
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Poly(2-ethyl-2-oxazoline-co-N-propylethylene imine)s by controlled partial reduction of poly(2-ethyl-2-oxazoline): synthesis, characterization and cytotoxicity†
Sebastian
Halupczok
*a,
Maria
Pfister‡
*a,
Annemarie
Ringhand
*a,
Corinna
Fetsch§
a,
Alevtina
Cubukova
b,
Antje
Appelt-Menzel
bc and
Robert
Luxenhofer
 ad
ad
aPolymer Functional Materials, Chair for Advanced Materials Synthesis, Department for Chemistry and Pharmacy, Julius-Maximilians-Universität Würzburg, Röntgenring 11, 97070 Würzburg, Germany. E-mail: robert.luxenhofer@uni-wuerzburg.de
bFraunhofer Institute for Silicate Research ISC, Translational Center Regenerative Therapies TLC-RT, Röntgenring 11, 97070 Würzburg, Germany
cUniversity Hospital Würzburg, Chair Tissue Engineering and Regenerative Medicine (TERM), Röntgenring 11, 97070 Würzburg, Germany
dSoft Matter Chemistry, Department of Chemistry, University of Helsinki, 00014 Helsinki, Finland
First published on 12th November 2020
Abstract
The partial reduction of poly(2-ethyl-2-oxazoline) was investigated. A series of poly(2-ethyl-2-oxazoline-co-N-propylethylene imine)s were synthesized by direct reduction using lithium aluminum hydride or borane/dimethylsulfide (BH3/DMS), respectively. It is shown that the degree of reduction can be readily controlled either by the reaction time when using an excess of LiAlH4 or by the stoichiometry of BH3/DMS, as was demonstrated by 1H-NMR spectroscopy. Differential scanning calorimetry revealed that the glass transition temperature of the products decreased with increasing degree of reduction up to 25% of reduction, above which no glass transition could be detected. Moreover, acid–base titration showed a very pronounced, reduction degree dependent buffering capacity of these polymers between pH 4 and 8, which is of great interest, e.g. in the context of endosomal escape. This control over the reduction allows to tailor the synthesis of partially cationic polymers on the basis of poly(2-oxazoline)s, which, in combination over the hydrophilic/lipophilic balance through the side chain length allows a tight control over materials properties. Such materials may be interesting, inter alia, for biomaterials or organic electronics.
Introduction
Poly(ethylene imine) (PEI) is a widely used cationic polymer that can be prepared in two different architectures, linear and branched (Scheme 1). PEI is commonly used as a flocculant and as a retention aid in paper production or in the textile industry. In the biomedical community, PEI is better known for its use as a transfection agent1,2 or as a protein/nucleic acid precipitation agent.3 | ||
| Scheme 1 a) Reaction scheme for the preparation of (a) branched and (b) linear poly(ethylene imine), which is well known from the literature. (c) Reaction scheme for the (partial) reduction of poly(2-alkyl-2-oxazoline)s to poly(N-alkyl ethylene imine)s via poly(2-alkyl-2-oxazoline-co-N-alkylethylene imine)s presented herein. | ||
While the branched PEI version is prepared by cationic polymerization of aziridine (Scheme 1a), the linear version is prepared in two steps. The first step is the cationic ring-opening polymerization of 2-oxazolines. The second step is the exhaustive hydrolysis of the intermediate non-ionic poly(2-oxazoline), which yields linear PEI (Scheme 1b). However, poly(2-oxazoline)s can also be transformed into cationic polymers by another strategy, which has been previously described, but much less investigated. The amide motif in the repeating unit can be reduced to a tertiary amine (Scheme 1c).4,5 The resulting polymers are known as poly(N-alkyl ethylene imine)s (PAEI). PAEI can also be obtained by alkylation of PEI employing the Leuckart–Wallach reaction. To date and the best of our knowledge, this has been realized only for methylation using the Eschweiler–Clarke reductive N-methylation. In fact this approach predates the amide reduction.6,7
Besides the difference of secondary (PEI) vs. tertiary amine (PAEI), the PAEI are much more versatile with respect to their physico-chemical properties by virtue of the N-substituent. Interestingly, while thousands of scientific reports deal with PEI one way or the other, hardly more than a handful of reports dealing with synthesis,4–7 properties4,8 and use8–10 of PAEI can be found in the literature and these few reports have received remarkably little attention. More recently, Fukuda et al. investigated the solution properties of poly(N-methylethylene imine) synthesized via the Eschweiler–Clarke methylation and claim this as an interesting water-soluble polycationic material.8
To the best of our knowledge, all previous reports in the context of PAEI discussed the synthesis and properties of PAEI homopolymers. However, we hypothesized that partial reduction of POx should lead to copolymers of POx and PAEI (P(Ox-co-AEI)), but this has not been described to date. This contribution investigates the partial reduction of POx using two different reducing agents, BH3 and LiAlH4, previously described for the exhaustive reduction of POx.4,5 The partially reduced POx were characterized via1H-NMR spectroscopy, differential scanning calorimetry and acid–base titration. Finally, selected samples were investigated also with respect to their cytotoxicity using human primary dermal fibroblasts.
Materials and methods
Materials
All chemicals and reagents were purchased from Sigma-Aldrich or Acros and used as received unless stated otherwise. The poly(2-ethyl-2-oxazoline) had a molar mass of 50 kg mol−1 and a dispersity of 3–4 (SiAl product number #372846).Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2). The polymer was precipitated in Et2O, dissolved in water and lyophilized.
:2). The polymer was precipitated in Et2O, dissolved in water and lyophilized.
NMR analysis of the reduction degree
NMR spectra were recorded on a Fourier 300 (1H; 300.12 MHz), Bruker Biospin (Rheinstetten, Germany) at 298 K. The spectra were calibrated to the signal of residual protonated solvent (CDCl3: 7.26 ppm). For the determination of the degree of reduction, the ratio of the integrals of signals at 1.12 and 0.85 ppm was used.Differential scanning calorimetry (DSC)
For DSC studies, samples were placed into flat-bottom aluminum pans with crimped-on lids and heated/cooled on a calibrated DSC 204 F1 Phoenix equipped with a CC200 F1 Controller, (NETZSCH, Selb, Germany). The dynamic scans were recorded in nitrogen atmosphere with a heating rate of 5 °C min−1 (0°–180 °C).Titration
Titration curves were recorded on 905 Titrando (Metrohm, Filderstadt, Germany). The samples were dissolved in 0.1 M HCl und were titrated with a 0.1 M NaOH standard solution at an average rate of 0.5 mL per minute.Cell viability tests
Human primary dermal fibroblasts (hDF) were isolated from skin tissue biopsies (University Hospital Würzburg, local ethics approval: 182/10, 25.11.2015) and maintained in DMEM (Gibco, 32430-027) + 10% FCS (PAN-Biotech, P30-3306) in adherent culture as previously described.12 HDF were applied for viability studies from passage 2 to 4. Cells were seeded in a cell density of 1 × 104 cells per well (A = 0.34 cm2) into 96-well plates (Greiner Bio-one, 655983) and pre-cultured in a total medium (DMEM + 10% FCS) volume of 200 μL per well for 24 h. As a positive control 1% SDS (Carl Roth GmbH + Co. KG, 4260) solution was used to induce cytotoxic effects. Cytotoxicity of the test substances was evaluated after 48 h of incubation for concentrations covering a wide range from 0.05 to 2000 mg L−1 (for P(EtOx0.40-co-NPrEI0.60) only up to 200 mg L−1 because of its limited solubility). After treatment, CellTiter-Glo® Luminescent Cell Viability Assay (Promega, Mannheim, Germany, G7571) was performed according to the manufacturer's instructions. Luminescence was measured with an Infinite M200 fluorescence reader (Tecan Group, Switzerland). Cytotoxicity studies were performed in triplicates of three independent biological replicates, results are presented as means ± SEM.Results and discussion
Different reagents can be used for the reduction of tertiary amides to amines. For the reduction of POx to PAEI, Saegusa5 and Hoogenboom4 described the use of LiAlH4 and BH3/DMS, respectively. In these reports, the POx was always fully reduced to the PAEI. In contrast, the purpose of the present study was to investigate the partial reduction of poly(2-ethyl-2-oxazoline) (PEtOx) to access novel poly(2-ethyl-2-oxazoline-co-N-propylethylene imine)s (P(EtOx-co-NPrEI)) (Scheme 2). | ||
| Scheme 2 Reaction scheme for the partial reduction of poly(2-ethyl-2-oxazoline) with either LiAlH4 or BH3/DMS as reducing agent in cyclic ethers at refluxing temperature. | ||
The degree of reduction was determined using 1H-NMR spectroscopy. The 1H-NMR spectra of PEtOx are characterized by three main signal groups, the backbone protons give rise to a signal around δ = 3.4–3.5 ppm, the side chain methylene group at δ = 2.2–2.5 ppm and the methyl group at δ = 1.0–1.2 ppm (Fig. 1, signals A, B and C). The PNPrEI gives rise to signals at δ = 2.6 ppm attributed to the backbone protons, at δ = 2.4 ppm from the side chain methylene group adjacent to the nitrogen while at δ = 1.4 ppm appear the signals attributed to the central methylene group of the propyl side chain (Fig. 1, signals D, E and F). Finally, the side chain methyl group gives rise to signals at δ = 0.8 ppm (Fig. 1, signal G). As can be expected, the partially reduced samples show more complex 1H-NMR spectra (Fig. 1) which could complicate analysis of the reduction degree. Fortunately, however, two signals, one per respective repeat unit, remain relatively isolated at various reduction degrees and can be used for the determination of the degree of reduction. These signals are the two signals of the methyl groups in the side chains at δ = 1.1 ppm (Fig. 1, signal C) and δ = 0.8 ppm (Fig. 1, signal G). Accordingly, the degree of reduction was calculated as the ratio of the integrals IG/(IG + IC). The absence of the sharp signal D until rather high degrees of reduction suggests that the reduction is occurring randomly along the polymer chain instead of in a block-like fashion.
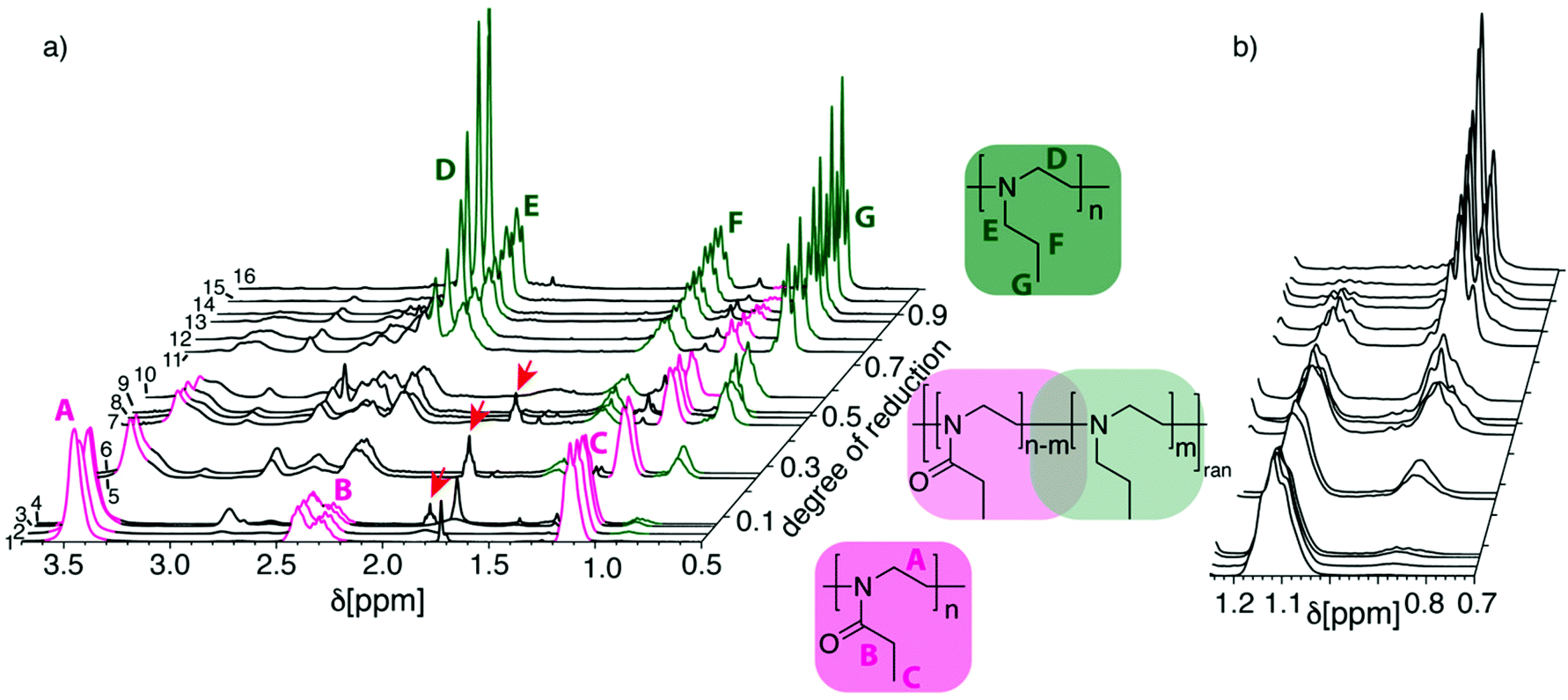 | ||
| Fig. 1 (a) Waterfall plot of NMR spectra in CDCl3 of reactions with LiAlH4 depicted in Table 1. (b) Detailed view of the two signals at 1.12 ppm and 0.85 ppm, which were used for the determination of the degree of reduction. The numeration of traces in (a) corresponds to numeration of experiments in Table 1. | ||
We decided to investigate the reduction of PEtOx with both previously reported reducing agents. Initially, we studied whether the reduction could be controlled by the stoichiometry of the added LiAlH4. Important to note, one eq. of LiAlH4 should be able to reduce 2 eq. of amide.13 As it turned out, this strategy does allow partial reduction of POx, but only with limited degree of control. In five different reactions, we added 30%, 30%, 50%, 70% and 150% of reduction equivalents (with respect to amide groups in the polymer) of LiAlH4. After 68 h at 313 K, the degrees of reduction, determined by 1H-NMR were 3%, 6%, 25%, 46% and 100%, respectively (Table 1). In the first experiment carried out in THF, we observed phase separation of a gel-like material during the reaction. This can be attributed to the reduced solubility of the partially reduced POx. Therefore, and in accordance to earlier reports, the following experiments were carried out in dichloromethane. However, also in the first experiment with DCM, we observed precipitation, presumably because the concentration was too high. Subsequent experiments were carried out at a lower concentration and no precipitation was observed.
The precipitation presumably is also the reason for the very low degree of reduction observed in the first experiments, which was much lower than expected from the stoichiometry. However, also in the absence of precipitation, the determined degree of reduction did not correspond satisfyingly with the stoichiometry. Nevertheless, these first experiments clearly showed that partial reduction of POx can be used to obtain the desired P(EtOx-co-NPrEI) copolymers.
It is known from literature that sub-stoichiometric use of LiAlH4 can lead to C–N bond scission and formation of alcohols or aldehydes, which, in our case is synonymous with side chain scission.14 Indeed, in the NMR of the latter two reactions, we do see some evidence of the corresponding side product 1-propanol in the 1H-NMR spectra (Fig. 1, red arrowheads). However, in the majority of cases, we found no evidence of this side reaction.
In a second set of experiments, we added an excess of LiAlH4 (1.5 eq. per amide) and quenched the reaction after predetermined times (313 K, 0.5, 1, 2, 4, 24 and 68 h) by addition of an excess of water. After workup, NMR spectroscopy was again used to determine the degree of reduction. We realized that even after only 30 min the reduction was almost quantitative and after 1 h only signals of the fully reduced PNPrEI were observed (data not shown). Interestingly, Micovic and Mihailovic already reported in 1955 that the reduction of tertiary amides is very fast in spite of a majority of researchers conducting the reaction for prolonged times.13
Therefore, we investigated the kinetics of the reduction by conducting the PEtOx reduction directly in the NMR tube at 295 K. For this, 5 mg of LiAlH4 (1.3 eq./[amide]) were dispersed in 0.1 mL of THF-d8 in a screwcap NMR tube and 10 mg of PEtOx dissolved in 0.6 mL dichloromethane were added. Immediately, NMR spectra were acquired over the course of 300 min. Under these conditions, again, a very fast reduction of the polymer was observed. Even at the earliest measurement (≈3 min), the degree of reduction was already more than 20% and after 60 min, the degree of reduction reached 90% (Fig. 2). For 50% reduction, the reaction time under these conditions was approx. 10 min. Notably, formation of 1-propanol was not observed.
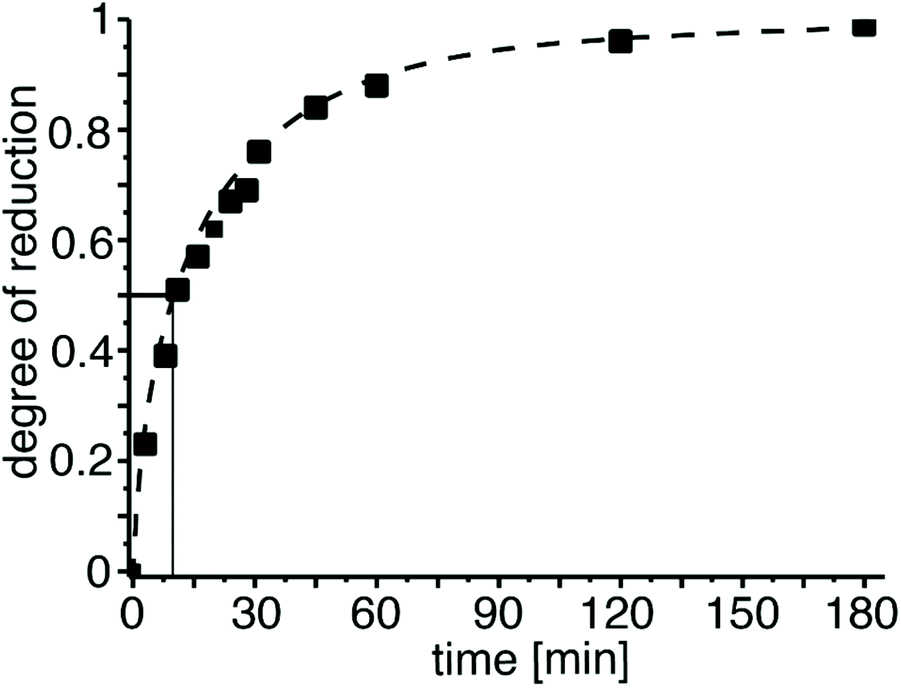 | ||
| Fig. 2 1H-NMR kinetic investigation of the reduction of PEtOx using an excess of LiAlH4 at 298 K. The degree of reduction was determined using the signals of the terminal methyl groups in the side chains at 1.1 ppm (PEtOx) and 0.8 ppm (PNPrEI), respectively. | ||
Hoogenboom and co-workers reported on the exhaustive reduction of POx, including PEtOx using borane/dimethylsulfide (BH3/DMS)4 following a procedure reported by Perner and Schulz.11 Accordingly, we investigated the partial reduction using this reagent. Subsequent refluxing in MeOH transforms the remaining reducing agent into the more volatile trimethoxyborane, which can easily be removed under reduced pressure. Similar as with LiAlH4, we investigated the use of substoichiometric amounts of reducing agent to control the partial reduction of POx (Table 1). Again, the products were characterized by 1H-NMR (Fig. 3). In contrast to the situation with LiAlH4, BH3/DMS enables an improved control of the partial reduction via the stoichiometry. The calculated degrees of reduction corroborated reasonably well with the targeted values. Thus, the degree of partial reduction could be readily controlled from 10% to about 80%.
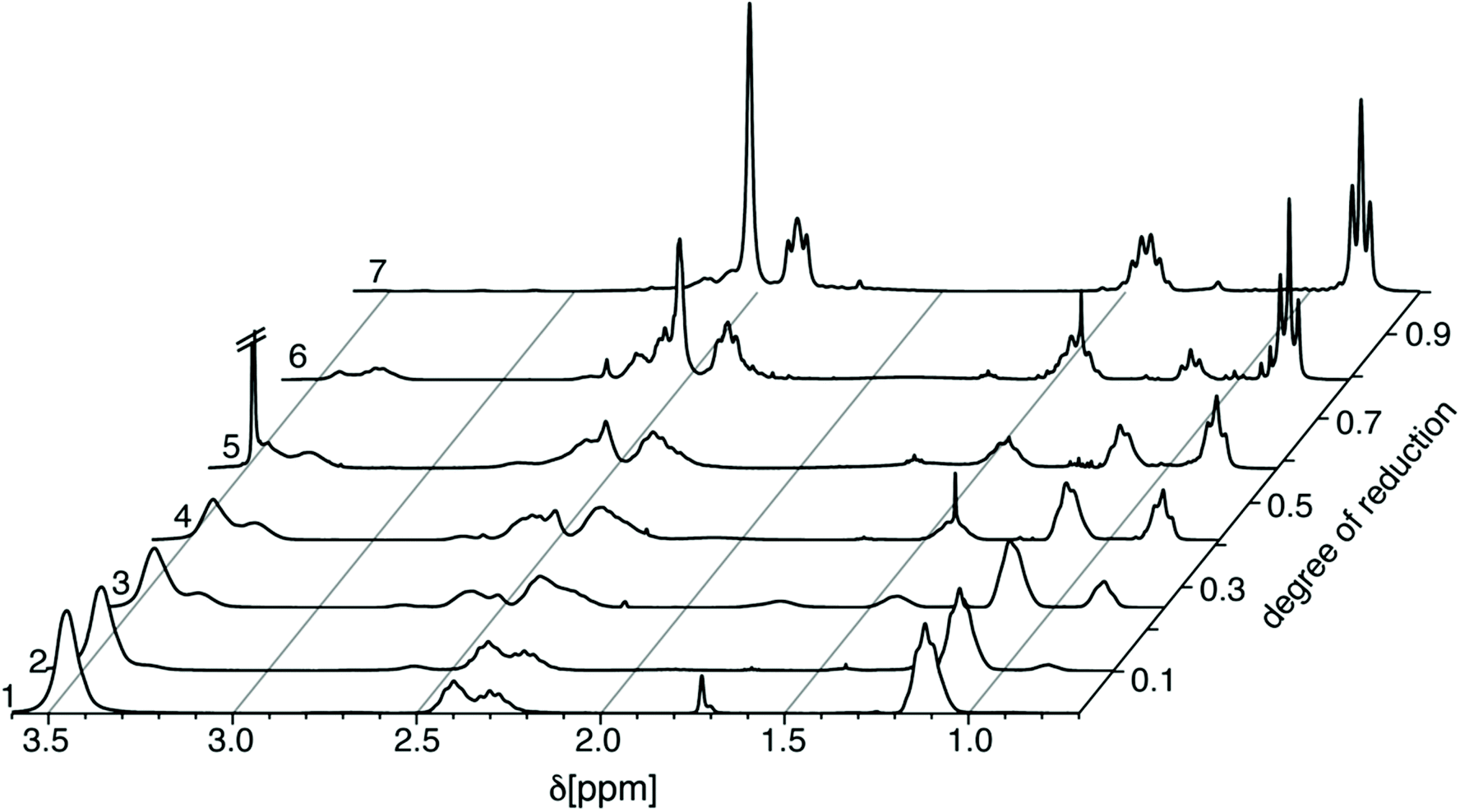 | ||
| Fig. 3 1H-NMR spectra of PEtOx (trace 1), partially (P(EtOx-co-NPrEI, traces 2–6) and fully reduced PEtOx (PNPrEI, trace 7) with BH3/DMS at 343 K. | ||
Hoogenboom and co-workers also compared the thermal properties of POx and PAEI by means of differential scanning calorimetry.4 In the case of fully reduced PEtOx, i.e. PNPrEI, they reported that neither Tg nor Tm could be detected. Since PEtOx is well known to have a Tg of around 330 K, we were curious how the Tg changes with the degree of reduction. We found that at 10% reduced side chains, the Tg shifted to 313 K (Fig. 4). Increasing the degree of reduction further to 25%, the Tg decreased to 293 K. Samples with an even higher degree of reduction (≥40%) did not show a Tg in our experimental setup, which agrees well with the observation by Hoogenboom and co-workers.
 | ||
| Fig. 4 A DSC heating curves of poly(2-ethyl-2-oxazoline) (PEtOx) and partially reduced samples with a degree of reduction of 0.1, i.e. P(EtOx0.9-co-NPrEI0.1) and 0.25, i.e. P(EtOx0.75-co-NPrEI0.25). | ||
The water solubility of several POx shows a dependence on the temperature.15–18 Regarding the molar mass and the polymer architecture, the lower critical solution temperature (LCST) of PEtOx ranges between 343 to about 370 K. Hoogenboom et al. reported that PNPrEI is insoluble in water, which shows the influence and importance of the tertiary amide motif in POx for the solubility. It will be interesting to study the influence of partial reduction on the LCST, but this is outside the scope of the current study. Here, the focus is on studying the buffering capacity of the partially reduced POx. This is a relevant property for a number of applications, for example for the complexation of proteins in nanozymes during the formation of IPECs (“interpolyelectrolyte complexes” formed with two polyelectrolytes of opposite charge) or the complexation with nucleic acids (polyplexes).1,19–24 Accordingly, we investigated the acid–base titration of solution of PEtOx (as control) as well as the partially P(EtOx1−x-co-NPrEIx) copolymers. The polymers were dissolved in 0.1 M HCl and titrated against 0.1 M NaOH (Fig. 5). Important to note, the polymer with 73% reduction was fully soluble at 2 mg mL−1 in 0.1 M HCl but precipitation was observed starting at a pH between 7 and 8 (ESI, Fig. S1†). Compared with 5 mg mL−1 weight concentration, the titration curve shows an abrupt change and drastic increase of the slope starting at pH 7.5, which also indicates precipitation of the polymer with higher reduction degrees, which was confirmed visually. Therefore, the 2-oxazoline repeating units are important for the solubility in aqueous medium and a more detailed investigation into the solubility of partially reduced PEtOx at different temperatures and pH values is warranted.
 | ||
| Fig. 5 Titration curves at different degrees of reduction, ranging between 17–73%. The titrations were performed at the weight concentration of 5 mg mL−1. Polymers were dissolved in 0.1 M HCl and titrated with 0.1 M NaOH. The titrations were carried out in duplicate or triplicate. The reference titration is in dotted lines, the individual titration curves are shown in black solid lines while the average is orange solid lines. The horizontal grey segments define the best pH buffer range of the respective copolymers. | ||
As can be expected, PEtOx does not show any effect in the acid–base titration (used as reference in titrations, Fig. 5 and 6) as tertiary amides are not ionized between pH 2 and 12. We are aware that Hsiue and co-workers and a few other authors repeatedly suggested a pH responsive character of POx,25,26 but this seems unlikely to be due to any protonation of tertiary amides at this low acid molarity. We suspect that the effect observed may have been due to end-groups or other impurities. In contrast to POx, all reduced samples clearly show buffering properties during the potentiometric pH titration. Interestingly, the buffer capacity appears to be strongly influenced by the degree of reduction, clearly hinting at cooperative effects, or neighbor group effects27 within the polymer as has been described in different polyelectrolytes including poly(vinylamine).28,29 Additionally, the buffering capacity is dependent on the weight concentrations.
 | ||
| Fig. 6 Averaged titration curves of the copolymers with a degree of reduction of 25% (left) and 50% (right), respectively (orange curves). The green lines show the gradient of the averaged titration curves. The buffer area is located between the two maxima of the gradient and is indicated by the dotted vertical lines. The grey horizontal box highlights the pH-buffer range as determined by a reduced slope of the titration curve. The enlarged section is added to highlight the near-linear behavior of the titration curve inside the buffer range without the first derivative. | ||
The first derivative of the titration curves can be used to determine the pH-buffer range, which is localized between the two maxima of the first derivative (Fig. 6). The lower slope and change of direction indeed show the buffer capacity of the polymers. The broader buffer volume also proves the strong influence of the degree of reduction (Fig. 6, double-head arrows). The titration curve reflects to some extent the titration curve of branched PEI as reported by Andresen and co-workers, with a buffering starting already at pH 4–5.20 This is particularly interesting, as at the highest degrees of reduction the buffering starts and is most dominant only at higher pH values (Fig. S2†). The preliminary results presented here warrant a much more detailed investigation of the apparent buffer capacities at different degrees of reduction.
The polymer with every second repeat unit (50%, statistically) reduced shows a very interesting titration behavior showing a particularly linear behavior between pH 4 and 8.5 (Fig. 6). It is immediately apparent that such broad buffering capacity of P(EtOx0.5-co-NPrEI0.5) could be very interesting for a range of applications, including non-viral vectors for gene therapy. For this, an important fact in the endosomal pathway is the pH value range between 7.4 and 4.6 along the endocytic pathway.30,31 Particularly interesting in this context is that the broad buffering capacity is observed at intermediate charge densities. This may have major implications on the cytotoxicity of the corresponding materials.
Accordingly, the cytocompatibility of partially reduced P(EtOx-co-NPrEI) was investigated using human primary dermal fibroblast cells (hDF) and treatment for 48 h. The cells were exposed to concentrations of up to 2 g L−1, which was the approximate limit of solubility for the polymers with the highest degree of reduction at physiological pH. Cell viability was assessed with CellTiter-Glo® assay (Fig. 7).
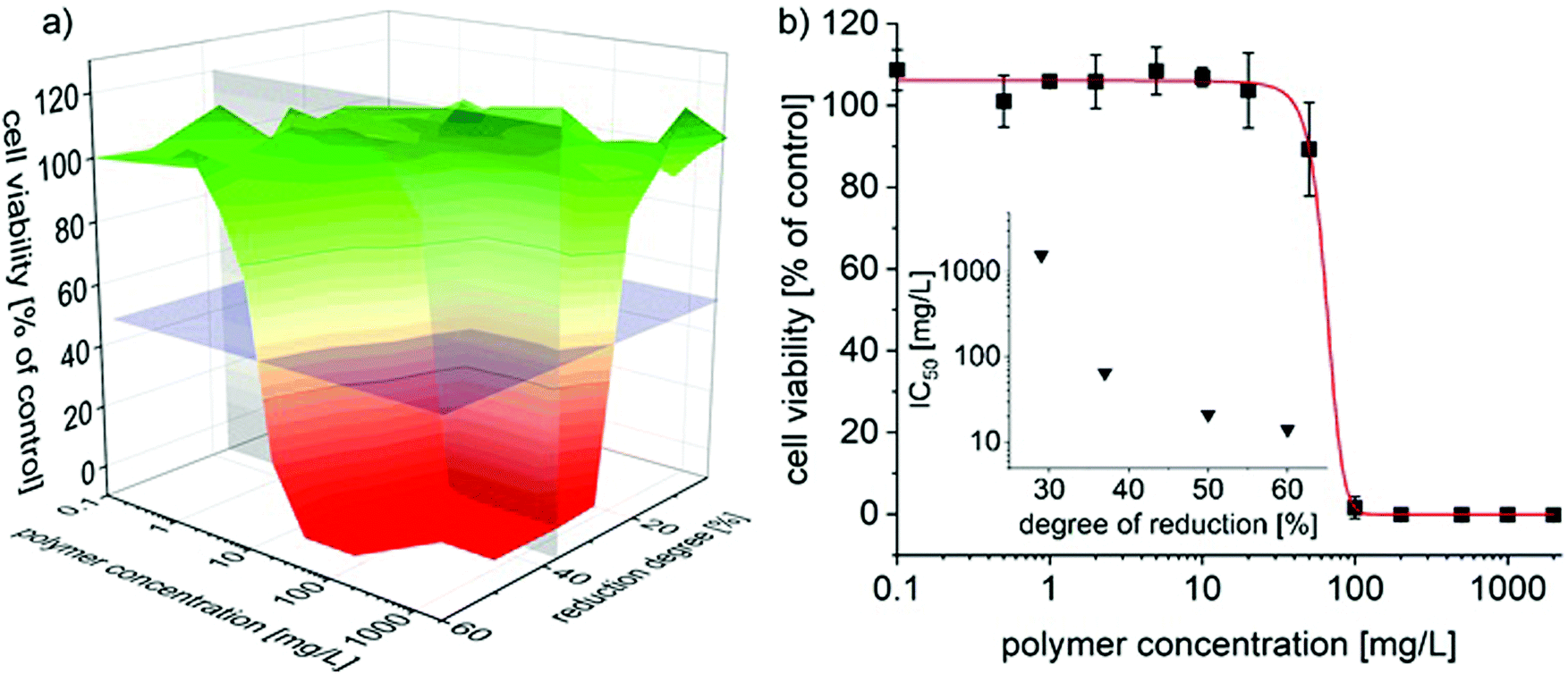 | ||
| Fig. 7 (a) 3D plot of the concentration and reduction degree dependent cell viability of human dermal fibroblasts (hDF) after 48 h of exposure with partially reduced PEtOx500. The blue-gray horizontal plane with the z-intercept 0.5 visualizes the IC50 values. The light gray vertical plane intersects at a reduction degree of 37%. Only at reduction degrees exceeding 20% considerable cytotoxicity is observed at the investigated concentrations and times. (b) Concentration dependent cell viability for P(EtOx0.63-co-NPrEI0.37), with experimental data fitted using a Boltzmann fit (red line). The inset shows that between 25 and 40% reduction degree, the IC50 values drop sharply and appears to level-off thereafter. | ||
For reduction degrees below 25%, no effect on the cell viability was found in the investigated concentration range and incubation time. However, in the case of P(EtOx0.77-co-NPrEI0.23), morphological changes were observed by light microscopy at the highest concentration, even though no significant cytotoxic effect was observed at or below 2 g L−1. It can be assumed that at higher concentrations cytotoxicity will emerge. In contrast, P(EtOx0.71-co-NPrEI0.29) induced severe cytotoxicity at 2 g L−1 (Fig. S3†). Further increase in the degree of reduction results in a very profound decrease of the IC50 values. P(EtOx0.63-co-NPrEI0.37) already results in an estimated IC50 of 50 mg L−1 (Fig. 7b) and the IC50 values of P(EtOx0.5-co-NPrEI0.5) and P(EtOx0.4-co-NPrEI0.6) range between 10 and 20 mg L−1. Hsiue et al.,32 Kronek33 as well Hoogenboom34et al. reported that partially hydrolyzed PEtOx exhibits much lower cytotoxicity compared to PEI and apparently also compared to the presently studied partially reduced POx. For the partially hydrolyzed PEtOx, only hydrolysis degrees of 50% and more lead to considerable cytotoxicity. Hoogenboom et al. also investigated the cytotoxicity of partially hydrolyzed poly(2-n-propyl-2-oxazoline).35 The comparison with these results is particularly interesting, as poly(2-n-propyl-2-oxazoline) is more hydrophobic compared to PEtOx as the partially reduced PEtOx introduced here. The poly(2-n-propyl-2-oxazoline) was hydrolyzed by only 10% and different chain lengths were investigated. Interestingly, irrespective of the chain length, even these very low hydrolysis degrees elicited a very pronounced cytotoxicity in three independent biological assays. IC50 values were in the range of 10–20 μM or <20 mg L−1, albeit at 72 h of incubation time. In comparison, IC50 values of P(EtOx-co-NPrEI) were of an order of magnitude higher for the degrees of reduction of up to 29% and only reached similar values at a reduction degree of 50% or more. Rangelov et al. presented the cytocompatibility of hydrolyzed poly(2-iso-propyl-2-oxazoline)s where the IC50 value of P(iPrOx0.47-co-NEI0.53) is similar to the value observed for P(EtOx0.5-co-NPrEI0.5).36 Unfortunately, the authors did not report cytotoxicity for other hydrolysis degrees. As mentioned before, it might be concluded that the cytocompatibility decreased with the higher hydrophobicity of the 2-oxazoline side chains.
Poly(2-oxazoline)s as a very diverse polymer family have seen a tremendous increase of interest for the preparation of biomaterials.37 Recent years have seen the first-in-man clinical trials for POx-based polymer drug conjugates,38 very significant advances in preclinical studies of POx based micellar drug delivery systems,39–48 and hydrogels for various biomedical applications.49–53 The presented results add another layer of chemical versatility to this already multifarious polymer family, and are particular interesting for potential application for the complexation of oppositely charged biomacromolecules such as proteins and polynucleic acids.
Conclusion
In conclusion, we could show that the controlled reduction of poly(2-ethyl-2-oxazoline)s can be used to prepare poly(2-ethyl-2-oxazoline-co-N-propylethylene imine)s as well as poly(N-propylethylene imine)s after exhaustive reduction. We found that borane/dimethylsulfide complex is a better reagent for this purpose compared to LiAlH4. This is the first report showing the synthesis and some properties of such copolymers. Considering the side chain variability of POx, we believe that our approach can be used to access a large variety of different copolymers varying in hydrophilicity and hydrophobicity and cationic charge density. Such materials could be very interesting in a large variety of applications, including applications as biomaterials or in organic electronics showing acceptable cell viability at moderate degrees of reduction. In contrast to the partially hydrolyzed PEtOx, the partially reduced polymers cause increasing cytotoxicity, which may be due to the stronger interactions between the cell membrane and the polymers with their increasing lipophilic character. However, compared to partially hydrolyzed PnPrOx, the partially reduced PEtOx exhibit an improved cytocompatibility, as up to 17% degree of reduction, the polymers exhibited no cytotoxicity at the highest analyzed concentrations. This highlights the potential to influence physicochemical properties and cytocompatibility utilizing the rich chemistry of poly(2-oxazoline)s.Future studies will have to elucidate the effect of side chains on the rate of the reduction and cytotoxicity, similar to studies investigating the effects of POx side chains on the hydrolysis. Also the reduction of poly(2-oxazine)s should be investigated. Moreover, the investigation of the influence of the degree of reduction on the lower critical solution temperature of the polymers will be interesting to study.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the Free State of Bavaria. Start-up funding for R. L. by the University Würzburg and the German Plastics Center SKZ is gratefully acknowledged.References
- A. Akinc, M. Thomas, A. M. Klibanov and R. Langer, J. Gene Med., 2005, 7, 657–663 CrossRef CAS.
- W. T. Godbey, K. K. Wu and A. G. Mikos, J. Controlled Release, 1999, 60, 149–160 CAS.
- R. R. Burgess, Methods Enzymol., 2009, 463, 331–342 CAS.
- H. M. L. Lambermont-Thijs, L. Bonami, F. E. Du Prez and R. Hoogenboom, Polym. Chem., 2010, 1, 747–754 CAS.
- T. Saegusa, A. Yamada, H. Taoda and S. Kobayashi, Macromolecules, 1978, 11, 435–436 CrossRef CAS.
- R. Tanaka, I. Ueoka, Y. Takaki, K. Kataoka and S. Saito, Macromolecules, 1983, 16, 849–853 CrossRef CAS.
- R. Tanaka, M. Koike, T. Tsutsui and T. Tanaka, J. Polym. Sci., Polym. Lett. Ed., 1978, 16, 13–19 CrossRef CAS.
- Y. Fukuda, D. Abe, Y. Tanaka, J. Uchida, N. Suzuki, T. Miyai and Y. Sasanuma, Polym. J., 2016, 48, 1065–1072 CrossRef CAS.
- R. Tanaka, T. Fujita, H. Nishibayashi and S. Saito, Solid State Ionics, 1993, 60, 119–123 CrossRef CAS.
- T. Saegusa, S. Kobayashi, A. Yamada and S. Kashimura, Polym. J., 1979, 11, 1–6 CrossRef CAS.
- T. Perner and R. C. Schulz, Br. Polym. J., 1987, 19, 181–188 CrossRef CAS.
- F. Karl, M. Wussmann, L. Kress, T. Malzacher, P. Fey, F. Groeber-Becker and N. Uceyler, Ann. Clin. Transl. Neurol., 2019, 6, 1797–1806 CrossRef CAS.
- V. M. Micovic and M. L. Mihailovic, J. Org. Chem., 1955, 18, 1190–1200 CrossRef.
- M. A. Ruttenberg, L. C. Craig and T. P. King, Biochemistry, 1964, 3, 758–764 CrossRef CAS.
- A. Zahoranova, M. Mrlik, K. Tomanova, J. Kronek and R. Luxenhofer, Macromol. Chem. Phys., 2017, 218, 1700031 CrossRef.
- N. Zhang, R. Luxenhofer and R. Jordan, Macromol. Chem. Phys., 2012, 213, 1963–1969 CrossRef CAS.
- N. Zhang, R. Luxenhofer and R. Jordan, Macromol. Chem. Phys., 2012, 213, 973–981 CrossRef CAS.
- R. Hoogenboom and H. Schlaad, Polym. Chem., 2017, 8, 24–40 RSC.
- H. Wei and E. K. Wang, Chem. Soc. Rev., 2013, 42, 6060–6093 RSC.
- R. V. Benjaminsen, M. A. Mattebjerg, J. R. Henriksen, S. M. Moghimi and T. L. Andresen, Mol. Ther., 2013, 21, 149–157 CrossRef CAS.
- D. V. Pergushov, A. H. E. Müller and F. H. Schacher, Chem. Soc. Rev., 2012, 41, 6888–6901 RSC.
- D. S. Manickam, A. M. Brynskikh, J. L. Kopanic, P. L. Sorgen, N. L. Klyachko, E. V. Batrakova, T. K. Bronich and A. V. Kabanov, J. Controlled Release, 2012, 162, 636–645 CrossRef CAS.
- E. V. Batrakova, S. Li, A. D. Reynolds, R. L. Mosley, T. K. Bronich, A. V. Kabanov and H. E. Gendelman, Bioconjugate Chem., 2007, 18, 1498–1506 CrossRef CAS.
- A. Harada and K. Kataoka, Macromolecules, 1998, 31, 288–294 CrossRef CAS.
- C. H. Wang, C. H. Wang and G. H. Hsiue, J. Controlled Release, 2005, 108, 140–149 CrossRef CAS.
- X. Qu, Y. Zou, Y. Jin, Y. Zhou, Z. Wang, C. He, Y. Deng, X. Li, Y. Zhou and Y. Liu, J. Chin. Pharm. Sci., 2017, 26, 582–588 CAS.
- S. Lifson and B. Kaufman, J. Chem. Phys., 1957, 27, 1356–1362 CrossRef CAS.
- C. Chang, F. Fish, D. D. Muccio and T. St. Pierre, Macromolecules, 1987, 20, 621–625 CrossRef CAS.
- A. Katchalsky, J. Mazur and P. Spitnik, J. Polym. Sci., 1957, 23, 513–532 CrossRef CAS.
- C. C. Scott and J. Gruenberg, Bioessays, 2011, 33, 103–110 CrossRef CAS.
- G. Sahay, D. Y. Alakhova and A. V. Kabanov, J. Controlled Release, 2010, 145, 182–195 CrossRef CAS.
- G.-H. Hsiue, H.-Z. Chiang, C.-H. Wang and T.-M. Juang, Bioconjugate Chem., 2006, 17, 781–786 CrossRef CAS.
- R. Shah, Z. Kronekova, A. Zahoranova, L. Roller, N. Saha, P. Saha and J. Kronek, J. Mater. Sci.: Mater. Med., 2015, 26, 157 CrossRef.
- H. P. Van Kuringen, J. Lenoir, E. Adriaens, J. Bender, B. G. De Geest and R. Hoogenboom, Macromol. Biosci., 2012, 12, 1114–1123 CrossRef CAS.
- M. Mees, E. Haladjova, D. Momekova, G. Momekov, P. S. Shestakova, C. B. Tsvetanov, R. Hoogenboom and S. Rangelov, Biomacromolecules, 2016, 17, 3580–3590 CrossRef CAS.
- N. Toncheva-Moncheva, E. Veleva-Kostadinova, C. Tsvetanov, D. Momekova and S. Rangelov, Polymer, 2017, 111, 156–167 CrossRef CAS.
- T. Lorson, M. M. Lübtow, E. Wegener, M. S. Haider, S. Borova, D. Nahm, R. Jordan, M. Sokolski-Papkov, A. V. Kabanov and R. Luxenhofer, Biomaterials, 2018, 178, 204–280 CrossRef CAS.
- R. W. Moreadith, T. X. Viegas, M. D. Bentley, J. M. Harris, Z. H. Fang, K. Yoon, B. Dizman, R. Weimer, B. P. Rae, X. L. Li, C. Rader, D. Standaert and W. Olanow, Eur. Polym. J., 2017, 88, 524–552 CrossRef CAS.
- N. Vinod, D. Hwang, S. H. Azam, A. E. D. Van Swearingen, E. Wayne, S. C. Fussell, M. Sokolsky-Papkov, C. V. Pecot and A. V. Kabanov, Sci. Adv., 2020, 6, eaba5542 CrossRef CAS.
- D. Hwang, T. Dismuke, A. Tikunov, E. P. Rosen, J. R. Kagel, J. D. Ramsey, C. Lim, W. Zamboni, A. V. Kabanov, T. R. Gershon and M. Sokolsky-Papkov, bioRxiv, 2020 DOI:10.15761/BTT.1000104.
- A. Zinger, L. Koren, O. Adir, M. Poley, M. Alyan, Z. Yaari, N. Noor, N. Krinsky, A. Simon, H. Gibori, M. Krayem, Y. Mumblat, S. Kasten, S. Ofir, E. Fridman, N. Milman, M. M. Lübtow, L. Liba, J. Shklover, J. Shainsky-Roitman, Y. Binenbaum, D. Hershkovitz, Z. Gil, T. Dvir, R. Luxenhofer, R. Satchi-Fainaro and A. Schroeder, ACS Nano, 2019, 13, 11008–11021 CrossRef CAS.
- X. M. Wan, J. J. Beaudoin, N. Vinod, Y. Z. Min, N. Makita, H. Bludau, R. Jordan, A. Wang, M. Sokolsky and A. V. Kabanov, Biomaterials, 2019, 192, 1–14 CrossRef CAS.
- A. C. Pöppler, M. M. Lübtow, J. Schlauersbach, J. Wiest, L. Meinel and R. Luxenhofer, Angew. Chem., Int. Ed., 2019, 58, 18540–18546 CrossRef.
- M. M. Lübtow, L. C. Nelke, J. Seifert, J. Kühnemundt, G. Sahay, G. Dandekar, S. L. Nietzer and R. Luxenhofer, J. Controlled Release, 2019, 303, 162–180 CrossRef.
- M. M. Lübtow, M. S. Haider, M. Kirsch, S. Klisch and R. Luxenhofer, Biomacromolecules, 2019, 20, 3041–3056 CrossRef.
- X. M. Wan, Y. Z. Min, H. Bludau, A. Keith, S. S. Sheiko, R. Jordan, A. Z. Wang, M. Sokolsky-Papkov and A. V. Kabanov, ACS Nano, 2018, 12, 2426–2439 CrossRef CAS.
- M. M. Lübtow, L. Hahn, M. S. Haider and R. Luxenhofer, J. Am. Chem. Soc., 2017, 139, 10980–10983 CrossRef.
- Z. J. He, X. M. Wan, A. Schulz, H. Bludau, M. A. Dobrovolskaia, S. T. Stern, S. A. Montgomery, H. Yuan, Z. B. Li, D. Alakhova, M. Sokolsky, D. B. Darr, C. M. Perou, R. Jordan, R. Luxenhofer and A. V. Kabanov, Biomaterials, 2016, 101, 296–309 CrossRef CAS.
- M. M. Lübtow, T. Lorson, T. Finger, F. K. Groeber-Becker and R. Luxenhofer, Macromol. Chem. Phys., 2020, 221, 1900341 CrossRef.
- L. Hahn, M. Maier, P. Stahlhut, M. Beudert, V. Flegler, S. Forster, A. Altmann, F. Töppke, K. Fischer, S. Seiffert, B. Böttcher, T. Lühmann and R. Luxenhofer, ACS Appl. Mater. Interfaces, 2020, 12, 12445–12456 CrossRef.
- T. R. Dargaville, J. R. Park and R. Hoogenboom, Macromol. Biosci., 2018, 18, 1800070 CrossRef.
- D. Van Der Heide, B. Verbraeken, R. Hoogenboom, T. R. Dargaville and D. Hickey, Biomater. Tissue Technol., 2017, 1, 1–5 Search PubMed.
- T. Lorson, S. Jaksch, M. M. Lübtow, T. Jungst, J. Groll, T. Lühmann and R. Luxenhofer, Biomacromolecules, 2017, 18, 2161–2171 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py01258k |
| ‡ Current address: Element Logic Germany GmbH, Hanns-Martin-Schleyer Straße 3, 74177 Bad Friedrichshall, Germany. |
| § Current address: Emil Kiessling GmbH, Am Ohlenberg 8, 64390 Erzhausen, Germany. |
| This journal is © The Royal Society of Chemistry 2021 |