@F@C (Fig. 4c) exhibited peak intensities of ∼7.09 mA h g−1 V−1, ∼1.84 mA h g−1 V−1, and ∼4.50 mA h g−1 V−1, respectively, indicating more stable lithium storage with higher FeSiF6 content. This indicates that a higher FeSiF6 content better inhibits the formation of c-Li15Si4. This inhibition can be attributed to two main factors: first, the addition of FeSiF6 promotes the formation of a stable SEI film, which reduces the consumption of lithium ions through side reactions, thereby stabilizing the interface structure of the silicon anode and inhibiting the formation of c-Li15Si4;18,65 second, the inclusion of FeSiF6 can alter the lithium ion insertion pathways and diffusion rates, encouraging the formation of an amorphous phase and thus having a suppressive effect.66–69 Additionally, SEI film modifications were noted in Fig. 4c, especially the emergence of a broad peak around 1.5 V linked to fluorine incorporation from FeSiF6,18 enhancing SEI stability. To verify the reliability of the experimental results, the first cycle (Fig. 4e) and the second cycle (Fig. 4f) CV curves of the five materials were plotted at a scan rate of 0.1 mV s−1, along with the CV curve of FeSiF6 alone (Fig. S13†). The comparison reveals that after compounding the materials, the peak of Li15Si4 at around 0.5 V is smaller than that of the original Fe25Si75 material, indicating that the reactivity of the composite material at this potential has decreased.70 By comparing the CV curves of different electrodes in the first cycle, it can be seen that the range of 0.15–0.75 V corresponds to the SEI formation process for the Fe–Si alloy anode material.71,72 For the CV curve of the electrode with added FeSiF6, a new broad peak appears between 1.33 V and 1.70 V. The position of this peak shows a certain deviation from the SEI formation position in the CV curve of pure FeSiF6, indicating that this peak results from the combined effects of the composite material. Furthermore, this peak disappears in the second cycle, which may indicate that the composite material forms a relatively stable SEI film during the initial cycle and creates an effective passivation layer on the surface.73 By comparing with the literature, this phenomenon may be due to the decomposition of FeSiF6, where some substances (presumably F−) react with the electrolyte, resulting in the formation of a more stable and robust SEI film.18 The consistent performance and the presence of F-ions, as analyzed in CV curves and supported by the literature, imply that FeSiF6 significantly contributes to SEI film stability.
 | ||
| Fig. 4 Differential capacity curves of Fe25Si75 at (a) the 1st cycle and (b) 5th cycle at 0.1 A g−1. The differential capacity curves for Fe–Si@F@C anodes at (c) the 1st cycle and (d) 5th cycles at 0.1 A g−1. CV profiles of (e) the 1st cycle and (f) 2nd cycle for different electrodes at a scan rate of 0.1 mV s−1. | ||
CV analysis of Fe25Si75, FeSiF6, and Fe–Si@F@C electrodes after multiple cycles at 1 A g−1 (Fig. 5a–c) revealed distinct behaviors. Fe25Si75 shows reduced peak intensities for a-LixSi and c-Li15Si4 transitions (∼0.3–0.5 V) with cycling (Fig. 5a), suggesting a decrease in reaction activity. In contrast, FeSiF6 (Fig. S13†) exhibited increased peak intensities across cycles, indicating stable specific capacity due to the reversible Li+ insertion/extraction, enhanced by electrolyte permeation facilitating Li+ migration.74 Fe–Si@F@C (Fig. 5b) displayed stabilized peaks related to a-LixSi and c-Li15Si4 transitions after ∼50 cycles, suggesting different protective mechanisms. Graphite improves mechanical stability and mitigates structural stress from volume expansion, while FeSiF6's protective effect, akin to FeSi2's role, warrants further investigation to elucidate its specific contributions to silicon-based anodes.
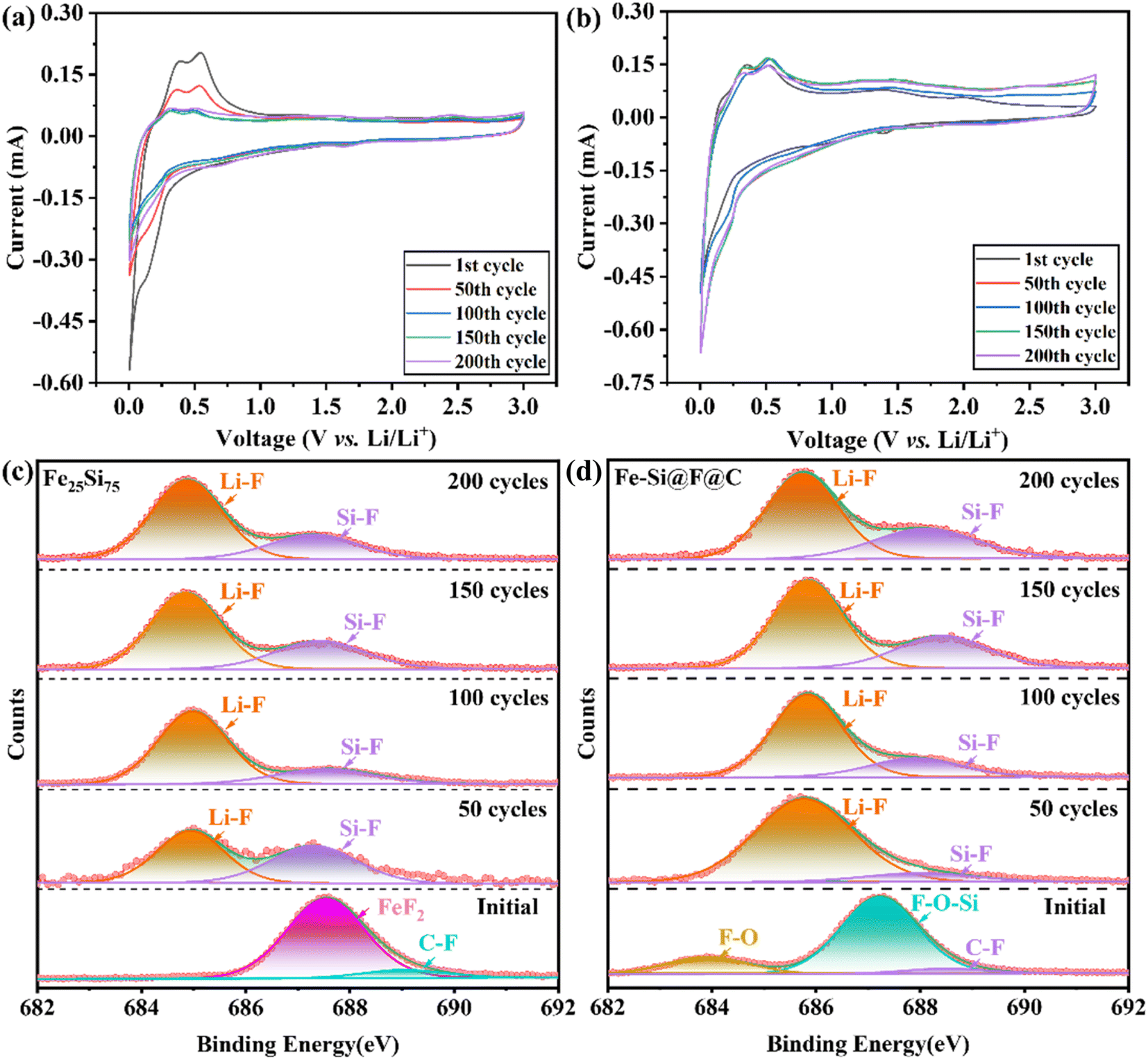 | ||
| Fig. 5 Cyclic voltammetry profiles of (a) Fe25Si75 and (b) Fe–Si@F@C composite anode in the first five cycles at a sweep rate of 0.5 mV s−1. High-resolution XPS spectra of F 1s of (c) Fe25Si75 and (d) Fe–Si@F@C under different cycle times. | ||
The test results for the F element are shown in Fig. 5c and d below. It is evident from the graph that at 50 cycles, the Li–F (685 eV) peak intensity of Fe25Si75 is significantly lower,75 and the SEI film contains a large amount of Si–F (687.4 eV).76 In contrast, the composite material exhibits higher Li–F content with less Si–F. This further confirms that the presence of FeSiF6 provides a substantial amount of F-containing bonds on the material's surface. During the formation of the SEI film, the Si–O–F and F–O bonds on the surface break, allowing more F ions to combine with Li and thereby generating more Li–F bonds within the SEI film. As the number of cycles increases, the binding energy of the F element in both cases becomes nearly identical. Moreover, both materials exhibit good stability in terms of capacity with increasing cycle numbers. This indicates a preference for forming LiF-rich SEI films on the anode surface, which enhances Li+ transport and minimizes side reactions and electrolyte consumption. The presence of LiF correlates with decreased impedance, highlighting its role in mitigating volume expansion and stabilizing electrode structures during cycling.77 Nevertheless, Fe25Si75 exhibited a rapid decline in capacity during the first few cycles, likely due to its inability to fully counteract the micron-scale volumetric expansion of the silicon anode.78 Conversely, the Fe–Si@F@C, with FeSiF6, not only strengthens the SEI film but also mitigates volume expansion more effectively, leading to superior cycling stability.79 This indicates the promoting effect of FeSiF6 on the generation of LiF within the SEI film, thereby enhancing the electrochemical stability of the material.
XPS analysis of silicon-based anodes modified with FeSiF6 revealed changes in the SEI film composition during cycling (Fig. S14a–c†). Initial C 1s spectra for all samples showed peaks typical of organic solvent molecules, including C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–H, and C–C, mainly from graphite, carbon black, and PVDF binder, notably including C–F bonds. An increase in C–C/C–H bonds at 284.8 eV was observed across spectra, indicating electrolyte decomposition and SEI formation.80 After 50 cycles, new peaks at 288.5 eV for ROCO2Li and 289.6 eV for Li2CO3 appeared, representing major SEI components.18 Fe25Si75 showed increased ROCO2Li and Li2CO3 (Fig. 5e), suggesting a thicker SEI film compared to the thinner films on the other materials, which correlated with their stable performance. After 200 cycles, the ROCO2Li and Li2CO3 intensities decreased in Fe25Si75, remained stable in FeSiF6, and increased in Fe–Si@F@C, likely due to SEI film thickening from cycling in Fe–Si@F@C. In contrast, Fe25Si75's fluctuating peak intensities could be due to SEI film rupture and regeneration caused by volume expansion, whereas FeSiF6 showed stability. The incorporation of FeSiF6 into the Fe–Si alloy is inferred to strengthen the SEI film's toughness,81 with its thickness accumulating over cycles due to silicon's volume expansion. Fig. S15† presents the Si 1s spectra for the three electrode materials with varying cycle numbers, initially showing peaks for Si–Si (99.4 eV), Fe–Si (99.8 eV), and Si–C (102 eV) bonds.82 Notably, the composite electrode exhibits both Si–Si and Fe–Si bonds. Post-cycling, these peaks shift to SiOx (∼101 eV) and LixSiOy (102 eV), indicating Si–O bond involvement in reactions with the SEI film.83,84 Fe25Si75 shows Si–Si/Fe–Si bond peaks at 150 and 200 cycles, contrasting with the more stable peak intensities of FeSiF6 and Fe–Si@F@C. This stability discrepancy is attributed to Fe25Si75's SEI film rupture from volume expansion, exposing internal materials, in line with existing research. Fig. S16† showcases the Li 1s spectra of the three materials at various cycles, revealing similar peak positions for them all, suggesting comparable Li-containing components in their SEI films, including Li2O (54.3 eV), ROCO2Li and LiCO3 (54.8 eV), and LiF (55.7 eV). It also demonstrated the formation of an SEI film rich in LiF on the anode material surface, corroborated by the analysis of the F element.
O, C–H, and C–C, mainly from graphite, carbon black, and PVDF binder, notably including C–F bonds. An increase in C–C/C–H bonds at 284.8 eV was observed across spectra, indicating electrolyte decomposition and SEI formation.80 After 50 cycles, new peaks at 288.5 eV for ROCO2Li and 289.6 eV for Li2CO3 appeared, representing major SEI components.18 Fe25Si75 showed increased ROCO2Li and Li2CO3 (Fig. 5e), suggesting a thicker SEI film compared to the thinner films on the other materials, which correlated with their stable performance. After 200 cycles, the ROCO2Li and Li2CO3 intensities decreased in Fe25Si75, remained stable in FeSiF6, and increased in Fe–Si@F@C, likely due to SEI film thickening from cycling in Fe–Si@F@C. In contrast, Fe25Si75's fluctuating peak intensities could be due to SEI film rupture and regeneration caused by volume expansion, whereas FeSiF6 showed stability. The incorporation of FeSiF6 into the Fe–Si alloy is inferred to strengthen the SEI film's toughness,81 with its thickness accumulating over cycles due to silicon's volume expansion. Fig. S15† presents the Si 1s spectra for the three electrode materials with varying cycle numbers, initially showing peaks for Si–Si (99.4 eV), Fe–Si (99.8 eV), and Si–C (102 eV) bonds.82 Notably, the composite electrode exhibits both Si–Si and Fe–Si bonds. Post-cycling, these peaks shift to SiOx (∼101 eV) and LixSiOy (102 eV), indicating Si–O bond involvement in reactions with the SEI film.83,84 Fe25Si75 shows Si–Si/Fe–Si bond peaks at 150 and 200 cycles, contrasting with the more stable peak intensities of FeSiF6 and Fe–Si@F@C. This stability discrepancy is attributed to Fe25Si75's SEI film rupture from volume expansion, exposing internal materials, in line with existing research. Fig. S16† showcases the Li 1s spectra of the three materials at various cycles, revealing similar peak positions for them all, suggesting comparable Li-containing components in their SEI films, including Li2O (54.3 eV), ROCO2Li and LiCO3 (54.8 eV), and LiF (55.7 eV). It also demonstrated the formation of an SEI film rich in LiF on the anode material surface, corroborated by the analysis of the F element.
The cycling stability and volume expansion mitigation of FeSiF6 in silicon-based anodes were evaluated through cross-sectional morphology characterization before and after cycling (Fig. 6). The Fe25Si75 electrode expanded significantly from 6.15 µm to 21.89 µm, a ∼256% increase, after 200 cycles, exhibiting electrode fracture and detachment (Fig. 6a). In contrast, the FeSiF6 electrode showed a minimal volume expansion of ∼34%, correlating with stable cycling performance (Fig. 6b). The Fe–Si@F@C composite electrode, incorporating FeSiF6 and graphite, demonstrated controlled volume expansion of ∼84% after 200 cycles (Fig. 6c), indicating that FeSiF6 effectively reduces volume expansion in silicon alloy anodes.
 | ||
| Fig. 6 Cross-sectional morphologies of the electrodes: (a) Fe25Si75, (b) FeSiF6, and (c) Fe–Si@F@C after different number of cycles (initial state, 100 cycles and 200 cycles). | ||
EIS measurements on Fe25Si75 and Fe–Si@F@C electrodes after various cycling stages (Fig. 7) showed a rapid reduction in charge transfer resistance (Rct) for Fe25Si75, dropping from an initial 462 Ω to 93 Ω within fifty cycles and stabilizing at about 50 Ω after 100 cycles. This change is attributed to LiF accumulation in the SEI film, as identified by XPS, which improves Li+ migration and lowers impedance. Similarly, the Fe–Si@F@C electrode, benefiting from FeSiF6 incorporation, shows a marked initial impedance decrease, with impedance fluctuating based on LiF content in the SEI film before stabilizing at approximately 39 Ω after 150 cycles. The integration of FeSiF6 and graphite into silicon-based electrodes significantly improves interface contact stability and electronic transport to the copper foil. Our research identifies mechanisms for enhanced cycling stability of these electrodes (Fig. 7c),18,28,74,81 focusing on three key findings: (1) dQ/dV and SEM analyses show FeSiF6's role in stabilizing the c-Li15Si4 alloy and mitigating volume expansion; (2) CV and dQ/dV investigations, supported by XPS, reveal that FeSiF6 leads to a thinner, more stable, and flexible SEI film in early cycles, enhancing electrode–electrolyte interaction; and (3) the analysis suggests that FeSiF6 may inhibit the reaction between the Si material and electrolyte to form Li2SiF6, thereby reducing Li+ consumption and volume changes. Comparing our results with the existing literature (Table S1 and Fig. S17†), our composite anodes demonstrate competitive capacities and stabilities, highlighting the effectiveness of adding FeSiF6 and graphite in improving silicon-based anodes' performance.
 | ||
| Fig. 7 EIS spectra of (a) Fe25Si75 and (b) Fe–Si@F@C with a scan rate of 0.1 mV s−1 under different cycle times. (c) Schematic diagram of the performance improvement mechanism in the Fe–Si@F@C anode. | ||
4. Conclusions
This research presents a novel anode material modifier, FeSiF6, through a straightforward chemical reaction, exploring its enhancement effects and lithium storage mechanisms on silicon-based composite anode materials. Three composites were synthesized, displaying stable performance, with Fe–Si–T@C achieving a high initial discharge capacity of 3250.8 mA h g−1 at 0.1 A g−1 and an initial CE of 86.88%. Despite its lower capacity retention of 52% after 200 cycles at 1 A g−1, the other two materials maintained over 94% retention. All composites showed robust rate performance, with Fe–Si@C–T sustaining a capacity of 534.4 mA h g−1 even at 4 A g−1. The analysis identified FeSiF6's key roles: (1) suppression of Li15Si4 formation, offering a transitional L2(a-Si) area to stabilize cycling performance; (2) formation of a LiF-rich SEI film, mitigating volume expansion and enhancing Li+ transport through LiF inclusion; and (3) potential reduction in irreversible Li2SiF6 formation, improving capacity and stability in graphite–silicon composites. This study introduces a viable, environmentally friendly silicon-based anode composite material, highlighting its unique lithium storage mechanism and potential for advancing silicon anode commercialization. Further investigation into this material could significantly benefit the development of next-generation high-performance LIBs.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Jichang Sun: writing – original draft, data curation, investigation, and visualization. Xiaoyi Liu: validation, data curation, and investigation. Penglun Zheng: conceptualization and writing – review & editing. Yang Zhao: writing – review & editing. Yun Zheng: funding acquisition, data curation, visualization, and writing – review & editing. Jingchao Chai: data curation and visualization. Zhihong Liu: funding acquisition and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate the funding support of the Excellent Discipline Cultivation Project by JHUN (No. 2023XKZ008), State Key Laboratory of Precision Blasting, Jianghan University (No. PBSKL2022102), and the Open Project of Civil Aircraft Fire Science and Safety Engineering Key Laboratory of Sichuan Province (No. MZ2023KF03).References
- J. R. Szczech and S. Jin, Energy Environ. Sci., 2011, 4, 56–72 RSC.
- P. Guan, W. Zhang, C. Li, N. Han, X. Wang, Q. Li, G. Song, Z. Peng, J. Li, L. Zhang and X. Zhu, J. Colloid Interface Sci., 2020, 575, 150–157 CrossRef CAS PubMed.
- A. Mukhopadhyay and B. W. Sheldon, Prog. Mater. Sci., 2014, 63, 58–116 CrossRef CAS.
- X. Su, Q. Wu, J. Li, X. Xiao, A. Lott, W. Lu, B. W. Sheldon and J. Wu, Adv. Energy Mater., 2014, 4, 1300882 CrossRef.
- N. Kim, Y. Kim, J. Sung and J. Cho, Nat. Energy, 2023, 8, 921–933 CrossRef CAS.
- W. Luo, X. Chen, Y. Xia, M. Chen, L. Wang, Q. Wang, W. Li and J. Yang, Adv. Energy Mater., 2017, 7, 1701083 CrossRef.
- G. G. Eshetu and E. Figgemeier, ChemSusChem, 2019, 12, 2515–2539 CrossRef CAS PubMed.
- L. Deng, Y. Zheng, X. Zheng, T. Or, Q. Ma, L. Qian, Y. Deng, A. Yu, J. Li and Z. Chen, Adv. Energy Mater., 2022, 12, 2200850 CrossRef CAS.
- J. Sun, C. Liu, P. Zheng, A. Chaturvedi, K.-H. Nam, Z. Li, Q. Liang, S. Huang, D. Fang, J. Chai, Q. Liu, Z. Liu, E. H. T. Teo, Z. Jian, W. Shu, Y. Zheng and C.-M. Park, Next Mater., 2023, 1, 100053 CrossRef.
- P. Zheng, J. Sun, H. Liu, R. Wang, C. Liu, Y. Zhao, J. Li, Y. Zheng and X. Rui, Batteries Supercaps, 2023, 6, e202200481 CrossRef CAS.
- D. S. M. Iaboni and M. N. Obrovac, J. Electrochem. Soc., 2016, 163, A255 CrossRef CAS.
- M. A. Rahman, G. Song, A. I. Bhatt, Y. C. Wong and C. Wen, Adv. Funct. Mater., 2016, 26, 647–678 CrossRef CAS.
- M. R. Zamfir, H. T. Nguyen, E. Moyen, Y. H. Lee and D. Pribat, J. Mater. Chem. A, 2013, 1, 9566–9586 RSC.
- X. H. Liu, L. Zhong, S. Huang, S. X. Mao, T. Zhu and J. Y. Huang, ACS Nano, 2012, 6, 1522–1531 CrossRef CAS PubMed.
- P. Li, H. Kim, S.-T. Myung and Y.-K. Sun, Energy Storage Mater., 2021, 35, 550–576 CrossRef.
- Y. Sun, N. Liu and Y. Cui, Nat. Energy, 2016, 1, 16071 CrossRef CAS.
- W. Luo, X. Chen, Y. Xia, M. Chen, L. Wang, Q. Wang, W. Li and J. Yang, Adv. Energy Mater., 2017, 7, 1701083 CrossRef.
- C. Xu, F. Lindgren, B. Philippe, M. Gorgoi, F. Björefors, K. Edström and T. Gustafsson, Chem. Mater., 2015, 27, 2591–2599 CrossRef CAS.
- H.-J. Shin, J.-Y. Hwang, H. J. Kwon, W.-J. Kwak, S.-O. Kim, H.-S. Kim and H.-G. Jung, ACS Sustainable Chem. Eng., 2020, 8, 14150–14158 CrossRef CAS.
- Y. Zhao, L. Zhang, J. Liu, K. Adair, F. Zhao, Y. Sun, T. Wu, X. Bi, K. Amine, J. Lu and X. Sun, Chem. Soc. Rev., 2021, 50, 3889–3956 RSC.
- J. Shi, X. Jiang, J. Sun, B. Ban, J. Li and J. Chen, J. Colloid Interface Sci., 2021, 588, 737–748 CrossRef CAS PubMed.
- Y. Jin, N.-J. H. Kneusels, L. E. Marbella, E. Castillo-Martínez, P. C. M. M. Magusin, R. S. Weatherup, E. Jónsson, T. Liu, S. Paul and C. P. Grey, J. Am. Chem. Soc., 2018, 140, 9854–9867 CrossRef CAS PubMed.
- S. Yamazaki, R. Tatara, H. Mizuta, K. Kawano, S. Yasuno and S. Komaba, J. Phys. Chem. C, 2023, 127, 14030–14040 CrossRef CAS.
- K. Kim, I. Park, S.-Y. Ha, Y. Kim, M.-H. Woo, M.-H. Jeong, W. C. Shin, M. Ue, S. Y. Hong and N.-S. Choi, Electrochim. Acta, 2017, 225, 358–368 CrossRef CAS.
- A. Schiele, B. Breitung, T. Hatsukade, B. B. Berkes, P. Hartmann, J. Janek and T. Brezesinski, ACS Energy Lett., 2017, 2, 2228–2233 CrossRef CAS.
- F. Dou, L. Shi, G. Chen and D. Zhang, Electrochem. Energy Rev., 2019, 2, 149–198 CrossRef CAS.
- Q. Liu, Y. Hu, X. Yu, Y. Qin, T. Meng and X. Hu, Nano Res. Energy, 2022, 1, e9120037 CrossRef.
- A. Casimir, H. Zhang, O. Ogoke, J. C. Amine, J. Lu and G. Wu, Nano Energy, 2016, 27, 359–376 CrossRef CAS.
- J. D. McBrayer, M.-T. F. Rodrigues, M. C. Schulze, D. P. Abraham, C. A. Apblett, I. Bloom, G. M. Carroll, A. M. Colclasure, C. Fang and K. L. Harrison, Nat. Energy, 2021, 6, 866–872 CrossRef CAS.
- H. Jo, J. Kim, D.-T. Nguyen, K. K. Kang, D.-M. Jeon, A. R. Yang and S.-W. Song, J. Phys. Chem. C, 2016, 120, 22466–22475 CrossRef CAS.
- S. Fan, H. Wang, J. Qian, Y. Cao, H. Yang, X. Ai and F. Zhong, ACS Appl. Mater. Interfaces, 2020, 12, 16411–16416 CrossRef CAS PubMed.
- Y. M. Yang, C. Loka, D. P. Kim, S. Y. Joo, S. W. Moon, Y. S. Choi, J. H. Park and K.-S. Lee, Met. Mater. Int., 2017, 23, 610–617 CrossRef CAS.
- Z. Wen, J. Yang, B. Wang, K. Wang and Y. Liu, Electrochem. Commun., 2003, 5, 165–168 CrossRef CAS.
- F. Dou, L. Shi, G. Chen and D. Zhang, Electrochem. Energy Rev., 2019, 2, 149–198 CrossRef CAS.
- X. Li, K. Li, M. Yuan, J. Zhang, H. Liu, A. Li, X. Chen and H. Song, J. Colloid Interface Sci., 2024, 667, 470–477 CrossRef CAS PubMed.
- Y. Liu, Z. Wen, X. Wang, A. Hirano, N. Imanishi and Y. Takeda, J. Power Sources, 2009, 189, 733–737 CrossRef CAS.
- M. Han, D. Zheng, P. Song and Y. Ding, Chem. Phys. Lett., 2021, 771, 138538 CrossRef CAS.
- C. Yu, X. Chen, Z. Xiao, C. Lei, C. Zhang, X. Lin, B. Shen, R. Zhang and F. Wei, Nano Lett., 2019, 19, 5124–5132 CrossRef CAS PubMed.
- A. Pastushenko and V. Lysenko, RSC Adv., 2016, 6, 8093–8095 RSC.
- Y. Zhang, Q. Zhang, X. He, L. Wang, J. Wang, L. Dong, Y. Xie and Y. Hao, Materials, 2023, 16, 1437 CrossRef CAS PubMed.
- J. Wu, Y. Cao, H. Zhao, J. Mao and Z. Guo, Carbon Energy, 2019, 1, 57–76 CrossRef CAS.
- M. Ruttert, V. Siozios, M. Winter and T. Placke, ACS Appl. Energy Mater., 2019, 3, 743–758 CrossRef.
- S. Chen, Z. Song, L. Wang, H. Chen, S. Zhang, F. Pan and L. Yang, Acc. Chem. Res., 2022, 55, 2088–2102 CrossRef CAS PubMed.
- E. Radvanyi, E. De Vito, W. Porcher, J. Danet, P. Desbois, J.-F. Colin and S. J. S. Larbi, J. Mater. Chem. A, 2013, 1, 4956–4965 RSC.
- H. Lin, K. Uosaki and H. Noguchi, Appl. Surf. Sci., 2021, 569, 151040 CrossRef CAS.
- H. Gao, L. Xiao, I. Plümel, G.-L. Xu, Y. Ren, X. Zuo, Y. Liu, C. Schulz, H. Wiggers, K. Amine and Z. Chen, Nano Lett., 2017, 17, 1512–1519 CrossRef CAS PubMed.
- M. N. Obrovac and L. J. Krause, J. Electrochem. Soc., 2007, 154, A103 CrossRef CAS.
- H.-T. Kwon, A.-R. Park, S.-S. Lee, H. Cho, H. Jung and C.-M. Park, J. Electrochem. Soc., 2019, 166, A2221 CrossRef CAS.
- V. L. Chevrier, G. Hautier, S. P. Ong, R. E. Doe and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 094118 CrossRef.
- Y. Su, J. Chen, H. Li, H. Sun, T. Yang, Q. Liu, S. Ichikawa, X. Zhang, D. Zhu and J. Zhao, Adv. Sci., 2022, 9, 2201419 CrossRef CAS PubMed.
- Y. Su, S. Liu, D. Zhu, Y. Luo, X. Zhang, J. Yan, J. Chen, L. Geng, B. Guo and H. Li, Energy Storage Mater., 2023, 59, 102779 CrossRef.
- H.-J. Zhang, Q.-C. Jia and L.-B. Kong, ACS Appl. Nano Mater., 2021, 4, 12514–12526 CrossRef CAS.
- B. Gattu, A Fundamental Study of Nanostructured Si Anodes for Lithium Ion Batteries, University of Pittsburgh, 2020 Search PubMed.
- R. Shao, J. Niu, F. Zhu, M. Dou, Z. Zhang and F. Wang, Nano Energy, 2019, 63, 103824 CrossRef CAS.
- S. Sekar, Y. Lee, D. Y. Kim and S. Lee, Nanomaterials, 2019, 9, 871 CrossRef CAS PubMed.
- Y. Zhang, L. Yue, H. Ding, Z. Xiong, L. Bai, M. Xu and Y. Qi, Nano Energy, 2024, 127, 109696 CrossRef CAS.
- S. Dong, Y. Song, M. Su, G. Wang, Y. Gao, K. Zhu and D. Cao, Chem. Eng. J., 2024, 481, 147988 CrossRef CAS.
- V. Subramanian, T. Karabacak, C. Masarapu, R. Teki, T.-M. Lu and B. Wei, J. Power Sources, 2010, 195, 2044–2049 CrossRef CAS.
- Y. Zhao, J. Wang, Q. He, J. Shi, Z. Zhang, X. Men, D. Yan and H. Wang, ACS Nano, 2019, 13, 5602–5610 CrossRef CAS PubMed.
- H. Kim, W. Choi, J. Yoon, J. H. Um, W. Lee, J. Kim, J. Cabana and W.-S. Yoon, Chem. Rev., 2020, 120, 6934–6976 CrossRef CAS PubMed.
- X. Gao, W. Lu and J. Xu, ACS Appl. Mater. Interfaces, 2021, 13, 21362–21370 CrossRef CAS PubMed.
- J. Tao, L. Liu, J. Han, J. Peng, Y. Chen, Y. Yang, H.-r. Yao, J. Li, Z. Huang and Y. Lin, Energy Storage Mater., 2023, 60, 102809 CrossRef.
- D. Wang, M. Gao, H. Pan, J. Wang and Y. Liu, J. Power Sources, 2014, 256, 190–199 CrossRef CAS.
- M. Obrovac and L. Krause, J. Electrochem. Soc., 2006, 154, A103 CrossRef.
- P. Bärmann, B. Krueger, S. Casino, M. Winter, T. Placke and G. Wittstock, ACS Appl. Mater. Interfaces, 2020, 12, 55903–55912 CrossRef PubMed.
- Y. Liu, W. Sun, X. Lan, R. Hu, J. Cui, J. Liu, J. Liu, Y. Zhang and M. Zhu, ACS Appl. Mater. Interfaces, 2019, 11, 38727–38736 CrossRef CAS PubMed.
- L. Gu, J. Han, M. Chen, W. Zhou, X. Wang, M. Xu, H. Lin, H. Liu, H. Chen and J. Chen, Energy Storage Mater., 2022, 52, 547–561 CrossRef.
- C. Gan, C. Zhang, W. Wen, Y. Liu, J. Chen, Q. Xie and X. Luo, ACS Appl. Mater. Interfaces, 2019, 11, 35809–35819 CrossRef CAS PubMed.
- J. Sung, N. Kim, J. Ma, J. H. Lee, S. H. Joo, T. Lee, S. Chae, M. Yoon, Y. Lee and J. Hwang, Nat. Energy, 2021, 6, 1164–1175 CrossRef.
- G. Li, G. Fan, X. Zhang, J. Han, Y. Wang, Y. Liu, L. Jia, B. Guo, C. Zhu and M. He, eTransportation, 2024, 20, 100315 CrossRef.
- M. Gu, Z. Wang, J. G. Connell, D. E. Perea, L. J. Lauhon, F. Gao and C. Wang, ACS Nano, 2013, 7, 6303–6309 CrossRef CAS PubMed.
- Q. Huang, K. Turcheniuk, X. Ren, A. Magasinski, D. Gordon, N. Bensalah and G. Yushin, Adv. Energy Mater., 2019, 9, 1803323 CrossRef.
- J. Zhu, M. S. D. Darma, M. Knapp, D. R. Sørensen, M. Heere, Q. Fang, X. Wang, H. Dai, L. Mereacre and A. Senyshyn, J. Power Sources, 2020, 448, 227575 CrossRef CAS.
- Y. She, X. Li, Y. Zheng, D. Chen, X. Rui, X. Lin and Y. Qin, Energy Environ. Mater., 2023, e12538 Search PubMed.
- M. Nie, J. Demeaux, B. T. Young, D. R. Heskett, Y. Chen, A. Bose, J. C. Woicik and B. L. Lucht, J. Electrochem. Soc., 2015, 162, A7008 CrossRef CAS.
- E. Markevich, G. Salitra, M. Afri, J. Kulisch, P. Hartmann and D. Aurbach, J. Electrochem. Soc., 2019, 166, A1685 CrossRef CAS.
- L.-B. Huang, G. Li, Z.-Y. Lu, J.-Y. Li, L. Zhao, Y. Zhang, X.-D. Zhang, K.-C. Jiang, Q. Xu and Y.-G. Guo, ACS Appl. Mater. Interfaces, 2021, 13, 24916–24924 CrossRef CAS PubMed.
- N. M. Johnson, Z. Yang, Q. Liu and Z. Zhang, J. Electrochem. Soc., 2022, 169, 040527 CrossRef CAS.
- J. Lin, L. Wang, Q. Xie, Q. Luo, D. L. Peng, C. Buddie Mullins and A. Heller, Angew. Chem., Int. Ed., 2023, 135, e202216557 CrossRef.
- M. Yu, E. Temeche, S. Indris, W. Lai and R. M. Laine, Green Chem., 2022, 24, 4061–4070 RSC.
- K. Schroder, J. Alvarado, T. A. Yersak, J. Li, N. Dudney, L. J. Webb, Y. S. Meng and K. J. Stevenson, Chem. Mater., 2015, 27, 5531–5542 CrossRef CAS.
- Q. Ma, Y. Zhao, Z. Hu, J. Qu, Z. Zhao, H. Xie, P. Xing, D. Wang and H. Yin, Mater. Today Energy, 2021, 21, 100817 CrossRef CAS.
- C. Li, Z. Liang, Z. Li, D. Cao, D. Zuo, J. Chang, J. Wang, Y. Deng, K. Liu and X. Kong, Nano Lett., 2023, 23, 4014–4022 CrossRef CAS PubMed.
- M. Nie, D. P. Abraham, Y. Chen, A. Bose and B. L. Lucht, J. Phys. Chem. C, 2013, 117, 13403–13412 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta02532f |
| This journal is © The Royal Society of Chemistry 2024 |