
Photo-switching in multi-stimuli-responsive low Z′-high Z′ co-crystal polymorphs†
Ishtiyaq
Ahmad
a,
Siriyara Jagannatha
Prathapa
b and
Aijaz A.
Dar
 *a
*a
aCrystal Engineering Laboratory, Department of chemistry, University of Kashmir, Srinagar, Hazratbal-190006, Jammu & Kashmir, India. E-mail: aijazku2015@gmail.com; daraijaz@uok.edu.in
bSCXRD Bangalore, Bruker India Scientific Pvt. Ltd., India
First published on 28th April 2025
Abstract
Emission switching in crystalline materials, a phenomenon uncommon for multi-component polymorphic crystals, is an intriguing with a wide range of applications in optics and optoelectronics. We reported co-crystal polymorphs obtained by the crystallisation of o-arsanilic acid (2-ABAA-2H) and 4,4′-bipyridyl (4,4′-BPY) in different solvent systems. Structural studies established that the crystal forms exist as low Z′ (=1), Z′′ (=3) [(4,4′-BPY-2H)2+(2-ABAA-H)2−] (1) and its high Z′ (=6), Z′′ (=18) [(4,4′-BPY-2H)62+(ABAA-H)12−] (2) forms and undergo proton transfer between crystal forms to exist as ionic solids. Form 1 is a green emitter (λmax 512 nm, ϕ 2.6%, τ 2.5 ns), while 2 is non-emissive. Mechanochromic studies establish the retention of green emission in the 1G form and emission turn-on in the 2G form. The ground forms respond to basic ammonia fumes by undergoing emission tuning to cyan and intense blue in 1-NH3 and 2-NH3, respectively. Thin films of both 1 and 2 exhibit green emission when prepared in a range of solvents, implying the retention of their microcrystalline phases upon dissolution. Aggregation-induced emission (AIE) studies of the products are reported and supported through FE-SEM and DLS studies. Structural studies indicate that slipped π–π interactions in the lattice of 1, form J-type aggregates, and are responsible for its solid-state emission, while 2, devoid of notable π–π interactions, is non-emissive due to loose crystal packing, plausibly leading to vibrational quenching.
Introduction
Luminescence switching in π-conjugated solids mainly arises due to differences in stacking modes, and the materials provide immense opportunities for the development of optical and optoelectronic devices with a wide range of applications.1,2 The establishment and study of structure–property correlations in luminescent molecular solids are significant for understanding the underlying mechanisms and developing design strategies. Crystal engineering focuses on understanding intermolecular interactions and utilizing this understanding to design materials, offering an opportunity to correlate structure with properties.3–5 Polymorphism, i.e. crystallization of the same compound in different crystal forms with different properties, is an intriguing phenomenon of immense significance and provides a unique opportunity to establish structure–function correlations and understand molecular interconversions through external stimuli.6–12 The differences in physicochemical properties between polymorphs range from mild to striking, particularly in solubility, mechanical behaviour, stability, conductivity, magnetism, luminescence, etc.13Research efforts for the development of organic functional materials are fuelled by a recent shift toward the development of organic alternatives to inorganic functional materials, including luminescent solids.14 Solid-state luminescence, an important material property, is, however, limited by challenges such as aggregation-caused quenching (ACQ) and its late exploration in organic solids.15–19 The materials provide more opportunities and convenience for device processing, development, fabrication and have been exploited for the development of organic light-emitting diodes (OLEDs),20,21 optoelectronics,22,23 optical waveguides (OWGs),24,25 organic solid-state lasers (OSSLs),26,27 organic light-emitting transistors (OLETs),28,29 sensors,30,31 biotags,32,33 stimuli-responsive materials (SRMs),34,35 bio-imaging,36,37 and up-conversion emission.38–41
Crystal engineering of luminescent organic crystalline materials has been reported through crystallochromism, polymorphism and coformer variation.42–45 Emission tuning and the realisation of multicolour emission through polymorphism, in particular, are well reported for single-component crystals but are rare in multi-component systems. ON/OFF emission switching is even more rarely observed in organic crystals. For example, Shimei and coworkers have realised emission switching in a pyrene dimer. The pristine crystal is non-emissive due to the ordered and long-range π stacking and undergoes reversible emission turn-on upon grinding/UV irradiation exposure, due to dimerisation.46,47 Similarly, Zhenguo and coworkers designed a donor–acceptor-based photo-switchable thermally activated delayed emission (TADF) luminogen and utilised it for the development of sequential and combinational single-component logic gate systems.48 Our exhaustive survey indicated “on–off” luminescent switching is unreported in co-crystal polymorphs.
Polymorphism in molecular crystals can result from various factors and is typically classified as conformational, packing or synthon polymorphism.49 High Z′ and low Z′ crystal forms of molecules, where Z′ represents the number of molecules or formulae units in an asymmetric unit, represent another rare form of polymorphism. Well reported for single-component crystals, high Z′-low Z′ co-crystal polymorphs are sparse [Table S1, ESI†] and have not been investigated for property studies, including luminescence.50
In this study, we report exceptionally high Z′-low Z′ co-crystal polymorphs 1 and 2, with unique “ON/OFF” emission switching arising from differences in π-stacking interactions. The non-emissive polymorph exhibits a striking emission turn-on upon grinding, and the grounded forms of both crystal forms exhibit a vapo-fluorochromic response upon exposure to base fumes. AIE studies of both forms have also been reported.
Results and discussion
We observed intriguing properties, including optics, in materials based on sulfonate-pyridinium supramolecular synthon. Therefore, we extended the design of materials to the structural analogue of organo-sulfonates, i.e., organo-arsenate, and tested its reactions with 2,2′-bipyridyl. Co-crystallization of equimolar mixtures of o-arsanilic acid and bipyridyl in acetone led to the formation of [(4,4′-BPY-2H)2+(2-ABAA-H)2−] 1 as a pale-yellow solid in a couple of days after slow evaporation at ambient conditions, while keeping the conditions the same, change in solvent system to methanol/water (30/70 v/v) led to the isolation of intense orange high Z′ polymorph [(4,4′-BPY-2H)62+(ABAA-H)12−] 2. The color polymorphs also exhibit different crystal morphologies and rare emission on–off switching. For example, 1 exhibits intense green emission, whereas the emission of 2 is quenched (Scheme 1).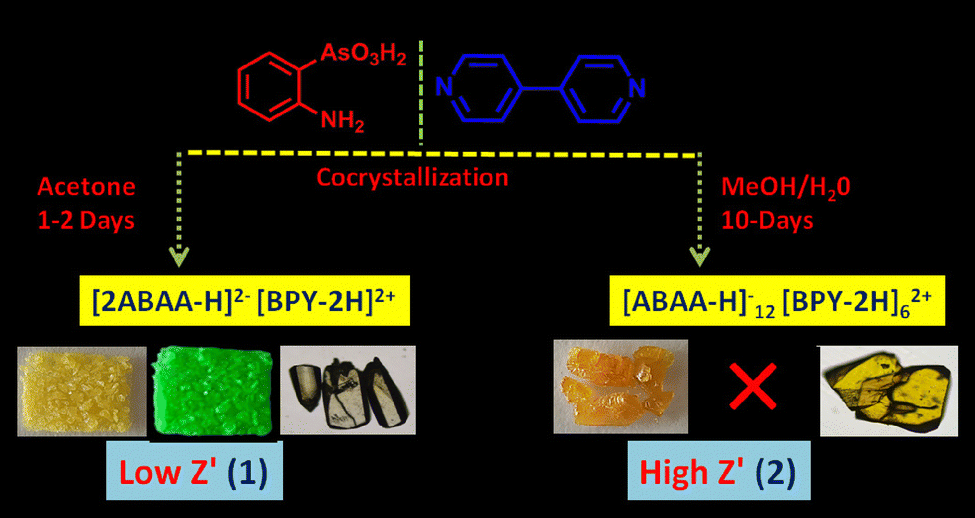 | ||
| Scheme 1 Chemical composition and synthetic scheme of molecular salts 1 and 2. | ||
The formation of the polymorphs was supported by Fourier transform infrared (FT-IR) spectroscopy: the absorption bands at around 3400 cm−1 and 3300 cm−1 correspond to the symmetric and asymmetric stretching of ν (N–H) of the amine groups, while the peak at around 3100 cm−1 corresponds to the N+–H stretching, indicating proton transfer between acid–base precursors. The bands at 915 and 715 cm−1 correspond to the symmetric and asymmetric stretching of the As-OH group, (Fig. 1a). Powder X-ray diffraction studies validate the crystalline nature of the bulk solids and establish the phase purity and stability of the bulk solid of the high Z′ polymorph, whereas minor discrepancies in the diffraction pattern of the computed crystal form and bulk solid plausibly arise due to the packing changes upon conversion to the microcrystalline state, (Fig. 1b and c). Both 1H and 13C NMR spectra were recorded, and the observed chemical shifts and peak area ratios validated the formation of 1 and 2, (Fig. S1–S4, ESI†)
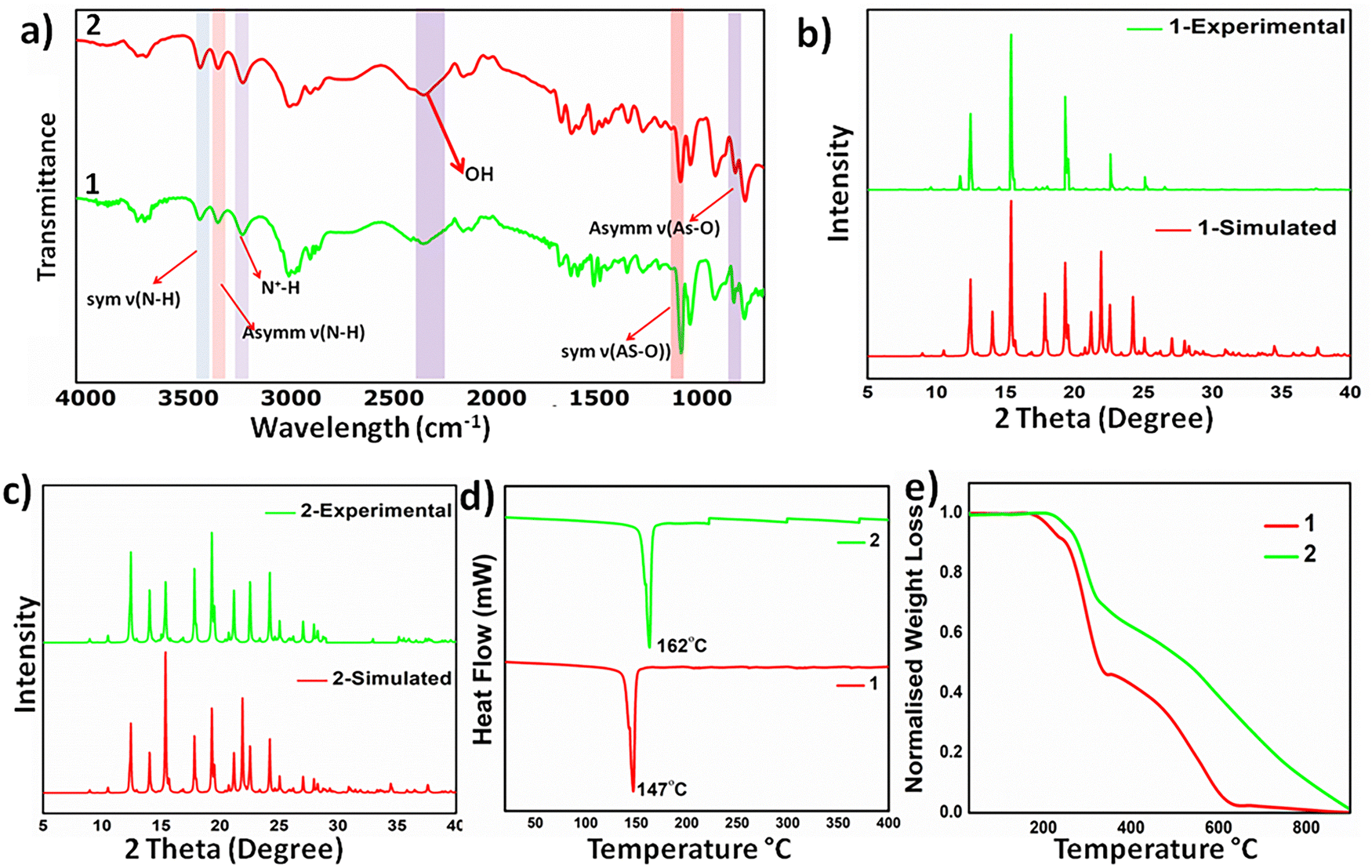 | ||
| Fig. 1 Characterisation data of 1 and 2: (a) FT-IR spectra, (b) and (c) powder X-ray diffraction data compared with their simulated data, (d) DSC plots and (e) TGA plots. | ||
Molecular salts exhibit moderate thermal stability and melt at 145–148 °C (1) and 159–162 °C (2). Thermogravimetric analysis (TGA) shows a higher thermal stability of 2, as the weight loss starts at 210 °C, compared to 1, for which the weight loss initiates at 196 °C, (Fig. 1d). Differential scanning calorimetry (DSC) analyses further substantiate the thermal behavior as sharp endothermic dips at 147 °C and 162 °C, corresponding to their melting points, observed in 1 and 2, respectively, (Fig. 1e). Careful analyses of the DSC curves rule out any phase transformation before melting points, particularly for high Z′ polymorphs, ruling out inter-conversion of the polymorphic forms.
Optical properties
Notable colour and luminescence differences in the two polymorphic forms prompted us to study their optical properties in detail. Solution-phase absorption studies of the polymorphic forms indicated the absence of any type of intramolecular charge transfer interaction in their aqueous solutions, as no absorption activity was observed beyond 400 cm−1, (Fig. S5 and S6, ESI†). Colour difference in 1 and 2 is corroborated by their diffuse reflectance studies, as the absorption spectra are significantly red-shifted, implying stronger intermolecular charge transfer interactions in the latter.Solvent-dependent absorption and emission studies were nearly identical for both 1 and 2, but the emission behavior varied depending on the nature of the solvent, (Fig. 2). Aqueous solutions of both forms are non-emissive, whereas emission is blue shifted in ethanol compared with methanolic solutions with a shift in the λmax values by 51 nm. This 51 nm shift can be attributed to a change in the molecular packing, aggregation effects and solute–solvent interactions (Fig. S7, ESI†). Solutions of both polymorphs in other solvents: dimethylformamide (DMF), tetrahydrofuran (THF), acetonitrile (MeCN) and acetone (Ace) were green emitting with subtle variations in the λmax, emission intensity, and quantum yield values (Tables S2 and S3, ESI†). Nearly identical solution-phase optical properties of the two forms hint toward the possible inter-conversion of the high Z′ form to its low Z′ form. The dissociation of co-crystals/salts on dissolution was ruled out based on comparative studies with starting materials, which was also supported by the retention of green emission on dissolution (Fig. 2 and Fig. S8, ESI†). Dynamic light scattering (DLS) studies indicate different extents of particle aggregation in different solvents, and the emission variation possibly arises due to differences in particle sizes (Table S4, ESI†).
 | ||
| Fig. 2 (a) and (b) Optical images of solutions of 1 and 2 in different solvents under 365 nm UV light; (c) and (d) PL spectra of 1 and 2 in different solvents at λex 365 nm and a concentration of 10−3 M. | ||
Solvent and particle size-dependent luminescence studies of 1 and 2 prompted us to investigate them further for their aggregation-induced emissions (AIE). The AIE of non-emissive aqueous solutions was performed against ethanol. Aqueous solutions of both 1 and 2 exhibit AIE with ethanol as a bad solvent, and the emission of AIEgens of 2 is more intense and red shifted by nearly 40 nm compared to 1, which shows structured emission bands of lower intensity (Fig. 3a and b). I/Io, where I represents the emission intensity of the solution in water only and Io is the emission intensity for a solution of definite fH2O/EtOH fractions, also validates a better AIE response of 2 (Fig. 3c and d).
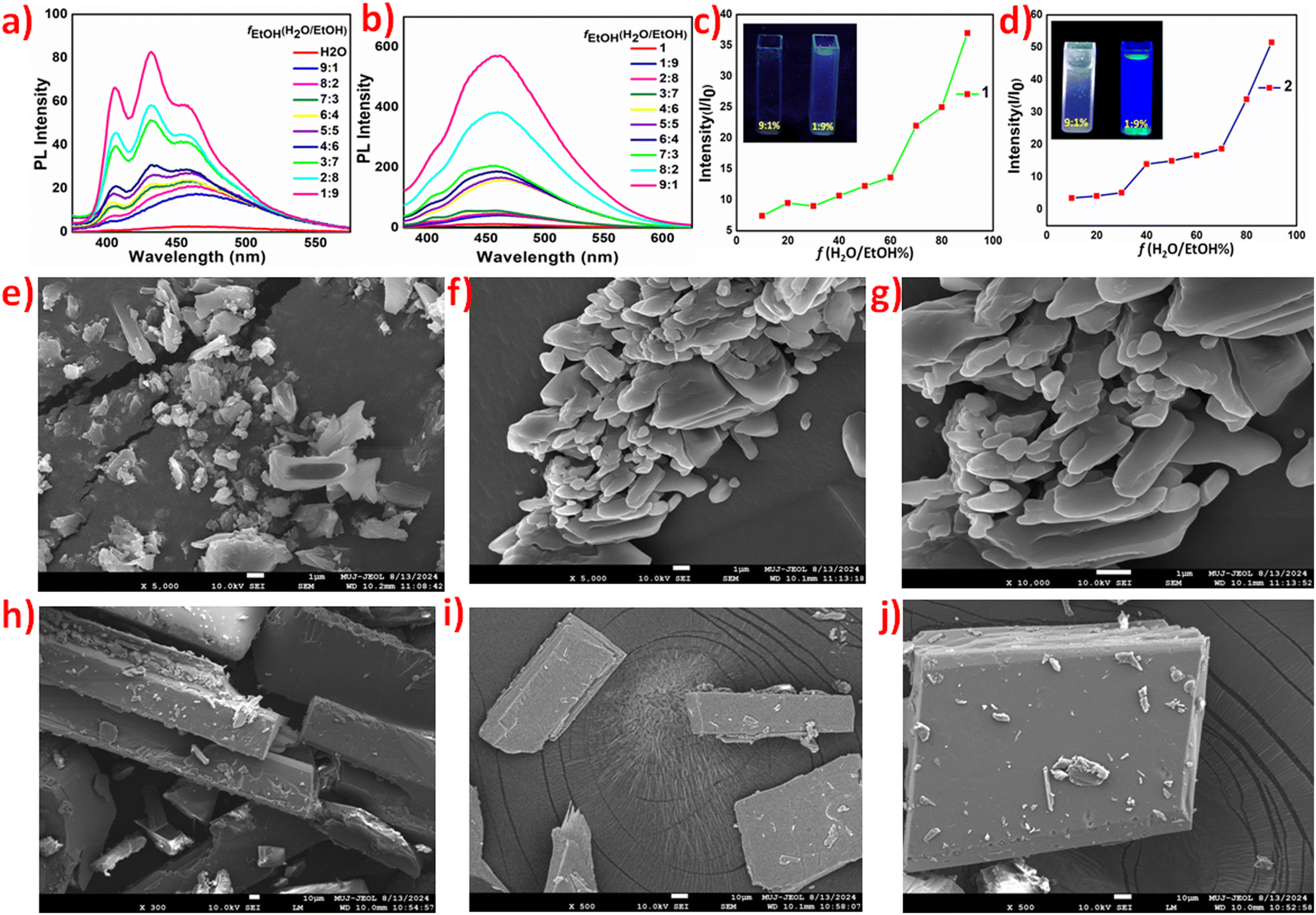 | ||
Fig. 3 AIE studies of 1 and 2. Photoluminescence (PL) spectra in different fH2O/EtOH fractions of 1 (a) and 2 (b) recorded for 10−3 M solutions; relative I/Io PL intensity plot at different fH2O/EtOH values for 1 (c) and 2 (d), with optical images of vials under a 365 nm UV lamp; FE-SEM images of AIE fractions at fH2O/EtOH values of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7, 7:3 and 9:1, respectively, for 1 (e)–(g) and 2 (h)–(j). :7, 7:3 and 9:1, respectively, for 1 (e)–(g) and 2 (h)–(j). | ||
With the gradual addition of ethanol, the emission of aqueous solutions of polymorphs 1 and 2 turns on until the maximum intensity is observed at an fH2O/EtOH value of 1:9, i.e., 10% of the water fraction (fw), with quantum efficiency values of 53.79 and 67.41% for 1 and 2, respectively. DLS studies validate the particle size increase with the addition of ethanol; hence, emission can be attributed to an increase in aggregation (Fig. S9, ESI†). Field emission-scanning electron microscopy (FE-SEM) analysis also confirmed aggregation and a gradual increase in particle size as the ethanol concentration increased (Fig. 3(e)–(j)). Fe-SEM images of 1 and 2 in pure water and pure ethanol are provided in the ESI† (Fig. S10).51
The solid-state PL-spectra of 1 shows an intense emission peak with λmax at 512 nm, corresponding to the observed green emission, with a quantum efficiency of 2.6% and CIE chromaticity coordinate values of 0.25 and 0.48, while 2 is non-emissive with a flat emission spectrum (Fig. 4). Fluorescence decay of the crystalline solid form of 1 using time-resolved photoluminescence (TRPL) and time-correlated single photon counting (TCSPC) method calculated the average lifetime value of 2.5 ns, (Fig. S11 and Table S5, ESI†).
 | ||
| Fig. 4 (a) Optical images of 1 and 2 crystals in daylight, bulk solid under UV light and their ground forms, vapochromic forms and piezochromic forms captured under a 365 nm UV light source; (b) and (c) DR-UV spectra of different forms of 1 and 2; (d) and (e) solid-state PL spectra of different forms of 1 and 2 at λex 365 nm. | ||
Thin films
The practical applications of luminescent materials are realized through their fabrication into thin films, which provide operational convenience. Thin film formation involves slurry coating, which may result in phase changes, hence varied optical properties. Therefore, thin film optical studies on polymorphic forms have been conducted. Interestingly, the thin films of both 1 and 2 prepared in different solvent systems yield only green emitting films (Fig. S12, ESI†). Diffuse reflectance, absorption and emission plots of the thin films are provided as ESI† (Fig. S13 and S14). These studies indicate possible phase changes of 2 upon dissolution and, as established by solution phase optical studies, its possible conversion to phase 1 upon dissolution.52Multi-stimuli responsive studies
Polymorphic systems are suitable for investigating stimuli-responsive behavior because they tend to interconvert on the application of external energy. The co-crystal polymorphs 1 and 2, though not inter-convertible, respond to mechanical grinding and exposure to base fumes. The mechano-fluorochromic response in 1 is not remarkable, as the shift in λmax is of 6 nm accompanied by a nearly 10% decrease in emission intensity,53 but is striking in 2, which undergoes emission turn on and the grounded form has brilliant green emission with λmax at 521 nm (Fig. 4d and e). Diffuse reflectance studies of the ground forms of 1G and 2G also indicated a blue shift in the absorption behavior (Fig. 4b and c). The emission variation and switching may be attributed to phase changes in the molecular solids upon grinding, which are evident in the powder X-ray diffraction pattern (Fig. 5). | ||
| Fig. 5 Powder X-ray diffraction patterns of 1 and 2 compared with their forms obtained after mechanical grinding of 1-G and 2-G and vapochromism of 1-NH3 and 2-NH3. | ||
The piezochromic studies, carried out by applying pressure up to 30 kPa, using a hydraulic IR-pellet press for compression of the crystalline forms of 1 and 2, resulted in a red-shifted emission of 9 nm in 1 and emission turn-on in 2 with a red-shift of 8 nm from its ground form 2G (Fig. 4 and Fig. S15, S16, ESI†). The results agree with the tight-binding model, which explains the compression-induced red-shift due to the closer approach of π-rings.
Both polymorphs, upon exposure to ammonia fumes under ambient conditions, exhibit a remarkable fluoro-chromic change. As visualized under UV exposure, the ammoniated solid forms 1-NH3 and 2-NH3 exhibit bright blue emissions, which were validated by solid-state fluorimetry. The emission spectra of 1-NH3 (λmax 478 nm) and 2-NH3 (λmax 525 nm) is significantly blue shifted compared to its ground form (Fig. 4 and Fig. S17, S18, ESI†). The FT-IR spectra of 1-NH3 and 2-NH3 shows new vibration peaks, which substantiate the incorporation of NH3 into the materials. Ammonia-treated solids show asymmetric Vasym (NH3) and symmetric Vsym (NH3) stretching modes for 1 at 3321 and 3185 cm−1, while for 2, these bands are present at 3311 and 3171 cm−1, respectively. The peak broadening in the 3400–2500 cm−1 region may be attributed to hydrogen bonding and the overlap of ν (N−H) and ν (O−H) absorptions of amine and water, respectively (Fig. S19, ESI†).
The diffuse reflectance spectra also indicate blue shifts in the absorption spectrum of ammoniated samples. Powder X-ray diffraction patterns show prominent changes in the phases of ammoniated forms (Fig. 5), resulting in emission tuning of the ammoniated solid forms.
Structural studies of the polymorphic forms, discussed vide infra, provide essential insights into the structure–property relationship. Of the two polymorphs, 2 forms a head-to-tail π-dimer in its crystal lattice, with slipped π-type stacking (centroid–centroid distance 3.636 and 3.748 Å, slip angle 76.84° and 86.24°, pitch angle 149.1°) (Fig. S20, ESI†). While there is no significant π–π interactions observed in 1, as the π centroid distances were too long (greater than 4 Å), (Fig. 6a, b). The observations agree with the observed properties, as π–π interactions are primarily responsible for the aggregation-caused quenching of emission, rendering 2 non-emissive in crystalline form, while 1, devoid of π-interactions, is emissive. The emission turn-on in 2 on grinding or compression is plausibly triggered by the displacement of the π-stacked rings, resulting in reduced interactions.
 | ||
| Fig. 6 Depiction of π-interactions in 1 (a) and 2 (b). | ||
Crystallographic studies
1 crystallizes in the monoclinic space group P21/c. The asymmetric unit consists of two anions of arsenate and a cation of bis-pyridinium, validating its non-stoichiometric composition of [(4,4′-BPY-2H)2+(2-ABAA-H)2−]. Crystal components are associated by direct ionic arsenate-pyridinium interaction: O3⋯H4–N4 [D–H⋯A: 170.23(3)°; D⋯A: 1.673(2) Å] and O4⋯H3–N3 [168.88(3)°; 1.733(2) Å], (Fig. 7a). | ||
| Fig. 7 (a) Molecular structure fragment of 1, (b) and (c) unit cell of 1 along a-axis represented in capped stick and space-fill models and (d) packing fragment of 1. | ||
The hydroxyl group and oxygen atom of the arsenate group are involved in the formation of cyclic centrosymmetric R22(8) synthons: O6⋯H5–O5 [112.64(3)°; 2.112(2) Å] and O1⋯H2–O2 [120.37(3)°; 1.891(2) Å], about the non-equivalent arsenate acids. The amine groups in two non-equivalent arsenate acids also exhibit the same pattern of intermolecular interactions i.e. one of the two amine hydrogens is involved in an intramolecular interaction with adjacent oxygen atoms: N2–H2B⋯O3 [78.25(3)°; 2.866(2) Å] and N1–H1A⋯O4 [142.94(3)°; 2.221(2) Å], while other hydrogens form intermolecular bonds, N2–H2A⋯O1 [18.70(3)°; 2.016(2) Å] and N1–H1B⋯O6 [169.24(3)°; 2.046(2) Å]. Interestingly, there are no other significant intermolecular interactions in the crystal lattice of 1, and the hydrogen bonding interactions lead to the formation of a supramolecule of two interpenetrated hydrogen-bonded lattices, as shown in Fig. 7b–d.
2 crystallizes in the monoclinic space group P21/c. The asymmetric unit consists of twelve anions of organo arsenate and six cations of bis-pyridinium, validating the non-stoichiometric composition of [(4,4′-BPY-2H)62+(ABAA-H)12−] and the empirical formula same as 1. All organo-arsenates were mono-deprotonated and the pyridyl centers were protonated, and there are direct ionic N+–H⋯O interactions observed between the two types of charged centers (Fig. 8a and Table S6, ESI†). The bipyridyl cations in 1 are planar, but of the six non-equivalent bipyridyl cations in 2, five are twisted with torsion angles ranging from −140° to 180° (Table S7, ESI†). The planar cation with a torsion value of 5.18° appeared twice with half occupancy in the asymmetric unit.
 | ||
| Fig. 8 (a) and (b) Molecular structure fragments of 2, (c) and (d) depiction of the unit cell of 2 along the c-axis and a-axis in capped stick and space-fill models. | ||
Interestingly, supramolecular aggregation through non-covalent interaction in all organo arsenate anions is similar as the arsenate groups engage in the formation of R22(8) homo-synthon, while third oxygen atoms are involved in the formation of direct ionic arsenate-pyridinium (N+–H⋯O) interactions, in addition to intramolecular interaction with one of the protons of the amine group. The other proton of the amine group forms intermolecular hydrogen bonds with one of the arsenate oxygen atoms involved in the formation of the homo-synthon. With these interactions, the arsenate anions grow into an independent 2-dimensional network (Fig. S21, ESI†), which is linked by bipyridyl cations into a three-dimensional network (Fig. 8b) and (Fig. S22, ESI†). Through weak C–H⋯π and π⋯π interactions, there is multiple-fold interpenetration of the hydrogen-bonded networks, resulting in the formation of a 3-dimensional solid. The structure and packing diagrams of 2 are shown in Fig. 8.
Hirshfeld analysis
The Hirshfeld surface analysis (graphical representations) and two-dimensional Hirshfeld surface fingerprint plots (HSFP) were calculated using Crystal Explorer.54 The Hirshfeld surface (dnorm) is given in (Fig. 9a and b), with transparency obtained using the ball-and-stick. Hirshfeld surfaces were generated and analyzed based on color coding: red, blue, and white. Red identifies contacts closer than the sum of their van der Waals radii, blue surfaces indicate distances longer than the sum of their van der Waals radii and white surfaces indicate contacts equal to the sum of their van der Waals radii. Atoms involved in short contacts or H-bonding interactions are displayed by red spots on the surface, which are mostly visualized over electronegative oxygen and arsenic atoms generated over dnorm surfaces in the case of both 1–2. Hirshfeld surface analysis fingerprint plots quantify the contribution of various intermolecular contacts toward crystal packing and suggest that H–H (37.8%) is the predominant contact in 1, while in 2, O–H (39.9%) is the most predominant contact. C–H contacts representing C–H⋯π interactions are the third and second most important contacts in 1 and 2, respectively. The contributions of various other contacts are depicted in (Fig. 9e).55 Predominantly higher contribution of O–H contacts, corresponding to hydrogen bonding interactions in non-emissive form 2, possibly promotes non-radiative vibration pathways resulting in emission quenching. | ||
| Fig. 9 Hirshfeld analysis of 1 and 2: (a) dnorm surface of 1, (b) dnorm surface of 2, (c) electrostatic potential map of 1, (d) electrostatic potential map of 2 and (e) fingerprint analysis of 1–2, plotted as a bar chart. | ||
π–π stacking is not a dominant interaction in the polymorphs and is further specified by constructing the HS over the shape index, whereas the curvedness provides information about planarity provided in (Fig. S23 and S24, ESI†). The existence of consecutive red and blue triangular regions around the phenyl rings on the shape index is an indication of π–π stacking.
The quantification of interactions is also useful when structural differences are very subtle and may be misleading, as in the case of polymorphs. To study the distribution of charge and nature of potentials, electrostatic potential maps were generated, which depict varying potential according to their colour differences as shown by blue. The negative electrostatic potential is shown by an intense red region in both 1–2 and is covered by oxygen and arsenic atoms, whereas the positive electrostatic potential is covered by bipyridine and the phenyl ring of arsenate, (Fig. 9c and d).
The application part is mentioned in ref. 56 and 57 (Fig. S25 and Table S8, ESI†).
Experimental section
Methods and materials
o-Arsanilic acid (PAA-2H) (99%, Sigma-Aldrich) and 4,4′-bipyridyl (99%, Sigma-Aldrich) were used as received. Distilled methanol and water were used for co-crystallization. Crystallization was performed using the slow evaporation method. Melting points were determined using an MP70 melting point system capillary apparatus (Mettler Toledo) in closed-end capillaries. Infrared spectroscopic data for molecular solids and their cocrystals were obtained using an Agilent Technologies Cary 630 FT-IR (4000–400 cm−1) made in Malaysia. DR-UV-vis studies (both absorbance and reflectance) were carried out on a Shimadzu 2600 spectrometer in BaSO4 medium. Fluorescence emission spectra were recorded on an Agilent spectrophotometer at different exciting wavelengths in liquid and solid phases. Thermal gravimetric and differential thermal analyses of the samples were performed on a simultaneous thermal analyzer-STA (LINSEIS, USA 6807/8835/16) using an alumina crucible at a heating rate of 10 °C. Data collection and refinement of single crystal analysis: single-crystal data were collected on a Rigaku Saturn 724+ CCD diffractometer using a graphite monochromator (Mo Kα, λ = 0.71075 Å). The selected crystals were mounted on the tip of a glass pin using mineral oil and placed in the cold flow of nitrogen gas. Complete hemispheres of data were collected using ω and φ scans (0.3°, 16 s per frame). Integrated intensities were obtained using Rigaku Crystal Clear-SM Expert 2.1 software and corrected for absorption correction. Structure solution and refinement were performed using the SHELX package. The structures were solved by direct methods and were completed by iterative cycles of ΔF synthesis and full-matrix least-squares refinement against F. The crystallographic table is shown in (Table S9, ESI†).Hirschfield studies
Hirshfeld analysis was performed using the CrystalExplorer 17.5 software. Color coding mapped on the dnorm surface represents contacts that indicate short (red color), intermediate (blue color), and long contacts (white color) compared to the sum of the van der Waals interactions.PXRD studies
Powder X-ray diffraction studies were carried out on an X-ray diffractometer (Rigaku Japan D/max 2500) using Cu-Kα radiation having λ = 1.54178 Å with a 2θ ranging from 5° to 50° at a scanning rate of 5° per minute in a step size range of 0.05° per second.Vapochromic studies
The vapochromic tests in 1–2 were carried out at room temperature by exposing the two compounds to volatile organic vapours of amine, i.e., ammonia, showing emission tuning upon different exposures. At room temperature, compounds were kept in an uncapped glass vial (3 mL) containing 10 mg of the compound, which was placed inside a 100 mL glass beaker containing 20 mL of ammonia. The vial was then sealed and allowed to form saturated solvent vapor. The vapochromic properties in the solid state were evaluated by UV-Vis diffuse reflectance spectroscopy and fluorescence spectroscopy.Mechanochromic studies
The mechanochromic studies in 1–2 were carried out by grinding the compounds in a mortar and pestle for 20 minutes. The products underwent mechanochromic changes between 3 and 5 minutes, after which no further changes in the optical properties were observed. The resultant change in colour and emission was evaluated by UV/vis diffuse reflectance and fluorescence spectroscopy.Quantum yield
The quantum yield of 1–2 in the solution phase was calculated following the standard protocol using quinine sulfate as a reference (0.1 M H2SO4, η = 1.33, Φ 0.54%), as reported in the literature. The absorbance and the emission of 1–4 were recorded for quinine sulfate at the same excitation wavelength. The solution of 1–4 was prepared in different solvents, and the calculations were done as per the equation given below:where “S” represents sample 1–2 and “R” represents reference (quinine sulphate, ΦR = 0.54) respectively, ΦS = quantum yield of the sample, Ab is the absorbance, A designates area under emission and η represents the refractive index of the solvent.

Florescence lifetime and quantum yield studies
The fluorescence quantum yields of the powders were measured using a calibrated Horiba integrating sphere combined with a Horiba JOBIN YVON Fluoromax-4 spectrometer. The time-resolved photoluminescence lifetimes (TRPL) were investigated using an Edinburgh Life Spec II instrument. Temperature-dependent (100–300 K) photoluminescence (PL) and delayed fluorescence lifetime measurements were conducted using a liquid nitrogen-cooled optical cryostat (Optistat, Oxford Instruments) in conjunction with an Edinburgh FSP-920 instrument.Field emission scanning electron microscopy
SEM analysis was carried out on a JEOL 10.0 kV instrument with a CCD camera. The instrument has a resolution of 0.36 mm (point to point) and can be magnified up to 6 lakh times to determine the morphology of aggregation-induced emission-generating species at different fractions.Synthesis of 1
Compound 1 was synthesized by the in situ reaction of o-arsanilic acid (217 mg, 1 mmol) and 4,4′-bipyridyl (156 mg, 1 mmol) in Acetone solvent, resulting in light yellow colour blocks formed after 7 days by the slow evaporation method. Yield: 300 mg; 40.1%; MP: >250 °C; pH: 4.25; FT-IR, ν (neat): 3410 (s), 3343 (s), 3000 (s), 2301 (br), 2107 (br), 1660 (s), 1320 (vs), 1,140 (vs), 900 (s). UV-visible = 334 nm. 1H NMR δ, 500 MHz, DMSO-d6, ppm: 8.7 (dd, J = 5 Hz, 1H), 7.8 (dd, J = 5 Hz, 1H), 7.3 (m, 1H), 6.6 (m, 1H), 4.6 (s, 8H). 13C NMR δ, 101 MHz, DMSO-d6, ppm: 150.39, 150.20, 143.99, 133.44, 130.83, 120. 96, 115.65, 115.46, and 112.50.Synthesis of 2
Compound 2 was synthesized by the in situ reaction of o-arsanilic acid (217 mg, 1 mmol) and 4,4′-bipyridyl (156 mg, 1 mmol) in MeOH/H2O (30/70 v/v) solvent system, resulting in the formation of dark yellow colour blocks after 10 days using the slow evaporation method. Yield: 300 mg; 40.1%; MP: >250 °C; pH: 4.25; FT-IR, ν (neat): 3410 (s), 3343 (s), 3000 (s), 2301 (br), 2107 (br), 1660 (s), 1320 (vs), 1140 (vs), 900 (s). UV-visible = 382 nm. 1H NMR δ, 500 MHz, DMSO-d6, ppm: 8.7 (dd, J = 5 Hz, 1H), 7.8 (t, 1H), 7.2 (m, 1H), 6.6 (m, 1H), 6.54 (s, 8H). 13C NMR δ, 101 MHz, DMSO-d6, ppm: 150.99, 150.80, 144.65, 134.17, 131.48, 121.60, 116.36, 116.17 and 112.99.Conclusion
Unusual low Z′-high Z′ cocrystal polymorphs with unique optical properties have been reported and thoroughly investigated. High Z′ polymorphs show significant thermal stability with no phase change before their melting point; however, they undergo striking emission turn-on upon grinding. The mechanochromic change in the low Z′ polymorph is mild, whereas the grounded forms of both polymorphs exhibit notable vapochromism upon exposure to base fumes. Powder diffraction studies establish multi-stimuli responses of polymorphic forms due to phase changes. Solvent dependent, AIE and thin film studies of the polymorphic forms have been reported. Diffraction studies were conducted to understand structure–property correlations. Single-crystal diffraction analysis indicates that on/off emission switching between the two polymorphic forms arises due to variation in π-stackings, which are not significant in emissive polymorph 1 and prominent in non-emissive polymorph 2.Author contributions
Ishtiyaq Ahmad carried out the experiments, collected data and compiled the manuscript. Siriyara Jagannatha supported the crystal refinement and analysis. Aijaz A. Dar conceived the problem, supervised the work and compiled the manuscript.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for the new products have been deposited at the CCDC under reference numbers 2421864 and 2421865.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
A. A. D. acknowledges DST-SERB, New Delhi, for funding under the Core Research Grant (CRG/2022/003693). I. A. thanks the Department of Chemistry and the University of Kashmir for their support and facilities. We also acknowledge the anonymous reviewers, whose constructive comments have helped improve the manuscript.References
- A. Haque, K. M. Alenezi, M. S. Khan, W.-Y. Wong and P. R. Raithby, Chem. Soc. Rev., 2023, 52, 454–472 RSC
.
- Y. Xiao, L. Liu, P. Xu, F. Sun, F. Li, X. Liu, Y. Yin, J. Leng, F. Zhang and S. Jin, Adv. Opt. Mater., 2024, 12, 2400747 CrossRef CAS
- A. A. Ganie, A. A. Ahangar, A. Dhir, A. K. Gupta and A. A. Dar, J. Phys. Chem. C, 2023, 127, 9257–9267 CrossRef CAS
- I. Ahmad, A. A. Ganie and A. A. Dar, CrystEngComm, 2020, 22, 3933–3942 RSC
- A. A. Ganie and A. A. Dar, Cryst. Growth Des., 2021, 21, 3014–3023 CrossRef CAS
- A. A. Malik, Z. M. Saeed, I. Ahmad, T. Alkhidir, P. B. Managutti, S. Mohamed and A. A. Dar, ACS Appl. Opt. Mater., 2024, 2, 1709–1720 CrossRef CAS
- D. Gentili, M. Gazzano, M. Melucci, D. Jones and M. Cavallini, Chem. Soc. Rev., 2019, 48, 2502–2517 RSC
- B. P. Krishnan and K. M. Sureshan, J. Am. Chem. Soc., 2015, 137, 1692–1696 CrossRef CAS PubMed
- J. Yang, Z. Ren, B. Chen, M. Fang, Z. Zhao, B. Z. Tang, Q. Peng and Z. Li, J. Mater. Chem. C, 2017, 5, 9242–9246 RSC
- Y. Yang, A. Li, Z. Ma, J. Liu, W. Xu, Z. Ma and X. Jia, Dyes Pigm., 2020, 181, 108575 CrossRef CAS
- Y. Liu, A. Li, S. Xu, W. Xu, Y. Liu, W. Tian and B. Xu, Angew. Chem., Int. Ed., 2020, 59, 15098–15103 Search PubMed
- B. Huang, D. Jiang, Y. Feng, W.-C. Chen, Y. Zhang, C. Cao, D. Shen, Y. Ji, C. Wang and C.-S. Lee, J. Mater. Chem. C, 2019, 7, 9808–9812 RSC
- B. K. Saha, N. K. Nath and R. Thakuria, Chem. Rec., 2023, 23, e202200173 CrossRef CAS PubMed
- S. Garain, S. N. Ansari, A. A. Kongasseri, B. C. Garain, S. K. Pati and S. J. George, Chem. Sci., 2022, 13, 10011–10019 RSC
- A. H. Kohli, A. A. Malik, A. A. Ganie and A. A. Dar, New J. Chem., 2024, 48, 11090–11098 RSC
- A. A. Dar, A. A. Ahangar, C. Femina, A. A. Malik, J. V. Parambil and P. K. Sajith, J. Phys. Chem. C, 2024, 128, 18901–18912 CrossRef CAS
- M. D. Malla, A. A. Malik, A. A. Ahangar, S. Dey, N. Sharma, D. A. Jose, D. Chopra and A. A. Dar, ChemistrySelect, 2024, 9, e202404488 CrossRef CAS
- S. Wu, B. Zhou and D. Yan, Adv. Opt. Mater., 2021, 9, 2001768 CrossRef CAS
- C. Y. Ke, M. N. Chen, Y. C. Chiu and G. S. Liou, Adv. Electron. Mater., 2021, 7, 2001076 CrossRef CAS
- J.-H. Tan, J.-M. Jin, W.-C. Chen, C. Cao, R. Wang, Z.-L. Zhu, Y. Huo and C.-S. Lee, ACS Appl. Mater. Interfaces, 2022, 14, 53120–53128 CrossRef CAS PubMed
- U. Balijapalli, R. Nagata, N. Yamada, H. Nakanotani, M. Tanaka, A. D'Aléo, V. Placide, M. Mamada, Y. Tsuchiya and C. Adachi, Angew. Chem., Int. Ed., 2021, 60, 8477–8482 CrossRef CAS PubMed
- X. Zhang, H. Dong and W. Hu, Adv. Mater., 2018, 30, 1801048 CrossRef PubMed
- A. A. Dar and S. Rashid, CrystEngComm, 2021, 23, 8007–8026 RSC
- M. Annadhasan, S. Basak, N. Chandrasekhar and R. Chandrasekar, Adv. Opt. Mater., 2020, 8, 2000959 CrossRef CAS
- D. P. Karothu, G. Dushaq, E. Ahmed, L. Catalano, S. Polavaram, R. Ferreira, L. Li, S. Mohamed, M. Rasras and P. Naumov, Nat. Commun., 2021, 12, 1326 CrossRef CAS PubMed
- X. Wang, Z. Z. Li, M. P. Zhuo, Y. Wu, S. Chen, J. Yao and H. Fu, Adv. Funct. Mater., 2017, 27, 1703470 CrossRef
- Y. Jiang, Y.-Y. Liu, X. Liu, H. Lin, K. Gao, W.-Y. Lai and W. Huang, Chem. Soc. Rev., 2020, 49, 5885–5944 RSC
- S. K. Park, S. Varghese, J. H. Kim, S.-J. Yoon, O. K. Kwon, B.-K. An, J. Gierschner and S. Y. Park, J. Am. Chem. Soc., 2013, 135, 4757–4764 CrossRef CAS PubMed
- M. McCarthy, B. Liu, E. Donoghue, I. Kravchenko, D. Kim, F. So and A. Rinzler, Science, 2011, 332, 570–573 CrossRef CAS
- S. Yousuf, J. Mahmoud Halabi, I. Tahir, E. Ahmed, R. Rezgui, L. Li, P. Laws, M. Daqaq and P. Naumov, Angew. Chem., Int. Ed., 2023, 135, e202217329 CrossRef
- A. Vinod Kumar, M. Rohullah, M. Chosenyah, J. Ravi, U. Venkataramudu and R. Chandrasekar, Angew. Chem., Int. Ed., 2023, 62, e202300046 CrossRef CAS PubMed
- K. Youssef, A. Gasonoo, M. Allain, H. Melville, L. Sanguinet, G. C. Welch and F. Gohier, ACS Appl. Opt. Mater., 2024, 2, 1610–1618 CrossRef CAS
- Y. Dai, J. Chen, C. Zhao, L. Feng and X. Qu, Angew. Chem., Int. Ed., 2022, 61, e202211822 CrossRef CAS PubMed
- I. Ahmad, A. A. Malik and A. A. Dar, Cryst. Growth Des., 2022, 22, 6483–6492 CrossRef CAS
- I. Ahmad, A. A. Ganie, S. Ahmad, A. A. Ahangar, C. M. Reddy and A. A. Dar, CrystEngComm, 2023, 25, 3164–3170 RSC
- Y. Huang, J. Xing, Q. Gong, L.-C. Chen, G. Liu, C. Yao, Z. Wang, H.-L. Zhang, Z. Chen and Q. Zhang, Nat. Commun., 2019, 10, 169 CrossRef
- T. Tian, Y. Fang, W. Wang, M. Yang, Y. Tan, C. Xu, S. Zhang, Y. Chen, M. Xu and B. Cai, Nat. Commun., 2023, 14, 4429 CrossRef CAS PubMed
- L. Sun, W. Zhu, W. Wang, F. Yang, C. Zhang, S. Wang, X. Zhang, R. Li, H. Dong and W. Hu, Angew. Chem., Int. Ed., 2017, 56, 7831–7835 CrossRef CAS
- Y.-L. Shi, M.-P. Zhuo, X.-D. Wang and L.-S. Liao, ACS Appl. Nano Mater., 2020, 3, 1080–1097 CrossRef CAS
- X.-G. Yang, Z.-M. Zhai, X.-M. Lu, L.-F. Ma and D. Yan, ACS Cent. Sci., 2020, 6, 1169–1178 CrossRef CAS
- Y. Wang, H. Wu, L. O. Jones, M. A. Mosquera, C. L. Stern, G. C. Schatz and J. F. Stoddart, J. Am. Chem. Soc., 2023, 145, 1855–1865 CrossRef CAS
- I. Ahmad and A. A. Dar, J. Phys. Chem. C, 2023, 127, 18684–18693 CrossRef CAS
- A. A. Dar, S. H. Lone, I. Ahmad, A. A. Ahangar, A. A. Ganie and C. Femina, Mater. Adv., 2024, 5, 1056–1064 RSC
- I. Ahmad, A. A. Malik, S. Ahmad and A. A. Dar, Cryst. Growth Des., 2024, 24, 4790–4800 CrossRef CAS
- A. A. Dar and A. A. Malik, J. Mater. Chem. C, 2024, 12, 9888–9913 RSC
- W. Zhao, Z. Ding, Z. Yang, T. Lu, B. Yang and S. Jiang, Chem. – Eur. J., 2024, 30, e202303202 CrossRef CAS PubMed
- R. Kubota, S. Takahashi, S. Nagai and S. Ito, Chem. – Asian J., 2023, 18, e202300124 CrossRef CAS PubMed
- S. K. B. Mane, Y. Mu, Z. Yang, E. Ubba, N. Shaishta and Z. Chi, J. Mater. Chem. C, 2019, 7, 3522–3528 RSC
- P. N. Zolotarev and N. A. Nekrasova, Cryst. Growth Des., 2020, 20, 7152–7162 CrossRef CAS
- S. Aitipamula, P. S. Chow and R. B. Tan, CrystEngComm, 2014, 16, 3451–3465 RSC
- D. Barman, M. Annadhasan, A. P. Bidkar, P. Rajamalli, D. Barman, S. S. Ghosh, R. Chandrasekar and P. K. Iyer, Nat. Commun., 2023, 14, 6648 CrossRef CAS PubMed
- B. Prusti, P. Sarkar, S. K. Pati and M. Chakravarty, J. Mater. Chem. C, 2021, 9, 9555–9570 RSC
- B. Prusti and M. Chakravarty, ChemPlusChem, 2020, 85, 2652–2656 CrossRef CAS PubMed
-
M. Turner, J. McKinnon, S. Wolff, D. Grimwood, P. Spackman, D. Jayatilaka and M. Spackman, CrystalExplorer17, University of Western Australia, 2017 Search PubMed
- B. Roy, M. C. Reddy and P. Hazra, Chem. Sci., 2018, 9, 3592–3606 RSC
- A. A. Ahangar, I. Ahmad and A. A. Dar, New J. Chem., 2023, 47, 4775–4783 RSC
- T. Samanta and R. Shunmugam, Mater. Adv., 2021, 2, 64–95 RSC
Footnote |
| † Electronic supplementary information (ESI) available: NMR, hydrogen bonding parameters, Hirshfeld surfaces, crystal structure diagrams, solution phase studies, mercury sensing. CCDC 2421864 and 2421865. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5tc00492f |
| This journal is © The Royal Society of Chemistry 2025 |