
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diastereoselective synthesis of trisubstituted olefins using a silicon-tether ring-closing metathesis strategy†
Stéphane
Wittmann‡
,
Alexander F.
Tiniakos
and
Joëlle
Prunet
 *
*
WestCHEM, School of Chemistry, University of Glasgow, Joseph Black Building, University Avenue, Glasgow G12 8QQ, UK. E-mail: joelle.prunet@glasgow.ac.uk
First published on 5th March 2020
Abstract
The diastereoselective synthesis of trisubstituted olefins with concomitant C–C bond formation is still a difficult challenge, and olefin metathesis reactions for the formation of such alkenes are usually not high yielding or/and diastereoselective. Herein we report an efficient and diastereoselective synthesis of trisubstituted olefins flanked by an allylic alcohol, by a silicon-tether ring-closing metathesis strategy. Both E- and Z-trisubstituted alkenes were synthesised, depending on the method employed to cleave the silicon tether. Furthermore, this methodology features a novel Peterson olefination for the synthesis of allyldimethylsilanes. These versatile intermediates were also converted into the corresponding allylchlorodimethylsilanes, which are not easily accessible in high yields by other methods.
Introduction
The trisubstituted E olefin motif with a methyl substituent is present in numerous polyketide natural products. Among these, callipeltoside A1 and dolabelide C2 possess another common feature: an allylic alkoxy group on the lone substituent of the trisusbstituted olefin (highlighted in red in Scheme 1). During our previous approaches to the synthesis of dolabelide C,3 we had envisaged appending the side chain by a cross-metathesis (CM) reaction between an allylic alcohol and a gem-disubstituted olefin to form the C24–C25 trisubstituted olefin. Unfortunately, all attempts to perform CM between diverse allylic alcohols (protected or not) and the required gem-disubstituted olefin for dolabelide C led to dimerisation of the allylic alcohol partner.4 Silicon-tether ring-closing metathesis (RCM) reactions with O–Si–O linkages have been widely used for the formation of alkenes, when the corresponding CM reactions are not high yielding or/and diastereoselective.5 Previous works by Kobayashi et al.6 and Evans et al.7 illustrate the use of such linkage for the formation of E and Z trisubstituted olefins, respectively, via eight-membered rings (Scheme 2). | ||
| Scheme 1 Structures of callipeltoside A and dolabelide C, highlighting the trisubstituted olefin motif, adapted from ref. 10. | ||
 | ||
| Scheme 2 Differences between previous RCM with silicon tethers leading to trisubstituted olefins in previous works and our work. | ||
However, there are much fewer examples of RCM reactions with O–Si–C tethers. The reported formations of trisubstituted olefins from allyl silanes lead to bicyclic products,8 and the others focus on vinyl silanes.9 We propose a strategy for an efficient and diastereoselective synthesis of trisubstituted olefins flanked by an allylic alcohol, which involves a RCM reaction with a O–Si–C tether. In our preliminary communication,10 the metathesis precursors C were prepared by dehydrogenative coupling of allylic alcohols A and allyldimethylsilanes B, and the metathesis products D were converted into the E olefin (Scheme 2). Herein, we report our preliminary attempts to synthesise compounds C and a synthesis of versatile allylchorodimethylsilanes from silanes B. Furthermore, the Z olefin isomer can also be accessed by choosing the appropriate method to cleave the tether (Scheme 2).
Results and discussion
The preparation of the RCM substrates C we first envisioned is shown in Scheme 3: it would entail silyl ether formation between allylic alcohols A and allylchlorodimethylsilanes B′.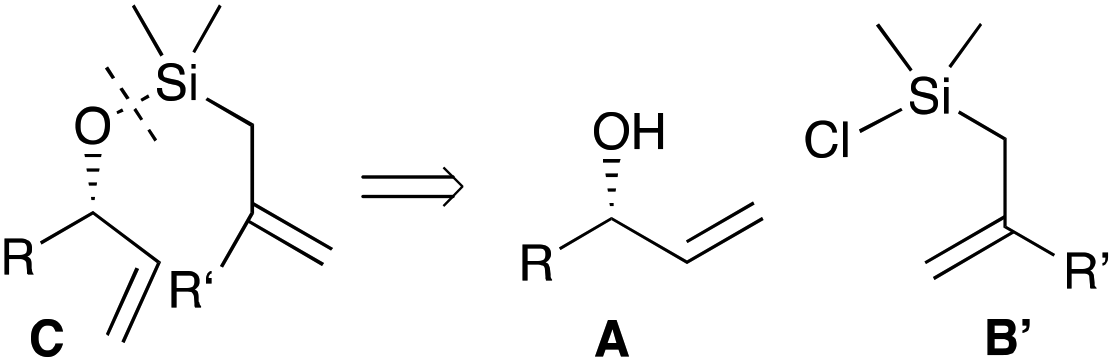 | ||
| Scheme 3 First route for the preparation of RCM substrates C. | ||
The first issue to address was the preparation of compounds B′. Allylchlorodimethylsilanes can be prepared by reaction of the corresponding allyl Grignard reagents with dichlorodimethylsilane, but the yields are usually low. For example, allylchlorodimethylsilane was synthesised in 44% yield11 using this method, and the methallyl derivative in 39% yield.12 Organoindium derivatives have been reported to favour monoaddition of allyl nucleophiles, thus leading to better yields of allylchlorosilanes.13 As a preliminary trial, allylbromide, when treated with indium and dichlorodimethylsilane in 1,3-dimethyl-2-imidazolidinone (DMI), led to allylchlorodimethylsilane, which was not isolated but directly reacted with alcohol 1 in the presence of triethylamine and DMAP to furnish the desired silyl ether 2 in 56% yield (Scheme 4). RCM reaction of compound 2 with Grubbs 2 catalyst proceeded smoothly14 to furnish cyclic silyl ether 3 in 74% yield, thus validating our strategy.
 | ||
| Scheme 4 First attempt at allylchlorosilane formation using indium, followed by silicon-tether RCM reaction. | ||
We then prepared several allylic bromides with a gem-disubstituted olefin, which are models or racemic versions of the side chain of dolabelide C, by a known method.15 Esters 4a–c10 were subjected to trimethylsilylmethylmagnesium chloride in the presence of anhydrous cerium trichloride, and the resulting β-hydroxysilanes were stirred with silica gel in dichloromethane to furnish trimethylsilanes 5a–c in 61–91% yield (Scheme 5). Bromination of the Peterson olefination products 5a–c was performed with pyrrolidone hydrotribromide 6 in THF to give the allylic bromides 7a–c in 88–91% yield.
 | ||
| Scheme 5 Preparation of allylic bromides by a Peterson olefination/bromination sequence. | ||
The allylchlorodimethylsilane formed in situ from 7a and dichlorodimethylsilane in the presence of indium, was treated with alcohol 1 to furnish silyl ether 8a in 58% yield (Scheme 6). Unfortunately, this reaction was not reproducible, and variable yields were obtained after further attempts. Moreover, compounds 8b and 8d could not be prepared by this method. These poor results might be due to the additional steric hindrance present in 7b and 7d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16 (R = OTES and OPMB, respectively), which renders the indium derivatives less reactive.17 When magnesium was employed instead of indium with compound 7b, silyl ether 8b was produced in only 4% yield, along with several side-products resulting from the cleavage of the triethylsilyl ether. The same reaction was tried on the PMB derivative 7d; however, in this case, only the Würtz coupling product was obtained. This Barbier-type reaction was also attempted with zinc,16,18 but the desired silyl ethers 8a,b,d were never observed.
16 (R = OTES and OPMB, respectively), which renders the indium derivatives less reactive.17 When magnesium was employed instead of indium with compound 7b, silyl ether 8b was produced in only 4% yield, along with several side-products resulting from the cleavage of the triethylsilyl ether. The same reaction was tried on the PMB derivative 7d; however, in this case, only the Würtz coupling product was obtained. This Barbier-type reaction was also attempted with zinc,16,18 but the desired silyl ethers 8a,b,d were never observed.
 | ||
| Scheme 6 Synthesis of RCM precursors using indium. | ||
Inspired by a report from Maruoka and coworkers who described the preparation of bromosilanes from the corresponding silanes,19 we converted allylic bromides 7a–c into silanes 9a–c in 81–86% yield, by reaction of the Grignard reagents derived from 7a–c with chlorodimethylsilane (Scheme 7). It was found that slow addition of a solution of the bromide compound to a solution of 3.5 equivalents of magnesium and 1 equivalent of chlorodimethylsilane was necessary to reduce the formation of the Würtz coupling products. Maruoka et al. used bromine for the conversion of the silanes to the bromosilanes, but these conditions are not compatible with the alkene present in 9a–c. Several chlorination conditions were employed – CuCl2, catalytic CuI;20 FeCl3, acetyl chloride21 – without success. Finally, the use of hexachloroethane and catalytic palladium dichloride furnished the chlorosilane intermediates, which were treated with allylic alcohol 1 in the presence of triethylamine and DMAP to give the desired chlorosilanes 8a–c in 47–77% yield.22
 | ||
| Scheme 7 Synthesis of RCM precursors via silanes. | ||
At this point, we were satisfied to have found a reliable method for the synthesis of RCM precursors 8a–cvia the chlorosilanes, but the overall sequence was too long to be practical, so we sought to shorten the route in two different ways. Silyl ethers 8a–c could be prepared directly from silanes 9a–c by a copper-catalysed dehydrogenative coupling,23 bypassing one step and the need to deal with sensitive chlorosilanes. Furthermore, we envisioned a novel Peterson olefination for the synthesis of allyldimethylsilanes 9a–c from esters 4a–c, which would proceed in an analogous fashion as the formation of allyltrimethylsilanes 5a–c (Scheme 5). As previously reported in our communication,10 both steps proceeded uneventfully (Scheme 8). It is worth noting that this new olefination reaction provides an easy access to allyldimethylsilanes, but also to the corresponding chlorosilanes (see Scheme 7).
 | ||
| Scheme 8 Synthesis of RCM precursors by Peterson olefination and dehydrogenative coupling, adapted from ref. 10, showing an improved yield of 9c. | ||
RCM reactions of silyl ethers 8a–c in the presence of the Grubbs 2 catalyst in refluxing dichloromethane gave the cyclic products 10a–c in 63–94% yield. These cyclic silyl ethers are unstable on silica gel, and the yield of 10a could be improved from 63% (ref. 10) to 83% simply by performing the purification by a quick filtration through a silica plug instead of a column chromatography. As previously reported, direct cleavage was attempted on 10b with diverse fluoride sources and Lewis acids, but without success, leading to isomerisation product 13b′ or elimination products 14b and 14b′ (see Table 1 for structures of these compounds).10 We thus resorted to a two-step process, starting with cleavage of the silicon–oxygen bond with methyllithium to give allyltrimethylsilanes 11a–c (Scheme 9).
 | ||
| Scheme 9 RCM reaction and tether cleavage.10 | ||

Removal of the trimethylsilyl group was then optimised on compound 11b (Table 1). Employing Lewis acids such as BF3·OEt2 or Sc(OTf)3 (entries 1 and 2) only led to elimination product 14b′, with the TES group still in place. When 11b was treated with TBAF under mild conditions (entry 3), the starting compound was recovered in full. Product 12b, resulting from direct protodesilylation of the TMS group was obtained under more forcing conditions (entry 4), along with isomerisation product 13b and elimination product 14b; in all those compounds, the TES group had been removed. In order to reduce the amount of elimination product and favor the reprotonation, water was added to the reaction mixture (entry 5); in this case, the desired olefin 12b was produced in 35% yield, along with 35% of the isomerised olefin 13b. Although these conditions were not ideal, they were applied to 11a and 11c, for which the TMS removal would not be disrupted by the TES group cleavage. The desired products were obtained in 68% and 64% yields, respectively, along with 28% of the corresponding isomerisation product in each case (Scheme 9).
Trimethylsilyl group removal was further optimised on compound 11a by varying the fluoride source (Scheme 10). When using solid TBAF hydrate, the reaction was complete after 30 min at low temperature, and the amount of isomerised product 13a was reduced. With TBAF(t-BuOH)4, the amount of isomerised product was further decreased, but the yield of the desired product 12a was only moderate.
 | ||
| Scheme 10 Influence of fluoride source in trimethylsilyl group removal. | ||
As previously reported in our communication,10 this new synthetic route was applied to the synthesis of the C16–C30 fragment of dolabelide C.
We then reasoned that this methodology could also be employed for the construction of Z-trisubstituted olefins, if the silyl substituent was used to extend the chain (Scheme 11). Alcohol 1 was reacted with known methallyldimethylchlorosilane12 in the presence of imidazole and DMAP to furnish silyl ether 15 in quantitative yield.24 Ring-closing metathesis of 15 with the Grubbs 2 catalyst gave cyclic silyl ether 16, which was subjected without purification to Tamao–Fleming oxidation, and diol 17 was obtained in excellent yield for the two steps. In this manner, a terminal alkene (in red in 1) can be converted into a Z-trisubstituted olefin (in red in 17) in three high yielding steps, and the primary alcohol is a convenient handle for subsequent elongation of the chain.
 | ||
| Scheme 11 Formation of Z-trisubstituted olefins by silicon-tether RCM. | ||
Conclusions
In conclusion, we have developed a new methodology for the construction of E-trisubstituted olefins flanked by an allylic alcohol, featuring a silicon-tether ring-closing metathesis reaction. In the course of synthesising the metathesis substrates, we designed a novel Peterson olefination reaction that enabled direct access to allyldimethylsilanes. These allysilanes are versatile intermediates that can be converted into the corresponding chlorosilanes, which are otherwise difficult to obtain in good yields. This methodology was applied to the synthesis of the C16–C30 fragment of dolabelide C, showcasing its practical aspect. Furthermore, Z-trisubstituted olefins could also be prepared when Tamao–Fleming oxidation was used to cleave the silicon tether.Experimental section
General methods
Unless otherwise noted all reactions were carried out under an argon atmosphere. The glassware used was flame-dried under vacuum (∼0.6 mbar) using a heat gun. Air- and moisture-sensitive liquids and solutions were transferred via syringe or stainless steel cannula flushed with argon prior to use. Unless otherwise noted all reagents were obtained from commercial suppliers and used without further purification. All reactions were stirred with a magnetic stirrer. Temperatures refer to bath temperatures. Reaction solvents were collected from an in-house solvent purification system (THF, CH2Cl2, Et2O, CH3CN). Dry ethanol, dry methanol and dry DMF were used from commercial bottles. NMR spectra were recorded on a Bruker DPX-400 spectrometer (1H NMR at 400 MHz and 13C NMR at 100 MHz) or a Bruker DPX-500 spectrometer (1H NMR at 500 MHz and 13C NMR at 126 MHz). Chemical shifts are reported in ppm. 1H NMR spectra were recorded with CDCl3 as the solvent using residual CHCl3 (δ = 7.26) as internal standard, and for 13C NMR spectra the chemical shifts are reported relative to the central resonance of CDCl3 (δ = 77.16). Signals in NMR spectra are described as singlet (s), doublet (d), triplet (t), quartet (q), quintet (quint), septet (sept), multiplet (m), broad (br) or combination of these, which refers to the spin–spin coupling pattern observed. Spin–spin coupling constants reported are uncorrected. Two-dimensional (COSY, HSQC, HMBC, NOESY) NMR spectroscopy was used where appropriate to assist the assignment of signals in the 1H and 13C NMR spectra. IR spectra were obtained employing a Shimadzu FTIR-8400 instrument with a Golden Gate™ attachment that uses a type IIa diamond as a single reflection element so that the IR spectrum of the compound (solid or liquid) could be detected directly (thin layer). High resolution mass spectra were recorded under ESI and CI conditions by the analytical services at the University of Glasgow. Flash chromatography was executed under forced flow conditions, using the indicated solvent system and EMD Guduran silica gel 60 as solid support. Thin layer chromatography (TLC) was carried out on Merck silica gel 60 covered aluminum sheets, and monitored by UV-light or by staining with a solution of anisaldehyde or potassium permanganate.2,2-Dimethyl-6-phenethyl-3,6-dihydro-2H-1,2-oxasiline 3
Allyl bromide (0.70 g, 5.8 mmol, 1.8 equiv.) was added to a stirred solution of indium (0.44 g, 3.8 mmol, 1.2 equiv.) in DMI (7.5 mL) at room temperature under argon. The mixture was stirred for 2 h at room temperature under argon. Dichlorodimethylsilane (0.48 g, 3.8 mmol, 1.2 equiv.) was added to the mixture and the reaction was stirred for 3 h at 70 °C under argon. The mixture was allowed to cool down and was added dropwise to a stirred solution of 1 (0.50 g, 3.1 mmol), triethylamine (0.78 g, 7.7 mmol, 2.5 equiv.) and DMAP (0.19 g, 1.5 mmol, 0.50 equiv.) in THF (10 mL) at 0 °C under argon. The reaction mixture was stirred overnight at room temperature under argon. The reaction mixture was then quenched with a saturated aqueous solution of sodium hydrogen carbonate (10 mL) and extracted with ether (3 × 10 mL). The ether extracts were combined and washed with brine (2 × 20 mL), dried (MgSO4), filtered and concentrated in vacuo to deliver a yellow oil. Purification by column chromatography on silica gel (petroleum ether–ether 99/1) gave silyl ether 2 (0.29 g, 56%) as a clear oil. 1H NMR (400 MHz, CDCl3) δ 7.30–7.26 (m, 2H), 7.20–7.16 (m, 3H), 5.89–5.74 (m, 2H), 5.18 (ddd, J = 17.1, 1.5, 1.4 Hz, 1H), 5.08 (ddd, J = 10.4, 1.5, 1.3 Hz, 1H), 4.92–4.84 (m, 2H), 4.14 (m, 1H), 2.73–2.57 (m, 2H), 1.89–1.74 (m, 2H), 1.63 (d, J = 8.1 Hz, 2H), 0.13 (s, 3H), 0.12 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 142.3, 141.3, 134.3, 128.5, 128.5, 125.9, 114.5, 113.8, 73.7, 39.6, 31.8, 25.2, −1.5, −1.7. IR (neat, cm−1) 3078, 3027, 2949, 2915, 2860, 1631, 1605, 1497, 1454, 1422, 1252, 1083, 1030.To a solution of silane 2 (0.15 g, 0.58 mmol) in dichloromethane (11 mL) was added second-generation Grubbs’ catalyst (25 mg, 0.030 mmol, 5.0 mol%) under argon. The solution was stirred at 45 °C (reflux) for 2 h and monitored by TLC. The reaction mixture was then concentrated in vacuo. Purification of the crude compound by column chromatography on silica gel (petroleum ether–ether 98/2) gave the product 3 (99 mg, 74%) as a clear oil. 1H NMR (400 MHz, CDCl3) δ 7.29–7.26 (m, 2H), 7.22–7.16 (m, 3H), 5.91–5.85 (m, 1H), 5.57 (dq, J = 10.8, 2.2 Hz, 1H), 4.45–4.39 (m, 1H), 2.81–2.66 (m, 2H), 1.91–1.75 (m, 2H), 1.30 (ddt, J = 17.5, 4.7, 2.2 Hz, 1H), 1.20 (dddd, J = 17.5, 5.6, 2.7, 2.2 Hz, 1H), 0.21 (s, 3H), 0.18 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 142.6, 132.5, 128.7, 128.4, 125.8, 124.4, 71.6, 40.0, 31.5, 12.4, 0.57, −0.5. IR (neat, cm−1) 3026, 2958, 2936, 2877, 1642, 1604, 1497, 1454, 1257, 1030. HRMS (EI, m/z) calcd for (C14H20OSi)+ 232.1283, found 232.1280.
Trimethyl(2-methyleneheptyl)silane 5a
Cerium(III) chloride heptahydrate (27.8 g, 74.5 mmol, 3.50 equiv.) was added to a 500 mL three-necked round-bottomed flask and dried under vacuum at 120 °C for 2 h, 140 °C for 2 h and then at 160 °C for 2 h. The flask was allowed to cool down and was purged with argon for 10 min, and dry THF (200 mL) was added. The mixture was stirred for 20 h at room temperature under argon to give the cerium(III) chloride–THF complex as a white precipitate. A trimethylsilylmethylmagnesium chloride solution (1.30 M in THF, 57.0 mL, 74.5 mmol, 3.50 equiv.) was added dropwise at −78 °C under argon. The gray suspension was stirred for 30 min, and then ethyl caproate (3.07 g, 21.3 mmol) in dry THF (15 mL) was added at −78 °C. The reaction was stirred at −78 °C for 2 h, the flask was then removed from the cold bath, and the mixture was stirred at room temperature for 15 h. The reaction mixture was cooled to 0 °C, a saturated aqueous solution of ammonium chloride (200 mL) was added at 0 °C, and the mixture was stirred for 20 min. Water was added, and the mixture was extracted with ether (3 × 150 mL). The combined ether extracts were washed with water and brine (2 × 300 mL), then dried (Na2SO4), filtered and concentrated in vacuo to give a yellow oil. The oil was then stirred overnight with silica in dichloromethane. Filtration, followed by concentration in vacuo gave the allylic silane 5a (3.59 g, 91%) as a clear oil. 1H NMR (400 MHz, CDCl3) δ 4.58–4.57 (m, 1H), 4.51–4.49 (m, 1H), 1.94 (t, J = 7.6 Hz, 2H), 1.52 (d, J = 1.0 Hz, 2H), 1.47–1.39 (m, 2H), 1.35–1.22 (m, 4H), 0.89 (t, J = 7.0 Hz, 3H), 0.01 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 148.2, 106.8, 38.4, 31.8, 27.7, 27.0, 22.8, 14.2, −1.1. IR (neat, cm−1) 2956, 2929, 2861, 1633, 1467, 1418, 1248, 1157. HRMS (CI, m/z) calcd for (C11H25Si)+ 185.1726, found 185.1722.Triethyl(2-((trimethylsilyl)methyl)hept-1-en-4-yloxy)silane 5b
The crude silane 5b was obtained from the corresponding ester 4b (4.94 g, 18.0 mmol) according to the procedure described above for silane 5a. The crude oil was purified by column chromatography on silica gel (petroleum ether–ether 98/2) and the resulting oil was stirred overnight with silica in dichloromethane. The mixture was then filtered and concentrated in vacuo to give the allylic silane 5b (4.81 g, 85%) as a clear oil. 1H NMR (400 MHz, CDCl3) δ 4.61–4.59 (m, 1H), 4.56–4.54 (m, 1H), 3.82–3.76 (m, 1H), 2.14 (ddd, J = 13.6, 5.8, 0.8 Hz, 1H), 2.06 (ddd, J = 13.6, 7.1, 0.5 Hz, 1H), 1.52 (s, 2H), 1.51–1.26 (m, 4H), 0.96 (t, J = 8.1 Hz, 9H), 0.89 (t, J = 7.1 Hz, 3H), 0.60 (q, J = 7.8 Hz, 6H), 0.02 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 144.7, 109.8, 71.3, 46.8, 39.5, 27.3, 18.8, 14.4, 7.1, 5.3, −1.2. IR (neat, cm−1) 2955, 2910, 2876, 1634, 1458, 1416, 1248, 1099, 1074, 1040, 1005. HRMS (ESI, m/z) calcd for (C17H38OSi2Na)+ 337.2353, found 337.2327.(4-(Benzyloxy)-2-methyleneheptyl)trimethylsilane 5c
Cerium(III) chloride heptahydrate (21.3 g, 57.1 mmol, 3.50 equiv.) was dried under vacuum at 120 °C for 2 h, 140 °C for 2 h and then at 160 °C for 2 h. The flask was allowed to cool down to room temperature and was purged with argon for 10 min, and dry THF (190 mL) was added. The mixture was stirred for 20 h at room temperature under argon to give the cerium(III) chloride–THF complex as a white precipitate. To a stirred solution of magnesium (1.4 g, 57 mmol, 3.5 equiv.) and 1,2-dibromoethane (3 drops) in THF (48 mL) at 65 °C was added slowly a solution of chloromethyltrimethylsilane (8.0 mL, 58 mmol, 3.5 equiv.) in THF (12 mL). The solution was then stirred for 2 h at room temperature. The above solution was then added dropwise (over 1 h) at −78 °C to the cerium. The gray suspension was stirred for 30 min, and then ester 4c (4.09 g, 16.3 mmol) in dry THF (20 mL) was added at −78 °C. The reaction was stirred at −78 °C for 2 h, the flask was then removed from the cold bath, and the mixture was stirred at room temperature for 15 h. A saturated aqueous solution of ammonium chloride (200 mL) was added at 0 °C, and the mixture was stirred for 20 min. Water was added, and the mixture was extracted with diethyl ether (3 × 200 mL). The combined ether extracts were washed with water and brine (2 × 300 mL), then dried (Na2SO4), filtered and concentrated in vacuo to give a yellow oil. The oil was stirred one day with silica gel in dichloromethane. The mixture was then filtered and concentrated in vacuo and the resulting crude oil was purified by column chromatography on silica gel (petroleum ether–ether 98/2) to give the allylic silane 5c (2.90 g, 61%) as a clear oil. 1H NMR (500 MHz, CDCl3) δ 7.37–7.26 (m, 5H), 4.68–4.66 (m, 1H), 4.61–4.59 (m, 1H), 4.57 (d, J = 11.5 Hz, 1H), 4.50 (d, J = 11.5 Hz, 1H), 3.57–3.51 (m, 1H), 2.33 (dd, J = 14.0, 6.2 Hz, 1H), 2.09 (dd, J = 14.0, 6.6 Hz, 1H), 1.55 (d, J = 2.6 Hz, 2H), 1.53–1.47 (m, 3H), 1.40–1.32 (m, 1H), 0.91 (t, J = 7.1 Hz, 3H), 0.03 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 144.8, 139.2, 128.4, 127.9, 127.5, 109.8, 77.9, 71.2, 43.3, 36.6, 27.2, 18.9, 14.4, −1.1. IR (neat, cm−1) 2957, 2934, 2872, 1497, 1454, 1248, 1090, 1069, 1028. HRMS (ESI, m/z) calcd for (C18H30OSiNa)+ 313.1958, found 313.1949.2-(Bromomethyl)hept-1-ene 7a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) 25
25
Pyrrolidone hydrotribromide (4.1 g, 7.8 mmol, 1.0 equiv.) was added to a stirred solution of the allylic silane 5a (1.44 g, 7.82 mmol) and pyridine (4.1 mL, 51 mmol, 6.5 equiv.) in dry THF (322 mL) at −10 °C under argon. The mixture was stirred for 2 h and allowed to warm to room temperature during this period. A saturated aqueous solution of sodium thiosulfate (250 mL) was added and the resulting mixture was stirred for 5 min. The mixture was extracted with ether (3 × 200 mL). The ether extracts were combined and washed with a saturated aqueous solution of copper sulfate (2 × 300 mL) and brine (2 × 300 mL), then dried (Na2SO4), filtered and concentrated in vacuo to deliver a yellow oil. Purification by column chromatography on silica gel (petroleum ether–ether 99/1) gave the bromide 7a (1.36 g, 91%) as a clear oil. Obtained data in accordance with previously reported data.25
(2-(Bromomethyl)hept-1-en-4-yloxy)triethylsilane 7b
Bromide 7b (2.35 g, 92%) was obtained as a clear oil according to the procedure for bromination described above from the corresponding allylic silane 5b (2.50 g, 7.95 mmol). 1H NMR (400 MHz, CDCl3) δ 5.24–5.23 (m, 1H), 4.99–4.98 (m, 1H), 4.06 (dd, J = 10.0, 0.9 Hz, 1H), 3.99 (dd, J = 10.0, 0.9 Hz, 1H), 3.83 (app quint, J = 5.9 Hz, 1H), 2.40 (ddd, J = 14.1, 5.9, 1.0 Hz, 1H), 2.34 (ddd, J = 14.1, 5.9, 1.0 Hz, 1H), 1.45–1.27 (m, 4H), 0.96 (t, J = 8.0 Hz, 9H), 0.90 (t, J = 7.0 Hz, 3H), 0.60 (q, J = 8.0 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 143.2, 117.9, 71.0, 41.2, 39.4, 37.6, 18.8, 14.4, 7.1, 5.2. IR (neat, cm−1) 2957, 2936, 2913, 2876, 1458, 1414, 1377, 1238, 1209, 1105, 1074, 1040, 1005. HRMS (CI, m/z) calcd for (C14H3079BrOSi)+ 321.1249, found 321.1248.(((2-(Bromomethyl)hept-1-en-4-yl)oxy)methyl)benzene 7c
Bromide 7c (2.61 g, 88%) was obtained as a clear oil according to the procedure for bromination described above from the corresponding allylic silane 5c (2.90 g, 10.0 mmol). (Purification by column chromatography on silica gel (petroleum ether–ether 98/2).) 1H NMR (500 MHz, CDCl3) δ 7.37–7.25 (m, 5H), 5.25 (s, 1H), 5.04 (s, 1H), 4.57 (d, J = 11.5 Hz, 1H), 4.53 (d, J = 11.5 Hz, 1H), 4.03 (d, J = 10.0 Hz, 1H), 3.98 (d, J = 10.0 Hz, 1H), 3.59 (app quint, J = 5.8 Hz, 1H), 2.51 (dd, J = 14.7, 5.6 Hz, 1H), 2.46 (dd, J = 14.7, 6.6 Hz, 1H), 1.62–1.35 (m, 4H), 0.92 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 143.1, 138.8, 128.5, 127.9, 127.7, 117.8, 77.4, 71.1, 37.9, 37.5, 36.3, 18.8, 14.3. IR (neat, cm−1) 2957, 2932, 2870, 1497, 1454, 1352, 1209, 1067, 1028. HRMS (ESI, m/z) calcd for (C15H21O79BrNa)+ 319.0668, found 319.0660.1-((2-(Bromomethyl)hept-1-en-4-yloxy)methyl)-4-methoxybenzene 7d
A solution of bromide 7b (0.99 g, 3.1 mmol) in 5/95 HF/CH3CN (20 mL) was stirred for 2 h at room temperature. A saturated aqueous solution of calcium chloride (40 mL) was added to the reaction mixture. The mixture was extracted with diethyl ether (3 × 20 mL) and the combined organic layers were washed with a saturated aqueous solution of sodium bicarbonate (20 mL), brine (50 mL), dried (Na2SO4), filtered and concentrated in vacuo to give the alcohol as a clear oil (0.63 g, 98%), which was used without further purification. To a stirred solution of sodium hydride (48.0 mg, 1.14 mmol) in diethyl ether (2 mL) was added dropwise a solution of 4-methoxybenzyl alcohol (1.10 g, 7.56 mmol) in diethyl ether (2 mL). The mixture was stirred for 20 min at room temperature. Freshly distilled trichloroacetonitrile (0.800 mL, 7.56 mmol) was then added dropwise at 0 °C and the resulting mixture was stirred for 1 h and the temperature was allowed to warm up during this period. The solution was then concentrated in vacuo and the crude oil was washed with a solution of pentane (5 mL) and methanol (0.6 mL), filtered and concentrated in vacuo. This process washing/filtration/concentration was repeated three times to give 4-methoxybenzyl trichloroacetimidate (1.9 g, 89%) as a clear oil. To a stirred solution of the alcohol (717 mg, 3.46 mmol) in dichloromethane (7.5 mL) was added the previously formed 4-methoxybenzyl trichloroacetimidate in dichloromethane (0.5 mL), followed by camphorsulfonic acid (81 mg, 0.35 mmol). The mixture was then stirred for one day at room temperature. A saturated aqueous solution of sodium bicarbonate (15 mL) was then added. The mixture was extracted with diethyl ether (3 × 10 mL) and the combined organic layers were washed with brine (30 mL), dried (Na2SO4), filtered and concentrated in vacuo to deliver an oil. Purification by column chromatography (dry column) on silica gel (petroleum ether–ether 9/1) gave the protected alcohol 7d (962 mg, 85%) as a clear oil. 1H NMR (400 MHz, CDCl3) δ 7.26 (d, J = 8.5 Hz, 2H), 6.87 (d, J = 8.5 Hz, 2H), 5.24 (s, 1H), 5.03–5.02 (m, 1H), 4.49 (d, J = 11.1 Hz, 1H), 4.45 (d, J = 11.1 Hz, 1H), 4.02 (dd, J = 10.0, 0.8 Hz, 1H), 3.97 (d, J = 10.0, 0.8 Hz, 1H), 3.80 (s, 3H), 3.58–3.53 (m, 1H), 2.51–2.41 (m, 2H), 1.58–1.32 (m, 4H), 0.91 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 159.3, 143.1, 130.9, 129.5, 117.8, 113.9, 77.0, 70.7, 55.4, 37.8, 37.6, 36.3, 18.8, 14.3. IR (neat, cm−1) 2955, 2934, 2870, 1613, 1512, 1464, 1302, 1244, 1209, 1173, 1034. HRMS (ESI, m/z) calcd for (C16H2379BrO2Na)+ 349.0774, found 349.0764.Dimethyl(2-methyleneheptyl)silane 9a10
To a stirred solution of magnesium (150 mg, 6.22 mmol, 3.30 equiv.) and chlorodimethylsilane (0.210 mL, 1.86 mmol, 1.00 equiv.) in THF (1 mL) was added dropwise (over 1 h) at room temperature a solution of bromide 7a (355 mg, 1.86 mmol) in THF (1.8 mL). The reaction mixture was stirred at room temperature for 2 h. A saturated aqueous solution of ammonium chloride (5 mL) was then added and the mixture was extracted with ether (3 × 5 mL). The combined organic layers were washed with brine (20 mL), dried (Na2SO4), filtered and concentrated in vacuo to deliver silane 9a (257 mg, 81%) as a clear oil. 1H NMR (400 MHz, CDCl3) δ 4.61–4.60 (m, 1H), 4.56–4.54 (m, 1H), 3.90 (non, J = 3.5 Hz, 1H), 1.98 (t, J = 7.6 Hz, 2H), 1.59 (dd, J = 3.5, 1.0 Hz, 2H), 1.47–1.40 (m, 2H), 1.35–1.24 (m, 4H), 0.89 (t, J = 7.1 Hz, 3H), 0.09 (d, J = 3.5 Hz, 6H). Obtained data in accordance with previously reported data.10(2-((Dimethylsilyl)methyl)hept-1-en-4-yloxy)triethylsilane 9b10
Hydrosilane 9b (804 mg, 86%) was obtained as a clear oil from the corresponding bromide 7b (999 mg, 3.11 mmol) according to the procedure described above for hydrosilane 9a. In this case, the reaction time was increased to 5 h. (Purification by column chromatography on silica gel (petroleum ether).) 1H NMR (400 MHz, CDCl3) δ 4.64–4.62 (m, 1H), 4.62–4.60 (m, 1H), 3.91 (non, J = 3.6 Hz, 1H), 3.83–3.77 (m, 1H), 2.19 (ddd, J = 13.6, 5.8, 1.1 Hz, 1H), 2.11 (ddd, J = 13.6, 7.2, 0.9 Hz, 1H), 1.62–1.57 (m, 2H), 1.51–1.25 (m, 4H), 0.96 (t, J = 8.0 Hz, 9H), 0.89 (t, J = 6.9 Hz, 3H), 0.60 (q, J = 8.0 Hz, 6H), 0.10 (d, J = 3.6 Hz, 3H), 0.09 (d, J = 3.6 Hz, 3H). Obtained data in accordance with previously reported data.10(4-(Benzyloxy)-2-methyleneheptyl)dimethylsilane 9c10
Hydrosilane 9c (1.95 g, 81%) was obtained as a clear oil from the corresponding bromide 7c (2.59 g, 8.71 mmol) according to the procedure described above for hydrosilane 9a. In this case, the reaction time was increased to 5 h. (Purification by column chromatography on silica gel (petroleum ether–ether 97/3).) 1H NMR (400 MHz, CDCl3) δ 7.37–7.24 (m, 5H), 4.70–4.69 (m, 1H), 4.66–4.65 (m, 1H), 4.57 (d, J = 11.5 Hz, 1H), 4.49 (d, J = 11.5 Hz, 1H), 3.93 (non, J = 3.6 Hz, 1H), 3.58–3.52 (m, 1H), 2.37 (ddd, J = 14.1, 6.1, 1.2 Hz, 1H), 2.14 (ddd, J = 14.1, 6.5, 1.0 Hz, 1H), 1.66–1.57 (m, 2H), 1.53–1.31 (m, 4H), 0.90 (t, J = 7.1 Hz, 3H), 0.10 (d, J = 3.6 Hz, 3H), 0.09 (d, J = 3.6 Hz, 3H). Obtained data in accordance with previously reported data.10Dimethyl(2-methyleneheptyl)(5-phenylpent-1-en-3-yloxy)silane 8a10
To a stirred solution of silane 9a (36 mg, 0.21 mmol, 1.2 equiv.) in THF (0.05 mL) were added palladium dichloride (0.40 mg, 0.0020 mmol, 1.2 mol%) and hexachloroethane (25 mg, 0.10 mmol, 0.60 equiv.). The reaction mixture was stirred for 1 h at room temperature. The reaction mixture was then added to a stirred solution of alcohol 1 (28 mg, 0.18 mmol), triethylamine (0.060 mL, 0.44 mmol, 2.5 equiv.) and DMAP (11 mg, 0.090 mmol, 0.50 equiv.) in THF (0.4 mL) at 0 °C. The reaction mixture was stirred for 1 h at room temperature. A saturated aqueous solution of sodium bicarbonate (3 mL) was added and the mixture was extracted with ether (3 × 3 mL). The combined organic layers were washed with brine (10 mL), dried (Na2SO4), filtered and concentrated in vacuo to deliver an oil. Purification by column chromatography on silica gel (petroleum ether–ether 99.5/0.5) gave the silane 8a (27 mg, 47%) as a clear oil. 1H NMR (400 MHz, CDCl3) δ 7.31–7.27 (m, 2H), 7.20–7.17 (m, 3H), 5.84 (ddd, J = 17.0, 10.3, 6.3 Hz, 1H), 5.17 (dt, J = 17.0, 1.7 Hz, 1H), 5.08 (ddd, J = 10.3, 1.7, 1.1 Hz, 1H), 4.62–4.61 (m, 1H), 4.57–4.56 (m, 1H), 4.16–4.10 (m, 1H), 2.72–2.57 (m, 2H), 1.99 (t, J = 7.7 Hz, 2H), 1.88–1.74 (m, 2H), 1.63 (s, 2H), 1.47–1.39 (m, 2H), 1.35–1.24 (m, 4H), 0.89 (t, J = 7.0 Hz, 3H), 0.13 (s, 3H), 0.12 (s, 3H). Obtained data in accordance with previously reported data.103,3-Diethyl-9,9-dimethyl-7-methylene-11-phenethyl-5-propyl-4,10-dioxa-3,9-disilatridec-12-ene 8b10
Silane 8b (490 mg, 77%) was obtained as a clear oil and as a 1:1 mixture of diastereomers from the corresponding silane 9b and alcohol 1 (224 mg, 1.38 mmol) according to the procedure described above for silane 8a. (Purification by column chromatography on silica gel (petroleum ether–ether 99.5/0.5).) 1H NMR (400 MHz, CDCl3) δ 7.30–7.26 (m, 2H), 7.19–7.15 (m, 3H), 5.83 (ddd, J = 17.0, 10.3, 6.3 Hz, 1H), 5.16 (dt, J = 17.0, 1.5 Hz, 1H), 5.07 (ddd, J = 10.3, 1.8, 1.1 Hz, 1H), 4.66–4.62 (m, 2H), 4.12 (app q, J = 6.3 Hz, 1H), 3.82–3.77 (m, 1H), 2.72–2.56 (m, 2H), 2.22–2.08 (m, 2H), 1.88–1.75 (m, 2H), 1.67–1.59 (m, 2H), 1.50–1.26 (m, 4H), 0.95 (t, J = 7.9 Hz, 9H), 0.89 (t, J = 6.8 Hz, 3H), 0.60 (q, J = 7.9 Hz, 6H), 0.15–0.11 (m, 6H). Obtained data in accordance with previously reported data.10
(4-(Benzyloxy)-2-methyleneheptyl)dimethyl((5-phenylpent-1-en-3-yl)oxy)silane 8c10
Silane 8c (1.85 g, 60%) was obtained as a clear oil and as a 1:1 mixture of diastereomers from the corresponding silane 9c and alcohol 1 (1.14 g, 7.06 mmol) according to the procedure described above for silane 8a. (Purification by column chromatography on silica gel (petroleum ether–ether 99/1).) 1H NMR (400 MHz, CDCl3) δ 7.37–7.26 (m, 7H), 7.20–7.17 (m, 3H), 5.85 (dddd, J = 17.0, 10.3, 6.4, 1.7 Hz, 1H), 5.18 (ddt, J = 17.0, 3.0, 1.5 Hz, 1H), 5.11–5.07 (m, 1H), 4.75–4.72 (m, 1H), 4.71–4.68 (m, 1H), 4.59 (d, J = 11.5 Hz, 1H), 4.51 (d, J = 11.5 Hz, 1H), 4.14 (q, J = 6.4 Hz, 1H), 3.58 (quint, J = 6.1 Hz, 1H), 2.73–2.58 (m, 2H), 2.45–2.38 (m, 1H), 2.21–2.15 (m, 1H), 1.90–1.77 (m, 2H), 1.72–1.64 (m, 2H), 1.56–1.33 (m, 4H), 0.92 (t, J = 6.9 Hz, 3H), 0.16 (s, 3H), 0.15 (s, 3H). Obtained data in accordance with previously reported data.10
(Z)-1-Phenyl-7-(triethylsilyloxy)-5-((trimethylsilyl)methyl)dec-4-en-3-ol 11b
To a stirred solution of oxysilane 10b (351 mg, 0.810 mmol) in diethyl ether (30 mL) at −78 °C was added dropwise methyllithium (1.6 M solution in diethyl ether, 1.5 mL, 2.4 mmol, 3.0 equiv.). The reaction was stirred 1 h at −78 °C and was allowed to warm up to room temperature for 1 h. The mixture was quenched by addition of a saturated aqueous solution of ammonium chloride (50 mL) and was extracted with diethyl ether (3 × 30 mL). The combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. Purification by filtration through a silica plug gave allylic alcohol 11b (364 mg, quant.) as a clear oil and as a 1:1 mixture of diastereomers. 1H NMR (500 MHz, CDCl3) δ 7.29–7.16 (m, 5H), 5.14 (d, 1H, J = 8.9 Hz), 4.27–4.20 (m, 1H), 3.82–3.76 (m, 1H), 2.81–2.63 (m, 2H), 2.15–2.10 (m, 1H), 2.04 (td, J = 13.6, 6.9 Hz, 1H), 1.92–1.83 (m, 1H), 1.78–1.69 (m, 1H), 1.64 (d, 0.5H, J = 13.4 Hz), 1.57–1.52 (m, 1H), 1.55 (d, 0.5H, J = 13.4 Hz), 1.49–1.28 (m, 4H), 1.19 (d, 0.5H, J = 3.6 Hz), 1.18 (d, 0.5H, J = 3.7 Hz), 0.96 (t, 4.5H, J = 7.9 Hz), 0.96 (t, 4.5H, J = 7.9 Hz), 0.89 (t, 3H, J = 6.8 Hz), 0.60 (q, 6H, J = 7.9 Hz), 0.01 (s, 4.5H), 0.00 (s, 4.5H). 13C NMR (126 MHz, CDCl3) δ 142.3, 139.0, 138.6, 128.6, 128.5, 127.4, 127.1, 125.9, 71.9, 71.4, 68.2, 68.1, 47.4, 47.0, 39.5, 39.4, 32.1, 32.0, 23.0, 22.5, 18.8, 18.5, 14.5, 14.4, 7.1, 5.3, −0.6, −0.7. IR (neat, cm−1) 3421, 2955, 2935, 2876, 1496, 1456, 1415, 1249, 1156, 1124, 1101, 1039, 1006. HRMS (ESI, m/z) calcd for (C26H48O2Si2Na)+ 471.3085, found 471.3095.
(E)-5-Methyl-1-phenyldec-4-ene-3,7-diol 12b and 5-methylene-1-phenyldecane-3,7-diol 13b10
To a stirred solution of allylic silane 11b (35.9 mg, 0.080 mmol) in DMF (1.7 mL) were added water (9 μL, 0.47 mmol, 6.0 equiv.) and tetrabutylammonium fluoride (1.0 M solution in THF, 0.48 mL, 0.48 mmol, 6.0 equiv.). The solution was stirred at 65 °C for 1 h and was then quenched by addition of a saturated aqueous solution of ammonium chloride (2 mL). The mixture was then extracted with diethyl ether (3 × 2 mL) and the combined organic layers were washed with brine (3 × 5 mL), dried (Na2SO4), filtered and concentrated in vacuo. Purification by column chromatography on silica gel (petroleum ether–ethyl acetate 4/6) gave alcohol 12b (7.3 mg, 35%) as a clear oil and alcohol 13b (7.3 mg, 35%) as a clear oil (1:1 mixtures of diastereomers). 12b: 1H NMR (400 MHz, CDCl3) δ 7.31–7.26 (m, 2H), 7.20–7.16 (m, 3H), 5.35–5.31 (m, 1H), 4.44–4.38 (m, 1H), 3.76–3.69 (m, 1H), 2.75–2.62 (m, 2H), 2.21 (dd, J = 13.5, 3.3 Hz, 1H), 2.06 (ddd, J = 13.5, 9.3, 0.8 Hz, 1H), 1.99–1.90 (m, 1H), 1.82–1.73 (m, 1H), 1.69 (d, J = 1.2 Hz, 3H), 1.51–1.41 (m, 4H), 0.94 (t, J = 7.1 Hz, 3H). 13b: 1H NMR (400 MHz, CDCl3) δ 7.31–7.17 (m, 5H), 5.01 (s, 1H), 4.99 (s, 1H), 3.82–3.71 (m, 2H), 2.86–2.79 (m, 1H), 2.75–2.66 (m, 1H), 2.34–2.02 (m, 4H), 1.85–1.76 (m, 2H), 1.50–1.34 (m, 4H), 0.96–0.92 (m, 3H). Obtained data in accordance with previously reported data.10
(E)-5-Methyl-1-phenyldec-4-en-3-ol 12a and 5-methylene-1-phenyldecan-3-ol 13a10
To a stirred solution of allylic silane 11a (38.0 mg, 0.12 mmol) in DMF (2.5 mL) at 0 °C were added TBAF(H2O)3 (99.0 mg, 0.315 mmol, 2.50 equiv.). The solution was then stirred at 0 °C for 30 min and was then quenched by addition of a saturated aqueous solution of ammonium chloride (3 mL). The mixture was then extracted with diethyl ether (3 × 5 mL) and the combined organic layers were washed with brine (3 × 5 mL), dried (MgSO4), filtered and concentrated in vacuo. Purification by column chromatography on silica gel (petroleum ether:ether 3:2) gave alcohol 12a (22.0 mg, 71%) as a clear oil and alcohol 13a (5.0 mg, 16%) as a clear oil. Following the same procedure 12a and 13a were prepared in 54% and 10% yield respectively using TBAF(t-BuOH)4 (133 mg, 0.24 mmol, 2 equiv.). 12a: 1H NMR (400 MHz, CDCl3) δ 7.29–7.27 (m, 2H), 7.24–7.14 (m, 3H), 5.23 (ddd, J = 8.7, 2.5, 1.2 Hz, 1H), 4.40 (dtd, J = 9.9, 6.6, 3.5 Hz, 1H), 2.69–2.67 (m, 2H), 2.00 (m, 2H), 1.98–1.89 (m, 1H), 1.82–1.72 (m, 1H), 1.65 (d, J = 1.3 Hz, 3H), 1.48–1.39 (m, 2H), 1.37–1.25 (m, 5H), 0.90 (s, 3H). 13a: 1H NMR (400 MHz, CDCl3) δ 7.32–7.28 (m, 2H), 7.24–7.17 (m, 3H), 4.90 (m,1H), 4.84 (s, 1H), 3.79–3.68 (m, 1H), 2.92–2.80 (m, 1H), 2.71 (dt, J = 13.8, 8.1 Hz, 1H), 2.28 (dd, J = 13.5, 3.3 Hz, 1H), 2.11 (dd, J = 13.8, 9.4 Hz, 1H), 2.01 (t, J = 7.7 Hz, 2H), 1.84–1.74 (m, 3H), 1.46–1.26 (m, 6H), 0.90 (t, J = 7.0 Hz, 3H). Obtained data in accordance with previously reported data.10
Chlorodimethyl(2-methylallyl)silane12
To a 3-neck flask fitted with a reflux condenser was added pre-dried magnesium turnings (4.86 g, 200 mmol, 2.00 equiv.), diethyl ether (230 mL) and 3 drops of 1,2-dibromoethane. Then, a mixture of dimethyldichlorosilane (12.1 mL, 100 mmol) and 3-chloro-2-methyl-1-propene (14.7 mL, 150 mmol, 1.50 equiv.) in diethyl ether (20 mL) was added dropwise over 4 h, maintaining gentle reflux. The reaction mixture was stirred for a further 12 h at room temperature at which point a white precipitate had formed. After filtering off the white solid, the filtrate was concentrated at 20 °C, 250 mbar. The crude product was then purified by vacuum distillation at 50 °C, 20 mbar furnishing chlorodimethyl(2-methylallyl)silane (4.99 g, 34 mmol, 34%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 4.60 (s, 1H), 4.50 (s, 1H), 1.73 (m, 3H), 1.57 (s, 2H), 0.10 (2s, 6H). Obtained data in accordance with previously reported data.12Dimethyl(7-methylallyl)((5-phenylpent-1-en-3-yl)oxy)silane 15
To a stirred solution of allylic alcohol 1 (500 mg, 3.08 mmol) and chlorodimethyl(2-methylallyl)silane (687 mg, 4.62 mmol, 1.50 equiv.) in dichloromethane (6 mL) at 0 °C was added imidazole (630 mg, 9.25 mmol, 3.00 equiv.) and DMAP (10 mg, 0.08 mmol, 0.025 equiv.). The reaction mixture was then left to warm to room temperature and stirred for further 2 h. Then, water (5 mL) was added and the mixture was extracted with diethyl ether (3 × 5 mL), dried (MgSO4), filtered and concentrated in vacuo. Purification by column chromatography on silica gel (petroleum ether:ether 99:1) gave 15 (845 mg, 3.08 mmol, quant.) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.31–7.28 (m, 3H), 7.21–7.18 (m, 2H), 5.85 (ddt, J = 16.9, 10.3, 6.4 Hz, 1H), 5.19 (dd, J = 17.1, 0.9 Hz, 1H), 5.10 (dd, J = 10.3, 0.8 Hz, 1H), 4.64 (s, 1H), 4.55 (s, 1H), 4.18–4.14 (m, 1H), 2.73–2.60 (m, 2H), 1.90–1.80 (m, 2H), 1.78 (s, 3H), 1.66 (s, 2H), 0.16 (s, 3H), 0.15 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 142.8, 142.2, 141.2, 128.4, 128.3, 125.7, 114.4, 108.9, 73.6, 39.5, 31.6, 28.9, 25.3, −1.1, −1.4. IR (neat, cm−1) 2959, 2914, 2940, 1638, 1454, 1252, 1078. HRMS (ESI, m/z) calcd for (C17H28OSiNa)+ 297.1651, found 297.1637.
5,8,8-Trimethyl-3-phenethyl-3,9-dihydro-2H-8,9-oxasiline 16
Diene 15 (260 mg, 0.95 mmol) and Grubbs II (20.0 mg, 0.024 mmol, 0.025 equiv.) in degassed dichloromethane (9.5 mL) were refluxed for 2 h. The solvent was then removed by concentrating in vacuo. Petroleum ether (5 mL) and diethyl ether (5 mL) were then added and the diethyl ether was removed by concentrating in vacuo. The resultant solution that had specs of dark solid was passed through a plug of silica and rinsed with petroleum ether, thus giving the desired silacycle 16 as colorless oil that was immediately used in the next step with no further purification. 1H NMR (400 MHz, CDCl3) δ 7.30–7.25 (m, 2H), 7.24–7.14 (m, 3H), 5.35–5.34 (m, 1H), 5.19 (ddd, J = 9.1, 4.5, 2.2 Hz, 1H), 2.80–2.64 (m, 2H), 1.89–1.73 (m, 5H), 1.27–1.11 (m, 2H), 0.19 (s, 3H), 0.16 (s, 3H).13C NMR (101 MHz, CDCl3) δ 142.6, 132.4, 128.5, 128.2, 126.0, 125.6, 71.4, 40.2, 31.4, 28.2, 17.6, 0.3, −0.8. IR (neat, cm−1) 3028, 2959, 1715, 1454, 1250, 1140, 1030. HRMS (ESI, m/z) calcd for (C15H22OSiNa)+ 269.1338, found 269.1328.(Z)-2-Methyl-6-phenylhex-2-ene-1,4-diol 17
The above silacycle 16 was dissolved in a mixture of MeOH (9.5 mL) and THF (9.5 mL), then KF (275 mg, 4.75 mmol, 5.00 equiv.) and KHCO3 (219 mg, 2.19 mmol, 2.30 equiv.) were added followed by the dropwise addition of a 30% aqueous solution of H2O2 (4.6 mL, 35 mmol, 37 equiv.). The reaction mixture was left to stir at room temperature for 16 h and was quenched with a saturated aqueous solution of sodium thiosulfate (20 mL). NaCl was then added until the mixture became saturated followed by extraction of the product with ethyl acetate (5 × 20 mL). The combined organic layers were dried (MgSO4), filtered and concentrated in vacuo. Purification by column chromatography on silica gel (petroleum ether:ether 1:1) gave diol 17 (168 mg, 0.81 mmol, 86% over 2 steps) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.30–7.26 (m, 2H), 7.22–7.15 (m, 3H), 5.40 (d, J = 8.3 Hz, 1H), 4.46–4.41 (m, 1H), 4.22 (d, J = 12.3 Hz, 1H), 4.00 (d, J = 12.3 Hz, 1H), 2.74–2.63 (m, 2H), 2.04–1.90 (m, 3H), 1.83 (m, 3H) 1.81–1.76 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 141.7, 138.7, 130.9, 128.4, 128.4, 125.9, 67.3, 62.1, 39.1, 31.7, 21.8. IR (neat, cm−1) 3024, 2920, 2851, 1724, 1601, 1493, 1454, 1373, 1261, 1072. HRMS (ESI, m/z) calcd for (C13H18OSiNa)+ 269.1199, found 269.1190.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the University of Glasgow and the EPSRC (Doctoral Training Allocation for A. F. T. EP/M508056/1).References
- A. Zampella, M. V. D'Auria, L. Minale, C. Debitus and C. Roussakis, J. Am. Chem. Soc., 1996, 118, 11085 CrossRef CAS.
- K. Suenaga, T. Nagoya, T. Shibata, H. Kigoshi and K. Yamada, J. Nat. Prod., 1997, 60, 155 CrossRef CAS.
- (a) A. Vincent and J. Prunet, Synlett, 2006, 2269 CAS; (b) L. Grimaud, R. de Mesmay and J. Prunet, Org. Lett., 2002, 4, 419 CrossRef CAS PubMed; (c) L. Grimaud, D. Rotulo, R. Ros-Perez, L. Guitry-Azam and J. Prunet, Tetrahedron Lett., 2002, 43, 7477 CrossRef CAS; (d) D. Rotulo-Sims and J. Prunet, Org. Lett., 2007, 9, 4147 CrossRef CAS PubMed.
- M.-G. Braun, A. Vincent, M. Boumediene and J. Prunet, J. Org. Chem., 2011, 76, 4921 CrossRef CAS PubMed.
- (a) P. A. Evans, Metathesis in Natural Product Synthesis, ed. J. Cossy, S. Arseniyadis and C. Meyer, Wiley, New York, 2010, p. 225 Search PubMed; (b) A. Čusak, Chem. – Eur. J., 2012, 18, 5800 CrossRef PubMed; (c) J. H. Jun, S. Javed, C. N. Ndi and P. R. Hanson, Synthesis of Heterocycles by Metathesis Reactions, ed. J. Prunet, Springer, 2017, p. 319 Search PubMed.
- R. Matsui, K. Seto, Y. Sato, T. Suzuki, A. Nakazaki and S. Kobayashi, Angew. Chem., Int. Ed., 2011, 50, 680 CrossRef CAS PubMed.
- P. A. Evans, A. Čusak, A. Grisin, M. J. Lawler and M. Pink, Synthesis, 2016, 48, 2402 CrossRef CAS.
- (a) F. Li and M. J. Miller, J. Org. Chem., 2006, 71, 5221 CrossRef CAS PubMed; (b) J. C. Tung, W. Chen, B. C. Noll, R. E. Taylor, S. C. Fields, W. H. Dent III and F. R. Green, Synthesis, 2007, 2388 CAS.
- See for example: C. Rodríguez-Escrich, F. Urpí and J. Vilarrasa, Org. Lett., 2008, 10, 5191 CrossRef PubMed.
- For a preliminary account of this work, see: A. F. Tiniakos, S. Wittmann, A. Audic and J. Prunet, Org. Lett., 2019, 21, 589 CrossRef CAS PubMed , https://pubs.acs.org/doi/10.1021/acs.orglett.8b03552. Further permissions related to the material excerpted should be directed to the ACS.
- A. Meijere, D. Faber, U. Heinecke, R. Walsh, T. Müller and Y. Apeloig, Eur. J. Org. Chem., 2001, 663 CrossRef.
- A. Fürstner and D. Voigtländer, Synthesis, 2000, 959 CrossRef.
- Z. Li, C. Yang, H. Zheng, H. Qiu and G. Lai, J. Organomet. Chem., 2008, 693, 3771 CrossRef CAS.
- C. Meyer and J. Cossy, Tetrahedron Lett., 1997, 38, 7861 CrossRef CAS.
- J. S. Clark, C. A. Baxter, A. G. Dossetter, S. Poigny, J. L. Castro and W. G. Whittingham, J. Org. Chem., 2008, 73, 1040 CrossRef CAS PubMed.
- Compound 7d was obtained by cleavage of the silyl ether of 7b followed by reprotection of the resulting alcohol as its p-methoxybenzyl ether, see Experimetal section for details.
- J. Legros, F. Meyer, M. Coliboeuf, B. Crousse, D. Bonnet-Delpon and J.-P. Bégué, J. Org. Chem., 2003, 68, 6444 CrossRef CAS PubMed.
- Y. Zhang, X. Jia and J.-X. Wang, Eur. J. Org. Chem., 2009, 2983 CrossRef CAS.
- A. Shibato, Y. Itagaki, E. Tayama, Y. Hokke, N. Asao and K. Maruoka, Tetrahedron, 2000, 56, 5373 CrossRef CAS.
- A. Kunai and J. Ohshita, J. Organomet. Chem., 2003, 686, 3 CrossRef CAS.
- R. Savela, W. Zawartka and R. Leino, Organometallics, 2012, 31, 3199 CrossRef CAS.
- P. Pongkittiphan, E. A. Theodorakis and W. Chavasiri, Tetrahedron Lett., 2009, 50, 5080 CrossRef PubMed.
- H. Ito, A. Watanabe and M. Sawamura, Org. Lett., 2005, 7, 1869 CrossRef CAS PubMed.
- Methallyldimethylsilane was too volatile to be isolated in good yield from the Peterson olefination.
- L. E. Overman and D. Lesuisse, Tetrahedron Lett., 1985, 26, 4167 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ob02563d |
| ‡ Present address: Institut de Chimie Moléculaire et des Matériaux d'Orsay, CNRS UMR 8182, Université Paris-Sud, Université Paris-Saclay, Bâtiment 420, 91405 Orsay cedex, France. |
| This journal is © The Royal Society of Chemistry 2020 |