
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
PPX/PXP-type ligands (X = O and S) and their transition metal complexes: synthesis, properties and applications
Franziska
Flecken
 and
Schirin
Hanf
*
and
Schirin
Hanf
*
Institute for Inorganic Chemistry, Karlsruhe Institute of Technology, Engesserstr. 15, 76131 Karlsruhe, Germany. E-mail: schirin.hanf@kit.edu
First published on 1st October 2024
Abstract
Short-bite diphosphines of the form R2P–X–PR2 (PXP; X = O, S; R = aryl, alkyl), incorporating an oxygen or sulphur atom as bridging unit X, are widely underexplored compared to their N- and C-containing PNP- and PCP-type counterparts. However, these PXP ligands undergo an interesting phosphorotropic equilibrium with the PPX (R2P(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X)–PR2) tautomer, which opens up a very versatile coordination chemistry. This article covers the impact of the ligand backbone in short-bite ligands on their coordination chemistry, reactivity and applications. Especially in PXP-type complexes, metallophillic interactions can be induced in the case of coinage metals, which lead to fascinating photo-optical properties. Furthermore, PPX/PXP-type complexes are believed to exhibit a promising behavior in catalysis, due to the potential hemilability of the ligand and the therewith involved availability of free active sites for substrate binding.
X)–PR2) tautomer, which opens up a very versatile coordination chemistry. This article covers the impact of the ligand backbone in short-bite ligands on their coordination chemistry, reactivity and applications. Especially in PXP-type complexes, metallophillic interactions can be induced in the case of coinage metals, which lead to fascinating photo-optical properties. Furthermore, PPX/PXP-type complexes are believed to exhibit a promising behavior in catalysis, due to the potential hemilability of the ligand and the therewith involved availability of free active sites for substrate binding.
Introduction
Bidentate diphosphines are prominent phosphorus-based ligands in coordination chemistry, which have been widely recognized in the past based on their flexible coordination towards transition metals and their ability to be easily modified electronically and sterically.1,2 These steric and electronic modifications can be achieved through the tuning of the residues attached to the phosphorus atoms or via the modification of the ligand backbone, which bridges the two phosphorus units. Major tools are hereby the adjustment of the number as well as the nature of the bridging atoms.2–4 A plethora of ligand sets has therefore been developed in the last decades, including ligands with larger bridging units, such as bis[(2-diphenylphosphino)phenyl] ether (DPEphos), 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP) or PNP-type pincer ligands, which are even capable of coordinating a transition metal in a tridentate fashion. Down this road also short-bite ligands, which only inherit one central bridging atom, such as a CH2 or a NH/NR unit, have attracted attention. Prominent examples for such ligands are bis(diphenylphosphino)methane (Ph2P–CH2−PPh2, dppm)3 and bis(diphenylphosphino)amine (Ph2P–N(H)–PPh2, dppa),5,6 which both have shown to be capable of coordinating a range of transition metals. Compared to dppm and dppa, as lead examples in the field of short-bite ligands, oxygen- and sulphur-bridged diphosphines, such as POP- (R2P–O–PR2) and PSP-based (R2P–S–PR2) ligands, are still largely underrepresented (Fig. 1). The only review concerning such so-called inorganic backbone phosphines appeared in 2002 by Woollins and Appleby.2 To shed more light on these interesting short-bite ligands, the scope of this article is to highlight recent advances in the field of POP- and PSP-based diphosphine ligands in general, their coordination chemistry and their application in photo-optics and catalysis. | ||
| Fig. 1 Representative diphosphines with larger ligand backbones (left) and candidates with a central monoatomic bridge (right). As members of the group of short-bite diphosphines, dppm and dppa have widely been investigated, whereas their POP- and PSP-based analogues have been largely overlooked. | ||
Phosphorotropic tautomerism of PPX/PXP ligands
Similarly to related secondary phosphine oxides and sulphides (Ph2P(X)H; X = S, O), PPX/PXP-type ligands have been reported to undergo an unique tautomeric equilibrium between the PPX (R2P(X)–PR2; X = O, S; R = aryl, alkyl) and the PXP (R2P–X–PR2; X = O, S; R = aryl, alkyl) tautomer. In the PXP tautomer both P atoms are assumed to have a formal oxidation state of +III. This changes upon shift of the equilibrium resulting in a +IV state of the P(X) and a +II state of the P atom within the PPh2 fragment (Fig. 2). This equilibrium, also known as phosphorotropic tautomerism, strongly relies on the heteroatom incorporated in the ligand backbone and on the steric and electronic properties of the rests attached to the phosphorus atoms.7–11 If various phosphorus residues are present (R2P(X)–PR′2; R ≠ R′), different forms of the PPX tautomer can be observed through rearrangement processes, whereas the number of possible tautomers decreases if all residues are identical (Fig. 2).10
 | ||
| Fig. 2 Phosphorotropic tautomerism of the PXP/PPX ligand with the oxidation states of the phosphorus atoms given in red. With sterically more demanding or electron withdrawing substituents on the phosphorus atoms, the PXP-based tautomer is favoured, whereas in the case of sterically less demanding or less electron withdrawing groups the PPX tautomer dominates. | ||
The nature of the residues on the P atoms determines, if predominantly one tautomer or a tautomer mixture is present. The nature of the substituents also influences the extent to which this equilibrium can be affected by the reaction conditions, such as temperature, storage time, or the presence of additives.10 In general, electron withdrawing groups (EWG) attached to the P atoms stabilise the PXP tautomer, while electron donating groups favour a shift of the equilibrium towards the PPX side.2,11 Since the introduction of EWG's as P substituents lowers the nucleophilicity of the phosphorus atoms, a comparably higher nucleophilicity can be ascribed to the X atom, which then ultimately results in the formation of the PXP tautomer.2,11 Hereby a smaller effect of EWG's on the equilibrium is found for sulphur-bridged ligands compared to the oxygen-based analogues.10 This finding can be rationalised by DFT (density functional theory) calculations, carried out by Ogawa and coworkers,12 who have shown that the HOMO (highest occupied molecular orbital) is primarily localised on the PPS moiety, with minor contributions from the phosphorus substituents. In contrast, for the PPO counterpart, the HOMO is distributed across the entire ligand molecule. These findings not only suggest that the phosphorus substituents have a stronger influence on the phosphorotropic tautomerism in PPO than in PPS ligands, but also indicate that PPS ligands are more reactive than their PPO counterparts. Beyond the electronic modulation of the phosphorotropic tautomerism, steric factors play a crucial role, whereby sterically demanding groups were found to destabilise the monoxide and sulphide PPX (X = O, S) tautomer.2,11 In accordance with these requirements, examples of PPX-based compounds are Ph2P(X)–PPh2,12–14 Cy2P(X)–PCy2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11 or (EtO)2P(S)–P(OEt)2,11 whereas (tBu)2P–X–P(tBu)2,15 (CF3)2P–X–P(CF3)27 or Mes2P–S–PMes211 represent examples of the PXP tautomers (Cy = cyclohexyl, EtO = ethoxy, tBu = tert-butyl, Mes = mesityl, Fig. 2). In addition to the steric and electronic modification of the substituents on the phosphorus atoms, the phosphorotropic tautomerism can be further influenced by metal coordination and the formation of so-called coordination-stabilised tautomers, a phenomenon which will be discussed later.
11 or (EtO)2P(S)–P(OEt)2,11 whereas (tBu)2P–X–P(tBu)2,15 (CF3)2P–X–P(CF3)27 or Mes2P–S–PMes211 represent examples of the PXP tautomers (Cy = cyclohexyl, EtO = ethoxy, tBu = tert-butyl, Mes = mesityl, Fig. 2). In addition to the steric and electronic modification of the substituents on the phosphorus atoms, the phosphorotropic tautomerism can be further influenced by metal coordination and the formation of so-called coordination-stabilised tautomers, a phenomenon which will be discussed later.
Interestingly, PNP-based ligands show a similar tautomerism between iminodiphosphines (R2P–PR2= NR′, PPN) and diphosphonylamines (R2P–N–PR2, PNP). However, for the majority of these compounds, the equilibrium is shifted completely to the PNP side, which represents a widely explored ligand class, especially in terms of photo-optical16–18 and catalysis-related applications.19–21 For example, dppa, a classic PNP-type ligand, only exists in the PNP tautomeric form under ambient conditions, whereas in the oxygen- and sulphur-containing analogues (PPX/PXP, X = O, S, with R = Ph) the equilibrium shifts entirely to the PPX tautomer. In the case of PNP-based ligands, the equilibrium is tremendously affected by the nature of the nitrogen residue and less by phosphorus substituents, whereby only strong electron withdrawing groups, such as C6H4(o-CF3), were shown to be capable of stabilising the PPN tautomer.22,23 This is in contrast with short-bite POP- and PSP-based ligands, where EWG's shift the equilibrium strongly to the PXP side. In most cases, the PPN tautomer is highly unstable, resulting in P–P bond cleavage in presence of small molecules, such as methanol, H2O2 and water, or in an equilibrium shift to the PNP tautomer.22 Interestingly, some PNP compounds can convert to the PPN form reversibly through protonation/deprotonation mechanisms. Again, this conversion is strongly influenced by the nitrogen residue and requires a proton acceptor within this residue.23 PCP ligands do not show such a tautomeric behaviour.
Synthesis of PPX/PXP ligands
The synthesis of R2P(X)–PR2/R2P–X–PR2 (X = O, S) compounds dates back to the 1960s. The procedures for synthesising these ligands often depend on the nature of the phosphorus substituents, which can include (fluorinated) alkyl, aryl, OR, or NR2 groups. Compared to the formation of monoxide species, which are frequently obtained from side reactions or degradation products,24–28 the isolation of comparable sulphur analogues is less often described. Whereas most of the synthetic approaches are solely applicable for PPO/POP- or PPS/PSP-type compounds, method 4 and 6 can be utilised for both the oxygen- and sulphur-based short-bite ligands.
As shown in reaction (1) in Fig. 2, degradation reactions starting from PNP-based ligands, can lead to the formation of PPO/POP compounds through a hydrolytic cleavage of the P–N bond. However, the exact mechanism of these degradation reactions remains unknown and several potential pathways have been described in the literature. Some reports suggest the hydrolytic P–N bond cleavage of the coordinated PNP ligand and a subsequent recombination of the two phosphorus units.29 In other studies mechanisms are proposed, which include an initial equilibrium shift from the PNP to the PPN tautomer, followed by the PN bond cleavage. In both cases, an amine results as side product.28 Besides P–N bond cleavages, esters, such as Ph2P–O–C(O)R (R = Ph30 or CHCH2)31 were shown to be susceptible towards rearrangement reactions, yielding PPO and R–C(O)OC(O)R (Fig. 3, reaction (2)). Additionally, radical-based reactions have been reported. For example, Ogawa and coworkers described the reaction of Ph2P(O)˙ radicals with (Ph2P)2 to form PPO (Fig. 3, reaction (3)).32 PPO/POP- and PPS/PSP-type compounds can also be synthesised via the conversion of R2PCl with R2P(X)H,14,24,33 R2PXR′ (R′ = H, R, Na, K),34–36 Na2S,11 Li2S,4 R3SiONa,37 H2O,38,39 oxidation of R2P–PR240 or the reaction of R2PHal (Hal = I or Cl) with Ag2CO3 (Fig. 3, reactions (4)–(9)).41,42 It should be mentioned that reactions with water are usually observed as side reactions caused by trace amounts of water, as experiments involving R2PCl compounds are typically conducted under an inert gas atmosphere. As mentioned before, depending on the nature of the residues on the phosphorus atoms, either the PXP, the PPX or a mixture of both tautomers is formed.
 | ||
| Fig. 3 Various synthetic strategies for the formation of PPX/PXP-type ligands. R = (fluorinated) alkyl, aryl, OR, or NR2. | ||
Reactivity of PPX/PXP ligands
It is evident that the different tautomers (PPX/PXP; X = O, S) show different reactivities, as indicated by their distinct phosphorus oxidation states. The fundamental reactions of PPX compounds, which were reported before 2002, were already summarised by Appelby and Wollins.2 The majority of described reactions is based on the cleavage of the P–P bond, for example via acid hydrolysis forming R2POH and R2PCl2,41 or the electrophilic attack of methyl bromide at the P(II), which causes the formation of Ph2PMe and Ph2P(O)Br.43 Notably, the acidic hydrolysis was reported to be reversible through addition of trifluoro acetic acid in trifluoroacetic anhydride, if the POP ligand was chelating a metal ion.8
In the last years, Ogawa et al. extended the investigation of the reactivity of PPX compounds and explored, for example, the reversible P–P bond cleavage in presence of a radical starter or UV light. The highly active Ph2P(X)˙ radical is capable of attacking unsaturated C–C bonds forming a carbon-based radical, which again can react with PPO liberating a new Ph2P(X)˙ radical.44 Hereby, the presence of oxygen or sulphur is fundamental for this type of reactivity, since with Ph2PPPh2, similar reactions have not been observed. The reactivity of PPS is thereby much higher than for PPO. In the context of photocatalysis, Takano et al. investigated the photocatalytic ethylene insertion into the P–P bond of PPO and subsequent sulphur-oxidation of the unoxidized P atom.45 Beyond the photoinduced activation of the P–P bond, Yang et al. have reported the activation of C–F bonds and simultaneous phosphorylation by converting aryl fluorides with PPO in presence of a base and [Ni(COD)2] (COD = 1,5-cyclooctadiene) at elevated temperatures.46 The reactivity is initiated through the P–P bond cleavage of PPO. The application of PPO as a phosphorus source has also been attempted by the group of Ogawa in Pd- and Rh-catalysed hydrophosphination reactions of terminal alkynes. Hereby it has been shown that catalytic amounts of PPO can enhance the phosphorylation of other phosphine oxides as reagents.47,48
At this point, it should be mentioned, that contrary to the bridging units in dppm (CH2) and dppa (NR; R = alkyl, aryl, H), in POP- and PSP-type ligands no further ligand backbone modification is feasible. In PCP- and PNP-type ligands this has proven to be an effective modification strategy, especially for the formation of anionic ligands through deprotonation.19
PPX/PXP-based coordination compounds
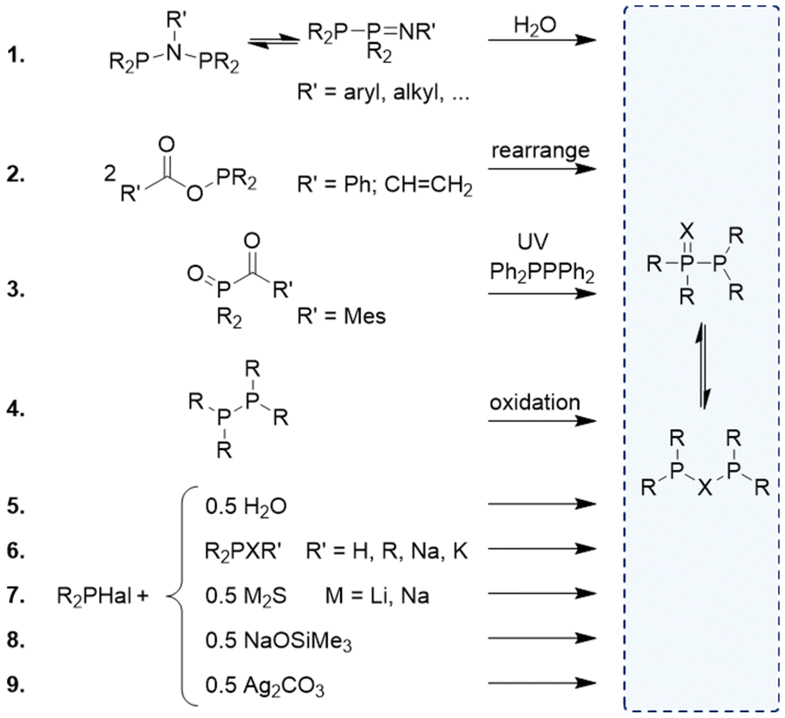
A unique feature of short-bite PPX/PXP-based ligands is their ability to adopt a variety of different coordination modes towards transition metals, which is mainly based on their occurrence in two different tautomeric forms (Fig. 4). Consequently, a wide range of metals in different oxidation states can be coordinated. The position of the phosphorotropic equilibrium and the resulting coordination mode are strongly influenced by the nature of the heteroatom X, the residues of the phosphorus atoms and the properties of the metal (Fig. 5).4,9,14,35,49 In addition, the choice of metal precursor and the presence of additional ligands also affect the coordination mode of the PPX/PXP ligands.9,29,50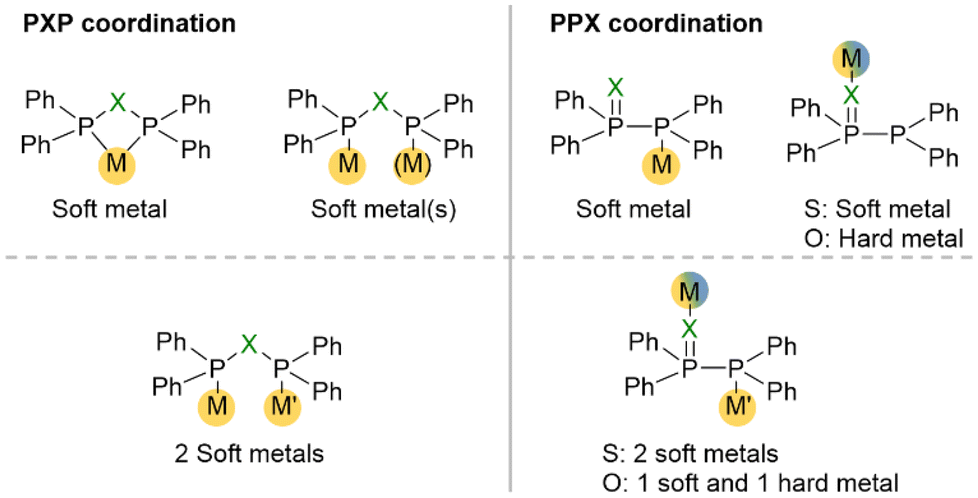 | ||
| Fig. 4 Potential coordination modes of the PXP (left) and PPX (right) tautomers. Additional ligands are omitted for clarity. With X = O, coordination of hard metals can be achieved, while the P and S donor sites tend to coordinate soft metals. Heterobimetallic complexes can be formed (bottom). | ||
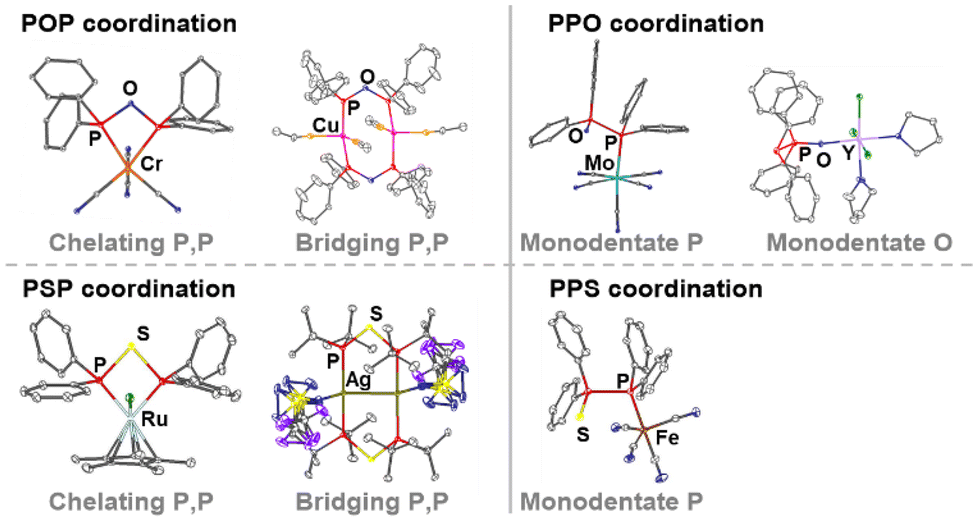 | ||
| Fig. 5 Selected examples of PXP- and PPX-based coordination compounds.4,8,9,11,14 Hard metals result in oxygen coordination and soft metals in phosphorus coordination. An equilibrium shift of the PPX to the PXP tautomer can occur, which results in the formation of coordination-stabilised tautomers, such as seen with the dinuclear copper complex and the mononuclear Ru- and Cr-based chelate complexes. | ||
The PPX tautomer features two different donor sites, namely the phosphorus atom, which possesses the lone electron pair [P(II)], and the oxygen or sulphur atom in the ligand backbone. The increased softness (based on the HSAB principle) of sulphur compared to oxygen provides two donor sites within the PPS ligand, which both coordinate preferably rather soft metals. However, there are only two reported examples of PPS-based Fe(0)11 and Cr(0)49 complexes, in which the coordination occurs via the P(II) atom. Contrary, oxygen presents a hard donor site enabling the coordination of harder metals, such as Ga(III),51 Zr(IV),52 Al(III),53 Y(III)14 and Fe(II),14 whereas softer metals, such as Cu(I),29,54 Mo(0),9 Co(0),55 or Fe(0)56–59 are coordinated through the P(II) atom of the PPO ligand. The PPX tautomer opens the possibility for the formation of heterobimetallic complexes via coordination of the two different donor sites. In this context, the combination of two different metals can enable cooperative effects, which can lead to outstanding properties of the bimetallic complexes in catalysis, photo-optics or magnetism. A chelating coordination of the P(II) and the X atom in PPX ligands9 is assumed to be highly unlikely, due to the high ring strain and has never been reported.
The synthesis of PXP-type complexes is more complicated compared to their PPX-type counterparts, due to the PPX tautomer being often more stable under ambient conditions. Therefore, initially several PXP-type complexes were only isolated as by-products, resulting from degradation reactions of other phosphine ligands.29,60,61 However, recently PXP-based complexes, such as molybdenum, copper or gold complexes, have been obtained from the direct reaction of metal precursors with the PPX/PXP-type ligand. This has proven to be a successful strategy, independent of whether the free ligand is present as PXP or PPX tautomer.8,14 PXP-based complexes, resulting from ligands, which are usually present as PPX tautomers under ambient conditions, are referred to as coordination-stabilised tautomers.
In PXP-type complexes, the ligand can either adopt a chelating or bridging coordination mode of one or two metals, respectively. The formation of dinuclear complexes, where the PXP ligand acts as a bridging ligand, often occurs to avoid the high strain associated with the four-membered P–X–P–M ring in chelate-type metal complexes.3 Similar observations have been reported for PCP-type ligands, where dppm as ligand forms majorly dinuclear complexes, while ligands with a higher number of carbon atoms in the backbone, such as in bis(diphenylphosphino)ethane (dppe), act as chelate ligands in mononuclear complexes. For POP ligands, examples of the bridging coordination mode include Au,14,62 Fe,63 Cr,64 Rh65 or Cu complexes,29,60,66,67 while for the chelating coordination mode Cr,8 Mo,68 W,9 Ru61,69 and Rh70,71 complexes have been reported. For PSP-type ligands, complexes, in which the PSP ligand adopts a bridging coordination, have been reported for Ag,11 W,72 Mn,73 Cu35 and Ni,7 whereas chelate complexes have been isolated in the case of Mo,74 Ru4 and Ni.50 The bridging coordination mode of the short-bite diphosphine ligands can bring two metal atoms in close proximity, which is particularly interesting if the metal–metal distance of d10 metals falls below the sum of the van der Waals radii and therefore in the range of metallophilic interactions.75 Such interactions can lead to extraordinary properties, especially in the field of photo-optics or catalytic reactions.3,76 Interestingly, to the best of our knowledge, no example is known of an isolated PXP-type complex, in which only one phosphorus atom binds to a metal centre, while the other phosphorus atom remains uncoordinated, as reported for PCP-77,78 and PNP-type3,79,80 complexes. Another potentially interesting compound class would be heterobimetallic PXP-based complexes, through which two different metal atoms would come in close proximity. Examples of such heterobimetallic complexes stabilised by the oxygen- or sulphur-containing PPX/PXP ligand have not been reported yet but are expected to show outstanding catalytic properties, in analogy to the PNP- and PCP-type counterparts, as seen in dppa-81 and dppm-82,83 based bimetallic complexes.
Influence of the ligand backbone in coordination compounds
The nature of the heteroatom in the backbone of short-bite ligands has a strong impact on the underlying coordination chemistry, due to the size and hard-/softness of the heteroatom involved and the corresponding electronegativity. Since the POP and PSP ligand (R = Ph) both do not exist in the PXP tautomer under ambient conditions, the comparison of the free PXP ligands is not possible. Therefore, two analogue transition metal complexes of the POP and PSP (with R = Ph) ligands, namely [Cu3(μ3-Cl)2(μ-PXP)3]+35 and [Mo(CO)4(PXP)] (X = O,8 S74), are being considered to study the influence of the ligand backbone in short-bite ligands on structural parameters of the coordination compounds. These complexes can easily be compared to the respective PCP (C = CH2, for Mo84 and Cu85) and PNP (N = H, for Mo79 and Cu86) analogues (Fig. 6).
 | ||
| Fig. 6 Comparison of different bridging atoms (C, N, O, S) within the backbone of short-bite diphosphine ligands using [Mo(CO)4(PXP)] and the cation [Cu3(μ3-Cl)2(μ-PXP)3]+ as examples. The electronegativity of the respective element is given in black and the covalent radii [pm] is given in grey (right). The hard/soft character of the bridging atoms are displayed by colours: yellow: soft, green: intermediate, blue: hard. | ||
Firstly, [Cu3(μ3-Cl)2(μ-PXP)3]+ complexes can give insights into the electronic situation caused by the different ligand backbones via31P{1H} NMR spectroscopy. The 31P chemical shift correlates strongly with the electronegativity of the bridging atoms, in the order of O > N > S > C, indicating the deshielding of the phosphorus atoms.
Further, the tuning of the ligand backbone clearly impacts the molecular structures of [Cu3(μ3-Cl)2(μ-PXP)3]+, as indicated by the differences in the P–X bond lengths, as well as the P–X–P bond angles (Table 1). The P–X bond increases in the order of POP ∼ PNP < PCP < PSP, whereby a bond elongation of 0.5 Å from POP to PSP is observed. Similar findings become obvious for the P–X–P bond angle, which decreases in the order of POP ∼ PNP > PCP > PSP. A tremendous difference of around 20° for the P–X–P bond angles of POP and PSP can be revealed (POP > PSP). Even the smallest P–O–P angle in reported chelate-type complexes70 is with 95.4(11)° still significantly larger than the reported P–S–P angles for the cationic [Cu3(μ3-Cl)2(μ-PSP)3]+ complex. The parameters are in accordance with the general conclusion in literature, that P–N–P angles are larger than the ones in PCP-type ligands.87
| Cu–P | P–X | Cu–Cu | P–X–P | δ | |
|---|---|---|---|---|---|
| POP35 | 2.222(2)–2.241(2) | 1.642(4)–1.653(4) | 2.950(1)–3.033(1) | 120.9(3)–124.8(2) | 101.7 |
| PSP35 | 2.246(2)–2.257(2) | 2.111(3)–2.118(3) | 2.992(2)–3.046(1) | 99.6(1)–101.6(1) | 23.9 |
| dppa86 | 2.236(4)–2.274(3) | 1.627–1.705 | 2.937(2)–3.040(2) | 124.66–124.95 | 37.2 |
| dppm85 | 2.248(3)–2.273(5) | 1.823–1.841 | 3.063(3)–3.328(2) | 112.1(5)–116.0(7) | –14.4 |
As discussed above, through the bridging coordination mode of PXP-type ligands, metallophilic interactions can be induced and the impact of PNP, PCP and POP ligands on these interactions can be discussed in the case of dinuclear Au and Cu complexes. Unfortunately, no analogue PSP complexes have been reported so far. For bimetallic [Au2(μ2-PXP)2]Y2 (Y = ClO4−, PF6−) complexes, the observed metal–metal distances are similar for PCP- (2.9258(9)88–3.120 Å89) and POP-based (2.9302(7) Å)14 compounds, while smaller metal–metal distances have been reported for PNP-coordinated Au2 complexes (2.7944(19)90–2.838(2)91 Å, NR = 2,6-iPr2C6H3, Et). All these distances are below the sum of the van der Waals radii and therefore provide the grounds for aurophilic interactions. Looking at the 3d coinage metal copper in [Cu2(MeCN)n(μ2-PXP)2]Y2 (Y = PF6−, BF4−, ClO4−), a different trend can be observed, since PCP-type complexes (C = CH2, dppm, 3.75592–3.7679(7)93 Å, n = 4) are not capable of bringing the metals in such a close proximity as PNP-(N = NH, dppa, 3.341(2) Å)94 and POP-type (3.371(3) Å)14 complexes. It should be underlined that the metals and the corresponding metal–metal distances are strongly influenced by additional ligands present on the metal centres. Hereby, a strong decrease of the Cu–Cu distance can be observed with a decreasing number of coordinating MeCN molecules. Whereas four coordinating MeCN molecules provide Cu–Cu distances of 3.75592–3.7679(7)93 Å (PCP, dppm), 3.340 Å94 (PNP, dppa) and 3.371(3) Å (POP) with three coordinating MeCN molecules the Cu–Cu distance is decreased and is found to be close to the sum of the van der Waals radii (PNP: 2.869(4) Å94, POP: 2.8965(24) Å). Also in the case of the dppm complex, the Cu–Cu distance is reduced to 3.3885(7) Å, when only three coordinating MeCN molecules are being present.93 For a POP-coordinated complex, the coordination of only one MeCN molecule per Cu atom leads to a drop in the Cu–Cu distance to 2.4408(23) Å. This unambiguously corresponds to cuprophilic interactions, which consequently facilitates remarkable photo-emission properties.14
Apart from the impact of the ligand backbone on the molecular structures of complexes with bridging PXP ligands, also chelate-type complexes, as in the case of [Mo(CO)4(PXP)] (Table 2), are clearly affected by the modification of the ligand backbone. In the chelate-type complexes, the bite angles increase in the order of POP < PNP < PCP < PSP according to the increasing P–P bond distances. The PSP bite angle is hereby closest to the dppe analogue with a CH2-CH2 bridging unit (80.44(8)°).95
From the before-mentioned parameters, it can be concluded that the P–X bond lengths as well as the P–X–P and P–M–P bond angles strongly correlate with the size of the bridging atom in the ligand backbone (O < N < C < S).
Tautomerism in PPX/PXP-based metal complexes
Another interesting feature, which has rather been neglected up to now, is the tautomerism of PPX/PXP ligand-coordinated transition metal complexes. Hereby, it came to light that in solution, PPX- and PXP-stabilised complexes can co-exist, as observed in the case of [Au2Cl2(μ2-POP)].14 Similar observations were made by Wong and co-workers, who reported a thermally initiated rearrangement of the PPO ligand (coordinated via the P) in [M(CO)5(PPO)] (M = Cr, Mo, W) complexes to the POP tautomer in [M(CO)4(μ2-POP)] under the loss of one CO ligand.9Photo-optical applications
By using diphosphines as ligands for transition metals, metallophilic interactions of d10 metals can emerge through a bridging coordination mode. These interactions are known to initiate interesting photo-optical properties, such as long luminescence lifetimes or thermochromism effects.14,75,96 A drastic impact of the bridging atom in the ligand backbone of short-bite ligands was hereby identified. Whereas dinuclear gold and copper complexes stabilised by PCP- and PNP-type ligands showed a bright photo-luminescence at room temperature,91,97–100 in the case of the POP-based counterparts photo-emission can only be observed at 77 K.14 To the best of our knowledge, the photo-optical properties of dinuclear PSP-based complexes have not yet been investigated. A similar finding was reported for trinuclear complexes. Again, trinuclear dppm-coordinated copper complexes show a bright luminescence at room temperature, which cannot be observed for the POP and the PSP counterparts.35,100 However, at 77 K POP-stabilised Cu3 complexes were shown to exhibit strong photo-optical emission with lifetimes exceeding 100 μs. Interestingly, with PSP-type analogues, no photo-emission could be observed at all. This outcome was attributed to the larger size of the sulphur atom in the ligand backbone, which causes a loss in rigidity of the PSP-containing complexes and therewith involved a higher number of non-radiative decays.35Potential catalytic applications
The unique dynamic tautomerism of the PXP/PPX (X = O, S) ligand, also within transition metal complexes, might be advantageous for the application of such metal complexes as homogeneous catalysts. In both tautomeric forms of the ligands, two donor sites are available, which opens the possibility to act as hemilabile ligand in metal complexes. These hemilabile properties are advantageous in catalytic applications to offer free coordination sites, which are available for substrate binding. This finding was already reported for other short-bite ligands, such as PCP-type ligands, for which superior catalytic behaviour of Ru complexes of dppm compared to dppe and dppp (TOF(dppm): 51 h−1 ≫ TOF(dppe): 30 h−1, TOF (dppp): 26 h−1) in the conversion of ethanol to n-butanol was attributed to a higher hemilability based on an enhanced ring strain.101 A similar behaviour can therefore be expected for the oxygen and sulphur analogues. Initial studies have already given the perspective, that free coordination sites at the active centre can reversibly be opened up in the presence of substrates. The vacancy can either result from de-coordination of one P(III)-atom of the PXP tautomer from the metal centre or from the PPX/PXP-specific equilibrium shift to the PPX form, where only one donor site coordinates the metal centre. The latter process would describe a unique type of hemilability.Conclusions and outlook
In this frontier article, highlights of the underestimated PPO/POP- and PPS/PSP-type short-bite ligands are summarised and compared to the well-studied PCP and PNP ligands. One key feature of these oxygen- and sulphur-bridged diphosphines is their phosphorotropic equilibrium between the PPX and PXP tautomer. As a result, PXP/PPX ligands (X = O, S) can adopt various coordination modes based on the different donor sites available in the two potential tautomers. Also, in presence of transition metals so-called coordination-stabilised tautomers can be formed.The incorporation of S and O into the backbone of short-bite ligands strongly impacts their electronic properties and the molecular structures of corresponding transition metal complexes. Whereas POP ligands show similar structural parameters as dppa, which can be attributed to similar sizes of the oxygen and nitrogen atoms in the ligand backbone, a great difference to PSP-type complexes is identified. The significantly larger size of the sulphur atom results in larger bond lengths and a higher flexibility within the backbone. Consequently, a very acute backbone can be found in PSP-based transition metal complexes. The effect of the increased flexibility can also be recognized in photo-optical properties of corresponding coinage metal complexes, since the loss in rigidity is accompanied by a higher number of non-radiative decays. To optimise POP- and PSP-based complexes for photo-optical applications, further modification of the phosphorus residues or combinations with other ligands on the metal must be conducted to counteract non-radiative decays.
The tautomeric equilibrium of the oxygen- and sulphur-containing short-bite ligands not only plays a role in the free PPX/PXP (X = O, S) ligands, but also in their transition metal complexes. Hereby, external factors can be utilised to shift this tautomeric equilibrium of the ligand within metal complexes, for example in the presence of other compounds. This behaviour opens interesting opportunities for catalytic applications and has to be investigated in detail as part of future studies.
Data availability
No primary research results, software or code have been included. For the comparison of crystal structures, crystallographic data from the CCDC has been analysed (https://www.ccdc.cam.ac.uk). The references to the original literature is given in the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank the Stiftung der deutschen Wirtschaft for a doctoral scholarship for F. F. The authors also gratefully acknowledge support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) through the Collaborative Research Centre “4f for Future” project A6 (CRC 1573, project number 471424360).References
- A. L. Clevenger, R. M. Stolley, J. Aderibigbe and J. Louie, Chem. Rev., 2020, 120, 6124–6196 CrossRef CAS PubMed.
- T. Appelby and J. D. Woollins, Coord. Chem. Rev., 2002, 235, 121–140 CrossRef.
- R. J. Puddephatt, Chem. Soc. Rev., 1983, 12, 99–127 RSC.
- P. E. Sues, A. J. Lough and R. H. Morris, Chem. Commun., 2014, 50, 4707–4710 RSC.
- H. Nöth and L. Meinel, Z. Anorg. Allg. Chem., 1967, 349, 225–240 CrossRef.
- O. Schmitz-DuMont, B. Ross and H. Klieber, Angew. Chem., Int. Ed. Engl., 1967, 6, 875–876 CrossRef CAS.
- H. Einspahr and J. Donohue, Inorg. Chem., 1974, 13, 1839–1843 CrossRef CAS.
- E. H. Wong, L. Prasad, E. J. Gabe and F. C. Bradley, J. Organomet. Chem., 1982, 236, 321–331 CrossRef CAS.
- E. H. Wong, R. M. Ravenelle, E. J. Gabe, F. L. Lee and L. Prasad, J. Organomet. Chem., 1982, 233, 321–331 CrossRef CAS.
- I. F. Lutsenko and V. L. Foss, Pure Appl. Chem., 1980, 52, 917–944 CrossRef CAS.
- S. Yogendra, S. S. Chitnis, F. Hennersdorf, M. Bodensteiner, R. Fischer, N. Burford and J. J. Weigand, Inorg. Chem., 2016, 55, 1854–1860 CrossRef CAS PubMed.
- Y. Sato, S.-i. Kawaguchi, A. Nomoto and A. Ogawa, Chem. – Eur. J., 2018, 25, 2295–2302 CrossRef PubMed.
- Y. Yamamoto, R. Tanaka, M. Ota, M. Nishimura, C. C. Tran, S.-i. Kawaguchi, S. Kodama, A. Nomoto and A. Ogawa, J. Org. Chem., 2020, 85, 14708–14719 CrossRef CAS PubMed.
- F. Flecken, T. Grell and S. Hanf, Dalton Trans., 2022, 51, 8975–8985 RSC.
- B. S. N. Huchenski and A. W. H. Speed, Chem. Commun., 2021, 57, 7128–7131 RSC.
- V. R. Naina, A. K. Singh, S. Shubham, F. Krätschmer, S. Lebedkin, M. M. Kappes and P. W. Roesky, Dalton Trans., 2023, 52, 12618–12622 RSC.
- V. W.-W. Yam, W. K.-M. Fung and M.-T. Wong, Organometallics, 1997, 16, 1772–1778 CrossRef CAS.
- S. Naik, S. Kumar, J. T. Mague and M. S. Balakrishna, Dalton Trans., 2016, 45, 18434–18437 RSC.
- C. Fliedel, A. Ghisolfi and P. Braunstein, Chem. Rev., 2016, 116, 9237–9304 CrossRef CAS PubMed.
- A. Ghisolfi, F. Condello, C. Fliedel, V. Rosa and P. Braunstein, Organometallics, 2014, 34, 2255–2260 CrossRef.
- K. Blann, A. Bollmann, J. T. Dixon, F. M. Hess, E. Killian, H. Maumela, D. H. Morgan, A. Neveling, S. Otto and M. J. Overett, Chem. Commun., 2005, 620–621 RSC.
- Z. Fei, R. Scopelliti and P. J. Dyson, Eur. J. Inorg. Chem., 2004, 2004, 530–537 CrossRef.
- Z. Fei, N. Biricik, D. Zhao, R. Scopelliti and P. J. Dyson, Inorg. Chem., 2004, 43, 2228–2230 CrossRef CAS PubMed.
- S. Jin, G. C. Haug, V. T. Nguyen, C. Flores-Hansen, H. D. Arman and O. V. Larionov, ACS Catal., 2019, 9, 9764–9774 CrossRef CAS.
- M. Gruber, P. G. Jones and R. Schmutzler, Chem. Ber., 1990, 123, 1313–1317 CrossRef CAS.
- M. Aydemir, A. Baysal and B. Gümgüm, J. Organomet. Chem., 2008, 693, 3810–3814 CrossRef CAS.
- S. Priya, M. S. Balakrishna and S. M. Mobin, Polyhedron, 2005, 24, 1641–1650 CrossRef CAS.
- T. Posset, F. Rominger and J. Blümel, Chem. Mater., 2005, 17, 586–595 CrossRef CAS.
- K. Naktode, R. K. Kottalanka, H. Adimulam and T. K. Panda, J. Coord. Chem., 2014, 67, 3042–3053 CrossRef CAS.
- R. S. Davidson, R. A. Sheldon and S. Trippett, J. Chem. Soc. C, 1967, 1547–1552 RSC.
- D. J. Irvine, C. Glidewell, D. J. Cole-Hamilton, J. C. Barnes and A. Howie, J. Chem. Soc., Dalton Trans., 1991, 1756–1772 Search PubMed.
- Y. Sato, S.-i. Kawaguchi, A. Nomoto and A. Ogawa, Synthesis, 2017, 49, 3558–3567 CrossRef CAS.
- D. Hunter, J. K. Michie and W. Stewart, Phosphorus Sulfur Relat. Elem., 1981, 10, 267–270 CrossRef CAS.
- V. L. Foss, V. A. Solodenko and I. F. Lutsenko, Zh. Obshch. Khim., 1979, 49, 2418–2428 CAS.
- F. Flecken, A. Knapp, T. Grell, C. Dreßler and S. Hanf, Inorg. Chem., 2023, 62, 13038–13049 CrossRef CAS PubMed.
- S. Inokawa, Y. Tanaka, H. Yoshida and T. Ogata, Chem. Lett., 1972, 1, 469–470 CrossRef.
- K. M. Cooke, T. P. Kee, A. L. Langton and M. Thornton-Pett, J. Organomet. Chem., 1991, 419, 171–180 CrossRef CAS.
- C.-Y. Chen and R. J. W. L. Fèvre, J. Chem. Soc., 1965, 3473–3504 RSC.
- R. F. Hudson, R. J. G. Searle and F. H. Devitt, J. Chem. Soc. B, 1966, 789–792 RSC.
- J. Ellermann, M. Schütz, F. W. Heinemann, M. Moll and W. Bauer, Z. Naturforsch., B: J. Chem. Sci., 1997, 52, 795–800 CrossRef CAS.
- J. L. Virlichie and P. Dagnac, Rev. Chim. Miner., 1977, 14, 355–358 CAS.
- J. E. Griffiths and A. B. Burg, J. Am. Chem. Soc., 2002, 82, 1507–1508 CrossRef.
- W. Dabkowski, A. Ozarek, S. Olejniczak, M. Cypryk, J. Chojnowski and J. Michalski, Chem. – Eur. J., 2009, 15, 1747–1756 CrossRef CAS PubMed.
- Y. Sato, S.-i. Kawaguchi, A. Nomoto and A. Ogawa, Angew. Chem., Int. Ed., 2016, 55, 9700–9703 CrossRef CAS PubMed.
- H. Takano, H. Katsuyama, H. Hayashi, W. Kanna, Y. Harabuchi, S. Maeda and T. Mita, Nat. Commun., 2022, 13, 7034 CrossRef CAS PubMed.
- J. Yang, L. Fan, C. Chen, M. Wang, B. Sun, S. Wang, H. Zhong and Y. Zhou, Org. Biomol. Chem., 2023, 21, 494–498 RSC.
- S.-i. Kawaguchi, M. Kotani, T. Ohe, S. Nagata, A. Nomoto, M. Sonoda and A. Ogawa, Phosphorus, Sulfur Silicon Relat. Elem., 2010, 185, 1090–1097 CrossRef CAS.
- Y. Yamamoto, K. Fujiwara and A. Ogawa, Organometallics, 2023, 42, 2590–2597 CrossRef CAS.
- P. G. Jones, A. K. Fischer, M. Farkens and R. Schmutzler, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2002, 58, m478–m479 CrossRef CAS.
- F. Flecken, T. Grell and S. Hanf, PSP-coordinated nickel(II) complexes as Kumada coupling catalysts, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-fdgcn.
- M. R. Kopp and B. Neumüller, Z. Anorg. Allg. Chem., 1999, 625, 739–745 CrossRef CAS.
- T. Ogawa, Y. Kajita and H. Masuda, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m1129 CrossRef CAS PubMed.
- A. M. Lifschitz, N. A. Hirscher, H. B. Lee, J. A. Buss and T. Agapie, Organometallics, 2017, 36, 1640–1648 CrossRef CAS.
- T. S. Sukhikh, R. M. Khisamov and S. N. Konchenko, Molecules, 2021, 26, 2030 CrossRef CAS PubMed.
- S. G. Bott, J. C. Wang and M. G. Richmond, J. Chem. Crystallogr., 1999, 29, 603–608 CrossRef CAS.
- F.-Y. Chen, M.-Y. Hu, X.-L. Gu, X.-F. Liu and P.-H. Zhao, Transition Met. Chem., 2021, 46, 645–653 CrossRef CAS.
- L.-C. Song, M. Cao, Z.-Q. Du, Z.-H. Feng, Z. Ma and H.-B. Song, Eur. J. Inorg. Chem., 2014, 2014, 1886–1895 CrossRef CAS.
- Y.-F. Liu, Inorg. Chim. Acta, 2011, 378, 338–341 CrossRef.
- X.-F. Liu and X.-Y. Yu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, m1552 CrossRef CAS PubMed.
- A. Aloisi, J.-C. Berthet, C. Genre, P. Thuéry and T. Cantat, Dalton Trans., 2016, 45, 14774–14788 RSC.
- S. Pavlik, K. Mereiter, M. Puchberger and K. Kirchner, Organometallics, 2005, 24, 3561–3575 CrossRef CAS.
- I. Cano, M. A. Huertos, A. M. Chapman, G. Buntkowsky, T. Gutmann, P. B. Groszewicz and P. W. N. M. v. Leeuwen, J. Am. Chem. Soc., 2015, 137, 7718–7727 CrossRef CAS PubMed.
- E. H. Wong, F. C. Bradley, L. Prasad and E. J. Gabe, J. Organomet. Chem., 1984, 263, 167–177 CrossRef CAS.
- C. Zeiher, J. Mohyla, I. P. Lorenz and W. Hiller, J. Organomet. Chem., 1985, 286, 159–170 CrossRef CAS.
- A. D. Burrows, M. F. Mahon, M. T. Palmer and M. Varrone, Inorg. Chem., 2002, 41, 1695–1697 CrossRef CAS PubMed.
- Y. Zhao, Y. Zhou, T. Chen, S.-F. Yin and L.-B. Han, Inorg. Chim. Acta, 2014, 422, 36–39 CrossRef CAS.
- Y. Zhou, S. Yin, Y. Gao, Y. Zhao, M. Goto and L.-B. Han, Angew. Chem., Int. Ed., 2010, 49, 6852–6855 CrossRef CAS PubMed.
- F. C. Bradley, E. H. Wong, E. J. Gabe, F. L. Lee and Y. Lepage, Polyhedron, 1987, 6, 1103–1110 CrossRef CAS.
- J. Bravo, J. Castro, S. García-Fontán, M. C. Rodríguez-Martínez and P. Rodríguez-Seoane, Eur. J. Inorg. Chem., 2006, 2006, 3028–3040 CrossRef.
- D. J. Irvine, D. J. Cole-Hamilton, J. Barnes and P. K. G. Hodgson, Polyhedron, 1989, 8, 1575–1577 CrossRef CAS.
- E. Piras, B. Powietzka, F. Wurst, D. Neumann-Walter, H.-J. Grützmacher, T. Otto, T. Zevaco and O. Walter, Catal. Lett., 2013, 143, 673–680 CrossRef CAS.
- M. P. Duffy, Y. Lin, L. Y. Ting and F. Mathey, New J. Chem., 2011, 35, 2001–2003 RSC.
- S. Hoehne, E. Lindner and J.-P. Gumz, Chem. Ber., 1978, 111, 3818–3822 CrossRef CAS.
- F. A. Cotton, L. R. Falvello, M. Tomas, G. M. Gray and C. S. Krainhanzel, Inorg. Chim. Acta, 1984, 82, 129–139 CrossRef CAS.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- M. H. Pérez-Temprano, J. A. Casares, Á. R. d. Lera, R. Álvarez and P. Espinet, Angew. Chem., Int. Ed., 2012, 51, 4917–4920 CrossRef PubMed.
- F. Faraone, G. Bruno, S. L. Schiavo, G. Tresoldi and G. Bombieri, J. Chem. Soc., Dalton Trans., 1983, 433–438 RSC.
- M. M. Olmstead, C. L. Lee and A. L. Balch, Inorg. Chem., 2002, 21, 2712–2716 CrossRef.
- M. Knorr and C. Strohmann, Organometallics, 1999, 18, 248–257 CrossRef CAS.
- C. S. Browning and D. H. Farrar, J. Chem. Soc., Dalton Trans., 1995, 521–530 RSC.
- P. Braunstein, J. Durand, X. Morise, A. Tiripicchio and F. Ugozzoli, Organometallics, 2000, 19, 444–450 CrossRef CAS.
- S. D. Orth, M. R. Terry, K. A. Abboud, B. Dodson and L. McElwee-White, Inorg. Chem., 1996, 35, 916–922 CrossRef CAS PubMed.
- Y. Yang, K. A. Abboud and L. McElwee-White, Dalton Trans., 2003, 4288–4296 RSC.
- K. K. Cheung, T. F. Lai and K. S. Mok, J. Chem. Soc. A, 1971, 1644–1647 RSC.
- C. D. Nicola, Effendy, F. Fazaroh, C. Pettinari, B. W. Skelton, N. Somers and A. H. White, Inorg. Chim. Acta, 2005, 358, 720–734 CrossRef.
- Z. Yu, Q.-F. Zhang, Y. Song, W.-Y. Wong, A. Rothenberger and W.-H. Leung, Eur. J. Inorg. Chem., 2007, 2007, 2189–2197 CrossRef.
- S. Jamali, M. Rashidi, M. C. Jennings and R. J. Puddephatt, Dalton Trans., 2003, 2313–2317 RSC.
- Q.-Y. Cao, B. Yin and J.-H. Liu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m2730–m2731 CrossRef CAS.
- D. Li, X. Sun, N. Shao, G. Zhang, S. Li, H. Zhou, J. Wu and Y. Tian, Polyhedron, 2015, 93, 17–22 CrossRef CAS.
- S. Pal, N. Kathewad, R. Pant and S. Khan, Inorg. Chem., 2015, 54, 10172–10183 CrossRef CAS PubMed.
- J. S. Field, J. Grieve, R. J. Haines, N. May and M. M. Zulu, Polyhedron, 1998, 17, 3021–3029 CrossRef CAS.
- T.-H. Huang, M.-H. Zhang, M.-L. Tao and X.-J. Wang, Synth. React. Inorg. Met.-Org. Chem., 2014, 44, 986–990 CrossRef CAS.
- M. I. Bruce, B. K. Nicholson, B. W. Skelton, A. H. White and N. N. Zaitseva, Inorg. Chim. Acta, 2016, 453, 647–653 CrossRef CAS.
- H. Liu, M. J. Calhorda, M. G. B. Drew, V. Félix, J. Novosad, L. F. Veiros, F. F. d. Biani and P. Zanello, J. Chem. Soc., Dalton Trans., 2002, 4365–4374 RSC.
- K. Maitra and J. H. Nelson, Polyhedron, 1998, 18, 203–210 CrossRef.
- M. Dahlen, E. H. Hollesen, M. Kehry, M. T. Gamer, S. Lebedkin, D. Schooss, M. M. Kappes, W. Klopper and P. W. Roesky, Angew. Chem., Int. Ed., 2021, 60, 23365–23372 CrossRef CAS PubMed.
- C.-M. Che, H.-L. Kwong, V. W.-W. Yam and K.-C. Cho, J. Chem. Soc., Chem. Commun., 1989, 885–886 RSC.
- R. Provencher and P. D. Harvey, Inorg. Chem., 1996, 35, 2235–2241 CrossRef CAS PubMed.
- D. Li, C.-M. Che, W.-T. Wong, S.-J. Shieh and S.-M. Peng, J. Chem. Soc., Dalton Trans., 1993, 653–654 RSC.
- J. K. Bera, M. Nethaji and A. G. Samuelson, Inorg. Chem., 1999, 38, 218–228 CrossRef CAS.
- G. R. M. Dowson, M. F. Haddow, J. Lee, R. L. Wingad and D. F. Wass, Angew. Chem., Int. Ed., 2013, 52, 9005–9008 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |