
Catalytic dinitrogen silylation by tris(pyrazolyl)borate-supported titanium complexes
Chenrui
Liu†
a,
Ling-Ya
Peng†
b,
Yumeng
Chen†
a,
Jingyi
An
a,
Zhaoxin
Li
a,
Wenshuang
Huang
a,
Ganglong
Cui
 *ac and
Shaowei
Hu
*a
*ac and
Shaowei
Hu
*a
aCollege of Chemistry, Beijing Normal University, No. 19, Xin-wai street, Beijing 100875, P. R. China. E-mail: shu@bnu.edu.cn
bSchool of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi’an 710119, China
cKey Laboratory of Theoretical and Computational Photochemistry, Ministry of Education, College of Chemistry, Beijing Normal University, Beijing 100875, P. R. China. E-mail: ganglong.cui@bnu.edu.cn
First published on 2nd December 2025
Abstract
Titanium dinitrogen complexes supported by tris(pyrazolyl)borate and alkoxide/aryloxide ligands catalyse N2 silylation. Titanium silylimide and disilylamide model complexes represent the first well-defined group IV imide/amide catalysts for N2 reduction. Silylamine release and regeneration of the dinitrogen complex highlight the potential of early transition metals for N2 catalysis.
The activation and transformation of chemically inert dinitrogen (N2) is a fundamental challenge with significant implications for human society. In nature, certain microorganisms convert N2 to ammonia under ambient conditions using nitrogenases.1 In contrast, the industrial Haber–Bosch process, which converts N2 and H2 into NH3, requires high temperatures and pressures, making it highly energy-intensive.2
To better understand nitrogen fixation and develop catalysts for milder conditions, various studies on artificial N2 fixation with transition metal complexes have been conducted.3 Several complexes have been developed to catalyse the conversion of N2 to NH3 in the presence of electron and proton sources.4 Alternatively, the reduction of N2 to silylamines catalysed by transition metal complexes can be achieved in the presence of reducing and silylating agents, broadening the approaches to functionalizing dinitrogen.5 While significant progress has been made with middle and late transition metal dinitrogen complexes, well-defined group IV metal complexes capable of catalysing N2 conversion to ammonia or silylamines remain scarce.6 Furthermore, group IV metal imide or amide catalysts have not yet been reported.7 Here, we present a series of titanium dinitrogen complexes bearing tris(pyrazolyl)borate (Tp) and alkoxide or aryloxide ligands that mediate the catalytic reduction of N2 to silylamines. Additionally, separately synthesized titanium silylimide and disilylamide complexes represent unprecedented well-defined group IV imide/amide species capable of catalyzing N2 reduction. Key transformation steps, such as the release of silylamine and regeneration of the titanium dinitrogen complex, have also been elucidated.
The tris(pyrazolyl)borates (Tp) are attractive ligands for catalysts in various transformations and generally considered as formal analogues of the cyclopentadienyl ligand, owing to their similar charge, number of electrons donated and facial coordination geometry.8 Tp-ligated titanium alkyl and chloride complexes have shown notable activity in olefin polymerization,8,9 highlighting their ability to stabilize reactive titanium centres. Motivated by these properties, we explore Tp-supported titanium complexes as potential catalysts for N2 fixation. In view of the fact that transition metal complexes featuring Tp ligands capable of activating dinitrogen are relatively rare,10 the investigation of Tp-supported group IV metal complexes for dinitrogen fixation is obviously of great interest and importance.
A Tp* (Tp* = HB(3,5-Me2-pyrazolyl)3) supported titanium chloride complex Tp*TiCl2(THF) could be conveniently prepared through the reaction of TiCl3(THF)3 and Tp*K (Scheme 1A). Building on our recent discoveries in vanadium and molybdenum catalysis for dinitrogen reduction, where bulky alkoxide or aryloxide ligands were crucial in stabilizing complexes and enhancing catalytic activity,11 we introduced similar groups into the titanium complexes. Treatment of Tp*TiCl2(THF) with one equivalent of lithium aryloxide or alkoxide in toluene afforded Tp*Ti(L)Cl (L = OC6H3Me2-2,6, 1a, 53%; L = OC6H3iPr2-2,6, 1b, 56%; L = OC6H3tBu-2,6, 1c, 64%; L = OtBu, 1d, 63%). Complexes 1a, 1b, and 1d feature a coordinated THF ligand and adopt similar distorted octahedral geometries, whereas steric congestion imposed by the bulky 2,6-di-tert-butylphenoxide ligand in 1c precludes THF coordination, resulting in a slightly distorted square pyramidal geometry (τ = 0.36, see the SI).
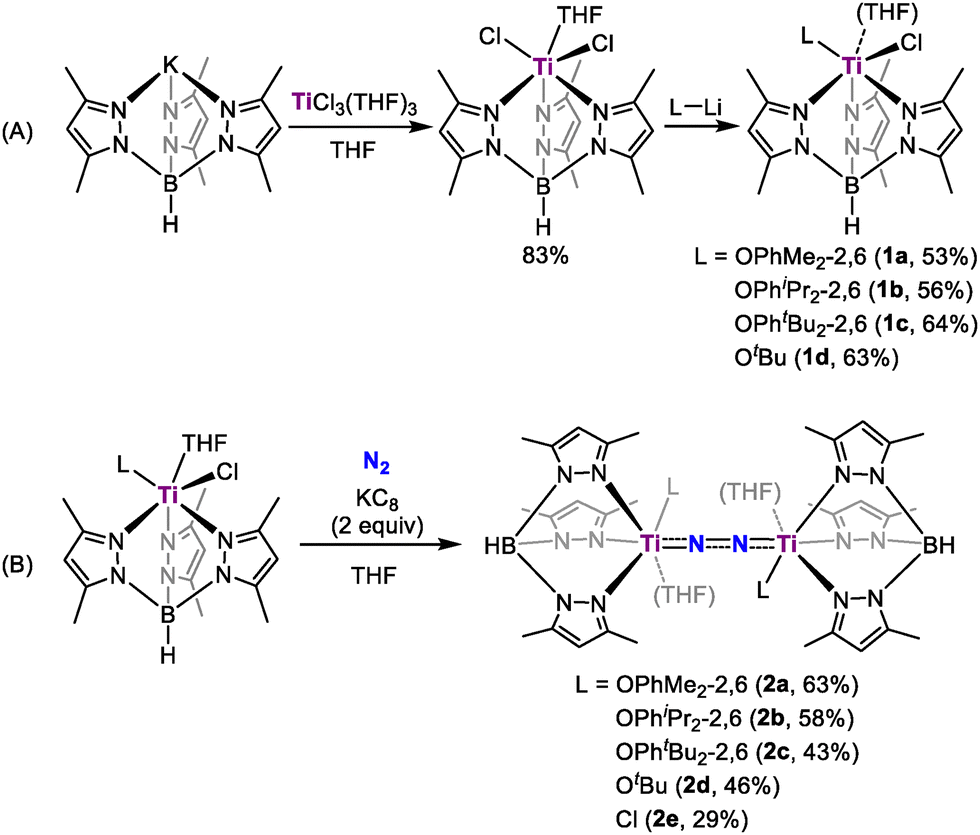 | ||
| Scheme 1 Synthesis of titanium chloride complexes (A) and their reduction to dinitrogen complexes (B) | ||
Reduction of 1a with two equivalents of KC8 in THF at room temperature under a N2 atmosphere afforded a dinitrogen-bridged dititanium complex [Tp*Ti(OC6H3Me2-2,6)]2(µ-η1:η1-N2)(THF)2 (2a) in 63% yield as dark green crystals (Scheme 1B). Complex 2a is stable both in the solid-state or in solution at room temperature. X-ray diffraction study revealed that the dinitrogen ligand is bonded to two Ti atoms in an end-on bridged manner, with each Ti atom also coordinated by a Tp* ligand, a THF molecule and a 2,6-dimethylphenoxide ligand (Fig. 1). 2a has a nearly linear Ti1–N1–N2–Ti2 topology, with Ti1–N1–N2 and Ti2–N2–N1 angles of 174.0(2)° and 173.2(1)°. The N–N bond length (1.237(2) Å) is significantly elongated compared to free N2 (1.098 Å), and slightly shorter than that of [(Me2N)C(NiPr)2]4Ti2(µ-N2) (1.28(1) Å)12 and {[(Me3Si)2NC-(NC6H11)2]4Ti}2(µ-N2) (1.278(3) Å),13 suggesting strong activation of N2. The Ti–N bond distances (Ti1–N1: 1.805(2) Å; Ti2–N2: 1.802(2) Å) are longer than Ti–Nimido (1.689(2) Å) in [(nacnac)Ti = NH(Ntol2)] (nacnac = [(2,6-iPr2C6H3)NC(CH3)]2CH, tol = 4-CH3C6H4),14 but shorter than the Ti–Namido (1.932(4) Å) bond distances in [(Me3Si)2C5H3]2Ti(NH2),15 indicating the existence of σ- and π-bonding in the Ti–N bonds. The 15N NMR spectrum of the isotopically labelled complex 2a-15N, prepared from the reaction of 1a with KC8 under 15N2, displays a singlet at δ 48.2 ppm. This substantial upfield shift from the value of 114.7 ppm reported for [(OO)Ti(C5H5N)]2(μ-η1:η1-N2) (OO = (1,4-(OC6H4MetBu)2C6H4))16 is indicative of increased electron density and a reduced bond order of the N–N unit, demonstrating a much higher degree of N2 activation in 2a.
 | ||
| Fig. 1 Molecular structure in the solid-state of 2a with thermal ellipsoids drawn at 30% probability. Hydrogen atoms were omitted for clarity. | ||
The computed Ti–N Mayer bond orders of 1.55 further support this bond interaction. The X-ray photoelectron spectroscopy (XPS) analysis of 2a reveals a binding energy of 458.06 eV (Fig. S16), which indicates that the oxidation states of the metal centres tend to be Ti(IV).13 The UV-vis spectroscopic studies of 2a exhibited absorption at 336 nm. The Raman spectrum of solid 2a displayed a peak at 1336 cm−1, which shifted to 1307 cm−1 in 2a-15N. This unusual low ν(N–N) stretching vibration indicates a significant reduction of N–N bond order and is consistent with the computational analysis. Solid-state magnetization measurements (SQUID) for 2a showed no paramagnetic signal, reflecting the diamagnetic nature of the complex.
The electronic structure of 2a was explored through theoretical calculations using the B3LYP/def2-SVP method. Complex 2a behaves as a closed-shell species. Analysis of the frontier orbitals reveals two π-orbital interactions between the π* orbitals of dinitrogen and dxy (dxz) orbitals of two titanium atoms (Fig. S29). In the HOMO, the main contributions come from the two titanium centres (20.3% and 20.2%) and dinitrogen (36.5%). While the HOMO−1 consists primarily of the 3d orbitals of titanium (26.2% and 26.1%) and the π* orbitals (37.8%) of dinitrogen. This suggests a double-bond character between each titanium and nitrogen atom. Natural bond orbital analysis further supports this observation (see the SI).
Other Tp* ligated titanium dinitrogen complexes were prepared using similar approaches. Complexes [Tp*Ti(OC6H3Me2-2,6)]2(µ-η1:η1-N2)(THF)2 (L = OC6H3iPr2-2,6, 2b; L = OtBu, 2d; L = Cl, 2e) were isolated in a yield of 58%, 46% and 29%, respectively. X-ray diffraction analysis confirmed that the structures of 2b, 2d, and 2e are similar to that of 2a (see the SI). In contrast, the 2,6-di-tert-butylphenoxide ligated titanium dinitrogen complex [Tp*Ti(OC6H3tBu2-2,6)]2(µ-η1:η1-N2) (2c) was prepared as a THF-free titanium dinitrogen complex, attributed to the increased steric hindrance around the metal centre (Fig. S24).
With a series of titanium dinitrogen complexes in hand, we have explored the catalytic reductions of dinitrogen into ammonia. However, an only slightly greater-than-stoichiometric amount of NH3 (2.7 ± 0.1) was detected when 2a was utilized as a catalyst with [H(Et2O)2][BArF4] (ArF = (3,5-(CF3)2C6H3)) as the acid and KC8 as the reductant (see the SI). Alternatively, we investigated the capabilities of titanium complexes in the catalytic silylation of dinitrogen using silicon and electron equivalents. The initial investigation of N2 reduction performance employed Me3SiCl as the silicon source and KC8 as the electron source. The generated tris(trimethylsilyl)amine was hydrolyzed with HCl, and the resulting NH4Cl was quantified by the indophenol method (Table 1).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | Reductant | R3SiCla | Solvent | TONb | Yieldc/% |
| Reactions were performed at −40 °C.a Stoichiometric quantities of Me3SiCl and reductant.b TON = equiv. of [N(SiMe3)3]/equiv of [Ti], and represents the average of ≥2 independent runs.c Yield = 3 × NH4+/reductant. | ||||||
| 1 | 2a | KC8 (100) | Me3SiCl (100) | THF | 5.9 ± 0.9 | 35 |
| 2 | 2b | KC8 (100) | Me3SiCl (100) | THF | 6.0 ± 0.3 | 36 |
| 3 | 2c | KC8 (100) | Me3SiCl (100) | THF | 5.9 ± 0.2 | 35 |
| 4 | 2d | KC8 (100) | Me3SiCl (100) | THF | 5.4 ± 1.2 | 32 |
| 5 | 2e | KC8 (100) | Me3SiCl (100) | THF | 5.1 ± 0.6 | 31 |
| 6 | – | KC8 (100) | Me3SiCl (100) | THF | 0.1 ± 0 | 0.2 |
| 7 | 3a | KC8 (100) | Me3SiCl (100) | THF | 5.9 ± 0.6 | 35 |
| 8 | 4a | KC8 (100) | Me3SiCl (100) | THF | 7.5 ± 0.7 | 45 |

The turnover number (TON) was calculated based on the amount of silylamine produced per Ti center. To ensure accurate quantification, the products from the same catalytic run, namely N(SiMe3)3 and NH4+ (from hydrolysis), were analysed via independent methods (1H NMR and GC-MS for the silylamine, and the indophenol method for ammonia), all of which showed consistent results. Under conditions of 100 equiv. of Me3SiCl and 100 equiv. of KC8 in THF at room temperature, both the dinitrogen complexes (2a–e) and their precursors (Tp*TiCl2(THF) and 1a–d) produced only a small amount of N(SiMe3)3 (1.9–4.1 equiv.) with yields spanning a range from 6% to 18% (Table S2). The catalytic reaction generated side products including Me3SiSiMe3 (13% yield) and Me3SiOC4H9 (∼1% yield), implicating trimethylsilyl radicals (Me3Si˙) in pathways involving dimerization and reaction with the THF solvent.17 As lower temperature can suppress side reactions,11b we conducted the catalysis with 2a at −40 °C, resulting in an increased production of silylamine (5.9 ± 0.9 equiv.) and an improved overall yield of 35% (entry 1). Similarly, 2b produced up to 6.0 ± 0.3 equiv. of N(SiMe3)3 with a yield of 36%. Besides, we have investigated the effectiveness of various reducing agents, including Li, Na, K, and KC8, finding that KC8 was the most effective (5.9 vs. 0.5–5.8 equiv., Table S6). To trace the nitrogen source, catalysis was conducted with complex 2a-15N under a 15N2 atmosphere. After acidic work-up, 15NH4+ was detected by 1H NMR spectroscopy (Fig. S15), unambiguously verifying dinitrogen gas as the nitrogen source.
Additionally, we explored the reactions in toluene, THF, DME, and Et2O, determining that the conditions in THF exhibited better conversion than those in other solvents (5.9 vs. 0.3–5.5, Table S4). The catalysis was further explored using other sterically hindered silicon reagents. However, lower amounts of silylamines were detected with iPr3SiCl and chlorodimethylvinylsilane (Me2(CH2CH)SiCl) (Table S5).
To gain more information about the catalytic process, stoichiometric reactions of 1a with Me3SiCl and KC8 were performed, but no intermediate complex was isolated, likely due to its high reactivity under the reaction conditions. On the other hand, we have also attempted to synthesize the titanium silylimide model complex by adding Me3SiN3 to a toluene solution of 2a. The reaction afforded complex Tp*Ti = NSiMe3(OC6H3Me2-2,6)(THF) (3a) in 59% yield, accompanied by the extrusion of N2 (Scheme 2A). X-ray diffraction of 3a shows a six-coordinate mononuclear framework with a silylimido ligand, an aryloxide ligand, a coordinated THF molecule, and a Tp* ligand, adopting a slightly distorted octahedral geometry (Fig. 2). The silylimido ligand is coordinated to the Ti atom in a near-linear manner (Ti1–N1–Si1: 178.3(1)°). The Ti–N distance in 3a (Ti1–N1: 1.725(2) Å) is longer than that of reported Ti–Nnitrido bonds (1.660(2) Å)18 and shorter than that of Ti–Namide bonds (1.929(3) Å).19
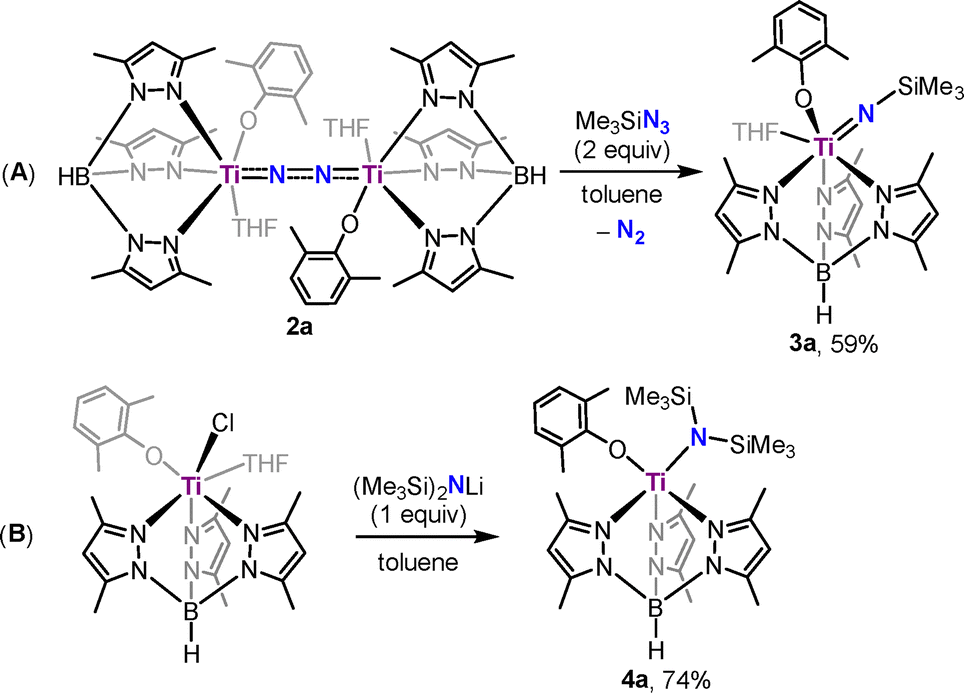 | ||
| Scheme 2 Synthesis of silylimide (3a, A) and disilylamide (4a, B) complexes. | ||
 | ||
| Fig. 2 Molecular structure in the solid-state of 3a with thermal ellipsoids drawn at 30% probability. Hydrogen atoms were omitted for clarity. | ||
Separately, reaction of 1a with lithium bis(trimethylsilyl)amide at room temperature afforded the titanium bis(trimethylsilyl)amide complex Tp*Ti[N(SiMe3)2](OC6H3Me2-2,6)(THF) (4a) in 74% yield as orange crystals, structurally confirmed by X-ray diffraction (Scheme 2B, for details see the SI).
Treatment of 3a with one equivalent of KC8 and Me3SiCl at −40 °C led to the release of N(SiMe3)3 in 28% yield, with recovery of unreacted 3a. In contrast, 4a under the same conditions afforded N(SiMe3)3 in 68% yield along with regeneration of the titanium dinitrogen complex 2a (33% yield), suggesting that 3a might first form 4a before converting to 2a. Under catalytic conditions (100 equiv. of KC8 and Me3SiCl at −40 °C under N2), 3a and 4a served as active catalysts for dinitrogen silylation (Table 1, entries 7 and 8). Notably, 4a exhibited the highest TON of 7.5 ± 0.7 with a yield of 45%.
Transition metal dinitrogen complexes are widely employed as catalysts for synthesizing ammonia and silylamines from N2, with imide and amide species recognized as key intermediates. However, effective imide and amide complex catalysts for N2 reduction are rare. While molybdenum and iron imide/amide complexes show notable silylation activity,7 group IV analogues remain unreported. Here titanium silylimide and disilylamide complexes provide the first such examples, displaying catalytic performance comparable to that of dinitrogen complexes.
Notably, the disilylamide complex liberated N(SiMe3)3 and regenerated the titanium dinitrogen complex in the present of KC8 and Me3SiCl, modelling the late-stage cycle. These results suggest a plausible mechanism where reductive silylation of the dititanium dinitrogen complex affords silylimide and disilylamide intermediates, which release N(SiMe3)3 and regenerate dinitrogen complexes upon further silylation. A pathway where N2(SiMe3)4 is released and subsequently converted to N(SiMe3)3 cannot be excluded.6a,b,20
In summary, this work demonstrates catalytic N2 silylation using Tp-supported titanium complexes and reports the first well-defined group IV silylimide/disilylamide catalysts for this reaction. The observed reactivity, particularly the disilylamide's ability to liberate silylamine and close the catalytic cycle by regenerating the dinitrogen complex, supports a plausible pathway involving silylation of bimetallic dinitrogen-bridged titanium species. These findings establish Tp-supported titanium complexes as a unique platform for dinitrogen functionalization, underscoring the broader potential of early transition metals in catalytic N2 reduction.
This work was supported by the National Natural Science Foundation of China (22371017), Startup Funding from Beijing Normal University (312232110), the Young Thousand Talents Program (110532107) and the Fundamental Research Funds for the Central Universities. We thank Prof. Haoling Sun (Beijing Normal University) for SQUID measurements, and Prof. Zhi Li and Ms Gangmei Li (Beijing Normal University) for the help with XPS analysis.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: synthetic procedures, NMR, UV-Vis and Raman spectra, crystallographic data, and computational details. See DOI: https://doi.org/10.1039/d5cc04879f.CCDC 2331148 (1a), 2331149 (1b), 2350473 (1c), 2216825 (1d), 2310979 (2a), 2311060 (2b), 2350469 (2c), 2311244 (2d), 2312824 (2e), 2334087 (3a) and 2331239 (4a) contain the supplementary crystallographic data for this paper.21a–k
Notes and references
- (a) B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS PubMed; (b) O. Einsle and D. C. Rees, Chem. Rev., 2020, 120, 4969–5004 CrossRef CAS PubMed.
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- (a) S. Kim, F. Loose and P. J. Chirik, Chem. Rev., 2020, 120, 5637–5681 CrossRef CAS PubMed; (b) D. Singh, W. R. Buratto, J. F. Torres and L. J. Murray, Chem. Rev., 2020, 120, 5517–5581 CrossRef CAS PubMed; (c) S. J. K. Forrest, B. Schluschaß, E. Y. Yuzik-Klimova and S. Schneider, Chem. Rev., 2021, 121, 6522–6587 CrossRef CAS PubMed; (d) Z.-J. Lv, J. Wei, W.-X. Zhang, P. Chen, D. Deng, Z.-J. Shi and Z. Xi, Natl. Sci. Rev., 2020, 7, 1564–1583 CrossRef CAS PubMed.
- (a) M. J. Chalkley, M. W. Drover and J. C. Peters, Chem. Rev., 2020, 120, 5582–5636 CrossRef CAS PubMed; (b) Y. Tanabe and Y. Nishibayashi, Coord. Chem. Rev., 2022, 472, 214783 CrossRef CAS.
- Y. Tanabe and Y. Nishibayashi, Coord. Chem. Rev., 2019, 389, 73–93 CrossRef CAS.
- (a) L. R. Doyle, A. J. Wooles, L. C. Jenkins, F. Tuna, E. J. L. McInnes and S. T. Liddle, Angew. Chem., Int. Ed., 2018, 57, 6314–6318 CrossRef CAS PubMed; (b) A. Wong, F. Y. T. Lam, M. Hernandez, J. Lara, T. M. Trinh, R. P. Kelly, T. Ochiai, G. Rao, R. D. Britt, N. Kaltsoyannis and P. L. Arnold, Chem Catal., 2024, 4, 100964 CAS; (c) P. Ghana, F. D. van Krüchten, T. P. Spaniol, J. van Leusen, P. Kögerler and J. Okuda, Chem. Commun., 2019, 55, 3231–3234 RSC; (d) M. Mori, J. Organomet. Chem., 2004, 689, 4210–4227 CrossRef CAS.
- (a) Q. Liao, N. Saffon-Merceron and N. Mézailles, Angew. Chem., Int. Ed., 2014, 53, 14206–14210 CrossRef CAS PubMed; (b) Q. Liao, N. Saffon-Merceron and N. Mézailles, ACS Catal., 2015, 5, 6902–6906 CrossRef CAS; (c) R. Araake, K. Sakadani, M. Tada, Y. Sakai and Y. Ohki, J. Am. Chem. Soc., 2017, 139, 5596–5606 CrossRef CAS PubMed.
- S. Trofimenko, Chem. Rev., 1993, 93, 943–980 CrossRef CAS.
- (a) L. E. Manzer, J. Organomet. Chem., 1975, 102, 167–174 CrossRef CAS; (b) S. Murtuza, O. L. Casagrande and R. F. Jordan, Organometallics, 2002, 21, 1882–1890 CrossRef CAS; (c) K. Itagaki, K. Kakinuki, S. Katao, T. Khamnaen, M. Fujiki, K. Nomura and S. Hasumi, Organometallics, 2009, 28, 1942–1949 CrossRef CAS.
- (a) D. C. Cummins, G. P. A. Yap and K. H. Theopold, Eur. J. Inorg. Chem., 2016, 2349–2356 CrossRef CAS; (b) S. Zhang, H. Fallah, E. J. Gardner, S. Kundu, J. A. Bertke, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2016, 55, 9927–9931 CrossRef CAS PubMed; (c) A. Reinholdt, D. Pividori, A. L. Laughlin, I. M. DiMucci, S. N. MacMillan, M. G. Jafari, M. R. Gau, P. J. Carroll, J. Krzystek, A. Ozarowski, J. Telser, K. M. Lancaster, K. Meyer and D. J. Mindiola, Inorg. Chem., 2020, 59, 17834–17850 CrossRef CAS PubMed.
- (a) W. Huang, L.-Y. Peng, J. Zhang, C. Liu, G. Song, J.-H. Su, W.-H. Fang, G. Cui and S. Hu, J. Am. Chem. Soc., 2023, 145, 811–821 CrossRef CAS PubMed; (b) Z. Li, C. Liu, J. An, X. Wang and S. Hu, ACS Catal., 2024, 14, 6558–6564 CrossRef CAS.
- S. M. Mullins, A. P. Duncan, R. G. Bergman and J. Arnold, Inorg. Chem., 2001, 40, 6952–6963 CrossRef CAS PubMed.
- B. Wu, R. Feng, Z.-B. Yin, H. Yan, X. Wang, G.-X. Wang, J. Wei and Z. Xi, Sci. China: Chem., 2023, 66, 755–759 CAS.
- B. L. Tran, M. P. Washington, D. A. Henckel, X. Gao, H. Park, M. Pink and D. J. Mindiola, Chem. Commun., 2012, 48, 1529–1531 RSC.
- C. D. Sofield, M. D. Walter and R. A. Andersen, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, m465–m466 CrossRef PubMed.
- T. Kurogi, Y. Ishida and H. Kawaguchi, Chem. Commun., 2013, 49, 11755–11757 RSC.
- (a) J. H. Sharp and M. C. R. Symons, J. Chem. Soc. A, 1970, 3084–3087, 10.1039/J19700003084; (b) L. E. Gusel'nikov, Y. P. Polyakov, E. A. Volnina and N. S. Nametkin, J. Organomet. Chem., 1985, 292, 189–203 CrossRef.
- M. E. Carroll, B. Pinter, P. J. Carroll and D. J. Mindiola, J. Am. Chem. Soc., 2015, 137, 8884–8887 CrossRef CAS PubMed.
- B. C. Bailey, F. Basuli, J. C. Huffman and D. J. Mindiola, Organometallics, 2006, 25, 2725–2728 CrossRef CAS.
- R. Feng, Y. Jiang, X. Shi, X. Wang, W. Chen, F. Xie, J. Su, J. Wei, S. Ye and Z. Xi, CCS Chem., 2023, 5, 2473–2481 CrossRef CAS.
- (a) CCDC 2331148: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2j7rbw; (b) CCDC 2331149: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2j7rcx; (c) CCDC 2350473: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2jwvq0; (d) CCDC 2216825: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2ddsh3; (e) CCDC 2310979: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2hkrqk; (f) CCDC 2311060: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2hkvb8; (g) CCDC 2350469: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2jwvlw; (h) CCDC 2311244: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2hl18f; (i) CCDC 2312824: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2hmp72; (j) CCDC 2334087: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2jbt4v; (k) CCDC 2331239: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2j7v8x.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |