
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Toward the stereochemical assignment and synthesis of hemicalide: DP4f GIAO-NMR analysis and synthesis of a reassigned C16–C28 subunit†
Callum I.
MacGregor
,
Bing Yuan
Han
,
Jonathan M.
Goodman
and
Ian
Paterson
*
University Chemical Laboratory, Lensfield Road, Cambridge, CB2 1EW, UK
First published on 1st March 2016
Abstract
Using the DP4f GIAO-NMR method, the stereochemistry of hemicalide was computationally analysed, resulting in a reassignment at C18 as supported by improved NMR shift correlations with a model C13–C25 fragment 23. An advanced C16–C28 subunit 6 of this potent anticancer agent was then synthesised with the revised 18,19-syn relationship.
Hemicalide (1, Fig. 1) is a potent cytotoxic polyketide recently isolated from the marine sponge Hemimycale sp. collected in the Torres archipelago, Vanuatu, in the South Pacific.1,2 It was reported to display impressive growth inhibitory activity against a panel of human cancer cell lines with picomolar IC50 values,2 acting through a novel tubulin-targeting antimitotic mechanism.3 Its complex planar structure 1, featuring an elaborate polyoxygenated carboxylic acid containing three separate olefinic regions and two δ-lactone rings, was deduced using extensive NMR spectroscopic analysis.
 | ||
| Fig. 1 The planar structure 1 of hemicalide and the relative configurations 2–4 previously reported for the C8–C13, C18–C24 and C36–C46 regions, with the reassigned relative configuration 5 for the C18–C24 region and the revised C16–C28 subunit 6 in this work. | ||
Given the limited amount of hemicalide isolated, elucidation of its full 3D structure has proved challenging. At the time of the patent disclosure by Massiot, no assignments were indicated at the 21 stereocentres, leading to over two million possible stereoisomers.2 To help alleviate this stereoconundrum, the relative configuration of the isolated stereohexad contained within the terminal C1–C15 region was assigned as shown in 2,4 based on synthetic model fragments and NMR spectroscopic correlations. This proposal by Ardisson was later independently supported by our group using computational DP4-based NMR shift analysis.5 More recently, the relative configurations of the isolated C18–C24 and C36–C42 stereoclusters were assigned as in 3 and 4 respectively, following the synthesis of various model fragments and NMR spectroscopic correlations.6,7 Using a combined computational and experimental NMR analysis, herein we reassign the configuration of one of these δ-lactone regions to that shown in 5, together with outlining a flexible and modular strategy for pursuing the total synthesis of hemicalide based on the construction of a suitably functionalised C16–C28 subunit 6 with the revised C18 stereochemistry.
Although the relative configuration of the C16–C25 δ-lactone region of hemicalide was previously assigned as shown in 3,6 assumptions made from considering a subset of diastereomeric model fragments might be misleading. In particular, we felt that the analysis of 1H NMR splitting patterns and 3J coupling constants made in these compounds, leading to the proposed 18,19-anti relationship in 3, did not provide incontrovertible evidence for this assignment.8 Inspection of the 1H NMR spectroscopic data for related δ-lactones suggests that the alternative syn relationship may also be reasonable.7a,c,9 To determine the stereochemistry with more confidence, further studies on all possible diastereomeric permutations within this region are desirable.
For this purpose, a computational DFT NMR analysis might enable the in silico screening of multiple stereoisomers, avoiding an otherwise lengthy synthetic campaign to prepare a library of model fragments for comparison. To this end, the DP4 parameter gives a probability that the calculated GIAO 1H and 13C NMR chemical shifts for a given molecular structure matches the experimental data.5 However, noting that there are over a million possible diastereomers to calculate and the time required for each diastereomer increases with both the number of atoms and the flexibility of the system, it was considered unrealistic to attempt to analyse the entire structure of hemicalide. Nevertheless, although originally intended for the structural elucidation of complete organic molecules, the DP4 method has been successfully extended in certain cases to the assignment of the relative stereochemistry within virtual fragments of natural products.5,10 As this NMR shift analysis now represents an extension of the applicability of the DP4 probability method, we distinguish it here by using the descriptor DP4f since it is fragment-based. Hence, we decided to approach the hemicalide problem by breaking down the complete structure 1 into more manageable virtual fragments featuring the characteristic stereoclusters.
Building on the protocol11–13 developed for the earlier DP4f-based configurational analysis of the open chain C1–C15 region, the two cyclic stereoclusters containing a δ-lactone ring were treated in isolation. The computational analysis of the α,β-dihydroxy-δ-lactone region (cf. red box in 1, Fig. 1) was tackled first. For completeness, we chose to analyse all possible 16 diastereomers of the virtual fragment 7 (Fig. 2). This incorporates an extended C13–C27 sequence with the associated level of substitution, unsaturation and oxygenation of hemicalide. The combined 1H and 13C DP4f GIAO-NMR shift analysis of 7 revealed some intriguing findings. Out of all possible 16 diastereomers (7A–P in the ESI†), 7M was predicted as the most likely candidate with 99% probability. Notably, 7M has a syn relationship between the adjacent methyl and acyloxy substituents at C18 and C19; otherwise the stereochemistry is defined as shown with respect to the substitution on the δ-lactone ring and the methyl-bearing stereocentre at C24.
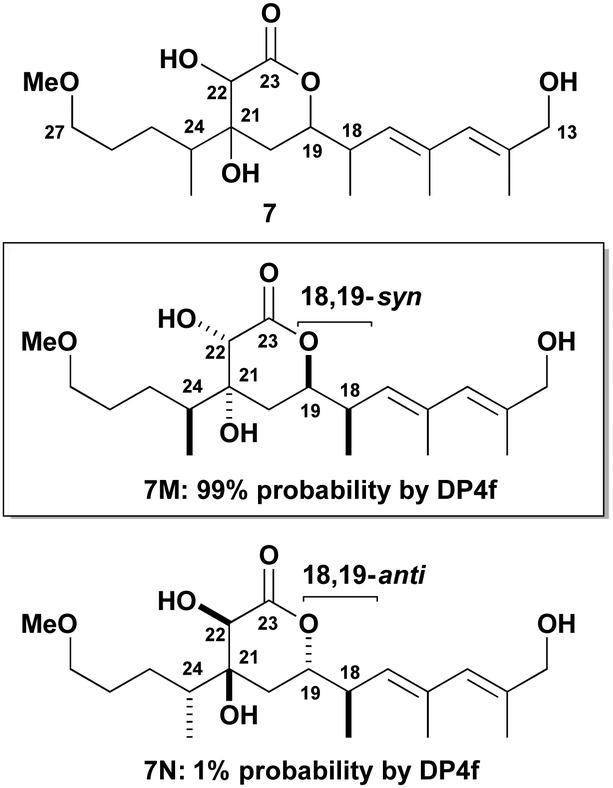 | ||
| Fig. 2 Summary of results for the combined 1H and 13C DP4f GIAO-NMR shift analysis of the α,β-dihydroxy-δ-lactone stereocluster of 1 (red box) based on the virtual C13–C27 fragment 7. | ||
As discussed in the ESI,† we have less confidence in this probability than we would for a normal DP4 calculation, but it is sufficiently high that it is likely that 7M possesses the natural product stereochemistry. By contrast, the 18,19-anti isomer 7N, corresponding to that favoured by the Ardisson group,6 was calculated as having a significantly lower probability of 1%. Supported by the evidence from the experimental NMR correlation studies discussed later, we propose that 7M (corresponding to 5 in Fig. 1) represents the most probable relative configuration for this region, where a syn relationship between C18 and C19 is preferred.
Moving forward, a DP4f-based configurational analysis on the C34–C44 region (cf. blue box in 1, Fig. 1) was required. This region of hemicalide also features five stereocentres and, in terms of substitution pattern, resembles that contained within the other δ-lactone stereocluster, except that a hydroxyl group is lacking at C39 compared to that at C21. In initiating this work, there was limited NMR data (excluding NOE correlations) available to us to steer the stereochemical assignment.2 For completeness, therefore, all 16 possible diastereomers (8A–P in the ESI†) of the virtual fragment 8 (Fig. 3) were considered, incorporating the C34–C44 region with the associated level of substitution, unsaturation and oxygenation. At the time, the preliminary results from this DP4f-based analysis predicted isomer 8M as the most likely candidate stereostructure with 84% probability, which was less certain than the previous assignment. However, the recent findings by Cossy and co-workers7b,c in the assignment of the stereochemistry as 4 (Fig. 1) within this same region, gave us cause to re-evaluate this initial DP4f prediction. Notably, a diagnostic NOE correlation was reported between H37 and H40 in the 1H NMR spectrum of hemicalide, indicating that these protons are likely positioned on the same face of the δ-lactone, i.e. suggesting a 1,4-cis-substituted ring rather than trans as in 8M. As the synthetic model fragments employed for these NMR correlation studies realistically embody the entire C34–C46 portion of hemicalide, including the hydroxyl-bearing stereocentre at C45, this additional structural feature was then incorporated to give a more elaborate virtual fragment 9 (Fig. 3).
 | ||
| Fig. 3 Summary of results for the combined 1H and 13C DP4f GIAO-NMR shift analysis of the C34–C44 and C34–C46 α-hydroxy-δ-lactone regions of 1 (blue box) based on virtual fragments 8 and 9. | ||
To reduce the computational time, it was decided to initially analyse a subset of six out of the now 32 possible diastereomers for 9, two of these incorporating the relative stereochemistry originally predicted by DP4f using 8 and four of the diastereomers considered by Cossy7c (see the ESI†) satisfying the experimental NOE constraints. Remarkably, this more focused DP4f treatment favoured isomer 9A with 99% probability, possibly because the extended side-chain incorporating the C45-hydroxyl affects the preferred conformation by hydrogen bonding. This prediction of 9A was in agreement with the Ardisson–Cossy team, where the indicated 1,4-anti relationship between the separated C45 and C42 stereocentres should be viewed as tentative (see the ESI†). The change in the preferred candidate with inversion of configuration at both C39 and C40, as a result of increasing the size of the virtual fragment, re-emphasised the additional uncertainty introduced in this approach when assigning the stereochemistry of a conformationally flexible structure. At this stage, these preliminary findings for a subset of C34–C46 stereoisomers, along with our ongoing DP4f-based NMR chemical shift analysis of other parts of the hemicalide structure, should be considered as a work in progress, although encouragingly support is added to the experimental NMR correlation work. Building on the increased confidence gained from these computational NMR studies, we embarked on our synthetic campaign toward hemicalide as described below.
Guided by the proposed reassignment for the C13–C27 region and other available (but incomplete) stereochemical information as summarised in structure 10 (Scheme 1), a suitably flexible and modular synthesis plan was devised. An sp2–sp2 cross-coupling reaction was envisaged to forge the C15–C16 bond and a stereocontrolled aldol addition to form the C28–C29 bond, leading back to the subunits 6, 11 and 12. We initially targeted the central C16–C28 subunit 6 with the required configuration of the δ-lactone ring, incorporating the methyl-bearing centre at C24 and 18,19-syn stereochemistry. A vinyl iodide moiety was introduced to enable a Stille cross-coupling with 12 to assemble the (14E,16E)-diene, followed by a complex aldol fragment union with 11 and ketone reduction to set the C29 and C27 stereocentres respectively. Bis-O-methylation and final deprotection would then deliver hemicalide (or stereoisomers thereof). Importantly, this planned aldol/reduction sequence14 was designed to selectively access any of the four possible stereoisomers.
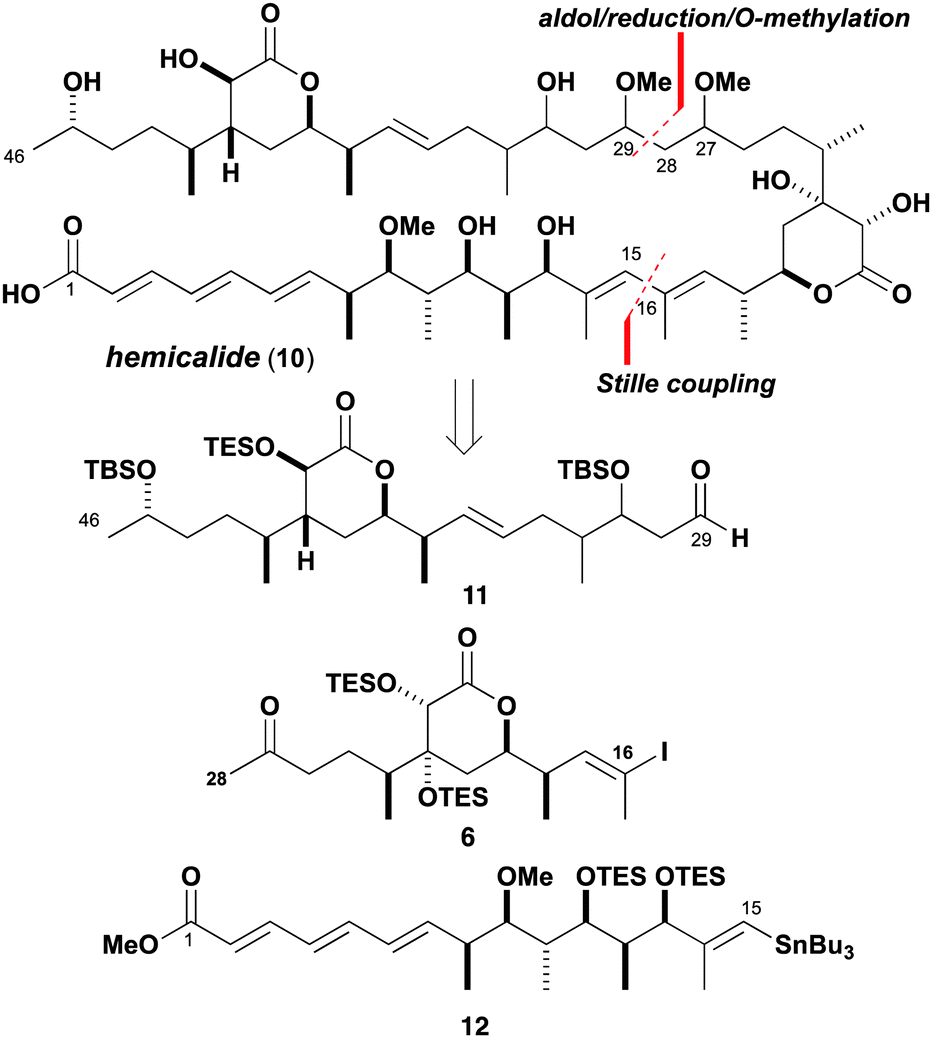 | ||
| Scheme 1 Partial stereochemical assignment 10 for hemicalide and synthesis plan involving key subunits 6, 11 and 12. Note that only the relative configuration within each of the three stereoclusters is suggested. | ||
Recognition of the 1,4-syn relationship between C19 and C24 in 6 (Scheme 2) guided the strategic use of an asymmetric boron-mediated aldol reaction15 between the methyl ketone 13 and the aldehyde 14, both readily obtained from (S)-Roche ester. Alkyne 1516 was prepared from 16 by a Corey–Fuchs reaction.17 Regioselective hydrozirconation18 and iodination of 15 gave the corresponding (Z)-vinyl iodide. DDQ-mediated cleavage of the PMB ether revealed the alcohol which was subsequently oxidised (DMP)19 to give 14. Using our standard conditions ((−)-Ipc2BCl, Et3N),15 methyl ketone 1320 underwent aldol addition with 14 to generate the 1,4-syn adduct 17 with high diastereoselectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr). To access the δ-lactone 18, a sequence of TBS ether formation and enolate addition (EtOAc, LDA) gave ester 19. Cleavage of the TBS ether then induced lactonisation and elimination gave 18. Guided by earlier work,6 a syn-dihydroxylation (OsO4) was performed to give the anticipated diol 20 as a single diastereomer. Following bis-TES ether formation, the PMB ether was cleaved (DDQ) and oxidised (DMP) to give aldehyde 21. Finally, chain extension was achieved by HWE olefination (Ba(OH)2)21 and Stryker reduction22 of the intermediate enone to give the completed C16–C28 subunit 6.
:1 dr). To access the δ-lactone 18, a sequence of TBS ether formation and enolate addition (EtOAc, LDA) gave ester 19. Cleavage of the TBS ether then induced lactonisation and elimination gave 18. Guided by earlier work,6 a syn-dihydroxylation (OsO4) was performed to give the anticipated diol 20 as a single diastereomer. Following bis-TES ether formation, the PMB ether was cleaved (DDQ) and oxidised (DMP) to give aldehyde 21. Finally, chain extension was achieved by HWE olefination (Ba(OH)2)21 and Stryker reduction22 of the intermediate enone to give the completed C16–C28 subunit 6.
 | ||
| Scheme 2 Synthesis of hemicalide subunit 6 and model fragment 23. Reagents and conditions: (a) CBr4, PPh3, CH2Cl2, −78 to 0 °C, 75%; (b) nBuLi, MeI, THF, −78 to 0 °C, 94%; (c) Cp2ZrCl2, DIBAL, THF, 0 °C to rt; I2, −78 °C, 88%; (d) DDQ, CH2Cl2, pH 7 buffer, 0 °C to rt, 90%; (e) DMP, NaHCO3, CH2Cl2, 0 °C to rt, 80%; (f) 13, (−)-Ipc2BCl, Et3N, Et2O, 0 °C; 14, −78 to −20 °C, 70% (>20:1 dr); (g) TBSOTf, 2,6-lutidine, CH2Cl2, −78 to 0 °C, 95%; (h) EtOAc, LDA, THF, −78 °C, 97%; (i) HF·pyr, THF, 0 °C to rt, 85%; (j) DMAP, Ac2O/pyr/PhH, reflux, 83%; (k) 2% K2OsO4·2H2O, NMO, citric acid, tBuOH/H2O/THF, rt, 85% brsm; (l) TESOTf, 2,6-lutidine, CH2Cl2, 0 °C, 88%; (m) DDQ, CH2Cl2, pH 7 buffer, 0 °C to rt, 80%; (n) DMP, NaHCO3, CH2Cl2; 0 °C to rt; (o) (MeO)2P(O)CH2COMe, Ba(OH)2, THF, 0 °C to rt, 80% (2 steps); (p) [Ph3PCuH]6, PhMe, rt, 67%; (q) 20% Pd(PPh3)4, CuTC, [Ph2PO2][NBu4], DMF, 0 °C, 74%. | ||
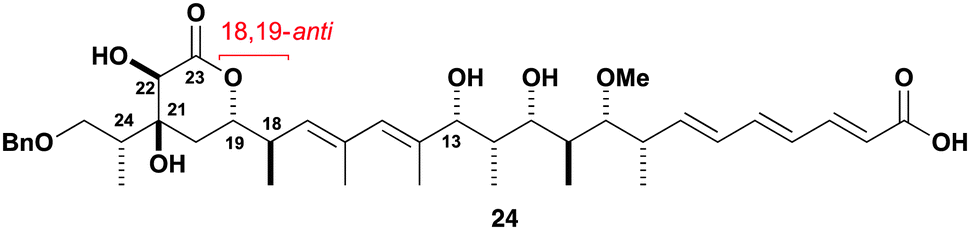
At this stage, we sought experimental support for our stereochemical prediction of 7M in Fig. 2. To access a suitable model diene for NMR correlations, 20 was submitted to Stille cross-coupling conditions23 with stannane 22 to give diene 23. Gratifyingly, this product showed improved24 correlations in 1H NMR data comparisons, especially for the oxymethines at H19 and H22 (|Δδ| = 0.01 and 0.04 ppm) that are considered as particularly diagnostic signals.6 In further support of the reassigned 18,19-syn stereochemistry, inspection of the resonance corresponding to H19 (δ 4.41, ddd, 3J = 11.6, 7.7, 3.7 Hz) in 23 revealed the same peak shape and comparable coupling constants to that in the natural product (δ 4.42, ddd, 3J = 11.3, 7.5, 3.5 Hz).2,7c
In conclusion, we have analysed the calculated GIAO 1H and 13C NMR chemical shifts of selected virtual fragments of hemicalide using the DP4f probability method. The central C16–C28 subunit 6 was then efficiently prepared with the predicted 18,19-syn relationship. In future work, we aim to more fully elucidate the stereochemistry of hemicalide as indicated in 10 (Scheme 1), and achieve a total synthesis of this promising anticancer agent and analogues based on the flexible route outlined here.
We thank the EPSRC for financial support (studentship to CIM) and the National Mass Spectrometry Centre (Swansea) for mass spectra.
Notes and references
- (a) S. M. Dalby and I. Paterson, Curr. Opin. Drug Discovery Dev., 2010, 13, 777 CAS; (b) G. M. Cragg and D. J. Newman, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 3670 CrossRef CAS PubMed; (c) I. Paterson and E. A. Anderson, Science, 2005, 310, 451 CrossRef PubMed.
- I. Carletti, G. Massiot and C. Debitus, Pat. Appl. Publ., WO2011051380 A1 (FR), 2011Chem. Abstr., 2011, 154, 513095 Search PubMed.
- K. H. Altmann and J. Gertsch, Nat. Prod. Rep., 2007, 24, 327 RSC.
- E. Fleury, M.-I. Lannou, O. Bistri, F. Sautel, G. Massiot, A. Pancrazi and J. Ardisson, J. Org. Chem., 2009, 74, 7034 CrossRef CAS PubMed.
- S. G. Smith and J. M. Goodman, J. Am. Chem. Soc., 2010, 132, 12946 CrossRef CAS PubMed.
- E. Fleury, G. Sorin, E. Prost, A. Pancrazi, F. Sautel, G. Massiot, M.-I. Lannou and J. Ardisson, J. Org. Chem., 2013, 78, 855 CrossRef CAS PubMed.
- (a) G. Sorin, E. Fleury, C. Tran, E. Prost, N. Molinier, F. Sautel, G. Massiot, S. Specklin, C. Meyer, J. Cossy, M.-I. Lannou and J. Ardisson, Org. Lett., 2013, 15, 4734 CrossRef CAS PubMed; (b) E. De Gussem, W. Herrebout, S. Specklin, C. Meyer, J. Cossy and P. Bultinck, Chem. – Eur. J., 2014, 20, 17385 CrossRef CAS PubMed; (c) S. Specklin, G. Boissonnat, C. Lecourt, G. Sorin, M.-I. Lannou, J. Ardisson, F. Sautel, G. Massiot, C. Meyer and J. Cossy, Org. Lett., 2015, 17, 2446 CrossRef CAS PubMed.
- During the Ardisson studies reported in ref. 6, it was observed that an anti relationship at C18–C19 in a model fragment resulted in a ddd for the 1H NMR oxymethine signal corresponding to H19 whilst a syn relationship gave an apparent dt. As hemicalide shows a ddd splitting pattern for H19, this steered the authors to their proposed assignment of an 18,19-anti relationship. However, the corresponding oxymethine signal in structurally related δ-lactones (ref. 7a, c and 9) with a syn relationship are all reported to have a ddd splitting pattern.
- J. A. Lafontaine, D. P. Provencal, C. Gardelli and J. W. Leahy, J. Org. Chem., 2003, 68, 4215 CrossRef CAS PubMed.
- I. Paterson, S. M. Dalby, J. C. Roberts, G. J. Naylor, E. A. Guzmán, R. Isbrucker, T. P. Pitts, P. Linley, D. Divlianska, J. K. Reed and A. E. Wright, Angew. Chem., Int. Ed., 2011, 50, 3219 CrossRef CAS PubMed.
- For a general procedure, see the ESI†. To begin with, a conformational search was carried out using MacroModel 9.9 (ref. 12) using a hybrid of Monte Carlo multiple-minimum (MCMM) (ref. 13)/low-mode sampling with the Merck Molecular Force field (MMFF) interfaced with Maestro 9.3 using CHCl3 as the solvent. NMR data for all conformers found within 10 kJ mol−1 of the global minima were calculated at the B3LYP6-31G(d,p) level using Jaguar 7.9.
- G. Chang, W. C. Guida and W. C. Still, J. Am. Chem. Soc., 1989, 111, 4379 CrossRef CAS.
- I. Kolossváry and W. C. Guida, J. Am. Chem. Soc., 1996, 118, 5011 CrossRef.
- For related applications of this aldol coupling strategy, see: (a) I. Paterson, K. Ashton, R. Britton, G. Cecere, G. Chouraqui, G. J. Florence, H. Knust and J. Stafford, Chem. – Asian J., 2008, 3, 367 CrossRef CAS PubMed; (b) I. Paterson, M. P. Housden, C. J. Cordier, P. M. Burton, F. A. Mühlthau and O. Loiseleur, Org. Biomol. Chem., 2015, 13, 5716 RSC.
- (a) I. Paterson and R. M. Oballa, Tetrahedron Lett., 1997, 38, 8241 CrossRef CAS; (b) I. Paterson, M. Donghi and K. Gerlach, Angew. Chem., Int. Ed., 2000, 39, 3315 CrossRef CAS; (c) I. Paterson, D. J. Wallace and K. R. Gibson, Tetrahedron Lett., 1997, 38, 8911 CrossRef CAS.
- A. B. Smith III and D. Lee, J. Am. Chem. Soc., 2007, 129, 10957 CrossRef PubMed.
- E. J. Corey and P. L. Fuchs, Tetrahedron Lett., 1972, 13, 3769 CrossRef.
- D. W. Hart, T. F. Blackburn and J. Schwartz, J. Am. Chem. Soc., 1975, 97, 679 CrossRef CAS.
- D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155 CrossRef CAS.
- I. Paterson, E. A. Anderson, S. M. Dalby, J. H. Lim, P. Maltas, O. Loiseleur, J. Genovino and C. Moessner, Org. Biomol. Chem., 2012, 10, 5861 CAS.
- I. Paterson, K.-S. Yeung and J. B. Smaill, Synlett, 1993, 774 CrossRef CAS.
- W. S. Mahoney, D. M. Brestensky and J. M. Stryker, J. Am. Chem. Soc., 1988, 110, 291 CrossRef CAS.
- A. Fürstner, J. A. Funel, M. Tremblay, L. C. Bouchez, C. Nevado, M. Waser, J. Ackerstaff and C. C. Stimson, Chem. Commun., 2008, 2873 RSC.
- See the ESI† for an NMR correlation comparison against a synthetic subunit 24 reported by the Ardisson–Cossy team (ref. 7a).
.
Footnote |
| † Electronic supplementary information (ESI) available: Computational and experimental details with copies of NMR spectra. See DOI: 10.1039/c6cc01074a |
| This journal is © The Royal Society of Chemistry 2016 |