Self-assembly of square-planar halide complexes of phosphine-stabilised stibenium salts
J. Wolfram
Wielandt†
,
Nathan L.
Kilah
,
Anthony C.
Willis
and
S. Bruce
Wild
*
Research School of Chemistry, Australian National University, Canberra, Australian Capital Territory 0200, Australia. E-mail: sbw@rsc.anu.edu.au; Fax: +61 2 6125 0750; Tel: +61 2 6125 4236
First published on 5th July 2006
Abstract
Chloride and bromide ions direct the self-assembly of supramolecular square-planar halide complexes in which four trimethylphosphine-stabilised diphenylstibenium ions surround the central halide ion in discrete centrosymmetrical structures of C4h symmetry.
Halide ions are versatile templates for inorganic and organic syntheses because of their spherical symmetry and ability to interact with a variety of molecular components through Lewis acid–base interactions and hydrogen bonding.1 Following the report in 1991 of the first halide-directed synthesis, a macrocyclic [12]mercuracarborand-4–chloride complex in which the chloride ion is encompassed within a square plane of mercury atoms,2 there have been described in the literature a variety of interesting finite, organometallic and transition metal halide complexes where the coordination geometries around the anions vary from square planar to polytopal spherical. Examples include a square planar organosamarium–chloride complex3 and pentagonal planar μ-hydroxolanthanoid–halide complexes,4 where the halide ions in each case are tightly held by Lewis acid–base interactions with the metal ions, and metallacages in which multiple hydrogen bonding from ligands associated with the transition metals augments the Lewis acid–base interactions between the halide and the transition metal atoms, as in the amidothiourea complex [Ni6(atu)8Cl]3+.5 In the macrocyclic complex [{P(μ-Nt-Bu)2}(μ-NH)]5(HCl), which is formed in a chloride-directed synthesis, hydrogen bonding alone holds the chloride ion in a pentagonal planar environment;6 it is this latter type of chelation of halide ions, in particular of chloride, that is being used increasingly to facilitate the syntheses of organic macrocycles, cages, double helices, and interlocked species.7
Here we report the spontaneous self-assembly and structural characterisation of the first discrete, square-planar halide complexes of a Group 15 element; in the tripositive cations of each complex, four phosphine-stabilised stibenium ions surround a chloride or bromide ion in a centrosymmetrical, square planar arrangement of C4h symmetry. The air- and water-stable complexes, [(Me3P→SbPh2)4X](PF6)3 (1, where X = Cl; 2, where X = Br), can be readily isolated from the organic layers following the reactions of four equiv chloro- or bromodiphenylstibine with four equiv trimethylphosphine in dichloromethane in the presence of aqueous ammonium hexafluorophosphate (Scheme 1).‡ In the electrospray mass spectra of 1 and 2 (Bruker Apex 3 FT-ICR spectrometer; ESI(+), capillary exit voltage 250 V) traces of the ions [(Me3P→SbPh2)4X]+ (X = Cl, Br) are observed at m/z 1443 and 1487 amu, respectively.
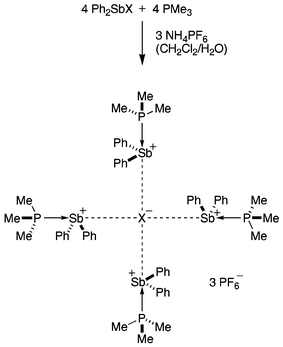 | ||
| Scheme 1 Synthesis of 1 (X = Cl) and 2 (X = Br). | ||
Colourless plates of the two complexes suitable for X-ray crystallography were obtained from dichloromethane–diethyl ether. The crystals of the two compounds are isomorphous (space group: P4/mnc). The molecular structure of the cation of the chloride complex 1 is shown in Fig. 1. The chloride ion in the tripositive cation is located at the centre of a supramolecular square of unipositive (trimethylphosphine)diphenylstibenium ions with Sb⋯Cl 3.1362(3) Å (sum of van der Waals radii for the two elements: 3.87 Å).8 The chloride ion in the complex is located at a site of crystallographic 4/m symmetry, and the Sb1 and P1 atoms lie on a mirror plane. The stereochemistry around antimony in each of the (Me3P→SbPh2)+ groups is trigonal pyramidal with the antimony atom at the centre of a trigonal plane containing the two ipso carbon atoms of the phenyl groups and the lone pair of electrons of the six-electron Ph2Sb+ group; the Me3P-P atom lies directly above the antimony and is orthogonal to the trigonal plane, viz.: P–Sb–C 89.42(8)°. The trigonal pyramidal geometry of the (Me3P→SbPh2)+ group corresponds closely with that of the cation in [Me3Sb→SbMe2]2[(MeSbBr3)2],9 and of the arsenic centres in the phosphine-stabilised arsenium salt [Ph3P→AsMePh]PF610 and similar compounds containing the arsenium ion derived from 10-chloro-5-hydrophenarsazine.11 The chlorine atom in the complex is trans to each of the four phosphorus atoms of each (Me3P→SbPh2)+ group (P–Sb⋯Cl 174.12(3)°; C–Sb⋯Cl 87.45(9)°), and completes, when the lone pair of electrons is included, the overall trigonal bipyramidal coordination geometry around each antimony atom. The neighbouring Sb⋯Sb distances of 4.4353(4) Å in the complex cation are not within binding range (sum of van der Waals radii: 4.24 Å).8 The salient interatomic distances for comparison in the isomorphous bromide complex 2 are the following: Sb⋯Br 3.2236(3) Å (sum of van der Waals radii: 3.99 Å), Sb⋯Sb 4.436(1) Å (sum of van der Waals radii: 4.24 Å).8 Attractive edge-to-face interactions between the eight phenyl groups above and below the square planes containing the halide ions could be contributing to the stabilisation of these supramolecular structures. The discrete nature of the binding of the chloride and bromide ions in the complexes is in contrast to the usual, short-and-long halide bridging observed in tertiary phosphine and arsine adducts of trivalent Group 15-element halides, which results in the crystallisation of halide-bridged oligomers.12
 | ||
| Fig. 1 ORTEP-3 structural representation of cation 1, hydrogens omitted for clarity (30% probability ellipsoids shown).13 | ||
The near linear binding of the halogen and phosphorus atoms to the trigonal planar diphenylstibenium cations in these novel complexes is now being exploited for the self-assembly of supramolecular ladders and grids.
Notes and references
- R. Vilar, Angew. Chem., Int. Ed., 2003, 42, 1460–1477 CrossRef CAS.
- X. Yang, C. B. Knobler and M. F. Hawthorne, Angew. Chem., Int. Ed. Engl., 1991, 30, 1507–1508 CrossRef; Z. Zheng, C. B. Knobler and M. F. Hawthorne, J. Am. Chem. Soc., 1995, 117, 5105–5113 CrossRef CAS.
- T. Dubé, S. Conoci, S. Gambarotta, G. P. A. Yap and G. Vasapollo, Angew. Chem., Int. Ed., 1999, 38, 3657–3659 CrossRef CAS.
- R. Wang, H. D. Selby, H. Liu, M. D. Carducci, T. Jin, Z. Zheng, J. W. Anthis and R. J. Staples, Inorg. Chem., 2002, 41, 278–286 CrossRef CAS; R. Wang, Z. Zheng, T. Jin and R. J. Staples, Angew. Chem., Int. Ed., 1999, 38, 1813–1815 CrossRef CAS.
- R. Vilar, D. M. P. Mingos, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1998, 37, 1258–1261 CrossRef CAS; R. Vilar, D. M. P. Mingos, A. J. P. White and D. J. Williams, Chem. Commun., 1999, 229–230 RSC; S.-T. Cheng, E. Doxiadi, R. Vilar, A. J. P. White and D. J. Williams, J. Chem. Soc., Dalton Trans., 2001, 2239–2244 RSC.
- M. J. Duer, F. García, R. A. Kowenicki, V. Naseri, M. McPartlin, M. L. Stead, R. S. Stein and D. S. Wright, Angew. Chem., Int. Ed., 2005, 44, 5729–5733 CrossRef CAS.
- R. Vilar, Struct. Bonding, 2004, 111, 85–137 CAS; P. D. Beer, M. R. Sambrook and D. Curiel, Chem. Commun., 2006, 2105–2117 RSC.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- H. Althaus, H. J. Breunig and E. Lorkl, Chem. Commun., 1999, 1971–1972 RSC.
- K. A. Porter, A. C. Willis, J. Zank and S. B. Wild, Inorg. Chem., 2002, 41, 6380–6386 CrossRef CAS.
- N. Burford, P. J. Ragogna, K. Sharp, R. McDonald and M. J. Ferguson, Inorg. Chem., 2005, 44, 9453–9460 CrossRef CAS.
- A. R. J. Genge, N. J. Hill, W. Levason and G. Reid, J. Chem. Soc., Dalton Trans., 2001, 1007–1012 RSC; S. Petrie, R. Stranger, A. D. Rae, A. C. Willis, X. Zhou and S. B. Wild, Organometallics, 2006, 25, 164–171 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- S. Mackay, C. J. Gilmore, C. Edwards, N. Stewart and K. Shankland, maXus Computer Program for the Solution and Refinement of Crystal Structures; Nonius, Delft, The Netherlands, MacScience, Japan and University of Glasgow, Glasgow, Scotland, 1999 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidari and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435 CrossRef.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
Footnotes |
| † Present address: Institut für Chemie, Karl-Franzens-Universität Graz, Graz, Austria. |
| ‡ Selected characterisation data: 1: Yield 54%. mp dec > 101 °C. 1H NMR (300.075 MHz, CDCl3, 298 K): δ 1.44 (36H, d, 2J (PH) = 10.0 Hz, PMe), 7.55–7.65 (40H, m, ArH); 31P{1H} NMR (121.47 MHz, CDCl3, 298 K); δ = −30.5 (s, Me3P), −143.6 (sept., 1J (PF) = 714.6 Hz, PF6−); MS (ESI): m/z: 1443 (M+, trace), 353 (C12H19PSb, 100); Found: C, 38.2; H, 4.0; Cl, 2.0. Calc. for C60H76ClF18P7Sb4: C, 38.4; H, 4.1; Cl, 1.9%.2: Yield 43%. mp dec > 97 °C. 1H NMR (300.075 MHz, CDCl3, 298 K): δ 1.42 (36H, d, 2J (PH) = 10.1 Hz, PMe), 7.40–7.75 (40H, m, ArH); 31P{1H} NMR (121.47 MHz, CDCl3, 298 K): δ = −30.0 (s, Me3P), −143.7 (sept., 1J (PF) = 713.0 Hz, PF6−); MS (ESI): m/z: 1489 (M+, trace), 353 (C12H19PSb, 100); Found: C, 37.7; H, 4.0; Br, 4.5. Calc. for C60H76BrF18P7Sb4: C, 37.5; H, 4.0; Br, 4.2%.Crystal data for 1: C60H76ClF18P7Sb4, M = 1878.53, 0.47 × 0.35 × 0.27 mm, tetragonal, space group P4/mnc, 2θmax = 60.06°, a = 15.4979(2) Å, c = 15.5355(3) Å, V = 3731.4(1) Å3, Z = 2, ρcalcd = 1.673 g cm−3, μ(MoKα) = 1.700 mm−1, F(000) = 1848, absorption correction by Gaussian integration method implemented in maXus14 (Tmin/Tmax 0.535/0.662), 56523 reflections measured, 2827 independent reflections (Rint = 0.036) and 124 parameters. Final R1 = 0.0288 (for 1623 obs. data, I > 3σ(I)), wR2 = 0.034, largest diff. peak and hole: 0.98 and −0.93 e Å−3. CCDC 600536.Crystal data for 2: C60H76BrF18P7Sb4, M = 1922.96, 0.36 × 0.22 × 0.05 mm, tetragonal, space group P4/mnc, 2θmax = 55.04°, a = 15.5328(2) Å, c = 15.5904(3) Å, V = 3761.4(1) Å3, Z = 2, ρcalcd = 1.698 g cm−3, μ(MoKα) = 2.181 mm−1, F(000) = 1866, absorption correction by Gaussian integration method implemented in maXus14 (Tmin/Tmax = 0.509/0.897), 44245 reflections measured, 2261 independent reflections (Rint = 0.07) and 133 parameters. Final R1 = 0.0171 (for 1108 obs. data, I > 3σ(I)), wR2 = 0.0200, largest diff. peak and hole: 0.47 and −0.32 e Å−3. CCDC 600535. Data for all structures were collected at 200 K with MoKα radiation (λ = 0.71073 Å). Hydrogen atoms were included at idealised positions and ride on the atoms to which they are bound. The structures were solved by direct methods (SIR92),15 and refined using the least-squares method on F.16 For crystallographic data in CIF or other electronic format see DOI: 10.1039/b607136h |
| This journal is © The Royal Society of Chemistry 2006 |