
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLabel-free fluorescence detection of kinase activity using a gold nanoparticle based indicator displacement assay
Cristian
Pezzato
a,
Davide
Zaramella
a,
Massimiliano
Martinelli
a,
Grégory
Pieters
a,
Mario A.
Pagano
*b and
Leonard J.
Prins
*a
aDepartment of Chemical Sciences, University of Padova, Via Marzolo 1, 35131 Padova, Italy. E-mail: leonard.prins@unipd.it
bDepartment of Pharmaceutical and Pharmacological Sciences, University of Padova, Via Marzolo 5, 35131, Padova, Italy. E-mail: mario.pagano@unipd.it
First published on 14th November 2014
Abstract
A straightforward indicator-displacement assay (IDA) has been developed for the quantitative analysis of ATP→ADP conversion. The IDA relies on the use of gold nanoparticles passivated with a monolayer of thiols terminating with a 1,4,7-triazacyclononane (TACN)·Zn2+ head group. The analytes ATP and ADP compete to a different extent with a fluorescent probe for binding to the monolayer surface. In the presence of ATP the fluorescent probe is free in solution, whereas in the presence of ADP the fluorescent probe is captured by the nanoparticles and its fluorescence is quenched. The linear response of the fluorescence signal towards different ratios of ATP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ADP permitted the detection of protein kinase activity simply by adding aliquots of the enzyme solution to the assay solution followed by measurement of the fluorescent intensity. The assay poses no restrictions on the target kinase nor does it require labeling of the kinase substrate. The assay was tested on the protein kinases PIM-1 and Src and validated through a direct comparison with the classical radiometric assay using the [γ-32P]-labeled ATP.
:ADP permitted the detection of protein kinase activity simply by adding aliquots of the enzyme solution to the assay solution followed by measurement of the fluorescent intensity. The assay poses no restrictions on the target kinase nor does it require labeling of the kinase substrate. The assay was tested on the protein kinases PIM-1 and Src and validated through a direct comparison with the classical radiometric assay using the [γ-32P]-labeled ATP.
Introduction
Protein kinases (PKs) are a class of enzymes that play critical roles in a variety of cellular functions including cell growth, development, differentiation, membrane transport, and cell death.1 Their function is exerted by transferring the γ-phosphate group from ATP to a serine, threonine or tyrosine residue in specific substrate proteins. These phosphorylation events modulate the activity of a vast number of proteins, including ion channels, transcription factors and phosphatases.2 PKs act primarily as components of signalling pathways in which extra- and intracellular stimuli are transduced by the cell into a series of phosphorylation events that ultimately bring about a cellular response, such as cell growth, development, differentiation, membrane transport, and cell death.3,4 Abnormalities in signalling pathways can lead to various pathological conditions including many forms of cancer. For this reason, PKs are highly important targets for both basic research and drug development.5Traditionally, PK assays rely on the use of 32P-labeled ATP ensuring a high sensitivity.6 However, the application of these assays is hampered by the generated radioactive waste and the short half-life of 32P-labeled ATP. Besides, the laborious protocols are not compatible with high-throughput screening. Therefore non-radioactive methods are rapidly gaining interest.7,8 In particular, various fluorescence techniques (fluorescence intensity endpoint measurement,9 fluorescence resonance energy transfer (FRET),10 fluorescence polarization (FP),11 and fluorescence lifetime imaging (FLIM)12) have emerged as alternatives. Nonetheless, for a large part these techniques rely on elaborate constructs involving fluorophores conjugated to enzymes or antibodies. Consequently, there is a strong drive to develop new kinase assays using simple and stable components.13–17
Previously, we have studied the interaction between small anionic molecules, such as nucleotides and peptides, and Au MPC 1·Zn2+ (d = 1.8 ± 0.4 nm), which are gold nanoparticles (d = 1.8 ± 0.4 nm) passivated with thiols terminating with a 1,4,7-triazacyclononane (TACN)·Zn2+ head group (Fig. 1).18,19 It was found that such molecules bind to the monolayer surface under saturation conditions even at low micromolar concentrations in aqueous buffer. Detailed studies have revealed that the interactions originate from a combination of electrostatic and hydrophobic interactions and coordination bonds, the relative contributions depending on the structure of the molecules.20 Recently, we have shown that such subtle changes in the binding affinity permitted discrimination of the eight di- and trinucleotides (XDP and XTP, with X = A, T, G, C) by Au MPC 1·Zn2+.21 In that study, fluorescence output signals were generated by the displacement of fluorescent probe molecules from the surface of Au MPC 1·Zn2+ upon the addition of analytes. The observed large difference in the affinity between ATP and ADP prompted us to investigate whether this sensing system would be able to serve as a PK assay by quantitatively reporting on the conversion of ATP into ADP (Fig. 1).
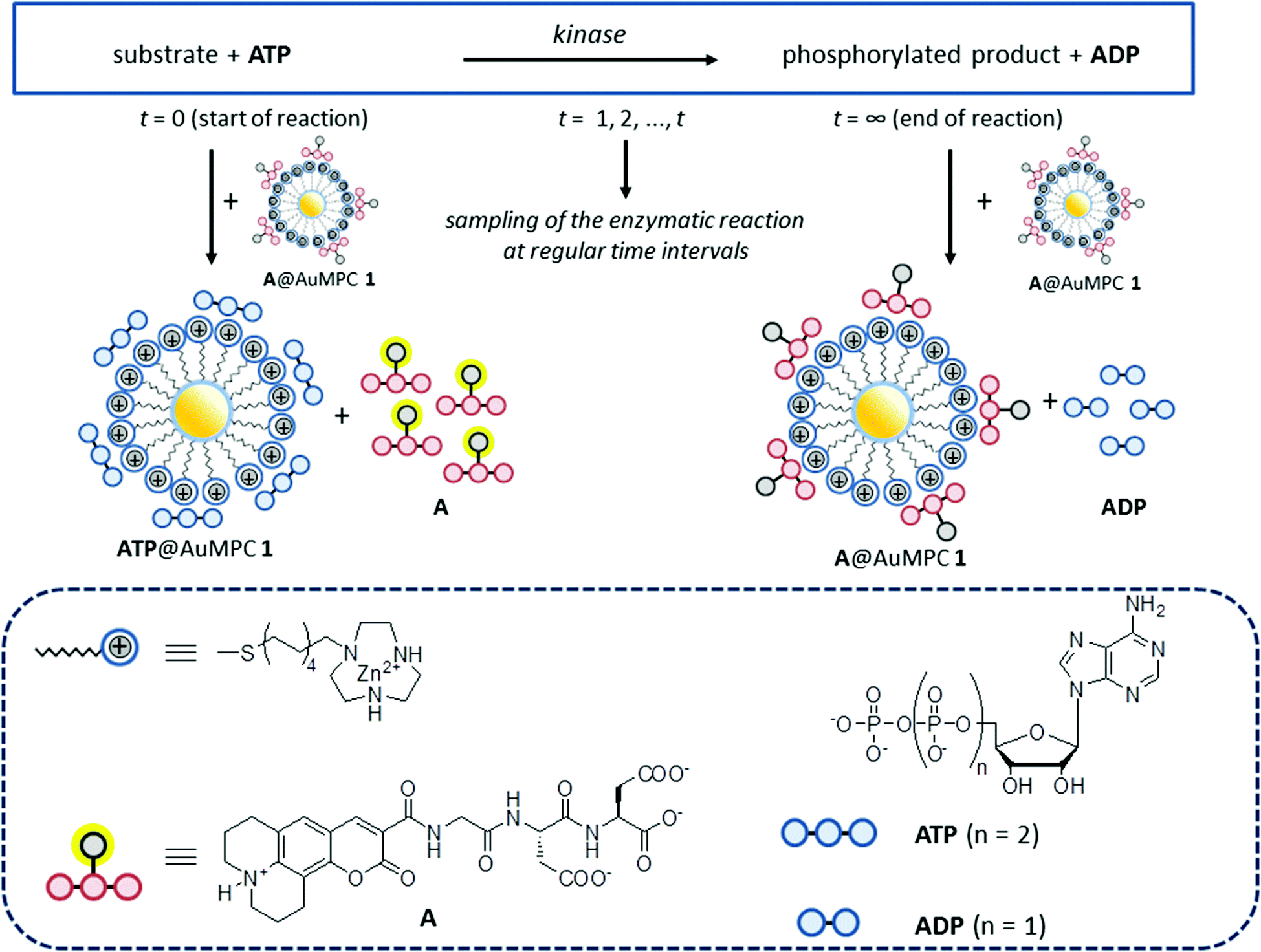 | ||
| Fig. 1 Working principle of the displacement assay. Protein kinase activity results in the transfer of a phosphate-group from ATP to the enzyme substrate yielding the phosphorylated product and ADP. ATP and ADP have different affinities for Au MPC 1 because of their different charges. ATP is able to displace the (quenched) fluorescent probe A from the surface of Au MPC 1 resulting in a turn ON of fluorescence, but ADP is less competitive and does not display A. Sampling the reaction over time gives aliquots with different ATP:ADP ratios leading to different displacement levels of probe A and, thus, different fluorescence intensities. | ||
Results and discussion
In order to set up the indicator-displacement assay (IDA)22 a fluorescent indicator was selected based on the following criteria: (1) excitation and emission wavelengths far away from the (near-) UV region to avoid interference with natural chromophores/fluorophores (e.g. Trp- or Tyr-residues), (2) the ability to bind Au MPC 1·Zn2+ under saturation conditions at low micromolar concentrations in aqueous buffer, and (3) the capability to respond in a very different manner to ATP and ADP in competition experiments. The fluorescent tripeptide A, equipped with coumarin 343, satisfied the first two points with λex = 450 nm and λem = 493 nm and a surface saturation concentration (SSC) equal to 3.7 ± 0.2 μM at a head group concentration (i.e. [TACN·Zn2+]) of 10 μM at pH 7.5. The third point was verified by performing two displacement experiments in which the fluorescence intensity of A was measured upon the addition of increasing amounts of ATP or ADP to a solution of A@Au MPC 1·Zn2+ (Fig. 2a). Initial fluorescent intensities in these experiments started at around 9% of the maximum signal expected for a full release of probe A, due to the presence of a small amount of free A in solution.23 In agreement with our previous results,21 much larger concentrations of ADP were required to displace the same amount of A from Au MPC 1·Zn2+ resulting from the lower number of negative charges in ADP compared to ATP. The maximum difference in signal intensity was observed at an analyte concentration of 3.0 μM.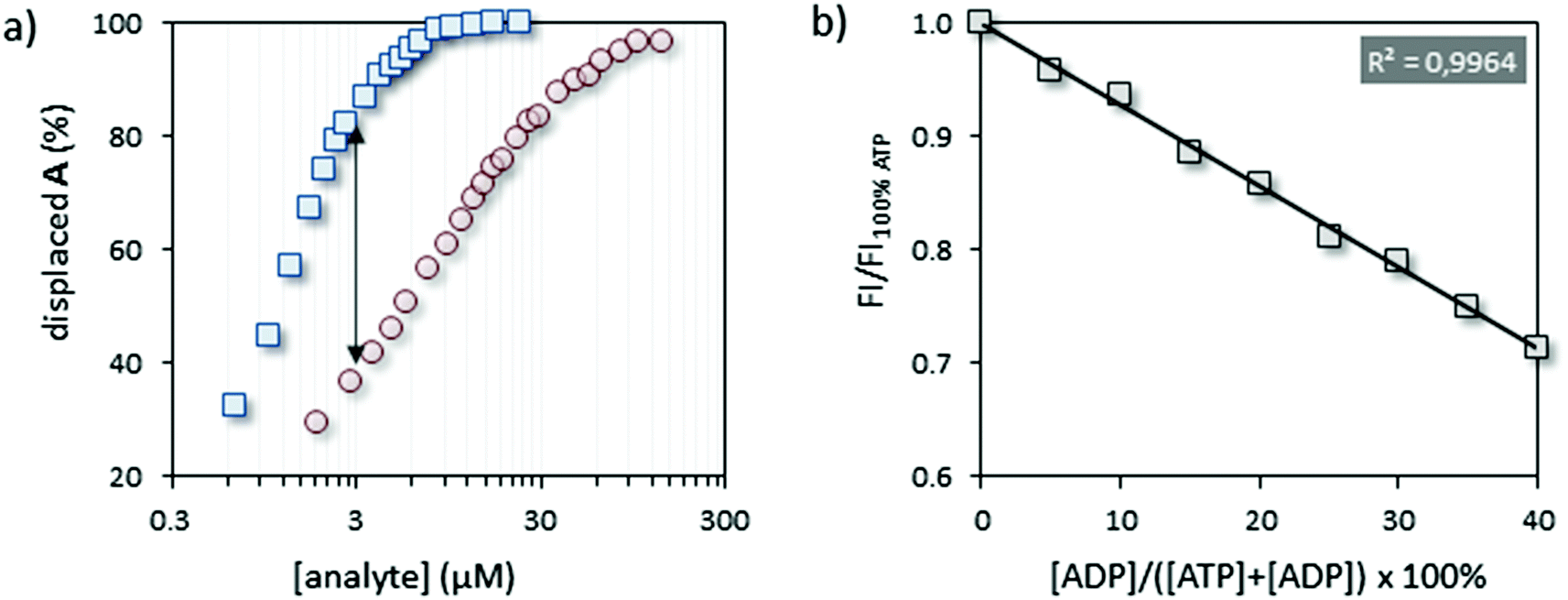 | ||
| Fig. 2 (a) Displacement of A from the surface of Au MPC 1·Zn2+ upon the addition of ATP (blue squares) or ADP (red circles). (b) Normalized fluorescence intensity at 493 nm as a function of the ratio ATP:ADP at a constant concentration of 3 μM. Experimental conditions: [TACN·Zn2+] = 10 μM, [A] = 3.7 μM, [HEPES] = 20 mM, pH = 7.5, T = 25 °C. | ||
Having confirmed the ability of the system to discriminate in a straightforward manner between ATP and ADP, we next investigated whether the system could respond quantitatively to different ratios of ATP and ADP. As a test case we chose Src, the namesake of the Src-family kinases (SFKs), a group of tyrosine kinases, physiologically involved in the early steps of signal transduction, the increased activity of which is associated with several types of cancer.24,25 Binding of ATP to Src is characterized by a KM-value of 8.6 μM.26 As a substrate, we used a specific peptide (here renamed Src-tide, H-KVEKIGEGTYGVVYK-H, 250 μM) derived from the cyclin-dependent kinases cdc2 (residue 6 through 20) and used in vitro for testing the activity of SFKs in the presence of the co-factors Mn2+ and Mg2+ (5 mM).27 In order to minimize the possible interferences of the enzyme, substrate and co-factors with the displacement assay we opted for a discontinuous approach in which the enzyme solution was sampled and diluted to the assay concentration for read out. Initially, we performed a calibration of the system by artificially simulating enzyme activity. Twelve independent solutions were prepared, each of them containing all the enzyme assay components except for the enzyme, but with different ratios of ATP:ADP at a constant overall concentration of 50 μM. Aliquots of these solutions were added to the IDA assay solution containing A@Au MPC 1·Zn2+ in 96-well plates to reach an overall concentration of ATP and ADP equal to 3.0 μM. The fluorescence intensities originating from displaced A were measured after a signal stabilization time of 15 minutes (Fig. 2b). After that time, the signal intensity remained constant indicating that the ATP:ADP ratio is not altered in the assay solution. Importantly, the fluorescence intensity decreased linearly as a function of the relative amount of ADP added (simulating up to 40% conversion). This illustrates that the ATP→ADP conversion can be quantitatively assessed and that the enzyme assay components do not interfere with the displacement assay.
At this point we validated the system as a true protein kinase assay by following the Src induced conversion of ATP to ADP as a function of time (Fig. 3a). In order to demonstrate unambiguously the reliability of the new assay, enzyme activity was also measured in parallel using a conventional radiometric assay with the [γ-32P]-labeled ATP. In order to assure right comparison a common reaction medium was prepared containing all the components (except for ATP) at the same concentrations used for the calibration, but this time with Src-kinase (30 μL of a stock solution with a specific activity of ≈2.5 pmol min−1 μL−1). The mixture was split into two portions, to one of which a small amount of [γ-32P]-ATP was added. To the second portion the same volume of mQ water was added. The resulting mixtures were incubated at 37 °C and the phosphorylation reaction was initiated by adding ATP (50 μM). Every 4 minutes samples were taken and quenched by pouring the aliquots in 0.1% phosphoric acid (radiometric assay) or by freezing them in dry ice (displacement assay). After a time course of 35 minutes the aliquots were processed using conventional techniques (radiometric assay) or the protocol described above (displacement assay). Both assays gave a nearly superimposable result in terms of ATP conversion as a function of time indicating the reliability of the displacement assay (Fig. 3b). Our assay measures ATP→ADP conversion which may, in principle, have origins other than kinase activity. However, the fact that the results of our assay match in a quantitative manner those of the conventional radiometric assay (which measures substrate conversion) indicates that other ATP consuming-processes do not occur in the enzyme solution.
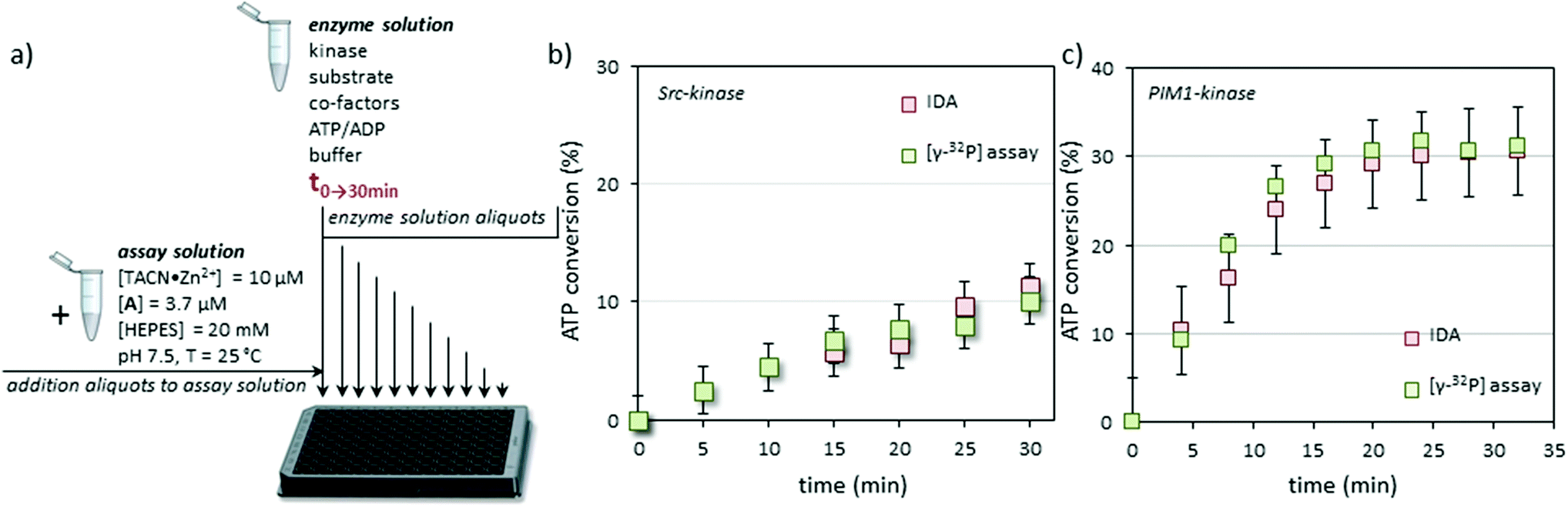 | ||
| Fig. 3 (a) Practical set up of the IDA: aliquots from the enzyme solution taken at regular time intervals are diluted into the assay solution to reach a final concentration of [ATP] + [ADP] = 3.0 μM. (b) Comparison of the results of the IDA and radiometric assay for Src-kinase. Experimental conditions for enzyme solution: Src-kinase (30 μL of a stock solution with a specific activity of ≈2.5 pmol min−1 μL−1), [Src-tide] = 250 μM, [Mg2+] = 5 mM, [Mn2+] = 5 mM, [ATP] = 50 μM, [HEPES] = 20 mM, pH = 7.5, 37 °C. (c) Comparison of the results of the IDA and radiometric assay for PIM1-kinase. Experimental conditions for enzyme solution: PIM1-kinase (50 μL of a stock solution with a specific activity of ≈10 pmol min−1 μL−1), [PIM-tide] = 100 μM, [Mg2+] = 10 mM, [ATP] = 100 μM, [HEPES] = 20 mM, pH = 7.5, 37 °C. | ||
Sampling of the enzyme solution and dilution of the aliquots in the assay buffer has the advantage that the initial experimental conditions for the enzyme (pH, buffer, substrate, cofactors, etc.) hardly affect signal generation by the displacement assay. This, together with the fact that signal generation is a consequence of ATP→ADP conversion rather than substrate-phosphorylation, makes it a universal assay for kinases. To demonstrate this, the protocol was repeated using PIM-1, the Proviral Integration site for Moloney murine leukemia virus kinase 1, a PK that is overexpressed in a number of tumors with a role in the resistance to apoptosis.28 PIM-1 binds ATP with a lower affinity compared to Src (KM = 90 μM vs. 8.6 μM).29 In contrast to Src, which requires one or more acidic amino acids in the vicinity of the phosphorylation site, PIM-1 targets serine or threonine residues surrounded by basic amino acids. The substrate used in the PIM-1 assay was synthesized according to Bullock et al. who had designed a highly specific substrate for PIM-1 (here renamed Pim-tide = H-ARKRRRHPSGPPTA-NH2, 100 μM) using a positional scanning peptide library approach.30 Also for this enzyme, both the radiometric assay and the displacement assay gave virtually identical results (Fig. 3c). For this enzyme, the decrease in the rate of ATP consumption as a function of time observed in either the radiometric or the displacement assay is ascribed to enzyme degradation under the experimental conditions.
The assay volume is an important issue for a practical application of the assay, since they determine how much enzyme is required to measure its activity. In the assays described above, considerably larger aliquots were taken for the displacement assay compared to the radiometric assay (30 vs. 5 μl, respectively). A straightforward way to reduce the required volume is by using 384-well plates rather than 96-well plates. For each well a working volume of 24 μl was used containing 23.2 μl of the assay solution (A@Au MPC 1·Zn2+) and just 0.8 μl of the enzyme solution. This allowed us to perform 4 readings from a single aliquot of 5 μl, which is similar to that used for the radiometric assay, maintaining a reliable detection of ATP→ADP conversion. With regard to the kinase detection limits, a 5% conversion of ATP in the enzyme solution is required in order to create a change in the fluorescence signal that can be accurately detected (see the calibration curve in Fig. 2b). For the Src- and PIM1-kinase assays discussed here, this implies a minimal ATP consumption of 2.5 and 5 μM, respectively, in the enzyme solution. In terms of enzyme activity these concentration changes imply a lower detection limit of the order of 0.1 pmol min−1 μL−1 (assuming a 30 minutes assay time).
Conclusions
We have developed a straightforward assay for the detection of kinase activity. The assay does not rely on antibodies, but instead, is based on easily accessible synthetic components. The assay is discontinuous: aliquots are taken from enzyme solutions and sampled in the ratio ATP:ADP by means of a fluorescence displacement assay. The assay was quantitatively validated by a comparison with the conventional radiometric assay based on the [γ-32P]-labeled ATP. It is to be noted that the assay-solution (composed of Au MPC 1 and A) gave the same response even after storage for 2 weeks at 4 °C. The fact that the assay relies on the detection of ATP→ADP makes it suitable for the detection of any kinase activity. We have shown that the new assay is fast, straightforward to use and gives reliable quantitative data. The ability to perform the assay in conventional microtiter plates allows for high-throughput screening. Indeed, our results point to the use of this assay in pharmaceutical studies aimed at discovering kinase inhibitors. Considering that the assay relies on the detection of ATP→ADP conversion and that signal generation results from a competition between negatively charged ATP/ADP with probe A for binding to Au NP 1, we consider application of the assay in its current form less useful for in vivo measurements of kinase activity.
Experimental section
Synthesis and characterization
The synthesis and characterization of Au MPC 1·Zn2+ (ref. 19) and probe A21 have been described previously. The enzyme substrates Src-tide (H-KVEKIGEGTYGVVYK-H) and PIM-tide (H-ARKRRRHPSGPPTA-NH2) were synthesized by conventional Fmoc-based SPPS using a Zinsser Sophas automated peptide synthesizer. The tyrosine kinase Src was a kind gift from Prof. A. M. Brunati at the University of Padova; PIM-1 was purchased from Sigma-Aldrich.Src-tide (H-KVEKIGEGTYGVVYK-H). HPLC (Agilent RRHD Zorbax Eclipse Plus C18 (2.1 × 150 mm 1.8 μm), gradient: 5–95% B (A: H2O + 0.1% HCOOH, B: CH3CN + 0.1% HCOOH) in 5 minutes, λ = 280 nm): 5.35 min. MS (ESI+, CH3CN + 0.1% HCOOH) m/z: 835.5 ([M + 2H]2+, calcd: 835.5), 557.4 ([M + 3H]3+, calcd: 557.3), 418.4 [M + 4H]4+, calcd: 418.2).
PIM-tide (H-ARKRRRHPSGPPTA-NH2). HPLC (Phenomenex RP Jupiter 4 μm Proteo 90 Å, gradient: 5–55%B (A: H2O + 0.1% TFA, B: CH3CN + 0.1% TFA) in 60 minutes, λ = 226 nm): 14.95 min. MS (ESI+, CH3CN + 0.1% HCOOH) m/z: 1628.0 ([M + H]+, calcd: 1628.5), 1651.0 ([M + Na]+, calcd: 1651.5).
Fluorescence measurements were performed on a TECAN M1000 PRO micro-plate reader using Greiner Bio-one microtiter plates (polystyrene 96-WELL*F and 384-WELL*F). LC-MS measurements were performed on an Agilent 1290 Infinity UPLC, equipped with a diode array detector (DAD), connected to an ESI-MS detector.
Assay solution
The assay solution (50 mL) was prepared by adding the following volumes to a volumetric flask: 2 mL of HEPES buffer (pH 7.5, stock: 0.5 M, final concentration: 20 mM), 108 μL of Au MPC 1·Zn2+ (stock: 4.62 mM, final concentration: 10 μM – referring to [TACN·Zn2+]), 0.5 mL of Zn(NO3)2 (stock: 1 mM, final concentration: 10 μM), and 68.5 μL of A (final concentration: 3.7 μM). The solution was stored at 4 °C and could be used for at least 2 weeks without observing alteration of the signal.Displacement studies
Displacement experiments were performed by adding consecutive amounts of a stock-solution of a competitor ([ATP] = 1.0 mM, or [ADP] = 5.6 mM) in mQ water to 3 mL of the assay solution. Each addition was followed kinetically, monitoring the emission of probe A at 493 nm.Src-kinase assay
:0 to 0.6:0.4 were prepared at a constant concentration of 50 μM in HEPES buffer (pH = 7.5, 20 mM). Separate microtiter plate wells were loaded with 329 μL of the assay solution and 21 μL of the calibration solution. Measurements were performed kinetically, monitoring the emission of probe A at 493 nm for 30 min. All the signals became stable after at least 15 min. Repeating the calibration with samples containing ATP, ADP and, additionally, MgCl2 (5 mM), MnCl2 (5 mM), Src-tide (0.25 mM) and HEPES (pH = 7.5, 20 mM) gave identical results, indicating that all the components do not compromise the IDA efficiency.
PIM1-kinase assay
:0 to 0.6:0.4 were prepared at a constant concentration of 100 μM. Additionally, each sample contained MgCl2 (10 mM), PIM-tide (0.1 mM), and HEPES (pH 7.5, 20 mM). Separate microtiter plate wells were loaded with 340 μL of the assay solution and 10.5 μL of the calibration solution. Measurements were performed by monitoring the emission of probe A at 493 nm for 30 min. Also here, signals stabilised after around 15 min.
The radiometric assay was performed using an identical reaction mixture, but in the presence of ATP [γ-32P] (3% v/v). In this case, the kinase reaction was quenched by pouring the aliquots (5 μL) into 0.1% phosphoric acid (25 μL), which in turn were spotted on a 2 cm2 piece of a Whatman P81 filtermat, washed three times for 4 minutes in 75 mM H3PO4, air dried, put in a vial with a scintillation fluid, and assayed for 32P in a beta-counter (Packard).
Acknowledgements
Financial support from the ERC (StG-239898) and the University of Padova (CPDR125182 and CPDA138148) is acknowledged.References
- L. A. Pinna, Acc. Chem. Res., 2003, 36, 378–384 CrossRef CAS PubMed.
- G. Manning, D. B. Whyte, R. Martinez, T. Hunter and S. Sudarsanam, Science, 2002, 298, 1912–1934 CrossRef CAS PubMed.
- P. Vandergeer, T. Hunter and R. A. Lindberg, Annu. Rev. Cell Biol., 1994, 10, 251–337 CrossRef CAS PubMed.
- T. Hunter, Cell, 2000, 100, 113–127 CrossRef CAS.
- P. Cohen, Nat. Rev. Drug Discovery, 2002, 1, 309–315 CrossRef CAS PubMed.
- C. J. Hastie, H. J. McLauchlan and P. Cohen, Nat. Protocols, 2006, 1, 968–971 CAS.
- H. C. Ma, S. Deacon and K. Horiuchi, Expert Opin. Drug Discovery, 2008, 3, 607–621 CrossRef CAS PubMed.
- Y. Li, W. Xie and G. Fang, Anal. Bioanal. Chem., 2008, 390, 2049–2057 CrossRef CAS PubMed.
- K. Kupcho, R. Somberg, B. Bulleit and S. A. Goueli, Anal. Biochem., 2003, 317, 210–217 CrossRef CAS.
- M. Sato and Y. Umezawa, Methods, 2004, 32, 451–455 CrossRef CAS PubMed.
- E. A. Gaudet, K. S. Huang, Y. Zhang, W. Huang, D. Mark and J. R. Sportsman, J. Biomol. Screening, 2003, 8, 164–175 CrossRef CAS PubMed.
- F. S. Wouters and P. I. H. Bastiaens, Curr. Biol., 1999, 9, 1127–1130 CrossRef CAS.
- T. Zhang, N. Y. Edwards, M. Bonizzoni and E. V. Anslyn, J. Am. Chem. Soc., 2009, 131, 11976–11984 CrossRef CAS PubMed.
- A. E. Hargrove, S. Nieto, T. Zhang, J. L. Sessler and E. V. Anslyn, Chem. Rev., 2011, 111, 6603–6782 CrossRef CAS PubMed.
- G. Ghale, V. Ramalingam, A. R. Urbach and W. M. Nau, J. Am. Chem. Soc., 2011, 133, 7528–7535 CrossRef CAS PubMed.
- J. Bai, C. Liu, T. Yang, F. Wang and Z. Li, Chem. Commun., 2013, 49, 3887–3889 RSC.
- S. Martic and H.-B. Kraatz, Chem. Sci., 2013, 4, 42–59 RSC.
- R. Bonomi, A. Cazzolaro, A. Sansone, P. Scrimin and L. J. Prins, Angew. Chem., Int. Ed., 2011, 50, 2307–2312 CrossRef CAS PubMed.
- G. Pieters, A. Cazzolaro, R. Bonomi and L. J. Prins, Chem. Commun., 2012, 48, 1916–1918 RSC.
- G. Pieters, C. Pezzato and L. J. Prins, Langmuir, 2013, 29, 7180–7185 CrossRef CAS PubMed.
- C. Pezzato, B. Lee, K. Severin and L. J. Prins, Chem. Commun., 2013, 49, 469–471 RSC.
- B. T. Nguyen and E. V. Anslyn, Coord. Chem. Rev., 2006, 250, 3118–3127 CrossRef CAS PubMed.
- By performing the displacement experiments exactly at the SSC rather than at 80% as in previous studies, the system is responsive also to the additions of small quantities of analyte.
- M. C. Frame, Biochim. Biophys. Acta, Rev. Cancer, 2002, 1602, 114–130 CrossRef CAS.
- D. L. Wheeler, M. Iida and E. F. Dunn, Oncologist, 2009, 14, 667–678 CrossRef CAS PubMed.
- R. Karni, S. Mizrachi, E. Reiss-Sklan, A. Gazit, O. Livnah and A. Levitzki, FEBS Lett., 2003, 537, 47–52 CrossRef CAS.
- H. C. Cheng, H. Nishio, O. Hatase, S. Ralph and J. H. Wang, J. Biol. Chem., 1992, 267, 9248–9256 CAS.
- N. S. Magnuson, Z. Wang, G. Ding and R. Reeves, Future Oncol., 2010, 6, 1461–1478 CrossRef CAS PubMed.
- M. A. Pagano, J. Bain, Z. Kazimierczuk, S. Sarno, M. Ruzzene, G. Di Maira, M. Elliott, A. Orzeszko, G. Cozza, F. Meggio and L. A. Pinna, Biochem. J., 2008, 415, 353–365 CrossRef CAS PubMed.
- A. N. Bullock, J. Debreczeni, A. L. Amos, S. Knapp and B. E. Turk, J. Biol. Chem., 2005, 280, 41675–41682 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |