
Sustained acid release from stimuli responsive organic crystals facilitates shape modulation in metal nanoparticle synthesis†
Khalid
Naim‡
a,
Prodipta
Samadder‡
 a,
Atikur
Rahman
a,
Subash Chandra
Sahoo
b and
Prakash P.
Neelakandan
*ac
a,
Atikur
Rahman
a,
Subash Chandra
Sahoo
b and
Prakash P.
Neelakandan
*ac
aInstitute of Nano Science and Technology, Sector 81, Mohali 140306, India. E-mail: ppn@inst.ac.in
bDepartment of Chemistry, Panjab University, Sector 14, Chandigarh 160014, India
cAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad 201002, India
First published on 24th February 2025
Abstract
Stimuli-responsive fluorescent materials are gaining significant attention in the development of smart materials, particularly for applications in sensing, drug delivery, and environmental monitoring. In this study, we demonstrate how molecular engineering can transform simple naphthalidenimine-boron complexes-known for their remarkable photophysical properties-into functional materials with pH- and temperature-sensitive luminescence. Detailed crystallographic and spectroscopic analyses reveal the critical role of the donor moiety in modulating charge-transfer interactions, which not only enhance the photophysical characteristics but also introduce stimuli-responsive behaviour. Additionally, the gradual, sustained proton release from these molecules facilitates metal nanoparticle synthesis, which aids in surface passivation and controls nanoparticle growth kinetics, resulting in well-defined shapes. This research underscores the potential of molecular engineering to design advanced materials with tailored, responsive properties, opening new avenues for applications where environmental adaptability is key.
Introduction
Stimuli-responsive materials have emerged as a promising materials class, capable of modulating their physico-chemical behaviour in response to external stimuli, including mechanical stress, light, temperature, and chemical environments. Due to their ability to alter their state or function in response to external conditions, they are of interest for a wide range of applications, including in sensors, actuators, drug delivery systems, self-healing materials, and adaptive coatings.1–6 Despite the growing interest and progress in this area, many of the current stimuli-responsive systems are based on complex molecular architectures, polymeric networks, or composite structures that can be difficult to synthesize and process. These systems often require specialized equipment or conditions for production, which limits their scalability and widespread use. Additionally, maintaining the desired responsiveness over multiple cycles and ensuring the long-term stability of such materials can be challenging.The difficulties in the development of stimuli-responsive materials can be addressed through crystal engineering, which offers a strategic approach for the rational design of materials with targeted properties.7–11 The physico-chemical properties of molecular crystals are governed not only by the intrinsic electronic nature of the constituent molecules but also by the supramolecular arrangements and intermolecular interactions within the crystal lattice.12–15 The interplay between molecular packing and external perturbations provides an opportunity to fine-tune various properties. In particular, this tuning is central to the development of stimuli-responsive luminescent switching compounds, where controlled emission modulation can be achieved through precise molecular design and crystal packing strategies. Through intelligent molecular design, the structural aspects such as molecular conformation, hydrogen bonding, π–π stacking, and van der Waals forces can be manipulated to develop adaptive luminescent crystals.16–18 These crystals would exhibit tuneable photophysical properties that could be switched using external triggers such as heat, mechanical force, or exposure to specific analytes. Furthermore, the incorporation of dynamic molecular features—such as rotational or conformational flexibility—within the crystal lattice would enhance stimuli responsiveness, leading to novel materials with superior adaptability for real-world applications.19,20
Boron-containing organic compounds have garnered significant attention due to their unique luminescent properties, photosensitization capabilities, and optoelectronic characteristics, making them valuable for a wide range of applications.21–26 However, many commonly used boron-containing dyes like BODIPY face challenges such as fluorescence quenching upon aggregation and small Stokes shifts.27,28 To overcome these limitations, it is crucial to develop new compounds with improved photophysical properties. This includes dyes with long-wavelength emission, which are particularly important for applications in biological imaging, where deep tissue penetration and minimal phototoxicity are desired. Large Stokes shifts are also highly sought after, as they enhance the contrast and signal-to-noise ratio in fluorescence-based applications.29–31
Boron complexes of salicylaldimines are emerging in this direction and show potential for optical and biological applications.32,33 A key strategy for improving the performance of these boron-containing dyes is the incorporation of large π-conjugated frameworks. These extended conjugated systems can effectively lower the energy gap between the excited and ground states, leading to longer emission wavelengths. The addition of strong donor–acceptor moieties further enhances these properties, creating a push–pull electronic effect that can promote more efficient charge transfer and facilitate the desired long-wavelength emission.34–36 Additionally, such modifications can impart stimuli-responsive behaviour to the compounds, enabling them to change their photophysical properties in response to external triggers such as pH, temperature, light, or mechanical force.
Recently, we investigated the thermosalience and luminescent switching behaviour of single crystals of two naphthalidenimine-boron complexes, designated as 1 and 2 (Chart 1).37 These compounds consisted of donor and acceptor groups connected via a phenyl spacer, which facilitated the delocalization of π-electrons from the aryl amine to the boryl group. This electronic interaction resulted in the formation of an intramolecular charge transfer (ICT) state, a critical feature that influenced their photophysical properties. It was observed that subtle molecular parameters played a critical role in modulating charge transfer interactions, which in turn imparted dynamic properties to the crystals. Herein, we further examine the photophysical properties of 1 and 2, as well as their protonated counterparts (denoted as 1H and 2H, respectively), under a range of experimental conditions. Notably, the single crystals of the protonated compounds displayed an interesting behaviour: they released HCl in a slow and controlled manner, which was utilized for the synthesis of metal nanoparticles. By controlling the release of HCl, we were able to fine-tune the reaction conditions for nanoparticle formation, showcasing the practical utility of these protonated boron complexes in materials chemistry.
 | ||
| Chart 1 Structures of 1, 2, 1H and 2H. | ||
Results and discussion
Photophysical properties
The synthesis and the luminescence properties of 1 and 2 in the single crystalline state were reported earlier.37 At the outset, the photophysical properties of 1 and 2 were studied in solvents of different polarity (Fig. 1 and Table 1). It was observed that the absorption characteristics of both molecules were not affected much with solvent polarity. For example, in a low polar solvent such as toluene, the absorption maxima of 1 and 2 were 451 and 449 nm, respectively, whereas in a polar solvent like DMSO, the maxima were observed at 457 and 455 nm, respectively. The insignificant changes in the absorption maxima at different solvent polarities indicate the non-polar nature of the ground state of these molecules. On the other hand, the emission properties of 1 and 2 were significantly influenced by the solvent polarity: in toluene and DMSO the emission maxima of 1 were 591 and 651 nm whereas for compound 2 the emission maxima were 581 and 632 nm, respectively. The dependence of the emission features of 1 and 2 on solvent polarity suggests the existence of intramolecular charge transfer (ICT) states wherein the aryl amine moiety functions as the donor and the boryl moiety as the acceptor. The ICT states were further established using Lippert–Mataga plots of Stokes shift versus orientation polarizabiltiy wherein we observed upward straight lines with a small slope (Fig. S5, ESI†).38 It was striking that both compounds exhibited large Stokes shifts (∼150 nm) in all solvents which could be ascribed to the unsymmetry in their molecular structure and the enhanced ICT effect resulting from the strong electron donating ability of the diethylamino group. | ||
| Fig. 1 Absorption (solid lines) and fluorescence (dashed lines) spectra of (a) 1 (20 μM) and (b) 2 (20 μM) in toluene and DMSO. (c) and (d) Photographs of 1 and 2, respectively, in toluene and DMSO in ambient and UV light (365 nm). DMSO is dimethylsulfoxide. | ||
| Solvent | 1 | 2 | ||||
|---|---|---|---|---|---|---|
| Absorption λmax (nm) | Emission λem (nm) | Quantum yield, Φf | Absorption λmax (nm) | Emission λem (nm) | Quantum yield, Φf | |
| DMF is dimethylformamide, DMSO is dimethylsulfoxide.a Negligible. | ||||||
| Toluene | 451 | 591 | 0.05 | 449 | 581 | 0.11 |
| Chloroform | 462 | 604 | 0.07 | 460 | 594 | 0.13 |
| Ethyl acetate | 447 | 610 | 0.03 | 445 | 596 | 0.06 |
| Acetone | 452 | 628 | 449 | 620 | ||
| Acetonitrile | 450 | 634 | 449 | 629 | ||
| DMF | 455 | 635 | 451 | 631 | ||
| DMSO | 457 | 651 | 455 | 632 | ||
| Formic acid | 402 | 476 | 0.18 | 415 | 511 | 0.63 |
| Trifluoroacetic acid | 411 | 480 | 0.06 | 447 | 529 | 0.21 |
| HCl | 407 | 467 | 0.12 | 328, 424 | 505 | 0.63 |
Aggregation
It is assumed that the phenyl spacer unit in 1 and 2 can rotate freely in the solution, the restriction of which could result in an enhancement in their fluorescence.37 Preparing aggregates of 1 and 2 is a viable method to restrict the rotation of the phenyl spacer unit which would validate our assumptions. To study their aggregation behaviour, we monitored the changes in the absorption and emission of both compounds in acetonitrile with varying amount of water. Compound 1 showed an absorption peak at 450 nm which exhibited hypochromic and bathochromic shifts to 460 nm upon increasing the water content above 80%. In the case of emission spectrum of 1, we observed a gradual decrease in emission intensity till 60% water content which was followed by a sudden increase and a hypsochromic shift from 650 to 560 nm when the water content was increased to 80%. A further increase in the water content to 90% resulted in a decrease in the emission (Fig. S6 and S7, ESI†). In contrast, the absorption spectrum of compound 2, which showed an absorption maximum at 450 nm in acetonitrile, exhibited a hypochromic shift at 40% water. A further increase in water content to 60% resulted in a decrease in intensity along with a bathochromic shift to 540 nm. Simultaneously, a decrease in emission followed by an enhancement was observed from 0–50% and then to 80%, respectively. The enhanced emission was also accompanied by a hypsochromic shift from 630 to 590 nm when the water content was increased to 80% (Fig. S8 and S9, ESI†). The observed changes in the absorption and emission spectra of 1 and 2 in acetonitrile–water mixtures could be attributed to aggregation followed by the agglomeration of the aggregates when the water content was increased above 60%. It is noteworthy that 2 formed aggregates at a lower water content of 60% than 1, which indicated that 1 is more polar than 2.Effect of temperature and acid
As these compounds have moieties that could respond to changes in the immediate environment, we investigated the changes in their fluorescence towards temperature and acid/base (Fig. S10 and S11, ESI†). While the solutions of 1 and 2 in polar solvents like acetone, acetonitrile, DMF and DMSO were negligibly emissive at room temperature, intense reddish-orange fluorescence was observed at −196 °C (Fig. S12 and S13, ESI†). The changes in the fluorescence of 1 and 2 at low temperature could be correlated to the changes observed in acetonitrile–water mixtures and thus could be attributed to restricted intramolecular rotation.39 The phenyl spacer connecting the donor and acceptor moieties is expected to rotate freely in the solution in room temperature thereby facilitating non-radiative decay of the excited states. On the other hand, under frozen or aggregated state, these rotations could be effectively restricted thereby allowing the radiative relaxation pathways resulting in the enhancement of fluorescence.To gain insights to the temperature dependent photophysical properties, variable temperature NMR studies were performed (Fig. S14 and S15, ESI†). We observed that upon decreasing the temperature, the chemical shift corresponding to the protons of compound 2 exhibited significant shifts whereas temperature had a negligible effect on the chemical shift of the protons of compound 1. In general, the protons associated with the aryl amine moiety of 2 were observed to shift upfield whereas the protons associated with the boryl moiety were deshielded. For instance, the chemical shift of the protons He′ and Hf′ shifted from 8.64 and 8.38 ppm to 8.60 and 8.42 ppm upon decreasing the temperature from +40 to −40 °C.
The observed temperature-dependent changes in the chemical shifts of compound 2, in contrast to the negligible effect on compound 1, can be attributed to differences in their molecular dynamics and electronic environments. In solution, the absorption maximum of 1 is red-shifted as compared to 2. Similarly, from the Lippert–Mataga plots, we calculated the difference in the dipole moments of 1 and 2 in the ground and excited states (μE − μG),40,41 which were found to be 3.604 and 3.560 D, respectively. Considering that 1 and 2 are structurally same except for the position of the functional groups, the different Δμ for 1 and 2 suggests that the ICT states are stable in 1 as compared to that in 2. Consequently, it is assumed that the temperature window of our experiments fails to perturb the charge transfer state in 1 due to its enhanced stability, resulting in negligible changes in chemical shift values. In contrast, in the case of 2, the relatively weak ICT exhibits noticeable changes in chemical shift values with varying temperature.
As the aromatic amino group has a Lewis-basic character, protonation could influence the photophysical properties of these compounds. To test this, we investigated the effect of acids on the fluorescence of 1 and 2. The typically low emissive pale-yellow solutions of 1 and 2 in organic solvents were observed to become highly emissive in solvents like formic acid and trifluoroacetic acid. In formic acid, the fluorescence emission maxima of 1 and 2 were observed in the blue and green regions with the emission maxima at 476 and 511 nm, respectively (Fig. S16, ESI†). Importantly, the fluorescence quantum yield of 1 and 2 were significantly high in formic acid with quantum yields of 0.18 and 0.63, respectively (Table 1). To gain insights in to the possible changes in the structure of 1 and 2 induced by acids, the protonation process was monitored by 1H NMR spectroscopy measurements (Fig. 2). The addition of one equivalent of trifluoroacetic acid to 1 and 2 resulted in a downfield shift in the chemical shift of the protons Hc and Hd in the case of 1 and Hc′ and Hd′ in the case of 2, which indicates protonation of the amino group on 1 and 2. The protonation process is thus inferred to effectively interrupt the donor–acceptor character thereby modulating the fluorescence of the molecules.
 | ||
| Fig. 2 1H NMR spectra of 1 (27 mM) and 2 (27 mM) (a), (c) before and (b), (d) after the addition of 1 equivalent of trifluoroacetic acid. | ||
Crystals of protonated compounds
To obtain direct evidence on the protonation of the diethylamino group of 1 and 2, we attempted to grow their crystals under an acidic environment. Even though our attempts with formic acid and trifluoroacetic acid were futile, we were successful in obtaining single crystals of 1 and 2 when hydrochloric acid was used. Needle shaped crystals of protonated 1 and 2 (denoted as 1H and 2H hereafter) were obtained by the slow evaporation method (Fig. 3). In contrast to the intensely coloured crystals of the parent compounds 1 and 2, the crystals of 1H and 2H were mildly coloured (Fig. 3) with absorption maxima at 408 and 433 nm, respectively. Similarly, the emission maxima of the acid co-crystals 1H and 2H were blue-shifted as compared to 1 and 2 with maxima at 484 and 542 nm, respectively (Fig. S17, ESI†). Surprisingly, the fluorescence quantum yields of the crystals of 1H and 2H were lower (0.06 and 0.04, respectively) as compared to their parent compounds 1 and 2 (0.12 and 0.21, respectively) (Table S1, ESI†). | ||
| Fig. 3 Digital photographs of the crystals of (a)–(d) 1, 2, 1H, and 2H, respectively, under 365 nm UV light. Scale bar is 200 μm. | ||
Stimuli-responsive behaviour of crystals
The blue shift in the emission of 1H and 2H as compared to 1 and 2 indicated the disappearance of CT interactions. Our investigations on 1 and 2 showed that the CT states could be perturbed using heat thereby resulting in thermosalient and thermochromic properties.37 We were thus interested to investigate if the crystals of 1H and 2H responded to heat. To test this, a few long rod-like light yellow-coloured crystals of 1H were gradually heated on a hot-stage microscope. At about 135 °C, a few crystals turned red whereas the yellow colour of the remaining crystals intensified (Fig. 4a and Video S1, ESI†). As the heating was continued gradually, the intense yellow-coloured crystals turned deep red at ∼208 °C and finally melted at 247 °C. Apart from the thermochromic behaviour, we observed evolution of gas bubbles above 247 °C which could be attributed to the release of HCl gas upon heating the crystals of 1H. Similar to the crystals of 1H, the crystals of 2H exhibited thermochromic behaviour. Upon heating, the transparent crystals of 2H initially turned reddish at about 107 °C, then became opaque at 260 °C and eventually melted at 283 °C (Fig. 4b and Video S2, ESI†). We further observed that the fluorescence of 1H and 2H exhibited substantial changes upon heating wherein the emission maxima shifted to longer wavelengths at higher temperatures (Fig. S18, ESI†). These observations suggested that 1H and 2H exhibited unique thermal responsiveness. The observed colour and fluorescence change upon heating indicated release of HCl from the protonated compounds. | ||
| Fig. 4 Hot stage microscopy images of the single crystals of (a) 1H and (b) 2H. | ||
To comprehend thermochromism and the liberation of HCl gas, differential scanning calorimetry (DSC) of the crystals were conducted (Fig. S19, ESI†). Compound 1H displayed a DSC profile featuring a broad peak from 180 to 243 °C indicative of a pre-melting phase transition and molecular rearrangements within the crystal lattice. Intriguingly, an exothermic peak emerged from 250 to 297 °C post-melting, signifying the release of HCl gas. Notably, no peaks were observed during the cooling cycle, indicating the irreversible nature of the observed phase transition. The DSC data for 2H revealed a broad peak from 158 to 210 °C, denoting a phase transition while a distinct sharp peak corresponding to the melting transition appeared from 257 to 274 °C.
To quantify the amount of acid released, we performed an acid–base titration using methyl orange as an indicator. For this, the crystals of 1H and 2H were added to water and stirred for 5 hours wherein we observed a change of colour of the crystals which suggested dissociation of acid from the crystals. The residual crystals were filtered out and upon analysis by fluorescence and NMR spectroscopy (Fig. S20–S22, ESI†) revealed the formation of the parent compounds 1 and 2, thereby suggesting the complete dissociation of HCl from the protonated crystals 1H and 2H. The solution containing acid was estimated using a solution of sodium carbonate, which showed that 2.10 and 1.85 mM of HCl was bound per milligram of 1H and 2H, respectively.
The observations from hot-stage microscopy indicated that HCl was released from the crystals of 1H and 2H. However, in the case of the crystals of 1H, bubbles were observed to evolve upon heating which was not observed in the case of the crystals of 2H. This suggests a difference in the rate at which HCl is released from the crystals of 1H and 2H. To investigate the difference in the rate at which HCl is released from the crystals of 1H and 2H, the kinetics of HCl release was studied by monitoring time dependent changes in the fluorescence of 1H at 480 nm and 2H at 542 nm (Fig. S23a and S24a, ESI†). The relative changes in emission intensity revealed distinct kinetic phases in the release process (Fig. S23b and S24b, ESI†). Initially, HCl release from 1H occurred at a slow rate, with a rate constant of 0.07 h−1, indicating slow diffusion. In contrast, 2H exhibited a more rapid initial release, with a rate constant of 0.27 h−1. After 5 hours for 1H and 3 hours for 2H, the acid release was significantly accelerated, with rate constants of 3.46 and 3.26 h−1, respectively. This shift suggests that that a critical threshold—likely linked to structural or environmental changes—has been reached, enabling more efficient acid release. Subsequently, after 6 hours, the release rate decelerated, with rate constants of 0.08 h−1 for 1H and 0.07 h−1 for 2H. The overall release pattern reflects a dynamic sequence of phases: an initial slow release governed by kinetic barriers, a rapid acceleration driven by HCl liberation, and a final deceleration as the majority of acid is released. In the case of 2H, the initial release was faster than that of 1H, but after overcoming kinetic barriers, both compounds exhibited a similar rapid-release phase, followed by a gradual slowdown as the majority acid had been released.
X-ray diffraction
To understand the differences in the thermal response of 1H and 2H compared to their parent compounds, single-crystal X-ray diffraction analysis was performed. It showed that the protonated compounds 1H and 2H belongs to the monoclinic P21/c and orthorhombic space group Pbca whereas the parent compounds 1 and 2 crystallized in a monoclinic crystal system with P21/c space group and triclinic crystal system P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, respectively (Table S2, ESI†). Solvent molecules were included within the crystal lattice of 2H whereas they were not included in the crystal lattice of 1H (Fig. 5). The protonation of the diethylamino group was unequivocally confirmed from the single crystal data wherein we observed the formation of a N–H bond with a bond length of 0.98 Å in both compounds thereby confirming the formation of a covalent bond. It is noteworthy that after protonation the dihedral angle between C18N2C20 plane and the phenyl spacer increased significantly to 88.46° and 87.95° for 1H and 2H, respectively, as compared to 12.29° and 5.07°, respectively for their parent compounds 1 and 2 (Fig. S25 and S26, ESI†). Furthermore, the phenyl moiety in 1H and 2H were observed to twist out of the C11N11H1 plane by an angle of 83.03° and 46.79°, respectively (Fig. S27, ESI†). The boron-containing six-membered ring was observed to be out of plane with respect to the plane of the other aromatic ring with a dihedral angle of 15.19° and 23.11° between the O1–B1–N1 and the C1–C10–C11 planes for 1H and 2H, respectively (Fig. S28, ESI†). The B1–O1 and B1–N1 distances were slightly longer in 2H (1.437 and 1.579 Å, respectively) than the corresponding distances in 1H (1.434 and 1.572 Å, respectively).
space group, respectively (Table S2, ESI†). Solvent molecules were included within the crystal lattice of 2H whereas they were not included in the crystal lattice of 1H (Fig. 5). The protonation of the diethylamino group was unequivocally confirmed from the single crystal data wherein we observed the formation of a N–H bond with a bond length of 0.98 Å in both compounds thereby confirming the formation of a covalent bond. It is noteworthy that after protonation the dihedral angle between C18N2C20 plane and the phenyl spacer increased significantly to 88.46° and 87.95° for 1H and 2H, respectively, as compared to 12.29° and 5.07°, respectively for their parent compounds 1 and 2 (Fig. S25 and S26, ESI†). Furthermore, the phenyl moiety in 1H and 2H were observed to twist out of the C11N11H1 plane by an angle of 83.03° and 46.79°, respectively (Fig. S27, ESI†). The boron-containing six-membered ring was observed to be out of plane with respect to the plane of the other aromatic ring with a dihedral angle of 15.19° and 23.11° between the O1–B1–N1 and the C1–C10–C11 planes for 1H and 2H, respectively (Fig. S28, ESI†). The B1–O1 and B1–N1 distances were slightly longer in 2H (1.437 and 1.579 Å, respectively) than the corresponding distances in 1H (1.434 and 1.572 Å, respectively).
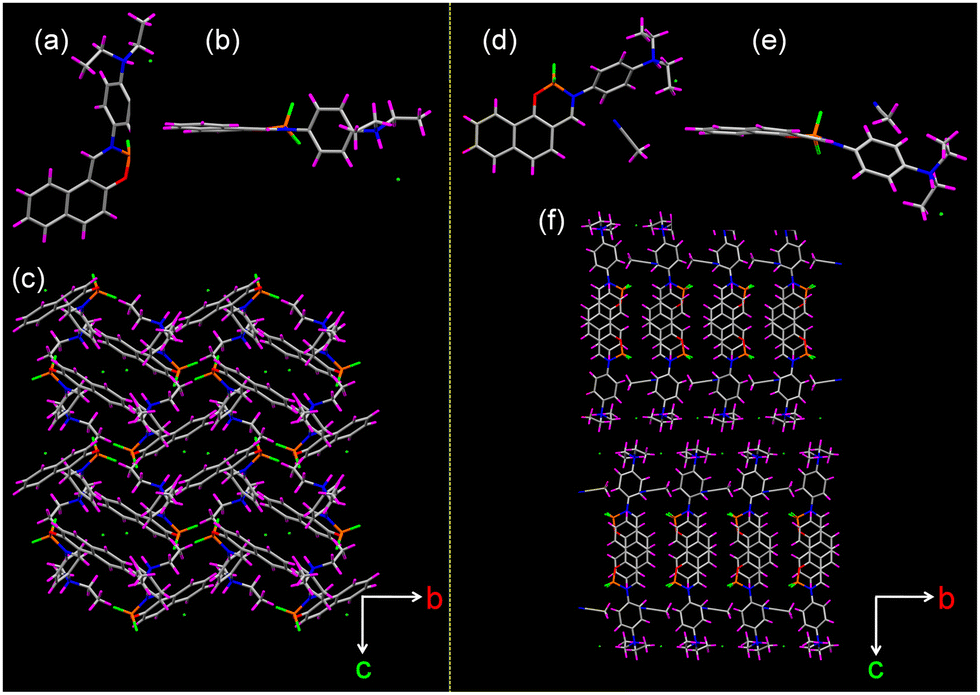 | ||
| Fig. 5 (a) and (d) Top, (b) and (e) side view of the single crystal structure of 1H and 2H. (c) and (f) Crystal packing of 1H and 2H viewed along the a-axis. | ||
Next, we employed Hirshfeld surface (HS) and two-dimensional finger-print analyses to probe the percentage of weak intermolecular interactions in 1H and 2H (Fig. S29, ESI†).42 Investigations of 2D-fingerprint plots derived from HS analyses showed that C⋯H, C⋯C, H⋯H, N⋯H, F⋯H and O⋯H interactions governed the packing in compounds 1H and 2H. The contribution of each of these interactions in 1H and 2H is listed in Table S3 (ESI†). It is known that the packing motif in the crystal states could be determined by estimating the ratio (ρ) between C⋯H and C⋯C interactions. Accordingly, 1H was deduced to display a herringbone packing (ρ > 4.5) that is typically observed in polyaromatic hydrocarbons whereas the close packing in 2H was observed as a 2D lamellar γ-motif (1.2 < ρ < 2.7).37
According to the theory of frontier molecular orbits, for 1 and 2, the highest occupied molecular orbital (HOMO) is mainly located on the (diethyl)amino group, while the lowest unoccupied molecular orbital (LUMO) is partially distributed over the whole molecule (Fig. S30, ESI†). In contrast, for 1H and 2H, the LUMO was spread over the whole molecule with decreased electron cloud density on aryl amine moiety. Since the protonation of its nitrogen atom reduces the electron donating ability of aryl amine, the LUMO should be less delocalized in the protonated molecules. Consequently, the lower delocalization of the LUMO stabilizes the excited molecule at a higher band gap, leading to the blue-shifts in the absorption and emission of the 1H and 2H molecules.
We next calculated the quinoid character (δr) in 1H and 2H using the bond lengths of the donor moiety which in turn helped in determining the efficiency of the charge transfer from the donor to the acceptor moiety (see Table S4, for the details of calculation, ESI†).43 The quinoid character (δr) was found to be −0.005 and −0.002 for 1H and 2H, respectively. The lower quinoid character of 1H and 2H, as compared to their parent compounds 1 and 2 (0.015 and 0.025, respectively), indicates higher single bond character to the C–N (C12–N1 and C15–N2) bonds. As a result of this protonation, the charge transfer phenomena are inferred to be weak in 1H and 2H as compared to their parent compounds.
The subtle differences in the molecular arrangements obtained from the single-crystal XRD data are key to understanding the different responses of 1H and 2H to temperature. The dihedral angles in 1H and 2H are notably larger than those in their parent compounds 1 and 2 (Table 2). Specifically, 1H displays a larger dihedral angle than 2H, which directly impacts their photophysical properties. The parent compounds 1 and 2 emit orange and red light, with maxima at 583 nm and 612 nm, respectively, while 1H and 2H emit in the blue and green regions, with maxima at 408 nm and 433 nm, respectively. These observations are consistent with the well-established trend that a reduced HOMO–LUMO energy gap leads to red-shifted emissions. Theoretical calculations further reinforce this trend, indicating that the HOMO–LUMO gap in 1 and 2 is smaller than in 1H and 2H. Moreover, the gap in 2H is slightly smaller than in 1H, which aligns with the observed emission patterns.
| S. no. | Parameter | 1 | 1H | 2 | 2H |
|---|---|---|---|---|---|
| 1 | Dihedral angle between C18N2C20 plane and the phenyl spacer (°) | 12.29 | 88.46 | 5.07 | 87.95 |
| 2 | Twist of the phenyl moiety out of the C11H11N1 plane (°) | 44.41 | 83.03 | 42.07 | 46.79 |
| 3 | Dihedral angle of boron-containing six-membered ring (O1–B1–N1 to C1–C10–C11) (°) | 32.17 | 15.19 | 25.39 | 23.11 |
| 4 | Quinoid character, δr | 0.015 | –0.005 | 0.025 | –0.001 |
| 5 | Et2N–H bond length (Å) | — | 0.980 | — | 0.980 |
HCl release for nanoparticle synthesis
The slow release of HCl from crystals holds potential for a range of applications, particularly in the synthesis of metal nanoparticles with unconventional shapes. For instance, cubic or triangular silver and gold nanoparticles are renowned for their unique surface plasmon resonance properties.44 Enhancing synthetic conditions without compromising yield, purity, monodispersity, or scalability could improve the commercial viability of these materials. While various chemical methods have been developed to synthesize these nanoparticles, many rely on environmentally harmful conditions, such as high temperatures, organic solvents, and prolonged reaction times. Importantly, most methods use acids to control nanoparticle size and shape by regulating pH, reducing metal ions, passivating surfaces, and managing growth kinetics to achieve well-defined shapes.45–47 A critical factor in these processes is the gradual and sustained release of acid, which enables selective growth of specific crystal faces. A sudden acid addition, on the other hand, may lead to metal salt precipitation or the formation of larger, spherical particles.48 Thus, HCl-releasing crystals present a promising alternative for nanoparticle synthesis.With this objective, we sought to synthesize silver and gold nanoparticles using 1H. The metal salts (AgNO3 and HAuCl4) were reduced to their respective nanoparticles with the aid of 1H in the presence of polyvinylpyrrolidone (PVP). Notably, the reaction was carried out in water under ambient conditions, without the need to control temperature, pressure, or pH. Remarkably, silver nanocubes and gold nanotriangles were obtained within five hours. The morphology and optical properties of the nanoparticles were examined using scanning electron microscopy (SEM). As shown in Fig. 6, silver nanocubes with an edge length of 150 ± 2 nm and equilateral gold nanotriangles with an edge length of 330 ± 1 nm were formed. Both morphologies were uniform, demonstrating the precision of the synthetic method. On the other hand, when the nanomaterials were synthesized under similar reaction conditions, except for the use of 1H, the resulting particles were of random shapes and sizes (Fig. S31, ESI†). This directly highlights the crucial role of 1H in modulating the morphology of silver and gold nanoparticles.
 | ||
| Fig. 6 Scanning electron microscopy images of (a) and (b) silver nanocubes and (c) and (d) gold nanotriangles. Scale bar 100 nm. | ||
The elemental composition of the nanoparticles were confirmed through energy-dispersive X-ray (EDAX) mapping and X-ray photoelectron spectroscopy (XPS) (Fig. S32–S34, ESI†). The observation of silver and gold in the EDAX mapping clearly suggests the presence of these elements. Further, the overall XPS survey spectrum of silver nanocubes showed signals at binding energies of 367.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) and 373.9eV corresponding to the 3d5/2 and 3d3/2 orbitals of Ag(0). In the case of gold nanotriangles, the XPS spectrum showed two peaks of Au(0), 4f5/2 and 4f7/2, with binding energies of 87.6 and 83.8eV, respectively. These values directly align with the literature and confirmed the formation of metal nanostructures.49,50 Next, the optical spectra of the silver nanocubes and gold nanotriangles were measured. We observed distinct localized surface plasmon resonance peaks at 404 and 559 nm for silver nanocubes and gold nanotriangles, respectively, which matched with those reported in the literature for similar morphologies (Fig. S35, ESI†).51,52 Therefore, by utilizing this novel acid-releasing crystal for nanoparticle synthesis, we successfully circumvented the need for three undesirable conditions: high temperature, the use of organic solvents, and prolonged reaction times.
and 373.9eV corresponding to the 3d5/2 and 3d3/2 orbitals of Ag(0). In the case of gold nanotriangles, the XPS spectrum showed two peaks of Au(0), 4f5/2 and 4f7/2, with binding energies of 87.6 and 83.8eV, respectively. These values directly align with the literature and confirmed the formation of metal nanostructures.49,50 Next, the optical spectra of the silver nanocubes and gold nanotriangles were measured. We observed distinct localized surface plasmon resonance peaks at 404 and 559 nm for silver nanocubes and gold nanotriangles, respectively, which matched with those reported in the literature for similar morphologies (Fig. S35, ESI†).51,52 Therefore, by utilizing this novel acid-releasing crystal for nanoparticle synthesis, we successfully circumvented the need for three undesirable conditions: high temperature, the use of organic solvents, and prolonged reaction times.
Conclusions
This study explored the optical properties of boron difluoride complexes 1 and 2 under varying solvent polarities and their protonated counterparts. Both 1 and 2 exhibited distinct dark orange to red fluorescence with significant Stokes shifts. Their emission behaviour was notably influenced by solvent polarity, primarily due to intramolecular charge transfer (ICT) states, with the aryl amine moiety acting as the electron donor and the boryl moiety as the acceptor. Additionally, the emission characteristics of these compounds were significantly modulated in the presence of acids. Spectral analysis and single-crystal X-ray diffraction revealed that the acid-responsive emission behaviour of 1 and 2 is linked to the protonation of the Lewis-basic nitrogen in the aromatic amino group. We also investigated the optical properties of the protonated forms, 1H and 2H, which displayed thermochromic behaviour and released HCl gas upon heating. Furthermore, the gradual release of acid from 1H was used to modulate the shape of metal nanoparticles. Given their fluorescence and stimuli-responsive properties, these compounds show great potential for applications in fluorescent sensing, labelling reagents, and advanced nanomaterials synthesis.Author contributions
K. N., P. S. and P. P. N. designed the experiments; K. N. and P. S. carried out synthesis, crystallization, photophysical and mechanical property studies. A. R. carried out aggregation studies. P. S. carried out nanoparticle synthesis. S. C. S. collected, solved, and refined the single-crystal X-ray diffraction data. K. N., P. S. and P. P. N. analysed the data and co-wrote the paper.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 1, 1H, 2 and 2H have been deposited at the Cambridge Crystallographic Data Centre under 2003922, 2361379, 2003921 and 2361403, and can be obtained from https://www.ccdc.cam.ac.uk.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Council of Scientific and Industrial Research, New Delhi (02(0438)/21/EMR-II) for financial support, the Sophisticated Analytical Instrumentation Facility, Panjab University, Chandigarh, for analytical facilities, and DST-FIST for single crystal facility at Panjab University, Chandigarh.Notes and references
- I. C. Hou, L. Li, H. Zhang and P. Naumov, Smart Mol., 2024, 2, e20230031 CrossRef.
- Y. Shymborska, A. Budkowski, J. Raczkowska, V. Donchak, Y. Melnyk, V. Vasiichuk and Y. Stetsyshyn, Chem. Rec., 2024, 24, e202300217 CrossRef CAS PubMed.
- M. Wei, Y. Gao, X. Li and M. J. Serpe, Polym. Chem., 2017, 8, 127–143 RSC.
- A. J. R. Amaral and G. Pasparakis, Polym. Chem., 2017, 8, 6464–6484 RSC.
- A. Jiménez-Sánchez, N. Farfán and R. Santillan, J. Phys. Chem. C, 2015, 119, 13814–13826 CrossRef.
- C. D. L. H. Alarcón, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- C. Yu, X. Jiang, M. B. Al-Handawi, P. Naumov, L. Li, Q. Yu and G. Wang, Angew. Chem., Int. Ed., 2024, 63, e202403397 CrossRef CAS PubMed.
- S. Das, L. Catalano and Y. Geerts, Small, 2024, 20, 2401317 CrossRef CAS PubMed.
- X. Pan, L. Lan, Q. Di, X. Yang and H. Zhang, Wearable Electron., 2024, 1, 111–118 CrossRef.
- M. Annadhasan, A. R. Agrawal, S. Bhunia, V. V. Pradeep, S. S. Zade, C. M. Reddy and R. Chandrasekar, Angew. Chem., Int. Ed., 2020, 59, 13852–13858 CrossRef CAS PubMed.
- E. Li, K. Jie, M. Liu, X. Sheng, W. Zhu and F. Huang, Chem. Soc. Rev., 2020, 49, 1517–1544 RSC.
- G. R. Krishna, R. Devarapalli, G. Lal and C. M. Reddy, J. Am. Chem. Soc., 2016, 138, 13561–13567 CrossRef CAS PubMed.
- W. M. Awad, D. W. Davies, D. Kitagawa, J. M. Halabi, M. B. Al-Handawi, I. Tahir, F. Tong, G. Campillo-Alvarado, A. G. Shtukenberg, T. Alkhidir, Y. Hagiwara, M. Almehairbi, L. Lan, S. Hasebe, D. P. Karothu, S. Mohamed, H. Koshima, S. Kobatake, Y. Diao, R. Chandrasekar, H. Zhang, C. C. Sun, C. Bardeen, R. O. Al-Kaysi, B. Kahr and P. Naumov, Chem. Soc. Rev., 2023, 52, 3098–3169 RSC.
- M. Annadhasan, D. P. Karothu, R. Chinnasamy, L. Catalano, E. Ahmed, S. Ghosh, P. Naumov and R. Chandrasekar, Angew. Chem., Int. Ed., 2020, 59, 13821–13830 CrossRef CAS PubMed.
- S. Bhunia, S. Chandel, S. K. Karan, S. Dey, A. Tiwari, S. Das, N. Kumar, R. Chowdhury, S. Mondal, I. Ghosh, A. Mondal, B. B. Khatua, N. Ghosh and C. M. Reddy, Science, 2021, 373, 321–327 CrossRef CAS PubMed.
- X. Yang, M. B. Al-Handawi, L. Li, P. Naumov and H. Zhang, Chem. Sci., 2024, 15, 2684–2696 RSC.
- X. Ding, C. Wei, L. Wang, J. Yang, W. Huang, Y. Chang, C. Ou, J. Lin and W. Huang, SmartMat., 2024, 5, e1213 CrossRef CAS.
- S. Wu, B. Zhou and D. Yan, Adv. Opt. Mater., 2021, 9, 2001768 CrossRef CAS.
- C. Wei, L. Li, Y. Zheng, L. Wang, J. Ma, M. Xu, J. Lin, L. Xie, P. Naumov, X. Ding, Q. Feng and W. Huang, Chem. Soc. Rev., 2024, 53, 3687–3713 RSC.
- A. J. Thompson, A. I. C. Orué, A. J. Nair, J. R. Price, J. McMurtrie and J. K. Clegg, Chem. Soc. Rev., 2021, 50, 11725–11740 RSC.
- R. J. Grams, W. L. Santos, I. R. Scorei, A. Abad-García, C. A. Rosenblum, A. Bita, H. Cerecetto, C. Viñas and M. A. Soriano-Ursúa, Chem. Rev., 2024, 124, 2441–2511 CrossRef CAS PubMed.
- K. Tanaka, M. Gon, S. Ito, J. Ochi and Y. Chujo, Coord. Chem. Rev., 2022, 472, 214779 CrossRef CAS.
- S. Shah, P. Marandi and P. P. Neelakandan, Front. Chem., 2021, 9, 708854 CrossRef CAS PubMed.
- S. Ito, M. Gon, K. Tanaka and Y. Chujo, Polym. Chem., 2021, 12, 6372–6380 RSC.
- M. Abdelrahman and H. Ali, J. Text. Color. Polym. Sci., 2020, 17, 43–55 Search PubMed.
- C. C. Vidyasagar, B. M. Muñoz Flores, V. M. Jiménez-Pérez and P. M. Gurubasavaraj, Mater. Today Chem., 2019, 11, 133–155 CrossRef CAS.
- M. Poddar and R. Misra, Coord. Chem. Rev., 2020, 421, 213462 CrossRef CAS.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- P. Kaur and K. Singh, J. Mater. Chem. C, 2019, 7, 11361–11405 RSC.
- H. Gao, X. Zhao and S. Chen, Molecules, 2018, 23, 419 CrossRef PubMed.
- J. Joseph, A. Cotruvo, A. T. Aron, K. M. Ramos-Torres and C. J. Chang, Chem. Soc. Rev., 2015, 44, 4400–4414 RSC.
- S. Yang, K. Lu and H. Xiao, Curr. Opin. Chem. Biol., 2024, 81, 102473 CrossRef CAS PubMed.
- J. Massue, D. Jacquemin and G. Ulrich, Organics, 2021, 2, 365–375 CrossRef CAS.
- D. Li, H. Zhang and Y. Wang, Chem. Soc. Rev., 2013, 42, 8416–8433 RSC.
- D. Frath, S. Azizi, G. Ulrich, P. Retailleau and R. Ziessel, Org. Lett., 2011, 13, 3414–3417 CrossRef CAS PubMed.
- X. Ren, F. Zhang, H. Luo, L. Liao, X. Song and W. Chen, Chem. Commun., 2020, 56, 2159–2162 RSC.
- K. Naim, S. C. Sahoo and P. P. Neelakandan, ACS Appl. Mater. Interfaces, 2022, 14, 22650–22657 CrossRef CAS PubMed.
- Z. Yang, W. Qin, J. W. Y. Lam, S. Chen, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Chem. Sci., 2013, 4, 3725–3730 RSC.
- F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 14192–14196 CrossRef PubMed.
- M. E. Vázquez, J. B. Blanco and B. Imperiali, J. Am. Chem. Soc., 2005, 127, 1300–1306 CrossRef PubMed.
- S. Mukherjee, A. Chattopadhyay, A. Samanta and T. Soujanya, J. Phys. Chem., 1994, 98, 2809–2812 CrossRef CAS.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- T. Michinobu, J. C. May, J. H. Lim, C. Boudon, J.-P. Gisselbrecht, P. Seiler, M. Gross, I. Biaggio and F. Diederich, Chem. Commun., 2005, 737–739 RSC.
- Y. Hang, A. Wang and N. Wu, Chem. Soc. Rev., 2024, 53, 2932–2971 RSC.
- V. Pawlik, S. Zhou, S. Zhou, D. Qin and Y. Xia, Chem. Mater., 2023, 35, 3427–3449 CrossRef CAS PubMed.
- S. Zhou, J. Li, K. D. Gilroy, J. Tao, C. Zhu, X. Yang, X. Sun and Y. Xia, ACS Nano, 2016, 10, 9861–9870 CrossRef CAS PubMed.
- S. H. Im, Y. T. Lee, B. Wiley and Y. Xia, Angew. Chem., Int. Ed., 2005, 44, 2154–2157 CrossRef CAS PubMed.
- H. J. Han, T. Yu, W.-S. Kim and S. H. Im, J. Cryst. Growth, 2017, 469, 48–53 CrossRef CAS.
- N. M. Vinita, U. Devan, S. Durgadevi, S. Anitha, D. Prabhu, S. Rajamanikandan, M. Govarthanan, A. Yuvaraj, M. Biruntha, A. Antony Joseph Velanganni, J. Jeyakanthan, P. A. Prakash, M. S. Mohamed Jaabir and P. Kumar, Sci. Rep., 2023, 13, 2230 CrossRef CAS PubMed.
- Y. Wang, H. Y. Zou and C. Z. Huang, Nanoscale, 2015, 7, 15209–15213 RSC.
- Sachin, S. Pal and R. Bokolia, Mater. Today Proc., 2022, 62, 3818–3822 CrossRef CAS.
- T.-H. Yang, J. Ahn, S. Shi and D. Qin, ACS Nano, 2021, 15, 14242–14252 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, crystal data and additional figures. CCDC 2003921, 2003922, 2361379, and 2361403. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5tc00047e |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |